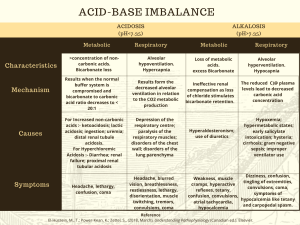
CHAPTER 6 Acid-Base Homeostasis in the Fetus and Newborn Timur Azhibekov, MD, Istvan Seri, MD, PhD, HonD 6 • In Addition to the Buffering Systems, Fetal Acid-Base Homeostasis Is Regulated by Maternal Respiratory and Renal Function and the Immature Fetal Kidneys • Fetal Acid-Base Homeostasis Affect Fetal Growth and May Have Long-Term Consequences • Certain Aspects of Obstetrical Management of Labor and Delivery (e.g., Prenatal Steroid Administration or the Practice of Delayed Cord Clamping) and Anesthesia During Labor and Delivery Also Affect, Albeit Transiently, Fetal and Postnatal Acid-Base Homeostasis Introduction This chapter addresses the regulation of fetal and neonatal acid-base balance with a focus on the elimination of the acid load by the placenta, lungs, and kidneys, briefly discusses the impact of acid-base disturbance on fetal and postnatal growth, and describes the effect of selected obstetrical management approaches on fetal and neonatal acid-base balance. Hydrogen ion concentration is tightly regulated by the intracellular and extracellular buffer systems and respiratory and renal compensatory mechanisms. The normal range of hydrogen ion concentration in the extracellular fluid is between 35 and 45 nEq/L (3.5–4.5 × 10−8 M), which translates to a pH range of 7.35 to 7.45 (pH = −log10 [H+]). Under physiologic circumstances, volatile and fixed acids generated by normal metabolism are excreted and the pH remains stable.1 Carbonic acid is the most common volatile acid produced and is readily excreted by the lungs in the form of carbon dioxide. Fixed acids, such as lactic acid, ketoacids, phosphoric acid, and sulfuric acid, are buffered principally by bicarbonate in the extracellular compartment. The bicarbonate used in this process is then regenerated by the kidneys in a series of transmembrane transport processes linked to the excretion of hydrogen ions in the form of titratable acids (phosphate and sulfate salts) and ammonium. Several aspects of the regulation of acid-base homeostasis are developmentally regulated in the fetus and neonate and thus differ from those in children and adults. These developmentally regulated differences of acid-base homeostasis and their impact on fetal and postnatal growth are reviewed in this chapter. Regulation of Acid-Base Homeostasis Respiratory Acidosis Unlike in fetal respiratory acidosis, in postnatal respiratory acidosis, immediate activation of the pulmonary compensatory mechanism leads to enhanced elimination of carbon dioxide, and the resulting fall in carbon dioxide concentration increases the pH toward normal (pH = [HCO3−] ÷ pCO2; where [HCO3−] = bicarbonate and pCO2 = carbon dioxide tension). The rapid activation of the respiratory compensatory 85 Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. Acid-Base Homeostasis in the Fetus and Newborn 85.e1 Abstract This chapter addresses the physiology and regulation of fetal and neonatal acid-base balance at the cellular and organ system levels, with attention to the clinical relevance of the complex interactions between the mother and the fetus ensuring the maintenance of fetal acid-base homeostasis and the physiology of acid-base homeostasis in the neonate following delivery. The physiologic processes regulating the elimination of the acid load by the placenta, the lungs, and the kidneys are reviewed in more detail along with the impact of acid-base disturbance on fetal and postnatal growth. Finally, the effects of selected obstetrical management approaches on fetal and neonatal acid-base balance are addressed. Keywords acid-base homeostasis mother fetus neonate respiratory acidosis metabolic acidosis maternal anesthesia Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. 6 86 Perinatal Mineral, Electrolyte, and Acid-Base Homeostasis mechanism is a result of the free movement of carbon dioxide across the blood-brain barrier,2 leading to instantaneous changes in cerebrospinal fluid (CSF) and cerebral interstitial fluid hydrogen ion concentrations. The degree of maturity of the central respiratory control system3 and pulmonary function determines the overall effectiveness of the respiratory compensatory mechanism. B Correction of Fetal Respiratory Acidosis Fetal respiratory acidosis develops when prolonged maternal hypoventilation occurs with maternal asthma, airway obstruction, narcotic overdosing, maternal anesthesia, severe hypokalemia, and magnesium sulfate toxicity. Fetal breathing movements increase, and the fetal kidneys exert a maturation-dependent limited response by reclaiming more bicarbonate in an attempt to restore the physiologic 20 : 1 ratio of bicarbonate to carbonic acid, resulting in a return of the pH toward normal.4 In the fetus, only the renal compensation has some limited physiologic significance when respiratory acidosis develops due to prolonged maternal hypoventilation. Rather, the placenta, coupled with maternal compensatory mechanisms, performs most of the effective compensatory functions. Correction of Postnatal Respiratory Acidosis In the clinical setting, acute neonatal respiratory acidosis develops most frequently in preterm infants with respiratory distress syndrome. Although stimulation of the respiratory center in the brain by the decrease in pH due to the elevated interstitial carbon dioxide concentration immediately increases respiratory rate and depth, carbon dioxide elimination by the lungs is usually limited because of immaturity and parenchymal disease. Importantly, in premature infants, increase in minute ventilation is achieved primarily by the increase in respiratory rate accompanied by only minor changes in tidal volume. On the other hand, term infants respond with increased tidal volume first.5 Respiratory compensation can be further affected by the decreased carbon dioxide sensitivity of the premature infant6 and the altered interaction between arterial oxygen tension (pO2) and