Molecular dynamics simulations of a bioactive glass nanoparticle
advertisement

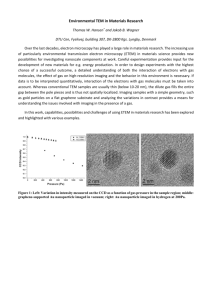
Journal of Materials Chemistry C Dynamic Article Links < Cite this: J. Mater. Chem., 2011, 21, 12660 PAPER www.rsc.org/materials Molecular dynamics simulations of a bioactive glass nanoparticle Antonio Tilocca* Received 2nd May 2011, Accepted 20th June 2011 DOI: 10.1039/c1jm11927c Molecular dynamics simulations of a Bioglass spherical nanoparticle (6 nm diameter) have been carried out to investigate the main structural features induced by the reduced size, which could play a specific role in the observed enhanced bioreactivity of these systems, in addition to the higher surface area. Compared to the bulk glass or to the extended flat surface, the simulations reveal that the most relevant effects of the reduced size are a further slight reduction in the already low silicate connectivity on the nanoparticle surface, together with a ring size distribution shifted towards three-membered rings, and a higher Na+/Ca2+ ratio in close proximity of the surface. The possible ways in which these effects can translate into higher bioreactivity are discussed. Introduction Due to their ability (‘‘bioactivity’’) to interact and rapidly form stable bonds with hard and soft tissue, modified silicate glass compositions such as 45S5 Bioglass (46.1SiO2; 24.4Na2O; 26.9CaO; 2.6P2O5 mol%) are widely employed in dental and orthopaedic clinical applications.1 Compact bioactive glasses (in solid blocks) are employed in low-load bearing applications, such as middle-ear implants and maxillofacial reconstruction,2 whereas current clinical applications in orthopaedics and dentistry mostly involve the use of glass microparticles as fillers to treat bone and periodontal defects.3–5 The superior performances of bioactive glasses in these applications compared to other materials have recently been associated with their ability to promote growth of new tissue, thanks to the osteogenic activity of the ionic dissolution products of the glass.6 The potential ability of bioactive glasses to stimulate regeneration of living tissues, rather than just replacing them, has triggered new research on these materials, aiming at exploiting their potential use in tissue engineering.7 A key factor to control both the tissuebonding and tissue-regeneration activity is the size of the glass particles. Reducing the particle size below the conventional micrometre scale leads to an enhancement in the bioreactivity of materials for medical applications. For instance, nanoscale features introduced on the surface improve the cellular response of titania implant substrates;8–10 protein adhesion and proliferation were significantly enhanced on the surface of hydroxyapatite nanocrystals compared to the flat surface;11 compared to microparticles, the presence of nanosized bioactive glass particles enhances protein adsorption on composites;12 nanometre-sized bioactive glass particles induce a higher dentin remineralisation rate, again compared to micrometre-sized particles,13 and it has Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: a.tilocca@ucl.ac.uk 12660 | J. Mater. Chem., 2011, 21, 12660–12667 also been proposed that the reduction of particle size could enhance the antibacterial properties of bioactive glasses.14 The biocompatibility and non-toxicity of sub-micron bioactive glass nanoparticles were also assessed and proven very recently.15 A combination of different factors is likely responsible for the superior bio-reactivity of sub-micron glass particles: the high surface area/volume ratio certainly contributes to enhance protein adsorption and favours release of the ionic products of glass dissolution; moreover, it has been suggested that the match between nanoscale features on the glass surface and nanometresized collagen and apatite molecular units improve the contact at the implant–tissue interface and enhances deposition and mineralisation of new living tissues.10 A deeper understanding of these factors is often hampered by the intrinsic difficulty to characterise systems of this size using experimental methods; on the other hand, atomistic computer simulations can nowadays approach nanosized systems in a relatively straightforward fashion.16 Molecular Dynamics (MD) computer simulations have recently shown their efficacy to reveal structural and dynamical features of bulk bioglasses (BGs) which are correlated to the glass bioreactivity.17 In particular, given the established role of the glass dissolution in starting and sustaining the bioactive process, the modelling studies of BGs have focused on identifying links between specific