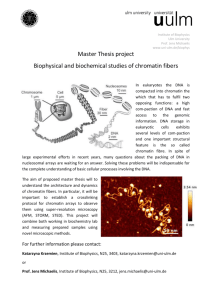
Crucial role of dynamic linker histone binding
advertisement

Published online 11 July 2012 Nucleic Acids Research, 2012, Vol. 40, No. 18 8803–8817 doi:10.1093/nar/gks600 Crucial role of dynamic linker histone binding and divalent ions for DNA accessibility and gene regulation revealed by mesoscale modeling of oligonucleosomes Rosana Collepardo-Guevara1 and Tamar Schlick1,2,* 1 Department of Chemistry, New York University, 100 Washington Square East, New York, NY 10003 and 2Courant Institute of Mathematical Sciences, New York University, 251 Mercer Street, New York, NY 10012, USA Received March 7, 2012; Revised May 15, 2012; Accepted May 28, 2012 ABSTRACT Monte Carlo simulations of a mesoscale model of oligonucleosomes are analyzed to examine the role of dynamic-linker histone (LH) binding/unbinding in high monovalent salt with divalent ions, and to further interpret noted chromatin fiber softening by dynamic LH in monovalent salt conditions. We find that divalent ions produce a fiber stiffening effect that competes with, but does not overshadow, the dramatic softening triggered by dynamic-LH behavior. Indeed, we find that in typical in vivo conditions, dynamic-LH binding/unbinding reduces fiber stiffening dramatically (by a factor of almost 5, as measured by the elasticity modulus) compared with rigidly fixed LH, and also the force needed to initiate chromatin unfolding, making it consistent with those of molecular motors. Our data also show that, during unfolding, divalent ions together with LHs induce linker-DNA bending and DNA–DNA repulsion screening, which guarantee formation of heteromorphic superbeads-on-a-string structures that combine regions of loose and compact fiber independently of the characteristics of the LH–core bond. These structures might be important for gene regulation as they expose regions of the DNA selectively. Dynamic control of LH binding/unbinding, either globally or locally, in the presence of divalent ions, might constitute a mechanism for regulation of gene expression. INTRODUCTION Understanding how chromatin fibers fold and unfold as well as details of their structure and dynamics on a range of spatial and temporal scales is important for interpreting fundamental template-directed processes such as DNA replication, transcription, and repair. Indeed, the tightly packed complex array of DNA with histone proteins undergoes continuous chemical modification and dynamic association of proteins, such as linker histones (LHs), which control the accessibility of the genetic material. Together with internal variations, such as the nucleosome repeat length (NRL) associated with the basic repeating unit of DNA wrapped around the nucleosome core (147 bp) plus the variable linker-DNA length, and external factors such as the ionic environment, these changes determine the shape of the chromatin fiber at different stages of the cell cycle. Although it is clear that LHs are essential for understanding chromatin compaction (1–3), many questions regarding the structure and behavior of LH, and its role in gene regulation remain open [for a thorough review, see (4)]. We have been intrigued by suggestions that the dynamic binding/unbinding of LHs in vivo (5,6) and in vitro (7) may function to alter chromatin organization by generating complex interaction networks that impart global changes from local rearrangements (5,8–11). Such networks are plausible because LHs, sandwiched between entering and exiting linker DNA, lead to LH/linker DNA association called DNA stems that rigidify chromatin; conversely, LH dissociation can disrupt these networks and trigger unfolding rearrangements. Growing evidence points to a key role for LH dynamic binding during regulation of chromatin structure and gene expression (12); that is in vivo LH dynamic binding behavior might allow remodeling factors to bind to temporarily available nucleosomal sites and induce chromatin structural changes to either activate or repress genes (13). In fact, lower H1 mobility, resulting for instance from LH dephosphorylation, is suggested to maintain chromatin in a compact structure and shut down gene expression, *To whom correspondence should be addressed. Tel: +1 212 998 3596; Fax: +1 212 995 4152; Email: schlick@nyu.edu ß The Author(s) 2012. Published by Oxford University Press. This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/ by-nc/3.0), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. 8804 Nucleic Acids Research, 2012, Vol. 40, No. 18 while enhanced H1 mobility is linked to undifferentiated cells that require flexible chromatin to enable transcription (14); this is consistent with the presence of LHs with lower mobility causing inhibition of stem cell differentiation (15) and higher LH mobility observed in pluripotent stem cells (14). Furthermore, experiments (14,16) and our previous modeling of chromatin in monovalent salt (17) have linked increased mobility of LH (e.g. induced by phosphorylation of LH or acetylation of core histones) to facile chromatin fiber opening. Our work (17) also suggested that fast and slow LH dynamic binding populations, found simultaneously in vivo (10,18), cooperate to promote chromatin unfolding with selective DNA exposure at low forces. However, it remains unclear how such local exchange processes occur and how they affect chromatin organization and accessibility at physiological ionic conditions (i.e. with divalent ions). Divalent ions enhance DNA compaction as they bring linker-DNA segments closer to one another by screening their electrostatic repulsion (19). Modeling and experiments have revealed that chromatin fibers with both LHs and magnesium ions adopt a compact heteromorphic architecture that combines straight and bent DNA linkers (19). Although it is clear that divalent ions favor structural variations in compact chromatin, it is unknown what the combined effects of divalent ions and LH dynamic binding/unbinding are during chromatin’s fiber opening. Fiber heteromorphicity in divalent conditions has led us to speculate that together divalent ions and dynamic-LH binding/unbinding might serve a fundamental function for chromatin unfolding: increasing fiber fluidity by accommodating more easily structural perturbations and multiple fiber forms. Recent chromatin applications, by atomic force microscopy (20), optical tweezers (21–25), and magnetic tweezers (26), measured the fiber’s stretching response using force– extension curves and provided information on chromatin’s unfolding behavior. Based on optical tweezers experiments of medium-NRL chromatin, the Bustamante group offered evidence for an irregular zigzag structure stabilized by nucleosome–nucleosome interactions <7 pN (21,27). The van Noort group studied chromatin with two different NRLs (short = 167 bp and medium = 197 bp) subject to stretching at physiological salt with magnesium ions and suggested that the response of the short-NRL array is consistent with a zigzag organization, while that of the medium-NRL arrays supports a solenoid topology (26). The latter study also showed that the presence of LHs does not change the stiffness of medium-NRL fibers when magnesium ions are present. Here, we develop and apply a computational approach to simulate dynamic-LH binding/unbinding under divalent salt conditions (see ‘Materials and Methods’ section) and study the stretching response of single chromatin fibers as a function of LH concentration and mobility. Our simulation approach is grounded on a mesoscale model derived from experimental data of the chromatin fiber and atomistic properties of its components. This model combines different coarse-grained strategies for the nucleosome core, histone tails, DNA, and LHs (Figure 1). Such a modeling strategy allows us Figure 1. Mesoscale model of the basic chromatin building block including: nucleosome core surface with wrapped DNA modeled as an irregularly shaped rigid body with uniformly distributed charges; linker DNA treated using the discrete worm-like chain model; and histone tails and linker histones coarse-grained as bead models. The solvent around the oligonucleosome is treated implicitly as a continuum. The screening of electrostatic interactions due to monovalent ions in solution (0.15 M NaCl) is treated using a Debye–Hückel potential. A low concentration of magnesium ions is considered by a firstorder approximation as developed in (32), by reducing the DNA bending persistence length and allowing the DNA beads to nearly touch one another. to capture the essential physics of the chromatin fiber (like its electrostatics, DNA and nucleosome mechanics, structural irregularity, and histone-tail flexibility), and at the same time to reduce the system dimensionality markedly and sample the chromatin conformational space exhaustively. This approach has been developed, validated against experiments, and refined over the past few years, as detailed extensively in (28–34). Our results suggest a dramatic softening effect of the chromatin fiber when LHs associate/dissociate dynamically compared with fixed (or static) LH species; this produces a stretching resistance compatible with unfolding at natural forces of molecular motors. Moreover, the detailed fiber configurations corresponding to each stage of the unfolding process reveal that divalent ions favor transitional states that combine regions of fully stretched fiber with prominent compact clusters or superbeads. These unusual ‘superbeads-on-a-string’ forms, observed experimentally at different cell types (35) or in the presence of low concentrations of magnesium ions (36), suggest mechanisms by which intrinsic and external factors, such as LH binding and divalent ions, lead to higher order fiber forms. Although related unfolding arrays emerged in our studies without divalent ions focusing on two binding/unbinding scenarios for LHs, the clumps were sparse (17). This work indicates that divalent ions are necessary to enhance nucleosome-cluster stability. Thus, LH mobility and divalent ions may