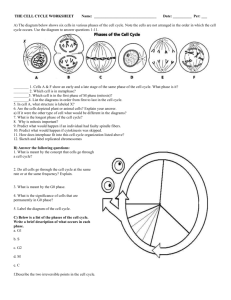
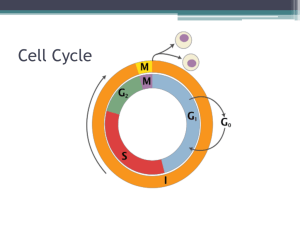
Chromosomal Instability and Tumorigenesis:
advertisement

Chromosomal Instability and Tumorigenesis: Genetic Analysis of the Murine Spindle Checkpoint gene Mpsl by Stephanie Zhi-Juan Xie B.A. Biology University of Chicago, 1996 SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN BIOLOGY AT THE MASSACHUSETTS INSTITUTE OF TECHNOLOGY FEBRUARY 2007 C 2007 Massachusetts Institute of Technology. All rights reserved. Signature of Author: Certified by: Peter K. Sorger Professor of Biology Thesis Supervisor Accepted by: __ • .. I _ _ _ = v Stephen P. Bell Professor of Biology Chairman, Biology Graduate Committee v OF TECHNOOGY FEB 2 2W007 SLIBRARP,•S.__ Chromosomal Instability and Tumorigenesis: Genetic Analysis of the Murine Spindle Checkpoint Gene Mpsl by Stephanie Zhi-Juan Xie Submitted to the Department of Biology on January 11, 2007 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biology ABSTRACT The ubiquity of aneuploidy in human cancers, particularly solid tumors, suggests a fundamental link between errors in chromosome segregation and tumorigenesis. The spindle checkpoint ensures accurate chromosome segregation by delaying anaphase onset until all kinetochores achieve bipolar attachment to the mitotic spindle. Abrogation of the mammalian spindle checkpoint can cause chromosomal instability (CIN), the frequent gain or loss of entire chromosomes during cell divisions. While CIN has been proposed to be sufficient to initiate tumorigenesis, no direct evidence has shown that CIN itself is a causal agent for human cancer. To determine if CIN can promote tumor formation, I generated mice containing a conditional mutant allele of the spindle checkpoint kinase Mpsl (MpslA) and induced its expression in thymocytes. I show that MpslA is a partial loss of function allele for spindle checkpoint activation which results in chromosome missegregation in MEFs and embryonic lethality inmice. Expression of MpslA in thymocytes causes sporadic lymphomas and a decrease in thymus size. Strikingly, when MpslA was introduced into a p53 heterozygous background, all mice developed thymomas due to a loss of heterozygosity of the wildtype p53 allele. In contrast, no p53 heterozygous micedeveloped thymomas in the same time frame when Mpsl is wildtype. Moreover, MpslA also accelerated tumor formation in p 19AF heterozygous thymocytes. I propose that MpslA-induced CIN activates p53, and results in apoptosis of the majority of pressure- for ;nactivation of strong selecptiove SpsI A- expressirn cells. Therefore, there- ;is the p53 pathway, and in those cells where p53 loss does occur, MpslA-induced CIN drives tumor development. In summary, I show that CIN caused by a weakened checkpoint is sufficient to facilitate tumorigenesis when the p53 pathway is also impaired. Thesis Supervisor: Peter K. Sorger Title: Professor of Biology, Professor of Biological Engineering · : · ·,. i. : · 'I- i . STEPHANIE ZHIJUAN XIE Education Ph.D. student in Biology at Massachusetts Institute of Biology, 2006 Laboratory of Peter K. Sorger B.A. degree in Biology, University of Chicago, 2000 Undergraduate thesis in the laboratory of Douglas Bishop Hunter College High School, NYC, 1996 Honors and Awards Paul and Cleo Schimmel Scholar, 2000-2004 Elected to Sigma Xi, 2000 Honorable Mention, National Science Foundation Graduate Fellowship, 2000 Honorable Mention, Howard Hughes Predoctoral Fellowship in Biological Sciences, 2000 Student Marshall, University of Chicago, 2000 Publications A partial loss of function mutation in the mitotic checkpoint causes CIN and drives tumorigenesis in mice. Xie, S. and Sorger, P.K. Intended for submission to Nature, Spring 2007 Chromosome segregation and genomic stability. Draviam, V.M., P.K. Curr Opin Genet Dev. 2004 Apr;14(2):120-5. S., Sorger, Poster Presentations AACR: Cancer Susceptibility and Cancer Susceptibility Syndromes 2006 ASCB Annual Meeting, San Francisco 2005 ISREC: Cell and Molecular Biology of Cancer, Lausanne 2005 ASCB Annual Meeting, Washington, D.C. 2004 BSCB/BACR: Cell Biology of Cancer 2003 ACKNOWLEDGEMENTS I would like to thank Peter for taking me into his lab and giving me the freedom to find my own way. I am grateful to you for your enthusiasm and support and for creating a wonderful research environment to go to these last five years. I would like to thank the members of my thesis committee, Tyler Jacks and Steve Bell, for your invaluable advice over the years. I would like to thank Jianzhu Chen and Randall King for joining my defense committee. I would like to thank all the members of the Sorger lab, past and present, especially Ying Yue, Aurora Burds Conner, Annagret Schulze-Lutum, Rob Hagan, John Albeck, Suzanne Gaudet, Viji Draviam, Patrick Meraldi, Irina Shapiro, Jessica Tytell and Alexa Turke. I am would also like to thank Margaret White. I am grateful for all your encouragement, support and intellectual advice. You made going to lab fun! To all the people that helped this yeast geneticist to successfully make the transition to becoming a mouse geneticist, many thanks! I would like to extend a special thanks to Tyler and Jianzhu for making the expertise in their labs accessible, and to the many members of their labs who took time out of their busy lives to listen to my questions. I would not have been able to complete much of my research without invaluable advice from Qing Ge and Chris Dillon, who were kind enough to teach a complete novice immunology. Thanks to all those at DCM and the CCR histology core - particularly Stephanie Haskell and Michael Brown. I would like to give a special thanks to all those kind enough to read the rough drafts of this thesis, particularly Ying Yue and Aurora Burds Conner, who prevented incoherence from late nights to be propagated. I would like to thank all my classmates from biograd 2000, especially Kevin Lai, Sunny Wong, Mimi Lee, Mandy Tam, David Doroquez and Mark Rosenzweig, for the not so tranquil (but very fun) first year and all the advice and support through the years. I would like to extend a special thanks to all my friends, particularly from Hunter, Tang and 69 and 71 Elm St, for all that you are. You know who you are and I won't embarrass you guys. Well, I'll embarrass some of you anyway, at least all the mahjong and dimsum buddies - Aaron, David, Due, Jenny, Justin, Kev, Lesley, Mandy, Mark, Mimi, Nate, Pat, Sal, Sarah, Shirley, Tony. Thanks Ina and Matthew for listening. Thank you Shirley for periodically reminding me that I'm a girl and girls should have fun. To Derek, thank you for all the love and encouragement, for sharing all the good and not so good times, and reminding me to "play safe". I love you. And to my family, mom, dad and bro, thank you for reminding me what is most important. I am so grateful for your love and support through these years. Table of Contents 9 Chapter 1 Introduction: The spindle checkpoint, chromosomal instability and cancer I. Chromomsomal Instability and Cancer 10 A. Introduction B. Searching for the molecular basis of CIN C. Spindle checkpoint deficiencies in tumors D. Senescence or tumorigenesis? 10 14 14 20 E. Connection between DNA damage and mitotic progression and cancer? 21 F. Conclusion 22 23 II. The spindle checkpoint A. Cell cycle and cell cycle checkpoints B. The canonical players of the spindle checkpoint C. Interactions between the spindle checkpoint and the cell cycle D. Spindle checkpoint signaling E. Mpsl: a spindle checkpoint kinase F. Meiosis and the spindle checkpoint 26 27 29 30 36 38 39 III. Tumor suppressor genes and oncogenes A. p53: DNA damage, aneuploidy and cancer 40 B. p53-dependent and p53-independent functions of p 9ARF 43 C. Oncogenic K-ras and lung cancer 45 IV. Conclusion 46 References 48 Chapter 2 A mutation in Mpsl causes chromosomal instability and accelerates tumorigenesis in mice 67 Abstract 68 Introduction 69 Results 70 Discussion 96 Methods 106 Acknowledgements 112 References 113 Chapter 3 Loss ofp53, not pl9ARF can rescue MpslA induced lethality in mouse embryonic fibroblasts 119 Abstract 120 Introduction 121 Results 125 MpslA decreases viability of MEFs, which is rescued by p53 inactivation 125 MpslA increases chromosome segregation defects, but not centrosome amplification in p53 null MEFs 129 Inactivating pl19ARF does not rescue MpsA induced cellular lethality 139 Mpsl"'; pl9 RF+; Lckcre+ mice develop tumors with an increase penetrance over MpslfEf; Lckcre+ mice 139 Discussion 142 Methods Methods 150 Acknowledgements 153 Acknowledgements References References 154 Chapter 4 siRNA depletion of Mpsl causes chromosome missegregation and accelerates mitotic timing in HeLa cells 160 Abstract 161 Introduction 162 Results 165 siRNA depletion of Mps abolishes kinetochore localization of Madl and Mad2 165 Complete siRNA depletion of Mps 1 by Mps 1-4 abolishes the checkpoint 169 Mpsl controls mitotic timing in addition to monitoring chromosome segregation 173 Discussion 178 Methods 184 Acknowledgements 185 185 Acknowledgements References 186 Chapter 5 Conclusions, Discussion and Future Directions 190 Conclusions and Discussion 192 MpslA is a partial loss of function mutation in the spindle checkpoint p53 inhibits CIN 192 194 The spindle checkpoint kinase Mpsl has multiple functions 197 CIN as a facilitator of tumorigenesis 198 Future Directions 199 MpslA as a universal facilitator of tumorigenesis? 199 What is the mechanism of Mps 1A induced chromosome missegregation? The intersection of p53 and the spindle checkpoint in tumorigenesis 200 202 Tissue specific effects of spindle checkpoint inactivation and genetic modifiers 204 Mpsl and human cancer 207 Summary 208 References 210 Appendix Determining the effect of MpslA in two post mitotic tissues: a conditional Kras lung cancer model and liver-specific deletion by Albumin-Cre 217 Introduction 218 Results and Discussion 220 MpslA causes a modest decrease in mouse mortality when expressed in the liver 220 MpslA has a modest effect on the tumor spectrum of K-rasG1 2 lungs 224 Methods 229 Acknowledgements 230 References 231 List of Figures Figure 1.1: Figure 2.1: Figure 2.2: Figure 2.3: 24 The canonical spindle checkpoint proteins. ...................................... MpslA is a truncation mutation that retains kinetochore localization. ......... 71 MpslA causes embryonic lethality in mice before embryonic day 10.5....... 73 MpslA facilitates tumorigenesis in p53-heterozgyous thymocytes by inducing LOH of p53 ............................................................... 77 Figure 2.4: MpslA accelerates lymphomagenesis in p53-null knockout thymocytes..... 