carbon dioxide sensitivity, particularly at the level of central receptors, during the neonatal period as compared with adults.7 As in the fetus, the kidneys reclaim more bicarbonate in response to respiratory acidosis. However, renal compensation is limited by the developmentally regulated immaturity of renal tubular functions, especially during the first few weeks of postnatal life. Regulation of Acid-Base Homeostasis Metabolic Acidosis As in respiratory acidosis, the pulmonary gas exchange serves as the immediate regulator of acid-base homeostasis when metabolic acidosis develops. However, because plasma bicarbonate does not readily cross the blood-brain barrier, CSF bicarbonate levels change only with a delay.8 Therefore full activation of the respiratory acid-base regulatory system occurs only hours after the development of metabolic acidosis. This is different from the previously described truly immediate activation of the respiratory acid-base regulatory system by respiratory acidosis. In the CSF, generation of bicarbonate via hydroxylation of the dissolved carbon dioxide during CSF formation is the primary source of bicarbonate.9 On the other hand and as mentioned earlier, changes in arterial carbon dioxide tension rapidly affect carbon dioxide tension in the CSF, even in the setting of primary metabolic acidosis. Changes in CSF carbon dioxide levels also readily affect CSF bicarbonate levels.10 In addition, the respiratory response to changes in CSF bicarbonate levels in metabolic acidosis appears to be not as robust as that triggered by changes in carbon dioxide tension.2 Finally, the respiratory compensatory mechanism is complemented by removal of the acid load by the kidneys. Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. Acid-Base Homeostasis in the Fetus and Newborn 87 Fetoplacental Elimination of Metabolic Acid Load Fetal respiratory and renal compensation in response to changes in fetal pH is limited by the level of maturity and the surrounding maternal environment. However, although the placentomaternal unit performs most compensatory functions,4 the fetal kidneys have some, although limited, ability to contribute to the maintenance of fetal acid-base balance. The most frequent cause of fetal metabolic acidosis is fetal hypoxemia owing to abnormalities of uteroplacental function or blood flow, or both. Primary maternal hypoxemia or maternal metabolic acidosis secondary to maternal diabetes mellitus, sepsis, or renal tubular abnormalities is an unusual cause of fetal metabolic acidosis. The pregnant woman, at least in late gestation, maintains a somewhat more alkaline plasma environment compared with that of nonpregnant controls. This pattern of acid-base regulation in pregnant women is present during both resting and after maximal exertion and may serve as a protective mechanism from sudden decreases in fetal pH. Maintenance of the less acidic environment during pregnancy appears to be achieved through reduced plasma carbon dioxide and weak acid concentrations.4,11 The placenta plays an essential role in the maintenance of fetal acid-base balance when metabolic acidosis develops. As mentioned earlier, fetal metabolic acidosis most frequently occurs when abnormal uteroplacental function or blood flow results in fetal hypoxemia. Fetal hypoxemia then causes a shift to anaerobic metabolism and large quantities of lactic acid accumulate. As hydrogen ions are buffered by the extracellular and intracellular buffering systems of the fetus, pH drops as plasma bicarbonate decreases. Because of the unhindered diffusion of carbon dioxide through the placenta,12 restoration of normal fetal pH initially occurs through elimination of the volatile element of the carbonic acid-bicarbonate system via the maternal lungs. However, as lactate and other fixed acids cross the placenta more slowly,4 the onset of maternal renal compensation of fetal metabolic acidosis is delayed. In addition, if fetal oxygenation improves, the products of anaerobic metabolism are also metabolized by the fetus. Because there is no physiologic significance to respiratory compensation of metabolic acidosis in utero, the finding that the respiratory control system in the fetus is much less sensitive to changes in pH than in the neonate13 has little practical importance. Yet, a decrease in the fetal pH stimulates breathing movements in the fetus.14,15 Finally, as for the role of the fetal kidneys in the maintenance of acid-base balance, available evidence indicates that the fetal kidneys excrete both inorganic16–18 and organic acids19 and are also able to reabsorb bicarbonate.20,21 Studies in fetal sheep have found age-dependent increases in glomerular filtration rate (GFR) and urinary titratable acid, ammonium, and net acid excretion.16 A positive relationship also exists between changes in GFR and bicarbonate, sodium, and chloride excretions.16,18 Yet, the adaptive capacity of the fetal kidney to changes in fetal acid-base balance is limited. In fetal sheep the hydrochloric acid infusion-induced metabolic acidosis results in increases in titratable acid, ammonium, and net acid excretion without significant changes in GFR or renal tubular bicarbonate absorption.18 However, as mentioned earlier, under certain conditions, such as volume depletion20 or recovery from mild hypocapnic hypoxia,21 the fetal kidney has the ability to increase bicarbonate reabsorption. It is also important to note that the vast majority of these data have been obtained in animal models and that there is only very limited information available concerning renal acidification by the human fetus.22 In addition, the physiologic importance of the adaptive fetal renal responses is limited compared with that in the postnatal period because the acid load excreted in the fetal urine remains within the immediate fetal environment and needs to be eliminated by the placenta or metabolized by the fetus. Indeed, amniotic fluid acid-base status and electrolyte composition have been shown to affect the fetus. When the effects of amnion infusion of physiologic saline with those of lactated Ringer were compared in the fetal sheep, significant increases Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. 