introduce a combined regulatory mechanism for gene expression through a localized transient exposure of chromosomal DNA. Nucleic Acids Research, 2012, Vol. 40, No. 18 8805 MATERIALS AND METHODS Because of the intrinsic complexity of the chromatin fiber in solution (i.e. the many atoms that constitute it, the multiple time and length scales involved in its dynamics, and the interplay of factors that intervene in its structural reorganization), simulating chromatin’s folding/unfolding represents a grand challenge for bimolecular modelers. On the one hand, the accuracy of atomistic models is desirable to capture the details of the chemical interactions, but on the other hand an all-atom study of oligonucleosomes is prohibitive due to the massive system dimensionality (e.g. a 50-nucleosome array without solvent already contains >1 million atoms). Coarse-graining represents one of the only alternatives to model chromatin, and indeed several other large-scale bimolecular systems [for an excellent review, see (37)], because it dramatically reduces the system size by averaging out many effects (e.g. protein/DNA sequence effects, hydrogen bonding, atomistic fluctuations, and solvation), while simultaneously maintains the essential physical and chemical information required to analyze its structural organization. This dimensionality reduction is a major advantage as it allows extensive sampling of the coarse-grained phase space. Coarse-grained modeling has been useful to complement experimental analyses and help dissect the factors that control the structure and dynamics of the chromatin fiber (38). Among the existing chromatin coarse-grained models, those that treat the DNA and nucleosome by geometric descriptions have provided insight on the physical properties of the chromatin polymer (27,39–44), while more realistic approaches based on all-atom studies and/ or experimental data have dissected the influence of LHs, nucleosome interactions, and ionic environment on chromatin structure (17,19,32–34,45,46). Indeed, in silico stretching experiments have been instrumental for analyzing chromatin’s unfolding behavior and its implications for DNA accessibility (17,27,40,44,47). In particular, they have shown that chromatin’s unfolding response is consistent with an irregular zigzag architecture (27), that the persistence length of medium-NRL fibers is strongly affected by the excluded volume of the nucleosomes (47), that chromatin is much more resistant to stretching than to bending (40), that fiber stiffness decreases with the NRL and increases in the presence of rigid DNA stems (17,40), and that simulated conformations aid the analysis of chromatin force extension experiments (17,44). Analytic approaches have also provided information on the mechanical properties of the chromatin fiber (48–50). Here, we refine our mesoscale model of chromatin, which uses different coarse-grained interpretations for the different oligonucleosome components, to analyze the effect of LH dynamic binding during force-induced fiber unfolding. Mesoscale model Our model has been recently described in detail in (32,34), and below we summarize the main features of the strategies used to treat each oligonocleosome element (Figure 1). cores: the nucleosome protein core, excluding histone tails, with wrapped DNA is modeled as a rigid irregular body with 300 Debye– Hückel charges uniformly distributed on the nucleosome molecular surface. The charges are optimized to reproduce the full atom electric field around the nucleosome core by the discrete surface charge optimization (DiSCO) algorithm (28), which solves the complete nonlinear Poisson–Boltzmann equation. . Histone tails: we model the 10 histone tails protruding out of each core (the N-termini of each H2A, H2B, H3, and H4, plus the C-termini of each H2A) as flexible chains of beads (each bead comprises five amino acids) with the first bead rigidly attached to the parent core. The stretching and bending flexibility constants of each tail inter-bead segment are modeled by harmonic potentials with parameters developed to mimic their atomistic flexibilities (32). The charges of the tail beads are also modeled to reproduce the atomistic properties of the amino acids it represents. . DNA linkers: the DNA that connects consecutive nucleosomes is treated as a chain of spherical beads that have a salt-dependent charge parameterized using the Stigter procedure (51). The mechanical properties of the linker-DNA chains are also considered and described with the worm-like chain model (52,53). The equilibrium DNA inter-bead segment is 3 nm (9 bp), thus to model an NRL of 209 bp, we use six DNA beads (seven segments) per linker. The exiting and entering DNA linkers attached to the nucleosome define an angle of 108 , which corresponds to the 147 DNA bp tightly wound 1.7 times around the core. . Linker histones: the LH proteins are modeled based on rat H1d LH. The family of eukaryotic LH proteins H1 are formed by a globular domain (80 amino acids) and two highly positively charged (lysine-rich) terminal tails (a short N-tail of 14 amino acids and a long C-tail of 100 amino acids) (54). We neglect the short relatively uncharged N-terminal domain and model the C-terminal domain by two charged beads, and the globular domain by a single bead. The LH beads are placed on the dyad axis of each nucleosome and are separated by a inter-bead distance of 2.6 nm, as suggested by Bharath et al. (55). Each LH bead is assigned a Debye–Hückel charge, also optimized with DiSCO. Here, we have refined our LH model by introducing moderate bending and stretching flexibility to the LH beads through semi-stiff harmonic potentials (with stretching and bending constants empirically set one order of magnitude greater than those describing histone-tail motion), and by allowing the LH beads to interact electrostatically with all chromatin components (see Supplementary Material). Through the ‘LH reorganization’ MC move, described below, we produce oligonucleosome ensembles with LH beads in optimized positions near the dyad axis. Dynamic-LH binding/unbinding is . Nucleosome 8806 Nucleic Acids Research, 2012, Vol. 40, No. 18 accounted for by the new ‘LH on and off’ move presented below. . Solvent and ionic environment: the water around the oligonucleosome is treated implicitly as a continuum. The screening of electrostatic interactions due to the presence of monovalent ions in solution (0.15 M NaCl) is treated using a Debye–Hückel potential (electrostatic screening length of 1.27 nm1) (32) and, as described above, with the charges on each component parameterized considering salt-dependent screening. A low concentration of magnesium ions is considered as developed in (32), by reducing the DNA bending persistence length (from 50 to 30 nm) and the repulsion among DNA linkers (DNA–DNA screening length of 2.5 nm1). Note that only the screening length of the DNA linkers is adjusted because this is a phenomenological approach based on the argument that, within compact chromatin, divalent ion screening allows linker DNAs to almost touch each other. The change in screening length for all interactions due to a low concentration of magnesium ions in solution is small (i.e. from 1.27 to 1.31 nm1) and does not modify the results (32). To prevent overlap among chromatin components, each nucleosome charge, linker-DNA bead, histone-tail bead, and LH bead are assigned an excluded volume. Specific expressions for the oligonuclesome energy are given in the Supplementary Material. Furthermore details and all values of parameters can be found in (17,32,34); in addition, Supplementary Table S1 provides a list of model parameters and the appropriate reference where their values can be found. Model limitations In the past years, innovative chromatin coarse-grained models, considering very different approximations, have sprouted [for a recent review, see (56)]. Our model can simulate moderate oligonucleosome sizes (i.e. 12–48 nucleosomes per fiber) and considers, among other features, the charged and contoured nucleosome surface, the flexibility of histone tails, and the presence of LH proteins. At the same time, compromises are made as follows. First, although our histone-tail treatment considers them as unstructured protein regions, some experiments (57–59) and modeling (60) have suggested the intermittent presence of local secondary structure motifs among the nucleosome histone tails. Our tail models have been parameterized to reproduce the average atomistic behavior of the tails. Second, we model the histone protein core plus DNA around it as a rigid entity, and thus omit the effects of nucleosome unwrapping and sliding. For chromatin under tension, nucleosome wrapping/unwrapping might become relevant at low forces [4.5 pN for no-LH chromatin in monovalent salt (44)]. Nonetheless, our coarse-grained model is suitable for capturing structural rearrangements at the level of whole chromatin fibers, which take place at longer time scales. Third, as discussed previously (32), our mesoscale model does not account for charge–charge correlation effects, specific protein–protein interactions, and desolvation effects. Although charge correlation effects become important in systems with highly charged surfaces and multivalent counterions, an accurate modeling of these effects requires explicit treatment of ions and solvent, and is not feasible for the large oligonucleosome systems studied here. Specific protein– protein interactions are needed to properly describe internucleosome interactions, but they are expected to be relatively weak when compared with the strong electrostatic interactions among chromatin components. Similarly, the desolvation effects are expected to be negligible in comparison with the electrostatic interactions. A comprehensive treatment of these effects is not yet feasible for large oligonucleosomes as it requires a detailed description of the nucleosome and explicit treatment of the solvent. Monte Carlo algorithm We sample oligonucleosome conformations at