81 Table 2.1: Karyotype analysis of MpslA/A; p53+/- thymic lymphomas..................... 85 Figure 2.5: MpslA decreases thymus size and increases heterogeneity in thymocytes. . 86 Figure 2.6: MpslA is a partial loss of function mutation in the spindle checkpoint that induces CIN and sporadic lymphomagenesis ...................................... ...... 88 Table 2.2: Number of cells in immune organs of mice at 6 weeks of age ..................... 92 Table 2.3: T cell development in thymi of mice at 6 weeks of age. ............................. 93 Table 2.4: Percentage circulating peripheral T cells of mice at 3-4 weeks of age ........ 94 Figure 2.7: MpslA accelerates tumorigenesis in p53-heterozygous conventional knockout m ice ........................................................................................................ 99 Table 2.5: MpslA does not induce centrosome duplication in thymocytes ................... 101 Figure 3.1: MpslA decreases MEF viability which is rescued by inactivating p53...... 126 Figure 3.2: MpslA is a partial loss of function mutation that increases chromosome 130 missegregation in p53-null MEFs ...................................... 132 Table 3.1: Analysis of mitotic timing in MEFs .................................... Table 3.2: MpslA does not induce centrosome duplication in MEFs ....................... 133 Figure 3.3: MpslA increases CIN in p53-null MEFs through multiple passages.......... 137 Figure 3.4: MpslA decreases viability of pl9ARF knockout MEFs, but increases tumorigenesis in pl 9ARF+/- mice....................................................................... 140 Table 4.1: Mpsl siRNA sequences ...................................... 166 Figure 4.1: siRNA depletion of Mps abolishes kinetochore localization of Mad2. ... 167 Figure 4.2: Complete siRNA depletion of Mpsl by Mpsl-4 abolishes the checkpoint. 170 Table 4.2: Analysis of mitotic timing in cells depleted of Mps 1......................... 172 174 Figure 4.3: Mps1 controls mitotic timing ..................................... 176 Figure 4.4: Mpsl depletion causes chromosome missegregation...................... Figure 4.5: A model for the dual functions of Mpsl during mitosis in the spindle checkpoint and mitotic timing. ....................................... 180 Figure Al: Mps lA causes a modest increase in mortality when expressed in the liver 221 Table Al: Mice from Alb-cre survival study that were found dead or found with liver tum ors. .................................................................................................................... 223 Figure A2: MpslA causes a modest shift in K-rasGl2D lungs from adenomas to papillom as............................................................................................................... 225 Chapter 1 Introduction: The spindle checkpoint, chromosomal instability and cancer Note: Sections of this chapter have been adapted, with permission, from the following review: Draviam, V. M., Xie, S., and Sorger, P. K. Chromosome segregation and genomic stability. Curr Opin Genet Dev. 2004. Apr; 14 (2):120-5. I. Chromomsomal Instability and Cancer The acquisition of genomic instability, an abnormal cell state associated with an increased rate of heritable alterations, is a crucial step in the development of human cancer. Genomic instability has multiple causes of which chromosomal instability (CIN), the frequent gain or loss of entire chromosomes during cell divisions, and microsatellite instability (MIN), associated with defects in DNA mismatch repair, have received the most attention. CIN has been observed in many human tumors and increased CIN is often correlated to the severity of the cancer (Loeb et al., 2003; Wilkens et al., 2004). Whereas the connection between a MIN phenotype and cancer is well established, the argument that CIN causes cancer remains circumstantial (Lengauer et al., 1997; Shibata et al., 1994). Nonetheless, the ubiquity of aneuploidy in human cancers, particularly solid tumors, suggests a fundamental link between errors in chromosome segregation and tumorigenesis. Current research in the field is focused on elucidating the molecular basis of CIN, including the possible roles of defects in the spindle checkpoint and defects in other regulators of mitosis. A. Introduction Since the late- 19 th century, abnormal chromosome number has been recognized as a nearly ubiquitous feature of human cancers (Hansemann, 1890). Careful study of colorectal cancers has shown that about 85% are aneuploid, and contain cells with an average of 60 to 90 chromosomes (reviewed in (Pellman, 2001). Moreover, tumors with higher clinical grades and poorer prognosis are typically associated with greater degrees of aneuploidy. Despite its long history and clinical relevance, the study of aneuploidy has yet to prove Boveri's postulate that abnormal chromosome number is a cause rather than a consequence of the cancerous state (Boveri, 1914). More precisely, it remains unclear whether aneuploidy arises early in tumorigenesis and plays a role in tumor development or whether it arises late and reflects a general breakdown of cell cycle control. Genomic instability is thought to be critical in the multi-step process by which cells accumulate the mutations characteristic of a cancerous state. For example, human colorectal cancers, which usually exhibit significant genomic instability, have been found to have 11,000 or more genetic alterations, and presumably, an ability to overcome a wide variety of negative controls on proliferation (Stoler et al., 1999). The mutator hypothesis posits that genomic instability arises early in tumorigenesis to increase subsequent occurrence of tumor-promoting mutations and genetic lesions, some of which will be tumor-promoting (Loeb et al., 2003; Nowell, 1976). Mutator genes, unlike oncogenes or tumor suppressor genes (TSGs), do not directly affect rate of cell growth or death, but instead increase the chance that oncogene and TSG mutations will appear (Baranovskaya et al., 2001). An important advance in the study of aneuploidy was the discovery by Vogelstein and colleagues that many cancer cell lines exhibit chromosome instability (CIN), a phenotype in which cell division is accompanied by an abnormally high rate of chromosome loss and gain (Lengauer et al., 1997). Cell fusion studies involving these cell lines also suggest that specific recessive mutations are responsible for CIN. These findings argue that aneuploidy is a consequence of GCIN-promoting mutations rather than of sporadic errors in mitosis. Thus, CIN can be considered as one form of genomic instability, along with elevated rates of mutation, errors in DNA repair and somatic hyper-recombination (reviewed in (Masuda and Takahashi, 2002), see Table 1.1). The potential consequences of CIN are best understood within the context of the mutator hypothesis, and modeling studies have shown that CIN is sufficiently powerful as a mutagen to drive tumor progression (Nowak et al., 2002). Indeed, the extreme view has been put forth that CIN alone is sufficient for cancer - in the absence of either oncogene or TSG mutation (Duesberg and Li, 2003). Table 1.1: Ways to acquire genomic instability Examples of dysregulated genes in the Examples of commonly associated disorders No. Types of genomic instability Biological processes crucial to maintain genomic stability 1 Mutations: Microsatellite instability (MIN) Mis-match repair (MMR) MSH2; PMS1 PMS2; MLH1 HNPCC (Narayan and Roy, 2003; Wei et al., 2002) Nuclear Excision Repair (NER) XPA-XPG; CSA; CSB Xeroderma pigmentosum Deletion, translocation DNA damage signaling ATM; ATR; BRCAl; P53 Ataxia telangiectasia; Breast carcinoma DSB repair Blm, Nbs, Wrn Bloom's syndrome; pathway 2 (Christmann et al., 2003; Jasin, 2002) Werner syndrome 2 DNA cross-link repair Sister chromatid cohesion and condensation Numerical changes: Chromosomal Spindle checkpoint instability (CIN) -Loss/gain of Centrosome numbers. chromosomes -Altered ploidy Cytokinesis (Christmann et al., 2003; Pellman, 2001) FANCA-FANCG PTTG Fanconi anemia Pituitary tumors Bub 1 Colorectal cancers Aurora A Colorectal tumors Breast carcinomas Cell death following prolonged mitotic arrest p53; BCl2 Other experiments challenge the idea that CIN is important for tumorigenesis. Studies in mice, for example, have shown that adenomas can develop without changes in karyotype or other obvious genomic instability (Haigis et al., 2002). The difficulty in proving a connection between CIN and cancer is that the molecular mechanisms of chromosome segregation, and the specific lesions that give rise to CIN, remain largely unknown. This situation contrasts with our current understanding of microsatellite instability (MIN), a type of genomic instability associated with errors in DNA mismatch repair (MMR) and a consequent 1000-fold increase in the rate of DNA mutation (reviewed in (Christmann et al., 2003)). Interestingly, CIN and MIN appear to be mutually exclusive in many tumor cells examined suggesting the mutations that lead to either form of instability are in different sets of genes (Lengauer et al., 1997; Loeb, 1991). Prior to the discovery that the cancer-susceptibility syndrome HNPCC results from mutations in the MMR system, attempts to show that tumor cells have a higher intrinsic mutation rate than normal cells were unsuccessful. The clinical significance of links between cancer and MMR is emphasized by data showing that mutations in MSH1 and MSH2 (MMR genes) are found not only in HNPCC, but also in sporadic human colorectal cancers (Narayan and Roy, 2003). Moreover, knockout studies with the murine MMR genes demonstrate that MMR defects directly promote tumorigenesis (Wei et al., 2002). Finally, biochemical and cell biological studies show that MMR defects directly increase the mutation rate. The argument that MIN plays a direct role in the development of cancer is therefore supported by three strong pieces of evidence: a connection to hereditary and sporadic tumors in humans, a cancer model in mice, and compelling biochemical data on the mechanism. As yet, none of these types of data are available for CIN. B. Searching for the molecular basis of CIN Aneuploidy is thought to arise from errors in chromosome segregation such as non-disjunction and loss. Accurate segregation is maintained both by the intrinsic properties of the mitotic machinery and by a spindle checkpoint (reviewed in (Yu, 2002) and described later in this chapter). Thus, in eukaryotes ranging from yeast to mice to humans, mutations in cell cycle regulators, checkpoint proteins and structural components of the mitotic spindle can cause CIN (Masuda