6 88 Perinatal Mineral, Electrolyte, and Acid-Base Homeostasis in fetal plasma sodium and chloride concentrations were noted only in the physiologic saline infusion group.23 In addition, fetal arterial pH decreased in the physiologic saline group and the change in the fetal pH was directly related to the changes in plasma chloride concentrations. However, despite the significant changes in plasma sodium and chloride concentrations and pH, fetal plasma electrolyte composition and acid-base balance remained in the physiologic range, leaving these findings with little clinical significance.23 B Postnatal Elimination of Metabolic Acid Load The most frequent causes of increased anion gap (lactic acid) metabolic acidosis in the neonate are hypoxemia or ischemia secondary to perinatal asphyxia; vasoregulatory disturbances and/or myocardial dysfunction caused by immaturity, sepsis, or asphyxia; severe lung disease with or without pulmonary hypertension; certain types of structural heart disease; and volume depletion. Severe metabolic acidosis caused by a neonatal metabolic disorder is rare but should always be considered. Preterm neonates frequently present with a mild to moderate normal anion gap acidosis, which almost always is the consequence of the low renal bicarbonate threshold of the premature kidney.24–27 However, the use of carbonic anhydrase inhibitors28 and parenteral alimentation, as well as the maturation-related decreased sensitivity to aldosterone, have also been suggested to contribute to the development of normal anion gap acidosis in the neonate.29–32 As mentioned earlier, in metabolic acidosis caused by the accumulation of lactic acid, hydrogen ions are buffered by the intracellular and extracellular buffering systems and plasma bicarbonate concentration decreases and pH drops. Restoration of pH toward normal initially occurs through elimination of the volatile element of the carbonic acid–bicarbonate system via the lungs. This process may be severely compromised in the sick preterm and term neonate with parenchymal lung disease. The principle mechanism of the renal compensation is the regulation of renal tubular bicarbonate and acid secretion in response to changes in extracellular pH. Although full activation of this system requires at least 2 to 3 days, changes in renal acidification may be seen as early as a few hours following the development of the acid-base disturbance. Although renal compensation is the ultimate mechanism that adjusts the hydrogen ion content of the body, this compensatory function is also affected by the immaturity of the neonatal kidneys.27,33 Both renal hemodynamic and tubular epithelial factors play a role in the limited renal compensatory capacity of the newborn. Renal blood flow (RBF) significantly increases after the immediate postnatal period, and some of the renal vasodilatory mechanisms are functionally mature as early as the 24th week of gestation.34 Similar to RBF, GFR is also low in the immediate postnatal period and increases as a function of both gestational and postnatal age.35–37 Indeed, the low GFR is considered the primary hemodynamic factor limiting the ability of the neonate to adequately handle an acid load.27,33 In addition, net renal acid excretion is regulated by several tubular epithelial functions.38 In the proximal tubule the following major transport mechanisms regulate active acid extrusion and transepithelial bicarbonate reabsorption: the H+-ATPase, the Na+,K+-ATPase–driven secondary active Na+/H+ antiporter in the apical membrane and the electrogenic Na+/3HCO3− cotransporter in the basolateral membrane, and the Na+,K+-ATPase–driven tertiary active Na+-coupled organic ion transporter.39 Because approximately 80% of the filtered bicarbonate is reabsorbed in the proximal tubule,40 the function of these proximal tubular transporters determines the renal threshold for bicarbonate reabsorption. The bicarbonate threshold is approximately 18 mEq/L in the premature and approximately 21 mEq/L in the term infant,24,41 and it reaches adult levels (24–26 mEq/L) only after the first postnatal year.25,26 However, in the extremely low gestational age neonate the renal bicarbonate threshold may be as low as 14 mEq/L. Because renal carbonic anhydrase is present and active during fetal life42 and because its activity is similar in the 26-week-old extremely immature neonate to that of the adult,43 a developmentally regulated immaturity Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. Acid-Base Homeostasis in the Fetus and Newborn 89 of the function of the previously described proximal tubular transporters is most likely responsible for the low bicarbonate threshold during early development. Indeed, both the activity and the hormonal responsiveness of the proximal tubular Na+,K+-ATPase are decreased in younger compared with older animals.44 Moreover, similar to the decrease in bicarbonate reabsorption in the proximal tubule,45 the activity of Na+/H+ antiporter is also decreased to approximately one-third of the adult level.46 In addition to immaturity, medications used in critically ill neonates may also affect proximal tubular bicarbonate reabsorption. For example, via inhibition of the proximal tubular Na+/H+ antiporter, dopamine may potentially decrease the low bicarbonate threshold of the neonate.47–49 Carbonic anhydrase inhibitors also decrease proximal tubular bicarbonate reabsorption by limiting bicarbonate formation and hydrogen ion availability for the Na+/H+ antiporter.50 By acting on several transport proteins along the nephron, furosemide directly increases urinary excretion of titratable acids (phosphate and sulfate