Coalescence Model for Crumpled Globules Formed in Polymer Collapse Please share
advertisement

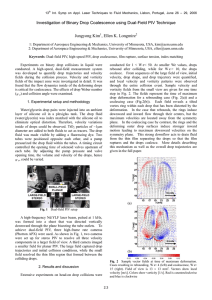
Coalescence Model for Crumpled Globules Formed in Polymer Collapse The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Bunin, Guy, and Mehran Kardar. "Coalescence Model for Crumpled Globules Formed in Polymer Collapse." Phys. Rev. Lett. 115, 088303 (August 2015). © 2015 American Physical Society As Published http://dx.doi.org/10.1103/PhysRevLett.115.088303 Publisher American Physical Society Version Final published version Accessed Thu May 26 18:38:13 EDT 2016 Citable Link http://hdl.handle.net/1721.1/98178 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Detailed Terms PRL 115, 088303 (2015) PHYSICAL REVIEW LETTERS week ending 21 AUGUST 2015 Coalescence Model for Crumpled Globules Formed in Polymer Collapse Guy Bunin and Mehran Kardar Department of Physics, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA (Received 29 March 2015; published 20 August 2015) The rapid collapse of a polymer, due to external forces or changes in solvent, yields a long-lived “crumpled globule.” The conjectured fractal structure shaped by hierarchical collapse dynamics has proved difficult to establish, even with large simulations. To unravel this puzzle, we study a coarse-grained model of in-falling spherical blobs that coalesce upon contact. Distances between pairs of monomers are assigned upon their initial coalescence, and do not “equilibrate” subsequently. Surprisingly, the model reproduces quantitatively the dependence of distance on segment length, suggesting that the slow approach to scaling is related to the wide distribution of blob sizes. DOI: 10.1103/PhysRevLett.115.088303 PACS numbers: 82.35.-x, 68.43.Jk, 82.35.Pq The rapid collapse of a polymer into a dense globule is a long-standing problem [1–12]. Such a collapse may be triggered by changes in solvent quality, causing the polymer to reduce its solvent–exposed surface area by forming a dense globule. A polymer may also be condensed by active forces, as in the rearrangement of DNA by proteins in the cell nucleus [2]. The rapid collapse does not allow sufficient time for formation of topological entanglements, which abound in an equilibrated compact globule [1–4,6,13]. It is suggested [1] that during collapse segments of the polymer initially condense to spherelike “blobs,” which coarsen upon contact to form larger blobs. At any given time during the process the state is then assumed to be characterized by a single length scale [1,7–9], e.g., the typical size of the blobs or the width of the tube connecting the blobs [see, e.g., Fig. 1(a) [14]]. A central assumption is that when two blobs coalesce they remain more or less segregated within the newly formed structure. This is due to the slow relaxation processes within blobs, and due to topological constraints that prevent polymer segments forming the blobs from freely mixing—unlike a melt of independent polymer segments with open ends [1,15–20]. The final configuration is thus predicted to be a constant density, self-similar, hierarchical structure, known as the “crumpled globule” or “fractal globule” [1–6,11]. The end-to-end distance rm of segments of length m in the resulting globule is predicted to scale as rm ∼ m1=d in d space dimensions (throughout the Letter d ¼ 3). This is in contrast to the equilibrium state reached at much later times [10], where rm ∼ m1=2 for small m, saturating at the globule size rmax ¼ N 1=d , where N is the length of the polymer [21]. These predictions have been tested in several simulations of polymer compaction [2,4–6,11], which generally confirm that the rapidly collapsed state is not entangled, and is indeed different from the equilibrium globule. However, they do not agree upon its fractal nature. In particular, the expected scaling rm ∼ m1=d has not been conclusively confirmed, even with the largest size simulations (recently 0031-9007=15=115(8)=088303(5) extended to polymers of up to 250 000 monomers [6,22]). This implies either a very large crossover scale before fractal behavior is clearly manifested, or else that the collapsed state is not strictly self-similar. The large finite-size effects are at least partly