and Takahashi, 2002). However, two potential causes of CIN have received particular attention: mutations in spindle checkpoint proteins and defects in the regulation of centrosome numbers (reviewed in (Nigg, 2002)). Centrosome amplification, observed in many tumor cells, can often cause multipolar mitosis and lead to aberrant chromosome segregation (Marx, 2001; Sato et al., 1999). By decoupling the structural events of the chromosome cycle from cell cycle progression, defects in the spindle checkpoint lead to non-disjunction and the gain or loss of chromosomes. C. Spindle checkpoint deficiencies in tumors The spindle checkpoint proteins form a signal transduction system containing at least three key functional entities: a sensor that monitors the state of kinetochoremicrotubule attachment, an amplifier that makes cell cycle progression sensitive to the mis-alignment of a single chromosome, and one or more regulators that control the activity of the anaphase promoting complex responsible for degrading securin and mitotic cyclins (Yu, 2002). The spindle checkpoint acts to delay the onset of anaphase until all pairs of duplicated (sister) chromatids have achieved bipolar attachment to the microtubules of the mitotic spindle. Bipolar attachment involves the binding of one kinetochore in a pair of sisters to microtubules emanating from one spindle pole and the binding of the other kinetochore to microtubules emanating from the opposite pole. The majority of checkpoint genes (Madl, Mad2, BubR1, Bubl, Bub3, Mpsl, and Aurora-B) have been highly conserved among higher and lower eukaryotes; although two genes (Rod and Zwl 0) are restricted to metazoans and one gene (Rae 1) appears to regulate the checkpoint in mice but not in yeast (Babu et al., 2003; Carmena and Earnshaw, 2003; Yu, 2002). Commonly used anti-neoplastic agents, such as paclitaxel and vincristine, affect microtubule dynamics and trigger the spindle checkpoint. These drugs cause mitotic arrest and cell death in checkpoint-proficient cancer cells. In checkpoint-impaired tumors, anti-microtubule drugs may increase chromosome missegregation and aggravate CIN (reviewed in (Mollinedo and Gajate, 2003)). Alternatively, a lack of checkpoint control may sensitize cells to chemotherapeutics because severe failure of cell division gives rise to inviable daughter cells. Thus, the spindle checkpoint status of a tumor is likely to be an important consideration for chemotherapy, but no systematic clinical studies investigating this possibility have been reported to date. A high percentage of solid tumors fail to arrest in response to microtubule poisons such as nocodazole, suggesting an impaired spindle checkpoint, and prompting a search in human tumors for mutations in checkpoint genes. Such mutations have been found in some CIN human colorectal cancer lines (Cahill et al., 1998) and the expression of a truncated form of the Bub kinase similar to that found in tumors has been shown to cause defects in chromosome segregation (Taylor et al., 1998). However, only a small fraction of CIN cancer cells appear to carry mutations in the Mad and Bub checkpoint genes (Tighe et al., 2001). To date, the strongest evidence for a direct role for aneuploidy as a direct cause in human cancer is the discovery of BubR1 mutations in several individuals with mosaic variegated aneuploidy (MVA), a rare disorder where individuals are prone to cancer (Hanks et al., 2004; Matsuura et al., 2006). Although one study was unsuccessful in detecting Mad2 mutations in 32 human primary gastric cancers, a recent report found Mad2 was mutated in 45% of the tested gastric cancer tissues (Kim et al., 2005b; Tanaka et al., 2001). Moreover, two Mad2 mutant alleles containing amino acid substitutions were identified which when overexpressed in HeLa cells and then challenged with nocadazole caused aneuploidy (Kim et al., 2005b). However, none of the checkpoint mutations found in human cells have been shown to be tumorigenic when reintroduced into mice. Sequestration of checkpoint proteins has also been proposed as a way in which to impair the spindle checkpoint (see below) in tumorigenesis. In adult Tcell leukemias, the Tax viral oncoprotein appears to bind and mislocalize Madl thereby inactivating the checkpoint and presumably aiding tumor progression (Kasai et al., 2002). Breast cancer-specific gene 1 (BCSG1) may also exert its oncogenic effect by binding to and reducing BubRl protein levels (Gupta et al., 2003). Furthermore, Breast cancer gene 1 (BRCA1) appears to regulate Mad2 through binding the transcription factor Octl and mutating Brcal in mice decreases Mad2 expression levels (Wang et al., 2004b). Future study of these phenomena may help to reconcile the observed high rate of spindle checkpoint inactivation with the infrequent mutation of the canonical checkpoint genes. The connection between inactivation of the spindle checkpoint and tumorigenesis has also been studied in mice. One complication of these studies is that the deletion of Mad and Bub genes in mice causes widespread apoptosis during early development and embryonic lethality (Babu et al., 2003; Dobles et al., 2000; Kalitsis et al., 2000; Wang et al., 2004a). Thus, it has not been possible to compare directly rates of tumor formation in wild-type and checkpoint-deficient animals. However, the loss of one copy of BubR1 or the conditional loss of CENP-E in mice cause abnormal chromosome number in both splenic and liver cells, respectively (Putkey et al., 2002; Wang et al., 2004a). Moreover, heterozygous Mad2 (Mad2 /') has been shown to promote the formation of non-lethal lung tumors (Michel et al., 2001). Similarly, Bub3 /- Rael+/ compound mice are characterized by spindle checkpoint dysfunction, chromosome missegregation, elevated rates of aneuploidy, and increased tumor formation following treatment with a carcinogen (Babu et al., 2003). BubR1+ - mice also exhibit enhanced carcinogen-induced tumor development (Dai et al., 2004). Furthermore, BubR1 haploinsufficiency increases the incidence of colonic tumors in ApcMirn+ mice (Rao et al., 2005). Overall, these data suggest a real but weak connection between tumor incidence and mutation of spindle checkpoint genes. Recent studies suggest that mitotic regulators other than Mads and Bubs may be promising candidates for genes whose mutation cause CIN. Aurora A has been implicated in mitotic entry and centrosome duplication (Giet et al., 2002; Hirota et al., 2003; Meraldi et al., 2002). The Aurora-A gene lies in a chromosomal region commonly amplified in human epithelial cancers, and Aurora-A mRNA is overexpressed in a wide variety of human cancers (Miyoshi et al., 2001). Overproduction of Aurora-A appears to disrupt the binding of BubR1 to Cdc20 (an activator of the anaphase promoting complex) and to abrogate the spindle checkpoint, thereby generating a CIN phenotype (Jiang et al., 2003). Adenomatous Polyposis Coli (APC), a microtubule associated protein localized to kinetochores, is another gene frequently mutated in human colorectal cancers. Dominant mutations in APC cause chromosome segregation errors, although the connection between APC and CIN in human tumors remains controversial (Green and Kaplan, 2003; Haigis et al., 2002; Kaplan et al., 2001; Sieber et al., 2002). Several other structural proteins with a role in spindle assembly are overexpressed in tumors (see Table 1.2), although their role in CIN is not yet known. To date, the study of human cancers and of mouse models has failed to identify a checkpoint gene whose mutation, like that of p53 in the DNA damage response, has a direct and widespread role in tumorigenesis. Why is this? One possibility is that the spindle checkpoint is simply not involved in CIN in human tumors. Alternatively, a wide variety of different genes -including Mads and Bubs- may play a role in CIN but each mutation may be found in only a small percentage of tumors (i.e. BubR1 and MVA). Another possibility is that we are not searching for mutations in the right spindle checkpoint gene or that alterations in the checkpoint need not be dramatic to exert an effect on tumor development. The mitotic delay established by the spindle checkpoint is seldom permanent and in the presence of an activated checkpoint, many cells eventually bypass the delay and eventually progress to G1, often as tetraploid cells (Rieder and Maiato, 2004; Shi and King, 2005). Both the biochemical signal from unattached kinetochores and the subsequent response by checkpoint protens are not all-or-none events, and can be weakened, particularly in partial depletion studies of spindle Table 1.2: Chromosome segregationgenes mutated in human cancers Genes Incidence in human tumors APC Familial Adenomatous Polyposis; other colorectal cancers (Wei et al., 2002) Ovarian adenocarcinomas (Rask et al., 2003) Human lung, colon, pancreas, prostate and breast cancers and high grade non-Hodgkin's lymphomas (Ambrosini et al., 1997) Hepatic tumor (Charrasse et al., 1995) Carcinoma cell lines (Chen et al., 1997) Pituitary adenomas (Saez et al., 1999) Binds to the plus end of microtubules and kinetochores Spindle assembly INCENP Colorectal cancer cell lines(Adams et al., 2001) Chromosome passenger protein Plkl Primary colorectal cancers had elevated expression (Takahashi et al., 2003) Primary colorectal tumors breast tumours (Bischoff et al., 1998; Zhou et al., 1998) Colorectal cancers (Ota et al., 2002) SK3-P Survivin Ch-TOGI HEC 1 hSecurin/PTTG AuroraA/Stk6/ STK15/BTAK/ Aurora2 AuroraB Madl Mad2 Bubl BubR1 Adult T cell leukemia (ATL) (Kasai et al., 2002) Ovarian cancer (Wang et al., 2002) Colorectal cancers (Cahill et al., 1998; Shichiri et al., 2002) Colon carcinomas (Shichiri et al., 2002) Attributed function Supportive evidences from cell- culture Mouse model studies studies (Green and Kaplan, 2003; Kaplan et al., 2001) N/A (Pellman, 2001; Su et al., 1992) N/A Chromosome passenger protein; anti-apoptotic factor (Temme et al., 2003) (Uren et al., 2000) Spindle assembly (Gergely et al., 2003) (Chen et al., 1997) N/A (Pei and Melmed, 1997; Yu et al., 2003) (Adams et al., 2001) (Mei et al., 2001) Mitotic entry Cytokinesis (Lee et al., 2003) N/A Spindle assembly; mitotic entry (Anand et al., 2003; EwartToland et al., 2003) (Ota et al., 2002) (EwartToland et al., 2003) N/A (Kasai et al., 2002) N/A Spindle checkpoint (Michel et al., 2001) Spindle checkpoint kinase Spindle checkpoint kinase (Lee, 2003; Lin et al., 2003) (Lee, 2003) (Dobles et al., 2000; Michel et al., 2001) (Lee et al., 1999) (Lee et al., 1999; Wang et al., 2004a) Kinetochore assembly Sister chromatid cohesion Chromosome passenger protein; Cytokinesis Spindle