constant temperature with five different Monte Carlo (MC) moves (global pivot, local translation, local rotation, tail regrowth, and LH reorganization) and one optional move to account for dynamic-LH binding/unbinding (LH on and off, described in ‘LH dynamic binding modeling’ below). . Pivot, translation, and rotation chain moves: the global pivot move is implemented by randomly choosing one linker-DNA bead or nucleosome core and a random axis passing through the chosen component. The shorter part of the oligonucleosome about this axis is rotated by an angle chosen from a uniform distribution within [0, 20 ]. The local translation and rotation moves also select randomly a oligonucleosome chain component (linker-DNA bead or core) and an axis passing through it. In the translation move, the component is moved along the axis by a distance sampled from a uniform distribution within [0, 0.6 nm]. In the rotation move, the component is rotated about the axis by an angle sampled from a uniform distribution in the range [0, 36 ]. All three MC moves are accepted or rejected based on the Metropolis criterion. . Tail regrowth move: the tail regrowth move is implemented to sample histone-tail conformations based on the configurational bias MC method (61,62). The move randomly selects a histone-tail chain and regrows it bead-by-bead using the Rosenbluth scheme (63). To prevent histone-tail beads from penetrating the nucleosome core, the volume enclosed within the nucleosome surface is discretized, and any trial configurations that place the beads within this volume are rejected automatically. . LH reorganization move: the LH reorganization move is implemented by randomly selecting one LH bead and an axis passing through it, and then translating the bead along that axis by a distance sampled from a uniform distribution within [0, 0.3 nm]. As done in the tail regrowth move, any trial configurations that place the LH beads within the nucleosome discretized volume are rejected automatically. The rest of the Nucleic Acids Research, 2012, Vol. 40, No. 18 8807 trial configurations are Metropolis criterion. selected based on the The pivot, translation, rotation, tail regrowth, and LH reorganization moves are attempted with probabilities of 0.2, 0.1, 0.1, 0.4, and 0.2, respectively. In simulations that consider LH dynamic binding, the probabilities for the pivot, translation, rotation, tail regrowth, LH reorganization, and LH on and off moves are 0.2, 0.1, 0.1, 0.4, 0.1, and 0.1, respectively. LH dynamic binding modeling Modeling LH’s behavior is intricate. In vivo, H1 binds dynamically to the cores, exchanging continuously among nucleosomal binding sites (5,6). To approximate the dynamic-LH binding behavior (5,6), we have developed an MC procedure that allows the LH proteins to continuously bind and unbind to different cores during the simulations. This ‘LH on and off move’ proceeds as follows: (1) One LH–core binding site is selected at random. (2) If the LH is bound to a core, a trial configuration is formed by either leaving the LH bound or with a probability Pd 2 (0, 1), dissociating it and diffusing it to infinity so that its contribution to the total energy is zero. If the LH is unbound, the trial configuration is formed by re-associating it to its core with a probability Pa 2 (0, 1). (3) The trial configuration is then accepted or rejected based on the Metropolis criterion. The values of Pd and Pa describe the dissociationand-diffusion and association probabilities of LH, respectively. They also measure the LH–core binding affinity and determine the average number of cores that have a LH bound to them at any given MC step. Throughout the simulations, each core can be bound to one or zero LH molecules, and there are always enough LH proteins to saturate the nucleosome array. Low LH concentrations thus reflect low LH–core binding affinities. The two parameters, Pd and Pa are determined by the LH–core binding affinity but are difficult to obtain experimentally due to the complexity of the chromatin fiber. As revealed by fluorescent recovery after photobleaching (FRAP) experiments, the complex and dynamic LH–chromatin interaction is controlled cooperatively by two DNA binding sites on LH’s globular domain and the C-terminal tail (10). The role of the N-tail of LH has not been clearly identified, and the exact mechanism by which LH’s globular domain and C-tail act during binding is still under debate. In addition, the binding properties of LH also depend on many factors, such as the specific subtype of LH [H1.1 and H1.2 have low affinity, H1.0 and H1.3 moderate affinity, and H1.4 and H1.5 high affinity (64)], the presence of post-translational modifications of LHs domains, replacement of core histones by variants, and the interaction of LH with other chromatin structural proteins (12). Thus, in this work, we consider a range of values that span possible binding scenarios (Table 1). Table 1. LH dynamic binding model parameters and resulting proportion of LH–core bonds and fiber resting length Case Pa Pd Fixed LH Effective diffusion (very high affinity): Pa Pd High affinity: Pa > Pd Moderate affinity: Pa = Pd Low affinity: Pa < Pd No LH 1 1 0 100 49 0.25 80.45 ± 8.09 53 1 1 0.5 0 0.5 1 1 1 %LH–core bonds ± SD 67.16 ± 9.54 50.29 ± 10.19 33.40 ± 9.64 0 Resting length (nm) 60 71 73 74 All fibers with LHs were modeled in high monovalent salt and low concentration of magnesium ions. Fibers without LH were modeled in high monovalent salt only. Our MC sampling of oligonucleosome conformations allows us to extract qualitative structural and mechanistic information of chromatin’s unfolding process. Clearly, our MC procedure is statistical and by ‘dynamic binding’ we mean a non-zero probability of binding and/ or unbinding. The different chromatin conformations in the resulting equilibrium ensemble have similar numbers of total LH–core bonds; however, the specific locations of the bound sites change among the different conformations. The resulting equilibrium ensemble thus mimics an array of chromatin fibers in which the LH proteins bind/unbind in a stop-and-go mode (5). Implementation of fiber stretching We mimic the extension experiments by fixing the geometric center position of the first nucleosome core to its initial position and applying a constant force to the last nucleosome in the oligonucleosome chain, as done previously by Aumann et al. (40). In practice, we add an stretching energy term (Epull) to the total oligonucleosome energy (see Supplementary Material); this term is proportional to the product of the stretching force (Fpull) and the distance in the z-direction between the geometric centers of the first (z1) and last (zNc ) nucleosomes, i.e. Epull ¼ Fpull jzNc z1 j. Simulation details Our simulations are performed at 293 K and high monovalent salt concentration (0.15 M of NaCl). Simulations for fibers with LH (dynamic and fixed) also consider a low concentration of magnesium ions. Every simulation set includes 12 trajectories that cover the mean DNA twist angle and two DNA twist deviations (±12 ) from the mean twist to mimic natural variations, as done previously (34). Each simulation trajectory was run for up to 50 million MC steps. Convergence of our simulations is reached well before 45 million MC steps as shown elsewhere (34). For statistical analysis, the last 5 million steps were used and only conformations spaced by 100 000 steps were considered. For our initial configurations, we use representative zigzag equilibrium conformations at zero pulling force obtained previously in the presence of fixed LHs and magnesium ions (34). 8808 Nucleic Acids Research, 2012, Vol. 40, No. 18 RESULTS AND DISCUSSION 1.5 (a) Dramatic effect of dynamic-LH binding on chromatin fiber’s elasticity Exploration of various LH dynamic binding scenarios In Figure 2b, we compare the stretching behavior of chromatin fibers without LH nor magnesium ions, with LHs and magnesium ions, and with LHs that bind/unbind dynamically with different affinities and magnesium ions; Supplementary Figure S1 presents additional data for fibers without LH and with magnesium ions. In Table 1, we present the values for the association and dissociation-and-difussion probabilities we use to model the different binding affinities. Each binding regime produces chromatin fibers with different average LH– core bond concentrations. In the table we provide the resulting LH–core bond concentrations at zero pulling force; for each combination of probabilities, similar concentrations are obtained at higher forces. The cases of Pd = 0 and Pa = 0 reproduce fixed-LH (100% LH bound) and no-LH (0% LH bound) behavior, respectively (red and blue curves in Figure 2b). Other probability combinations produce ensembles with intermediate concentrations (four other colored curves in Figure 2b). For example, the probability combination with PaPd (i.e. Pa = 1 and Pd = 0.25) mimics dynamic-LH chromatin fibers where LHs exhibit effective diffusion behavior (10). This means that, following dissociation, rebinding occurs much faster than diffusion to infinity. I(k) 0.5 0 0 2 4 6 8 10 12 k − nucl. num. 40 (b) +LH (fixed) +Mg 2+; 100% LH 35 +LH (dyn) +Mg 2+; ~80% LH +LH (dyn) +Mg 2+; ~67%LH 30 +LH (dyn) +Mg 2+; ~50% LH Force (pN) Using our mesoscale model, we analyze the unfolding behavior of 24-core 209-bp repeat oligonucleosomes with one LH permanently attached to each core and with LHs that are allowed to bind/unbind to their cores with different affinities (as described in Table 1). These simulations are performed in physiological conditions (high monovalent salt: 0.15 M NaCl, low concentration of magnesium ions, and room temperature) and are started from converged zigzag configurations. As reference, we perform additional simulations of 24-core 209-bp repeat arrays without LH with monovalent ions only; simulations analyzing the effect of the NRL, different ratios of fixed LH–core bonds, and two LH/core binding affinities in monovalent ions only are reported elsewhere (17), where longer NRLs were shown to soften the fiber and an interesting mechanism to facilitate controlled fiber unfolding by the simultaneous presence of fast and slow LH binding pools was described. The 209-bp repeat fiber is suitable to dissect the role of LH in chromatin unfolding; experiments (65) and modeling (17,34) have shown that LH’s compacting and structural effect is strongest for medium-NRL arrays [medium NRLs range roughly from 190 to 210 bp (34)]. In addition, the 209-bp repeat length is common in nature and close to the value of chicken erythrocyte chromatin used widely in many studies [(19,21,36)]. The stretching experiment is implemented by fixing the position of the first nucleosome core and applying a stretching force of up to 40 pN to the last core (Figure 2a). Such forces mimic cellular constraints exerted by molecular machines [e.g. RNA and DNA polymerases (66–70)]. 