structural features and the tendency of a glass composition to degrade and dissolve in an aqueous environment. Classical (employing empirical force fields) Molecular Dynamics (MD) simulations of the bulk and of the flat surface have helped to understand the role of several structural factors in the bioactivity, such as the silicate network connectivity,18,19 the presence of ring and chain fragments,20,21 and the tendency to form modifier (Na and Ca) cation-rich aggregates.22 Ab initio (parameter-free) MD simulations have examined the properties of the melt precursors23,24 and focused on the reactivity of the flat surface, highlighting the main adsorption sites and the mechanism of the initial dissolution This journal is ª The Royal Society of Chemistry 2011 steps of 45S5 Bioglass.25,26 All these studies have involved extended periodic systems, and no previous simulations have yet targeted bioactive glass nanoparticles, even though a number of recent studies27–33 has shown the efficacy of classical MD to model a wide range of structural and dynamical properties of crystalline and amorphous oxide particles with sizes up to 10 nm. It must be remarked that the much heavier computational demands of ab initio simulations do not currently allow their applications to model systems of this size, containing several thousands of atoms, even though the continuous developments in the performances of electronic structure codes make it now possible to tackle nanoclusters of smaller size, for instance in a bottom-up approach.16 In the present work, classical MD simulations are carried out to obtain the first realistic model of a bioactive glass nanoparticle: the particle diameter (6.5 nm) is representative of the size of small BG nanoparticles that can be prepared using experimental methods.34,35 The analysis is mainly focused on identifying specific features associated with the reduced size and high active surface area, which differ from the corresponding bulk-like properties, and could thus represent key factors in the special bioreactivity of nanosized BGs. Computational methodology Classical MD simulations were performed with the DL_POLY 2.20 code,36 using an ionic (full-charges) shell-model interatomic potential18,37 recently developed to model multicomponent phosphosilicate glasses. Oxygen atoms were represented as core– shell units, with a small mass (0.2 atomic units) assigned to the shells to allow them to follow the core motion adiabatically.38 Besides the harmonic interaction with the corresponding core, oxygen shells interact with each other and with all cations through short-range Buckingham potentials. Coulombic forces act between all species and three-body truncated† harmonic potentials are used to control the intra-tetrahedral O–Si–O and O–P–O angles. The MD time step was 0.2 fs; no periodic boundary conditions were applied, in order to model the isolated nanoparticle. Electrostatic forces were calculated through direct large enough to Coulomb sum, with a distance cut-off of 70 A, include all atomic pairs in the system. The cut-off for van der Waals interactions was set to 8 A. After a short relaxation at 300 K, the random sphere was heated up and held at 2500 K in a constant-temperature MD run of 240 ps, during which the total potential energy decreased and converged to a stable value. A further microcanonical run of 240 ps at an average temperature of 2500 K followed, during which the potential energy only showed thermal oscillations around the same constant value. Starting from this equilibrated melt, the particle was quenched to room temperature in four consecutive constant-temperature runs at 2000–1500–1000 and 300 K, respectively, lasting 55 ps each, corresponding to a nominal cooling rate of 10 K ps1, which is the standard for MD simulations of melt-derived glasses.41 A final microcanonical run of 240 ps at an average temperature of 300 K completed the simulation. The analysis presented in this manuscript was carried out on all configurations extracted from this last trajectory, sampled every 0.4 ps. Results A snapshot of the final structure of the 45S5 nanoparticle is shown in Fig. 1. The particle maintained a roughly spherical shape: the ratios I1/I2 and I1/I3 between the three calculated principal moments of inertia were 1.05 and 1.08, and the corresponding a, b and c ellipsoid parameters were 33.65, 32.66 and close to the 32 A radius of the initial sphere. 