checkpoint N/A (Cutts et al., 1999) checkpoint components by RNA interference (Martin-Lluesma et al., 2002; Meraldi et al., 2004; Rieder and Maiato, 2004). Thus, a weakened checkpoint would still cause CIN and possibly promote tumorigenesis, but not necessarily inactivate checkpoint signaling ((Baker et al., 2006) and Chapter 2). D. Senescence or tumorigenesis? Recent studies of mice with mutated spindle checkpoint genes suggest that although all spindle checkpoint mutations induce aneuploidy, CIN is as likely to cause cellular senescence and early aging as tumorigenesis (reviewed in (Baker et al., 2005)). BubR +/-mice, as indicated earlier, exhibit aneuploidy, but have little elevated predisposition to cancer (Wang et al., 2004a). Instead, decreased expression of BubR1 causes early onset aging phenotypes and decreased lifespan in mice as demonstrated with a series of BubR1 genetic mutants combining hypomorphic and knockout alleles (Baker et al., 2004). This effect can be directly attributed to increased CIN. In fact, wildtype mice show declining levels of BubR1 with age in several tissues, particularly in the testes. BubR1 mutant mice are also infertile due to meiotic defects. (Meiosis and the spindle checkpoint will be discussed further later in this chapter.) How do we reconcile this with the human BubR1 mutations in MVA? First, since MVA is quite rare, the primary phenotype for BubR1 inactivation may be early aging. Second, patients with MVA may exhibit early aging since they are prone to developing cataracts ((Furukawa et al., 2003; Matsuura et al., 2006). A link between early onset aging and checkpoint induced aneuploidy is further strengthened from a study of Bub3+/-; Rae+/-compound mice (Baker et al., 2006). Like BubR1 haploinsufficient mice, Bub3+/-; Rae+/-mice have a low propensity for spontaneous tumor formation (Babu et al., 2003). However, Bub3+'; Rae+/- compound mutant mice develop signs of aging earlier than either single mutant. Furthermore, Bub3 /-; Rae+/- mouse embryonic fibroblasts (MEFs) undergo premature senescence which appears to be dependent on the p53-regulated growth arrest pathway since several proteins in the pathway including pl 9ARF, p53, p21, and p16 are upregulated. In contrast, Mad2' /- MEFs do not display premature cellular senescence (Baker et al., 2006). Instead, Mad2 inactivation predisposes mice to tumor development ((Michel et al., 2001); Ying Yue, personal communication). These data suggest that inactivation of spindle checkpoint components can be assigned to at least two groups: 1) those that cause premature senescence and early aging and 2) those that cause cancer. The DNA damage and repair field is rampant with examples of mutations that can either cause diseases displaying either early aging or cancer (Dimri, 2005; Dolle et al., 2006; Fuss and Cooper, 2006). Evidence is accruing that links spindle checkpoint components to protective effects against both cancer and aging, but clear aetiological evidence to an extent similar to the DNA repair field remains to be shown. E. Connection between DNA damage and mitotic progression and cancer? MIN and CIN are often presented as alternative routes to genomic instability, but considerable evidence is accumulating that DNA damage pathways interact strongly with spindle checkpoint mutations. For example, whereas Mad2/1MEFs are inviable, Mad2 / p53 -/-double knockout MEFs are viable (Burds et al., 2005). This suggests an important link between the spindle checkpoint and the DNA damage pathway. Mpsl, a spindle checkpoint kinase, may link mitosis to the G2 DNA damage checkpoint. Mpsl has been shown to interact with Chk2 and phosphorylate Chk2 on threonine 68 (Wei et al., 2005). Recently, Mpsl has also been shown to phosphorylate the Bloom syndrome gene BLM during mitosis (Leng et al., 2006). Upon phosphorylation, BLM, a member of the RecQ family of helicases, can interact with the mitotic kinase Plkl (Leng et al., 2006). Rad5 1, another component of the DNA damage pathway, has also been shown to interact with the breast-cancer associated protein Brca2 (reviewed in (Jasin, 2002)). Mice harboring homozygous Brca2 truncations (Brca2Tr rTr) develop thymic lymphomas with an impaired spindle checkpoint and mutations in p53, Bubl or BubR1 (Lee et al., 1999). Intriguingly, /T expression of a truncated Bub 1 protein also appears to induce transformation of Brca2Trr MEFs (Lee et al., 1999). Moreover, BRCA2 has been reported to interact with PLK1 and the spindle checkpoint kinase hBubR1 in vitro (Futamura et al., 2000; Lee et al., 2003). These data offer the possibility that CIN may arise from a synergy between errors in mitosis and lesions in DNA repair pathways. F. Conclusion The evidence that genomic instability plays a critical role in tumorigenesis is strong, and extensive circumstantial data points to CIN as an important source of genomic instability. Chromosome missegregation resulting from the deregulation of the spindle checkpoint is thought to be one cause of CIN. However, evidence for this connection remains weak, and the molecular basis of CIN remains largely unknown. To better elucidate the connections between aneuploidy and tumorigenesis, it will be necessary to gain a better understanding of chromosome segregation events in normal cells and to identify target genes whose mutations might cause CIN in tumors. Sensitive assays for CIN in live animal cells will be necessary for this analysis (Meraldi et al., 2004; Rieder and Khodjakov, 2003). Such assays have already been shown in yeast to be essential for the identification and analysis of CIN-promoting mutations (Nigg, 2002). Finally, the effects of CIN on tumor formation must be investigated in mice. Only then will we be able to establish the role that errors in chromosome segregation play in genomic instability and the development of human cancer. II. The spindle checkpoint Chromosomal instability (CIN) refers to an abnormal cellular state associated with an increased rate of chromosome loss or gain (Draviam et al., 2004; Kops et al., 2005). During mitosis, a cell must properly attach all its chromosomes spindle microtubules and align them such that one daughter cell receives one complete set of chromosomes and the other daughter cells receives the second identical set. The spindle checkpoint, also known as the spindle assembly checkpoint or the mitotic checkpoint, monitors genomic stability by ensuring faithful transmission of chromosomes to the daughter cells during mitosis (Amon, 1999; Gardner and Burke, 2000; Shah and Cleveland, 2000; Wassmann and Benezra, 2001). The spindle checkpoint delays anaphase until all sister chromatids have made bipolar attachments to microtubules emanating from opposite poles of the mitotic spindle (Fig. 1.1). Chromosomes have specialized multi-protein complexes called kinetochores, that bind to their centromeres and attach the DNA to microtubules. It is crucial that the mitotic spindle only begin to pull sister chromatids apart once every kinetochore has properly bound microtubules. Strikingly, a single misattached kinetochore is sufficient to activate the spindle checkpoint and delay mitotic progression to allow additional time for chromosomes to become properly attached to the spindle (Li and Nicklas, 1995; Rieder et al., 1995). Abrogation of the spindle checkpoint would be detrimental to genomic stability as chromosome missegregation leads to CIN. In this section, I explain the paradigm of a cell Figure 1.1 kinase >eat Iprotein APC/C Metaphase r Y Anaphase Figure 1.1: The canonicalspindle checkpointproteins. Spindle checkpoint proteins localize to unattached kinetochores and prevent the metaphase to anaphase transition by inhibiting the activation of the APC/C. Mpsl is required for kinetochore localization of Madl and Mad2. Mad2 and BubR1 can directly as well as in combination with Bub3 and BubR1 inhibit Cdc20 from activating the APC/C, thus preventing ubiquitination of securin and mitotic progression. cycle checkpoint, describe the discovery of spindle checkpoint components and briefly examine checkpoint signaling and its importance in meiosis. A. Cell cycle and cell cycle checkpoints Cell duplication proceeds through a series of events called the cell cycle that occurs in a tightly regulated and temporally ordered fashion (Dash and El-Deiry, 2004; Kastan and Bartek, 2004). The cell must first accumulate the necessary nutrients for growth during Gl before it can replicate its DNA in S phase. In G2, a cell undergoes a further period of growth before it is ready to enter mitosis when the cell must first segregate its chromosomes before it undergoes cytokinesis. To ensure the cell cycle proceeds in the correct manner, checkpoints act to delay downstream events until upstream events have been properly transpired. For example, DNA damage surveillance mechanisms operate prior to DNA replication and mitosis in G1 and G2 to maintain genome fidelity by arresting cell cycle progression until the damage has been repaired (Hartwell and Weinert, 1989). The original checkpoint paradigm, RAD9, was identified in Saccharomyces cervisiae in a genetic screen for mutations that allow budding yeast to continue to undergo cell division despite radiation induced DNA damage (Weinert and Hartwell, 1988). Whereas wildtype yeast arrest in G2 upon exposure to low dosages of X-ray irradiation, rad9 cells fail to arrest and proceed to mitosis with DNA damage and may die. However, when rad9 cells are treated simultaneously with X-rays and nocodazole to arrest cells in mitosis, cells have sufficient time to repair DNA lesions and viability is restored. Furthermore, rad9 cells exhibit normal growth and viability in the absence of exposure to DNA damaging agents (Weinert and Hartwell, 1988). Thus, cell cycle checkpoints were proposed to be non-essential surveillance systems that monitor essential cell processes and respond in the event of a rare problem by inducing cell cycle arrest (Hartwell and Weinert, 1989; Weinert and Hartwell, 1988). B. The canonical players of the spindle checkpoint The spindle checkpoint delays the onset of anaphase by inhibiting the anaphase promoting complex/ cyclosome (APC/C), a multi-subunit E3 ubiquitin ligase, until all chromosomes have achieved stable microtubule attachments and congressed to the metaphase plate (Amon, 1999). Yeast with a wildtype spindle checkpoint undergo very accurate chromosome segregation as less than one missegregation event can be detected in 105 cell divisions (Hartwell et al., 1982). Like the DNA damage checkpoint, genetic screens in S. cervisiae identified the canonical components of the spindle checkpoint (Hoyt et al., 1991; Li