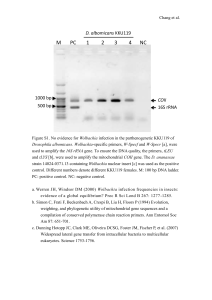
16S rRNA
advertisement

16S rRNA Muna Salah 16S rRNA gene sequencing • Highly conserved regions – Identical in all bacteria – Single PCR primer pair can amplify 16S rRNA genes from diverse bacteria • Highly variable regions – Conserved within species – Divergent g between species Image from Alimetrics.net 16S rRNA Sequences • The h genes coding di for f 16S rRNA, 23S rRNA, and d 5S rRNA are essential for existence of each bacterial cell due to its involvement in protein synthesis. • Their sequences are generally highly conserved amongg bacteria. • Some variable regions are present within these sequences. sequences • 16S rRNA is conserved at the strain level. Primer forward (Upper; 5’) Samp_A Samp_B Samp_C Samp_D Samp_E Samp_F Samp_G Primer reverse (Lower; 3’) 5’ATCGCTCGATCGTAAATCGCGTTAGCGGTAGCTATACGATTTATCGATCGTATACGTAGCGTAGCGTAATTTTAAAACCGT 5’.......................G................A........................................ 5’............................C............................G....................... 5’.......................G......................................................... 5’.......................................AC..........T............................. 5’...................................T............................................. 5’...................................T.........._..........C....................... Sequence BLAST 3’ 3’ 3’ 3’ 3’ 3’ 3’ Assay Workflow Culture, Biopsy Nucleic acid extraction Amplification of the TARGET GE region (PCR) DNA sequence Result ! - Identification at Species / Subspecies level Sequence alignment (BLAST – NCBI) General 16S rRNA sequencing strategy • IIsolate l t DNA • Amplify 16S rRNA genes using PCR and primers recognizing i i conserved d regions i • Perform NGS – usually Roche 454 • Use sequence data to identify types and abundances of bacterial “species” • Measure community diversity (alpha) • Compare diversity between communities, locations treatments, locations, treatments etc. etc (beta) Why use 16s rRNA gene sequencing The 16s rRNA gene sequence show a high degree of conservation among species This is assumed to result from the importance of the 16S rRNA as a species. critical component of cell function. Few other genes are as highly conserved as the 16S rRNA gene. gene Although the absolute rate of change in the 16S rRNA gene sequence is not known, it does mark evolutionary distance and relatedness of organisms 16s rRNA is present in almost all bacteria, often existing as a multigene family, or operons. The 16s rRNA gene (1,500 (1 500 bp) is large enough for informatics purposes. purposes 16s rRNA gene sequencing studies as opposed to the more cumbersome a pu at o s involving o g DNA-DNA hybridization yb d at o investigations. est gat o s manipulations 16s rRNA gene of various bacteria are extensively studied and is present in many databases Generating Phylogenetic Trees and Comparing Sequences • Comparisons are commonly shown as phylogenetic trees and linear alignments. • A phylogenetic tree is a tree‐structured graph used d in i computational i l biology bi l to visualize i li the h result of a hierarchical clustering calculation. • The result of a clustering is presented either as the distance(dissimilarity) or the similarity between the clustered rows or columns depending on the selected distance measure measure. Methods commonly used for generating phylogenetic trees NJ method (Neighboor (Neighboor-joining) joining), UPGMA method (Unweighted pair group method with arithmetic averages) WPGMA method ((Weighted g p pair g group p method with arithmetic averages) APPLICATIONS OF BACTERIAL IDENTIFICATION USING 16S rRNA SEQUENCING Identifying Id if i unidentified id ifi d b bacteria i or iisolates l with i h ambiguous bi profiles. • One of the most attractive potential uses of 16S rRNA gene sequence informatics is to provide genus and species identification for isolates that do not fit any recognized biochemical profiles, for strains generating only a “low likelihood” or “acceptable” identification according to commercial systems, systems or for taxa that are rarely associated with human infectious diseases. • The cumulative results from a limited number of studies to date suggest that 16S rRNA gene sequencing provides genus identification in most cases (>90%) but less so with regard to species (65 to 83%), with from 1 to 14% of g the isolates remainingg unidentified after testing. Conclusion • The traditional identification of bacteria on the basis of phenotypic characteristics is generally not as accurate as identification based on genotypic methods. • Comparison of the bacterial 16S rRNA gene sequence has emerged as a preferred genetic p g technique. q 16S rRNA ggene sequence q analysis y can better identify poorly described, rarely isolated, or phenotypically aberrant strains, can be routinely used for identification of mycobacteria, and can lead to the recognition of novel pathogens and noncultured bacteria. bacteria • Problems remain in that the sequences in some databases are not accurate, there is no consensus quantitative definition of genus or species based on 16S rRNA gene sequence data, Conclusion • The proliferation of species names based on minimal genetic and phenotypic differences raises communication difficulties, and micro heterogeneity in 16S rRNA gene sequence within a species is common common. • p its accuracy, y 16S rRNA ggene sequence q analysis y lacks widespread p Despite use beyond the large and reference laboratories because of technical and cost considerations. Thus, a future challenge is to translate information from 16S rRNA gene sequencing into convenient biochemical testing schemes, making the accuracy of the genotypic identification available to the smaller and routine clinical microbiology laboratories.