30.95 A, In order to characterise the nanoparticle, we have analysed several structural parameters, focusing in particular on how the structure changes moving from the internal to the surface region. Fig. 2 shows the total number, density and fraction profiles of each species i (i ¼ O, Si, Na, Ca, P) going from the centre to the surface of the nanoparticle. Given the approximately spherical shape of the nanoparticle, the profiles were calculated by dividing the volume in concentric spherical shells of thickness dR ¼ 1 A, located at distance (R dR) from the particle centre of mass. The atomic fraction and density in each shell were calculated as A standard approach involving melt-and-quench of a random mixture, which has proven effective to model a wide range of bulk17 and nanosized28,29,33,39 amorphous materials, was employed to generate a model of a 45S5 Bioglass nanoparticle of approximately 6 nm diameter. An initial mixture of 10 017 atoms at the correct composition (46.1SiO2; 24.4Na2O; 26.9CaO; 2.6P2O5 mol%) was inserted at random positions in a sphere of radius, yielding the experimental 45S5 Bioglass 31.725 A density of 2.702 g cm3.40 Besides the density, the only constraints applied in the initial structure generation were distance cutoffs to prevent particles to start unphysically close to each other. † We found that switching from the screened to the equivalent truncated form of the harmonic 3-body term in the original potential improves the stability of the trajectory and damping of core–shell motion is no longer needed in the MD simulations. This journal is ª The Royal Society of Chemistry 2011 Fig. 1 Structure of the Bioglass nanoparticle, extracted from the MD trajectory at 300 K. Silicate and phosphate groups are represented as balland-sticks, Na and Ca as spheres. Si, P, Na, Ca, O are coloured yellow, brown, blue, cyan and red, respectively. J. Mater. Chem., 2011, 21, 12660–12667 | 12661 Fig. 2 Total number ni(R) (top panel), number density ri(R) (middle) and fraction ci(R) (bottom panel) of different species as a function of their distance from the nanoparticle centre of mass. Dashed horizontal lines in the middle panel mark the corresponding number density of each species in the bulk. The larger statistical fluctuations seen towards the core reflect the decreasing number of atoms contained in volume elements for R / 0. ci(R) ¼ ni(R)/N(R) and ri(R) ¼ ni(R)/V(R), where ni and N are the time-averaged number of atoms of species i and of all species found in the shell, respectively, while V is the volume of the shell. The middle panel of Fig. 2 reveals significant departures from the bulk density of all species (bulk values are denoted by the dashed from the particle centre. These lines) starting beyond 30 A deviations can be used to define the surface region of the nano thick) where the distribution particle, as the external shell (5–6 A is significant different from the bulk. This same region is also highlighted by the relative atomic fractions shown in the bottom panel of the figure: the surface is characterised by a marked increase in the sodium fraction cNa, while the other fractions decrease. As a consequence, the nanoparticle surface is composed almost exclusively of Na and O, and significantly depleted in Si and Ca compared to the bulk. The time evolution of the atomic fractions in the core and surface regions during the full MD trajectory (Fig. 3) shows that Na enrichment at the surface mainly takes place during the cooling, after which the converged fractions oscillate around steady values, confirming that full structural relaxation has been achieved within the simulation time. Fig. 3 Time evolution of the atomic fractions in the core (top panel, from the centre) and surface including all atoms found at R < 15 A regions during the melt-and-quench process. (bottom panel, R > 30 A) characteristic nearest-neighbours distances) is not significantly affected by the reduced size. However, some interesting differences emerge in the peak heights. The lower intensity of the peaks in the surface region (red curves) obviously reflects the empty external region outside the nanoparticle, compared to a compact the g(r)’s bulk-like distribution. Below the surface (R < 30 A) normally overlap with their bulk counterpart, denoting full recovery of the bulk-like structure already in the internal regions directly under the nanoparticle boundary. The only notable exception is represented by the X–Na (X ¼ Si, Na, Ca) pairs: unlike the other curves, all the gX–Na(r) functions (central row of remain below the corresponding bulk values, Fig. 4) for R < 30 A Short-range structure The short range structure was analysed through the pair distri thick bution functions g(r) calculated separately in four 10 A (10, 20 A), (20, 30 A) and spherical shells spanning R ¼ (0, 10 A), where R is the distance from the particle centre of mass R > 30 A, (Fig. 