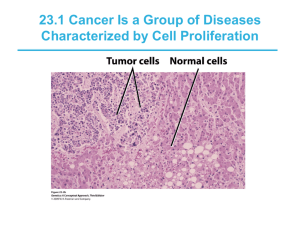
In vivo disease progression in hematopoietic malignancies
advertisement

In vivo pool-based shRNA screens to identify modulators of disease progression in hematopoietic malignancies by Corbin Elizabeth Meacham B.S. Biology University of Chicago, 2006 Submitted to the Department of Biology in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology at the Massachusetts Institute of Technology February 2012 © 2012 Massachusetts Institute of Technology. All rights reserved. Signature of Author: ________________________________________________ Department of Biology December 21, 2011 Certified by: ______________________________________________________ Michael T. Hemann Associate Professor of Biology Thesis Supervisor Accepted by: _____________________________________________________ Tania A. Baker E. C. Whitehead Professor of Biology Co-chairman, Biology Graduate Committee In vivo pool-based shRNA screens to identify modulators of disease progression in hematopoietic malignancies by Corbin Elizabeth Meacham Submitted to the Department of Biology on January 12, 2012 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biology Abstract shRNA screens have been very effective in identifying novel cancer genes in mammalian cells, but they have primarily been limited to in vitro applications in tumor cell lines. Whereas in vivo retroviral mutagenesis screens typically identify gain-of-function alterations in positive selection screens, shRNA screening approaches allow for the systematic interrogation of the impact of loss of function events across large gene sets. Using transplantable mouse models of Eµ-myc lymphoma and Bcr-Abl driven B-cell acute lymphoblastic leukemia, we have adapted shRNA screening approaches to in vivo applications and have performed large-scale loss of function genetic screens in tumors. Initial work suggested that screens with complex shRNA libraries could be performed in the in vivo setting using a model of Eµ-myc lymphoma. Here, the introduction of an shRNA library into lymphoma cells ex vivo, followed by transplantation, showed that as many as 1000 unique hairpins were represented in tumors in individual recipient animals. The set of genes targeted by shRNAs that negatively impacted lymphoma cell growth in vivo was highly enriched for regulators of cell motility, and suppression of these genes, which included Rac2, CrkL, and the poorly characterized actin monomer binding protein Twf, impaired lymphoma progression and tumor cell dissemination to sites of terminal metastatic disease. Additionally, suppression of Rac2 and Twf improved chemotherapeutic outcome, suggesting that lymphoma cell migration following therapy, from sites of minimal residual disease to sites of terminal disease, may be an important event in relapse. In a second screen using a BCR-Abl B-cell acute lymphoblastic leukemia model, we found that as many as 9,000 unique shRNAs could be identified in individual animals following tumor cell transplantation. Based on this result, we were able to perform an unbiased screen for modulators of leukemia cell growth in vivo using a genome-scale shRNA library composed of five pools of 10,000 shRNAs each. shRNAs that negatively impacted leukemia cell growth specifically in vivo were selected for validation, which included a set of hairpins targeting poorly characterized C2H2 zinc finger proteins. While neutral in the in vitro setting, a number of these shRNAs validated as single constructs in vivo. As an additional filter to select candidates for validation, we compared the set of genes targeted by candidate hairpins with genes that are transcriptionally upregulated in leukemia cells in vivo, and the leukemia genes Runx and Lmo2 met both of these criteria. Suppression of Runx and Lmo2 was selected against in tumors, suggesting that oncogenic pathways downstream of these genes may be used for leukemia cell growth or survival in vivo. We also compared the set of genes targeted by depleting hairpins with published DNA copy number alteration data from human ALL patients. In validation studies, we identified genes within regions of genomic alteration that impacted tumor cell 2 proliferation in vivo. One of these genes was the plant homeodomain finger protein Phf6. While inactivating mutations in Phf6 are a common alteration in T cell malignancies, we found that suppression of this gene negatively impacted the growth of B cell malignancies in vivo, indicative of lineage-specific role for Phf6 in hematopoietic tumors. Thesis advisor: Michael Hemann Title: Associate Professor of Biology 3 Table of Contents Abstract…………………………………………………………………………….……………..2 Table of Contents…………………………………………………………………….…….……4 Acknowledgements…………………………………………………………………….…….….6 Chapter 1: Introduction….………………………………………………………………….…...8 Insertional mutagenesis screening in heamtopoietic malignancies………………9 Transposon-based in vivo screens in hematopoietic and non-hematopoieitic tissues…………………….………………………………….….17 RNAi-based screens in mammalian cells……..…………………………..……..….22 In vivo pool-based shRNA screens in hematpoietic malignancies……..………...27 References……………………………………………………………………...….…..30 Chapter 2: In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression………………………………………..……39 Abstract…………………………………………….……………………………..…….40 Introduction………………………………………………...…………………...……...40 Results and Discussion…………………………………………………………….....42 Acknowledgements and author contributions…….………………………..…….48 References…………………………….………………………………………..…...49 Materials and methods………………….…………………………………..……...52 Figures……………………………………….………………………………..……..56 Tables………………………………………….………………………………..…...64 Supplementary Figures………………………...……………………………..…….65 Supplementary Tables……………………………...………………………..……..72 Chapter 3: Genome-scale in vivo screening reveals compensatory oncogenic pathways in B-cell leukemia………………………………….…………………………………….……..78 4 Abstract………………………………………….………………………….…......79 Introduction……………………………………….……...………………….….....79 Results and Discussion…………………………..…………………………..…..80 Acknowledgements and author contributions………..…………………..…….89 References………………………………………………..………………..……...90 Materials and methods…………………………………..………………..……...93 Figures……………………………………………………..…………..…………..95 Supplementary Figures…………………………….…………………..……….103 Supplementary Tables………………………………..………………..………..104 Chapter 4: Conclusions……………………………………………………………....……..109 5 Acknowledgements I would like to thank… Mike Hemann, for his support and guidance throughout graduate school. His ideas and critical approaches to scientific problems have been instrumental in moving projects forward. He has created a great lab environment that makes day-to-day lab life fun, and a lab culture that encourages interaction and collaborative scientific discussion. His enthusiasm for scientific discovery and genuine excitement about research have, and will continue, to impact my approach to science, and his sense of humor helps keep everything in perspective. The members of my thesis committee, Frank Gertler and Doug Lauffenburger, for their input on project direction and their scientific, as well as career, advice. In particular, I’d like to thank Frank for always being willing to discuss any scientific issues, as long as it was over a martini. The fantastic members of the Koch Institute core facilities, particularly Richard Cook, Alla Leshinsky, AJ Bhutkar, and Charlie Whittaker. Richard and Alla were responsible for the sequencing of our screening data, and AJ and Charlie developed and utilized bioinformatic pipelines to process these datasets. Their work has been extremely important in obtaining the results presented here. Luke Gilbert and Eleanor Cameron for their input and critical review of this document. Luke Gilbert, Hai Jiang, Jason Doles, and Justin Pritchard, who comprised the first crop of Hemann lab members. The days of sack playing, precision drinking, pool parties, sausage making, afternoons of loud 80’s music, and hot dog safaris, among other things, made graduate school a great experience. It goes without saying that scientific discussions with all of these individuals were also very helpful. My baymate, Holly Criscione. She patiently put up with my mid-day bay napping and shoeless science over the years. She has been key in keeping the Hemann lab running smoothly, especially when considering what she has to work with. All members of the Hemann lab, for being great colleagues. 6 My family, especially my mom and dad, who have been extremely supportive throughout graduate school. 7 Chapter 1: Introduction ______________________________________________________________________ 8 Insertional mutagenesis screening in hematopoietic malignancies Historically, genetic screens have been performed using the slow transforming viruses MuLV (murine leukemia virus) and MMTV (mouse mammary tumor virus) in the mouse hematopoietic system and mammary glands, respectively. Unlike acute transforming viruses, which contain viral oncogenes that rapidly induce disease in inoculated animals following infection, these viruses produce malignancies with a protracted latency, and their effect is mediated by viral alterations of host cell gene expression rather than by viral expression of an oncogene (Hayward et al., 1981; Jenkins et al., 1981; Neel et al., 1981; Varmus et al., 1981). In the case of MLV-induced disease, newborn mice are infected with the virus, which then propagates in the hematopoietic compartment (Rowe and Pincus, 1972). The virus stably integrates into the genome of host cells, and depending on the integration event, can either disrupt gene function, or enhance gene expression, with viral promoter or enhancer elements driving expression of proximal genes. When the integration event provides a selective advantage, or promotes transformation of the host cell, clonal outgrowth will occur, yielding hematopoietic tumors. Frequently, multiple clonal populations contribute to tumors in individual mice. Candidate transforming genes can then be identified based on their proximity to common insertion sites, or sites of viral integration in the genome that are found in multiple independent tumors at a frequency that is higher than expected by chance (Jonkers and Berns, 1996; Kool and Berns, 2009; Uren et al., 2005). Additionally, individual cells can undergo multiple rounds of infection, suggesting that sequential integration events near oncogenes at different stages of transformation may 9 contribute to disease development and progression (Cuypers et al., 1986; Kool and Berns, 2009; Selten et al., 1984). Insertional mutagenesis screens have effectively identified a number of key oncogenes in hematopoietic malignancies. Typically, Moloney MuLV infection of newborn mice produces T cell lymphomas; in these tumors, Myc (Corcoran et al., 1984; Selten et al., 1984) and Pim-1 (Cuypers et al., 1984; Selten et al., 1985) are frequently proximal to common insertion sites, resulting in upregulation of expression of these genes. In the case of Myc, this upregulation has been attributed to viral enhancer elements, whereas in the case of Pim-1, integration of the virus into the 3’ region of the gene is thought to truncate the 3’ UTR, stabilizing the transcript. Viruses can also induce gene expression by integrating into promoter regions. In a mouse strain predisposed to develop myeloid tumors (Mucenski et al., 1986), a common retroviral insertion site was identified in the promoter of Evi-1 (Mucenski et al., 1988). While Evi-1 is not normally expressed at high levels in hematopoietic tissues, viral activation of this zinc finger protein seems to negatively impact the ability to myeloid cell lines to terminally differentiate (Morishita et al., 1988). Alternatively, viral integrations can truncate cellular transcripts, resulting in the deletion of regulatory protein domains and producing gene products that are highly oncogenic. This type of viral alteration has been observed in Notch in T cell lymphomas, in which truncating viral insertions produce a gene product that lacks the 5’ ligandbinding extracellular domain, rendering Notch ligand independent. Additional insertions have also been observed in the carboxyl terminus of Notch, which results in increased stability of the Notch protein (Feldman et al., 2000; Girard et al., 1996; Hoemann et al., 2000). Interestingly, both of these alterations mimic mutations that are observed in a 10 large proportion of human T-cell acute lymphoblastic leukemia patients (Weng et al., 2004). In addition to identifying genes that promote tumor development, insertional mutagenesis screens have also been used to identify genes that cooperate with other oncogenes in accelerating tumor development. In these instances, insertional mutagenesis screens have been performed in transgenic mice that are predisposed to develop specific hematopoietic malignancies. Infection of these animals with a mutagenic virus reduces the latency to disease development, and cooperating oncogenes are found proximal to common insertion sites. In early studies with mice expressing high levels of the putative oncogene Pim-1, in which only a small percentage of animals develop T cell lymphomas, infection with MuLV accelerated disease onset, and 100% of animals developed malignancies. In the resulting tumors, either c-Myc or nMyc were activated by viral insertions, suggesting that Myc and Pim-1 cooperate in the development of T cell malignancies (van Lohuizen et al., 1989). In reciprocal insertional mutagenesis screens, Pim-1 was identified as a cooperating oncogene in Eµ-myc transgenic mice. In these mice, expression of high levels of Myc in the B cell compartment predisposes animals to develop B cell lymphomas (Adams et al., 1985), and infection of these animals with MuLV results in accelerated B cell, rather than T cell, malignancies, demonstrating that the Eu-myc transgenic background can alter the tumor spectrum of MuLV-induced tumors. Additionally, this data indicates that these Myc and Pim-1 can cooperate in the development of both B and T cell malignancies (Haupt et al., 1991; van Lohuizen et al., 1991). The oncogenic cooperation between Myc and Pim-1 was functionally validated in crosses between Eµ-Myc and Eµ-Pim-1 mice, where Pim-1 accelerates 11 lymphomagenesis in Eµ-Myc animals; Eµ-Myc Eµ-Pim-1 animals develop a prenatal Bcell lymphoblastic leukemia, whereas Eµ-Myc animals develop B cell malignancies with a latency of 2.5 to 12 months (Verbeek et al., 1991). In addition to identifying Pim-1, insertional mutagenesis screens have also identified a novel zinc finger protein and putative transcriptional regulator, Bmi-1, as cooperating with Myc in lymphoma development. Validation studies have shown that Bmi-1 can function as an oncogene; mice overexpressing Bmi-1 develop B and T cell malignancies, and elevated Bmi-1 expression accelerates Myc-mediated lymphomagenesis (Alkema et al., 1997; Haupt et al., 1993). Bmi-1 has since been shown to be a polycomb group protein that negatively regulates the Ink4a-Arf locus and thus cooperates with Myc by preventing Myc-mediated upregulation of this locus and the induction of apoptosis (Jacobs et al., 1999a; Jacobs et al., 1999b). More recent advances in sequencing technology, as well as annotation and assembly of the mouse genome, have facilitated the discovery of large numbers of novel common insertion sites and candidate oncogenes in insertional mutagenesis screens (Li et al., 1999; Lund et al., 2002; Mikkers et al., 2002; Suzuki et al., 2002). By PCR amplifying and sequencing the cellular DNA flanking viral insertion sites, the numerous independent insertions in individual tumors can be identified more comprehensively. Additionally, since integrated retroviruses can exert their effects over long distances due to enhancer elements, the assembled mouse genome allows for candidate genes that are distant to insertion site to be associated with that insertion event. Large-scale studies have now identified hundreds of unique insertion sites across panels of tumors, many of which are proximal to novel genes. 12 High-throughput insertional mutagenesis screens have been used to look for genotype-specific interactions (Lund et al., 2002; Uren et al., 2008), and to identify new genes involved in specific pathways (Mikkers et al., 2002). As an example of the latter, one screen was designed to identify genes involved in the Pim-1 and Pim-2 pathways. Previously, the authors had performed a screen in Eµ-Myc Pim-1-/- animals. Since Pim-1 is important in Eµ-Myc lymphomagenesis, insertion events that activate compensatory genes, either by genes downstream of Pim-1 or genes functioning in parallel pathways, would be expected to confer a selective advantage in tumor development. They found that retrovirus-mediated activation of the Pim-1 related gene, Pim-2, was observed in 80% of Eµ-Myc Pim-1-/- lymphomas (van Lohuizen et al., 1991). To identify genes that compensate for Pim-1 and Pim-2 loss, the authors performed an insertional mutagenesis screen in Eµ-Myc Pim-1-/- Pim-2-/- mice, and found that Pim-3, as well as a number of other genes, were specifically upregulated by viral integrations in the absence of Pim-1 and Pim-2, and therefore may function downstream or parallel to Pim-1 and Pim-2. Large-scale insertional mutagenesis screens have also been performed in Cdkn2a-/mice (Lund et al., 2002), as well as p53-/- and p19Arf-/- animals (van Lohuizen et al., 1991), to identify genes that are preferentially mutated on these genetic backgrounds. In the latter study, over 10,000 unique insertion sites from approximately 500 tumors were sequenced, with an average of over 20 unique insertions per mouse. With this number of independent insertion events, the authors were able to achieve the statistical power to necessary to determine if integration events occurred only in particular genetic backgrounds, or were found in the context of multiple genetic backgrounds. When only a subset of the integrations in each tumor are sequenced, integration sites that appear to be unique to a specific tumor genotype may actually be present in tumors of multiple genotypes, but may fail to be detected. Thus, improvements in sequencing technology 13 have facilitated the discovery of alterations that cooperate with specific genetic lesions in insertional mutagenesis screens. While insertional mutagenesis screens have been very effective in identifying important genes that promote the initiation or progression of hematopoietic malignancies, this approach does have a several limitations. Insertional mutagenesis screens require positive selection for a given phenotype, generally tumor development. Consequently, negative selection screens, or screens that identify genes, via gain or loss of function, that negatively impact a disease process, have not been performed. Additionally, gain of function mutations in oncogenes, rather than loss of function mutations in tumor suppressor genes, preferentially undergo positive selection in insertional mutagenesis screens. A single viral integration event proximal to an oncogene is sufficient to confer a selective advantage in these screens, whereas two independent viral integration events are required to disrupt both alleles of a tumor suppressor gene, and therefore, tumor suppressor genes are rarely identified in insertional mutagenesis screens. Only 10% of common viral insertion events were predicted to disrupt gene function in one large-scale screen (Suzuki et al., 2002), and in these cases, it was unclear if disrupted gene products were actually truncated oncogenic proteins lacking regulatory domains, rather than inactivated genes. In cases of monoallelic inactivation, disrupted genes may represent haploinsufficient tumor suppressors. One of the few instances where viral inactivation of a tumor suppressor has been observed is in Nf1; in these tumors, the wildtype Nf1 transcript could not be detected, suggesting that the gene had undergone bi-allelic inactivation, either by viral integration into both alleles, or by epigenetic silencing or mutation of the wildtype allele (Largaespada et al., 1995). To facilitate the identification of tumor suppressor genes, one 14 group performed an insertional mutagenesis screen in a Blm-deficient background. In this context, the frequency of heterozygous gene inactivation events becoming homozygous is increased, due to increased rates of mitotic recombination between nonsister chromatids. Even on this sensitized background, the majority of common insertion sites were predicted to activate, rather than disrupt, gene expression. However, the authors identified a number of viral integration events in the coding regions of genes that had become homozygous, and that were accompanied by decreased transcript levels of the target gene, strongly suggesting that these integration events had occurred in tumor suppressors (Suzuki et al., 2006). Additional challenges arise in identifying common insertion sites. Murine leukemia viruses preferentially insert into transcribed genes, near transcriptional start sites (Berry et al., 2006; Wu et al., 2003). When a background distribution of random viral integrations throughout the genome is assumed, clustering of insertion events in these genomic locations can result in false positives. Identification of putative oncogenes, or the genes that are impacted by common insertion events, is also difficult. Because viral enhancer elements can act over long distances (Lazo et al., 1990), viruses may increase the expression of distant genes, rather than the most proximal gene. To systematically identify virally deregulated genes, one group looked at the transcript levels of all genes within 100 kb of common insertion sites across a panel of tumors. For about 50% of insertion sites, they could not identify a deregulated gene within this window, and for other insertion sites, they found that multiple genes were often deregulated and that genes far from the insertion sites were overexpressed as frequently as proximal genes (Sauvageau et al., 2008). 