explained by partial mixing of short polymer segments, for which the topological constraints do not apply [1,15–20,22]. More generally, various protocols for constructing crumpled globules have been suggested [2,4–6,12,22], and it is not clear if the different procedures yield the same state. Settling these issues seems to require even larger simulations, or new theoretical insights. FIG. 1 (color online). The coalescence model combines a simulation of coalescing spherical drops (b),(d),(e), with estimates for distances between monomer pairs in the final structure. MD simulations (a),(c) with initial conditions identical to (b),(d) are shown. Initial conditions are SAW for (a),(b), RW for (c),(d), and 1D for (e). All panels show parts of the full systems. Colors show the position of monomers along the polymer, or the average position for drops. 088303-1 © 2015 American Physical Society PRL 115, 088303 (2015) PHYSICAL REVIEW LETTERS To this end, we propose a coarse-grained model for the crumpled globule formed by polymer collapse, focusing here on active compression. The evolution of blobs is modeled by drop coalescence, while distances between monomers in the final crumpled state are assigned without keeping track of their individual positions. This builds upon the topological segregation of blobs, and the slowness of subsequent internal rearrangements, and should apply at scales beyond those at which these conditions hold. The results highlight a different, dynamical source of slow convergence to a self-similar state, and draw connections to the physics of coagulation and drop coalescence. In particular, the assumption of a single length scale during the collapse may need to be amended, at least when the collapse is due to active compression. In the spirit of the blob theory [10], the basic entity in our model is a “drop,” an abstraction of the blob. A drop is a uniform density sphere that contains a subset of the monomers from the original polymer, without explicit position assignments within the drop. Initially, every monomer is a drop. Drops move (as detailed below) and coalesce into larger drops immediately upon collision, i.e., as soon as the spheres overlap in space. Drops do not break into smaller drops. Coalescence conserves volume, drops of volume v1 ; v2 forming a drop of volume v1 þ v2 . (With the monomer volume set to unity, the drop volume equals the number of monomers it contains.) The new drop is centered at the center of mass of the coalesced drops. The process terminates when all drops have merged to a single sphere. Example coalescence runs are compared with corresponding molecular dynamics (MD) runs in Fig. 1. Our main interest is the structure of the final collapsed state, as captured by the distances frm g. However, in the coarse-grained drop coalescence model we do not keep track of the internal structure of a drop. Instead we assign distance estimates to all pairs of monomers within a drop. Guided by the slow internal rearrangements in the blob picture, a distance estimate is assigned when a pair of monomers first comes together in a coalescence event, and is not changed thereafter. Upon coalescence of drops of volumes v1 and v2 , v1 × v2 pairs of monomers, one from each drop, are assigned a distance estimate. This distance is of order ðv1 þ v2 Þ1=d, the linear size of the new drop, which can be sampled from any distribution. As the results are highly insensitive to this choice, we simply assign all v1 v2 pairs the “distance” ðv1 þ v2 Þ1=d . The distance estimates satisfy triangle inequalities, and importantly are ultrametric, the tree structure reflecting the hierarchy of the collapse process [23]. At the end, when all monomers belong to a single drop, all monomer pairs have been assigned a distance estimate. To fully define the model, it remains to prescribe the drops’ motion between coalescence events, as well as the initial conditions. Here, we focus on a simple dynamics for active compression in which the drop velocity is week ending 21 AUGUST 2015 proportional to its distance from the origin, i.e., dR~ α =dt ¼ −R~ α , where R~ α is the position of the drop center. This corresponds to overdamped motion in a harmonic potential. It can also be viewed as a uniform