4). The good match of the distances corresponding to the main peak in each of the nanoparticle and the bulk g(r)’s suggests that the short-range structure (as represented by the 12662 | J. Mater. Chem., 2011, 21, 12660–12667 Fig. 4 Radial distribution functions g(r) calculated in four spherical shells located at different distances from the centre of mass of the BG represents the surface region); the g(r)’s of the nanoparticle (R > 30 A bulk BG19 are also shown. This journal is ª The Royal Society of Chemistry 2011 with a corresponding higher intensity in the gX–Na(r) functions at surface. This shows that the Na enrichment at the the (R > 30 A) surface highlighted above is balanced by an uniform perturbation which depletes the sodium coordination shells surrounding Si, Na, or Ca atoms throughout the nanoparticle. This uniform depletion reflects the homogeneous distribution of Na ions in the 45S5 BG bulk.17,18 Silicate network connectivity The Qn distributions (where Qn is a Si atom linked to another n network formers through n bridging oxygens) are shown in Fig. 5. The decrease in the Si and P density on the surface results in a reduced silicate network connectivity compared to the bulk. While the fraction of chain-like Q2 units does not markedly change there, the Q1 and Q3 curves intersect as a result of a surface depleted of branched Q3 units and richer in Q1 chain-end units. The average silicate network connectivity in the surface region (calculated from the Qn fractions there) turns out to be 1.83, compared to 2.07 in the bulk:18 on average, on the nanoparticle surface a Si atom forms 0.25 less Si–O–Si bridges than in the bulk. This small, but non-negligible difference is also reflected in the Si–Si running coordination numbers plotted in Fig. 6 (top panel), where again the contributions from the inner and outer spherical shells of the particle were distinguished by separately plotting the running coordination number n(r) of each shell. Coordination shell of the network-modifier ions The middle and bottom panels of Fig. 6 focus on another important feature of the nanoparticle surface: the reduced Ca–O and (especially) Na–O coordination. In particular, the number of nearest-neighbours oxygen atoms n(rc) (where rc is the distance of the first corresponding g(r) minimum) is reduced from 5.8 in the bulk to 4.0 in the surface for sodium, and from 6.1 to 5 for calcium. The more marked depletion in the oxygen coordination shell of exposed sodium atoms denotes that their increased fraction at the surface is not fully balanced by enough oxygen atoms, so that the latter are unable to complete the ideal pseudooctahedral coordination of Na in BGs.42 Fig. 5 Distribution of Qn silicate species going from the inner to the external (surface) regions of the nanoparticle: R is the distance from the particle centre of mass. The dashed lines denote the Qn distribution of the corresponding bulk model.19 This journal is ª The Royal Society of Chemistry 2011 Fig. 6 Running (cumulative) coordination number for Na–O, Ca–O and Si–Si pair interactions, calculated in different regions going from the to the inner regions (R < 30 A). The nanoparticle surface (R > 30 A) corresponding g(r) functions (dashed) and the bulk curves (blue) are also shown for reference. Rings Additional surface effects emerge from the distributions of intraand inter-tetrahedral angles shown in Fig. 7. The surface is characterised by significant distortion, especially in the Si–O–Si angles (Fig. 7, top panel), whose distribution is shifted to lower values compared to the bulk-like angle around 132 . Whereas significant deviations in the O–Si–O and Si–O–Si angles can result from three- and five-fold coordinated Si species,23 only a negligible amount of these defects (less than 0.1% of all Si atoms) was found in the nanoparticle. Another possible source of distortion in these angles is a different population of silicate rings compared to the bulk: Si atoms in 3-membered rings form smaller Si–O–Si angles compared to those found in 4-membered and larger rings which are dominant in bulk bioglasses.20 This is also suggested by the Fig. 7 O–Si–O and Si–O–Si angle distributions for Si atoms located in and inner regions of the nanoparticle. external (R $ 30 A) J. Mater. Chem., 2011, 21, 12660–12667 | 12663 Fig. 8 Si–Si–Si angle distributions for Si atoms located in external (R $ and inner regions of the nanoparticle. 