and Murray, 1991; Winey et al., 1991). Two genetic screens searching for mutations that would allow cells to bypass spindle damage induced by the microtubule destabilizing drug benomyl identified six genes: MADI, M4D2, and MAD3 for Mitotic Arrest Deficient, and BUB], BUB2, and BUB3 fo Budding Uninhibited by Benzimidazole (Hoyt et al., 1991; Li and Murray, 1991). In budding yeast, a mutation in any one of the six genes disables the ability of a cell to arrest in response to spindle damage suggesting these genes were part of a spindle assembly checkpoint (Hoyt et al., 1991; Li and Murray, 1991). BUB2 was subsequently shown to be required for mitotic exit, not the spindle checkpoint. Mutations in any of the MAD and BUB genes do not decrease viability in the absence of spindle damage, although mad mutations increase the chromosome loss rate 10-fold under normal growth conditions and up to 1000-fold in the presence of benomyl (Li and Murray, 1991). Two additional components were subsequently identified to be important for the spindle checkpoint: MPS 1 (Monopolar Spindle 1) and Cdc20. The Mads and Bubs were shown to target an activator of the APC/C: Cdc20 (Hwang et al., 1998). MPS1 was initially identified in a screen for mutants defective in spindle pole body (SPB) duplication in S. cerevisae, but was later shown to be essential for the spindle checkpoint (Weiss and Winey, 1996; Winey et al., 1991). The SPB duplication and spindle checkpoint functions of Mps lp are genetically separable; and it is the inactivation of Mpslp in SPB duplication that causes budding yeast inviability (Castillo et al., 2002; Schutz and Winey, 1998). Thus, the original definition of a cell cycle checkpoint, as an auxiliary system dispensable for normal growth, appears to hold true for the yeast spindle assembly checkpoint. The spindle assembly checkpoint has been functionally conserved in all eukaryotes (Wassmann and Benezra, 2001). All six budding yeast checkpoint components - MAD1-3, BUB1, BUB3, and MPSI - have metazoan homologues. However, BubR1, the metazoan homologue of Mad3p, is actually a hybrid of sequences from Mad3p and Bublp, with a Mad3p-like N-terminal domain and a C-terminal Bubllike kinase domain (Taylor et al., 1998). Like Bub and BubR1, Mps is also a kinase (Winey and Huneycutt, 2002). Madl is a coiled-coil protein (Sironi et al., 2002). Bub3 contains WD repeats and is also very similar to Rae 1, which functions in nucelocytoplasmic transport (Wang et al., 2001). Additional genes have been found to be required for the spindle checkpoint including Ipll/Aurora B kinase, Birlp/Survivin and Slil5p/INCENP; CENP-E (centromere-associated protein E); Rael; CMT2 (Caught by MAD Two); and also Rod and ZwlO which are unique to vertebrates (Adams et al., 2001; Babu et al., 2003; Chan et al., 2000; Cutts et al., 1999; Habu et al., 2002; Kallio et al., 2001; Putkey et al., 2002; Uren et al., 2000; Wang et al., 2001; Weaver et al., 2003). The canonical checkpoint components - Bubl, BubR1, Bub3, Madl, Mad2, and Mpsl - are part of a bona fide mammalian spindle checkpoint as inactivation of these proteins abrogates the checkpoint and results in chromosome missegregation, aneuploidy and failure to arrest in mitosis in response to microtubule destabilizing agents (Meraldi et al., 2004; Stucke et al., 2002; Taylor et al., 1998; Yu, 2002). C. Interactions between the spindle checkpoint and the cell cycle The importance of the spindle checkpoint for viability seems to increase with an organism's complexity. The loss of the checkpoint in S. cerevisiae leads to increased rate of chromosome loss, but is only lethal in the pressure of anti-microtubule drugs (Li and Murray, 1991). However, the checkpoint is essential in metazoans under normal growth conditions and inactivation of spindle checkpoint genes leads to embryonic lethality in mice (Dobles et al., 2000; Kalitsis et al., 2000; Wang et al., 2004a). In a multicellular organism like the mouse, where viability is dependent on the interaction of all of the cells, the apoptosis of aneuploid cells in G1 or mitotic catastrophe caused by impaired checkpoint function is disastrous for the embryo (Dobles et al., 2000). Furthermore, the spindle checkpoint is not required in mouse embryonic fibroblasts lacking p53, presumably because apoptosis is impaired (Burds et al., 2005). In unicellular organisms like S. cerevisiae, failure to undergo cell cycle arrest and transmission of genetic alterations either leads to death or adaptation to the mutations accumulated in the cell. Though these outcomes are tolerated in yeast, metazoans have evolved a more stringent set of cell cycle checkpoints that can induce apoptosis as well as cell cycle arrest to protect genome fidelity and prevent the propagation of cancerous cells. Expression levels of several spindle checkpoint components are regulated in a cell cycle dependent manner by E2F. The E2F family of transcription factors is crucial to the control of cell cycle progression by both tranactivating and repressing expression of genes integral to DNA replication, DNA repair and mitosis (Hansemann, 1890; Polager et al., 2002). The tumor suppressor pRB regulates the E2F proteins. Mad2 is a direct target of E2F 1 and cells with mutations in the Rb pathway have misregulated expression of Mad2 protein (Hernando et al., 2004). E2F1 and E2F3 were found to up-regulate the expression of several mitotic genes including BUBR1 (BUB B) and Aurora B (AIM1) by DNA microarray analysis in Rat cells (Polager et al., 2002). Another study utilizing a combination of chromatin immunoprecipitation and DNA microarray analysis in human cell lines found the repressive E2F4 efficiently binds to the promoters of hMAD2 (MAD2L1), hBUB3 and hMPS1 (TTK) (Ren et al., 2002). Moreover, the promoter of HEC 1,whose gene product Ndc80 is required for the kinetochore localization of both Mad2 and Mps 1, is regulated by both repressing and activating E2Fs (E2F4 and E2F 1) (Ren et al., 2002). Thus, one way the spindle checkpoint is integrated with the rest of the cell cycle is via regulation by the E2F family of transcription factors. D. Spindle checkpoint signaling To understand the mechanism of spindle checkpoint signaling, we must understand the signal for checkpoint activation, the signal transmission system, the mechanism of mitotic arrest and the cessation of checkpoint signaling that allows mitosis to continue. The underlying biochemistry of how the checkpoint is activated and then turned off is incomplete, but it is believed the "prevent anaphase" signal must originate from the kinetochores. The kinetochore is a complex structure built onto the centromere of each chromosome that mediates attachments to microtubules and encompasses as many as 60 proteins (De Wulf et al., 2003; McAinsh et al., 2003). The canonical mammalian spindle checkpoint proteins - Mpsl, Madl, Mad2, Bubl, BubR1, and Bub3 all localize to kinetochores during mitosis (Fisk and Winey, 2001; Jablonski et al., 1998; Kallio et al., 1998; Li and Benezra, 1996; Martinez-Exposito et al., 1999; Meraldi et al., 2004; Taylor et al., 1998; Taylor and McKeon, 1997). The timing of their arrival at kinetochores varies in mitosis, but all are present by nuclear envelope breakdown in late prophase and their abundance decreases following chromosome attachment, suggesting checkpoint components are monitoring kinetochore-microtubule interactions. Two models summarize an ongoing debate over the exact nature of the "prevent anaphase" signal sensed by spindle checkpoint components: 1)the spindle checkpoint components monitor the microtubule occupancy status on kinetochores; or 2) the checkpoint components monitor the tension generated across sister kinetochores by the bipolar attachment of sister chromatid pairs to microtubules emanating from opposite poles of the mitotic spindle. Many attempts have been made to uncouple kinetochoremicrotubule attachment from the tension generated across kinetochore pairs resulting from that attachment (Kapoor et al., 2000; Rieder et al., 1995; Stem and Murray, 2001). Support for the kinetochore-microtubule attachment model comes from experiments in PtK cells where anaphase initiates about 23 minutes after the last kinetochore captures microtubules (Rieder et al., 1994). Moreover, when the last unattached kinetochore is destroyed by laser ablation, cells are able to proceed to anaphase and undergo chromosome segregation (Rieder et al., 1995). Mad2 specifically localizes to unattached kinetochores in PtK cells, but those kinetochores that have lost tension due to microtubule stabilization by taxol treatment fail to recruit Mad2 (Waters et al., 1998). However, when DNA replication is prevented by a mutation in CDC6, an initiator of DNA replication in budding yeast, then chromatids can bind microtubules, but are not under tension since there is no sister and the spindle checkpoint is activated (Stem and Murray, 2001). Furthermore, when cells are exposed to monastrol, which inhibits the mitotic motor Eg5, synthelic (microtubules from the same pole) kinetochores attached to microtubules still retain Mad2 suggesting microtubule attachment alone is insufficient to satisfy the checkpoint (Kapoor et al., 2000). These experiments have shown that either the lack of microtubule attachment or tension can be sufficient for activation of the spindle checkpoint. For further complexity, the Iplil/Aurora B kinase has been implicated in spindle checkpoint function, specifically for the sensing and correction of synthelic attachments which do not produce tension (Hauf et al., 2003; Tanaka et al., 2002). However, the debate may never be resolved due to the interdependence of microtubule attachment and tension at kinetochores; kinetochore-microtubule attachment is stabilized by tension and both attachment and tension appear to be monitor by the spindle checkpoint (Nicklas et al., 2001). Nonetheless, the spindle checkpoint can monitor the status of sister chromatid bi-orientation and respond to any perturbations by arresting cells at the metaphase to anaphase transition. Checkpoint signaling is presumably transmitted by the three kinases in the canonical spindle checkpoint - Bubl, BubR1, and Mpsl. Yet, little is known about spindle checkpoint signal transmission since few targets have been described for these three checkpoint kinases. The kinase function of Mpsl is necessary for checkpoint activation, but whether the kinase domain of Bubl is required for the checkpoint is unclear (Abrieu et al., 2001; Chen, 2004; Warren et al., 2002). A truncated form of Bub1 lacking its C-terminal kinase domain is sufficient for checkpoint function in budding yeast, but