compression of space, as the distance R~ αβ ¼ R~ α − R~ β between two drops that have not yet coalesced changes as dR~ αβ =dt ¼ −R~ αβ , so all relative distances shrink by the same factor per unit time. Apart from coalescence, there is no additional interaction between drops. In particular, we do not impose polymeric bond interactions between sequential monomers. The initial conditions can be any configuration of the monomer positions. Here, we use random walk (RW), selfavoiding walk (SAW), and “one-dimensional” (1D) initial conditions. For the latter, monomers are positioned along a line as Rj ¼ jx̂ þ ηj , where x̂ is the unit vector in the x direction, and the ηj are independent Gaussian variables in d dimensions with hηj i ¼ 0, hη2j i ¼ c2 , and c a number of the order of the drop radius. Open polymers are used in all initial conditions. The SAW initial conditions are arguably the most natural for a polymer in a good solvent, but other initial conditions help in obtaining additional insight. In particular, 1D initial conditions are used only in the coalescence process, as the strong unidirectional compression will cause large internal rearrangements after blobs have formed within the MD. The coalescence model is compared with MD simulations where the polymer is composed of monomers with standard polymeric bond attraction forces Fnn , and repulsion Frep between all monomer pairs, see the Supplemental Material [24]. Together with the same external force as in the coalescence model, the monomers evolve as dRi =dt ¼ −Ri þ Fnn þ Frep . The MD runs are terminated when the polymer size stops decreasing. In both the coalescence and MD simulations we do not add noise to the dynamics, as the entire collapse process is fast (of order ln N, see below). Noise may further increase rearrangement processes in the MD simulations, which we try to minimize here. A longlived, metastable state is obtained, due to the time-scale separation between the fast collapse process and further rearrangements that are much slower. (As noted above, other crumpled globule metastable states may also exist [4–6,22], at later times.) Now, let dcoal be the distance estimate between monoij mers i; j in the coalescence model, and dMD ij ¼ ∥Ri − Rj ∥ the corresponding distance between the i; j monomers in the final state of the MD run. In what follows we apply MD the same analysis to dcoal ij , dij , referred jointly as dij . We define the ffi normalized end-to-end distance rm ≡ qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi C hd2ij iji−jj¼m , where the average h iji−jj¼m is over all monomer pairs separated along the polymer by m monomers, as well as over repeated runs of the two models, with initial conditions resampled for each run. The normalization C is chosen so that 088303-2 PRL 115, 088303 (2015) X m PHYSICAL REVIEW LETTERS N−m r2 ¼ 1. NðN − 1Þ=2 m (N − m is simply the number of pairs that are m units apart in an open polymer of length N). In this way the MD and coalescence models can be compared without any fitting parameters. Figure 2(a) compares rm from the coalescence model with MD simulations for SAW initial conditions. MD results are shown for different polymer lengths N. Strikingly, the MD and coalescence models agree quantitatively over the entire range where the MD results of different polymer sizes collapse, for 10−3 ≲ m=N ≤ 1. In the small m regime, where MD simulations with different N do not collapse, they show a different behavior (consistent with a random walk) not present in the coalescence model. This demonstrates that the coalescence process indeed represents a coarse-grained model, capturing large-scale behavior while incorporating different microscopic details. The absence of polymeric bonds in the coalescence model removes short-scale constraints that (as supported by the MD simulations) do not effect large-scale properties through the distance assignment. The m1=3 trend line in Fig. 2(a) indicates that the expected scaling is not present at the tested system sizes N ≤ 25 × 103 . To more carefully assess self-similarity, we study finite-size scaling in the standard form rm ¼ ðm=NÞ1=3 fðm=NÞ; ð1Þ expected to hold for 1 ≪ m. (The additional N −1=3 reflects the choice of normalization.) The scaling