30 A) Si–Si–Si angle distributions in Fig. 8, where a significant increase in the peak around 60 , characteristic of 3-membered rings,20 is observed for the Si atoms belonging to the surface of the nanoparticle, accompanied by a reduced intensity of those Si–Si–Si angles between 90 and 140 associated to 4- and 5-membered rings.20 The ring size distributions were explicitly calculated and shown in Fig. 9, confirming that three is indeed the dominant ring size of surface regions, whereas four-membered rings dominate the bulk-like core of the nanoparticle.20 Sodium mobility Having identified the main structural features of the nanoparticle, it is important to examine the dynamical behaviour of the system. In particular, it is interesting to focus on the dynamics of Na atoms because their local environment on the surface shows marked deviations from the bulk. Another important reason is that the high computational requirements of the present simulations, due to the large size and non-periodic nature of the system, limit the time window which can be explored to an interval where no significant ionic diffusion occurs at room temperature (atoms do diffuse in the melt, but it is the room temperature dynamics that is of interest here). Within this relatively static picture, sodium atoms are the most mobile and their dynamical behaviour is thus easier to characterise. The flat evolution of the Mean Square Displacement (MSD) of sodium in different regions (whose average MSD’s are also shown in Fig. 10 Fig. 9 Ring size distributions (number of rings per tetrahedral atom) and inner (R < 30 A) regions of the calculated in the surface (R > 30 A) nanoparticle. 12664 | J. Mater. Chem., 2011, 21, 12660–12667 Fig. 10 Mean Square Displacement of individual Na ions belonging to different regions of the nanoparticle, going from the R ¼ 0 core to the R > surface. The averaged MSD(Na) in each region (bold coloured lines) 30 A is also plotted. as coloured lines) confirms that no long-range diffusion occurs at room temperature on the simulated time window. However, the individual MSD curves denote a small but increasing fraction of more mobile Na ions as we move away from the centre of the nanoparticle (going from the bottom to the top panels in Fig. 10). Direct inspection of the MD trajectories of these atoms shows that their dynamics can be described as either a single jump or a shuttling motion between two adjacent sites. These local hops appear more frequently for Na atoms located close to the surface, as shown by the traces of the individual MD trajectories plotted in Fig. 11, only for the Na ions located near the centre and near the edge of the nanoparticle. The figure highlights small groups of adjacent Na ions on the surface whose red traces are generally more expanded. Because the migration of mobile Na atoms in bulk BGs involves correlated hops, in a vacancy-like Fig. 11 3D traces of the individual MD trajectories of Na cations located near the centre (blue) and near the surface (red) of the nanoparticle. Atoms found in between these two regions are not shown for clarity. Note that the red traces which apparently overlap with the blue ones in the centre actually mark the trajectory of atoms located on the (longitudinally) opposite external side of the sphere. This journal is ª The Royal Society of Chemistry 2011 mechanism,42 it is plausible to come across small aggregates of more mobile ions on the nanoparticle surface. Discussion Table 1 reports the concentrations of specific sites found on the surface of the nanoparticle and the flat surface.21 In general, the BG nanoparticle shows a rather regular structure, with surface The equilibrated effects mostly limited to the external 5–10 A. nanoparticle does not contain any structural defect: in particular, neither under-coordinate Si defects (Si3c) nor two-membered (2M) rings, which were found, albeit in small concentrations, on the flat 45S5 Bioglass surface,21,25 are present. This suggests that these specific defects do not contribute to the special bioreactivity of nanosized bioglasses: the main surface effects turn out to involve: (i) high Na+/Ca2+ and Si(Q1)/Si(Q3) ratios; (ii) a relevant fraction of three-membered rings; and (iii) enhanced mobility of Na+. In common with the flat surface, the dry surface of the ascreated BG nanoparticle is significantly enriched in sodium and depleted in calcium cation: Fig. 2 denotes a significant population of sodium atoms extending through the boundaries of the nanoparticle, accompanied by a corresponding slight but constantly lower sodium density in the internal regions of the nanoparticle, compared to the bulk glass (Fig. 4). We have shown that the coordination shell of these exposed sodium atoms is significantly depleted of oxygen atoms, compared to their pseudo-octahedral coordination in the bulk (Fig. 6). The marked under-coordination of exposed Na+ ions will enhance