1 +LH (dyn) +Mg 2+; ~33% LH 25 −LH −Mg2+; 0% LH 20 15 10 5 0 0 100 200 300 400 500 600 Extension (nm) Figure 2. Effect of LH dynamic binding in the force–extension response of 24-core 209-bp repeat oligonucleosomes. (a) Representation of the force–extension experiment (left) and internucleosome interaction pattern of our converged zigzag starting conformation characterized by dominant k ± 2 and moderate k ± 5 interactions (right). (b) Force– extension curves for fibers pulled below 40 pN at room temperature. Fibers with LHs (fixed or dynamic) are simulated in high monovalent salt (0.150 M NaCl) and low concentration of magnesium ions. Fibers without LH are simulated in high monovalent salt only. The curve labels show the average LH–core bond concentration in each binding regime; such concentrations are obtained with the binding probabilities given in Table 1. The force–extension curve is a plot of the magnitude of the applied force versus the ‘end-to-end’ extension or distance (in the direction of the stretching force) between the geometric centers of the first and last nucleosome cores. Error bars in the force–extension curve denote the standard deviation from the mean of the ensemble average. Dynamic LH binding dramatically reduces LHs stiffening effect and more so as the LH–core binding affinity decreases. Consequently, such parameters yield equilibrium ensemble values of oligonucleosomes in which the majority of LH proteins (80%) are bound to cores. On the other hand, probability combinations with Pa < Pd describe a low LH-core binding affinity and yield chromatin fibers with a reduced number of LH–core bonds. Table 1 also indicates that the individual conformations in the ensemble have a LH-core bond concentration that deviates 10% from the mean, reflecting a fluctuation in the number of LH-core bonds consistent with dynamic regimes. The force–extension curves in Figure 2b demonstrate marked differences between fibers with fixed LHs (red curve), dynamic LHs (green, turquoise, purple and magenta curves), and no LH (blue curve). Note that all dynamic-LH curves lie between the two extreme cases (fixed-LH, red and no-LH, blue). Fibers without LH (blue curve) require only 2 pN to produce a 2-fold extension from their resting length (equilibrium fiber length at zero pulling force; values given in Table 1). In comparison, Nucleic Acids Research, 2012, Vol. 40, No. 18 8809 Table 2. Force-extension slope during two force regimes Fiber Dynamic LH (effective diffusion) Fixed LH No LH Straightening regime Unfolding regime Force (pN) Slope ± SD (pN/nm) Force (pN) Slope ± SD (pN/nm) 0–6 0–25 – 0.1211 ± 0.0624 0.4478 ± 0.0966 – >6 >25 <4 0.0319 ± 0.0089 0.0611 ± 0.0160 0.01077 ± 0.0005 Fibers with LHs were modeled in high monovalent salt and low concentration of magnesium ions. Fibers without LH were modeled in high monovalent salt only. the force needed to double the resting length of fixed-LH fibers (red curve) is well above experimental values (21,26), reaching 25 pN; these fibers are extremely stiff. By considering dynamic-LH binding/unbinding, we observe a dramatic reduction of fiber stiffness. The forces needed to double the resting length of fibers with dynamic LH–core bonds approach the no-LH value as the binding affinity decreases; for example, the forces for high, moderate, and low affinities are 4.5, 4, and 3 pN, respectively. Even for dynamic-LH fibers with the highest binding affinity considered (effective diffusion case), we observe a remarkable softening with respect to fixed-LH fibers: the force needed to double their resting length is only 6 pN (compared with 25 pN for fixed-LH species). The high stiffness of fixed-LH fibers results from the presence of magnesium ions and the formation of rigid DNA stems—209-bp chromatin with fixed-LH in monovalent salt only doubles its resting length at a much lower value, 10 pN (17). Divalent ions increase chromatin stiffness significantly by (i) bending the DNA linkers and screening the DNA–DNA repulsion, which allows the nucleosomes to come closer together, and (ii) permitting the nucleosome cores to rearrange in a wider range of conformations that yield optimum contacts within an extended array; both effects favor strong internucleosome interactions and make unfolding more challenging. Rigid DNA stems further screen DNA repulsion and lock the chromatin fiber in a compact form. This observation suggests that DNA stems and divalent ions work together to stabilize chromatin compaction, increasing the forces needed to unfold the fiber. Dynamic LH with high binding affinity In the remainder of this article, we elaborate upon the behavior of fibers with LHs that exhibit effective diffusion (highest binding affinity) by comparing it with that of fibers without LH and with fixed LHs. Hereafter, we refer to the fibers with LH molecules that display effective diffusion as ‘dynamic-LH fibers’. Results from this regime are the most relevant biologically because the majority of LH molecules are bound to cores, as in vivo (10). Softening caused by this dynamic-LH binding behavior can also be noted by comparing the slopes of the force– extension curves. In Figure 2b, separate regimes (force ranges), in which the force–extension curves of fixed-LH (red curve), dynamic-LH (green curve), and no-LH (blue curve) fibers present linear extension behavior individually, can be recognized. For each fiber, the force regimes can be distinguished by their different slopes; the slope quantifies the fiber’s propensity to extend during such regime. The forces at which these regimes occur and the corresponding slopes of the force–extension curves are shown in Table 2. Fibers with fixed and dynamic LHs display two separate force regimes. The slopes in both regimes are much smaller for dynamic-LH fibers than for the rigid fixed-LH arrays. In the first force regime, both curves have their largest slopes, which suggests that the fibers straighten and then open partially, maintaining their original zero-force structure. During the second force regime, the slopes drastically decrease; a small force increase now produces a notable fiber extension, signaling fiber unfolding. The slope for dynamic-LH fibers in this second force regime is 0.03 pN/nm, in excellent agreement with the 0.02–0.028 pN/nm values estimated for medium-NRL chromatin with magnesium ions (26). In comparison, the force–extension curve of no-LH fibers shows three different force regimes. Instead of an initial straightening regime, the fiber unfolds (minimum slope) and the force–extension curve resembles a plateau; the slope in this regime is six times smaller than that for fixed-LH fibers (Table 2). A similar plateau at low forces was previously reported by Cui and Bustamante (21) for medium-NRL chromatin at low salt, where a loose structure is expected. At intermediate forces, the increased slope suggests reorganization into a stiffer structure. At high forces, nonlinear behavior due to DNA over-stretching occurs. This fiber is further analyzed in (17). To further quantify chromatin’s stiffness, we estimate the stretching elasticity modulus s, defined as the force needed to produce a 2-fold extension (71). Fixed-LH fibers show a stiff elasticity modulus of s & 25 pN, close to values calculated by Aumann et al. (40) for fibers with rigid DNA stems. In comparison, the softer dynamic-LH fibers have an elastic modulus of s & 6 pN, in agreement with the s = 5–8 pN experimental values (21,72). This remarkable stiffness reduction (by a factor of almost 5) caused by LHs dynamic binding behavior demonstrates that LH mobility plays an important role in facilitating force-induced chromatin unfolding. Note that our force–extension curves mimic the behavior of integral chromatin fibers, i.e. fibers in which all nucleosomes remain intact; the effects of nucleosome unwrapping and sliding are not included in our model. Experiments indicate that the presence of histone variants and post-translational modifications throughout the dyad DNA entry/exit region can induce nucleosome 8810 Nucleic Acids Research, 2012, Vol. 40, No. 18 unwrapping even at zero force (73). For instance, H3 lysine 56 is located in the dyad region and its acetylation encourages transient nucleosome unwrapping (74). Some remodeling complexes also function by favoring nucleosome unwrapping. For example, the SWI/SNF complex is thought to strip off a section of DNA around the dyad position (75). It has been speculated that nucleosome unwrapping creates a DNA bulge on the nucleosome surface close to the dyad that propagates sliding the cores (76), and as such might increase the degree of positional delocalization (or ‘fuzziness’) of nucleosomes across the genome (77). In addition, modeling of medium-NRL chromatin without LH and without divalent ions has suggested that in native nucleosomes unwrapping becomes significant above 4.5 pN (44). We expect individual nucleosome unwrapping to modify the electrostatic properties of the partially unwrapped cores. Multiple transient nucleosome unwrappings and enhanced fuzziness might favor fiber compaction and fiber stiffening. Note, however, that for the conditions analyzed here (divalent ions