function fðxÞ should go to a constant for x → 0, such that rm ∝ ðm=NÞ1=3 for 1 ≪ m ≪ N. Data for both MD and coalescence models, for different initial conditions and system sizes, are summarized in Fig. 2. The difference between MD and coalescence results for the RW initial conditions is larger FIG. 2 (color online). Comparison of the coalescence model and MD results. (a) Normalized end-to-end distance rm , with SAW initial conditions. Inset: rm =ðm=NÞ1=3 for the same rm . (b) rm =ðm=NÞ1=3 for RW initial conditions. (c) rm =ðm=NÞ1=3 for the coalescence model with different initial conditions and system sizes N. week ending 21 AUGUST 2015 than for the SAW. Importantly, for both SAW and RW initial conditions, and for both models, the expected condition, fðm=N → 0Þ ¼ const, is not seen clearly for tested values of N; in all cases, the maximum of fðm=NÞ ¼ rm =ðm=NÞ1=3 appears to grow with increasing N. This effect is almost absent for the 1D initial conditions, and largest in the RW case. Since the coalescence model includes only a minimal set of microscopic details, it is surprising to see such a slow approach to the expected scaling behavior. Understanding this trend in the coalescence model should provide insights into the more complex case of the collapsing polymer. While simplified, a full understanding of the coalescence model—including the distribution of distance estimates assigned as a function of the time t and separation m—is still difficult. Fortunately, some insights regarding scaling (or lack thereof) can be gained by examining the distribution of drop volumes as a function of time, even without making reference to the distance estimate assignments. In particular, the volume distributions already show a slow approach to scaling. Let ρt ðvÞ be the distribution ofR drop sizes at time t. Volume conservation implies that dvvρt ðvÞ ¼ N, while R dvρt ðvÞ is the number of drops at time t (averaged over repeated runs). Within the scaling picture, the dynamics of the collapse is described by a single typical drop size as a function of time, v̄ðtÞ, when 1 ≪ v̄ðtÞ ≪ N. For example, in the tube picture v̄1=d may be the thickness of the tube. In such a scaling regime, ρt ðvÞ should depend on time only through v̄ðtÞ as N v ρt ðvÞ ¼ : ð2Þ ϕ v̄ðtÞ v̄ðtÞ2 R The factor v̄−2 ensures that dvvρt ðvÞ remains constant. P 2 We measure P P 2the typical size via v̄ðtÞ ≡ h n vn i= n vn ¼ h n vn i=N, where the sums run over all drops at time t, and the average h i is over initial conditions. This definition, standard in the drop coalescence and coagulation literature [28–31], addresses possible divergences of ρt ðvÞ at small volumes (see below). To test Eq. (2), we plot ϕt ðvÞ ≡ N −1 v̄ðtÞ2 ρt ðvÞ against the normalized volume v=v̄ðtÞ, and check for data collapse at different values of t. The distributions, depicted in Figs. 3(a) and 3(b) for 1D and RW initial conditions, respectively, are quite different. The one in Fig. 3(a) (1D initial conditions) is concentrated in the region 0.4 ≲ v=v̄ ≲ 1.6, and is strongly suppressed outside this interval. Thus, at any given time all volumes are of the same order, with a ratio of about 4 between the largest and smallest. The distributions at different times collapse nicely when plotted against v=v̄ðtÞ. In contrast, the distributions for RW initial conditions in Fig. 3(b) are very wide (note the log-log scale), with possibly a diverging tail at small volumes, v=v̄ ≪ 1. (The dashed line, x−1 , is 088303-3 PRL 115, 088303 (2015) PHYSICAL REVIEW LETTERS week ending 21 AUGUST 2015 is perhaps most similar to our model. There, the distribution of drop sizes is highly polydisperse when v̄ðtÞ (in our notation) grows exponentially in time [28]. Aggregating systems are commonly studied through the (mean-field) Smoluchowski equation [30,31]: Z Z 1 0 ∂ t ρðvÞ ¼ dv Jt;v0 ;v−v0 − dv0 J t;v0 ;v : ð3Þ 2 FIG. 3 (color online). Drop volume distributions in the coalescence model. (a) 1D initial conditions. Dashed line: sequential collisions theory. (b) RW initial conditions. Dashed line: x−1 trend line. (c) SAW initial conditions. (d) v̄ðtÞ for the three models. Dashed lines: ewt for different cases (see text). included as an indication of such potential divergence.) Moreover, the