their Lewis acidity; combined with the marked Ca2+ depletion at the nanoparticle surface (Table 1 and Fig. 2), one can thus expect that once the glass particle is immersed in an aqueous medium, the initial interface will be dominated by Na+–water interactions. Because high bioactivity is associated with (and, to a large extent, depends on) a fast initial dissolution of the glass once immersed in a physiological medium,1 these observations suggest that a high Na+/Ca2+ ratio is an important feature of as-created BG surfaces. Whereas this feature applies to both nanosized and larger BG samples,21 Table 1 shows that the Na+/Ca2+ ratio at the BG nanoparticle surface (4.29) is even larger than that measured at the flat surface (3.04), with both being much larger than the bulk ratio of 1.81. This could represent an important effect of the reduced size, which could contribute to a faster initial dissolution of the nanoparticle. Experimental and simulation data have revealed a large excess of non-bridging oxygen and alkali ions on the freshly created surface of silicate glasses, compared to the bulk.43–48 It has been proposed that the increased alkali fraction shields the negative charge of the excess NBOs created upon exposure of a fresh surface, which is thus stabilized by the relocation of mobile alkali cations in the outermost regions.43,48 This appears to be the driving force behind sodium enrichment of the BG surface as well: Fig. 12 (bottom panel) highlights a net increase in the NBO : BO fraction in the surface region, with a corresponding increase in the number of NBO per Si in the same region, also accompanied by a decrease in the NBO : Na ratio (top panel of Fig. 12). In mixed alkali–alkaline earth silicate glasses, sodium and potassium cations showed a much stronger tendency to transfer to the surface compared to calcium.46–48 Both thermodynamic and kinetic factors appear to play a role in this effect: on one hand, the more mobile alkali cations can react more rapidly to the sudden change in the local field created by the exposure of the surface;46 on the other hand, a lower free energy environment has been shown to favour the preferential migration of sodium to the surface.48 Similar effects are presumably involved also in this case: Na cations are much more mobile than Ca in BGs.42 Moreover, despite their similar size, the different charge results in a higher field strength for Ca2+ than Na+ and in a higher site selectivity. Previous MD simulations42 have shown that Ca ions enforce stricter requirements on their local environment in BGs, whereas Na ions are more flexible in this respect, and can more easily accept lower coordination numbers than those they normally have in the bulk. Na is thus more stable in a non bulk-like environment with a decreasing density of O neighbours, such as that found on the nanoparticle surface. Even though no defects are present, the silicate network in the surface region of the nanoparticle appears significantly perturbed with respect to the bulk, both in terms of inter-tetrahedral angles (Fig. 7) and of ring sizes (Fig. 8 and 9). The dominance of threemembered rings, in particular, is important: exposed small rings Table 1 Concentration (nm3) of ionic sites found on the 45S5 BG nanoparticle surface and on the corresponding flat surfacea Site Flat surface21 Nanoparticle Na+ Ca2+ Si3c NBO 2MR 3MR 7.0 2.3 0.13 11.75 0.17 0.58 7.3 1.7 0 11.9 0 0.32 a In order to directly compare the site density with that calculated for the thick spherical shell located flat surface,21 we have considered the 4 A (see Fig. 2). between R ¼ 30 and R ¼ 34 A This journal is ª The Royal Society of Chemistry 2011 Fig. 12 (Bottom panel) NBO and BO relative fractions; (top panel) NBO : Si and NBO : Na ratios, as a function of the distance from the nanoparticle centre of mass. J. Mater. Chem., 2011, 21, 12660–12667 | 12665 have been associated with bioactivity, as they can represent nucleation sites for the deposition of calcium phosphates (CaP) in an aqueous environment at the conditions (neutral pH) found in a physiological medium.49,50 The relative stability of these sites on hydrated BG surfaces, highlighted by previous simulations,17,25,26 could in fact enable a fraction of the small rings initially present on the dry surface to resist hydrolytic opening upon contact with moisture and thus remain available to support CaP nucleation. Even though the low connectivity of the 45S5 silicate network reduces the density of rings that can be formed throughout the structure (compared to higher-silica compositions), the more marked presence of small rings in the external regions of the nanoparticle can