and LHs), the effects of nucleosome unwrapping and sliding are expected to be reduced because experiments show that unwrapping in divalent ion conditions occurs only at high forces [>15–25 pN, approximately (22,23)] and that LHinduced stems restrict nucleosome movement. Consistently, other experiments have also found that, in the presence of magnesium ions, chromatin unfolds by initially forming an open beads-on-a-string structure with most internucleosome contacts broken and no unwrapped nucleosomes (26). Unfolding mechanism of chromatin with dynamic LHs To describe how the internal organization of nucleosomes in the chromatin fiber changes with the pulling force, we complement the force–extension curves with an analysis of representative simulation snapshots and internucleosome interaction patterns (32,34) (Figure 3). These patterns measure the relative intensity of histone-tail mediated interactions between nucleosomes separated by k neighbors (i ± k). For example, dominant i ± 2 contacts are indicative of a compact two-start conformation (the zigzag), while dominant i ± 1 and i ± 6 interactions suggest a 6-nucleosomes-per-turn solenoid model (34). The combined information in Figure 3 reveals different fiber opening mechanisms. For dynamic-LH fibers (left panels), we have the following steps: (i) within 0–6 pN, the fiber straightens while maintaining a zigzag organization with DNA stems (dominant i ± 2 and moderate i ± 5 contacts) (ii) within 7–12 pN, individual linker-DNA stems rupture, causing significant fiber extension and yielding partially unfolded heteromorphic structures we term ‘superbeads-on-a-string’; these structures combine stretched fiber regions with ‘superbeads’, or compact zigzag clusters, in which the nucleosomes interact strongly with their zigzag neighbors (i±2) and moderately with other cores (i±1 and i±3); (iii) between 13 and 16 pN, the DNA stems continue to break reducing the presence of superbeads and their intense i±2 contacts; (iv) at 16 pN, most DNA stems have ruptured and the fiber forms a distinctive ‘single-stack’ conformation with dominant i±1 interactions; the nucleosomes in this structure are vertically aligned, but instead of stacking parallel on top of one another, they are irregularly oriented to maximize the strength of i±1 interactions; and (v) above 16 pN, the single-stack fiber stretches to form an open beads-on-a-string conformation with negligible internucleosome interactions. Unfolding of fixed-LH fibers (middle panels) proceeds though the same five steps described above for dynamic-LH fibers. However, the forces needed to initiate each unfolding step are much higher when LHs are fixed due to destabilization of the rigid DNA stems and an enhanced configurational heterogeneity. Indeed, dynamic-LH DNA stems start rupturing above 6 pN compared with fixed-LH stems at only 25 pN. These forces coincide with those at which the force–extension slopes change, confirming that these slope changes signal structural transitions. Without LH (panels at right), the three force regimes correspond to conformations of irregular opening, single-stack, and unfolded chain, respectively. In the single-stack organization (4–10 pN), the parallel alignment of nucleosomes facilitates optimum internucleosome interactions and explains the higher resistance to stretching (increased slope) in this force range. Although for fibers without LH this transition into a single-stack conformation coincides with an increased force–extension slope, for fibers with dynamic and fixed LH this transition does not alter the shape of the force–extension curve and is revealed only by our simulation analysis. This different correlation between the slope change and actual conformational transition shows that the interactions between immediate nucleosomes, which stabilize the single-stack conformation, stiffen the fiber without LH at low forces (4–10 pN) but can be ruptured easily at higher forces (16 and 36 pN for dynamic and fixed LH, respectively). Notably, these single-stack conformations emerge for simulations started from zigzag fibers and are not evidence of an initial solenoid fiber organization (26). This underscores the utility of modeling to extract structural information from stretching experiments. Formation of superbeads-on-a-string intermediates is observed for all fibers in divalent conditions, regardless of LH binding rate (see representative snapshots in Supplementary Figure S2). In contrast, in fibers without LH and without divalent ions, formation of such structures is negligible. Stabilization of these structures requires the presence of LHs and divalent ions to decrease DNA– DNA repulsion, increase DNA bending, and bring the nucleosomes in closer contact inside compact clumps. This is consistent with the observation that chromatin fibers with LHs and divalent ions are heteromorphic and exhibit zigzag features of straight linker DNA combined with moderate DNA bending (19). The stability of superbeads-on-a-string structures in the presence of LHs and divalent ions is supported by several experimental observations. First, stable structures combining unfolded fiber regions and small aggregates of nucleosomes have been visualized by atomic force microscopy of trypsin-digested chromatin with LHs and Nucleic Acids Research, 2012, Vol. 40, No. 18 8811 (a), (b) (c) Figure 3. Effect of dynamic LH binding on chromatin’s unfolding mechanism, as characterized by (a) force–extension curves, (b) patterns of internucleosome interactions, and (c) simulation snapshots (space filling models in which alternating nucleosomes are colored white and navy, DNA as red and LH turquoise). The three vertical panels are for 24-core 209-bp oligonucleosomes at 0.15 M monovalent salt with: dynamic LH and Mg2+ (with Pa=1 and Pd=0.25), fixed LH and Mg2+, and no LH and no Mg2+ (green, red, and blue curves in Figure 2, respectively). These snapshots are presented for different force regimes: low (yellow background), moderate (blue background), high (pink background) and very high (purple background). Some longer structures are only shown partially at bottom, as denoted by three dots. Fibers with dynamic LHs unfold via superbeads-on-a-string (SBS) structures at much lower forces than fibers with fixed LHs. magnesium ions (36). This is relevant to our study because trypsin digestion removes the N-terminal histone tails and triggers an equivalent effect to that of applying a pulling force: inducing fiber unfolding (78). Second, electron microscopy of digested chromatin fibers with NRLs of 200 and 212 bp, both with LHs and in high monovalent salt, has already revealed the formation of regular clumps of nucleosomes, or superbeads, measuring 32 and 35 nm, respectively (35). Simple estimation in our chromatin snapshots indicates similar dimensions (see green lines in Figure 3). Animations of the three unfolding mechanisms are available on http://www.biomath.nyu.edu/index/ gallery.html. Internucleosome interaction energy during fiber unfolding The folded state of the chromatin fiber is stabilized by electrostatic interactions between the positively charged and flexible histone tails, which extend from the nucleosome surface, and the charged surfaces of neighboring cores. During force-induced stretching, this stabilizing internucleosome interaction energy must be overcome before the fiber unfolds. Figure 4 plots this energy as a function of the pulling force. We define this energy as the average nucleosome/nucleosome electrostatic interaction energy per core, considering all nucleosome charges in the surface and histone tails. The maximum strength of the stabilizing internucleosome interaction energy occurs at zero pulling force, when the fiber is most compact. Fibers without LH have a loose zero-force structure stabilized by a low internucleosome energy of 1 kbT (i.e. mean energy per core ± standard deviation: 1.03 ± 0.11 kbT). Fibers with fixed and dynamic LHs have compact structures in the absence of pulling force maintained by internucleosome energies of 5 kbT (i.e. Fixed LH = 5.04 ± 0.21 kbT and dynamic LH = 4.72 ± 0.26 kbT). Equilibrium data in Supplementary Figure S3 confirm these compaction trends. Internucleosome energies of 1 kbT and 5 kbT for loose and compact chromatin, respectively, are reasonable, considering that a value of 3 kbT has been determined for medium-NRL fibers with LHs in the absence of magnesium ions, where fibers are expected to be moderately compact (21,29). Our internucleosome energy for compact chromatin is weak when compared with the 14 kbT estimate for 197-bp fibers with LHs and magnesium ions (26). However, as recently challenged by Lavelle et al. (79), such a high internucleosome energy was derived assuming a solenoid conformation and a model with five variable parameters. Although a high 14 kbT internucleosome interaction energy is necessary to stabilize a solenoid arrangement with highly bent linkers and strong DNA–DNA repulsion, a weaker 3 kbT-value can maintain a zigzag fiber with straight linkers (29). Our 8812 Nucleic Acids Research, 2012, Vol. 40, No. 18 experiments (19,34); and force–extension behavior in monovalent salt conditions (17). A comparison of chromatin force–extension curves from different studies is complex because the results are highly sensitive to the experimental conditions; importantly, the many studies available analyze chromatin fibers of different characteristics (e.g. NRL, number of nucleosomes, and LH presence/absence) and under varying experimental conditions (e.g. ionic environment and applied forces). Thus, no single chromatin force– extension experimental or modeling benchmark exists. Nonetheless, here we plot our results together with selected available data to interpret general trends. In Figure 5, we compare our force–extension curves with experimental and modeling studies of mediumNRL chromatin fibers (190–210 bp). Given that the end-to-end equilibrium extension is correlated with the number of cores in the chromatin fiber, to facilitate the comparison in Figure 5, we