15 Candidate genes identified in insertional mutagenesis screens require experimental validation to show that they are bona fide oncogenes, or more infrequently, tumor suppressors. Traditionally, this has been done by making transgenic mice, and showing that a gene accelerates tumor development (Alkema et al., 1997; Haupt et al., 1993; Verbeek et al., 1991). Now that individual screens can identify hundreds of common insertion sites, testing candidates on a gene-by-gene basis is becoming increasingly difficult. These screens rely on the re-identification of known oncogenes to validate the quality of the datasets, and then comparison of screening data, human mutational data, and tumor copy number alteration data to identify potentially interesting and novel genes. However, without assays to rapidly assess the phenotypic impact of overexpression of candidate genes, the identification of functionally important genes within these large datasets remains challenging. Additionally, viral tropisms limit the types of tumors that can be studied in insertional mutagenesis screens. To produce a tumor, a virus must be able to infect a tissue, be expressed at high levels, and replicate within that tissue. Even within the hematopoietic system, different leukemia viruses produce tumors in distinct cell lineages; Graffi-type viruses usually produce myeloid malignancies (Erkeland et al., 2004), whereas Moloney viruses produce T cell malignancies, unless screens are performed on genetic backgrounds that are predisposed to B cell malignancies. In addition to the hematopoietic system, insertional mutagenesis screens have also been performed in the mammary gland, which can be infected by the mouse mammary tumor virus (MMTV). In these tumors, insertions proximal to Fgf and Wnt family members are frequently observed (Dickson et al., 1984; Nusse and Varmus, 1982; Peters et al., 1983; Theodorou et al., 2007). In another solid tumor model, one group initiated gliomas by 16 infecting the brains of mice with a Moloney MLV engineered to express PDGF, and identified a number of candidate genes that may cooperate with PDGF in tumorigenesis based on common insertion sites (Johansson et al., 2004; Uhrbom et al., 1998). Thus, while used in small number of non-hematopoietic tumors, insertional mutagenesis screens have not been broadly adapted to solid tumors. Transposon-based in vivo screens in hematopoietic and non-hematopoieitic tissues More recently, systems to perform in vivo transposon-based insertional mutagenesis screens have been developed. Unlike retroviral insertional mutagenesis screens, transposon-based mutagenesis screens can be performed in a broad array of tissue types. These screens make use of a bipartite system, where transgenic mice are generated that express concatemers of a mobile element, the transposon, and, in trans, the transposase, which is an enzyme that can mobilize the transposon. Once mobilized, transposons will re-integrate throughout the genome and can alter gene function. The reactivation of an ancient Tcl/mariner transposon has made transposonbased screens possible. This family of transposons is one of the most widespread, which suggested that family members may be less dependent on species-specific host factors, and therefore, if reactivated, might function in a range of host cells. By comparing sequences of twelve inactive salmanoid-type transposons, which are the youngest and most recently active transposons in the Tcl/mariner family, a consensus sequence for the active transposon was derived. From this consensus sequence, an 17 active transposase, termed Sleeping Beauty, was engineered and found to function in mouse and human cells (Ivics et al., 1997). In transposon screens, mice expressing the active Sleeping Beauty transposase are crossed to animals that have germline-integrated concatemers of transposons. Transposons are designed to contain polyadenylation sequences and splice acceptors, so insertion events can disrupt the function of tumor suppressor genes, as well as a viral LTR and splice donor cassette, allowing these elements to drive the expression of proximal proto-oncogenes. By expressing multiple copies of transposons in animals, the frequency of tumorigenic reintegration events increases. Once tumors arise, transposon junctions are PCR amplified and sequenced, and genes that are altered by transposons in multiple tumors are considered candidate oncogenes or tumor suppressors. In one of the first in vivo transposon screens, crosses between transgenic mice ubiquitously expressing an active transposase and animals with concatemers of 150-300 copies of a mutagenic transposon produced offspring that rapidly developed malignancies. Hematopoietic malignancies predominated, the majority of which were T cell lymphomas, although a small number of neoplasms and tumors were observed in other tissues (Dupuy et al., 2005). In another screen published at the same time, mice harboring fewer copies (approximately 25) of a transposon were crossed to transpoaseexpressing animals. Although no tumors developed in wildtype mice, soft tissue sarcomas did develop on a sensitized Arf-/- background at an accelerated rate (Collier et al., 2005). These studies identified frequent insertions in known oncogenes; activating Notch mutations were observed in T cell malignancies, and activating Braf mutations were seen in sarcomas. 18 While this initial work demonstrated the feasibility of using transposons for in vivo screens, they either primarily produced hematopoietic malignancies, or failed to produce tumors on non-sensitized genetic backgrounds. Subsequent modifications to these screens expanded the spectrum of tumors that could be studied. Alterations to the viral promoter within transposons were sufficient to change the tumor spectrum when combined with a ubiquitously expressed transposase; by replacing the MSCV LTR, which is highly expressed in hematopoietic cells, with a CAG promoter, which is expressed in a variety of tissue types, squamous cell carcinomas and hepatocellular carcinomas developed, rather than T cell malignancies (Dupuy et al., 2009). To precisely control the tumor spectrum in transposon screens, conditional systems have been developed, where the Sleeping Beauty transposase is floxed by lox-stop-lox sites. These mice have been crossed with animals expressing a tissue specific Crerecomibinase, restricting transposase expression to target tissues. When villin-Cre was used to activate transposase expression in the gastrointestinal tract, intestinal neoplasias and adenomas developed, and the human colorectal cancer gene APC was frequently inactivated by transposon insertions (Starr et al., 2009). By crossing lox-stoplox transposase animals with mice expressing the hepatocyte-specific albumin-Cre, transposition events were limited to the liver, resulting in the formation of pre-neoplastic nodules in this organ. In these nodules, Egfr was frequently truncated by transposon insertions, and a number of novel common insertion sites were also identified (Keng et al., 2009). The moth transposon PiggyBac has also been reactivated and shown to be functional in mammalian cells and in transgenic mice (Ding et al., 2005). When PiggyBac transposase mice were crossed to a number of different transposon lines, which varied 19 both in transposon copy number and the viral promoter contained within the transposon, double transgenic offspring developed hematopoietic and solid tumors. Similar to previous studies, the viral promoter skewed the tumor distribution; tumors with transposons containing the MSCV LTR were primarily hematopoietic, and transposons with the CAG promoter produced solid tumors. When common insertion sites from PiggyBac or Sleeping Beauty hematopoietic tumors were compared, a large proportion of these sites were novel to the PiggyBac system, suggesting that these two transposons may have different integration preferences. Thus, the range of mutations identified in transposon screens may be expanded by using multiple transposition systems (Rad et al., 2010). Using transposon mutagenesis approaches, in vivo screens can be performed in a broad array of tissues. In addition to identifying genes that promote tumorigenesis in solid and hematopoietic malignancies, they have also been used to identify mutations that cooperate with established oncogenic lesions. For example, in the intestine, transposon screens have been performed in mice with APC mutations (March et al., 2011). One group showed that a mutation in the gene Nucleophosmin, which is mutated in human acute myeloid leukemia (AML), produces AML in mice with a protracted latency, and used transposon screens to identify cooperating alterations that accelerate disease development (Vassiliou et al., 2011). While these screens have been very effective, they share a number of limitations with insertional mutagenesis screens. Like insertional mutagenesis screens, transposon screens are primarily positive selection screens. They identify genes, that when deregulated, promote tumor development, rather than genes that can negatively impact this process. Thus, they are useful in elucidating novel pathways that are important for 20 tumorigenesis, but cannot directly identify genes whose loss can impair tumor growth and progression, particularly in established tumors. Additionally, while transposons are designed to be able to disrupt the function of tumor suppressors and enhance the expression of proto-oncogenes, they are still biased towards the identification of oncogenes, since gain of function alterations require integration in only one allele of a gene, whereas inactivation of tumor suppressors requires bi-allelic transposon insertions. In intestinal lesions, insertions in the knows tumor suppressors Pten, Smad4, and Bmpr1a have been identified, without inactivation of the other allele, indicating that these genes may be haploinsufficient tumor suppressors in this disease, or that the second allele is inactivated by an alternative mechanism (March et al., 2011). Additionally, bi-allelic inactivation of Pten has been observed hematopoietic tumors (Rad et al., 2010); however, these bi-allelic events are expected to be far less frequent than the activation of oncogenes. The Sleeping Beauty transposon was shown to have relatively little insertional bias when a large number of insertion sites were mapped. Excluding the chromosome carrying the concatemers of transposons, where local hopping events frequently occur, insertion sites were widely distributed throughout the genome, although there were local preferences for AT-rich regions. Insertion events were slightly biased towards transcribed genes, but significantly less so than viruses (Yant et al., 2005). Thus, the identification of common insertion sites should be simplified in these screens, since a relatively uniform background insertion rate can be assumed. However, the large number of non-overlapping common integration sites in PiggyBac and Sleeping Beauty hematopoietic tumors would suggest that some integration biases do exist, and these 21 are dependent on the specific transposon used, which could lead to the false identification of common insertion sites. Like insertional mutagenesis screens, transposon screens generate large numbers of putative hits. Often, the most frequently altered genes are known oncogenes. Using these genes as controls for the quality of the overall dataset, oncogenic pathways and networks are then built based on the set of putative hits identified, to reduce the complex gene sets to a coherent biology. Alternatively, these screens rely on comparisons with other datasets, like tumor mutational or transcriptional data, to suggest that less well-established candidate hits have a causative role in disease development. However, new genes are rarely functionally validated, particularly in in vivo tumor models, making it unclear if the majority of genes identified in these screens are important in tumorigenesis. RNAi-based screens in mammalian cells The more recent utilization of RNA interference (RNAi) in mammalian systems has allowed for the systematic interrogation of the impact of the loss of function of genes in mouse and human cell lines. Initially delivered as chemically synthesized 21mer siRNA duplexes, RNAi constructs were shown to effectively silence target genes in mammalian cells (Caplen et al., 2001; Elbashir et al., 2001). To achieve stable, rather than transient suppression of target genes, these duplexes were modified and expressed endogenously as short-hairpin RNAs composed of a stem-loop structure (Brummelkamp et al., 2002; Paddison et al., 2002; Sui et al., 2002; Yu et al., 2002). To simultaneously assess the impact of the loss of function of large sets of genes and aid in the discovery 22 of novel genes involved in disease processes, libraries of shRNAs were generated that targeted the human and mouse genomes in an unbiased manner. Initially, these libraries were composed of short shRNA duplexes expressed from the RNA polymerase III promoter (Berns et al., 2004; Brummelkamp et al., 2003; Paddison et al., 2004). Based on observations that shRNAs embedded within the full-length mir30 transcript were processed to mature stem-loop structures by endogenous microRNA machinery and effectively silenced their target genes (Zeng et al., 2002), and that expression of shRNAs within this context resulted in increased levels of mature shRNAs, new libraries were synthesized using this shRNA design (Silva et al., 2005). Additionally, by expressing these shRNAs from the RNA polymerase II promoter, single copy genomic integration events were found to be sufficient to silence target genes, and gene silencing was largely independent of the site of integration (Dickins et al., 2005; Stegmeier et al., 2005). shRNA screens can be performed using either pool-based or arrayed approaches. In pooled screens, populations of cells are infected with a pooled shRNA library, and the enrichment or depletion of particular shRNAs within this bulk infected population is used as a readout of the impact of gene suppression on a cellular process. Arrayed screens rely on the infection of individual populations of cells with single shRNA constructs, typically in 96 or 384 well plates. These shRNA-infected cells are then monitored for phenotypic alterations. In one of the first unbiased shRNA screens, a poolbased based approach was used to identify genes that can overcome p53-mediated growth arrest. Here, cells were infected with a large pool of shRNAs, and only cells containing hairpins that promoted the bypass of p53-mediated arrest were able to proliferate, suggesting that genes targeted by these hairpins function in the p53 pathway 23 (Berns et al., 2004). In other early shRNA screening work, arrayed screens were performed to validate large-scale shRNA libraries. These screens were designed to identify genes that are necessary for the proteosome-mediated degradation of proteins. Here, individual shRNAs were co-transfected with a fluorescently tagged protein containing a degradation signal, and shRNAs that interfered with this process were identified by an increase in florescence in infected cell populations (Paddison et al., 2004; Silva et al., 2005). The discovery of genes that are important in cancer has been aided by shRNA screening approaches. Because shRNAs mimic gene loss of function events, these screens can be used to identify novel tumor suppressors. One method of identifying tumor suppressors has been to perform pool-based screens in partially transformed cell lines. In one screen, an unbiased library of shRNAs was introduced into mammary epithelial cells that express hTERT, the large T antigen of SV40, and high levels of Myc. This set of genetic alterations is not sufficient for anchorage-independent growth, a property that is indicative of transformation, but the addition of hairpins targeting a tumor suppressor can cause cells to grow in an anchorage-independent fashion. Here, transduction of partially transformed cells with an shRNA library and sequencing of hairpins that promoted transformation identified the gene REST as a tumor suppressor (Westbrook et al., 2005). Another tumor suppressor screen was performed in a partially transformed fibroblast cell line. In this line, the expression of hTERT, small T antigen, oncogenic RasV12, and suppression of p53 and p16 causes transformation. By omitting RasV12, and introducing libraries of shRNAs, the authors identified a number of hairpins that could substitute for RasV12 expression in transformation, and identified PITX1 as a regulator of Ras signaliing (Kolfschoten et al., 2005). These screens demonstrate how 24 positive selection for a transformed phenotype can facilitate the identification of tumor suppressor genes in pool-based screens. Pool-based negative selection shRNA screens have been used to identify putative drug targets in cancer cells. In these screens, shRNAs that adversely impact a cellular process, like proliferation or survival, are identified by a relative decrease in the abundance of those shRNAs. Initially, the abundance of shRNAs was quantified using microarrays. Here, either a unique barcode was attached to each shRNA, or the unique sequence of each hairpin was used as a molecular tag, and this unique region was PCR amplified from genomic DNA and hybridized to custom arrays containing the bar-coded oligonucleotides (Berns et al., 2004; Ngo et al., 2006; Paddison et al., 2004; Westbrook et al., 2005). Now, these amplified shRNAs can be directly sequenced and quantified using high-throughput approaches. In one example of a sensitization screen, shRNAs that negatively impacted two types of diffuse large B cell lymphomas were compared. Here, a pool of inducible shRNAs targeting approximately 2500 genes were introduced into activated B-cell like lymphomas and germinal center B cell lymphomas, which have distinct molecular profiles and genetic dependencies. Following a period of in vitro proliferation, shRNA abundance was quantified and used to identify genes that specifically impaired activated B-cell like lymphomas. Here, CARD11 was shown to be a modulator of activated B-cell like lymphoma growth (Ngo et al., 2006). Other large-scale negative selection screens have been performed using libraries targeting signaling regulators and genes in cancer-related pathways (Schlabach et al., 2008), as well as unbiased shRNA sets (Silva et al., 2008). By comparing different cell lines, these screens have identified shRNAs that were generally required for cancer cell viability and growth, as well as cell-line specific genetic dependencies. 25 shRNA screens have also been used to devise novel therapeutic approaches and to exploit specific cancer cell vulnerabilities. In synthetic lethal screens, groups have identified shRNAs that are toxic in cell lines expressing oncogenic Ras, but not cell lines with wildtype Ras, which include hairpins targeting regulators of mitotic progression (Luo et al., 2009) and regulators of NFκB signaling (Barbie et al., 2009), and pharmacological inhibition of these gene products may represent a therapeutic strategy in Ras mutant tumors. shRNA screens have also been used to study chemotherapeutic response in cancer cells, both to identify genes that are necessary for the action of drugs (Brummelkamp et al., 2006; Swanton et al., 2007), and to identify genes whose suppression sensitizes cells to therapy (Whitehurst et al., 2007). Additionally, shRNA screens have been performed that examine more complex aspects of cancer progression, like migration and invasion (Simpson et al., 2008). One genome-wide pool based shRNA screen identified a set of hairpins that increased cell migration into an extracellular matrix, as well as cell survival and proliferation within this matrix, using a three-dimensional culture system, and found that candidate hits from this screen also increased lung metastasis in in vivo assays (Gobeil et al., 2008). Thus, shRNA screens have been used to identify genes that impact diverse processes in cancer cells. By using shRNA screens, the impact of the loss of function of large numbers of genes can be simultaneously assayed. While early pool-based screens were positive selection screens, relying on the outgrowth of rare cells under growth-inhibitory conditions, methods to deconvolute shRNA representation have made negative selection screens possible. Bulk populations of cells can be infected with large pools of shRNAs, and hairpins that increase or decrease in representation after a period of growth, relative to an input population, can be detected using microarray or high-throughput sequencing 26 approaches. Additionally, improvements in high-throughput sequencing technology are now providing sufficient resolution to detect more subtle changes in shRNA representation in these screens. In contrast to insertional mutagenesis screens, scoring candidates in shRNA screens can be rapidly functionally validated. By re-introducing scoring shRNAs into cells as a single constructs, as well as additional shRNAs targeting the same gene, it is possible to easily assess if suppression of candidate genes recapitulates the screening phenotype. This greatly facilitates assigning candidate hits roles in biological processes. In vivo pool-based shRNA screens in hematopoietic malignancies Unlike viral insertional mutagenesis and transposon screens, shRNA based screens have primarily been performed in vitro, rather than in vivo. While they have effectively identified a large number of genes that are important for cancer cell proliferation and survival, in vitro screens fail to accurately mimic the physiological context of normal tumor growth. Due to local cues that can alter the growth and survival requirements of tumor cells, the genetic dependencies of cancer cells in vivo might differ from those in vitro. Thus, devising in vivo shRNA screening strategies could reveal novel genes and pathways that impact tumor progression. The work presented in this thesis describes the adaptation of pool-based shRNA screening approaches to in vivo applications. Specifically, I have made use of transplantable models of hematopoietic malignancies to screen for modulators of tumor cell growth in vivo. Both normal cells of the hematopoietic system, as well as hematopoietic tumors, can be isolated from donor mice, manipulated ex vivo, and 27 transplanted into recipient animals, suggesting that cells from this system might be amenable to transduction with shRNA libraries. Historically, retroviral manipulations of hematopoietic cells have been used to mark and track stem and progenitor cell populations. Early work showed that it was possible to harvest whole bone marrow from donor animals and culture these cells under conditions that promoted retroviral infection of progenitor cells (Joyner et al., 1983). Following transplantation, infected progenitor cells gave rise to differentiated cells in both the myeloid and lymphoid lineages in recipient animals, suggesting that manipulated progenitor cells can both engraft and contribute to normal hematopoiesis (Capel et al., 1989; Dick et al., 1985; Keller et al., 1985; Lemischka et al., 1986; Miller et al., 1984; Williams et al., 1984). Retroviral infections of whole bone marrow have also been used to model hematopoietic diseases in mice; by transducing this cell population with a construct expressing the Bcr-Abl fusion protein and transplanting infected cells into recipient animals, it has been possible to produce a chronic myeloid leukemia-like disease (Daley et al., 1990; Pear et al., 1998). Similar approaches have been used to generate transplantable models of acute myeloid leukemia and acute lymphoblastic leukemia. Transplantation of fetal liver HSCs transduced with MLL fusion proteins or the AML/ETO fusion protein produces AML in recipient animals (Zuber et al., 2009), and transplantation of pre-B cells transduced with Bcr-Abl produces ALL in recipient mice (Williams et al., 2007; Williams et al., 2006). Cells from established hematopoietic tumors can also be infected ex vivo and then transplanted into secondary syngeneic recipient mice. Transplantation of primary Eµ-myc tumors, which are B cell lymphomas driven by high levels of Myc expression and model human Burkitt’s lymphoma (Adams et al., 1985), produces secondary tumors that are pathologically indistinguishable from the original tumors, suggesting that tumor 28 growth in transplanted disease can accurately recapitulate many aspects of the primary tumor. Importantly, retroviral manipulations of these tumors have allowed defined genetic alterations to be studied in the in vivo context. For example, ex vivo introduction of the anti-apoptotic protein Bcl2 produces chemoresistant Eµ-myc tumors upon transplantation (Schmitt et al., 2000), and transduction of tumor cells with Bcl2 or a dominant negative Caspase9 construct provides a selective growth advantage in vivo (Schmitt et al., 2002). Additionally, shRNA-mediated gene suppression can be used to effectively mimic gene loss of function events in tumors in vivo; transplantation of Eµmyc fetal liver cells transduced with hairpins targeting p53 accelerates tumor onset, mimicking the impact of p53 loss (Hemann et al., 2003), and infection and transplantation of primary tumor cells with shRNAs targeting p53, Topoisomerase1, and Topoisomerase2a impact chemotherapeutic outcome in secondary malignancies (Burgess et al., 2008). Together, these results suggested that, by introducing pools of shRNAs into lymphoma cells and transplanting transduced cells to recipient mice, it might be possible to assay the impact of large sets of genetic lesions on tumor growth and perform loss of function screens in vivo. 29 Adams, J.M., Harris, A.W., Pinkert, C.A., Corcoran, L.M., Alexander, W.S., Cory, S., Palmiter, R.D., and Brinster, R.L. (1985). The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533-538. Alkema, M.J., Jacobs, H., van Lohuizen, M., and Berns, A. (1997). Pertubation of B and T cell development and predisposition to lymphomagenesis in Emu Bmi1 transgenic mice require the Bmi1 RING finger. Oncogene 15, 899-910. Barbie, D.A., Tamayo, P., Boehm, J.S., Kim, S.Y., Moody, S.E., Dunn, I.F., Schinzel, A.C., Sandy, P., Meylan, E., Scholl, C., et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108-112. Berns, K., Hijmans, E.M., Mullenders, J., Brummelkamp, T.R., Velds, A., Heimerikx, M., Kerkhoven, R.M., Madiredjo, M., Nijkamp, W., Weigelt, B., et al. (2004). A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431-437. Berry, C., Hannenhalli, S., Leipzig, J., and Bushman, F.D. (2006). Selection of target sites for mobile DNA integration in the human genome. PLoS Comput Biol 2, e157. Brummelkamp, T.R., Bernards, R., and Agami, R. (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550-553. Brummelkamp, T.R., Fabius, A.W., Mullenders, J., Madiredjo, M., Velds, A., Kerkhoven, R.M., Bernards, R., and Beijersbergen, R.L. (2006). An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol 2, 202-206. Brummelkamp, T.R., Nijman, S.M., Dirac, A.M., and Bernards, R. (2003). Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424, 797-801. Burgess, D.J., Doles, J., Zender, L., Xue, W., Ma, B., McCombie, W.R., Hannon, G.J., Lowe, S.W., and Hemann, M.T. (2008). Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A 105, 9053-9058. Capel, B., Hawley, R., Covarrubias, L., Hawley, T., and Mintz, B. (1989). Clonal contributions of small numbers of retrovirally marked hematopoietic stem cells engrafted in unirradiated neonatal W/Wv mice. Proc Natl Acad Sci U S A 86, 4564-4568. Caplen, N.J., Parrish, S., Imani, F., Fire, A., and Morgan, R.A. (2001). Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci U S A 98, 9742-9747. Collier, L.S., Carlson, C.M., Ravimohan, S., Dupuy, A.J., and Largaespada, D.A. (2005). Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 436, 272-276. 30 Corcoran, L.M., Adams, J.M., Dunn, A.R., and Cory, S. (1984). Murine T lymphomas in which the cellular myc oncogene has been activated by retroviral insertion. Cell 37, 113122. Cuypers, H.T., Selten, G., Quint, W., Zijlstra, M., Maandag, E.R., Boelens, W., van Wezenbeek, P., Melief, C., and Berns, A. (1984). Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell 37, 141-150. Cuypers, H.T., Selten, G.C., Zijlstra, M., de Goede, R.E., Melief, C.J., and Berns, A.J. (1986). Tumor progression in murine leukemia virus-induced T-cell lymphomas: monitoring clonal selections with viral and cellular probes. J Virol 60, 230-241. Daley, G.Q., Van Etten, R.A., and Baltimore, D. (1990). Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247, 824-830. Dick, J.E., Magli, M.C., Huszar, D., Phillips, R.A., and Bernstein, A. (1985). Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/Wv mice. Cell 42, 71-79. Dickins, R.A., Hemann, M.T., Zilfou, J.T., Simpson, D.R., Ibarra, I., Hannon, G.J., and Lowe, S.W. (2005). Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 37, 1289-1295. Dickson, C., Smith, R., Brookes, S., and Peters, G. (1984). Tumorigenesis by mouse mammary tumor virus: proviral activation of a cellular gene in the common integration region int-2. Cell 37, 529-536. Ding, S., Wu, X., Li, G., Han, M., Zhuang, Y., and Xu, T. (2005). Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122, 473-483. Dupuy, A.J., Akagi, K., Largaespada, D.A., Copeland, N.G., and Jenkins, N.A. (2005). Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 436, 221-226. Dupuy, A.J., Rogers, L.M., Kim, J., Nannapaneni, K., Starr, T.K., Liu, P., Largaespada, D.A., Scheetz, T.E., Jenkins, N.A., and Copeland, N.G. (2009). A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res 69, 8150-8156. Elbashir, S.M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., and Tuschl, T. (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494-498. Erkeland, S.J., Valkhof, M., Heijmans-Antonissen, C., van Hoven-Beijen, A., Delwel, R., Hermans, M.H., and Touw, I.P. (2004). Large-scale identification of disease genes involved in acute myeloid leukemia. J Virol 78, 1971-1980. 31 Feldman, B.J., Hampton, T., and Cleary, M.L. (2000). A carboxy-terminal deletion mutant of Notch1 accelerates lymphoid oncogenesis in E2A-PBX1 transgenic mice. Blood 96, 1906-1913. Girard, L., Hanna, Z., Beaulieu, N., Hoemann, C.D., Simard, C., Kozak, C.A., and Jolicoeur, P. (1996). Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c-myc and Notch1 for oncogenesis. Genes Dev 10, 1930-1944. Gobeil, S., Zhu, X., Doillon, C.J., and Green, M.R. (2008). A genome-wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes Dev 22, 2932-2940. Haupt, Y., Alexander, W.S., Barri, G., Klinken, S.P., and Adams, J.M. (1991). Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 65, 753-763. Haupt, Y., Bath, M.L., Harris, A.W., and Adams, J.M. (1993). bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene 8, 3161-3164. Hayward, W.S., Neel, B.G., and Astrin, S.M. (1981). Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 290, 475-480. Hemann, M.T., Fridman, J.S., Zilfou, J.T., Hernando, E., Paddison, P.J., Cordon-Cardo, C., Hannon, G.J., and Lowe, S.W. (2003). An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet 33, 396400. Hoemann, C.D., Beaulieu, N., Girard, L., Rebai, N., and Jolicoeur, P. (2000). Two distinct Notch1 mutant alleles are involved in the induction of T-cell leukemia in c-myc transgenic mice. Mol Cell Biol 20, 3831-3842. Ivics, Z., Hackett, P.B., Plasterk, R.H., and Izsvak, Z. (1997). Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91, 501-510. Jacobs, J.J., Kieboom, K., Marino, S., DePinho, R.A., and van Lohuizen, M. (1999a). The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164-168. Jacobs, J.J., Scheijen, B., Voncken, J.W., Kieboom, K., Berns, A., and van Lohuizen, M. (1999b). Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev 13, 2678-2690. Jenkins, N.A., Copeland, N.G., Taylor, B.A., and Lee, B.K. (1981). Dilute (d) coat colour mutation of DBA/2J mice is associated with the site of integration of an ecotropic MuLV genome. Nature 293, 370-374. 32 Johansson, F.K., Brodd, J., Eklof, C., Ferletta, M., Hesselager, G., Tiger, C.F., Uhrbom, L., and Westermark, B. (2004). Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci U S A 101, 11334-11337. Jonkers, J., and Berns, A. (1996). Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim Biophys Acta 1287, 29-57. Joyner, A., Keller, G., Phillips, R.A., and Bernstein, A. (1983). Retrovirus transfer of a bacterial gene into mouse haematopoietic progenitor cells. Nature 305, 556-558. Keller, G., Paige, C., Gilboa, E., and Wagner, E.F. (1985). Expression of a foreign gene in myeloid and lymphoid cells derived from multipotent haematopoietic precursors. Nature 318, 149-154. Keng, V.W., Villanueva, A., Chiang, D.Y., Dupuy, A.J., Ryan, B.J., Matise, I., Silverstein, K.A., Sarver, A., Starr, T.K., Akagi, K., et al. (2009). A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol 27, 264-274. Kolfschoten, I.G., van Leeuwen, B., Berns, K., Mullenders, J., Beijersbergen, R.L., Bernards, R., Voorhoeve, P.M., and Agami, R. (2005). A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell 121, 849-858. Kool, J., and Berns, A. (2009). High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer 9, 389-399. Largaespada, D.A., Shaughnessy, J.D., Jr., Jenkins, N.A., and Copeland, N.G. (1995). Retroviral integration at the Evi-2 locus in BXH-2 myeloid leukemia cell lines disrupts Nf1 expression without changes in steady-state Ras-GTP levels. J Virol 69, 5095-5102. Lazo, P.A., Lee, J.S., and Tsichlis, P.N. (1990). Long-distance activation of the Myc protooncogene by provirus insertion in Mlvi-1 or Mlvi-4 in rat T-cell lymphomas. Proc Natl Acad Sci U S A 87, 170-173. Lemischka, I.R., Raulet, D.H., and Mulligan, R.C. (1986). Developmental potential and dynamic behavior of hematopoietic stem cells. Cell 45, 917-927. Li, J., Shen, H., Himmel, K.L., Dupuy, A.J., Largaespada, D.A., Nakamura, T., Shaughnessy, J.D., Jr., Jenkins, N.A., and Copeland, N.G. (1999). Leukaemia disease genes: large-scale cloning and pathway predictions. Nat Genet 23, 348-353. Lund, A.H., Turner, G., Trubetskoy, A., Verhoeven, E., Wientjens, E., Hulsman, D., Russell, R., DePinho, R.A., Lenz, J., and van Lohuizen, M. (2002). Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat Genet 32, 160-165. Luo, J., Emanuele, M.J., Li, D., Creighton, C.J., Schlabach, M.R., Westbrook, T.F., Wong, K.K., and Elledge, S.J. (2009). A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835-848. 33 March, H.N., Rust, A.G., Wright, N.A., Ten Hoeve, J., de Ridder, J., Eldridge, M., van der Weyden, L., Berns, A., Gadiot, J., Uren, A., et al. (2011). Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet 43, 1202-1209. Mikkers, H., Allen, J., Knipscheer, P., Romeijn, L., Hart, A., Vink, E., and Berns, A. (2002). High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet 32, 153-159. Miller, A.D., Eckner, R.J., Jolly, D.J., Friedmann, T., and Verma, I.M. (1984). Expression of a retrovirus encoding human HPRT in mice. Science 225, 630-632. Morishita, K., Parker, D.S., Mucenski, M.L., Jenkins, N.A., Copeland, N.G., and Ihle, J.N. (1988). Retroviral activation of a novel gene encoding a zinc finger protein in IL-3dependent myeloid leukemia cell lines. Cell 54, 831-840. Mucenski, M.L., Taylor, B.A., Ihle, J.N., Hartley, J.W., Morse, H.C., 3rd, Jenkins, N.A., and Copeland, N.G. (1988). Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors. Mol Cell Biol 8, 301-308. Mucenski, M.L., Taylor, B.A., Jenkins, N.A., and Copeland, N.G. (1986). AKXD recombinant inbred strains: models for studying the molecular genetic basis of murine lymphomas. Mol Cell Biol 6, 4236-4243. Neel, B.G., Hayward, W.S., Robinson, H.L., Fang, J., and Astrin, S.M. (1981). Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: oncogenesis by promoter insertion. Cell 23, 323-334. Ngo, V.N., Davis, R.E., Lamy, L., Yu, X., Zhao, H., Lenz, G., Lam, L.T., Dave, S., Yang, L., Powell, J., et al. (2006). A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441, 106-110. Nusse, R., and Varmus, H.E. (1982). Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31, 99-109. Paddison, P.J., Caudy, A.A., Bernstein, E., Hannon, G.J., and Conklin, D.S. (2002). Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 16, 948-958. Paddison, P.J., Silva, J.M., Conklin, D.S., Schlabach, M., Li, M., Aruleba, S., Balija, V., O'Shaughnessy, A., Gnoj, L., Scobie, K., et al. (2004). A resource for large-scale RNAinterference-based screens in mammals. Nature 428, 427-431. Pear, W.S., Miller, J.P., Xu, L., Pui, J.C., Soffer, B., Quackenbush, R.C., Pendergast, A.M., Bronson, R., Aster, J.C., Scott, M.L., et al. (1998). Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 92, 3780-3792. 34 Peters, G., Brookes, S., Smith, R., and Dickson, C. (1983). Tumorigenesis by mouse mammary tumor virus: evidence for a common region for provirus integration in mammary tumors. Cell 33, 369-377. Rad, R., Rad, L., Wang, W., Cadinanos, J., Vassiliou, G., Rice, S., Campos, L.S., Yusa, K., Banerjee, R., Li, M.A., et al. (2010). PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science 330, 1104-1107. Rowe, W.P., and Pincus, T. (1972). Quantitative studies of naturally occurring murine leukemia virus infection of AKR mice. J Exp Med 135, 429-436. Sauvageau, M., Miller, M., Lemieux, S., Lessard, J., Hebert, J., and Sauvageau, G. (2008). Quantitative expression profiling guided by common retroviral insertion sites reveals novel and cell type specific cancer genes in leukemia. Blood 111, 790-799. Schlabach, M.R., Luo, J., Solimini, N.L., Hu, G., Xu, Q., Li, M.Z., Zhao, Z., Smogorzewska, A., Sowa, M.E., Ang, X.L., et al. (2008). Cancer proliferation gene discovery through functional genomics. Science 319, 620-624. Schmitt, C.A., Fridman, J.S., Yang, M., Baranov, E., Hoffman, R.M., and Lowe, S.W. (2002). Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 1, 289-298. Schmitt, C.A., Rosenthal, C.T., and Lowe, S.W. (2000). Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med 6, 1029-1035. Selten, G., Cuypers, H.T., and Berns, A. (1985). Proviral activation of the putative oncogene Pim-1 in MuLV induced T-cell lymphomas. Embo J 4, 1793-1798. Selten, G., Cuypers, H.T., Zijlstra, M., Melief, C., and Berns, A. (1984). Involvement of cmyc in MuLV-induced T cell lymphomas in mice: frequency and mechanisms of activation. Embo J 3, 3215-3222. Silva, J.M., Li, M.Z., Chang, K., Ge, W., Golding, M.C., Rickles, R.J., Siolas, D., Hu, G., Paddison, P.J., Schlabach, M.R., et al. (2005). Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet 37, 1281-1288. Silva, J.M., Marran, K., Parker, J.S., Silva, J., Golding, M., Schlabach, M.R., Elledge, S.J., Hannon, G.J., and Chang, K. (2008). Profiling essential genes in human mammary cells by multiplex RNAi screening. Science 319, 617-620. Simpson, K.J., Selfors, L.M., Bui, J., Reynolds, A., Leake, D., Khvorova, A., and Brugge, J.S. (2008). Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol 10, 1027-1038. Starr, T.K., Allaei, R., Silverstein, K.A., Staggs, R.A., Sarver, A.L., Bergemann, T.L., Gupta, M., O'Sullivan, M.G., Matise, I., Dupuy, A.J., et al. (2009). A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 323, 17471750. 35 Stegmeier, F., Hu, G., Rickles, R.J., Hannon, G.J., and Elledge, S.J. (2005). A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A 102, 13212-13217. Sui, G., Soohoo, C., Affar el, B., Gay, F., Shi, Y., and Forrester, W.C. (2002). A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci U S A 99, 5515-5520. Suzuki, T., Minehata, K., Akagi, K., Jenkins, N.A., and Copeland, N.G. (2006). Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm-deficient mice. Embo J 25, 3422-3431. Suzuki, T., Shen, H., Akagi, K., Morse, H.C., Malley, J.D., Naiman, D.Q., Jenkins, N.A., and Copeland, N.G. (2002). New genes involved in cancer identified by retroviral tagging. Nat Genet 32, 166-174. Swanton, C., Marani, M., Pardo, O., Warne, P.H., Kelly, G., Sahai, E., Elustondo, F., Chang, J., Temple, J., Ahmed, A.A., et al. (2007). Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 11, 498-512. Theodorou, V., Kimm, M.A., Boer, M., Wessels, L., Theelen, W., Jonkers, J., and Hilkens, J. (2007). MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet 39, 759-769. Uhrbom, L., Hesselager, G., Nister, M., and Westermark, B. (1998). Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res 58, 5275-5279. Uren, A.G., Kool, J., Berns, A., and van Lohuizen, M. (2005). Retroviral insertional mutagenesis: past, present and future. Oncogene 24, 7656-7672. Uren, A.G., Kool, J., Matentzoglu, K., de Ridder, J., Mattison, J., van Uitert, M., Lagcher, W., Sie, D., Tanger, E., Cox, T., et al. (2008). Large-scale mutagenesis in p19(ARF)and p53-deficient mice identifies cancer genes and their collaborative networks. Cell 133, 727-741. van Lohuizen, M., Verbeek, S., Krimpenfort, P., Domen, J., Saris, C., Radaszkiewicz, T., and Berns, A. (1989). Predisposition to lymphomagenesis in pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell 56, 673-682. van Lohuizen, M., Verbeek, S., Scheijen, B., Wientjens, E., van der Gulden, H., and Berns, A. (1991). Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 65, 737-752. 36 Varmus, H.E., Quintrell, N., and Ortiz, S. (1981). Retroviruses as mutagens: insertion and excision of a nontransforming provirus alter expression of a resident transforming provirus. Cell 25, 23-36. Vassiliou, G.S., Cooper, J.L., Rad, R., Li, J., Rice, S., Uren, A., Rad, L., Ellis, P., Andrews, R., Banerjee, R., et al. (2011). Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet 43, 470-475. Verbeek, S., van Lohuizen, M., van der Valk, M., Domen, J., Kraal, G., and Berns, A. (1991). Mice bearing the E mu-myc and E mu-pim-1 transgenes develop pre-B-cell leukemia prenatally. Mol Cell Biol 11, 1176-1179. Weng, A.P., Ferrando, A.A., Lee, W., Morris, J.P.t., Silverman, L.B., Sanchez-Irizarry, C., Blacklow, S.C., Look, A.T., and Aster, J.C. (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269-271. Westbrook, T.F., Martin, E.S., Schlabach, M.R., Leng, Y., Liang, A.C., Feng, B., Zhao, J.J., Roberts, T.M., Mandel, G., Hannon, G.J., et al. (2005). A genetic screen for candidate tumor suppressors identifies REST. Cell 121, 837-848. Whitehurst, A.W., Bodemann, B.O., Cardenas, J., Ferguson, D., Girard, L., Peyton, M., Minna, J.D., Michnoff, C., Hao, W., Roth, M.G., et al. (2007). Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 446, 815-819. Williams, D.A., Lemischka, I.R., Nathan, D.G., and Mulligan, R.C. (1984). Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature 310, 476-480. Williams, R.T., den Besten, W., and Sherr, C.J. (2007). Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev 21, 22832287. Williams, R.T., Roussel, M.F., and Sherr, C.J. (2006). Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 103, 6688-6693. Wu, X., Li, Y., Crise, B., and Burgess, S.M. (2003). Transcription start regions in the human genome are favored targets for MLV integration. Science 300, 1749-1751. Yant, S.R., Wu, X., Huang, Y., Garrison, B., Burgess, S.M., and Kay, M.A. (2005). Highresolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol 25, 2085-2094. Yu, J.Y., DeRuiter, S.L., and Turner, D.L. (2002). RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A 99, 6047-6052. 37 Zeng, Y., Wagner, E.J., and Cullen, B.R. (2002). Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell 9, 1327-1333. Zuber, J., Radtke, I., Pardee, T.S., Zhao, Z., Rappaport, A.R., Luo, W., McCurrach, M.E., Yang, M.M., Dolan, M.E., Kogan, S.C., et al. (2009). Mouse models of human AML accurately predict chemotherapy response. Genes Dev 23, 877-889. 38 Chapter 2: In Vivo RNAi Screening Identifies Regulators of Actin Dynamics as Key Determinants of Lymphoma Progression ______________________________________________________________________ This work was originally published in Nature Genetics. Appropriate copyright permissions have been obtained. Meacham C.E., Ho E.E., Dubrovsky E., Gertler F.B., Hemann M.T. (2009). In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat Genet 41, 1133-1137. 39 Abstract Mouse models have dramatically improved our understanding of cancer development and tumor biology. However, these models have shown limited efficacy as tractable systems for unbiased genetic experimentation. Here, we report the adaptation of loss of function screening to mouse models of cancer. Specifically, we have been able to introduce a library of shRNAs into individual mice using transplantable Eµ-myc lymphoma cells. This approach has allowed us to screen nearly 1000 genetic alterations in the context of a single tumor-bearing mouse. Results from these experiments have identified a central role for regulators of actin dynamics and cell motility in lymphoma cell homeostasis in vivo, and validation experiments confirmed that these proteins represent bona fide lymphoma drug targets. Additionally, suppression of two of these targets, Rac2 and Twinfilin, potentiated the action of the front-line chemotherapeutic vincristine, suggesting a critical relationship between cell motility and tumor relapse in hematopoietic malignancies. Introduction Genetic screens in the mouse hematopoietic system have proven to be highly effective strategies for identifying novel and cooperating cancer genes. For example, Moloney-based retroviral insertional mutagenesis screens first characterized the potent oncogenes bmi-1 and pims 1-3 (Jonkers and Berns, 1996; van Lohuizen et al., 1991). Subsequently, these screens have been performed on numerous sensitized backgrounds, revealing significant insight into the relationship between specific oncogenic and tumor suppressor alterations (Uren et al., 2008). The more recent development of mice expressing active transposons has similarly facilitated the identification of novel cancer genes (Collier et al., 2005; Dupuy et al., 2005; Suzuki et al., 40 2006). While these approaches have been highly successful, they have several notable limitations. First, genes affected by insertional mutagenesis are identified as likely candidates based on proximity to the insertion site. Thus, numerous mice are required to identify “common insertion sites”, and affected genes need to be functionally validated. Second, despite the use of highly recombinant genetic backgrounds, the identification of relevant tumor suppressor genes by insertional mutagenesis is inefficient. Finally, insertional mutagenesis requires positive selection for a given phenotype, generally tumor development. Thus, sensitization screens based on the selective depletion of a given insertion site cannot be performed. We have recently adapted miRNA-based shRNA gene silencing to in vivo applications in the Eµ-myc lymphoma mouse, a well-established model of B cell lymphoma (Adams et al., 1985; Dickins et al., 2005; Hemann et al., 2003). This approach has subsequently been expanded to examine small sets of shRNAs in a cohort of mice (Zender et al., 2008). However, despite the disseminated and effective use of cell-based RNAi screens (Berns et al., 2004; Firestein et al., 2008; Grueneberg et al., 2008; Luo et al., 2008; Ngo et al., 2006; Schlabach et al., 2008; Westbrook et al., 2005), adaptation of these approaches to mouse models has been limited. Here, we use RNAi to interrogate loss of function phenotypes for large gene sets in the context of individual mice. Results from this study identify a set of genes involved in cytoskeletal organization and cell migration that are important for lymphoma progression in vivo. Importantly, this study demarcates critical determinants of tumor behavior in vivo, information that could not be obtained using conventional cell-based screening approaches. 