thus represent another key factors in its bioreactivity.25,21 Previous simulations21 had highlighted how the effective structural relaxation is able to restore the bulk-like network connectivity to a large extent on the flat BG surface, which did not differ significantly from the bulk in this respect. On the other hand, Fig. 5 and 13 show that the nanoparticle surface, while maintaining similar amounts of other Qn species, has a higher Q1/ Q3 ratio, and a correspondingly lower network connectivity, than the bulk and flat surface. This is another potentially critical factor leading to a faster release of soluble silica fragments from the nanoparticle into the contact medium and thus accelerating its dissolution and the release of ionic products which are deemed essential to trigger tissue regeneration.6 The higher mobility of Na cations at the surface, compared to the core, reflects the reduced number of oxygen atoms found in their coordination shell: previous simulations have shown that the diffusive hops of sodium between two sites in BGs involve intermediate species where a fraction of the Na–O interactions in the starting site has been lost.42 Due to the reduced Na–O coordination, the energetic cost of creating these intermediates will be reduced on the nanoparticle surface thus reducing the hopping barrier. Another factor to take into account is the reduced network density in the surface region: on one hand this could facilitate the local structural distortions needed to accommodate cations hopping to a new site,42 but on the other hand the lower density could also result in a reduced number of new favourable sites able to receive migrating Na. Additional simulations sampling much longer time scales would be needed to quantitatively address this important issue: the present results only give some indications that cation migration could be enhanced on the surface of a BG nanoparticle, which could favourably affect the cation release in solution. Conclusions The present simulations have analysed in detail the structure of an isolated, dry Bioglass nanoparticle. Upon immersion in an aqueous physiological medium, the surface will adsorb and react with water, and the hydrolysis will start the dissolution of the glass.1 Before explicitly studying these key processes, it is vital to understand the main features of the non-hydrated particle: because glass degradation will not proceed to any significant extent before the particle is set in a liquid environment, focusing on its dry form represents a suitable source of information and a reproducible reference to start exploring the properties of these systems (in the same spirit of other simulations of isolated/in vacuo nanoparticles28–33), which will be extended in the future with simulations of the nanoparticle/water interface. The analysis shows that the nanometre size maintains and enhances some of the properties of the BG bulk and flat surface most beneficial for the bioactive behaviour. The most important one is probably the high fragmentation of the silicate network: the network connectivity further decreases on the nanoparticle surface, with a Q1/Q3 ratio higher than in the bulk and in the flat surface, which could promote a faster release of soluble silica species (a key effect for the tissue-regeneration properties of these materials6) from nanosized BG particles. An important role is most likely played by three-membered silicate rings present in a higher fraction on the nanoparticle surface than in the bulk and whose association with the tissue-bonding bioreactivity as nucleation sites for calcium phosphate has been previously proposed.49 Finally, compared to the flat surface, the nanoparticle denotes an even more marked enrichment in Na+ ions with reduced oxygen coordination and (consequently) higher mobility: these features will accelerate the initial stages of the dissolution process, which involve exchange and release of these ions in the surrounding physiological medium. In addition and combined with the higher surface area, these specific effects could all contribute to the enhanced bioreactivity of sub-micron and nanosized Bioglass particles. In order to assess their relative weight, further simulations of the explicit interface between the nanoparticle and an aqueous medium are in progress. Acknowledgements The author would like to acknowledge financial support from Royal Society (University Research Fellowship). Dr K. Okhotnikov (Stockholm University) is acknowledged for prompting the modification of the 3-body function in the force field. Computer resources on the HECToR national high-performance computing service were provided through the EPSRC-funded Materials Chemistry Consortium (EP/F067496). Notes and references n 19 21 Fig. 13 Comparison of Q distributions of bulk, flat surface surface region). nanoparticle surface (averaged over the R > 30 A 12666 | J. Mater. Chem., 2011, 21, 12660–12667 and 1 L. L. Hench, J. Am. Ceram. Soc., 1998, 81, 1705. 