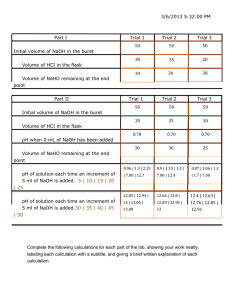
Cyano Type Blue Printing Cyanotype is a photographic printing process that produces a cyan-blue print. Engineers used the process well into the 20th century as a simple and low-cost process to produce copies of drawings, referred to as blueprints. Invented by Sir John Herschel in 1841, this simple process produces a continuous tone image of Prussian Blue using a sensitizing solution of ferric ammonium citrate and potassium ferricyanide. These iron salts, when exposed to natural or artificial ultraviolet light, are reduced to their ferrous state, producing a high contrast blue image when oxidised. The process was eminently suited to its traditional role in reproducing technical drawings, its most common use in engineering and architecture until the advent of modern photocopiers. However, it was a versatile process, and was used throughout the 19th century from Anna Atkins’ photograms of plants and seaweed for her books on botany (1843–55) to Henri LeSecq’s still life studies of the 1850s. Photographers at the end of the century used cyanotype paper for proofing negatives. Procedure: 1. Immerse pieces of bond paper one by one in the sensitizing solution and keep them immersed for 4-5 minutes. 2. Remove the wet pieces of paper and place them between sheets of filter paper. This should be done as quickly as possible and in a partially closed locker. Dry it for 10-15 minutes. 3. After the paper has dried, place an opaque object on top of the sensitizing paper, compress it between sheets of glass and expose to sunlight for 4-6 minutes. 4. Make 3-4 exposures, varying the time of exposure to optimize the best condition. 5. After the exposure, dip the paper into 0.1M ferric cyanide. It is important that the paper is immersed all at once, otherwise lines will appear on the blue field of the paper. 6. Remove the paper and dip it in 0.3M potassium dichromate solution for one minute. Afterwards, wash the paper first with 0.1M HCl and then tap water and dry. 7. Paste all the images in your lab notebook with their exposure times. Synthesis of Dibenzalacetone Aldol condensation is an important route of organic synthesis because it provides an efficient way to form carbon-carbon bond. In this condensation, an enol or enolate ion reacts with a c arbonyl compound to from a β-hydroxy ketone or β-hydroxy aldehyde, which is followed by dehydration. The reaction is used to manufacture solvents such as isophorone, used in printing inks, lacquers, adhesives and many other products. It is also used in the manufacture of α, β- unsaturated ketones known as chalcones. This condensation is generally used to create plasticizers which ae used to convert rigid plastic polyvinyl chloride into a soft, flexible elastic material. In today’s experiment, you will utilize this reaction to prepare dibenzalacetone. It is a common ingredient in sunscreens since it absorbs UV light. Dibenzalacetone was first prepared in 1881 by Claisen and Claparede. It is used as ligand for making organometallic complexes which are used as catalysts in coupling reactions. For example, Pd-DBA. O C O H NaOH O + H 3C + 2H2O CH3 Procedure: 1. Take 0.8 mL of benzaldehyde in a small conical flask. 2. Add 0.3 mL of acetone and 5 mL of ethanol to the above. Add 2 mL of NaOH solution to the mixture of benzaldehyde, acetone and ethanol mixture. 3. Mix and swirl the reaction mixture occasionally for 15 minutes. 4. A yellow flocculent precipitate should form. Filter the product using a Buchner funnel on a vacuum filtration set up. 5. Wash the obtained solid with 10-15 mL of water, followed by a washing with 8-10 mL of chilled ethanol. Recrystallization 1. Dissolve the crude product in 2 mL of ethyl acetate in a beaker and heat on water bath till you get a clear solution. 2. Allow the solution to cool slowly to room temperature and then cool it in an ice bath. Pure dibenzalacetone crystallizes. 3. Filter the pure product and allow to dry. Weigh the product and report the percentage yield. 