41 Results and Discussion Previous studies in the Eµ-myc system have suggested that Eµ-myc lymphomas are largely composed of cells with tumor initiating potential. Specifically, tumor cell dilution experiments have shown that as few as 10 tumor cells can produce tumors following tail vein injection into syngeneic recipient mice (Kelly et al., 2007). Thus, we reasoned that if nearly all tumor cells have the capacity to contribute to tumor formation following transplantation, then this system might accommodate the introduction of a diverse set of shRNA-infected cells into a given tumor. To test this, we performed a dilution experiment using lymphoma cell cultures that were partially transduced with a retroviral vector co-expressing an shRNA targeting Topoisomerase 2α (Top2A), an essential mediator of the cytotoxic effects of the chemotherapeutic doxorubicin (Burgess et al., 2008), and the gene encoding Green Fluorescent Protein (GFP) to mark infected cells. Viral multiplicity of infection (MOI) was titered, such that either 2.0% or 0.2% of lymphoma cells were infected. These partially transduced lymphoma cell populations were injected into syngeneic recipient mice, and mice were treated with doxorubicin at the time of lymphoma presentation. At both infection efficiencies, we saw a significant increase in the percentage of GFP-positive cells following treatment (Fig. 1a). These results showed that cells representing as little as 1/500th of the injected lymphoma cell population were retained at the time of doxorubicin treatment. These data are consistent with large numbers of lymphoma cells (at least 500) contributing to an individual tumor following transplantation in vivo, and suggest the Eµ-myc lymphoma model may be appropriate for in vivo, pool-based RNAi screens with complex shRNA libraries. 42 Based on these preliminary results, we performed an shRNA screen with a pool of approximately 2250 hairpins targeting 1000 genes with known or putative roles in cancer (Burgess et al., 2008). Retroviral plasmids expressing these shRNAs, as well as GFP, were pooled and packaged as a mixture of retroviruses. This retroviral pool was then used to infect lymphoma cells, and the resulting transduced cells were injected into recipient mice, maintained in culture, or collected immediately to serve as a reference sample (Fig. 1b). At the time of lymphoma presentation, tumors were harvested and genomic DNA was extracted from primary tumors and cultured cells. Following PCR amplification of shRNAs from genomic DNA using common primers (Fig. 1b), hairpin representation was analyzed by high throughput sequencing. Approximately 1600 of the original 2250 unique hairpins could be identified from each in vitro cultured sample, and, surprisingly, between 600 and 900 unique hairpins could be identified in lymphomas from individual mice (Fig. 1c). Thus, a large, diverse shRNA set can be introduced in vivo and a significant proportion of the initial library complexity can be maintained in this setting. Sequencing of shRNAs from genomic DNA derived from outgrown single cell clones showed that approximately 90% of cells were infected with only a single shRNA (Supplementary Fig. 1), suggesting that, at a minimum, 900 cells can contribute to the lymphoma burden in an individual mouse. Thus, not only can a large percentage of lymphoma cells give rise to a tumor following transplantation, but, in fact, many of these cells contribute to the growth of the resulting tumor. Unsupervised hierarchical clustering showed that the three in vivo samples were more similar to one another than to any of the cultured in vitro samples, based on the 43 hairpins that enriched or depleted in each setting (Fig. 1d). Thus, the set of genes that impacts cancer cell homeostasis in vitro is distinct from those that are central to tumor growth in vivo. Importantly, the number of sequencing reads obtained was sufficient to see both enrichment and depletion of shRNAs from the initial injected population. While many shRNAs displayed similar changes in representation in clustered samples, significant variation was also present in samples of the same type. This is likely due to the stochastic gain or loss of shRNAs following introduction into mice or serial re-plating in culture. Hairpins whose representation decreased at least 10-fold in all three replicates or enriched at least 5-fold in two out of three replicates, relative to their representation in the control cell population collected shortly after retroviral transduction, scored as candidate hits. The set of scoring shRNAs in vitro by this criteria was largely nonoverlapping with the set of shRNAs that scored in vivo (Fig. 1e, Supplementary Table 1), indicating that by performing shRNA screens in the context of a normal tumor microenviroment, we were able to identify a set of genes that exclusively impacts growth in a physiologically relevant setting. We focused our follow-up studies on shRNA sets that specifically affected tumor growth in vivo. As a more stringent criterion, we selected genes for which 2 or more cognate shRNAs depleted on average at least 10-fold in mice (Table 1). Based on gene ontology (GO) classifications and manual curation, shRNAs targeting genes involved in cell motility, including dynamic actin reorganization and cell adhesion, were highly represented in the set of hairpins that specifically depleted in vivo (8 out of 11). Several of these genes were chosen for validation (Supplementary Fig. 2a). These included 44 genes encoding Rac2, a hematopoetic-specific Rho GTPase important for the formation of lamellipodia during cell migration (Burridge and Wennerberg, 2004; Ridley et al., 1992), CrkL, an adaptor protein reported to be involved in the activation of Rac (Nishihara et al., 2002), and Twinfilin (Twf1), an actin monomer-binding and actin filament capping protein (Helfer et al., 2006). For these genes, multiple independent shRNAs (Fig. 2a) recapitulated the initial screening phenotype. Specifically, while cells grown in the presence of these shRNAs grew robustly in culture, they all showed a selective depletion in lymph nodes following tail vein injection (Fig. 2b). To further characterize the efficiency of our screen, we examined a non-motility gene targeted by two depleted shRNAs and two genes targeted by a single scoring shRNA. shRNAs targeting the genes encoding IL-6 (two depleted shRNAs in our screen) and Lyn kinase (one depleted shRNA in our screen) depleted following introduction in mice, while shRNAs targeting the gene encoding AIF-1 (one depleted shRNA in our screen) failed to deplete (Supplementary Fig. 2b and data not shown). Thus, shRNAs targeting four out of four genes from the gene set identified using our most stringent criteria validated as single constructs, while the validation rate for single scoring shRNAs was lower. To further characterize the role of genes identified in our screen in lymphoma homeostasis, we subjected shRNA-infected cells to secondary functional assays. Lymphoma cells suppressing Rac2, CrkL, or Twf1 showed motility defects in transwell migration assays, consistent with a role for these proteins in lymphoma cell migration (Fig. 2c and d and Supplementary Fig. 3a and b). Additionally, Rac2-deficient lymphoma cells showed defects in SDF-1α induced migration on fibronectin (Supplementary Movies 1 and 2). Suppression of Rac2 also resulted in impaired lymphoma cell migration in short term in vivo engraftment assays. Specifically, lymphoma cells suppressing Rac2 45 were depleted in the spleen and bone marrow two and twenty-four hours after tail vein injection (Fig. 3a). Similar to the effect Rac2 knockdown, lymphoma cells suppressing Wave2, an important mediator of cell migration known to function downstream of other Rac proteins (Smith and Li, 2004), showed chemotaxis defects in vitro and were selectively depleted in the lymph nodes at the time of disease presentation (Fig. 3b and Supplementary Fig. 3c and d). Suppression of Rac2 was also selected against in common sites of lymphoid metastasis, such as the liver, as seen by histology and by GFP enrichment analysis (Figs. 3c and d), suggesting that Rac2 might represent a meaningful lymphoma drug target. In fact, suppression of Rac2 in lymphoma cells extended both tumor free and overall survival following tail vein injection (Fig. 3e and f). Similarly, lymphoma-bearing mice treated with NSC23766 (Gao et al., 2004), an inhibitor of Rac1 and Rac2 (Supplementary Fig. 4a) (Cancelas et al., 2005), survived significantly longer than untreated controls (Fig. 4a). Notably, suppression of Twf1 also delayed tumor progression following lymphoma tail vein injection (Supplementary Fig. 4b). Thus, multiple proteins involved in actin reorganization and cell motility represent potential drug targets in B cell malignancies. Additionally, combinations of shRNAs produced synergistic effects on lymphoma growth (Supplementary Fig. 4c). Interestingly, when lymphomas were treated with the chemotherapeutic vincristine, knockdown of Rac2 or Twf1 extended animal survival following drug treatment (Fig. 4b and Supplementary Fig. 4d). Importantly, this effect was specific for therapeutic response in vivo, as suppression of Rac2 did not sensitize lymphoma cells to vincristine treatment in culture (data not shown). These results suggest that there is a 46 requirement for tumor cell mobilization in lymphoma relapse and that Rac2 and Twf1 activity is important in this mobilization (Fig. 4c). It further suggests that Rac2 or Twf1 inhibition, or inhibition of lymphoma cell motility by another mechanism, may synergize with conventional chemotherapeutics in the treatment of lymphoma. Using a well-established mouse model of B cell lymphoma, we have adapted RNAi screens to in vivo applications. With this system, we were able to screen over 900 unique hairpins in individual mice. While improvements in RNAi technology will be required to perform saturating loss of function studies, this work demonstrates the feasibility of using large, unbiased shRNA sets for in vivo screens. Importantly, unlike conventional microarray studies, this approach investigates gene function, rather than gene expression – implicating “scoring” genes as directly relevant to interrogated phenotypes. We can envision using a similar approach to examine modulators of therapeutic response or tissue-specific tumor dissemination. Importantly, identification of similarly tractable genetic systems may permit the adaptation of this screening methodology to study tissue development or the biology of solid tumors. Our screen identified modulators of lymphoma cell motility and chemotaxis as key determinants of tumor homeostasis. This group included known regulators of actinbased cell motility, as well as proteins, like Twf1, with known roles in actin dynamics but no established role in mammalian cell migration. Suppression of Rac2 and Twf1 in lymphoma cells impaired lymphoma cell migration to the lymph nodes and other organs that represent common sites of lymphoid metastasis. Additionally, pharmacological inhibition of Rac2 synergized with a conventional chemotherapeutic to extend the lifespan of tumor-bearing mice. These results highlight a potential therapeutic strategy in 47 hematopoietic cancers, suggesting that, in instances where there is minimal metastasis at the time of initial treatment with traditional chemotherapeutics, suppression of cell migration may improve therapeutic outcome. Acknowledgements and author contributions We would like to thank Ed van Veen for help with motility assays, Charlie Whittaker and Justin Pritchard for bioinformatic processing of sequencing data, Holly Thompson for expert technical assistance and members of the Hemann lab for helpful advice and discussions. Corbin Meacham and Michael Hemann designed the experiments and wrote the manuscript. Corbin Meacham and Emily Ho performed the screen and subsequent validation experiments. Ester Dubrovsky and Michael Hemann performed initial GFP dilution experiments. Frank Gertler designed motility experiments. 48 References Adams, J.M., Harris, A.W., Pinkert, C.A., Corcoran, L.M., Alexander, W.S., Cory, S., Palmiter, R.D., and Brinster, R.L. (1985). The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533-538. Berns, K., Hijmans, E.M., Mullenders, J., Brummelkamp, T.R., Velds, A., Heimerikx, M., Kerkhoven, R.M., Madiredjo, M., Nijkamp, W., Weigelt, B., et al. (2004). A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431-437. Burgess, D.J., Doles, J., Zender, L., Xue, W., Ma, B., McCombie, W.R., Hannon, G.J., Lowe, S.W., and Hemann, M.T. (2008). Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A 105, 9053-9058. Burridge, K., and Wennerberg, K. (2004). Rho and Rac take center stage. Cell 116, 167179. Cancelas, J.A., Lee, A.W., Prabhakar, R., Stringer, K.F., Zheng, Y., and Williams, D.A. (2005). Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med 11, 886-891. Collier, L.S., Carlson, C.M., Ravimohan, S., Dupuy, A.J., and Largaespada, D.A. (2005). Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 436, 272-276. Dickins, R.A., Hemann, M.T., Zilfou, J.T., Simpson, D.R., Ibarra, I., Hannon, G.J., and Lowe, S.W. (2005). Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 37, 1289-1295. Dupuy, A.J., Akagi, K., Largaespada, D.A., Copeland, N.G., and Jenkins, N.A. (2005). Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 436, 221-226. Firestein, R., Bass, A.J., Kim, S.Y., Dunn, I.F., Silver, S.J., Guney, I., Freed, E., Ligon, A.H., Vena, N., Ogino, S., et al. (2008). CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 455, 547-551. Gao, Y., Dickerson, J.B., Guo, F., Zheng, J., and Zheng, Y. (2004). Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 101, 7618-7623. Grueneberg, D.A., Li, W., Davies, J.E., Sawyer, J., Pearlberg, J., and Harlow, E. (2008). Kinase requirements in human cells: IV. Differential kinase requirements in cervical and renal human tumor cell lines. Proc Natl Acad Sci U S A 105, 16490-16495. Helfer, E., Nevalainen, E.M., Naumanen, P., Romero, S., Didry, D., Pantaloni, D., Lappalainen, P., and Carlier, M.F. (2006). Mammalian twinfilin sequesters ADP-G-actin and caps filament barbed ends: implications in motility. Embo J 25, 1184-1195. 49 Hemann, M.T., Fridman, J.S., Zilfou, J.T., Hernando, E., Paddison, P.J., Cordon-Cardo, C., Hannon, G.J., and Lowe, S.W. (2003). An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet 33, 396400. Jonkers, J., and Berns, A. (1996). Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim Biophys Acta 1287, 29-57. Kelly, P.N., Dakic, A., Adams, J.M., Nutt, S.L., and Strasser, A. (2007). Tumor growth need not be driven by rare cancer stem cells. Science 317, 337. Luo, B., Cheung, H.W., Subramanian, A., Sharifnia, T., Okamoto, M., Yang, X., Hinkle, G., Boehm, J.S., Beroukhim, R., Weir, B.A., et al. (2008). Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A 105, 20380-20385. Ngo, V.N., Davis, R.E., Lamy, L., Yu, X., Zhao, H., Lenz, G., Lam, L.T., Dave, S., Yang, L., Powell, J., et al. (2006). A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441, 106-110. Nishihara, H., Maeda, M., Oda, A., Tsuda, M., Sawa, H., Nagashima, K., and Tanaka, S. (2002). DOCK2 associates with CrkL and regulates Rac1 in human leukemia cell lines. Blood 100, 3968-3974. Ridley, A.J., Paterson, H.F., Johnston, C.L., Diekmann, D., and Hall, A. (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401-410. Schlabach, M.R., Luo, J., Solimini, N.L., Hu, G., Xu, Q., Li, M.Z., Zhao, Z., Smogorzewska, A., Sowa, M.E., Ang, X.L., et al. (2008). Cancer proliferation gene discovery through functional genomics. Science 319, 620-624. Smith, L.G., and Li, R. (2004). Actin polymerization: riding the wave. Curr Biol 14, R109111. Suzuki, T., Minehata, K., Akagi, K., Jenkins, N.A., and Copeland, N.G. (2006). Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm-deficient mice. Embo J 25, 3422-3431. Uren, A.G., Kool, J., Matentzoglu, K., de Ridder, J., Mattison, J., van Uitert, M., Lagcher, W., Sie, D., Tanger, E., Cox, T., et al. (2008). Large-scale mutagenesis in p19(ARF)and p53-deficient mice identifies cancer genes and their collaborative networks. Cell 133, 727-741. van Lohuizen, M., Verbeek, S., Scheijen, B., Wientjens, E., van der Gulden, H., and Berns, A. (1991). Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 65, 737-752. 50 Westbrook, T.F., Martin, E.S., Schlabach, M.R., Leng, Y., Liang, A.C., Feng, B., Zhao, J.J., Roberts, T.M., Mandel, G., Hannon, G.J., et al. (2005). A genetic screen for candidate tumor suppressors identifies REST. Cell 121, 837-848. Zender, L., Xue, W., Zuber, J., Semighini, C.P., Krasnitz, A., Ma, B., Zender, P., Kubicka, S., Luk, J.M., Schirmacher, P., et al. (2008). An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 135, 852-864. 51 Materials and Methods Cell culture and chemicals Eµ-Myc;Arf-/- mouse B-cell lymphomas were cultured in B cell medium (45% DMEM/45% IMDM/10% FBS, supplemented with 2 mM L-glutamine and 5µM β-mercaptoethanol). γirradiated NIH 3T3 cells were used as feeder cells. Vincristine, doxorubicin, and the Rac inhibitor NSC23766 were purchased from Calbiochem. For in vivo studies, drugs were dissolved in a 0.9% NaCl solution prior to IP injection. RNAi screen Lymphoma cells were infected with an shRNA library targeting the cancer 1000 gene set (≈2250 hairpins) to a final infection of ≈20%. 48 hours after infection, lymphoma cells were GFP sorted. Immediately after sorting, 2x106 lymphoma cells/mouse were injected into three syngeneic recipient mice by tail vein injection. Disease progression was monitored by lymph node palpation. Following the appearance of palpable lymphomas, approximately 14 days after tail-vein injection, lymphoma cells were harvested from the axillary, brachial, and cervical lymph nodes. For in vitro samples, infected lymphoma cells were plated in triplicate and maintained for two weeks. shRNAs were amplified from genomic DNA using primers that include adaptors for 454 sequencing (Supplementary Table 2). Following PCR amplification of hairpins, 454 sequencing was used to identify constituent shRNAs in each sample. Fold change in hairpin representation after proliferation in vitro or in vivo was determined by comparing shRNA representation in each sample to that in a control cell population collected immediately after cell sorting. Hairpins that depleted 10-fold in all three in vitro or in vivo samples, and hairpins that enriched 5-fold in two out of three samples, based on a normalized read numbers, were 52 considered candidate hits. Genes were not scored as candidate hits if multiple cognate hairpins showed opposing effects. All of the raw data from this screen has been deposited in the GEO database, accession number GSE16090. shRNA constructs shRNA constructs were designed and cloned as previously described (Dickins et al., 2005). Sequences targeted by shRNAs are provided in Supplementary Table 2. Western blotting and RT-qPCR For western blotting and RT-qPCR, protein or total RNA was isolated after retroviral infection and puromycin selection. Primers used for qPCR are available upon request. For western blotting, cell lysates were prepared in lysis buffer (1% sodium deoxycholine, 0.1% SDS, 1% triton-X, 10 mM Tris-HCl, pH 8.0, 140 mM NaCl) for 10 minutes. Lysates were cleared for 15 minutes at 14,000 rpm and mixed with 5x SDS sample buffer. Proteins were then run on a 12% SDS-PAGE gel, transferred to PVDF (Millipore) and detected with the following antibodies: anti-Rac2 (Proteintech Group, 1:200) and antitubulin (ECM Biosciences, 1:5000). Competition and survival assays For competition assays, lymphoma cells were partially infected with the indicated retroviruses and 4x106 cells were tail-vein injected into syngeneic recipient mice. Following the appearance of palpable lymphomas, lymphoma cells were harvested from the lymph nodes and the percentage of GFP+ cells was analyzed on a Becton Dickinson FACScan flow cytometer. In parallel, lymphoma cells were maintained in culture for two weeks to assess the effect of the indicated shRNA on lymphoma cell proliferation in vitro. 53 Dead cells were detected by propidium iodide incorporation (0.05 mg/mL) and were excluded from GFP analysis. For survival assays, cells were GFP sorted and 1 x 106 cells/mouse were injected by tail-vein injection. Mice were treated with the Rac inhibitor NSC23766 every 12 hours (2.5 mg/kg) starting 9 days after tail vein injection. Mice were treated with a single dose of vincristine (1.5 mg/kg) 11 days after tail vein injection. Tumor free survival was monitored by palpation and overall survival was based on body condition score. For short-term engraftment assays, cells were GFP or dsCherry sorted, mixed at an even ratio, and 1x106 cells/mouse were injected by tail-vein injection. Migration assays Recombinant murine SDF-1α (Peprotech) was used for migration assays. Lymphoma cells were serum starved in B cell media containing 2.5% FBS for 2 hours. 250,000 lymphoma cells resuspended in low serum B cell media were added to the upper chamber of 24-well transwell inserts (5 µm pore size, Millipore) and the indicated concentration of SDF was added to the lower chamber. The number of lymphoma cells that migrated to the lower chamber was quantified after 5 hours and is displayed as the fold change in cell number relative to control wells. For live imaging of lymphoma cell migration, glass bottom dishes (MatTek) were coated with fibronectin (Calbiochem). Lymphoma cells were resusupended in low serum B cell media at 2x106 cells/mL, plated on fibronectin-coated dishes, and allowed to adhere for 2 hours. Non-adherent cells were then removed by gentle washing and cells were imaged after stimulation with SDF (100 ng/mL). Statistical analysis 54 Statistical analysis was performed using GraphPad Prism4 software. Two-tailed Student’s t-tests, one sample t-tests, and one-way ANOVA were used, as indicated. For comparison of survival curves, a Mantel-Haenszel test was used. Error bars represent standard deviation. 55 Figures Figure 1 – In vivo RNAi screening strategy. (A) A diverse population of lymphoma cells is retained in tumors at the time of disease presentation. Cells were infected with a vector control or a retrovirus co-expressing GFP and an shRNA targeting Top2A. Partially transduced cell populations were injected into recipient mice. Palpable tumors 56 were treated with doxorubicin, and the percentage of GFP positive lymphoma cells was assayed at tumor relapse. (B) In vivo RNAi screening strategy. Lymphoma cells were infected with a pool of retroviruses containing 2250 distinct hairpins, and transduced lymphoma cell populations were injected into three recipient mice or passaged in three separate culture dishes. Lymphomas were harvested from tumor-bearing mice and shRNAs were PCR amplified from genomic DNA derived from tumors or from cultured cells. (C) The number of unique hairpins present in each sample after two weeks proliferation. (D) Clustering of samples based on shRNA enrichment or depletion. Color scale represents the mean normalized log2 of the fold change in shRNA read number relative to input cells. (E) Hairpins that scored as enriched or depleted in vivo are largely distinct those that enriched or depleted in vitro. Diagrams show the number of scoring genes in the in vivo and in vitro settings. 57 Figure 2 – Functional validation of shRNAs targeting putative cell motility genes. (A) shRNA-mediated stable knockdown of Rac2, CrkL, and Twf1. Target protein/gene expression was measured by immunoblotting or qPCR. For qPCR samples, n=2 and bar graphs represent mean and standard deviation. (B) In vivo GFP competition assay to functionally validate candidate hits. Lymphoma cell cultures, partially transduced with a vector coexpressing GFP and the indicated shRNA, were maintained in culture for two weeks or injected into recipient mice. The fold change in the percentage of GFP positive lymphoma cells, relative to cells at injection, is shown. p-values were determined by a 58 two-tailed Student’s t-test. (n=7 for vector control and shRac2-1, n=8 for shRac2-2 in left panel; n=4 for vector control, n=3 for shCrkL-1 and shCrkL-2 in middle panel; n=4 for vector control and shTwf-2, n=3 for shTwf-1 in right panel). (C and D) Rac2, CrkL, or Twf1 suppression causes chemotaxis defects in transwell migration assays. Cells expressing a control vector, shRac2 (C), shTwf1, or shCrkL (D) were stimulated with SDF-1α. The number of cells that migrated is displayed as a fold change relative to control wells. p-values were determined by one-way ANOVA or a two-tailed Student’s ttest. Bar graphs represent mean and standard deviation. n=2 for each experimental group. 