distributions fail to collapse in this tail. With SAW initial conditions [Fig. 3(c)] the results appear to be intermediate between the above two cases, with a distribution that is finite at v ¼ 0 (at least for tested N). The simulations in Figs. 3(a)–3(c) were carried out with N ¼ 2.5 × 104 , 5 × 104 , and 105 , respectively, to allow for better scaling in Figs. 3(b) and 3(c). The evolution of v̄ðtÞ is depicted in Fig. 3(d). For 1D and SAW initial conditions v̄ðtÞ ∝ ewt is a good fit at intermediate times, where w ¼ d=ðdν0 − 1Þ with ν0 describing ð0Þ the scaling rm ∼ mν0 in the initial condition. This form is explained as follows. At time t, segments of initial length v̄ðtÞ form drops of volume v̄ðtÞ, with diameter ½v̄ðtÞ1=d. The initial end-to-end distance of these segments scales as ½v̄ðtÞν0 . If this reduction in length follows the general exponential approach of free monomers [32], then ½v̄ðtÞν0 e−t ¼ ½v̄ðtÞ1=d , leading to the above relation [33]. For RW initial conditions the growth of v̄ðtÞ follows this form for a narrower interval, with wide crossover regions, probably related to the lack of scaling observed in Fig. 3(b). The difference between the three volume distributions in Fig. 3 can be directly observed in the coalescence and MD runs in Fig. 1; note especially the large number of small drops for RW initial conditions in Fig. 1(d). In the MD starting from the same initial conditions, these manifest as open segments of the polymer, alongside large condensed regions, see Fig. 1(c); the thickness of a putative “tube” is highly uneven. We now present theoretical approaches to analogous problems leading to the two very different volume distributions obtained in Figs. 3(a) and 3(b). Broad distributions, with power-law tails at small volumes, as in Fig. 3(b) for RW initial conditions, appear in related problems such as diffusion-limited aggregation [34], and drop coalescence [28,29]. Heterogeneous drop coalescence, where drops grow but no new drops are added, The first term is the change in density ρt ðvÞ due to creation of drops of size v, while the second term describes their removal due to coalescence. In the current context, this approximation postulates a rate Jt;v1 ;v2 ¼ K v1 ;v2 ρt ðv1 Þρt ðv2 Þ for collisions between drops of volumes v1 ; v2 , with a kernel K v1 ;v2 depending only on the volumes of the coalescing drops. Here, rather than explicitly constructing an approximate kernel, we refer to extensively studied scaling solutions of the Smoluchowski equation [30,31]. In particular, for homogeneous kernels such that K av1 ;av2 ¼ aλ K v1 ;v2 for a > 0, it is known that when v̄ðtÞ ∝ ewt , as in our case, the scaling function ϕðxÞ in Eq. (2) has the following properties: it is strongly (exponentially) decaying for x ≫ 1, and has a diverging powerlaw tail ϕðxÞ ∼ x−τ for 1 ≫ x, with 1 ≤ τ < 2 (the value of τ depends on the kernel). For example, the kernel K v1 ;v2 ¼ v1 þ 2 ffi admits an exact scaling solution ϕðxÞ ¼ pvffiffiffiffiffi ð1= 2π Þx−3=2 e−x=2 with v̄ ¼ e2t , so that τ ¼ 3=2. Interestingly, a slow approach to the asymptotic scaling is well documented for certain cases with such smallvolume tails [31,35–37]. The nature of this slow approach is still not well understood, and might be sensitive to the kernel form and initial conditions. It is intriguing to speculate on its relation to the present problem. In the case of 1D initial conditions in Fig. 3(a), essentially all drops at a given time have volumes of the order of v̄ðtÞ. Unlike RW initial conditions, all collisions here are sequential, i.e., between drops composed of adjacent segments along the polymer. Here, geometry matters: after a collision gaps are formed on both sides of the newly constructed drop, greatly reducing its probability of coalescing again before other drops have time to grow. Smaller drops leave smaller gaps, and have an increased probability of additional collisions. These effects are discussed in a quantitative way in the Supplemental Material [24]. An approximate evolution equation is derived that admits the scaling solution shown in Fig. 3(a) (dashed line), which strongly decays outside a narrow interval of v=v̄, just like the results from the full coalescence model. The coalescence model proposed