plot the force versus the extension-per-core. Considering the expected effects of the different conditions used in each study, the figure confirms that our simulated curves are consistent with previous works. Figure 4. Top: internucleosome interaction energy per nucleosome versus force for 24-core 209-bp oligonucleosomes at 0.15 M monovalent salt: fixed LH and Mg2+ (red) and dynamic LH and Mg2+ (green), versus no LH and no Mg2+ (blue). Error bars denote the standard deviation from the mean of the ensemble average. For visualization purposes, the low-force–low-energy region is expanded on the right. Bottom: space-filling models based on MC simulations of fibers with dynamic LHs and Mg2+ show DNA in red and alternating nucleosomes in white and blue. The zero-force energy is similar for fibers with fixed and dynamic LHs. The different force regions in which the curve with dynamic LHs has a constant slope correspond to: (1) low force where compact zigzag fibers form; (2) moderate force where superbeadson-a-string arrangements occur; (3) high force where a one-start structure is stable and (4) very high force where a fully extended array forms with no internucleosome interactions (zero energy). intermediate internucleosome energy of 5 kbT agrees well with the compact hetermorphic chromatin structure observed in divalent conditions, which combines straight and bent DNA linkers (19). Comparison with previous modeling and experimental works Our integrated chromatin coarse-grained model has been extensively validated against different available experimental data (32,34). In past work, we have shown that our model reproduces the following experimental results: salt-dependent sedimentation coefficients and packing ratios of 12-unit oligonucleosomes of chicken erythrocyte chromatin over a broad range of monovalent salt concentrations, with/without magnesium ions, and with/without LHs (32); diffusion and salt-dependent behavior of mononucleosomes, dinucleosomes, and trinucleosomes (30); salt-dependent extension of histone tails (30); the irregular zigzag and solenoid topologies of chromatin fibers (29,34), and their enhanced compaction upon LH binding (32,34); linker crossing orientations (32); internucleosome interaction patterns consistent with cross-linking and EM Fixed-LH curve Our stiffer fixed-LH curve lies closely below to that of no-LH fibers modeled by Aumann et al. (40). Even though Aumann’s fibers were simulated in the absence of LH (a softening factor), their relatively high stiffness can be explained by their much shorter NRL (190 bp versus 209 bp in our work). Decreased NRLs have been shown to significantly stiffen the chromatin fiber (17,26,40), because they hinder reorganization into extended arrays and favor intense internucleosome interactions (17). No-LH curve At low forces (<4 pN, see figure inset), the force–extension curve of our no-LH fibers lies closely above that of no-LH fibers simulated by Kepper et al. (44). This low force range is most relevant for analyzing unfolding behavior of our no-LH fibers, given that all but some i±1 internucleosome interactions are broken above such force. Compared with our arrays, the Kepper fibers have a slightly shorter NRL (199 bp versus 209 bp in this work) but were simulated at lower monovalent salt concentration (0.10 M NaCl versus 0.15 M in this work); these differences produce competing stiffening and softening effects in the stretching response, and explain the similitude between curves. This agreement supports both modeling approaches, which consider different approximations but nonetheless yield similar chromatin behavior. At low forces, our no-LH curve is also consistent with the experimental curve for 210-bp fiber of Cui and Bustamante (21). Although Cui and Bustamante’s curve is for chromatin with LHs, this curve is only moderately stiffer than our no-LH result because the former was handled at lower monovalent salt concentration (0.04 M NaCl versus 0.15 M in this work), which produces fiber loosening (34) similar to that observed in the absence of LH. At low forces, our no-LH curve also runs under the no-LH Nucleic Acids Research, 2012, Vol. 40, No. 18 8813 Figure 5. Comparison of our results with previous experimental (Exp.) and modeling (Mod.) force–extension curves. Bottom: force-extension curves of the various works. Top: low-force region expanded for visualization purposes. Results from this work are shown in solid lines (red = fixed LH, green = dynamic LH with Pa=1 and Pd=025, blue = no LH), results from other works are shown in dashed lines. The references and experimental/modeling conditions for each work are summarized in the legend. Data shown for other works has been taken directly from the different paper figures. For comparison purposes, instead of the extension, the extension-per-core (extension divided by the total number of cores) is used. curve of Kruithof et al. (26); this is also reasonable because the experimental fibers have a slightly shorter NRL (197 bp) and, unlike our no-LH fibers, were manipulated in the presence of magnesium ions, which induce compact folding and fiber stiffening. Considering the effects of nucleosome unwrapping might be important to model behavior of these no-LH fibers without divalent ions at higher forces (>4 pN). Dynamic-LH curve Our dynamic-LH fiber has an intermediate stiffness between the extremes of fixed-LH and no-LH curves. As expected, it is softer than Aumann’s 190-bp fiber (40)— consistently with softening induced by longer NRLs (17,26,40)—and stiffer than the low-salt fiber of Cui and Bustamante (21) and the no-LH fibers of Kepper et al. (44) and Kruithof et al. (26). Moreover, our results offer an explanation to Kruithof’s observation that LH molecules do not affect the stiffness of the chromatin fiber: as shown in Figure 2, fibers with highly mobile LH molecules (low affinity) can exhibit the same stiffness as fibers without LH. High LH mobility is consistent with the experiment because the strain induced in the fiber by the stretching process can destabilize the LH–core bonds. CONCLUSIONS Our suggestion for a critical role for dynamic linker histone binding/unbinding during force-induced chromatin fiber unfolding at divalent ion conditions is based on analyses of the force–extension curves, internucleosome interaction patterns, simulation snapshots, and internucleosome energy. These data also indicate that (i) fixed-LH DNA stems require forces >25 pN to break; (ii) dynamic-LH binding/unbinding behavior destabilizes the rigid DNA stems, reducing dramatically chromatin’s stiffness (stretching modulus) and the forces needed to initiate unfolding to 6 pN; (iii) in the presence of divalent ions and LHs, chromatin unfolding proceeds through irregular superbeads-on-a-string intermediate structures that combine fully extended DNA regions 8814 Nucleic Acids Research, 2012, Vol. 40, No. 18 with compact clumps; (iv) compact chromatin fibers with LHs and divalent ions are stabilized by an average internucleosome interaction energy of 5 kbT, consistent with a heteromorphic structure; and (v) zigzag fibers adopt a single-stack structure just before unfolding into a beads-on-a-string array. As discussed below, these observations have important biological implications. Although related softening due to dynamic-LH binding and heteromorphic intermediates emerged from our recent work with monovalent ions only (17), our present work suggests the important role of divalent ions and provides further insights into chromatin unfolding at physiological conditions. First, the added compaction introduced by divalent ions is not sufficiently strong to eliminate, or substantially attenuate, the dramatic softening produced by dynamic, compared with static, LH binding. Second, divalent ions induce unfolding through heteromorphic superbeads-on-a-string intermediates in all LH binding modes examined. In particular, such conformations appear in fibers with low affinity LH–core bonds, unlike the case for monovalent ions (17). The heteromorphic superbeads-on-a-string structures have characteristics and dimensions similar to those of experimentally observed chromatin lumps (35,36). Such structures might be important for gene regulation, as they selectively expose certain regions of the genome and maintain others protected inside compact clumps. The global stability of superbead structures in vivo depends on the ability of divalent ions to screen repulsion among DNA linkers, enhance DNA bending, and accommodate multiple internucleosome interactions (19). In addition, LH binding at a specific DNA entry/exit point would further screen the DNA linker repulsion locally. Therefore, modulation of the LH–core binding affinity, across a fiber, by post-translational modifications of LH and its binding sites (9,80) suggests a mechanism to guide the superbead and stretched segment locations and control DNA access. Thus, the combined effects of LH mobility and DNA bending might play a role for gene regulation through stabilization of softer fiber arrangements accessible to molecular motors and unfolding through heteromorphic conformations that facilitate selective DNA expression. These superbeads-on-a-string structures may also represent transitional states between different chromatin forms present at the various stages of the cell cycle. During cell-cycle progression, chromatin structure suffers large modifications that lead the dynamical changes in gene expression (81). In addition, modifications of chromatin structure during the cell cycle could act as signals to activate checkpoint pathways (82). It has also been suggested that chromatin structural changes could contribute to DNA repair itself, by facilitating lesion accessibility (82); which is consistent with chromatin transitioning between forms through superbeads-on-a-string structures. Thus, the high heterogeneity we observe in partially unfolded/transitional chromatin might indicate that divalent ions and dynamic LHs are essential facilitators of the chromatin structural plasticity