59 Figure 3 – Rac2 suppression impairs lymphoma cell migration and extends animal survival. (A) Rac2 suppression causes defects in short term engraftment. Lymphoma cells expressing dsCherry or coexpressing GFP and shRac2 were mixed, injected into 60 recipient mice, and assessed after 2 or 24 hours. p-values were determined by one-way ANOVA or a one-sample t-test. Bar graphs represent mean and standard deviation. n=1 for injected cells, n=3 for all other experimental groups. (B) Lymphoma cells suppressing Wave2 were depleted in an in vivo GFP competition assay. p-values were determined by a two-tailed Student’s t-test. (n=7 for vector control, n=4 for shWave2-1). (C) Suppression of Rac2 impairs lymphoma cell migration to the lymph nodes and liver. 14 days after transplantation, shRac2 recipients show markedly reduced tumor dissemination. (D) Partially-transduced lymphoma cells were harvested from the liver at the time of disease presentation and the percentage of GFP positive cells was assessed. p-values were determined by one-way ANOVA. (n=7 for vector control, n=4 for shRac2-1 and shRac2-2) (E and F) Suppression of Rac2 in lymphoma cells delays disease progression. GFP sorted lymphoma cells were injected into recipient mice. Survival is displayed in Kaplan-Meier format (n=5 mice per group for tumor free survival and n=10 mice per group for overall survival). 61 Figure 4 – Suppression of Rac activity delays disease progression and potentiates the action of the chemotherapeutic vincristine. (A) Pharmacological inhibition of Rac2 extends animal lifespan. Mice were treated with the Rac inhibitor NSC23776 every 12 hours starting nine days after injection of lymphoma cells. Results are shown in KaplanMeier format (n=5 for each experimental group). (B) Suppression of Rac2 activity extends animal survival following vincristine treatment. Mice bearing vector control or shRac2 tumors were treated with vincristine 11 days after injection of lymphoma cells, and overall survival was monitored (n=11 for each experimental group. Data was compiled from three independent experiments). (C) A model for the role of Rac2 in 62 relapse following vincristine treatment. The appearance of terminal disease following vincristine treatment may require tumor cell migration from sites of residual disease to metastatic sites, including liver, lung, and brain. 63 Table 1 – Genes targeted by at least 2 depleted shRNAs (on average >10-fold) in vivo Intended shRNA target cdkn1b crkl* cyr61 ddx11 il-6* map2k3 nek4 opn5 Protein name Cyclin-dependent kinase inhibitor p27 v-Crk sarcoma virus CT10 oncogene homolog Cysteine-rich angiogenic inducer 61 RAS-related C3 botulinum substrate 2 Interleukin 6 Map kinase kinase 3 NIMA (never in mitosis gene a)related kinase 4 Neuropsin rac2* RAS-related C3 botulinum substrate 2 twf1*1 Twinfilin, actin-binding protein, homolog 1 (Drosophila) v-Yes-1 Yamaguchi sarcoma viral oncogene homolog 1 yes1 Putative role in motility A cell cycle independent function of p27 regulates cell adhesion and migration via interaction with RhoA. An adaptor protein reported to be involved in the activation of Rac A secreted protein that mediates focal adhesions and is involved in cell attachment and migration. Operates upstream of p38 MAPk to regulate cell adhesion. A secreted protein that cleaves fibronectin, inhibiting alpha5 integrin-mediated adhesion. A hematopoetic-specific Rho GTPase important for the formation of lamellipodia during cell migration. An actin monomer-binding and actin filament capping protein. Signals downstream of CD95 to promote cell invasion in vivo. * - Subject to further validation 1 – Targeted by 3 shRNAs depleted by an average of >10-fold 64 Supplementary figures Supplementary figure 1. Determination of the number of viral integrations per tumor cell. Single cell clones were grown from tumor samples. Hairpins were amplified from genomic DNA derived from single cell clones using common primers, and the resulting PCR product was sequenced. A representative sequencing trace of a PCR product from a single cell clone is shown. 9/10 clones had one shRNA present in the PCR product, indicated by a single trace in the hairpin antisense sequence, and 1/10 clones contained two shRNAs (data not shown). 65 Supplementary Figure 2. Validation strategy. (A) A schematic representation of the GFP competition assay. Approximately 20-40% of the starting lymphoma cell population is transduced with a specific retroviral shRNA construct co-expressing GFP, and this partially transduced lymphoma cell population is injected into recipient mice. At the time of disease presentation, tumors are harvested and the percentage of GFP positive cells is assayed by flow cytometry. shRNAs that provide a growth advantage, disadvantage, or are neutral are predicted to lead to an increase, decrease, or no change in the percentage of GFP positive cells, respectively. (B) Lymphoma cells were partially transduced with the indicated shRNA and maintained in culture or injected into recipient 66 mice. Palpable tumors were harvested and the percentage of GFP positive lymphoma cells was assessed. The fold change in the percentage of GFP positive cells, compared cells at injection, is shown. p-values were determined by one-way ANOVA. (n=7 for vector control, n=3 for shIL-6, and n=4 for shLyn) 67 Supplementary figure 3. Validation of motility defects in cells expressing scoring shRNAs. (A) Suppression of candidate genes with additional shRNAs causes chemotaxis defects in transwell migration assays. Cells infected with the indicated shRNA were stimulated with SDF-1α. p-values were determined by a two-tailed Student’s t-test and bar graphs represent mean and standard deviation. n=2 for each experimental group. (B) Re-expression of Rac2 rescues chemotaxis defects in transwell migration assays. A mixed population of lymphoma cells was generated containing uninfected cells, cells infected with shRac2 co-expressing GFP, and cells infected with both shRac2 co-expressing GFP and an shRNA-resistant Rac2 construct co-expressing the fluorescent protein dsCherry. Following stimulation with SDF-1α, cells suppressing 68 Rac2 depleted in cells that had migrated, whereas, the percentage of cells expressing a Rac2 cDNA did not decrease. (C) shRNA-mediated stable knockdown of Wave2. Lymphoma cells were infected with an shRNA targeting Wave2 and target gene expression was measured by qPCR. Bar graphs represent mean and standard deviation. n=2 for all experimental groups. (D) Cells suppressing Wave2 show chemotaxis defects in transwell migration assays following stimulation with SDF-1α. pvalues were determined by a two-tailed Student’s t-test and bar graphs represent mean and standard deviation. n=2 for all experimental groups. 69 Supplementary Figure 4. Functional characterization of scoring shRNAs. (A) Inhibition of Rac activity impairs lymphoma cell migration in transwell migration assays. Lymphoma cells were pre-treated with the Rac inhibitor NSC23766 for two hours and were then stimulated with SDF-1α in the presence of NSC23766. p-values were determined by a two-tailed Student’s t-test and bar graphs represent mean and standard deviation. n=2 for all experimental groups. (B) Suppression of Twf1 in lymphoma cells extends animal lifespan. shTwf1 infected lymphoma cells were injected into recipient mice and survival was monitored. (n=12 for vector control, n=24 for Twf1) (C) Knockdown of multiple candidate genes has a synergistic effect in vivo. Pure populations of shTwf1 infected cells were partially transduced with an shRNA targeting Lyn, Rac2, or a different shRNA targeting Twf1 (all co-expressing the fluorescent protein Cherry) and 70 were injected into recipient mice. Lymphomas were harvested and the percentage of Cherry-positive cells was assessed. The fold change in the percentage of Cherrypositive cells, relative to cells at injection, is shown. p-values were determined by oneway ANOVA (n=3 for all experimental groups). (D) Suppression of Twf1 extends animal survival following vincristine treatment. Mice bearing shTwf1 tumors were treated with vincristine, and survival was monitored. (n=5 for vector control, n=4 for shTwf1). 71 Supplementary tables Supplementary Table 1 – Scoring shRNAs shRNAs enriched in vitro and in vivo Intended shRNA target Lima1 Orc4l Gene name LIM domain and actin binding 1 origin recognition complex, subunit 4-like (S. cerevisiae) shRNAs enriched specifically in vitro Intended shRNA target 4921537P18Rik Actb Akr1b7 Akt2 Apaf1 Cd177 Ctso Cyp19a1 Cyp2c29 Dck Elk3 Fosl2 Grb7 Grb8 Grb9 Hoxb6 Hpca Klk10 Klk1b26 Mcm10 Mmp13 Nefl Notch4 Opn4 Oxtr Polr2b Prkdc Rad51c Rarb Rfc4 Rhoa Serpinb5 Gene name RIKEN cDNA 4921537P18 gene actin, beta aldo-keto reductase family 1, member B7 thymoma viral proto-oncogene 2 apoptotic peptidase activating factor 1 CD177 antigen cathepsin O cytochrome P450, family 19, subfamily a, polypeptide 1 cytochrome P450, family 2, subfamily c, polypeptide 29 deoxycytidine kinase ELK3, member of ETS oncogene family fos-like antigen 2 growth factor receptor bound protein 7 S100 calcium binding protein A4 nuclear receptor coactivator 1 homeo box B6 hippocalcin kallikrein related-peptidase 10 kallikrein 1-related petidase b26 minichromosome maintenance deficient 10 (S. cerevisiae) matrix metallopeptidase 13 neurofilament, light polypeptide Notch gene homolog 4 (Drosophila) opsin 4 (melanopsin) oxytocin receptor polymerase (RNA) II (DNA directed) polypeptide B protein kinase, DNA activated, catalytic polypeptide RAD51 homolog c (S. cerevisiae) retinoic acid receptor, beta replication factor C (activator 1) 4 ras homolog gene family, member A serine (or cysteine) peptidase inhibitor, clade B, member 5 72 Stx5a Tcf4 Tnfrsf10b Tram1 Trp53 Twf1 Twf1 Vwa5a* syntaxin 5A transcription factor 4 tumor necrosis factor receptor superfamily, member 10b translocating chain-associating membrane protein 1 transformation related protein 53 twinfilin, actin-binding protein, homolog 1 (Drosophila) twinfilin, actin-binding protein, homolog 1 (Drosophila) von Willebrand factor A domain containing 5A shRNAs enriched specifically in vivo Intended shRNA target 4921537P18Rik Acss2 Appbp2 Arg2 Bard1 Cdk6 Cldn1 Col1a2 Cpa2 Ddr2 F3 Gas8 Gsn Hgf Igf1 Itgav Krit1 Krit1 Mc3r Mcm10 Mllt11 Nme1 Nrg2 Olfr1350 Olfr313 Pparg Ppm1j Prkd3 Prlr Rad17 Rarg Strap Vegfc Gene name RIKEN cDNA 4921537P18 gene acyl-CoA synthetase short-chain family member 2 amyloid beta precursor protein binding protein 2 arginase type II BRCA1 associated RING domain 1 cyclin-dependent kinase 6 claudin 1 collagen, type I, alpha 2 carboxypeptidase A2, pancreatic discoidin domain receptor family, member 2 coagulation factor III growth arrest specific 8 gelsolin hepatocyte growth factor insulin-like growth factor 1 integrin alpha V KRIT1, ankyrin repeat containing KRIT1, ankyrin repeat containing melanocortin 3 receptor minichromosome maintenance deficient 10 (S. cerevisiae) myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 11 non-metastatic cells 1, protein (NM23A) expressed in neuregulin 2 olfactory receptor 1350 olfactory receptor 313 peroxisome proliferator activated receptor gamma protein phosphatase 1J protein kinase D3 prolactin receptor RAD17 homolog (S. pombe) retinoic acid receptor, gamma serine/threonine kinase receptor associated protein vascular endothelial growth factor C 73 Vwa5a* Wnt5a Wwox von Willebrand factor A domain containing 5A wingless-related MMTV integration site 5A WW domain-containing oxidoreductase * - distinct Vwa5a shRNAs scored in vitro and in vivo shRNAs depleted in vitro and in vivo Intended shRNA target Map2k6 Top2a Vdr Gene name mitogen-activated protein kinase kinase 6 topoisomerase (DNA) II alpha vitamin D receptor shRNAs depleted specifically in vitro Intended shRNA target Cdkn3 Map2k3 Pdcd1 Gene name cyclin-dependent kinase inhibitor 3 mitogen-activated protein kinase kinase 3 programmed cell death 1 shRNAs depleted specifically in vivo Intended shRNA target Abcb4 Abcg2 Actr8 Aif1∫ Akr1b8 Akt3 Anxa1 Apaf1 Bmp10 Casp3 Cat Ccnd1 Ccnf Col1a1 Crkl * Crkl Ctsb Ctse Cyp24a1 Gene name ATP-binding cassette, sub-family B (MDR/TAP), member 4 ATP-binding cassette, sub-family G (WHITE), member 2 ARP8 actin-related protein 8 homolog (S. cerevisiae) allograft inflammatory factor 1 aldo-keto reductase family 1, member B8 thymoma viral proto-oncogene 3 annexin A1 apoptotic peptidase activating factor 1 bone morphogenetic protein 10 caspase 3 catalase cyclin D1 cyclin F collagen, type I, alpha 1 v-crk sarcoma virus CT10 oncogene homolog (avian)-like v-crk sarcoma virus CT10 oncogene homolog (avian)-like cathepsin B cathepsin E cytochrome P450, family 24, subfam. a, polypeptide 1 74 Ddx11 Ddx11 Dnajc3 Ehbp1l1 Epcam Erbb4 Ets2 Fgf5 Fgf7 Fgfr4 Hmgb1 Hmmr Igf1r Il13 Il6 * Il6 Insr Ivns1abp Krt15 Lyn * Map2k1 Map2k4 Map2k5 Mmp7 Mx2 Nek4 Nr2e1 Olfr116 Olfr295 Opn5 Opn5 Oxtr Perp Pfn1 Pglyrp4 Ppp2r4 Psen2 Psg17 Rac2 * Rac2 Rad51 Rasa1 Rb1 Skp2 DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 11 (CHL1-like helicase homolog, S. cerevisiae) DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 11 (CHL1-like helicase homolog, S. cerevisiae) DnaJ (Hsp40) homolog, subfamily C, member 3 EH domain binding protein 1-like 1 epithelial cell adhesion molecule v-erb-a erythroblastic leukemia viral oncogene homolog 4 (avian) E26 avian leukemia oncogene 2, 3' domain fibroblast growth factor 5 fibroblast growth factor 7 fibroblast growth factor receptor 4 high mobility group box 1 hyaluronan mediated motility receptor (RHAMM) insulin-like growth factor I receptor interleukin 13 interleukin 6 interleukin 6 insulin receptor influenza virus NS1A binding protein keratin 15 Yamaguchi sarcoma viral (v-yes-1) oncogene homol. mitogen-activated protein kinase kinase 1 mitogen-activated protein kinase kinase 4 mitogen-activated protein kinase kinase 5 matrix metallopeptidase 7 myxovirus (influenza virus) resistance 2 NIMA (never in mitosis gene a)-related expressed kinase 4 nuclear receptor subfamily 2, group E, member 1 olfactory receptor 116 olfactory receptor 295 opsin 5 opsin 5 oxytocin receptor PERP, TP53 apoptosis effector profilin 1 peptidoglycan recognition protein 4 protein phosphatase 2A, regulatory subunit B (PR 53) presenilin 2 pregnancy specific glycoprotein 17 RAS-related C3 botulinum substrate 2 RAS-related C3 botulinum substrate 2 RAD51 homolog (S. cerevisiae) RAS p21 protein activator 1 retinoblastoma 1 S-phase kinase-associated protein 2 (p45) 75 Stk17b Stx5a Tcf4 Timp4 Tm4sf1 Tpd52 Ttc1 Twf1 * Tyk2 Vegfa Xpc Yes1 Yes1 serine/threonine kinase 17b (apoptosis-inducing) syntaxin 5A transcription factor 4 tissue inhibitor of metalloproteinase 4 transmembrane 4 superfamily member 1 tumor protein D52 tetratricopeptide repeat domain 1 twinfilin, actin-binding protein, homol. 1 (Drosophila) tyrosine kinase 2 vascular endothelial growth factor A xeroderma pigmentosum, complementation group C Yamaguchi sarcoma viral (v-yes) oncogene homol. 1 Yamaguchi sarcoma viral (v-yes) oncogene homol. 1 * - validated in subsequent experiments ∫ - failed to validate in subsequent experiments 76 Supplementary Table 2 – Primer Sequences PCR primers used to amplify shRNAs from genomic DNA Primer Sequence Primer A 5’-GCCTCCCTCGCGCCATCAGCAGAAGGCTCGAGAAG GTATATTGCTGTTGACAGTGAGCG-3’ Primer B 5’-GCCTTGCCAGCCCGCTCAGCTAAAGTAGCCCCTTGA ATTCCGAGGCAGTAGGCA-3’. Sequences targeted by shRNAs shRNA Sequence targeted construct Rac2-1 5’-CCAAAGGGAGAGATGTGGAAA-3’ Rac2-2 5’-CAGGACCCGATCCATAGCAAA-3’ CrkL-1 5’-CCTGGAGTTTGCTTTATGTAA-3’ CrkL-2 5’-CAGGCTAAGTTCTATAGTAAA-3’ Twf-1 5’-CGTTACCATTTCTTTCTGTAT-3’ Twf-2 5’-GCAGTTGGAAATAGATATAAA-3’ Additional shRNA sequences are available upon request. 77 Chapter 3: Genome-scale in vivo screening reveals compensatory oncogenic pathways in B-cell leukemia __________________________________________________________________ 78 Abstract RNAi screening approaches have been extensively used in vitro to identify novel genes that impact tumor cell biology. However, these approaches have not been broadly adapted to in vivo systems. Here, we have used a transplantable model of B-cell acute lymphoblastic leukemia to perform a genome-scale shRNA screen for modulators of tumor progression in vivo. We have identified a large set of genes that uniquely impact leukemia cell growth in the in vivo setting, including a number of established leukemia genes, like Runx, Lmo2, and Phf6. While inactivating mutations in Phf6 are commonly observed in human T-cell acute lymphoblastic leukemia, we have found that suppression of this gene in B-cell malignancies impairs tumor progression, suggesting that Phf6 may play a lineage-specific role in hematopoietic malignancies. Introduction Retroviral insertional mutagenesis screens (Lund et al., 2002; Mikkers et al., 2002; Suzuki et al., 2002) and transposon-based screens (Collier et al., 2005; Dupuy et al., 2005) have been used as a tool for genetic discovery in hematopoietic and solid tumors. These screens have identified a number of novel genes that drive tumor initiation and progression, but are biased towards the identification of gain-of-function mutations and are primarily limited to positive selection screens. Using RNAi based screening approaches, it is possible to examine loss of function phenotypes in mammalian cells (Berns et al., 2004; Ngo et al., 2006; Paddison et al., 2004; Schlabach et al., 2008; Silva et al., 2008). These screens have been effective in identifying genes necessary for the proliferation and survival of transformed cells, but they have primarily 79 been limited to in vitro systems. Recently, we, and others, have adapted pool-based shRNA screening approaches to in vivo applications in transplantable tumors. Lowcomplexity shRNA pools were used to screen for tumor suppressors in mouse models of liver cancer (Zender et al., 2008) and B-cell lymphoma (Bric et al., 2009). Using a moderately-sized shRNA library, we performed an in vivo screen for genes that impacted disease progression in established B-cell lymphomas, and were able to introduce nearly 1000 genetic lesions into individual animals (Meacham et al., 2009). A similar number of genetic alterations were also introduced into a transplantable model of acute myeloid leukemia using an inducible shRNA system (Zuber et al., 2011). Thus far, no genome-scale shRNA screens have been performed in vivo. Here, we report the first example of such a screen. We have used five pools of 10,000 hairpins each to screen for genes that impact leukemia growth in a transplantable model of B-cell acute lymphoblastic leukemia. Importantly, in this model, we can identify as many as 9,000 unique shRNAs in the tumor burden from individual animals, and thus, are able to simultaneously assay the effect of thousands of genetic alterations in vivo. We performed a parallel screen in vitro, and found that many shRNAs uniquely impacted leukemia cell proliferation in the in vitro or in vivo settings. This approach has allowed us to identify genetic dependencies that are unique to tumor cells growing in the in vivo context. Results and Discussion Here, we have utilized a transplantable model of B-cell acute lymphoblastic leukemia (ALL) to perform a genome-scale screen for modulators of disease progression 80 in vivo. Specifically, in this model, transplantation of Arf-/- pre-B cells that express the p185 Bcr-Abl fusion protein into syngeneic recipient mice produces a B-cell leukemia within approximately two weeks (Williams et al., 2007; Williams et al., 2006). Importantly, since leukemias are transplanted into syngeneic mice, tumor cell growth occurs within the context of normal microenvironmental cues of the hematopoietic system. In this model, transplantation of as few as 20 leukemia cells is sufficient to produce disease in 100% of recipient animals (Williams et al., 2007). This, along with the relatively short latency in which disease arises, suggests that large numbers of leukemia cells are contributing to disease following transplantation. Therefore, we reasoned that this model might accommodate the introduction of diverse shRNA sets into transplanted tumors. Initially, to assess our ability to maintain library diversity in vivo, we introduced a moderately-sized shRNA library into transplanted tumor cells. Specifically, leukemia cells were transduced ex vivo with a pooled shRNA library containing approximately 2200 shRNAs that co-express the fluorescent protein GFP. We then sorted shRNA infected cells, and injected this cell population into recipient animals. At the time of terminal disease, tumor cells were harvested from the blood of mice, shRNAs were PCR amplified from tumors, and high throughput sequencing was used to assess hairpin representation. We could identify between 1800 and 1900 unique shRNAs in tumor cells derived from individual mice, suggesting that, starting with this library of 2200 shRNAs, the majority of library complexity was maintained in the in vivo setting (Supplementary figure 1). In previous work, we have found that we could represent, at most, 900 unique hairpins in vivo in a transplantable lymphoma model (Meacham et al., 2009), likely due to limited numbers of cells seeding disease in this system. Here, the identification of 81 1900 unique shRNAs in a single mouse indicates that large numbers of transplanted cells are contributing to tumor burden, and, therefore, that very large shRNA sets could potentially be introduced in vivo. Based on this finding, we decided to use a genomescale shRNA library, composed of five pools containing approximately 10,000 shRNAs each, to screen for modulators of leukemia growth in the in vivo setting. Leukemia cells were transduced with single pools of 10,000 shRNAs, and infected cells were injected into recipient mice or, in parallel, placed in culture (Figure 1a). Growth in both contexts allowed us to compare shRNAs that impact leukemia cell proliferation in vitro and in vivo. Once overt disease developed, leukemia cells were harvested from the blood of recipient animals and from cultured samples, shRNAs were PCR amplified from genomic DNA, and shRNA pool composition was deconvoluted using high-throughput sequencing. With injected cell populations containing 10,000 distinct shRNAs, we could detect, on average, between 7000 and 8000 unique hairpins in tumors derived from individual mice (Figure 1b). Unlike previous pool-based in vivo shRNA screens, where the number of unique hairpins represented in tumors has been limited to hundreds of shRNAs, in this model, thousands of shRNAs can be represented in vivo. In some mice, only 5000 to 6000 unique shRNAs could be identified, either due to smaller numbers of cells seeding disease in those tumors, or due to the clonal outgrowth of subsets of tumor cells, which can reduce the representation of other shRNA infected leukemia cells. However, even in these mice, we are still able to represent significantly more hairpins than has been