here does away with several microscopic details present in the collapsing polymer that could delay the asymptotic approach to scaling. The lack of simple scaling in coalescence thus points to deeper problems with the simple model of hierarchical collapse, in particular in the assumption of a sharply 088303-4 PRL 115, 088303 (2015) PHYSICAL REVIEW LETTERS defined blob scale. The coalescence model is interesting in its own right; it is closely related to widely studied problems of coarsening of growing water drops, but differs in the initial (polymeric) distribution of droplets. As demonstrated, different such initial conditions lead to widely dissimilar probability distributions. Probing the role of fractal initial conditions in coalescence problems should thus be worthy of further exploration. We thank Bernard Derrida, Maxim Imakaev, Leonid Mirny, and Adam Nahum for valuable discussions. This research was supported by the NSF through Grant No. DMR-12-06323. G. B. acknowledges the support of the Pappalardo Fellowship in Physics. [1] A. Yu. Grosberg, S. K. Nechaev, and E. I. Shakhnovich, J. Phys. (Paris) 49, 2095 (1988). [2] E. Lieberman-Aiden et al., Science 326, 289 (2009). [3] L. A. Mirny, Chromosome Res. 19, 37 (2011). [4] R. D. Schram, G. T. Barkema, and H. Schiessel, J. Chem. Phys. 138, 224901(2013). [5] A. Chertovich and P. Kos, J. Chem. Phys. 141, 134903 (2014). [6] M. V. Tamm, L. I. Nazarov, A. A. Gavrilov, and A. V. Chertovich, Phys. Rev. Lett. 114, 178102 (2015). [7] E. Pitard and H. Orland, Europhys. Lett. 41, 467 (1998). [8] G. E. Crooks, B. Ostrovsky, and Y. Bar-Yam, Phys. Rev. E 60, 4559 (1999). [9] C. F. Abrams, N.-K. Lee, and S. P. Obukhov, Europhys. Lett. 59, 391 (2002). [10] P.-G. De Gennes, Scaling Concepts in Polymer Physics (Cornell University Press, Ithaca, 1979). [11] A. Lappala and E. M. Terentjev, Macromolecules 46, 1239 (2013). [12] J. Smrek and A. Yu. Grosberg, Physica (Amsterdam) 392A, 6375 (2013). [13] P. Virnau, Y. Kantor, and M. Kardar, J. Am. Chem. Soc. 127, 15102 (2005). [14] Unlike Fig. 1(a), collapse in a poor solvent leads to distinct spherical “pearls,” connected by tenuous stretched segments of the polymer [8,9]. [15] T. Sakaue, Phys. Rev. Lett. 106, 167802 (2011). week ending 21 AUGUST 2015 [16] M. Cates and J. Deutsch, J. Phys. (Paris) 47, 2121 (1986). [17] A. Rosa and R. Everaers, Phys. Rev. Lett. 112, 118302 (2014). [18] A. Yu. Grosberg, Soft Matter 10, 560 (2014). [19] J. D. Halverson, W. B. Lee, G. S. Grest, A. Y. Grosberg, and K. Kremer, J. Chem. Phys. 134, 204904 (2011). [20] T. Vettorel, A. Yu. Grosberg, and K. Kremer, Phys. Biol. 6, 025013 (2009). [21] Another widely studied property, the monomer contact probability as a function of polymeric separation [2,3], is not addressed here. [22] M. Imakaev, K. Tchourine, S. Nechaev, and L. Mirny, Soft Matter 11, 665 (2015). [23] V. A. Avetisov, L. Nazarov, S. K. Nechaev, and M. V. Tamm, Soft Matter 11, 1019 (2015). [24] See Supplemental Material at http://link.aps.org/ supplemental/10.1103/PhysRevLett.115.088303 for a detailed description of MD simulations and a theory for drop evolution on a line, which includes Refs. [25–27]. [25] G. S. Grest and K. Kremer, Phys. Rev. A 33, 3628 (1986); K. Kremer and G. S. Grest, J. Chem. Phys. 92, 5057 (1990). [26] B. Derrida, C. Godreche, and I. Yekutieli, Europhys. Lett. 12, 385 (1990). [27] B. Derrida, C. Godreche, and I. Yekutieli, Phys. Rev. A 44, 6241 (1991). [28] F. Family and P. Meakin, Phys. Rev. A 40, 3836 (1989). [29] F. Family and P. Meakin, Phys. Rev. Lett. 61, 428 (1988). [30] P. G. J. Van Dongen and M. H. Ernst, J. Stat. Phys. 50, 295 (1988). [31] F. Leyvraz, Phys. Rep. 383, 95 (2003). [32] In the absence of coalescence events, dRij =dr ¼ −Rij implies Rij ðtÞ ¼ Rij ð0Þe−t . [33] In terms of ξ ≡ e−t , the linear compression of space, w is a dynamic scaling exponent, v̄ ∼ ξw , similar to the one in coalescence models [28–31]. [34] E. K. O. Hellén, P. E. Salmi, and M. J. Alava, Europhys. Lett. 59, 186 (2002). [35] K. Kang, S. Redner, P. Meakin, and F. Leyvraz, Phys. Rev. A 33, 1171 (1986). [36] A. B. Boehm, C. Poor, and S. B. Grant, J. Phys. A 31. 9241 (1998). [37] J. Blaschke, T. Lapp, B. Hof, and J. Vollmer, Phys. Rev. Lett. 109, 068701 (2012). 088303-5