required to trigger and signal vital cell-cycle events. The dramatic softening of fiber resistance to pulling by dynamic-LH binding in divalent conditions provides a novel explanation to two surprising recent observations: that (i) the experimental force–extension curves of fibers with and without LH are equivalent at low forces when magnesium ions are present (26), and (ii) transcription occurs within chromatin fibers non-uniformly compacted above the 30-nm chromatin level (83). Indeed, our results suggest that transcribed chromatin would be unfolded locally, by the pulling force generated by RNA polymerase to allow transcription, but remain compact at distal gene segments. Fiber softening correlates with unfolding at lower forces. This force reduction might be crucial as eukaryotic molecular motors that operate in chromatin, function within the low force range in which dynamic-LH fiber unfolding is observed in this work. For instance, a single-molecule study revealed that RNA polymerase II ceases to transcribe at 7.5 ± 2 pN (compared with 35 pN for its prokaryotic counterpart) or at 16.9 ± 3.4 pN in the presence of TFIIS, a transcription elongation factor that facilitates transcription (70). In addition, FRAP experiments suggest that the globular and C-terminal domains of H1 form the LH-chromatin bound state through alternative pathways involving several different partially bound intermediates that are susceptible to competition by other binding factors (10). Hence, LH dynamic binding/unbinding, together with binding factors might provide the opportunity for molecular motors to achieve chromatin stretching and gain access to the DNA at natural forces without disrupting the global chromatin organization. Furthermore, during gene expression, the mechanical constraints induced in the fiber structure by the pulling machinery may destabilize the LH–core bound state, enhancing LH mobility. Enhanced LH mobility in the presence of mechanical constraints is consistent with an optical tweezers experiment of chromatin with LH B4; it suggests that, during force-induced stretching, the LH– core bond is broken before nucleosome disruption occurs (24). Enhanced LH mobility may in fact distinguish active from silent chromatin and constitute a necessary factor to trigger fiber unfolding. Finally, our modeling snapshots and internucleosome patterns confirm that zigzag fibers unfold through a single stack conformation. A single-stack is thus not evidence for a solenoid structure (26). The solenoidal explanation in (26) is based on the observation that 197-bp arrays stretch with a constant slope up to a maximum length which is consistent with a regularly folded single stack of perpendicular nucleosomes and on the assumption that a zigzag fiber is associated instead with a shorter two-stack structure. As we observe, however, zigzag fibers with LHs conserve the force–extension slope past the two-stack size limit. The unfolding process could be further attenuated by the ability of the nucleosomal DNA to transiently peel-off the histone core. As discussed in Lavelle et al. (79), the experiments in (26) have already been re-interpreted to support a zigzag organization (79). The results presented here suggest a fascinating biological feature to regulate fiber structure and hence DNA Nucleic Acids Research, 2012, Vol. 40, No. 18 8815 accessibility, cell-cycle progression, and cell-cycle checkpoints, by divalent ions effects and a dynamic LH binding/unbinding mechanism. The possible crucial role that dynamic LH binding has in facilitating transient chromatin unfolding also underscores the importance of considering the highly dynamic nature of chromatin to decipher the external and internal factors that drive and alter gene expression. Recent data suggest that the pulling force created during transcription may organize the global architecture of an extended chromosomal domain in situ (84). Other architectural factors may include HMG proteins that accelerate LH mobility (9) or chromatincondensing proteins such as HP1 (85) and MENT (86) that may polymerize on the nucleosomes to inhibit transient unfolding of the chromatin fiber. Together with other variables that affect chromatin compaction and configurational transitions, such proteins suggest how small local differences can have profound global effects. SUPPLEMENTARY DATA Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–3 and Supplementary Methods. ACKNOWLEDGEMENTS The authors thank Dr Sergei Grigoryev for his invaluable comments and insights concerning this work. Computing support from the NYU HPC USQ and Cardiac clusters is also acknowledged. FUNDING National Science Foundation [MCB-0316771 to T.S.]; National Institutes of Health (NIH) [R01 GM55164 to T.S.]; American Chemical Society [PRF39225-AC4]; Petroleum Research Fund (to T.S.); Philip Morris USA (to T.S.); Philip Morris International (to T.S.); Schlumberger Faculty for the Future Program (to R.C.-G.). Funding for open accees charge: National Science Foundation [MCB-0316771 to T.S.]; NIH [R01 GM55164 to T.S.]. Conflict of interest statement. None declared. REFERENCES 1. Finch,J.T. and Klug,A. (1976) Solenoidal model for superstructure in chromatin. Proc. Natl Acad. Sci. USA, 73, 1897–1901. 2. Worcel,A., Strogatz,S. and Riley,D. (1981) Structure of chromatin and the linking number of DNA. Proc. Natl Acad. Sci. USA, 78, 1461–1465. 3. Woodcock,C.L., Frado,L.L. and Rattner,J.B. (1984) The higher-order structure of chromatin: evidence for a helicăl ribbon arrangement. J. Cell Biol., 99, 42–52. 4. McBryant,S.J., Lu,X. and Hansen,J.C. (2010) Multifunctionality of the linker histones: an emerging role for protein-protein interactions. Cell Res., 20, 519–528. 5. Misteli,T., Gunjan,A., Hock,R., Bustin,M. and Brown,D.T. (2000) Dynamic binding of histone H1 to chromatin in living cells. Nature, 408, 877–881. 6. Lever,M.A., Th’ng,J.P., Sun,X. and Hendzel,M.J. (2000) Rapid exchange of histone H1.1 on chromatin in living human cells. Nature, 408, 873–876. 7. Caron,F. and Thomas,J.O. (1981) Exchange of histone H1 between segments of chromatin. J. Mol. Biol., 146, 513–537. 8. Brown,D.T. (2003) Histone H1 and the dynamic regulation of chromatin function. Biochem. Cell Biol., 81, 221–227. 9. Catez,F., Ueda,T. and Bustin,M. (2006) Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol., 13, 305–310. 10. Stasevich,T.J., Mueller,F., Brown,D.T. and McNally,J.G. (2010) Dissecting the binding mechanism of the linker histone in live cells: an integrated FRAP analysis. EMBO J., 29, 1225–1234. 11. Caterino,T.L., Fang,H. and Hayes,J.J. (2011) Nucleosome linker DNA contacts and induces specific folding of the intrinsically disordered H1 carboxyl terminal domain. Mol. Cell. Biol., 31, 2341–2348. 12. Happel,N. and Doenecke,D. (2008) Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene, 431, 1–12. 13. Zlatanova,J., Caiafa,P. and van Holde,K. (2000) Linker histone binding and displacement: versatile mechanism for transcriptional regulation. FASEB J., 14, 1697–1704. 14. Yellajoshyula,D. and Brown,D.T. (2006) Global modulation of chromatin dynamics mediated by dephosphorylation of linker histone H1 is necessary for erythroid differentiation. Proc. Natl Acad. Sci. USA, 103, 18568–18573. 15. Meshorer,E., Yellajoshula,D., George,E., Scambler,P.J., Brown,D.T. and Misteli,T. (2006) Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell, 10, 105–116. 16. Dou,Y., Bowen,J., Liu,Y. and Gorovsky,M.A. (2002) Phosphorylation and an ATP-dependent process increase the dynamic exchange of H1 in chromatin. J. Cell Biol., 158, 1161–1170. 17. Collepardo-Guevara,R. and Schlick,T. (2011) The effect of linker histone’s nucleosome binding affinity on chromatin unfolding mechanisms. Biophys. J., 101, 1670–1680. 18. Carrero,G., Crawford,E., Hendzel,M.J. and de Vries,G. (2004) Characterizing fluorescence recovery curves for nuclear proteins undergoing binding events. Bull. Math. Biol., 66, 1515–1545. 19. Grigoryev,S.A., Arya,G., Correll,S., Woodcock,C.L. and Schlick,T. (2009) Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl Acad. Sci. USA, 106, 13317–13322. 20. Leuba,S.H., Yang,G., Robert,C., Samori,B., van Holde,K., Zlatanova,J. and Bustamante,C. (1994) Three-dimensional structure of extended chromatin fibers as revealed by tapping-mode scanning force microscopy. Proc. Natl Acad. Sci. USA, 91, 11621–11625. 21. Cui,Y. and Bustamante,C. (2000) Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl Acad. Sci. USA, 97, 127–132. 22. Bennink,M.L., Leuba,S.H., Leno,G.H., Zlatanova,J., de Grooth,B.G. and Greve,J. (2001) Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nat. Struct. Biol0, 8, 606–610. 23. Brower-Toland,B.D., Smith,C.L., Yeh,R.C., Lis,J.T., Peterson,C.L. and Wang,M.D. (2002) Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl Acad. Sci. USA, 99, 1960–1965. 24. Pope,L.H., Bennink,M.L., van Leijenhorst-Groener,K.A., Nikova,D., Greve,J. and Marko,J.F. (2005) Single chromatin fiber stretching reveals physically distinct populations of disassembly events. Biophys. J., 88, 3572–3583. 25. Roopa,T. and Shivashankar,G.V. (2006) Direct measurement of local chromatin fluidity using optical trap modulation force spectroscopy. Biophys. J., 91, 4632–4637. 26. Kruithof,M., Chien,F.-T., Routh,A., Logie,C., Rhodes,D. and van Noort,J. (2009) Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nat. Struct. Mol. Biol., 16, 534–540. 8816 Nucleic Acids Research, 2012, Vol. 40, No. 18 27. Katritch,V., Bustamante,C. and Olson,W.K. (2000) Pulling chromatin fibers: computer simulations of direct physical micromanipulations. J. Mol. Biol., 295, 29–40. 28. Zhang,Q., Beard,D.A. and Schlick,T. (2003) Constructing irregular surfaces to enclose macromolecular complexes for mesoscale modeling using the Discrete Surface Charge Optimization (DiSCO) algorithm. J. Compt. Chem., 24, 2063–2074. 29. Arya,G. and Schlick,T. (2006) Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl Acad. Sci. USA, 103, 16236–16241. 30. Arya,G., Zhang,Q. and Schlick,T. (2006) Flexible histone tails in a new mesoscopic oligonucleosome model. Biophys. J., 91, 133–150. 