4. Determine the melting point of the product. Synthesis of Aspirin Aspirin (acetylsalicylic acid) is a synthetic organic derived from salicylic acid. Salicylic acid is a natural product found in the bark of the willow tree and was used by the ancient Greeks and Native Americans, among others, to counter fever and pain. However, salicylic acid is bitter and irritates the stomach. In a Bayer laboratory in Wuppertal, Germany, young scientist Dr. Felix Hoffmann was the first to succeed in synthesizing a chemically pure and stable form of acetylsalicylic acid (ASA), which becomes the active ingredient in Aspirin. Aspirin is the most frequently sold pain reliever in the world, has been the subject of a Nobel prize (1982), and has been termed the ‘wonder drug’ of the century. It is singlehandedly responsible for the foundation and success of Bayer Pharmaceuticals (2019 revenue: 49 billion US dollars).Acetylation of Salicylic Acid O C OH + OH Salicylic Acid O O H3C O O C catalyst CH3 Acetic Anhydride H 3C O C O OH + H3C OH O Procedure: 1. Take a petri dish or watch glass and weigh x g of salicylic acid (Mol. Wt. 138.12 g/mol). 2. Transfer the weighed salicylic acid to a dry 150 mL conical flask. 3. Add 2.7 equivalents of acetic anhydride (Mol. Wt. 102.08 g/mol; Density 1.08 g/mL) using a measuring cylinder to the salicylic acid. Now add 5-6 drops of concentrated sulfuric acid and stir until all salicylic acid is dissolved. 4. Leave the reaction mixture undisturbed for 15-20 minutes. 5. Add 50 mL of water to the flask and swirl for two minutes and filter. 6. Collect the solid from the filter paper. Recrystallization 1. Dissolve the crude product in 7 mL of ethanol in a beaker and add 15 mL of distilled water. Heat on water bath till you get a clear solution. 2. Allow the solution to cool in an ice bath without disturbing. Pure acetylsalicylic acid crystallizes. 3. Filter the pure product and dry it by placing in between sheets of filter paper. Report the percentage yield. 4. Dissolve a f ew crystals of the dry compound in 0.5 mL methanol and a dd 2 d rops of FeCl3 solution. Note the color change. Repeat the above test with similar amount of salicylic acid and note the color change. 5. Determine the melting point of acetylsalicylic acid. Estimation of Iodine in iodized common salt using iodometry Iodine is an essential element for life and one of the heaviest elements required by living organisms. However, around one third of the world’s population lives in areas of iodine deficiency. The practice of adding iodine to salt is a safe, easy and effective way of overcoming iodine deficiency in our diet. Globally two chemical forms of iodine are used for iodization; Iodates (IO3-) and Iodides (I-). The iodides degrade more readily in presence of impurities, exposure to sunlight, moisture and exposure to heat, whereas the iodates remain stable under extremes of weather and handling. USA uses potassium iodide (77 mg/Kg) while Germany and India use potassium iodate (25-20 mg/Kg) for iodine fortification. Today you will use iodometry to estimate the amount of iodine in a salt sample. Procedure: Rapid test to determine the nature of iodizing reagent Take a pinch of common salt on a watch glass and divide into two parts: Test for iodate solution Moisten the first part with 2-3 drops of the given solution (mixture of A, D and E). If iodate is present the salt will turn blue/grey and the colour will be retained for several minutes before turning brown. Test for iodide solution Moisten the second part with 2-3 drops of the given solution (mixture of A, B and C). If iodide is present the salt will turn blue and remain blue for several minutes before fading. Determination of iodate content (if test A is positive) 1. Weigh X g of the given salt sample in a watch glass or petri dish. 2. Transfer the weighed salt into a 250 mL conical flask and dissolve it in 50 mL water. 3. Add 1 mL 2N H2SO4 then add 5 mL of 3 % KI solution using a measuring cylinder. The solution will turn yellow. Wrap the mouth of conical flask with a piece of filter paper and keep it in cupboard for 10 minutes. 4. Take 60-80 mL solution of (approx.) 0.005 M Na2S2O3 in a plastic beaker and use for titration. Rinse burette and fill it in and adjust zero level. 5. Remove flask from cupboard and titrate with Na2S2O3 solution until the solution turns pale yellow. Now add approx. 