2 I. Kinnunen, K. Aitasalo, M. Pollonen and M. Varpula, J. Craniomaxillofac. Surg., 2000, 28, 229. 3 J. Wilson and S. B. Low, J. Appl. Biomater., 1992, 3, 123. This journal is ª The Royal Society of Chemistry 2011 4 I. Allan, H. Newsam and M. Wilson, Biomaterials, 2001, 22, 1683. 5 M. J. Peltola, K. M. J. Aitasalo, A. J. Aho, T. Tirri and J. T. K. Suonp€ a€ a, J. Oral Maxillofacial Surg., 2008, 66, 1699. 6 I. D. Xynos, A. J. Edgar, L. D. K. Buttery, L. L. Hench and J. M. Polak, J. Biomed. Mater. Res., 2001, 55, 151. 7 L. L. Hench and J. M. Polak, Science, 2002, 295, 1014. 8 R. A. Gittens, T. McLachlan, R. Olivares-Navarrete, Y. Cai, S. Berner, R. Tannenbaum, Z. Schwartz, K. H. Sandhage and B. D. Boyan, Biomaterials, 2011, 32, 3395. 9 G. Mendonca, D. B. S. Mendonca, L. G. P. Simoes, A. L. Araujo, E. R. Leite, W. R. Duarte, F. J. L. Aragao and L. F. Cooper, Biomaterials, 2009, 30, 4053. 10 E. Palin, H. Liu and T. J. Webster, Nanotechnology, 2005, 16, 1828. 11 S. Okada, A. Nagai, Y. Oaki, J. Komotori and H. Imai, Acta Biomater., 2011, 7, 1290. 12 S. K. Misra, D. Mohn, T. J. Brunner, W. J. Stark, S. E. Philip, I. Roy, V. Salih, J. C. Knowles and A. R. Boccaccini, Biomaterials, 2008, 29, 1750. 13 M. Vollenweider, T. J. Brunner, S. Knecht, R. N. Grass, M. Zehnder, T. Imfeld and W. J. Stark, Acta Biomater., 2007, 3, 936. 14 V. Mortazavi, M. Mehdikhani Nahrkhalaji, M. H. Fathi, S. B. Mousavi and B. N. Esfahani, J. Biomed. Mater. Res., Part A, 2010, 94, 160. 15 S. Labbaf, J. R. Jones, O. Tsigkou, K. H. M€ uller, M. M. Stevens and A. E. Porter, Biomaterials, 2011, 32, 1010. 16 S. T. Bromley, I. P. R. Moreira, K. M. Neyman and F. Illas, Chem. Soc. Rev., 2009, 38, 2657. 17 A. Tilocca, J. Mater. Chem., 2010, 20, 6848. 18 A. Tilocca, A. N. Cormack and N. H. de Leeuw, Chem. Mater., 2007, 19, 95. 19 A. Tilocca, J. Chem. Phys., 2008, 129, 084504. 20 A. Tilocca, A. N. Cormack and N. H. de Leeuw, Faraday Discuss., 2007, 136, 45. 21 A. Tilocca and A. N. Cormack, Langmuir, 2010, 26, 545. 22 J. K. Christie and A. Tilocca, Chem. Mater., 2010, 22, 3725. 23 A. Tilocca, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 224202. 24 J. K. Christie, A. Pedone, M. C. Menziani and A. Tilocca, J. Phys. Chem. B, 2011, 115, 2038. 25 A. Tilocca and A. N. Cormack, J. Phys. Chem. C, 2008, 112, 11936. This journal is ª The Royal Society of Chemistry 2011 26 A. Tilocca and A. N. Cormack, ACS Appl. Mater. Interfaces, 2009, 1, 1324. 27 H. Zhang, B. Gilbert, F. Huang and J. F. Banfield, Nature, 2003, 424, 1025. 28 A. Roder, W. Kob and K. Binder, J. Chem. Phys., 2001, 114, 7602. 29 V. Van Hoang, J. Phys. Chem. B, 2007, 111, 12649. 30 T. X. T. Sayle, S. C. Parker and D. C. Sayle, Chem. Commun., 2004, 2438. 31 D. Spagnoli, J. C. Banfield and S. C. Parker, J. Phys. Chem. C, 2008, 112, 14731. 32 V. N. Koparde and P. T. Cummings, J. Phys. Chem. B, 2005, 109, 24280. 33 I. V. Schweigert, K. E. J. Lehtinen, M. J. Carrier and M. R. Zachariah, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 235410. 34 X. Chen, C. Guo and N. Zhao, Appl. Surf. Sci., 2008, 255, 466. 35 S. Lin, C. Ionescu, K. J. Pike, M. E. Smith and J. R. Jones, J. Mater. Chem., 2009, 19, 1276. 36 W. Smith and T. R. Forester, J. Mol. Graphics, 1996, 14, 136. 37 A. Tilocca, N. H. de Leeuw and A. N. Cormack, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 104209. 38 P. J. Mitchell and D. J. Fincham, J. Phys.: Condens. Matter, 1993, 5, 1031. 39 V. Van Hoang and B. T. H. Khanh, J. Phys.: Condens. Matter, 2009, 21, 075103. 40 C. C. Lin, L. C. Huang and P. Shen, J. Non-Cryst. Solids, 2005, 351, 3195. 41 K. Vollmayr, W. Kob and K. Binder, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 15808. 42 A. Tilocca, J. Chem. Phys., 2010, 133, 014701. 43 J. J. Kelso, C. G. Pantano and S. H. Garofalini, Surf. Sci., 1983, 134, L543. 44 S. H. Garofalini and S. M. Levine, J. Am. Ceram. Soc., 1985, 68, 376. 45 D. M. Zirl and S. H. Garofalini, J. Am. Ceram. Soc., 1992, 75, 2353. 46 O. Gedeon and J. Zemek, J. Non-Cryst. Solids, 2003, 320, 177. 47 A. Abbas, J. M. Delaye, D. Chaleb and G. Calas, J. Non-Cryst. Solids, 2003, 315, 187. 48 L. R. Corrales and J. Ju, J. Am. Ceram. Soc., 2006, 89, 36. 49 N. Sahai and M. Anseau, Biomaterials, 2005, 26, 5763. 50 V. Bolis, C. Busco, V. Aina, C. Morterra and P. Ugliengo, J. Phys. Chem. C, 2008, 112, 16879. J. Mater. Chem., 2011, 21, 12660–12667 | 12667