salts) and ammonium.51 On the other hand, by inhibition of the activation of aldosterone receptors, spironolactone indirectly decreases hydrogen ion excretion in the distal tubule. Under physiologic circumstances, the thick ascending loop of Henle and the distal tubule reabsorb the remaining filtered bicarbonate via transport mechanisms similar to those of the proximal tubule. In addition, by transporting bicarbonate across the membrane,39 the distal nephron-specific HCO3−/Cl− antiporter located in the basolateral membrane contributes to the reabsorption of bicarbonate. Hydrogen ions are excreted in the urine in the form of titratable acids (phosphate and sulfate salts) and as ammonium salts, which are formed by the combination of hydrogen with ammonia.39 Because the major constituent of titratable acid in the urine is H2PO4−, drugs that decrease proximal tubular phosphate reabsorption and thus increase the delivery of phosphate to the distal nephron may increase the renal acidification capacity of the neonate. Indeed, by inhibiting proximal tubular phosphate reabsorption, dopamine has been shown to increase the excretion of titratable acids in preterm infants.52 Furthermore, net hydrogen ion secretion in the distal nephron continues after the reabsorption of virtually all bicarbonate by active extrusion of hydrogen and via the ability of the distal tubular epithelium to maintain large transepithelial concentration gradients for hydrogen and bicarbonate.39 Urinary excretion of titratable acid and ammonium increases as a function of gestational and postnatal age.27,37 However, because effective urinary acidification is usually acquired by the age of 1 month, even in premature infants, postnatal distal tubular hydrogen ion secretion is inducible independent of the gestational age at birth.53,54 Aldosterone is one of the most important hormones influencing distal tubular acidification. By affecting the function of several different transport mechanisms, aldosterone stimulates net hydrogen ion excretion in the distal nephron.39 Of note is that the premature neonate has a developmentally regulated relative insensitivity to aldosterone.30,32 In summary, the renal response to metabolic acidosis in the immediate postnatal period consists of attenuated increases in GFR, proximal tubular bicarbonate reabsorption, and distal tubular net acid secretion. However, a significant improvement in the overall renal response occurs after the first postnatal month even in the premature infant.33,53,54 Regulation of Acid-Base Homeostasis Respiratory Alkalosis Correction of Fetal Respiratory Alkalosis Rather than causing fetal respiratory alkalosis, acute maternal hyperventilation may lead to the development fetal hypoxia and metabolic acidosis.55 The fetal acidosis under these circumstances is thought to be the consequence of the acute decrease in placental blood flow caused by the maternal hypocapnia-induced significant uterine Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. 6 90 Perinatal Mineral, Electrolyte, and Acid-Base Homeostasis vasoconstriction.56 In these cases, restoration of maternal carbon dioxide levels rapidly corrects both the abnormal uterine blood flow and the acid-base abnormality in the fetus. The physiologic hyperventilation of the pregnant woman causes a compensatory decrease in her serum bicarbonate concentration to approximately 22 mM4 without any apparent effect on the fetus (see earlier). B Correction of Postnatal Respiratory Alkalosis Neonatal respiratory alkalosis occurs most often in the febrile nonventilated neonate or in cases with iatrogenic hyperventilation of the intubated preterm or term infant. Rarely, respiratory alkalosis may be the presenting sign of a urea cycle disorder during the first days of postnatal life because the rising ammonia level may initially stimulate the respiratory center in the brain. As for the renal compensation of respiratory alkalosis, both urinary bicarbonate reabsorption and distal tubular net acid excretion decrease and thus extracellular pH tends to return toward normal. This renal compensation plays an important although somewhat limited role in neonatal respiratory alkalosis. Regulation of Acid-Base Homeostasis Metabolic Alkalosis Correction of Fetal Metabolic Alkalosis Although metabolic alkalosis is a very rare fetal condition, it may occur in hyperemesis gravidarum. As a result of the significant and lasting hydrogen chloride losses, maternal renal compensation results in retention of bicarbonate to maintain maternal anionic balance. Because bicarbonate is transported slowly across the placenta, the development of fetal metabolic alkalosis lags behind that of the mother. On the other hand, the maternal respiratory compensation (hypoventilation with the ensuing hypercapnia) tends to restore normal pH in the fetus as carbon dioxide is rapidly transported across the placenta. Correction of Postnatal Metabolic Alkalosis Metabolic alkalosis most frequently develops in the preterm neonate receiving prolonged diuretic treatment for bronchopulmonary dysplasia. Although there is little evidence that chronic diuretic management results in improved medium- or long-term pulmonary outcome, many neonatologists use this treatment modality, at least intermittently. If total body chloride and potassium content is not appropriately maintained during chronic diuretic administration, severe metabolic “contraction” alkalosis may develop, which also results in poor growth. The respiratory response is a decrease in the rate and depth of breathing to increase carbon dioxide retention. This response may be interpreted as a sign of worsening pulmonary condition in the ventilated preterm neonate and may inappropriately trigger an increase in ventilatory support. Thus respiratory compensation of metabolic alkalosis may be ineffective if the intubated neonate is subjected to iatrogenic overventilation on the mechanical ventilator. As for the