31. Arya,G. and Schlick,T. (2007) Efficient global biopolymer sampling with end-transfer configurational bias Monte Carlo. J. Chem. Phys., 126, 044107. 32. Arya,G. and Schlick,T. (2009) A tale of tails: How histone tails mediate chromatin compaction in different salt and linker histone environments. J. Phys. Chem. A, 113, 4045–4059. 33. Schlick,T. and Perišić,O. (2009) Mesoscale simulations of two nucleosome-repeat length oligonucleosomes. Phys. Chem. Chem. Phys., 11, 10729–10737. 34. Perišić,O., Collepardo-Guevara,R. and Schlick,T. (2010) Modeling studies of chromatin fiber structure as a function of DNA linker length. J. Mol. Biol., 403, 777–802. 35. Zentgraf,H. and Franke,W.W. (1984) Differences of supranucleosomal organization in different kinds of chromatin: cell type-specific globular subunits containing different numbers of nucleosomes. J. Cell. Biol., 99, 272–286. 36. Caño,S., Caravaca,J.M., Martı́n,M. and Daban,J.-R. (2006) Highly compact folding of chromatin induced by cellular cation concentrations. Evidence from atomic force microscopy studies in aqueous solution. Eur. Biophys. J., 35, 495–501. 37. Hyeon,C. and Thirumalai,D. (2011) Capturing the essence of folding and functions of biomolecules using coarse-grained models. Nat. Commun., 2, 1–11. 38. Schlick,T., Hayes,J.J. and Grigoryev,S. (2012) Toward convergence of experimental studies and theoretical modeling of the chromatin fiber. J. Biol. Chem., 287, 5183–5191. 39. Woodcock,C.L., Grigoryev,S.A., Horowitz,R.A. and Whitaker,N.A. (1993) Chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc. Natl Acad. Sci. USA, 90, 9021–9025. 40. Aumann,F., Lankas,F., Caudron,M. and Langowski,J. (2006) Monte Carlo simulation of chromatin stretching. Phys. Rev. E, 73, 041927. 41. Wu,C., Bassett,A. and Travers,A. (2007) A variable topology for the 30-nm chromatin fibre. EMBO Rep., 8, 1129–1134. 42. Kepper,N., Foethke,D., Stehr,R., Wedemann,G. and Rippe,K. (2008) Nucleosome geometry and internucleosomal interactions control the chromatin fiber conformation. Biophys. J., 95, 3692–3705. 43. Aumann,F., Sühnel,J., Langowski,J. and Diekmann,S. (2010) Rigid assembly and Monte Carlo models of stable and unstable chromatin structures: the effect of nucleosomal spacing. Theor. Chem. Acc., 125, 217–231. 44. Kepper,N., Ettig,R., Stehr,R., Marnach,S., Wedemann,G. and Rippe,K. (2011) Force spectroscopy of chromatin fibers: extracting energetics and structural information from Monte Carlo simulations. Biopolymers, 95, 435–447. 45. Mozziconacci,J., Wong,H. and Victor,J.M. (2007) All-atom model of the chromatin fiber containing linker histones: a versatile structure tuned by the repeat length. Febs. J., 274, 78. 46. Voltz,K., Trylska,J., Tozzini,V., Kurkal-Siebert,V., Langowski,J. and Smith,J. (2008) Coarse-grained force field for the nucleosome from self-consistent multiscaling. J. Comp. Chem., 29, 1429–1439. 47. Mergell,B., Everaers,R. and Schiessel,H. (2004) Nucleosome interactions in chromatin: fiber stiffening and hairpin formation. Phys. Rev. E., 70, 011915. 48. Schiessel,H., Gelbart,W.M. and Bruinsma,R. (2001) DNA folding: Structural and mechanical properties of the two-angle model for chromatin. Biophys. J., 80, 1940–1956. 49. Ben-Haı̈m,E., Lesne,A. and Victor,J.-M. (2001) Chromatin: a tunable spring at work inside chromosomes. Phys. Rev. E., 64, 051921. 50. Stehr,R., Kepper,N., Rippe,K. and Wedemann,G. (2008) The effect of internucleosomal interaction on folding of the chromatin fiber. Biophys. J., 95, 3677–3691. 51. Stigter,D. (1977) Interactions of highly charged colloidal cylinders with applications to double-stranded. Biopolymers, 16, 1435–1448. 52. Allison,S., Austin,R. and Hogan,M. (1989) Bending and twisting dynamics of short linear DNAs. Analysis of the triplet anisotropy decay of a 209 base pair fragment by Brownian simulation. J. Chem. Phys., 90, 3843–3854. 53. Heath,P.J., Gebe,J.A., Allison,S.A. and Schurr,J.M. (1996) Comparison of analytical theory with Brownian dynamics simulations for small linear and circular DNAs. Macromolecules, 29, 3583–3596. 54. Allan,J., Staynov,D. and Gould,H. (1980) Reversible dissociation of linker histone from chromatin with preservation of internucleosomal repeat. Proc. Natl Acad. Sci. USA, 77, 885–889. 55. Bharath,M.M., Chandra,N.R. and Rao,M.R. (2002) Prediction of an HMG-box fold in the C-terminal domain of histone H1: insights into its role in DNA condensation. Proteins, 49, 71–81. 56. Korolev,N., Yanping,F., Lyubartsev,A.P. and Nordenskiöld,L. (2012) Modelling chromatin structure and dynamics: Status and prospects. Curr. Opio. Struct. Biol., 22, 1–9. 57. Banères,J., Martin,A. and Parello,J. (1997) The N tails of histones H3 and H4 adopt a highly structured conformation in the nucleosome. J. Mol. Biol., 273, 503–508. 58. Wang,X., Moore,S.C., Laszckzak,M. and Ausi,J. (2000) Protein synthesis post-translation modification and degradation. J. Biol. Chem., 275, 35013–35020. 59. Kato,H., Gruschus,J., Ghirlando,R., Tjandra,N. and Bai,Y. (2009) Characterization of the N-terminal tail domain of histone H3 in condensed nucleosome arrays by hydrogen exchange and NMR. J. Am. Chem. Soc., 131, 15104–15105. 60. Potoyan,D.A. and Papoian,G.A. (2011) Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J. Am. Chem. Soc., 133, 7405–7415. 61. Frenkel,D., Mooij,G.C.A. and Smit,B. (1992) Novel scheme to study structural and thermal-properties of continuously deformable molecules. J. Phy. Condens. Matte., 4, 3053–3076. 62. de Pablo,J.J., Laso,M. and Suter,U.W. (1992) Simulation of polyethylene above and below the melting point. J. Chem. Phys., 96, 2395–2403. 63. Rosenbluth,M.N. and Rosenbluth,A.W. (1955) Monte Carlo calculation of the average extension of molecular chains. J. Chem. Phys., 23, 356–359. 64. Th’ng,R., Sung,M.Y. and Hendzel,M.J. (2005) H1 family histones in the nucleus. Control of binding and localization by the C-terminal domain. J. Biol. Chem., 280, 27809–27814. 65. Routh,A., Sandin,S. and Rhodes,D. (2008) Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc. Natl Acad. Sci. USA, 105, 8872–8877. 66. Yin,H., Wang,M.D., Svoboda,K., Landick,R., Block,S.M. and Gelles,J. (1995) Transcription against an applied force. Science, 270, 1653–1657. 67. Wang,M.D., Schnitzer,M.J., Yin,H., Landick,R., Gelles,J. and Block,S.M. (1998) Force and velocity measured for single molecules of RNA polymerase. Science, 282, 902–907. 68. Wuite,G.J., Smith,S.B., Young,M., Kelle,D. and Bustamante,C. (2000) Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature, 404, 103–106. 69. Maier,B., Bensimon,D. and Croquette,V. (2000) Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc. Natl Acad. Sci. USA, 97, 12002–12007. 70. Galburt,E.A., Grill,S.W., Wiedmann,A., Lubkowska,L., Choy,J., Nogales,E., Kashlev,M. and Bustamante,C. (2007) Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature, 446, 820–823. 71. Smith,S.B., Cui,Y. and Bustamante,C. (1996) Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded. Science, 271, 795–799. 72. Bancaud,A., Conde e Silva,N., Barbi,M., Wagner,G., Allemand,J.F., Mozziconacci,J., Lavelle,C., Croquette,V., Nucleic Acids Research, 2012, Vol. 40, No. 18 8817 Victor,J.M., Prunell,A. et al. (2006) Structural plasticity of single chromatin fibers revealed by torsional manipulation. Nat. Struct. Mol. Biol., 13, 444–450. 73. Simon,M., North,J.A., Shimko,J.C., Forties,R.A., Ferdinand,M.B., Manohar,M., Zhang,M., Fishel,R., Ottesen,J.J. and Poirier,M.G. (2011) Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl Acad. Sci. USA, 108, 12711–12716. 74. Neumann,H., Hancock,S.M., Buning,R., Routh,A., Chapman,L., Somers,J., Owen-Hughes,T., van Noort,J., Rhodes,D. and Chin,J.W. (2009) A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell, 36, 153–163. 75. Dechassa,M.L., Zhang,B., Horowitz-Scherer,R., Persinger,J., Woodcock,C.L., Peterson,C.L. and Bartholomew,B. (2008) Architecture of the SWI/SNF-nucleosome complex. Mol. Cell. Biol., 28, 6010–6021. 76. Clapier,C.R. and Cairns,B.R. (2009) The biology of chromatin remodeling complexes. Annu. Rev. Biochem., 78, 273–304. 77. Mavrich,T.N., Ioshikhes,I.P., Venters,B.J., Jiang,C., Tomsho,L.P., Qi,J., Schuster,S.C., Albert,I. and Pugh,B.F. (2008) A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res., 18, 1073–1083. 78. Grigoryev,S.A. and Krasheninnikov,I.A. (1982) Transient unfolding of trypsin-digested chromatin core particles. Eur. J. Biochem., 129, 119–125. 79. Lavelle,C., Victor,J.-M. and Zlatanova,J. (2010) Chromatin fiber dynamics under tension and torsion. Int. J. Mol. Sci., 11, 1557–1579. 80. Shukla,M.S., Syed,S.H., Goutte-Gattat,D., Richard,J.L.C., Montel,F., Hamiche,A., Travers,A., Faivre-Moskalenko,C., Bednar,J., Hayes,J.J. et al. (2011) The docking domain of histone H2A is required for H1 binding and RSC-mediated nucleosome remodeling. Nucleic Acids Res., 39, 2559–2570. 81. Cremer,T. and Cremer,C. (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet., 2, 292–301. 82. Koundrioukoff,S., Polo,S. and Almouzni,G. (2004) Interplay between chromatin and cell cycle checkpoints in the context of ATR/ATM-dependent checkpoints. DNA Repair, 3, 969–978. 83. Hu,Y., Kireev,I., Plutz,M., Ashourian,N. and Belmont,A.S. (2009) Large-scale chromatin structure of inducible genes: transcription on a condensed, linear template. J. Cell. Biol., 185, 87–100. 84. Baù,D., Sanya,A., Lajoie,B.R., Capriotti,E., Byron,M., Lawrence,J.B., Dekker,J. and Marti-Renom,M.A. (2011) The three-dimensional folding of the a-globin gene domain reveals formation of chromatin globules. Nat. Struct. Mol. Biol., 18, 107–114. 85. Canzio,D., Chang,E.Y., Shankar,S., Kuchenbecker,K.M., Simon,M.D., Madhani,H.D., Narlikar,G.J. and ADN Al-Sady,B. (2011) Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell, 41, 67–81. 86. McGowan,S., Buckle,A.M., Irving,J.A., Ong,P.C., BashtannykPuhalovich,T.A., Kan,W.-T., Henderson,K.N., Bulynko,Y.A., Popova,E.Y., Smith,I. et al. (2006) X-ray crystal structure of MENT: evidence for functional loop-sheet polymers in chromatin condensation. EMBO J., 25, 3144–3155.