10 drops of starch indicator. The solution will turn dark purple. Continue titrating until the solution becomes colourless. 6. Record the volume of titrant (Na2S2O3 solution) used and calculate the amount of iodine present in part per million (ppm). Repeat three times to get average value. Standardization of Sodium Thiosulfate 1. Pipette out 10 mL copper sulfate of concentration 0.005 M in a conical flask and add 5 mL of 3% KI solution. The solution will turn yellow in colour. 2. Titrate with Na2S2O3 solution until the solution turns pale yellow. Add 7-8 drops starch indicator solution at this stage. 3. Continue the titration until the purple colour fades, then add 5-6 drops of KSCN solution and titrate again. The end point gives a colourless solution. CAUTION: To ensure that you have obtained the true end point, stir the flask for 20 seconds and then wait for 20 seconds to make sure that the purple colour does not reappear. 4. Repeat the titration to get concordant readings. 5. Calculate the molarity of the given sodium thiosulfate solution. Conductometric Titration of HCl vs NaOH Several compounds wholly or partially dissociate into ions when dissolved in water. The conductance of such electrolytes depends on the concentration of the ions, temperature of the medium and nature of the ion. The nature of ion becomes a variable with respect to the charge and mobility of the particular ion. The electrolyte solutions obey Ohm’s law. Today a conductometry cell will be used to perform a titration of HCl vs NaOH. Procedure: CAUTION: THE ELECTRODES ARE FRAGILE INSTRUMENTS. HANDLE THEM GENTLY AND CAREFULLY. DO NOT ADJUST THE KNOBS ON THE INSTRUMENT. THE INSTRUMENT IS CALIBRATED. 1. Take NaOH (0.1 N) solution in a 50 mL burette and adjust zero reading. 2. Pipette out 25 mL of the given HCI solution in a 100/150 mL beaker. Add 25 mL of water to this. 3. Now add the NaOH solution from the burette in (2 drops) 0.2 mL increments and record the conductance after mixing the solution. 4. Continue the titration till you reach the initial conductance. 5. Repeat the experiment twice. 6. Plot the graph of volume of NaOH vs conductance and determine the equivalance point of the titration. 7. Calculate the normality of HCl solution. Determine the pI of a glycine using pH metry Glycine (means ‘sweet tasting’ in Greek) is the simplest amino acid with a single hydrogen atom as its side chain. In aqueous solution, glycine itself is amphoteric: at low pH the molecule can be protonated and at high pH it loses a proton. Today you will find the isoelectric point for glycine, which is the pH at which a molecule or surface carries no net electrical charge. At this pH, the molecule will not show any motion in an electric field. For a molecule to have a sharp isoelectric point, it should be amphoteric. Proteins and amino acids are common molecules that meet this requirement. For an amino acid with only one amine and one carboxyl group, the pI can be calculated from the pKa values of this molecule. You will perform a potentiometric titration for this study. Procedure: CAUTION: ELECTRODES ARE FRAGILE INSTRUMENTS. HANDLE THEM GENTLY AND CAREFULLY. DO NOT ADJUST THE KNOBS ON THE INSTRUMENT. THE INSTRUMENT IS CALIBRATED. 1. Take 50 mL of NaOH (0.1 M) solution in a 50 mL burette and adjust zero reading. 2. Pipette out 25 mL of the given solution of amino acid in a 250 mL of beaker and add 25 mL of distilled water to the amino acid solution using a pipette. 3. Insert the cleaned pH electrode into the beaker solution and record the initial pH of the solution. 4. Add NaOH in 0.5 mL increments from the burette. Stir the solution and mix it well. 5. Record the corresponding pH values until the pH starts increasing drastically. At this time, add 0.1 mL increments of NaOH till the pH stabilizes around 8. 6. After reaching pH 8, continue adding 0.5 mL increments of NaOH till you reach pH 11. 7. Plot the graph of pH vs volume of NaOH solution. 8. The two almost horizontal parts of the graph give the values of pKa1 and pKa2 for glycine. Use mid-points of these regions to get the values. 