neonatal renal compensation for metabolic alkalosis, urinary bicarbonate reabsorption and distal tubular net acid excretion fall, resulting in a return of the extracellular pH toward normal. Finally, metabolic alkalosis can also result from a nondiuretic administration– related loss of extracellular fluid containing disproportionally more chloride than bicarbonate. During the diuretic phase of normal postnatal adaptation, preterm and term newborns tend to retain relatively more bicarbonate than chloride.57 The obvious benefits of allowing this physiologic extracellular volume contraction to occur clearly outweigh the clinical importance of a mild contraction alkalosis developing during postnatal adaptation. Thus no specific treatment is needed in these cases, especially because with the stabilization of the extracellular volume status and the renal function with time, acid-base balance rapidly returns to normal. Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. Acid-Base Homeostasis in the Fetus and Newborn 91 Normal Acid-Base Balance and Growth Growth is most accelerated during fetal life. The normal fetus grows from a weight of 0.22 g at the eighth week of gestation to 3400 g at 40 weeks’ completed gestation.58 The estimated energy density of each gram of body weight gained (or lost) is 23 kJ (5.6 kcal). However, in premature infants, especially if they are critically ill and/or growth retarded, the energy density of the new tissue is estimated to be higher than 5.6 kcal/g.59 For instance, in small-for-gestational-age infants at approximately 5 weeks after birth, the total energy expenditure is estimated to be 20% greater than in appropriate-for-gestational-age controls.60 Fetal growth can be negatively affected by several fetal and placentomaternal conditions. Proven fetal conditions affecting fetal growth include certain genetic conditions and infection of the fetus.61 Placentomaternal conditions with demonstrated influence on fetal growth are primary placental insufficiency and maternal diseases, nutritional status or substance abuse leading to secondary placental insufficiency, decreased fetal nutrient availability or direct fetal toxicity, or a combination of these harmful effects on fetal well-being.61 Although direct evidence demonstrating an impact of chronic fetal acid-base abnormality on fetal growth is very limited, a mild shift in the fetal acid-base status has been suggested as the primary pathologic factor for intrauterine growth restriction caused by placental insufficiency of any etiology.62 From 18 weeks’ postconception, growth-retarded fetuses exhibit a greater degree of mild acidemia than their appropriately growing counterparts.63 This acidemia is attributed to the reduced perfusion and mild hypoxemia the growth-retarded fetus faces as a result of the placental insufficiency. According to the above hypothesis, the small initial reduction in the pH negatively affects nitric oxide production in the fetus, and it is the decreased availability of nitric oxide that then plays a major role in the ensuing growth restriction.62 The following findings are in support of this hypothesis. Because locally formed nitric oxide regulates tissue perfusion and thus oxygen delivery and tissue growth itself, it has been suggested to play a pivotal role in regulation of growth in the fetus.61,62 In addition to its effect on oxygen delivery to the tissues, nitric oxide is an anabolic factor. Indeed, it is necessary for normal growth of several tissues, including the bone and muscle, and for the action of different hormones, such as the parathyroid hormone, vitamin D, and estrogen, known to be of importance in fetal growth and development.64,65 Interestingly, the enzyme responsible for generating nitric oxide from L-arginine, the constitutive nitric oxide synthase (cNOS), is sensitive to changes in pH, and its activity decreases even with a mild shift in the pH toward acidosis.66 Thus a vicious cycle may develop in growth-retarded fetuses because the initial decrease in blood flow and pH caused by placental insufficiency may lead to decreased cNOS activity and thus nitric oxide production. Decreased nitric oxide production, in turn, leads to further decreases in tissue perfusion and thus in pH, exacerbating the decrease in local nitric oxide production.62 In addition to being the source of locally generated nitric oxide, L-arginine also serves as the source of polyamines and L-proline. These compounds are generated by the arginase enzyme and are important when growth and tissue repair processes predominate. The function of this enzyme is also pH dependent,67 and the proposed decrease in its activity in the growth-retarded fetus may contribute to further impairment of fetal growth. Based on this information, it seems that elevating the pH in the fetus toward normal and supplementing L-arginine to the mother may be a plausible approach to attenuate the impact on fetal growth of the placental insufficiency–induced decreased fetal oxygen delivery. However, due to the inherent difficulties associated with attempts to effectively control fetal pH, no clinical trial has as yet attempted this combined approach. As for the neonate, the syndrome of late metabolic acidosis of prematurity is an example how postnatal growth can be affected by alterations in the acid-base balance. This entity was first described in the 1960s, in which otherwise healthy Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. 