Full Text - BioTechniques
advertisement

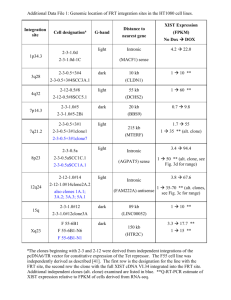
LY Benchmark W IT H P E R Keywords: recombination; cloning; clonase; one-pot reaction A) B) R Method summary: We developed efficient, cost and time saving protocols that enable the isolation of Gateway entry and expression clones using a single consolidated reaction capable of simultaneously performing BP and LR recombination steps. Vol. 55 | No. 5 | 2013 attB1 Gene Gene Expression Clone 1 attP1 attB2 Expression Clone Gene Expression Clone 6 attP2 Donor Vector + R (Amp ) ccdB R (Kan ) Gene Gene Expression Clone 2 Entry Clone Gene Expression Clone 3 Gene Expression Clone 5 Gene Expression Clone 4 LR Clonase® (Int + IHF + Xis) BP Clonase® (Int + IHF) attL1 attR1 attL2 Entry Clone E P R IN T The Gateway recombination system is characterized by its ability to transfer DNA sequences back and forth between an intermediate clone (the entry clone) and a variety of destination vectors. However, a number of applications do not need to exploit the advantages offered by the entry clone. Here we report reaction conditions for cloning DNA fragments into destination vectors in a single step reaction, thus reducing the cost and overall time needed to obtain an expression clone from three days to one. IO N S BioTechniques 55:265-268 (November 2013) doi 10.2144/000114101 thereby reducing the time needed to obtain a final expression clone from three days to one. The rationale for our approach stems from the fact that the main difference between the BP and LR reactions is the presence of the lambda excisionase (Xis) in the LR Clonase blend, which drives the recombination dynamics toward phage excision (3). Therefore, there should be an excisionase level where both reactions occur at similar rate. To facilitate the fine tuning of the BP/LR combined reaction, we employed a DNA fragment harboring the lacZα reporter gene. The linear double-stranded DNA fragment was used as delivered by a gene synthesis provider, without precloning or prior PCR amplification. Reactions were set up using different ratios of BP and LR Clonase in the presence of the reporter sequence as well as donor and destination vectors (see Supplementary Material). Successful recombination events into both vectors were represented as blue colonies selected on X-gal agar plates supplemented with the corresponding antibiotics. IS Life Technologies, Carlsbad, CA The Gateway cloning system exploits the site-specific recombination system utilized by bacteriophage lambda to shuttle sequences between plasmids bearing flanking compatible recombination attachment (att) sites (1). Once captured as an entry clone, a DNA fragment can be recombined into a variety of destination vectors resulting in expression clones geared to specific applications (Figure 1A). The recombination reactions are driven by two enzyme blends known by their commercial names: BP Clonase and LR Clonase (Figure 1B). Further details on the Gateway system can be found in an earlier review (2). One of the drawbacks of the system is that, starting from a linear PCR fragment, it takes two bacterial transformation steps (or three days) to obtain the final expression clone. For those applications that do not require the isolation of an entry clone, the workflow adds two days to the overall cloning timeframe compared with other cloning approaches. Here we describe a new approach combining the two Gateway steps, BP and LR recombination, into a single reaction, M Xiquan Liang, Lansha Peng, Chang-Ho Baek, and Federico Katzen O N Single step BP/LR combined Gateway reactions (KanR) + ccdB attR2 Destination Vector (AmpR) Figure 1. The Gateway recombination system. (A) A DNA fragment containing the gene of interest is recombined into a donor vector using the BP reaction (single-headed arrow) resulting in an entry clone. This clone can be used in an LR reaction (double-headed arrows) to transfer the fragment into multiple destination vectors, generating expression clones. The gene of interest in the expression clones may be transferred back by a BP reaction to generate an entry clone (double-headed arrows). (B) The BP Clonase contains the phage lambda integrase (int) and the E. coli integration host factor (IHF) and transfers DNA fragments flanked by attB sequences into a donor vector, which has the counterselectable marker ccdB flanked by attP sequences. The LR Clonase blend has the same enzyme components as the BP Clonase blend, plus the lambda excisionase (Xis), promoting the opposite reaction where a fragment is transferred from an entry clone into a destination vector generating the expression clone. AmpR, ampicillin resistance; KanR, kanamycin resistance. 