9. The average of these values (pKa1 and pKa2) gives the pI of glycine. To Determination the Energy of Activation In today’s experiment you will study the reaction between potassium permanganate and dilute oxalic acid at different temperatures. The permanganate ion MnO4- reduces to MnO2 changing the colour from bright purple/pink to yellow/brown. You will find the rate constant for this reaction at five different temperatures and then determine the activation energy for the reaction. PRECAUTION: DO NOT TOUCH THE WATER IN THE WATER BATH Procedure: 1. Using burettes, place 20 mL oxalic acid (0.25 M) in a conical flask and 10 mL KMnO4 (approximately 0.01 M) in a test tube. The exact concentration of KMnO4 should be noted from the blackboard. 2. Place both the vessels in a water bath to equilibrate for at least 5 minutes. 3. Mix the reactants in the conical flask and start the stop watch. 4. Swirl the reaction mixture regularly without taking it out of the water bath. 5. Record the time it takes for the mixture to turn yellow/brown. 6. Repeat the procedure with another sample at this temperature. 7. Repeat steps 1 to 6 for three other temperatures. 8. For the reading at 0 degree Celsius, time taken is 2215 seconds. Use this information as the fifth temperature reading in your experiment. 9. Determine the activation energy by plotting ln k Vs 1/T. (Temperature in degree kelvin) Data collection should consist of a table similar to: [KMnO4] = [Oxalic Acid] = Observation and Calculations: S. Temp Temp 1/T Time No. ( ͦ C) (K) for trial 1 (s) 1 2 3 4 5 6 7 8 9 10 Time Average Rate= k ln for Time [KMnO4]/time =Rate/[KMnO4][Oxalic] (k) trial (s) 2 (s) Show sample calculation for determining the rate: Rate = [KMnO4]/time. Show sample calculation for determining k: k =Rate/[KMnO4][Oxalic]. Plot of ln k Vs 1/T and determine Ea in kJ/mol from the slope. Preparation of [Ni (NH3)6] 2+ and its Analysis by Complexometric Titration (A) Preparation of Hexamminenickel (Il) chloride 1. Take 10 mL solution of nickel chloride hexahydrate (contains 3g of NiC12) in a 250 mL beaker. 2. Take 10 mL solution of aqueous ammonia in a measuring cylinder. 3. Add the ammonia solution drop wise to the solution of nickel chloride with constant stirring till the colour of the solution has changed from pale green to intense violet. 4. Allow the solution to stand at room temperature for 5 minutes, cover with watch glass. Then cool it in an ice bath for about 15 minutes. 5. Filter the solution and wash the crystals with 3-5 mL ammonia solution. 6. Dry the crystals using filter paper. 7. Report the weight of the dried complex. (B) Estimation of nickel(ll) by EDTA 1. Take 80 mL of 0.01M EDTA solution in a 250/500 ml plastic beaker and fill it in a clean burette up to the mark. 2. Weigh accurately 0.23 g of [Ni(NH3)6]Cl2 complex and transfer this to a 100 mL volumetric flask. Now add 50 mL of 1 N H2SO4 to dissolve it and makeup the solution to the mark with distilled water. 3. Pipette out 10 mL of the complex solution in a 250 mL conical flask and dilute it with 15 mL of distilled water. 4. Add 2-3 drops of murexide indicator and 5 mL NH4Cl solution (0.5M) to the conical flask. Now add ammonia solution (7-10 drops) to maintain a pH 7 (light green colour of the solution). 5. Titrate it with EDTA solution till the endpoint is near, add 3ml of ammonia solution and continue the titration till the endpoint (bluish violet colour appears). 6. Repeat the titration and get concordant values. 7. Calculate the amount of Ni present in the complex. 8. Put the remaining Ni complex, [Ni(NH3)6]Cl2, in a paper sachet, write your Roll Number on it and submit the sample in the box labelled ‘Product’. (C) Estimation of nickel by spectrophotometry 1. 2. 3. Several solutions (of NiCl2) of known concentration and one solution of unknown concentration will be provided to you. Measure absorbance of all the solutions at 395 nm using a UV-Visible spectrophotometer. Plot absorbance versus mg/mL of nickel. Determine the concentration of nickel present in the unknown solution in g/L. Extraction and Identification of DNA DNA or Deoxyribonucleic acid contains all genetic information necessary for growth, functioning and reproduction of almost all living organisms. DNA molecules consist of two biopolymer strands coiled around each other to form a double helix. The chemical and molecular structure