6 92 B Perinatal Mineral, Electrolyte, and Acid-Base Homeostasis premature infants after a few weeks developed mild to moderate anion gap acidosis and decreased growth rate. All of these infants were receiving high-protein cow’s milk formulas and demonstrated increased net acid excretion compared with controls. This type of late metabolic acidosis is now rarely seen, probably because of the use of special premature formulas and the changes made to regular formulas now containing a decreased casein-to-whey ratio and lower fixed acid loads. The diuretic administration–induced hypochloremic metabolic alkalosis is another example of the impact of acid-base balance on postnatal growth. This phenomenon is also associated with growth failure and may be a contributing factor of poor outcome in infants with bronchopulmonary dysplasia.68 Indeed, in addition to mild chronic hypoxemia and inadequate nutrition, the decrease in cell proliferation and diminished DNA and protein synthesis in response to intracellular alkalosis may contribute to the growth failure of infants with severe bronchopulmonary dysplasia.69 Chronic decrease in total body sodium resulting in a negative sodium balance may further hinder the growth of these infants.70 Aggressive chloride and potassium supplementation with relatively limited sodium supplementation decreases the risk for the development of clinically significant severe contraction alkalosis associated with chronic diuretic use in these patients. Obstetrical Management and Fetal and Neonatal Acid-Base Balance Evidence has accumulated on the impact of certain aspects of obstetrical management of labor and delivery on fetal and neonatal acid-base homeostasis. These effects are transient, and it is unclear whether they have an independent impact on clinically relevant neonatal outcome measures. Maternal betamethasone administration has been associated with a transient decrease in fetal movements including fetal breathing71 and fetal heart rate variability.71,72 Although the exact pathomechanism of these effects of antenatal betamethasone administration is unclear, animal73 and human74 data suggest that transient fetal acidosis following maternal steroid administration may, at least in part, explain the findings. It has been suggested that fetal acidosis is a consequence of the demonstrated fetal cardiovascular, endocrine, and metabolic effects of maternal steroid administration.74,75 However, in growth-restricted fetuses, cardiovascular responses to maternal steroid administration differ from those of fetuses with normal growth76,77 and may lead to significant acidosis and poor outcome.76 Delayed cord clamping also appears to have an impact on neonatal acid-base balance immediately after delivery, resulting in changes consistent with mixed acidosis during the first 45 seconds followed by primarily a metabolic acidosis by 90 seconds with delayed cord clamping.78 The metabolic acidosis immediately after delivery likely occurs because the normally produced and accumulated anaerobic metabolites in nonvital organs during labor and delivery are washed out faster in the larger blood volume of neonates with delayed cord clamping, and thus they exert a smaller effect on blood pH in these infants later on.78 Because the potential clinical relevance of fetal and immediate postnatal acidosis depends on the severity of hypoxemia and the associated acidosis, it may have importance only in the neonate with prolonged labor and difficult delivery.79 Observations in preterm infants suggest that delaying clamping of the cord even for 30 seconds is associated with a decrease in metabolic acidosis shortly after birth.80 Finally, the type of anesthesia during delivery via C-section also appears to have an impact on the fetal acid-base status. Findings of a meta-analysis of 27 studies found lower cord pH and larger base deficit in newborns of mothers who received spinal anesthesia compared with those who received general and epidural anesthesia.81 A more recent prospective observational cohort study examined the relationship between the type of anesthesia provided during C-section and fetal acid-base balance and neonatal condition upon delivery in 900 women with uncomplicated singleton pregnancies.82 The study found that epidural anesthesia was associated with higher Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. Acid-Base Homeostasis in the Fetus and Newborn 93 venous cord pH (7.30 ± 7.26–7.34) than general (7.25 ± 7.21–7.26) or spinal (7.23 ± 7.19–7.26) anesthesia and that neonatal well-being was negatively affected primarily by general anesthesia. These findings highlight the importance of taking the type of anesthesia into consideration when evaluating a neonate with metabolic acidosis in the immediate postnatal period following delivery via cesarean section. Summary This chapter has reviewed the available information and the gaps in our knowledge on how fetal and neonatal acid-base balance is regulated and the impact of alterations in acid-base balance on some aspects of fetal and postnatal growth, as well as how selected obstetrical management approaches affect fetal and neonatal acid-base balance. In the future, a better understanding of the role of growth factors and their interaction with the fetal acid-base status may result in improved early management of the growth-retarded fetus. This, in turn, may decrease the negative impact of growth retardation on brain and other organ development. REFERENCES 1. Masoro EJ. An overview of hydrogen ion regulation. Arch Intern Med. 1982;142:1019. 2. Sorensen SC. The chemical control of ventilation. Acta Physiol Scand. 1971;361:1. 3. Darnall RA. The role of CO2 and central chemoreception in the control of breathing in the fetus and the neonate. Respir Physiol Neurobiol. 2010;173:201. 4. Blechner JN. Maternal-fetal acid-base, physiology. Clin Obstet Gynecol. 1993;36:3. 5. Bodegård G. Control of respiration in newborn babies. III. Developmental changes of respiratory depth and rate responses to CO2. Acta Paediatr Scand. 1975;64:684. 6. Abu-Shaweesh JM. Maturation of respiratory reflex responses in the fetus and neonate. Semin Neonatol. 2004;9:169. 7. Wolsink JG, Berkenbosch A, DeGoede J, Olievier CN. The effects of hypoxia on the ventilatory response to sudden changes in CO2 in newborn piglets. J Physiol. 1992;456:39. 8. Nattie EE, Romer L. CSF HCO3− regulation in isosmotic conditions: the role of brain PCO2 and plasma HCO3. Respir Physiol. 