reported in other in vivo screens. To assess the upper limit of the number of hairpins that can be detected in individual tumors, we also combined all five pools of shRNAs and introduced this set of 50,000 hairpins into tumor cells prior to transplantation, and found that we could identify as many as 30,000 unique shRNAs in tumors from individual animals at the time of terminal disease (Supplementary figure 1). Thus, in this model of 82 B-cell ALL, we can systematically examine the impact of the loss of function of tens of thousands of genes on tumor cell growth in vivo using large, unbiased shRNA libraries. When samples were clustered based on the enrichment and depletion of shRNAs within each sample, the in vivo samples clustered together, as did the in vitro samples, suggesting that differential selective pressure causes hairpins to behave in distinct ways in these two settings, and that this behavior is sufficient to distinguish in vitro samples from in vivo samples. (Figure 1c). Additionally, the behavior of hairpins in vitro is poorly correlated with the behavior of hairpins in vivo (Figure 1d and 2a), and the set of shRNAs that deplete, on average, at least four-fold in the in vivo setting is largely nonoverlapping with the set of shRNAs that depleted to the same extent in vitro (Figure 2b). Together, these results indicate that shRNA-mediated suppression of target genes may differentially impact tumor cell growth in vitro and in vivo and suggest that leukemia cells may have unique genetic dependencies in the in vivo setting, due to factors like altered requirements for growth and survival in the presence of local microenvironmental cues. Thus, performing screens in vivo may allow context-specific determinants of disease progression to be identified. For validation studies, we focused on shRNAs that depleted specifically in the in vivo setting. Hairpins that depleted an average of four-fold in vivo and did not deplete more that 25% in vitro were considered candidates for validation. The set of scoring shRNAs by these criteria contained approximately 1700 hairpins that target annotated genes, predicted genes, and predicted proteins. Functional categorization of the genes targeted by scoring shRNAs (Huang da et al., 2009a, b) based on structural features revealed that these genes were enriched for a number of common protein domains (Figure 2c). These domains included C2H2 type zinc finger protein domains, which 83 primarily regulate gene expression (Iuchi, 2001), Kelch and BTB/POZ domains, which are involved in protein-protein interactions and regulate diverse cellular processes (Collins et al., 2001), and plant homeodomain (PHD) fingers, which recognize specific histone modifications and are part of multimeric complexes that can activate or repress gene transcription (Musselman and Kutateladze, 2011; Sanchez and Zhou, 2011). To validate candidate hairpins, we made use of in vitro and in vivo GFP competition assays, where leukemia cells were partially transduced with individual shRNA constructs coexpressing GFP, and were then injected into recipient mice or placed in culture. To assess the impact of a given hairpin on tumor cell growth, leukemia cells were harvested after a period of in vitro or in vivo proliferation, and the percentage of GFP positive cells was assayed by FACs (Figure 2d). In total, out of the eighteen shRNAs tested targeting genes containing Kelch and C2H2 zinc finger domains, eight depleted in vivo as single constructs (Figure 2e), suggesting a high validation rate for this dataset. Importantly, these shRNAs differentially impacted leukemia cell growth in vivo and in vitro; whereas these hairpins were neutral or, in some instances, provided a growth advantage in the in vitro context, they depleted in tumor cells harvested from mice. By performing screens in vivo, we have identified a number of shRNAs that negatively impact tumor cell proliferation specifically in the in vivo setting. As another filter to select candidate shRNAs for validation, we also generated transcriptional data from leukemia cells that were growing in vitro and in vivo. Like the screening data, these transcriptional profiles were sufficient to distinguish in vitro samples from in vivo samples using unsupervised clustering approaches (Figure 3a), and a large number of genes were significantly differentially expressed between the two settings. To identify genes that might be functionally important in the in vivo setting, we 84 compared the genes that were transcriptionally upregulated in vivo to those that were targeted by hairpins that depleted specifically in vivo, which revealed that the overlap between these gene sets was limited (Figure 3b and Supplementary table 1). Before individually testing candidate hairpins targeting this overlapping gene set, we built a small, high coverage validation library that contained six independent shRNAs directed at each of the 133 genes in this set. As a rapid and cost effective method of generating a validation library, shRNA oligonucelotides were synthesized on a chip, eluted, PCR amplified and bulk cloned into our shRNA expression vector. This bulk-cloned library was directly used to re-screen for modulators of leukemia cell growth in vitro and in vivo (Figure 3c). Only genes that had at least two independent shRNAs deplete specifically in vivo in this secondary screen (Supplementary table 2) were considered candidates for individual validation studies. When candidate hairpins were validated as individual constructs, five shRNAs depleted both in vitro and in vivo (Figure 3d), eight shRNAs depleted specifically in the in vivo setting, including two shRNAs targeting the C2H2 zinc finger proteins PogZ and Zfp28 (Figure 2e and 3e), and two shRNAs failed to validate. Two of the shRNAs that depleted specifically in the in vivo setting targeted Lmo2 and Runx1, genes with established roles in hematopoietic malignancies. Lmo2 translocations, resulting in overexpression of the full length Lmo2 protein, are found in T cell malignancies and have been suggested, at least in part, to promote tumorigenesis by blocking T cell development (Curtis and McCormack, 2010; McCormack et al., 2010). Thus far, a role for Lmo2 in B cell malignancies has not been well established. Runx translocations have been identified in B cell and myeloid malignancies. Whereas translocations result in the truncation of Runx in myeloid malignancies, full-length Runx is expressed in B-cell 85 malignancies (Golub et al., 1995; Romana et al., 1995), and it is unclear if Runx plays an oncogenic or tumor suppressor role in B-cell diseases. Supporting an oncogenic role for Runx, activating Runx mutations have been identified in insertional mutagenesis screens in B and T cell malignancies (Stewart et al., 1997; Wotton et al., 2002) and Runx gene overexpression has been observed in human B-cell ALL (Niini et al., 2002). In our datasets, both Runx and Lmo2 are transcriptionally upregulated in the in vivo setting, and hairpins targeting these genes confer a growth disadvantage, which indicates that Runx and Lmo2 are functionally important in vivo and that they may play a prooncogenic role in B cell malignancies. However, neither of these genes are involved in signaling downstream of Bcr-Abl, suggesting that leukemia cells may be utilizing alternative oncogenic pathways involving of Runx and Lmo2 for growth or survival in vivo. As a final means of filtering our shRNA screening data using existing biological datasets, we compared genes targeted by shRNAs that depleted in vivo with genes contained within amplicons found in human ALL patients (Beroukhim et al., 2010; Kuiper et al., 2007; Mullighan et al., 2008), which may facilitate the identification of genes that are functionally important within these large regions of amplification, as well as genes whose suppression has more pronounced phenotypic consequences. We mapped the genomic locations of the human orthologs of genes targeted by scoring shRNAs, and looked for genes that fell within amplified regions. A number of shRNAs targeting genes contained within ALL amplicons validated as single constructs (Figure 4a), including hairpins targeting the plant homeodomain finger protein, Phf6. Inactivating Phf6 mutations are frequently found in T-cell acute lymphoblastic leukemia (Zuber et al., 2009), and less frequently, acute myeloid leukemia (Van Vlierberghe et al., 2011), but 86 mutations have not been detected in B-lineage malignancies (Zuber et al., 2009). In contrast to T cell and myeloid malignancies, where inactivation of Phf6 promotes tumor development, our data suggests that suppression of Phf6 in B-cell leukemias may negatively impact disease progression, specifically in the in vivo setting. In all hematopoietic organs tested, including the spleen and bone marrow, where the majority of tumor cell proliferation occurs, the suppression of Phf6 was selected against, and in GFP competition assays, the percentage of cells suppressing Phf6 decreased progressively after transplantation, indicative of a role for this gene in leukemia cell growth or survival in vivo, rather than engraftment following transplantation. Suppression of Phf6 in pure populations of transplanted leukemia cells also significantly reduced peripheral leukemia burden in recipient animals, relative to a vector control (Figure 4B). In a transplantable model of Burkitt’s lymphoma, another B-cell malignancy, suppression of Phf6 was also specifically selected against in the in vivo context (Figure 4c), indicating the Phf6 suppression negatively impacts tumor growth in multiple B-cell diseases. When we looked at the effect of this hairpin in a transplantable T cell lymphoma, we found that hairpin-mediated suppression of Phf6 was neutral (Figure 4e), suggesting differential requirements for Phf6 gene function in B and T cell lineages. Additionally, cDNA-mediated Phf6 overexpression was potently selected against in T cell lymphomas (Figure 4e), whereas it was neutral in B-cell ALLs in vivo (Figure 4d). Together, these results indicate that Phf6 may play a lineage-specific role in hematopoietic malignancies, where overexpression of Phf6 impairs tumor cell growth in T cell malignancies, and suppression of Phf6 hinders disease progression in B cell malignancies. Importantly, these effects were observed only in the in vivo context, 87 suggesting that the in vivo tumor microenvironment influences the genetic dependencies of tumor cells. Here, in a transplantable model of B-cell acute lymphoblastic leukemia, we have reported the first example of a large-scale unbiased shRNA screen in vivo. This system permits the introduction of thousands of shRNAs into individual animals, allowing for the simultaneous assessment of the impact of loss-of-function events in tumors. By performing parallel screens in vitro and in vivo, we were able to compare the set of shRNAs that impacted tumor cell growth in these two settings, and identified a set of hairpins that uniquely influenced tumor cell growth in vivo. To select shRNAs for validation, genes targeted by this set of hairpins were functionally categorized, and were also filtered using transcriptional datasets and human copy number alteration datasets. The suppression of a number of genes impaired leukemia cell growth only in the in vivo setting in validation studies, suggesting that differential selective pressures in vivo and in vitro influence the genetic dependencies of tumor cells. We also found that a number of established leukemia genes had unexpected roles in leukemia cell proliferation in vivo. In this Bcr-Abl B-ALL model, the suppression of Runx and Lmo2 impaired disease progression, suggesting that Bcr-Abl driven B-cell leukemias may rely on growth and survival signals downstream of these genes. In other malignancies, these genes are activated by translocation events, and they act as the driving oncogenic lesion in tumors. Here, dependence on these genes occurs in the context of enforced expression of the oncogenic Bcr-Abl fusion protein, indicating that Bcr-Abl driven leukemia cells engage alternative oncogenic pathways in the in vivo setting. Suppression of the plant homeodomain protein Phf6 also impaired leukemia cell growth in vivo in B cell tumors; in contrast, human mutational data indicates that Phf6 88 loss is a key event in tumor progression in a subset of T-cell ALLs. Thus, Phf6 may differentially impact hematopoietic malignancies in a lineage-specific manner. Acknowledgments and author contributions We would like to thank AJ Bhutkar for bioinformatic processing of sequencing and microarray data, Alla Leshinsky and Richard Cook for sequencing expertise, and Manlin Luo for performing microarrays. We would also like to thank Luke Gilbert and Francisco Sanchez-Rivera for lymphoma cell lines, Luke Gilbert and Eleanor Cameron for critical reading of the manuscript, and all members of the Hemann lab for helpful advice and discussions. Corbin Meacham and Michael Hemann designed the screen and experiments. Corbin Meacham preformed the screen and all subsequent experiments and wrote the manuscript. 89 References Berns, K., Hijmans, E.M., Mullenders, J., Brummelkamp, T.R., Velds, A., Heimerikx, M., Kerkhoven, R.M., Madiredjo, M., Nijkamp, W., Weigelt, B., et al. (2004). A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431-437. Beroukhim, R., Mermel, C.H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., Barretina, J., Boehm, J.S., Dobson, J., Urashima, M., et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899-905. Bric, A., Miething, C., Bialucha, C.U., Scuoppo, C., Zender, L., Krasnitz, A., Xuan, Z., Zuber, J., Wigler, M., Hicks, J., et al. (2009). Functional identification of tumorsuppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 16, 324-335. Collier, L.S., Carlson, C.M., Ravimohan, S., Dupuy, A.J., and Largaespada, D.A. (2005). Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 436, 272-276. Collins, T., Stone, J.R., and Williams, A.J. (2001). All in the family: the BTB/POZ, KRAB, and SCAN domains. Mol Cell Biol 21, 3609-3615. Curtis, D.J., and McCormack, M.P. (2010). The molecular basis of Lmo2-induced T-cell acute lymphoblastic leukemia. Clin Cancer Res 16, 5618-5623. Dickins, R.A., Hemann, M.T., Zilfou, J.T., Simpson, D.R., Ibarra, I., Hannon, G.J., and Lowe, S.W. (2005). Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 37, 1289-1295. Dupuy, A.J., Akagi, K., Largaespada, D.A., Copeland, N.G., and Jenkins, N.A. (2005). Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 436, 221-226. Golub, T.R., Barker, G.F., Bohlander, S.K., Hiebert, S.W., Ward, D.C., Bray-Ward, P., Morgan, E., Raimondi, S.C., Rowley, J.D., and Gilliland, D.G. (1995). Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 92, 4917-4921. Huang da, W., Sherman, B.T., and Lempicki, R.A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1-13. Huang da, W., Sherman, B.T., and Lempicki, R.A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44-57. Iuchi, S. (2001). Three classes of C2H2 zinc finger proteins. Cell Mol Life Sci 58, 625635. Kuiper, R.P., Schoenmakers, E.F., van Reijmersdal, S.V., Hehir-Kwa, J.Y., van Kessel, A.G., van Leeuwen, F.N., and Hoogerbrugge, P.M. (2007). High-resolution genomic 90 profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 21, 12581266. Lund, A.H., Turner, G., Trubetskoy, A., Verhoeven, E., Wientjens, E., Hulsman, D., Russell, R., DePinho, R.A., Lenz, J., and van Lohuizen, M. (2002). Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat Genet 32, 160-165. McCormack, M.P., Young, L.F., Vasudevan, S., de Graaf, C.A., Codrington, R., Rabbitts, T.H., Jane, S.M., and Curtis, D.J. (2010). The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science 327, 879-883. Meacham, C.E., Ho, E.E., Dubrovsky, E., Gertler, F.B., and Hemann, M.T. (2009). In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat Genet 41, 1133-1137. Mikkers, H., Allen, J., Knipscheer, P., Romeijn, L., Hart, A., Vink, E., and Berns, A. (2002). High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet 32, 153-159. Mullighan, C.G., Miller, C.B., Radtke, I., Phillips, L.A., Dalton, J., Ma, J., White, D., Hughes, T.P., Le Beau, M.M., Pui, C.H., et al. (2008). BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453, 110-114. Musselman, C.A., and Kutateladze, T.G. (2011). Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res 39, 9061-9071. Ngo, V.N., Davis, R.E., Lamy, L., Yu, X., Zhao, H., Lenz, G., Lam, L.T., Dave, S., Yang, L., Powell, J., et al. (2006). A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441, 106-110. Niini, T., Vettenranta, K., Hollmen, J., Larramendy, M.L., Aalto, Y., Wikman, H., Nagy, B., Seppanen, J.K., Ferrer Salvador, A., Mannila, H., et al. (2002). Expression of myeloid-specific genes in childhood acute lymphoblastic leukemia - a cDNA array study. Leukemia 16, 2213-2221. Paddison, P.J., Silva, J.M., Conklin, D.S., Schlabach, M., Li, M., Aruleba, S., Balija, V., O'Shaughnessy, A., Gnoj, L., Scobie, K., et al. (2004). A resource for large-scale RNAinterference-based screens in mammals. Nature 428, 427-431. Romana, S.P., Poirel, H., Leconiat, M., Flexor, M.A., Mauchauffe, M., Jonveaux, P., Macintyre, E.A., Berger, R., and Bernard, O.A. (1995). High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood 86, 4263-4269. Sanchez, R., and Zhou, M.M. (2011). The PHD finger: a versatile epigenome reader. Trends Biochem Sci 36, 364-372. Schlabach, M.R., Luo, J., Solimini, N.L., Hu, G., Xu, Q., Li, M.Z., Zhao, Z., Smogorzewska, A., Sowa, M.E., Ang, X.L., et al. (2008). Cancer proliferation gene discovery through functional genomics. Science 319, 620-624. 91 Silva, J.M., Marran, K., Parker, J.S., Silva, J., Golding, M., Schlabach, M.R., Elledge, S.J., Hannon, G.J., and Chang, K. (2008). Profiling essential genes in human mammary cells by multiplex RNAi screening. Science 319, 617-620. Stewart, M., Terry, A., Hu, M., O'Hara, M., Blyth, K., Baxter, E., Cameron, E., Onions, D.E., and Neil, J.C. (1997). Proviral insertions induce the expression of bone-specific isoforms of PEBP2alphaA (CBFA1): evidence for a new myc collaborating oncogene. Proc Natl Acad Sci U S A 94, 8646-8651. Suzuki, T., Shen, H., Akagi, K., Morse, H.C., Malley, J.D., Naiman, D.Q., Jenkins, N.A., and Copeland, N.G. (2002). New genes involved in cancer identified by retroviral tagging. Nat Genet 32, 166-174. Van Vlierberghe, P., Patel, J., Abdel-Wahab, O., Lobry, C., Hedvat, C.V., Balbin, M., Nicolas, C., Payer, A.R., Fernandez, H.F., Tallman, M.S., et al. (2011). PHF6 mutations in adult acute myeloid leukemia. Leukemia 25, 130-134. Williams, R.T., den Besten, W., and Sherr, C.J. (2007). Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev 21, 22832287. Williams, R.T., Roussel, M.F., and Sherr, C.J. (2006). Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 103, 6688-6693. Wotton, S., Stewart, M., Blyth, K., Vaillant, F., Kilbey, A., Neil, J.C., and Cameron, E.R. (2002). Proviral insertion indicates a dominant oncogenic role for Runx1/AML-1 in T-cell lymphoma. Cancer Res 62, 7181-7185. Zender, L., Xue, W., Zuber, J., Semighini, C.P., Krasnitz, A., Ma, B., Zender, P., Kubicka, S., Luk, J.M., Schirmacher, P., et al. (2008). An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 135, 852-864. Zuber, J., McJunkin, K., Fellmann, C., Dow, L.E., Taylor, M.J., Hannon, G.J., and Lowe, S.W. (2011). Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 29, 79-83. Zuber, J., Radtke, I., Pardee, T.S., Zhao, Z., Rappaport, A.R., Luo, W., McCurrach, M.E., Yang, M.M., Dolan, M.E., Kogan, S.C., et al. (2009). Mouse models of human AML accurately predict chemotherapy response. Genes Dev 23, 877-889. 92 Materials and Methods Cell culture Bcr-Abl mouse acute lymphoblastic leukemia cells were cultured in RPMI (supplemented with 10% FBS, 4 mM L-glutamine and 5µM β-mercaptoethanol) RNAi screen To preserve library complexity, a minimum of 200-fold coverage of shRNA libraries was maintained at all steps in the screen. Leukemia cells were infected with individual pools of 10,000 shRNAs each to a final infection percent of 5-10%. 48 hours after infection, lymphoma cells were GFP sorted. 24-48 hours after sorting, 2x106 leukemia cells/mouse were injected into three syngeneic recipient mice by tail vein injection. At the time of injection, 4 million cells were collected to serve as the input sample. Disease progression was monitored by peripheral blood counts. Following the appearance of overt leukemias, approximately 12 days after tail-vein injection, leukemia cells were harvested from the blood of mice. For in vitro samples, infected leukemia cells were plated in triplicate and maintained for two weeks. Genomic DNA was isolated by proteinase K digestion and isopropanol precipitation. The antisense strand of shRNAs was amplified from genomic DNA using primers that include 1 basepair mutations to barcode individual samples (unmutated primers 5’ loop primer; NNNNNTAGTGAAGCCACAGATGTA; unmutated 3’ primer NNNNTTGAATTCCGAGGCAGTAGG). All PCR steps were performed in a UV hood to avoid cross-contamination between samples. Hairpins were amplified in multiple 50 uL reactions using HotStar Taq (Qiagen). After PCR amplification, samples were pooled and prepared for sequencing with Illumina’s genomic adaptor kit. At least 41 bases of the PCR product were sequenced with an Illumina HiSeq 2000 machine. 93 shRNAs with less than 100 reads in the input sample were excluded from further analysis, and read numbers for each shRNA were normalized to the total read numbers per sample to allow for cross-comparison between samples. Validation assays For GFP competition assays, pure populations of mCherry positive ALL cells were partially transduced with single shRNA constructs co-expressing GFP, and were infected to a final percentage of 40-60% GFP positive cells. 1x10^6 cells were injected into 6wk C57/BL6 females, and at the time of terminal disease, determined by body condition score, leukemia cells were harvested from the blood of mice. The percentage of GFP+ cells was analyzed on a Becton Dickinson LSRII. Microarray analysis Microarray studies were performed using Affymetrix Mouse Genome 430 2.0 arrays Validation library cloning shRNAs oligos were designed using siScales (http://gesteland.genetics.utah.edu/siRNA_scales/) and were bulk synthesized by LC Sciences (LC Sciences OligoMix). Eluted oligos were PCR amplified using the following primers: XhoI 5' primer; 5' CAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG 3' EcoRI 3' primer; 5' CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCA 3' and were batch cloned into a mir30 retroviral vector (Dickins et al., 2005). 