of the DNA is illustrated below. Chromosomal DNA, exists in the well known X shape and is bound by proteins into a supercoil. DNA was first isolated by Friedrich Miescher in 1869. Its molecular structure was first identified by James Watson and Francis Crick at the Cavendish Laboratory within the University of Cambridge in 1953. In 1960, Nirenberg and Har Gobind Khorana decoded DNA. Today you will extract DNA from peas and then identify it using UV-Visible spectroscopy and a chemical test. Procedure: 1. Take 5 mL of the (pea/onion) extract in a boiling tube and add 1.0 mL of the SDS solution and gently swirl. Let the mixture stand for 10 minutes in ice. 2. Add 4 drops of papain extract to the mixture and stir gently. 3. Now hold the boiling tube at an angle and pour very slowly 15 mL of ice cold ethanol down the wall of the test tube so that it forms a layer above the extract layer. 4. Allow the boiling tube to stand straight for a few minutes. 5. Some stringy white substance comes in the alcohol layer. This is DNA. 6. Use a hooked glass rod and place it such that its end is just below the alcohol layer. Now try to spool the DNA out of the tube. Identification of DNA Diphenylamine Test 1. In a test tube, add a small amount of crude DNA and 2 mL of 4% sodium chloride solution. Add 2 mL of diphenyl amine reagent and mix. 2. Place the test tube in boiling water bath for one hour and record changes. UV-Vis Absorption 1. Dissolve DNA in 2-3 mL of TE buffer solution and determine the ratio of absorption at 260 & 280 nm. Amount of Calcium in Milk Calcium is one of the minerals that the body needs daily. Milk and milk products have the best reserves of calcium. Interestingly, calcium seems to come in fifth place wherever it goes: it is the fifth most abundant element by mass in the Earth's crust (after oxygen, silicon, aluminum and iron); the fifth most abundant dissolved ion in seawater (after sodium, chloride, magnesium and sulfate); and the fifth most abundant element in the human body (after oxygen, carbon, hydrogen and nitrogen). It is, however, the most abundant metallic element in the human body, 99 percent of which can be found in our bones and teeth. Today you will find out the amount of calcium in milk by doing a complexometric titration with ethylenediaminetetraacetate (EDTAsodium salt). Procedure STEP A: Preparation of Mg-EDTA Indicator Solution 1. Take 50 mL of Mg-EDTA solution in a 250 mL glass beaker. (This Mg-EDTA solution contains 0.74 g of EDTA and 0.49 g of MgSO4 in 100 mL of water.) 2. Add 2-3 drops of phenolphthalein indicator to the Mg-EDTA solution. Fill a burette with 0.1 M NaOH solution. 3. Keep a white tile below the beaker containing Mg-EDTA solution. 4. Add the NaOH solution dropwise from the burette to the Mg-EDTA solution till it becomes light pink. Note the volume of NaOH required for this process and discard this solution in chemical waste bucket. 5. Take again 50 mL of Mg-EDTA solution in a 250 mL beaker and add the same volume of 0.1 M NaOH solution as noted in step 4. 6. Add distilled water such that the total volume; Mg-EDTA + NaOH + Water = 95 mL 7. Add 2 mL of pH 10 buffer solution and 8 drops of Eriochrome black-T indicator to the above solution. At this stage there are two possibilities. 7.1 If the solution is red in color, add 0.01M EDTA solution dropwise until the solution turns blue. 7.2 If the solution is blue in color add 0.01M MgSO4 solution dropwise until the solution turns red then add 0.01M EDTA solution dropwise until the solution turns blue again. 8. This solution will be used as the Mg-EDTA indicator solution in part B of the experiment. STEP B: Estimation of Calcium 1. Remove NaOH from the burette. Rinse the burette with 0.01 M EDTA and fill it with 0.01 M EDTA. 2. Pipette out 25 mL of the milk solution into a 250 mL conical flask. 3. Add 2 mL of pH 10 buffer, 10 mL of Mg-EDTA indicator solution (Prepared in step A) and 3 drops of Eriochrome black-T indicator. 4. Titrate it with the standard 0.01M EDTA (exact concentration on blackboard) solution until the color change is red to blue. 5. Repeat the titration to get concordant value. 6. Calculate the amount of calcium in the given sample in percentage or grams/liter.