1978;33:177. 9. Vogh BP, Maren TH. Sodium, chloride, and bicarbonate movement from plasma to cerebrospinal fluid in cats. Am J Physiol. 1975;228:673. 10. Javaheri S, Nardell EA, Kazemi H. Role of PCO2 as determinant of CSF [HCO-3] in metabolic acidosis. Respir Physiol. 1979;36:155. 11. Kemp JG, Greer FA, Wolfe LA. Acid-base regulation after maximal exercise testing in late gestation. J Appl Physiol. 1997;83:644. 12. Blechner JN, Meshia G, Barron DH. A study of the acid-base balance of fetal sheep and goats. Q J Exp Physiol. 1960;45:60. 13. Connors G, Hunse C, Carmichael L, et al. Control of fetal breathing in the human fetus between 24 and 34 weeks’ gestation. Am J Obstet Gynecol. 1989;160:932. 14. Jansen A, Shernick V. Fetal breathing and development of control of breathing. J Appl Physiol. 1991;70:143L. 15. Molteni RA, Melmed MH, Sheldon RE, et al. Induction of fetal breathing by metabolic acidemia and its effect on blood flow to the respiratory muscles. Am J Obstet Gynecol. 1980;136:609. 16. Kesby GJ, Lumbers ER. Factors affecting renal handling of sodium, hydrogen ions, and bicarbonate in the fetus. Am J Physiol. 1986;25l:F226. 17. Hill KJ, Lumbers ER. Renal function in adult and fetal sheep. J Dev Physiol. 1988;10:149. 18. Kesby GJ, Lumbers ER. The effects of metabolic acidosis on renal function of fetal sheep. J Physiol. 1988;396:65. 19. Elbourne I, Lumbers ER, Hill KJ. The secretion of organic acids and bases by the ovine fetal kidney. Exp Physiol. 1990;75:211. 20. Robillard JE, Sessions C, Burmeister L, Smith FG. Influence of fetal extracellular volume contraction on renal reabsorption of bicarbonate in fetal lambs. Pediatr Res. 1977;11:649. 21. Gibson KJ, McMullen JR, Lumbers ER. Renal acid-base and sodium handling in hypoxia and subsequent mild metabolic acidosis in fetal sheep. Clin Exp Pharmacol Physiol. 2000;27:67. 22. Blechner JN, Stenger VG, Eitzman DV, Prystowsky H. Effects of maternal metabolic acidosis on the human fetus and newborn infant. Am J Obstet Gynecol. 1967;9:46. 23. Shields LE, Moore TR, Brace RA. Fetal electrolyte and acid-base responses to amnioinfusion: lactated Ringer’s versus normal saline in the ovine fetus. J Soc Gynecol Investig. 1995;2:602. 24. Svenningsen NW. Renal acid-base titration studies in infants with and without metabolic acidosis in the postneonatal period. Pediatr Res. 1974;8:659. 25. Edelmann CM, Soriano JR, Boichis H, et al. Renal bicarbonate reabsorption and hydrogen ion excretion in normal infants. J Clin Invest. 1967;46:1309. 26. Avner ED. Normal neonates and the maturational development of homeostatic mechanisms. In: Ichikawa I, ed. Pediatric Textbook of Fluids and Electrolytes. Baltimore: Williams & Wilkins; 1990:107–118. Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. 6 94 B Perinatal Mineral, Electrolyte, and Acid-Base Homeostasis 27. Jones DP, Chesney RW. Development of tubular function. Clin Perinatol. 1992;19:33. 28. Tam B, Chhay A, Yen L, et al. Acetazolamide for the management of chronic metabolic alkalosis in neonates and infants. Am J Ther. 2014;21:477. 29. Trivedi A, Sinn JKH. Early versus late administration of amino acids in preterm infants receiving parenteral nutrition. Cochrane Database Syst Rev. 2013;(7):CD008771. 30. Sulyok E, Nemeth M, Tenyi I, et al. Relationship between maturity, electrolyte balance and the function of the renin-angiotensin-aldosterone system in newborn infants. Biol Neonate. 1979;35:60. 31. Brewer ED. Disorders of acid-base balance. Pediatr Clin North Am. 1990;37:429. 32. Stephenson G, Hammet M, Hadaway G, Funder JW. Ontogeny of renal mineralocorticoid receptors and urinary electrolyte responses in the rat. Am J Physiol. 1984;247:F665. 33. Guignard JP, John EG. Renal function in the tiny premature infant. Clin Permatol. 1986;13:377. 34. Seri I, Abbasi S, Wood DC, Gerdes JS. Regional hemodynamic effects of dopamine in the sick preterm infant. J Pediatr. 1998;133:728. 35. Fawer CL, Torrado A, Guignard J. Maturation of renal function in full-term and premature neonates. Helv Paediatr Acta. 1979;34:11. 36. Guignard JP, Torrado A, Da Cunha O, Gautier E. Glomerular filtration rate in the first three weeks of life. J Pediatr. 1975;87:268. 37. Arant BS. Developmental patterns of renal functional maturation compared in the human neonate. J Pediatr. 1978;92:705. 38. Baum M, Gattineni J, Satlin LM. Postnatal renal development. In: Alpern RJ, Moe OW, Caplan M, eds. Seldin and Giebisch’s The Kidney. 5th ed. Academic Press; 2013:911–931. 39. Hamm LL, Alpern RJ, Preisig PA. Cellular mechanisms of renal tubular acidification. In: Alpern RJ, Moe OW, Caplan M, eds. Seldin and Giebisch’s The Kidney. 5th ed. Academic Press; 2013:1917–1978. 40. Quigley R, Baum M. Neonatal acid base balance and disturbances. Semin Perinatol. 2004;28:97. 41. Schwartz GJ, Haycock GB, Edelmann CM, Spitzer A. Late metabolic acidosis: a reassessment of the definition. J Pediatr. 1979;95:102. 42. Robillard JP, Sessions C, Smith FG. In vivo demonstration of renal carbonic anhydrase activity in the fetal lamb. Biol Neonate. 1978;34:253. 43. Lonnerholm C, Wistrand PJ. Carbonic anhydrase in the human fetal kidney. Pediatr Res. 1983;17:390. 44. Fryckstedt J, Svensson LB, Linden M, Aperia A. The effect of dopamine on adenylate cyclase and Na+,K+-ATPase activity in the developing rat renal cortical and medullary tubule cells. Pediatr Res. 1993;34:308. 45. Schwartz GJ, Evan AP. Development of solute transport in rabbit proximal tubule. I. HCO-3 and glucose absorption. Am J Physiol. 1983;245:F382. 46. Baum M. Neonatal rabbit juxtamedullary proximal convoluted tubule acidification. J Clin Invest. 1990;85:499. 47. Felder CC, Campbell T, Albrecht F, Jose PA. Dopamine inhibits Na+/H+ exchanger activity in renal BBMV by stimulation of adenylate cyclase. Am J Physiol. 1990;259:F297. 48. Seri I. Cardiovascular, renal, and endocrine actions of dopamine in neonates and children. J Pediatr. 