265 www.BioTechniques.com Benchmark B) AmpR colonies - Expression clones (LacZ-α) 100 80 10000 80 60 1000 40 100 20 10 1 10000 1000 60 100 40 10 20 0 LR Clonase® (%) 0 0* 0 25 50 75 100 0 LR Clonase® (%) 0 0* 0 25 50 75 100 BP Clonase® (%) 0 100* 100 75 50 25 0 BP Clonase® (%) 0 100* 100 75 50 25 0 CE (% Blue colonies) 1 CE (% Blue colonies) CFU/plate D) KanR colonies - Entry clones (GFP) 1000000 80 100000 10000 60 1000 40 100 20 CFU/plate AmpR colonies - Expression clones (GFP) 100 CE (% fluorescent colonies) 100 CFU/plate CE (%pENTR+ clones) C) 10 10000 80 1000 60 100 40 10 20 0 1 1 0 LR Clonase® (%) 0 0 75 100 LR Clonase® (%) 0 0 75 100 BP Clonase® (%) 0 100* 25 0 BP Clonase® (%) 0 100* 25 0 CE (% pENTR+ clones) CE (% fluorescent colonies) CFU/plate pENTR CFU/plate 100000 CE (% blue colonies) 100 CFU/plate KanR colonies - Entry clones (LacZ-α) CFU/plate CE (% blue colonies) A) CFU/plate Figure 2. Efficiencies of consolidated BP and LR reactions. Reactions were set up using 100 ng of a linear synthesized fragment (A and B) or a PCR fragment (C and D), pDONR-221 (donor vector), and pEXP1-Dest (destination vector). Enzymes were added in the indicated proportion. Total reaction volume was kept at 10 µL. (A and C) Cloning efficiency (CE) and colony count (CFU/plate) on LB kanamycin X-gal agar plates. (B and D) CE and CFU/plate on LB ampicillin X-gal agar plates. Asterisks represent reactions where pEXP1-Dest DNA was purposely omitted. For further details see Supplementary Material. The number of green fluorescent colonies in (D) was determined by placing the plates on top of a UV transilluminator. Entry clones containing the PTAC-GFP fragment in (C) exhibited very little fluorescence and were virtually indistinguishable from truly non-fluorescent colonies. The presence of the insert in this case was validated by colony-PCR assays using M13-forward and reverse oligonucleotides. Clones with the expected insert are indicated as “pENTR+ clones.” AmpR, ampicillin resistance; KanR, kanamycin resistance. Vol. 55 | No. 5 | 2013 266 www.BioTechniques.com Benchmark The results showed that at elevated LR/BP ratios the product is enriched in the expression clone, whereas opposite proportions produce an enrichment of the entry clone (Figure 2A and 2B). The cloning efficiencies depicted in Figure 2A and 2B are based exclusively on the percentage of blue colonies, and therefore underestimate the real recombination efficiency. For example, DNA sequencing of four random kanamycin resistant white colonies revealed that all four contain the right insert with internal single nucleotide mutations, reflecting errors during DNA synthesis. On the other hand, the sequence of 10 random ampicillin resistant white colonies showed that half contained similar errors as above. Equivalent results were obtained when a PCR fragment encoding the GFP gene preceded by the tac promoter was used instead of the lacZα gene (Figure 2C and D), suggesting that the method is generally applicable. Overall our results showed that BP and LR recombination can proceed at high efficiencies in a single tube without the need for subsequent steps. If the exclusive goal is to obtain an expression clone, the use of LR Clonase alone is the preferred option, as it can perform both recombination events, producing thousands of expression clones. Under these conditions, when the excisionase protein is present, BP Vol. 55 | No. 5 | 2013 recombination operates at a lower rate and, as expected, a reduced number of entry clones are obtained. This number, although sufficiently large for most cases, may fluctuate depending on the nature and quality of the insert and vectors. Methods to further increase the chances of simultaneous entry clone selection include (i) using electroporation rather than chemical transformation protocols, or (ii) raising the BP:LR Clonase ratio to 1:3. The strategy presented here could be expanded to cover the assembly of multiple fragments into a single vector as proposed previously (4). However, the isolation of individual entry clones should be subjected to screening as all of them will likely provide the same antibiotic resistance. Finally, our approach can also be applied to transfer an insert from an expression clone to a second destination vector in a single step. In this case a linearization of the expression clone will be necessary to reduce the background, unless the antibiotic resistance markers of the two episomes are different. Author contributions XL, CHB, and LP performed the experiments. FK conceived the idea and wrote the manuscript. 268 Competing interests The authors hold financial interests in Life Technologies. References 1. Hartley, J.L., G.F. Temple, and M.A. Brasch. 2000. DNA cloning using in vitro site-specific recombination. Genome Res. 10:1788-1795. 2. Katzen, F. 2007. Gateway recombinational cloning: a biological operating system. Expert Opin Drug Discov. 2:571-589. 3. Landy, A. 1989. Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu. Rev. Biochem. 58:913949. 4. Cheo, D.L., S.A. Titus, D.R. Byrd, J.L. Hartley, G.F. Temple, and M.A. Brasch. 2004. Concerted assembly and cloning of multiple DNA segments using in vitro site-specific recombination: functional analysis of multi-segment expression clones. Genome Res. 14:2111-2120. Received 29 August 2013; accepted 16 October 2013. Address correspondence to Federico Katzen, Life Technologies, Carlsbad, CA. E-mail: federico.katzen@lifetech.com To purchase reprints of this article, contact: biotechniques@fosterprinting.com www.BioTechniques.com