94 se se ou ou 4 2 5 1.5 1 0.5 0 -0.5 -1 -1.5 -2 Log2 average fold change in vivo In pu t pl Sa e m 1 p Sa le m 2 pl e 3 In pu M ou t s M e1 ou s M e2 ou se 3 C m Sa PCR amplify hairpins High throughput sequencing Number of unique shRNA reads In vitro 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 2 D 95 -6 -4 t In vivo In vitro -2 0 Sa In vivo Sa e m 1 pl e m 2 pl e 3 In pu M ou t s M e1 ou s M e2 ou se 3 pu Pool 3 In 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 pl m In pu t pl Sa e m 1 p Sa le m 2 pl e 3 In pu M ou t s M e1 ou s M e2 ou se 3 Sa In Number of unique shRNA reads Pool 1 m In vitro Number of unique shRNA reads m pu t p Sa le 1 m p Sa le m 2 pl e 3 In pu M ou t s M e1 ou s M e2 ou se 3 Sa Number of unique shRNA reads 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 Sa In Sa pu t m pl Sa e m 1 p Sa le m 2 pl e 3 In pu M ou t s M e1 ou s M e2 ou se 3 Number of unique shRNA reads Sort infected cells M M se 1 Infect leukemia cells MOI ~1 3 (tagged with GFP) ou se 5 1 3 Figure 1 A Pool of ~10,000 shRNAs B M se ou ou M 2 4 3’ mir30 tro vi tro tro vi vi tro tro shRNA M In In In vi vi 5’ mir30 In In (relative to input) Log2 fold change in shRNA representation Figures 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 Pool 2 In vitro 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 Pool 4 In vivo In vitro In vivo Pool 5 In vivo 6 4 2 0 -2 -4 r =.383 Log2 average fold change in vitro 2 4 6 Figure 1 Genome-scale in vivo RNAi screening in a transplantable model of acute lymphoblastic leukemia (A) In vivo RNAi screening strategy. Leukemia cells were transduced with pooled retroviral shRNA libraries containing 10,000 distinct shRNAs coexpressing GFP. Following infection, cells were sorted for GFP, and injected into recipient mice or maintained in culture. Leukemia cells were harvested from tumorbearing animals, hairpins were amplified from genomic DNA, and shRNA representation was deconvoluted using high throughput sequencing. (B) shRNA library complexity is maintained in the in vivo setting. The number of unique hairpins detected in each sample is shown (C) Clustering of samples based on the enrichment or depletion of shRNAs. Data is displayed as the log2 of the fold change for each shRNA, and the fold change has been normalized the median fold change across all shRNAs in a sample. For this analysis, leukemia cells were transduced with the combined set of 50,000 shRNAs prior to screening, and hairpins with less than 100 reads in the input sample were excluded (D) Scatterplot showing the correlation between the average behavior of hairpins in vitro and in vivo for leukemia cells transduced with the combined set of 50,000 shRNAs. rvalue represents a Spearman correlation coefficient. 96 Figure 2 Characterization of shRNAs that deplete in vivo. (A) Abundance of shRNAs in tumors harvested from mice or cells maintained in culture. Data is displayed as the 97 average of the log2 fold change of in vivo (red) or in vitro (blue) samples relative to the input population. shRNAs are rank ordered based on the average fold change in vivo (left) or in vitro (right). Data is combined from five screens using individual pools of 10,000 shRNAs in each screen. (B) Unique sets of hairpins score as depleted in vivo and in vitro. Venn diagrams show the number of shRNAs that were four-fold depleted, on average, in vitro and in vivo. The fold change for each shRNA was normalized to the median fold change across all shRNAs in the in vivo or in vitro settings. (C) shRNAs that deplete in vivo are enriched for a number of common protein domains. shRNAs that depleted an average of 4-fold in vivo and less than 25% in vitro were functionally categorized using the DAVID functional annotation program (D) Validation strategy for individual shRNAs. Cells were partially transduced with single shRNA constructs coexpressing GFP, and injected into mice or placed in culture. After a period of in vitro or in vivo proliferation (approximately two weeks) the percentage of GFP positive cells was quantified by FACs. (E) Suppression of genes containing Kelch and C2H2 zinc finger protein domains is selected against in vivo. Results from in vitro and in vivo GFP competition assays are displayed as the fold change in the percentage of GFP positive cells relative to an input cell population. p-values were calculated using a two-tailed student’s T-test. 98 Figure 3 Validation of hairpins targeting genes that are transcriptionally altered in leukemia cells in vivo. (A) Clustering of tumor cells harvested from leukemic mice or culture based on their transcriptional profiles. (B) Overlap between genes that are induced in vivo and genes targeted by shRNAs that deplete in our screening data. (C) Secondary screening strategy. Six hairpins targeting all genes that overlapped between microarray and screening datasets were bulk synthesized and cloned into a retroviral expression vector, and this high-coverage library was used to re-screen for genes that depleted in vivo and were targeted by multiple independent hairpins (D) Suppression of established leukemia genes impairs B-ALL growth in vivo. shRNAs targeting genes that scored as depleted in the secondary screen were validated as single constructs in GFP competition assays. Results are displayed as the fold change in the percentage of GFP positive cells relative to an input population for shRNAs that depleted in the in vitro and 99 in vivo settings (left panel) or hairpins that depleted specifically in vivo (center and right panel). Hairpins targeting Runx and Lmo2 depleted specifically in vivo 100 control B cell B cell lymphoma-A lymphoma-B shPhf6 shPhf6 B-ALL control 101 B-ALL Phf6 cDNA control shPhf6 vo vi 0 N.S. In 0.2 d 0.4 re 0.6 1.4 vo 0.8 f6 sh Ph 60 ul tu E vi 1.0 C N.S. In D tro l on C Days post-injection re d 1.2 12 vo 10 tu 0.2 vi 0.4 ul shPhf6 In 1.0 Peripheral leukemia burden (x10^3 cells/ uL) control C 8 re d 6 tu 0.6 Fold change (% GFP+ cells) 0.8 ul 4 p=.005 C 0 2 vo 1.2 1.0 0.8 0.6 0.4 0.2 1.2 vi p=.0001 0 p=.0001 In p=.0001 p=.0002 re d C 0 p=.0001 tu 0 ul 0.2 C 0.4 vo 0.6 vi 0.8 In 1.0 d Fold change (% GFP+ cells) p=.0003 ul tu re 1.2 Fold change (% GFP+ cells) C on tro C l cu on lt sh tro ure Its l in d n2 v sh cu ivo I sh ts ltu D n2 re py in d v sh 30 D cu ivo sh py ltu So 30 red x1 in sh 1 c viv So ul o sh x1 tur M 1 e pz in d l vi 3 sh vo M cu sh zp ltu r D l3 ed gc in sh r2 c viv o D u sh gc ltu Ph r2 re d f6 in vi sh -1 Ph cu vo sh f6 ltu Ph -1 re f6 in d v sh -2 Ph cu ivo sh f6 ltu r Sl ik 2 in ed rk v sh 4 c iv o Sl ul itr tu k4 re in d vi vo p=.0008 Fold change (% GFP+ cells) Fold change (% GFP+ cells) 1.0 C d vo vi re vo o S od ne ple m en ar ro w Bl o Bo S od ne ple M en ar ro w Bo Bl 0.8 In tu ul C d 1.8 1.6 1.4 vi re Fold change (% GFP+ cells) control In d vo vi tu ul C In re tu ul C Figure 4 A 1.2 N.S. p=.0009 p=.0001 0.6 0.4 0.2 0 B p=.0026 50 40 30 20 10 0 shPhf6 p=.0001 1.2 0.8 1.0 0.4 0.6 0.2 0 Phf6 cDNA T cell lymphoma Figure 4 Characterization of Phf6 suppression in vivo in B and T cell malignancies. (A) Validation of shRNAs that target genes found in regions of amplification of human ALL patients. (B) Characterization of Phf6 suppression in ALL cells. An shRNA targeting Phf6 selectively depletes in vivo in the blood, spleen, and bone marrow in a GFP competition assay (left panel), and the percentage of cells suppressing Phf6 progressively decreases over time following tail vein injection (center panel). Suppression of Phf6 in pure populations of leukemia cells decreases peripheral leukemia burden (right panel). (C) Suppression of Phf6 is selected against in vivo in transplanted Eµ-myc tumors. Two independent primary lymphomas were used in this experiment. (D) Phf6 overexpression does not impact B-cell ALL growth in vivo. Leukemia cells were partially transduced with the mouse Phf6 cDNA co-expressing GFP and injected into recipient mice in a GFP competition assay. Results are displayed as the fold change in the percentage of GFP positive cells relative to the input population. (E) Phf6 suppression differentially impacts B and T cell malignancies. A GFP competition assays shows the effect of Phf6 suppression and overexpression in transplanted T cell lymphomas. 102 Supplementary Figure Supplementary Figure 1 Library complexity is maintained in transplanted tumors in a mouse model of Bcr-Abl acute lymphoblastic leukemia (A) Screening strategy to identify the number of unique shRNAs that can be represented in vivo. Leukemia cells were infected with a pool of 2200 shRNAs coexpressing GFP, sorted for GFP, and injected into mice. At the time of terminal disease, leukemia cells were harvested and hairpin representation was assessed by high throughput sequencing. (B) Number of unique shRNAs identified in the tumor burden from individual mice when a library of 2200 shRNAs was introduced into leukemia cells prior to transplantation. (C) Number of unique shRNAs identified in individual mice when a library of 50,000 shRNAs was introduced into leukemia cells prior to transplantation. A minimum of 10 reads was required for an shRNA to be included in the unique shRNA count. 103 Supplementary tables – Supplementary table 1 Supplementary table 1 Overlapping set of genes from transcriptional data and screening data. Genes that were significantly transcriptionally upregulated in leukemia cells harvested from the blood of mice relative to leukemia cells maintained in culture, and genes that were targeted by hairpins that depleted at least four-fold in vivo and less than 25% in vitro. Adam19 a disintegrin and metallopeptidase domain 19 Abhd1 abhydrolase domain containing 1 Acox3 acyl-Coenzyme A oxidase 3, pristanoyl Arf2 ADP-ribosylation factor 2 Aff1 AF4/FMR2 family, member 1 Arrb1 arrestin, beta 1 Atg2a ATG2 autophagy related 2 homolog A (S. cerevisiae) Atp13a2 ATPase type 13A2 Atp7a ATPase, Cu++ transporting, alpha polypeptide Btg1 B-cell translocation gene 1, anti-proliferative Bbs4 Bardet-Biedl syndrome 4 (human) Baz2b bromodomain adjacent to zinc finger domain, 2B Cabin1 calcineurin binding protein 1 Cabin1 calcineurin binding protein 1 Cask calcium/calmodulin-dependent serine protein kinase (MAGUK family) Ctsc cathepsin C Cd177 CD177 antigen BC006779 cDNA sequence BC006779 Ccrl1 chemokine (C-C motif) receptor-like 1 Chrnb1 cholinergic receptor, nicotinic, beta polypeptide 1 (muscle) Chd8 chromodomain helicase DNA binding protein 8 Cobll1 Cobl-like 1 Ccdc84 coiled-coil domain containing 84 CTD (carboxy-terminal domain, RNA polymerase II, polypeptide A) small Ctdsp2 phosphatase 2 BC035295 cysteine-serine-rich nuclear protein 2 Cyth2 cytohesin 2 Dcaf8 DDB1 and CUL4 associated factor 8 Diablo diablo homolog (Drosophila) Dvl1 dishevelled, dsh homolog 1 (Drosophila) Dnajb9 DnaJ (Hsp40) homolog, subfamily B, member 9 Emr4 Efna1 Fam160a2 Fam193a Glb1l EGF-like module containing, mucin-like, hormone receptor-like sequence 4 ephrin A1 family with sequence similarity 160, member A2 family with sequence similarity 193, member A galactosidase, beta 1-like Gpcpd1 Gvin1 H2afj Hspa1b Herc3 Hps5 glycerophosphocholine phosphodiesterase GDE1 homolog (S. cerevisiae) GTPase, very large interferon inducible 1 H2A histone family, member J heat shock protein 1B hect domain and RLD 3 Hermansky-Pudlak syndrome 5 homolog (human) 104 Hmha1 Ikbip Itpkb Itm2b Klhl17 Klhl6 L3mbtl3 Lrrn3 Lif Lnx2 Lmo2 Mmp9 Ms4a6c Morc3 Mgst1 Myh9 Myh9 Nfe2l1 Nfe2l1 Ostm1 Pdrg1 Pon2 Pdzd2 Pex1 Phactr2 Ppap2a Pik3r3 Plac8 Pafah1b3 Plekhn1 Pogz Parp6 Pan2 Prkar2b Ptprs Plp2 Rab2b Trp53inp2 Rin3 Rassf5 Rasal3 Rragc Reep2 Arhgap25 Arhgap26 1100001G20Rik 1700020O03Rik 1700109H08Rik 2610008E11Rik 2700081O15Rik 4930470P17Rik 4931406C07Rik 4933437N03Rik 5133401N09Rik 5730508B09Rik 9130008F23Rik histocompatibility (minor) HA-1 IKBKB interacting protein inositol 1,4,5-trisphosphate 3-kinase B integral membrane protein 2B kelch-like 17 (Drosophila) kelch-like 6 (Drosophila) l(3)mbt-like 3 (Drosophila) leucine rich repeat protein 3, neuronal leukemia inhibitory factor ligand of numb-protein X 2 LIM domain only 2 matrix metallopeptidase 9 membrane-spanning 4-domains, subfamily A, member 6C microrchidia 3 microsomal glutathione S-transferase 1 myosin, heavy polypeptide 9, non-muscle myosin, heavy polypeptide 9, non-muscle nuclear factor, erythroid derived 2,-like 1 nuclear factor, erythroid derived 2,-like 1 osteopetrosis associated transmembrane protein 1 p53 and DNA damage regulated 1 paraoxonase 2 PDZ domain containing 2 peroxisomal biogenesis factor 1 phosphatase and actin regulator 2 phosphatidic acid phosphatase type 2A phosphatidylinositol 3 kinase, regulatory subunit, polypeptide 3 (p55) placenta-specific 8 platelet-activating factor acetylhydrolase, isoform 1b, subunit 3 pleckstrin homology domain containing, family N member 1 pogo transposable element with ZNF domain poly (ADP-ribose) polymerase family, member 6 polyA specific ribonuclease subunit homolog (S. cerevisiae) protein kinase, cAMP dependent regulatory, type II beta protein tyrosine phosphatase, receptor type, S proteolipid protein 2 RAB2B, member RAS oncogene family ransformation related protein 53 inducible nuclear protein 2 Ras and Rab interactor 3 Ras association (RalGDS/AF-6) domain family member 5 RAS protein activator like 3 Ras-related GTP binding C receptor accessory protein 2 Rho GTPase activating protein 25 Rho GTPase activating protein 26 RIKEN cDNA 1100001G20 gene RIKEN cDNA 1700020O03 gene RIKEN cDNA 1700109H08 gene RIKEN cDNA 2610008E11 gene RIKEN cDNA 2700081O15 gene RIKEN cDNA 4930470P17 gene RIKEN cDNA 4931406C07 gene RIKEN cDNA 4933437N03 gene RIKEN cDNA 5133401N09 gene RIKEN cDNA 5730508B09 gene RIKEN cDNA 9130008F23 gene 105 A630001G21Rik E130309D02Rik Rnf114 Robo3 Runx1 Sesn3 Sik3 Ssbp2 Sirt5 RIKEN cDNA A630001G21 gene RIKEN cDNA E130309D02 gene ring finger protein 114 roundabout homolog 3 (Drosophila) runt related transcription factor 1 sestrin 3 SIK family kinase 3 single-stranded DNA binding protein 2 sirtuin 5 Smg1 Slc39a1 Slc9a9 Spnb2 Tagap Tnks2 Sin3a Tgm2 Timm8a2 Tmcc2 Tsc22d3 Ube2h Unkl Usp6nl SMG1 homolog, phosphatidylinositol 3-kinase-related kinase (C. elegans) solute carrier family 39 (zinc transporter), member 1 solute carrier family 9 (sodium/hydrogen exchanger), member 9 spectrin beta 2 T-cell activation Rho GTPase-activating protein tankyrase, TRF1-interacting ankyrin-related ADP-ribose polymerase 2 transcriptional regulator, SIN3A (yeast) transglutaminase 2, C polypeptide translocase of inner mitochondrial membrane 8 homolog a2 (yeast) transmembrane and coiled-coil domains 2 TSC22 domain family, member 3 ubiquitin-conjugating enzyme E2H unkempt-like (Drosophila) USP6 N-terminal like Mycn Vamp5 Wdr24 Wdr26 Ythdf2 Zbtb4 Zbtb48 Zfp161 Zfp28 Zfp263 Zfp408 Zfp598 Zfyve9 v-myc myelocytomatosis viral related oncogene, neuroblastoma derived (avian) vesicle-associated membrane protein 5 WD repeat domain 24 WD repeat domain 26 YTH domain family 2 zinc finger and BTB domain containing 4 zinc finger and BTB domain containing 48 zinc finger protein 161 zinc finger protein 2 zinc finger protein 263 zinc finger protein 408 zinc finger protein 598 zinc finger, FYVE domain containing 9 106 Supplementary tables – Supplementary table 2 Supplementary table 2 Genes from the microarray overlap validation screen that were targeted by two or more depleting shRNAs. The average fold change for each shRNA in vivo (relative to the input cell population) was divided by the average fold change of that shRNA in vitro to identify hairpins that specifically depleted in vivo. shRNAs were rank ordered based on this value, and the bottom 30% of shRNAs were considered depleting shRNAs Number of independent shRNAs Gene symbol Gene name 4 Chd8 chromodomain helicase DNA binding protein 8 4 Klhl17 kelch-like 17 (Drosophila) 4 Lmo2 LIM domain only 2 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 1700020O03Rik 1700109H08Rik 2700081O15Rik 4931406C07Rik Acox3 Adam19 Arf2 Baz2b Ctsc Dcaf8 Dvl1 E130309D02Rik Herc3 Hmha1 L3mbtl3 Lnx2 Lrrn3 3 3 3 Ms4a6c Myh9 Pdzd2 3 3 3 Plekhn1 Reep2 Rragc 3 3 3 Smg1 Tagap Tmcc2 3 3 3 3 3 Tnks2 Ube2h Zbtb48 Zfp161 Zfp408 RIKEN cDNA 1700020O03 gene RIKEN cDNA 1700109H08 gene RIKEN cDNA 2700081O15 gene RIKEN cDNA 4931406C07 gene acyl-Coenzyme A oxidase 3, pristanoyl a disintegrin and metallopeptidase domain 19 ADP-ribosylation factor 2 bromodomain adjacent to zinc finger domain, 2B cathepsin C DDB1 and CUL4 associated factor 8 dishevelled, dsh homolog 1 (Drosophila) RIKEN cDNA E130309D02 gene hect domain and RLD 3 histocompatibility (minor) HA-1 l(3)mbt-like 3 (Drosophila) ligand of numb-protein X 2 leucine rich repeat protein 3, neuronal membrane-spanning 4-domains, subfamily A, member 6C myosin, heavy polypeptide 9, non-muscle PDZ domain containing 2 pleckstrin homology domain containing, family N member 1 receptor accessory protein 2 Ras-related GTP binding C SMG1 homolog, phosphatidylinositol 3-kinase-related kinase (C. elegans) T-cell activation Rho GTPase-activating protein transmembrane and coiled-coil domains 2 tankyrase, TRF1-interacting ankyrin-related ADP-ribose polymerase 2 ubiquitin-conjugating enzyme E2H zinc finger and BTB domain containing 48 zinc finger protein 161 zinc finger protein 408 2 2 2 2 4930470P17Rik Atp7a BC006779 Cabin1 RIKEN cDNA 4930470P17 gene ATPase, Cu++ transporting, alpha polypeptide cDNA sequence BC006779 calcineurin binding protein 1 107 2 2 2 2 Ccdc84 Ccrl1 Cd177 BC035295 2 2 2 2 2 2 2 2 2 Ctdsp2 Diablo Dnajb9 H2afj Hps5 Ikbip Itpkb Klhl6 Morc3 2 2 2 Mycn Nfe2l1 Ostm1 2 2 2 Pafah1b3 Pdrg1 Phactr2 2 2 2 2 Pik3r3 Pogz Pon2 A430107D22Rik 2 2 2 2 2 2 Rassf5 Rin3 Runx1 Sik3 Sirt5 Slc39a1 2 2 Slc9a9 Spnb2 2 Timm8a2 2 2 2 2 2 2 2 Trp53inp2 Tsc22d3 Unkl Vamp5 Wdr26 Ythdf2 Zfp28 coiled-coil domain containing 84 chemokine (C-C motif) receptor-like 1 CD177 antigen cysteine-serine-rich nuclear protein 2 CTD (carboxy-terminal domain, RNA polymerase II, polypeptide A) small phosphatase 2 diablo homolog (Drosophila) DnaJ (Hsp40) homolog, subfamily B, member 9 H2A histone family, member J Hermansky-Pudlak syndrome 5 homolog (human) IKBKB interacting protein inositol 1,4,5-trisphosphate 3-kinase B kelch-like 6 (Drosophila) microrchidia 3 v-myc myelocytomatosis viral related oncogene, neuroblastoma derived (avian) nuclear factor, erythroid derived 2,-like 1 osteopetrosis associated transmembrane protein 1 platelet-activating factor acetylhydrolase, isoform 1b, subunit 3 p53 and DNA damage regulated 1 phosphatase and actin regulator 2 phosphatidylinositol 3 kinase, regulatory subunit, polypeptide 3 (p55) pogo transposable element with ZNF domain paraoxonase 2 RAS protein activator like 3 Ras association (RalGDS/AF-6) domain family member 5 Ras and Rab interactor 3 runt related transcription factor 1 SIK family kinase 3 sirtuin 5 solute carrier family 39 (zinc transporter), member 1 solute carrier family 9 (sodium/hydrogen exchanger), member 9 spectrin beta 2 translocase of inner mitochondrial membrane 8 homolog a2 (yeast) transformation related protein 53 inducible nuclear protein 2 TSC22 domain family, member 3 unkempt-like (Drosophila) vesicle-associated membrane protein 5 WD repeat domain 26 YTH domain family 2 zinc finger protein 2 108 Chapter 4: Conclusions ______________________________________________________________________ 109 This work describes the adaptation of pool-based shRNA screening approaches to in vivo models of hematopoietic malignancies. In mammalian systems, both insertional mutagenesis screens and shRNA screens have been widely used for cancer gene discovery. While retroviral insertional mutagenesis screens have effectively identified genes that promote tumorigenesis in vivo, these screens are biased towards the identification of oncogenes and rarely identify tumor suppressors. It is also difficult to identify genes that negatively impact a disease process in these screens, because they rely on the positive selection of a given phenotype. Additionally, it is not possible to use these screening methodologies to systematically test the impact of a defined set of genetic lesions on tumor progression. shRNA screens can be used to assess the effect of suppression of large sets of genes on tumor cell growth. Genes that either positively or negatively impact tumor cell proliferation can be identified, based on the enrichment or depletion of shRNAs targeting those genes. In insertional mutagenesis screens, positive selection only occurs for insertion events that are sufficient to promote transformation, strongly cooperate with other genetic alterations, or significantly accelerate transformation on sensitized backgrounds, whereas shRNA screens can identify genes whose suppression have more subtle phenotypic impacts. However, in contrast to insertional mutagenesis screens, shRNA screens have primarily been performed in vitro in tumor cell lines. In instances where local environmental cues influence dependency on certain genes and pathways, the identification of these context-specific determinants of tumor progression may require that screens be performed within a physiologically relevant context. Thus, we were interested in developing approaches to perform shRNA screens in vivo. 110 In previous work, it has been shown that genetic manipulations to hematopoietic tumors were sufficient to alter disease progression in vivo. For example, transplantation of chemoresponsive primary Eµ-myc lymphomas into secondary recipient mice after ex vivo transduction with a construct expressing Bcl2 produced chemoresistant tumors (Schmitt et al., 2000). Importantly, these tumors were transplanted into immunocompetent syngeneic recipient mice and transplanted tumors were pathologically indistinguishable from primary tumors, suggesting that the impact of individual genetic alterations on tumor progression can be studied in the context of a normal immune system and normal tumor microenvironment. Mimicking the impact of p53 loss in tumor development, shRNA-mediated suppression of p53 or Puma in transplanted Eµ-myc fetal stem cells was shown to accelerate lymphoma onset in recipient mice, indicating that shRNAs can be used in vivo to study loss of function phenotypes (Hemann et al., 2003; Hemann et al., 2004). In transplanted secondary Eµmyc lymphomas, suppression of p53, Topoisomerase1, or Topoisomerase2a by shRNAs were shown to impact chemotherapeutic outcome, further suggesting that shRNAs can effectively be used to examine the impact of specific genetic alterations in vivo (Burgess et al., 2008). To identify novel genetic modulators of tumor cell growth in Eµ-myc lymphomas, we were interested in introducing a pooled shRNA library into primary lymphomas, and transplanting this transduced population into syngeneic recipient animals. Preliminary dilution experiments indicated that a minimum of 500 lymphoma cells were seeding disease following transplantation of primary lymphomas, suggesting that this model might accommodate the introduction of a relatively complex shRNA library. Therefore, we infected lymphoma cells with an shRNA library composed of 2200 shRNAs targeting 1000 genes with known or putative roles in cancer. Lymphoma cells were either injected 111 into recipient animals or maintained in culture, allowing us to compare shRNAs that impacted lymphoma cell growth in the in vitro and in vivo settings. At the time of terminal disease, or after a comparable amount of time in culture, lymphoma cells were harvested and library pool composition was deconvoluted by high throughput sequencing. In this screen, we found that we could represent up to 900 unique shRNAs in the tumors of an individual animal, suggesting that a sufficient number of cells are seeding disease after transplantation to maintain library diversity, and therefore, screens using moderately-sized shRNA libraries can be performed in vivo. Additionally, when we compared the sets of shRNAs that enriched or depleted relative to the input cell population in vitro or in vivo, we found that scoring shRNAs were largely non-overlapping between these two settings. This suggests that context-specific genetic modulators of disease progression can be identified using in vivo screening approaches. In validation studies, we showed that a number of regulators of cell migration influenced lymphoma progression specifically in the in vivo setting, including Rac2, CrkL, and the relatively poorly