inhibition of Bub 1 kinase activity in Xenopus extracts causes partially compromises the checkpoint (Chen, 2004; Warren et al., 2002). Mps1 and Bub 1 have been shown to phosphorylate yeast Madl in vitro, but this has not been observed in vivo (Hardwick et al., 1996; Seeley et al., 1999). Surprisingly, the kinase domain of BubR1 is not required for inhibition of the APC/C in vitro (Tang et al., 2001). However, BubR1 kinase activity, which is activated by binding to CENP-E, has been shown to be essential for checkpoint signaling in MEFs (Putkey et al., 2002; Weaver et al., 2003). Checkpoint signal transmission does require multiple complexes of checkpoint proteins. Bub3 must be in a complex with Bubl or BubR1 to bind to kinetochores (Taylor et al., 1998;). Interaction with Bub3 is mediated by a GLEBs motif located in Bubl and BubR1/Mad3 (Wang et al., 2001). Madl and Mad2 require Mpsl for kinetochore localization in mammals (Liu et al., 2003; Martin-Lluesma et al., 2002). Mad2 also requires the Rod/ZwlO0 complex to localize to unattached kinetochores (Buffin et al., 2005). Mad 1 forms a homodimer via its coiled-coiled domain and recruits Mad2 to unattached kinetochores. Once there, Mad2 can bind and inhibit Cdc20, a WD domain protein that localizes to the kinetochore, by binding to an N-terminal 12-residue stretch of Cdc20 (Luo et al., 2002; Sironi et al., 2001). Mad2 may only bind Madl and be recruited to the kinetochore in an unphosphorylated form (Wassmann et al., 2003a). Crystal structures of the Madl-Mad2 complex reveal a tetramer of two Madl -Mad2 subcomplexes with the Mad2 C-terminal tails as flexible elements that undergo comformational changes to act as molecular 'safety belts' around Madl or Cdc20 (Sironi et al., 2002). Both Mad2 and BubR1 can independently interact directly with Cdc20 and inhibit the APC/C in vitro (Chan et al., 1999; Fang, 2002; Kallio et al., 1998; Sudakin et al., 2001; Tang et al., 2001). BubR1 and Mad2 inhibition of Cdc20 may also be cooperative. A mitotic checkpoint complex (MCC) purified from HeLa cells that contained BubR1, Bub3, Cdc20 and Mad2 was found to be - 3000-fold more potent at inhibiting the APC/C than recombinant Mad2 alone (Sudakin et al., 2001). The isolation of these complexes suggests that the spindle checkpoint functions as a complex multiprotein machine in contrast to a standard signal transduction cascade, but conflicting data suggests the individual molecular interactions require additional elucidation. What has been well characterized is the ultimate target of the checkpoint: Cdc20 and the APC/C (reviewed in Zachariae and Nasmyth, 1999). The APC/C is a highly regulated multi-subunit E3 ubiquitan ligase whose ubiquitination activity controls the timing of sister chromatid separation and mitotic exit via its regulators Cdc20 and Cdhl (Morgan, 1999; Visintin et al., 1997). Newly replicated sister chromatids are physically held together by a protein complex called cohesin composed of Smcl, Smc3, Sccl and Scc3 (Uhlmann, 2003). Conserved throughout evolution, cohesins localize to the centromeres and along the arms of sister chromatids following DNA replication. However, chromosomes cannot segregate until sister chromatid cohesion is lost following cleavage of the cohesin subunit Scc by Esplp/separase. Separase is bound to and inhibited by Pdslp/securin until the APC/C is activated by Cdc20 to ubiquitinate securin, targeting it for degradation by the 26S proteosome (Cohen-Fix et al., 1996; Coux et al., 1996). Mad2 and BubR1 physically interact with Cdc20 and prevent activation of the APC/C to prevent premature chromosome segregation (Chan et al., 1999; Fang, 2002; Kallio et al., 1998; Sudakin et al., 2001; Tang et al., 2001). Once all chromosomes have achieved bipolar attachment to the mitotic spindle, Cdc20 is released by the spindle checkpoint to activate the APC/C to dissolve sister chromatid cohesion (Kallio et al., 1998; Tavormina and Burke, 1998). Thus, when chromosomes are not properly attached or under proper tension during mitosis, a signal is generated to activate the spindle checkpoint machinery to prevent the degradation of securin, maintaining chromosome cohesion and preventing mitotic progression. Silencing of the spindle checkpoint to allow anaphase entry appears to be an active process. Cdc20 must be released by Mad2 to activate the APC/C for chromosome segregation to occur. Recently, a model for Mad2 activation has been proposed that suggests Mad2 exists in two conformations: an unbound open state that does not bind Cdc20 and a closed conformation when bound to Madl or Cdc20 (De Antoni et al., 2005; Nezi et al., 2006). Checkpoint activation promotes the conversion of cytosolic open Mad2 to the Madl/kinetochore bound closed conformation facilitating binding to and inhibiting Cdc20. When all kinetochores have properly bound to microtubules, Mad2 also interacts with an additional protein - CMT2/p31comet (Habu et al., 2002; Mapelli et al., 2006). CMT2 binds to Cdc20/Mad2 complexes and promotes the dissociation of Mad2 from Cdc20 (Habu et al., 2002). Robert Hagan and colleagues in the Sorger group have shown that CMT2 is a kinetochore-associated protein critical for exit from mitosis. RNAi depletion of CMT2 arrests cells in mitosis even though spindle checkpoint components have left kinetochores and this mitotic arrest is dependent on Mad2 (Robert Hagan, personal communication). Thus, mammalian cells require the active inhibition of a Mad2-dependent spindle checkpoint by CMT2 for normal mitotic progression. E. Mpsl: a spindle checkpoint kinase Mps1 is a dual-specificity kinase that is unique among the checkpoint proteins in S. cerevisiaebecause it is required for the spindle assembly checkpoint and SPB duplication, equivalent to the mammalian centrosome (Winey and Huneycutt, 2002). MPS 1 is the only essential checkpoint gene in budding yeast, but this is due to its role in SPB duplication (Castillo et al., 2002; Schutz and Winey, 1998). Mps lp interacts with yeast Damlp, an essential component of the DASH microtubule ring complex, which links kinetochores to microtubules thereby facilitating chromosome segregation (Jones et al., 1999; Li et al., 2002; Miranda et al., 2005). In S. pombe however, spMpslp/Mphlp acts exclusively in the spindle checkpoint (He et al., 1998). Epistatic analysis of the budding yeast checkpoint genes has placed scMPS 1 as the most upstream component of the checkpoint pathway (Weiss and Winey, 1996). Overexpression of Mps lp constitutively activates the spindle checkpoint, in the absence of spindle damage, which is dependent on the Mads and Bubs (Hardwick et al., 1996; He et al., 1998). Although scMpslp has been reported to phosphorylate scMadlp, this interaction has not been recapitulated in vertebrates (Hardwick et al., 1996). Nonetheless, Mps 1 appears to be upstream of other checkpoint components in metazoans as well since Mad2 and Madl localization to the kinetochore requires Mpsl (Abrieu et al., 2001; Liu et al., 2003; Martin-Lluesma et al., 2002). Homology between scMpslp and the vertebrate orthologs is confined to the C-terminal kinase region (Winey and Huneycutt, 2002). The mouse and human homologs of Mps 1, ESK and TTK, were first described in the literature as dual specificity kinases able to phosphorylate both tyrosine and serine/threonine residues and highly expressed in tissues with a high proliferation rate such as the thymus and the testis (Douville et al., 1992; Hogg et al., 1994; Mills et al., 1992). Subsequently, ESK and TTK have definitely been shown to be the Mpsl functional homolog in mouse and human; homologs have also been found in the fly, frog, zebrafish (Abrieu et al., 2001; Fischer et al., 2004; Fisk and Winey, 2001; Poss et al., 2002; Stucke et al., 2002). Four findings hold true for Mps 1 throughout evolution: 1)Mps1 localizes to kinetochores, 2) the kinase function of Mps I is required for the checkpoint, 3) Mps likely acts early in the checkpoint pathway and 4) Mpsl protein and mRNA levels are regulated during the cell cycle. Although the checkpoint function of Mps 1 has been conserved in vertebrates, there is conflicting data on the role of Mps1 in centrosome duplication. Although mouse Mps 1 has been described to be required for centrosome duplication, three reports have both argued for and against human Mpsl localizing to centrosomes and regulating their duplication (Fisk et al., 2003; Fisk and Winey, 2001; Liu et al., 2003; Stucke et al., 2004; Stucke et al., 2002; Winey and Huneycutt, 2002). Moreover, drosophila Mpsl does localize to centrosomes, but Mps 1 mutants do not have any centrosome defects (Fischer et al., 2004). Although the existence of a role for mammalian Mps 1 in centrosome duplication remains controversial, recent reports describing interactions with Chk2 and Blm suggest hMps may have other functions outside of the spindle checkpoint in the DNA damage response (see section IE) (Leng et al., 2006; Wei et al., 2005). Thus, Mps is a mitotic kinase essential for the spindle checkpoint from yeast to man and may have acquired additional functions that link the DNA damage response to accurate chromosome segregation. F. Meiosis and the spindle checkpoint In humans, aneuploidy has been linked to spontaneous abortions and developmental defects as well to many cancers (Hassold and Hunt, 2001). A trisomy or monosomy event has been identified in at least 5% of human pregnancies (Hassold and Hunt, 2001). Although aneuploidy almost always causes embryonic lethality and results in spontaneous abortions, 1 in 300 infants are born with aneuploidy, usually in a sex chromosome or with an additional chromosome 13, 18, or 21. The latter event causes Down syndrome, a developmental disorder in which individuals show decreased mental capacitity and physical defects (reviewed in (Scarbrough et al., 1982)). The strongest causal basis for aneuploidy in humans is increasing maternal age. Women over 40 have at least a one in three probability of a pregnancy complicated by abnormal chromosome numbers (Hassold and Hunt, 2001; Thomas et al., 2001). The basis of this age effect is unclear, although it is intriguing to speculate that a decrease in spindle checkpoint function plays a role (Baker et al., 2005; Ma et al., 2005). BubR1 hypomorphic mice undergo meiotic chromosome missegregation and have decreased fertility (Baker et al., 2004). A Mad2-dependent spindle checkpoint has been shown to exist during the first meiotic division in mouse oocytes, and overexpression of Mad2 causes a metaphase I arrest (Wassmann et al., 2003b). Moreover, Mad2 ÷/ - mouse oocytes undergo meiosis with abnormal mitotic timing and chromosome