1995;126:333. 49. Bobulescu IA, Quiñones H, Gisler SM, et al. Acute regulation of renal Na+/H+ exchanger NHE3 by dopamine: role of protein phosphatase 2A. Am J Physiol Renal Physiol. 2010;298:F1205. 50. Ellison DH. Physiology and pathophysiology of diuretic action. In: Alpern RJ, Moe OW, Caplan M, eds. Seldin and Giebisch’s The Kidney. 5th ed. Academic Press; 2013:1353–1404. 51. Hropot M, Fowler N, Karlmark B, Giebisch G. Tubular action of diuretics: distal effects on electrolyte transport and acidification. Kidney Int. 1985;28:477. 52. Seri I, Rudas G, Bors ZS, et al. Effects of low-dose dopamine on cardiovascular and renal functions, cerebral blood flow, and plasma catecholamine levels in sick preterm neonates. Pediatr Res. 1993;34:742. 53. Kerpel-Fronius E, Heim T, Sulyok E. The development of the renal acidifying processes and their relation to acidosis in low-birth-weight infants. Biol Neonate. 1970;15:156. 54. Sulyok E, Heim T. Assessment of maximal urinary acidification in premature infants. Biol Neonate. 1971;19:200. 55. Motoyama EK, Rivard G, Acheson F, Cook CD. Adverse effect of maternal hyperventilation on the foetus. Lancet. 1966;1:286. 56. Moya F, Morishima HO, Shnider SM, James L. Influence of maternal hypoventilation on the newborn infant. Am J Obstet Gynecol. 1965;91:76. 57. Ramiro-Tolentino SB, Markarian K, Kleinman LI. Renal bicarbonate excretion in extremely low birth weight infants. Pediatrics. 1996;98:256–261. 58. Appendix 2. Taeusch HW, Ballard RA, Gleason CA, eds. Avery’s Diseases of the Newborn. 8th ed. 2005:1574. 59. Davies PSW. Energy requirements for growth and development in infancy. Am J Clin Nutr. 1998;68:939S. 60. Davies PSW, Clough H, Bishop N, et al. Total energy expenditure in small-for-gestational-age infants. Arch Dis Child. 1996;74:F208. 61. Hay WW, Catz CS, Grave GD, Yaffe SJ. Workshop summary: fetal growth: its regulation and disorders. Pediatrics. 1997;99:585. 62. Stearns MR, Jackson CGR, Landauer JA, et al. Small for gestational age: a new insight? Med Hypotheses. 1999;53:186. 63. Nicolaides KH, Economides DL, Soothill PW. Blood gasses, pH and lactate in appropriate- and small-for-gestational-age fetuses. Am J Obstet Gynecol. 1989;161:996. 64. Kaiser FE, Dirighi M, Muchnick J, et al. Regulation of gonadotropins and parathyroid hormone by nitric oxide. Life Sci. 1996;59:987. Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. Acid-Base Homeostasis in the Fetus and Newborn 95 65. Evans CH, Stefanovic-Racic M, Lancaster J. Nitric oxide and it role in orthopedic disease. Clin Orthop Relat Res. 1995;312:275. 66. Fleming I, Hecker M, Busse R. Intracellular alkalinization induced by bradykinin sustains activation of the constitutive nitric oxide synthase in endothelial cells. Circ Res. 1994;74:1220. 67. Kuhn NJ, Ward S, Piponski M, Young TW. Purification of human hepatic arginase and its manganese (II)-dependent and pH-dependent interconversion between active and inactive forms: a possible pH-sensing function of the enzyme on the ornithine cycle. Arch Biochem Biophys. 1995;320:24. 68. Perlman JF, Moore V, Siegel MJ, Dawson J. Is chloride depletion an important contributing cause of death in infants with bronchopulmonary dysplasia? Pediatrics. 1986;77:212. 69. Heinly MM, Wassner SJ. The effect of isolated chloride depletion on growth and protein turnover in young rats. Pediatr Nephrol. 1994;8:555. 70. Sulyok E, Kovacs L, Lichardus B, et al. Late hyponatremia in premature infants: role of aldosterone and arginine vasopressin. J Pediatr. 1985;106:990. 71. Rotmensch S, Liberati M, Celentano C, et al. The effect of betamethasone on fetal biophysical activities and Doppler velocimetry of umbilical and middle cerebral arteries. Acta Obstet Gynecol Scand. 1999;78:768. 72. Senat MV, Minoui S, Multon O, et al. Effect of dexamethasone and betamethasone on fetal heart rate variability in preterm labour: a randomized study. Br J Obstet Gynaecol. 1998;105:749. 73. Bennett L, Kozuma S, McGarrigle HH, Hanson MA. Temporal changes in fetal cardiovascular, behavioral, metabolic and endocrine responses to maternally administered dexamethasone in the late gestation fetal sheep. Br J Obstet Gynaecol. 1999;106:331. 74. Shenhav S, Volodarsky M, Anteby EY, Gemer O. Fetal acid-base balance after betamethasone administration: relation to fetal heart rate variability. Arch Gynecol Obstet. 2008;278:333. 75. Derks JB, Mulder EJ, Visser GH. The effects of maternal betamethasone administration on the fetus. Br J Obstet Gynaecol. 1995;102:40. 76. Simchen MJ, Alkazaleh F, Adamson SL, et al. The fetal cardiovascular response to antenatal steroids in severe early-onset intrauterine growth restriction. Am J Obstet Gynecol. 2004;190:296. 77. Miller SL, Supramaniam VG, Jenkin G, et al. Cardiovascular responses to maternal betamethasone administration in the intrauterine growth-restricted ovine fetus. Am J Obstet Gynecol. 2009;201: 613.e1. 78. Wiberg N, Källén K, Olofsson P. Delayed umbilical cord clamping at birth has effects on arterial and venous blood gases and lactate concentrations. BJOG. 2008;115:697. 79. Hutchon DJ. Immediate cord clamping may increase neonatal acidaemia. BJOG. 2008;115:1190. 80. Ruangkit C, Moroney V, Viswanathan S, Bhola M. Safety and efficacy of delayed umbilical cord clamping in multiple and singleton premature infants—a quality improvement study. J Neonatal Perinatal Med. 2015;8:393. 81. Reynolds F, Seed PT. Anaesthesia for Caesarean section and neonatal acid-base status: a meta-analysis. Anaesthesia. 2005;60:636. 82. Tonni G, Ferrari B, De Felice C, Ventura A. Fetal acid-base and neonatal status after general and neuraxial anesthesia for elective cesarean section. Int J Gynaecol Obstet. 2007;97:143. Downloaded for Anonymous User (n/a) at UH Cleveland Medical Center from ClinicalKey.com by Elsevier on May 03, 2023. For personal use only. No other uses without permission. Copyright ©2023. Elsevier Inc. All rights reserved. 6