characterized actin binding protein Twinfillin. Suppression of these genes negatively impacted lymphoma cell dissemination, both to the lymph nodes and to sites of terminal metastatic disease, like the liver, lungs, and brain. Additionally, shRNA mediated suppression of Rac2 and Twinfillin in pure populations of lymphoma cells extended animal lifespan, at least in part by impairing these metastatic events. Furthermore, we found that shRNA-mediated suppression of Rac2 in lymphoma cells prolonged animal survival following treatment with the frontline chemotherapeutic vincristine, suggesting that sites of minimal residual disease following treatment may differ from sites of terminal disease, and the onset of morbidity after treatment requires Rac2-mediated cell migration to sites of terminal disease. Together, this data suggests 112 that modulators of cell migration may influence chemotherapeutic outcome in this lymphoma model. We then extended this in vivo screening approach to a transplantable model of Bcr-Abl positive B-cell acute lymphoblastic leukemia. Here, when we introduced a library of 2200 shRNAs into transplanted tumors, we found that we could identify up to 1900 unique shRNAs in an individual mouse, suggesting that very large numbers of transplanted cells are seeding disease in this model, and that this model might accommodate the introduction of complex shRNA libraries. Therefore, we decided to perform an in vivo screen for modulators of tumor growth using a genome-scale shRNA library, containing 5 pools of approximately 10,000 shRNAs each. Single pools of 10,000 shRNAs were introduced into leukemia cells prior to transplantation, and transduced cells were either injected into syngeneic recipient mice or placed in culture. When leukemia cells were harvested from animals at the time of terminal disease, we found that, on average, we could identify between 7000 and 8000 unique shRNAs in the context of an individual mouse. Thus, this ALL model represents a system in which shRNA representation of a genome-scale library can be robustly maintained in the in vivo setting, allowing us to perform large-scale, unbiased screens. Whereas previous in vivo screens have been limited to assaying the impact of hundreds of shRNAs, here we are able to screen using tens of thousands of hairpins. As in our previous work, we found that a large proportion of the sets of scoring shRNAs in the in vitro and in vivo settings were non-overlapping, suggesting that we are able to identify a subset of genes whose suppression may uniquely impact leukemia cell growth in vivo. Unsupervised hierarchical clustering of these samples patterns of enrichment or depletion of shRNAs were sufficient to distinguish cultured samples from in vivo samples. We focused our validation efforts on the set of hairpins that depleted 113 specifically in vivo, and to select candidates for validation we took a number of different approaches, including functionally categorizing the set of genes targeted by depleting hairpins, and filtering this dataset using both transcriptional data and human copy number alteration data. Functional categorization of genes targeted by candidate hairpins showed that the products of these genes were enriched for a number of common protein domains, including C2H2-type zinc finger domains. A set of hairpins targeting C2H2 zinc finger proteins validated as single constructs, and we found that suppression of several poorly characterized C2H2 zinc finger proteins was selected against in leukemia cells specifically in the in vivo setting. In the course of our validation studies, we were able to identify numerous examples of genes whose suppression negatively impacts tumor cell growth in vivo, but is neutral or has the opposite effect in the in vitro setting, indicating that the in vivo tumor environment may alter the genetic dependencies of tumor cells. By performing shRNA screens in vivo, we can identify genes that are important specifically within this setting. When the transcriptional profiles of leukemia cells harvested from mice were compared with those from leukemia cells proliferating in culture, we were again able to distinguish in vitro samples from in vivo samples using unsupervised hierarchical clustering, and found that a large number of genes were significantly differentially expressed in leukemia cells in vivo, relative to in vitro. This suggests that gene expression patterns, as well as the functional genetic dependencies of leukemia cells, may be altered by growth in the in vivo environment. However, when we compared the set of genes that were transcriptionally upregulated in vivo to genes targeted by shRNAs that depleted specifically in vivo, we found these sets to be primarily non-redundant. While this observation is consistent with data in yeast (Yeger-Lotem et al., 2009) it 114 suggests that, at least in this specific instance, transcriptional alterations are not well correlated with genes that are functionally important in disease progression. Other comparisons between matched microarray and screening datasets may show greater overlap between genes that are transcriptionally altered and genes targeted by hairpins that enrich or deplete in shRNA screens. Runx and Lmo2 were contained in the limited set of genes that were both transcriptionally upregulated in leukemia cells in vivo and that were targeted by shRNAs that depleted in vivo in our screen, and in independent validation experiments, suppression of these genes was selected against in the in vivo setting. Given that the driving oncogenic lesion in this ALL model is the Bcr-Abl fusion protein, it was somewhat unexpected that these genes, which can drive transformation in other types of leukemia, would be functionally important in these tumors. Lmo2 translocations are found in a subset of T cell malignancies, and high levels of Lmo2 expression promote tumor development by blocking T cell differentiation and increasing thymocyte self renewal (Curtis and McCormack, 2010; McCormack et al., 2010). While Lmo2 expression has been measured in B-cell malignancies and was found to vary by subtype and genetic alteration (Malumbres et al., 2011; Natkunam et al., 2007), a functional role for this gene in B-lineage tumors has not been established. Runx translocations, found in 20-25% of B-cell ALLs, result in overexpression of the full-length protein (Blyth et al., 2005; Golub et al., 1995; Romana et al., 1995), and transcriptional upregulation of Runx has been observed in human B-ALL patients (Niini et al., 2002), suggesting that this gene may be oncogenic in B cell malignancies. Our data indicates that both Runx and Lmo2 promote tumor progression in Bcr-Abl driven tumors, and that activation of oncogenic pathways downstream of these genes may promote leukemia cell growth or survival specifically in the in vivo setting. 115 When we mapped the location of the human orthologs of genes targeted by shRNAs that depleted specifically in the in vivo setting, we found that a number of these genes, including the plant homeodomain containing protein Phf6, fell within regions of amplification in human ALL patients. Plant homeodomain containing proteins recognize and bind to specific modifications on the lyseine tails of histones and are frequently part of multiprotein complexes that can activate or repress gene transcription (Musselman and Kutateladze, 2011; Sanchez and Zhou, 2011). Inactivating Phf6 mutations are frequently found in T-cell ALL (Van Vlierberghe et al., 2010), and less frequently, in AML (Van Vlierberghe et al., 2011) but have not been observed in B-cell malignancies (Van Vlierberghe et al., 2010). As a single constructs, multiple independent hairpins targeting Phf6 depleted specifically in BCR-Abl B-cell ALLs, and suppression of this gene was also selected against in Eµ-myc B cell lymphomas in vivo. This data suggests that, in contrast to T-cell ALLs, suppression of Phf6 in B cell malignancies impairs tumor progression. Whereas Phf6 suppression had no impact on tumor growth in T cell lymphomas, we found that Phf6 overexpression was selected against in these T cell tumors. This data is consistent with a model where Phf6 is necessary for the growth or survival of tumor cells in B cell malignancies, but expression of high levels of this gene impairs tumor progression in T cell malignancies. Additionally, this model is supported by data from human T cell malignancies, where loss of Phf6 in human patients, via mutation, is seen in a significant portion of T-cell ALLs. We have shown have shown that moderate to large scale shRNA screens can be performed in vivo in mouse models of hematopoietic malignancies. By using transplantable models of established tumors that can be manipulated ex vivo, we can introduce libraries of shRNAs into tumor cells, transplant transduced cells into recipient animals, and compare shRNA representation in the resulting tumors to representation in 116 an input cell population in order to identify shRNAs that impact leukemia or lymphoma cell growth in vivo. Other groups have used similar strategies to identify genes that accelerate tumor development in Eµ-myc tumors (Bric et al., 2009) and hepatocelluar carcinoma (Zender et al., 2008). In these screens, low complexity pools of approximately 50 shRNAs each were introduced into cells prior to transplantation. More recently, a screen using a pool of nearly 1000 shRNAs was performed in an inducible, transplantable model of AML, and in this work control shRNAs targeting an essential gene consistently depleted in transplanted tumors (Zuber et al., 2011). To our knowledge, our screening work in acute lymphoblastic leukemia represents the first unbiased, genome-scale screen that has been performed in vivo. Importantly, screens in vivo yield different sets of scoring shRNAs when compared to matched in vitro screens. Thus, these screens are not simply recapitulating the results of in vitro screens, but may help identify novel modulators of tumor progression. A subset of the scoring shRNAs are shared between in vitro and in vivo screens, and these are expected to target essential genes and key downstream modulators of oncogenic signals. In vivo cues may provide a unique signaling environment and thus alter the genes and pathways that are required for tumor cell proliferation and survival. Therefore, in vivo screens may help identify the unique genetic dependencies of tumor cells within the normal tumor microenvironment, and ultimately, could aid in the development of therapeutic strategies that exploit these dependencies. While it is currently difficult to assign a function to many of the shRNAs that specifically impact tumor cell growth in vivo, and derive a coherent biology from this set of scoring hairpins, continued study of these genes in the in vivo context may help clarify their role in disease progression. 117 A number of challenges accompany in vivo screens. One challenge that is shared with in vitro screening approaches is the selection of candidate hits for validation. Screens with genome-scale libraries produce large numbers of candidate hits, especially in screens that have the resolution to identify hairpins with intermediate phenotypes. The cost of systematically validating all of these candidate hits as individual constructs is prohibitive, particularly when validation assay are in vivo, but selectively validating a very limited number of hairpins without a clear rationale disregards any value or insight that can be obtained from the dataset as a whole. One approach to selecting candidate hits for validation is identifying pathways or functional categories that are overrepresented in genes targeted by scoring shRNAs, although programs designed for this purpose necessarily exclude uncharacterized or poorly characterized genes, and well-studied genes and pathways are often over-represented. Comparisons with human tumor gene expression data, mutational data, and copy number alteration data provide another means of filtering candidate hits for validation. Another method of rapidly validating sets of candidate shRNAs is the construction of high-coverage validation libraries. In our ALL screen, we identified a set of approximately 130 genes that were both transcriptionally upregulated in vivo and targeted by depleting shRNAs. Rather than selecting a subset of these for validation, we designed six independent shRNAs that targeted each gene, bulk synthesized and cloned this library, and directly used this bulk-cloned product to repeat our initial screen. Without sequence verifying individual constructs and pooling sequence-verified clones, we estimate that about 25% of this bulk-cloned library contains oligonucleotides that were mis-synthesized, and therefore non-functional. Because each hairpin is represented by many oligonucleotides, for any given hairpin, the library contains both correct and incorrect oligonucleotides. Mis-synthesized hairpins should behave as neutral constructs 118 in the validation screens, and if functional and non-functional hairpins cannot be resolved in the quantification of pool representation, they may dampen the apparent enrichment or depletion of functional hairpins. However, using this approach, the considerable savings in cost and time may make the construction of small custom libraries an effective validation strategy. Genes with multiple independent hairpins that score in secondary screens can then be selected for validation as individual constructs. Secondary screens may be especially useful when libraries that contain only 2-3 hairpins per gene were used for primary screens, or in cases where the hairpin coverage of each gene was highly variable. Another challenge in performing in vivo screens is the identification of models that are appropriate for screening purposes. Since the delivery of pools of shRNAs to tissues is challenging, transplantable tumors are the simplest models for in vivo screens. Thus far, our work has been limited to models of hematopoietic malignancies, although in vivo screens have also been performed in a transplantable liver model, and it should be possible to use these approaches in transplantable models of other solid tumors. One key consideration in the selection of tumor models for screens is the number of cells that seed disease following tumor cell transplantation. If only small numbers of cells are seeding disease following transplantation, the representation of shRNA libraries will be severely limited. The number of cells seeding disease can be estimated by infecting cells with a hairpin that has known and identifiable phenotype prior to transplantation, and seeing if that hairpin can be reproducibly identified in transplanted tumors. For example, if a hairpin that is infected to 0.5% is consistently found in transplanted tumors, this indicates that at least 200 cells are seeding disease in tumors. Alternatively, libraries of shRNAs can be introduced into cells prior to transplantation, and the number of unique 119 hairpins found in individual tumors can be quantified as a proxy for the number of cells seeding disease. An approximation of the number of hairpins seeding disease is important when selecting the appropriate shRNA pool size to use for screens. Generally, it is preferable that the number of cells seeding disease exceeds the number of hairpins in a library. Otherwise, some proportion of shRNAs will stochastically drop out of each tumor, because cells containing those hairpins don’t contribute to the population of cells that seed disease. If the same shRNAs stochastically drop out of all tumors in an experimental group, these hairpins will appear to deplete, but may actually have no biological impact. In instances where smaller numbers of cells are seeding disease in transplanted tumors, it is still possible to perform shRNA screens, but with less complex shRNA pools. In these cases, in vivo screens may be more appropriate for testing targeted shRNA libraries that contain sets of genes that are involved in a particular process of interest. In both of the in vivo screens we have performed, we have consistently observed that the variability between samples was higher for in vivo samples than cultured samples. We think the tumor microenvironment may cause fluctuations in the representation of tumor cells containing neutral hairpins, and differential numbers of cell containing particular hairpins seeding disease may also contribute to this variation between samples. Our group has shown that the coefficient of variation between samples is smaller in validating hairpins, relative to the cumulative behavior of all hairpins in vivo, suggesting that we may be able to accurately identify hairpins that are likely to validate as individual constructs by using cutoffs that select shRNAs exhibiting low variability between samples (Pritchard et al., 2011). 120 Whereas in vivo validation of hits from insertional mutagenesis screens or transposon screens requires generating transgenic animals and directly testing the impact of a specific alteration on tumor development, hits from in vivo shRNA screens can be rapidly functionally validated. Tumor cells can be partially transduced with single shRNA constructs coexpressing GFP, and assaying for a change in the proportion of cells that contain GFP provides a sensitive means to detect alterations in growth and survival conferred by the presence of shRNAs. Here, the rate of validation of candidate hits can be used as a measure of the quality of the screening dataset, and genes whose loss has a functional impact on tumor cell growth can easily be identified. Given the success of performing pool-based shRNA screens in vivo, we think that these approaches may be used to examine other aspects of tumor cell biology, such as resistance to chemotherapeutics. Clinical resistance to the targeted chemotherapeutic dasatanib in BCR-Abl B-cell acute lymphoblastic leukemia is a serious challenge, and a portion of this resistance occurs without mutations in the Abl kinase domain that prevent drug binding. Therefore, an in vivo screen for shRNAs that sensitize leukemia cells to dasatinib treatment may elucidate pathways that are important for drug resistance in vivo, and may also provide potential strategies to increase the efficacy of drug treatment. Similarily, screens with conventional chemotherapeutics may also yield insights to mechanisms of resistances in the in vivo microenvironment. In vivo shRNA-based screens could also be used to identify modulators of metastasis. In hematopoietic cancers, metastasis to the central nervous system represents a clinical challenge, and mechanisms of leukemia or lymphoma cell migration to the brain are poorly understood. Thus, an in vivo screen comparing shRNA representation in the blood to shRNA representation in leukemia cells in the brain might be used to identify modulators of CNS metastasis. As in vivo screening approaches are 121 extended to other models of solid tumors, genes that are involved in metastasis in these diseases could be identified in a similar manner. In vivo screening approaches can be a useful tool for unbiased cancer gene discovery, and as described here, may aid in the future identification of genes that are important in many stages of tumor progression. 122 References Blyth, K., Cameron, E.R., and Neil, J.C. (2005). The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 5, 376-387. Bric, A., Miething, C., Bialucha, C.U., Scuoppo, C., Zender, L., Krasnitz, A., Xuan, Z., Zuber, J., Wigler, M., Hicks, J., et al. (2009). Functional identification of tumorsuppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 16, 324-335. Burgess, D.J., Doles, J., Zender, L., Xue, W., Ma, B., McCombie, W.R., Hannon, G.J., Lowe, S.W., and Hemann, M.T. (2008). Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A 105, 9053-9058. Curtis, D.J., and McCormack, M.P. (2010). The molecular basis of Lmo2-induced T-cell acute lymphoblastic leukemia. Clin Cancer Res 16, 5618-5623. Golub, T.R., Barker, G.F., Bohlander, S.K., Hiebert, S.W., Ward, D.C., Bray-Ward, P., Morgan, E., Raimondi, S.C., Rowley, J.D., and Gilliland, D.G. (1995). Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 92, 4917-4921. Hemann, M.T., Fridman, J.S., Zilfou, J.T., Hernando, E., Paddison, P.J., Cordon-Cardo, C., Hannon, G.J., and Lowe, S.W. (2003). An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet 33, 396400. Hemann, M.T., Zilfou, J.T., Zhao, Z., Burgess, D.J., Hannon, G.J., and Lowe, S.W. (2004). Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci U S A 101, 9333-9338. Malumbres, R., Fresquet, V., Roman-Gomez, J., Bobadilla, M., Robles, E.F., Altobelli, G.G., Calasanz, M.J., Smeland, E.B., Aznar, M.A., Agirre, X., et al. (2011). LMO2 expression reflects the different stages of blast maturation and genetic features in B-cell acute lymphoblastic leukemia and predicts clinical outcome. Haematologica 96, 980-986. McCormack, M.P., Young, L.F., Vasudevan, S., de Graaf, C.A., Codrington, R., Rabbitts, T.H., Jane, S.M., and Curtis, D.J. (2010). The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science 327, 879-883. Musselman, C.A., and Kutateladze, T.G. (2011). Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res 39, 9061-9071. Natkunam, Y., Zhao, S., Mason, D.Y., Chen, J., Taidi, B., Jones, M., Hammer, A.S., Hamilton Dutoit, S., Lossos, I.S., and Levy, R. (2007). The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood 109, 1636-1642. 123 Niini, T., Vettenranta, K., Hollmen, J., Larramendy, M.L., Aalto, Y., Wikman, H., Nagy, B., Seppanen, J.K., Ferrer Salvador, A., Mannila, H., et al. (2002). Expression of myeloidspecific genes in childhood acute lymphoblastic leukemia - a cDNA array study. Leukemia 16, 2213-2221. Pritchard, J.R., Gilbert, L.A., Meacham, C.E., Ricks, J.L., Jiang, H., Lauffenburger, D.A., and Hemann, M.T. (2011). Bcl-2 family genetic profiling reveals microenvironmentspecific determinants of chemotherapeutic response. Cancer Res 71, 5850-5858. Romana, S.P., Poirel, H., Leconiat, M., Flexor, M.A., Mauchauffe, M., Jonveaux, P., Macintyre, E.A., Berger, R., and Bernard, O.A. (1995). High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood 86, 4263-4269. Sanchez, R., and Zhou, M.M. (2011). The PHD finger: a versatile epigenome reader. Trends Biochem Sci 36, 364-372. Schmitt, C.A., Rosenthal, C.T., and Lowe, S.W. (2000). Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med 6, 1029-1035. Van Vlierberghe, P., Palomero, T., Khiabanian, H., Van der Meulen, J., Castillo, M., Van Roy, N., De Moerloose, B., Philippe, J., Gonzalez-Garcia, S., Toribio, M.L., et al. (2010). PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet 42, 338-342. Van Vlierberghe, P., Patel, J., Abdel-Wahab, O., Lobry, C., Hedvat, C.V., Balbin, M., Nicolas, C., Payer, A.R., Fernandez, H.F., Tallman, M.S., et al. (2011). PHF6 mutations in adult acute myeloid leukemia. Leukemia 25, 130-134. Yeger-Lotem, E., Riva, L., Su, L.J., Gitler, A.D., Cashikar, A.G., King, O.D., Auluck, P.K., Geddie, M.L., Valastyan, J.S., Karger, D.R., et al. (2009). Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat Genet 41, 316-323. Zender, L., Xue, W., Zuber, J., Semighini, C.P., Krasnitz, A., Ma, B., Zender, P., Kubicka, S., Luk, J.M., Schirmacher, P., et al. (2008). An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 135, 852-864. Zuber, J., McJunkin, K., Fellmann, C., Dow, L.E., Taylor, M.J., Hannon, G.J., and Lowe, S.W. (2011). Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 29, 79-83. 124