segregation defects (Katja Wassmann, personal communication). Mad2 downregulation by siRNA interference in mouse oocytes also decreased the length of meiosis I (Wang et al., 2006). Mpsl mutations in zebrafish and fruit flies cause aneuploidy associated meiotic defects as well (Gilliland et al., 2005; Poss et al., 2004). Interestingly, while gaining or losing a single chromosome is a lethal event, gaining an entire set of chromosomes is less detrimental. Although extremely rare, triploidy babies, those that have a 3N complement of the genome instead of the normal 2N, can survive to birth and possibly live for an additional few hours after birth (Leisti et al., 1974). Moreover, a cell often chooses to forgo cytokinesis and become polyploid after a chromosome missegregation event instead of gaining or losing a single chromosome (Shi and King, 2005). This suggests that balancing gene expression is vital to a cell and therefore, understanding those cell cycle checkpoints that monitor genome stability and maintain proper gene dosage such as the spindle checkpoint is critical to preventing and curing human disease. III. Tumor suppressor genes and oncogenes It was Theodore Boveri who first drew attention to the abnormal number of chromosomes in human tumors and hypothesized that the act of losing or gaining chromosomes may be sufficient to cause a cancerous state (Boveri, 1914). Although the role of aneuploidy in tumorigenesis is still debated, the gain or loss of a few genes called tumor suppressor genes (TSPs) or oncogenes are firmly established as causes of human tumorigenesis (Balmain, 2001; Bertwistle et al., 2004; Duesberg and Li, 2003). Disregulation of TSPs and oncogenes desensitizes tumor cells to both proliferative signals and cell cycle checkpoints. Although current research is still trying to establish a clear relationship between components of the spindle assembly checkpoint and tumorigenesis, more than two decades of scientific literature clearly link the DNA damage checkpoint to human cancer (Christmann et al., 2003; Draviam et al., 2004; Kops et al., 2005; Loeb and Cheng, 1990; Sancar et al., 2004; West, 2003). Two of the key players in regulating the cell cycle in response to DNA damage are the TSPs TP53 and pl 9 ARF (Latonen and Laiho, 2005; Moore et al., 2003). A critical oncogene in human cancer is K-ras, part of a signal transduction pathway involved in reading extracellular cues to regulate cell growth, differentiation, and survival (Friday and Adjei, 2005; PerezMancera and Tuveson, 2005). In this section, I briefly describe their discovery, the functions of these genes, and their roles in cancer. A. p53: DNA damage, aneuploidy and cancer TP53 is an extensively studied tumor suppressor gene found to be mutated in over 50% of human cancers. The protein product of TP53, p53 was first detected in mouse tumor cells and to bind SV 40 Large T antigen in viral transformation studies (DeLeo et al., 1979; Linzer and Levine, 1979). TP53 is mutated in the germline of patients with LiFraumeni syndrome, an inherited disorder with a predisposition to cancer, particularly of the breast (Pierotti and Dragani, 1992). Described as the "gatekeeper" of the genome, p53 is a transcription factor that responds to a multitude of cellular stress including DNA damage, hypoxia, and nucleotide imbalance that impinges on accurate DNA replication and cell division (Levine, 1997). Normally, p53 protein levels in a cell are tightly regulated in a number of positive and negative feedback loops primarily through ubiquitin-mediated proteolysis (Harris and Levine, 2005). Upon the presence of cellular stress, p53 protein undergoes post-translational modification that increases protein stability and allows for transactivation of genes involved in cell cycle arrest, DNA repair or apoptosis. Two key modulators of p53 protein level are Mdm2, an E3 ubiquitin ligase, and pl 9 ARF , an inhibitor of Mdm2. The majority of TP53 mutations found in human cancer cluster to the DNA-binding domain of p53, suggesting tumor cells are selecting for the prevention of p53-dependent gene expression. What is the molecular mechanism of p53-dependent tumor suppression? One hypothesis for the high mutation rate of TP53 in human cancer is that the constitutive activation of the DNA damage checkpoint by DNA double strand breaks (DSBs) in tumor cells strongly selects for the inactivation of p53 (Halazonetis, 2004). The author argues cell carcinogenesis is intrinsically linked to the formation of DNA DSBs which in turn activates the DNA damage checkpoint during every cell cycle. Intriguingly, p53 inactivation predisposes the majority of mice to spontaneous formation of lymphomas, possibly because immune cells are highly prone to DSB formation from VDJ recombination (Bassing and Alt, 2004; Donehower et al., 1992; Jacks et al., 1994; Purdie et al., 1994). When inactivation of p53 and a DNA repair gene are combined, tumor onset is often accelerated and clonal translocations are observed strengthening the hypothesis that DNA DSBs favor a tumorigenic outcome (Bassing et al., 2003; Cheung et al., 2002; Jonkers et al., 2001; Morales et al., 2006). Although tumors lacking functional p53 are highly aneuploid, spectral karyotyping data from p53 null mouse lymphoma cells do not suggest DSBS play a direct role per se in these tumors since no clonal translocations are observed (Liao et al., 1998; Morales et al., 2006). Therefore, the strong selective pressure against a functional p53 and the presence of DNA DSBs may be coincidental. DNA DSBs, if left unrepaired, would induce p53-dependent apoptosis. The selection may be against the ability of p53 to induce cellular suicide when DNA damage is present in tumor cells. Aneuploidy is fundamentally linked to p53 inactivation suggesting p53 also monitors ploidy. Centrosomes have been found to be amplified in p53 null MEF lines resulting in chromosome missegregation defects (Fukasawa et al., 1996). Initial experiments suggested p53 may be maintaining diploidy as a component in the spindle checkpoint, because p53 -/ MEFs would exit mitosis without undergoing cytokinesis following treatment with microtubule poisons (Cross et al., 1995). However, subsequent careful examination in p53 -/-MEFs suggests p53 does not appear to play a role in the spindle checkpoint (Lanni and Jacks, 1998). Instead, p53 appears to be required to arrest tetraploid cells that fail to undergo cytokinesis. Furthermore, in a study examining the levels of CIN and mitotic checkpoint activity in nine human breast cancer cell lines, p53 mutation status did not correlate with CIN levels and spindle checkpoint deficiency (Yoon et al., 2002). Mammalian cells may have evolved a p53-dependent GI tetraploidy checkpoint as yet another mechanism to prevent propagation of potential cancerous cells that experience mitotic errors (Andreassen et al., 2001). Moreover, an epidemiological link between p53 loss and tetraploidy exists in human patients with Barrett's esophagus. Barrett's esophageal biopsies show that patients with tetraploid cell populations are predisposed to progression to aneuploidy and is interdependent on inactivation of TP53 (Galipeau et al., 1996). Recently, two studies have directly address this hypothesis in both p53-null human cells and in the mouse in vivo and propose one route to aneuploidy and cancer may be through an intermediate tetraploid state (Fujiwara et al., 2005; Shi and King, 2005). Shi and King found chromosome nondisjunction prevents cytokinesis and results in tetraploid, not aneuploid, cells. They further suggest that aneuploidy may arise from multipolar mitosis in the resultant tetraploid cell (Shi and King, 2005). Furthermore, Fujiwara et al. show that by blocking cytokinesis in p53 -'- cells and causing tetraploidy, cells can be transformed in vitro by exposure to a carcinogen, and even in the absence of carcinogen, p53 -'1tetraploid cells give rise to malignant cancers in vivo when transplanted into nude mice (Fujiwara et al., 2005) Thus, CIN resulting from chromosome segregation errors and loss of the ploidy sensing mechanism via p53 inactivation may constitute a major route towards tumorigenesis. B. p53-dependent and p53-independent functions of p 1 9 ARF The INK4a/ARF locus is frequently mutated in human cancer. This locus encodes two gene products required for cell proliferation: p 16 NK4a, a cyclin-dependent kinase inhibitor, and an alternative reading frame (ARF) encoding p 14 ARF in humans or pl9 A RF in mice (Lowe and Sherr, 2003; Quelle et al., 1995; Sherr, 2001). Although pl 9 ARF and p16 NK4a are both tumor suppressor genes and are often codeleted in tumor cells, inactivating pl 9 ARF alone in mice is sufficient to promote cancer (Kamijo et al., 1999; Kamijo et al., 1997). The primary function of Arf is to increase p53 protein stability by antagonizing Mdm2 function in response to cellular stress (Kamijo et al., 1998; Pomerantz et al., 1998; Weber et al., 1999). Although the half-life of p53 is typically about 15 minutes in MEFs, overexpression of pl 9 ARF significantly increases this to 73 minutes (Kamijo et al., 1998). Arf inhibits the ability of Mdm2 to ubiquitinate and target p53 for protein degradation by binding and sequestering Mdm2 to the nucleolus (Weber et al., 1999). Arf is normally silenced in adult tissues as Arf expression results in cellular senescence. However, upon elevated and sustained mitogenic growth signals such as through Myc overexpression and oncogenic ras signaling, p 9A RF expression is induced to prevent inappropriate growth by causing cell cycle arrest or apoptosis via p53 (Palmero et al., 1998; Zindy et al., 1998). Although tumor suppression by p19 ARF occurs primarily mediated by p53, Arf also has p53-independent functions (Moore et al., 2003; Weber et al., 2000). Inactivating Arf does not recapitulate p53 inactivation in mice as only 80% of Arf deficient mice develop tumors by 1 year of age compared to 100% tumor development by 10 months of age in p53 deficient mice mice (Donehower et al., 1992; Jacks et al., 1994; Kamijo et al., 1999; Purdie et al., 1994). Nonetheless, Arf deficient mice display a slightly different tumor spectrum from p53 /- mice, acquiring more sarcomas than lymphomas and additional tumor types such as carcinomas and nervous system tumors uncommon to p53-null mice (Kamijo et al., 1999). Moreover, mice where p19 A RF ,Mdm2 and p53 are triply inactivated display a broader tumor spectrum than p53-null mice alone (Weber et al., 2000). Arf-null mice also become blind shortly after birth, a phenotype not found in p53 null mice, due to improper eye vasculature regression during embryogenesis (McKeller et al., 2002). Gene expression profiling has shown that Arf induces the expression of both p53-dependent and p53independent anti-proliferative genes (Sugimoto et al., 2003). These p53-independent functions of p 1 9 ARF may be mediated through its functions in sumolyation and/or inhibiting ribosomal RNA processing (Bertwistle et al., 2004; Sugimoto et al., 2003; Tago et al., 2005). Interestingly, inactivating one copy of some ribosomal RNA genes in zebrafish predisposes animals to tumor formation possibly through reducing protein synthesis (Amsterdam et al., 2004). Thus, Arf inactivation may promote tumorigenesis by directly inhibiting the p53 pathway or indirectly by broadly altering protein expression levels. C. Oncogenic K-ras and lung cancer The Ras family of monomeric G proteins, H-ras, N-ras and K-ras, are "molecular switches" linking extracellular growth signals to intracellular signaling pathways controlling cell growth, differentiation and survival (Ellis and Clark, 2000). Ras proteins undergo extensive post-translational lipid modification to target these small 21 kDa proteins to the cellular membrane. Ras cycles between a GFP bound "off"' state to a GTP bound "on" state in response to the activation of various membrane bound receptors such as EGF receptor (Friday and Adjei, 2005). Activated Ras regulates multiple signal transduction pathways by interacting with three main effectors: Raf kinase in the mitogen activated protein kinase (MAPK) pathway, Phosphoinositide 3'-kinase (PI3K), and the GTPase Ral (Marshall, 1996). Ras proteins have intrinsically low GTPase activity and require interaction with GTPase activating proteins (GAPs) to stimulate hydrolysis of GTP to GDP and reverse Ras activation. H-ras and K-ras are analogous to the proteins responsible for transformation in the Harvey and Kirsten sarcoma viruses respectively, and N-ras was identified to be an oncogene in neuroblastoma (Chang et al., 1982; Hall et al., 1983; Shimizu et al., 1983; Taparowsky et al., 1983). Ras mutations are found in 30% of human tumors, with K-ras being the most frequently mutated (Ellis and Clark, 2000). The majority are point mutations in codons 12, 13 and 61 that abolishes the interaction of Ras with GAPs, thereby constitutively locking the Ras proteins in an activated GTP state. Lung cancer is a deadly disease that causes the highest rate of cancer associated deaths worldwide. Oncogenic K-ras mutations are found in 15% to 50% of human lung cancers (Friday and Adjei, 2005). This frequency increases to >90% in model models of lung cancer (Malkinson, 1998). A mouse lung cancer model, where the oncogenic allele K-ras G 12D is conditionally activated by intranasal delivery of Cre recombinase to the lungs of mice, has been used successfully to study the early steps of adenocarcinoma initiation and progression (Jackson et al., 2001; Reimann et al., 2001). K-ras induced adenocarcinoma in the mouse appears to originate in a group of stem cells at the bronchioalveolar duct junction (Kim et al., 2005a). However, oncogenic ras requires additional genetic events for tumor initiation. Sustained mitogenic signaling by the expression of oncogenic ras in normal primary cells induces premature cellular senescence and is dependent on p53, p 1 9 ARF , and p 161NK4A (Lin et al., 1998; Palmero et al., 1998; Serrano et al., 1997). When mutations in p53 are combined with K-ras G12D in the mouse lung, animals develop advanced lung adenocarcinoma that closely mimics advanced human lung cancer and is significantly more malignant than with oncogenic Kras alone (Jackson et al., 2005; Jackson et al., 2001). Thus, many safeguards have evolved to suppress tumor formation and multiple mutations must occur for a tumor to arise and develop to malignancy. IV. Conclusion Multiple levels of regulation have evolved to maintain genome stability and prevent tumorigenesis in mammalian cells. Mps 1 and other spindle assembly checkpoint components maintain euploidy by monitoring chromosome segregation. TP53, p 9ARF and the DNA damage checkpoint halt cell cycle progression to allow for DNA repair or programmed cell death when repair is unsuccessful. Signaling networks involving K-ras and other proto-oncogenes monitor external stimuli and control cell growth. Inactivating these and other genes in mice has been a fruitful method for understanding cancer biology. Recent advances in mouse genetic engineering utilizing Cre/LoxP site specific recombination have generated conditional mice with various mutations in p53 and an activating allele of K-ras G12D, further adding to the repertoire of genetic mouse models for the study of tumor formation. Spindle checkpoint inactivation in mice leads to embryonic lethality. No conditional genetic mouse models of spindle checkpoint genes have been described yet to address the role of chromosomal instability by checkpoint abrogation in cancer. Cancer has been shown to require the inactivation of multiple cellular safeguards. CIN would cause global changes in gene expression. Thus, instead of complete checkpoint inactivation, a weakened spindle checkpoint allele may ultimately be the best facilitator of an environment favorable for tumorigenesis. References Abrieu, A., L. Magnaghi-Jaulin, J.A. Kahana, M. Peter, A. Castro, S. Vigneron, T. Lorca, D.W. Cleveland, and J.C. Labbe. 2001. Mpsl is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 106:83-93. Adams, R.R., D.M. Eckley, P. Vagnarelli, S.P. Wheatley, D.L. Gerloff, A.M. Mackay, P.A. Svingen, S.H. Kaufmann, and W.C. Earnshaw. 2001. Human INCENP colocalizes with the Aurora-B/AIRK2 kinase on chromosomes and is overexpressed in tumour cells. Chromosoma. 110:65-74. Ambrosini, G., C. Adida, and D.C. Altieri. 1997. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 3:917-21. Amon, A. 1999. The spindle checkpoint. Curr Opin Genet Dev. 9:69-75. Amsterdam, A., K.C. Sadler, K. Lai, S. Farrington, R.T. Bronson, J.A. Lees, and N. Hopkins. 2004. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2:E139. Anand, S., S. Penrhyn-Lowe, and A.R. Venkitaraman. 2003. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 3:51-62. Andreassen, P.R., O.D. Lohez, F.B. Lacroix, and R.L. Margolis. 2001. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in Gl. Mol Biol Cell. 12:1315-28. Babu, J.R., K.B. Jeganathan, D.J. Baker, X. Wu, N. Kang-Decker, and J.M. van Deursen. 2003. Rae I is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 160:341-53. Baker, D.J., J. Chen, and J.M. van Deursen. 2005. The mitotic checkpoint in cancer and aging: what have mice taught us? Curr Opin Cell Biol. 17:583-9. Baker, D.J., K.B. Jeganathan, J.D. Cameron, M. Thompson, S. Juneja, A. Kopecka, R. Kumar, R.B. Jenkins, P.C. De Groen, P. Roche, and J.M. Van Deursen. 2004. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. Baker, D.J., K.B. Jeganathan, L. Malureanu, C. Perez-Terzic, A. Terzic, and J.M. van Deursen. 2006. Early aging-associated phenotypes in Bub3/Rael haploinsufficient mice. J Cell Biol. 172:529-40. Balmain, A. 2001. Cancer genetics: from Boveri and Mendel to microarrays. Nat Rev Cancer. 1:77-82. Baranovskaya, S., J.L. Soto, M. Perucho, and S.R. Malkhosyan. 2001. Functional significance of concomitant inactivation of hMLHI and hMSH6 in tumor cells of the microsatellite mutator phenotype. Proc NatlAcad Sci U S A. 98:15107-12. Bassing, C.H., and F.W. Alt. 2004. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst). 3:781-96. Bassing, C.H., H. Suh, D.O. Ferguson, K.F. Chua, J. Manis, M. Eckersdorff, M. Gleason, R. Bronson, C. Lee, and F.W. Alt. 2003. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 114:359-70. Bertwistle, D., M. Sugimoto, and C.J. Sherr. 2004. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 24:98596. Bischoff, J.R., L. Anderson, Y. Zhu, K. Mossie, L. Ng, B. Souza, B. Schryver, P. Flanagan, F. Clairvoyant, C. Ginther, C.S. Chan, M. Novotny, D.J. Slamon, and G.D. Plowman. 1998. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. Embo J. 17:3052-65. Boveri, T. .1914. Zur Frage der Entstehung maligner Tumoren. Fischer. Buffin, E., C. Lefebvre, J. Huang, M.E. Gagou, and R.E. Karess. 2005. Recruitment of Mad2 to the kinetochore requires the Rod/Zwl0 complex. Curr Biol. 15:856-61. Burds, A.A., A.S. Lutum, and P.K. Sorger. 2005. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl Acad Sci U S A. 102:11296-301. Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300-3. Carmena, M., and W.C. Earnshaw. 2003. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 4:842-54. Castillo, A.R., J.B. Meehl, G. Morgan, A. Schutz-Geschwender, and M. Winey. 2002. The yeast protein kinase Mpslp is required for assembly of the integral spindle pole body component Spc42p. J Cell Biol. 156:453-65. Chan, G.K., S.A. Jablonski, D.A. Starr, M.L. Goldberg, and T.J. Yen. 2000. Human Zwl0 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat Cell Biol. 2:944-7. Chan, G.K., S.A. Jablonski, V. Sudakin, J.C. Hittle, and T.J. Yen. 1999. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol. 146:941-54. Chang, E.H., M.A. Gonda, R.W. Ellis, E.M. Scolnick, and D.R. Lowy. 1982. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci U S A. 79:4848-52. Charrasse, S., M. Mazel, S. Taviaux, P. Berta, T. Chow, and C. Larroque. 1995. Characterization of the cDNA and pattern of expression of a new gene overexpressed in human hepatomas and colonic tumors. Eur JBiochem. 234:406-13. Chen, R.H. 2004. Phosphorylation and activation of Bub 1 on unattached chromosomes facilitate the spindle checkpoint. Embo J. 23:3113-21. Chen, Y., D.J. Riley, P.L. Chen, and W.H. Lee. 1997. HEC, a novel nuclear protein rich in leucine heptad repeats specifically involved in mitosis. Mol Cell Biol. 17:604956. Cheung, A.M., M.P. Hande, F. Jalali, M.S. Tsao, B. Skinnider, A. Hirao, J.P. McPherson, J. Karaskova, A. Suzuki, A. Wakeham, A. You-Ten, A. Elia, J. Squire, R. Bristow, R. Hakem, and T.W. Mak. 2002. Loss of Brca2 and p53 synergistically promotes genomic instability and deregulation of T-cell apoptosis. Cancer Res. 62:6194-204. Christmann, M., M.T. Tomicic, W.P. Roos, and B. Kaina. 2003. Mechanisms of human DNA repair: an update. Toxicology. 193:3-34. Cohen-Fix, O., J.M. Peters, M.W. Kirschner, and D. Koshland. 1996. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pdslp. Genes Dev. 10:3081-93. Coux, O., K. Tanaka, and A.L. Goldberg. 1996. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 65:801-47. Cross, S.M., C.A. Sanchez, C.A. Morgan, M.K. Schimke, S. Ramel, R.L. Idzerda, W.H. Raskind, and B.J. Reid. 1995. A p53-dependent mouse spindle checkpoint. Science. 267:1353-6. Cutts, S.M., K.J. Fowler, B.T. Kile, L.L. Hii, R.A. O'Dowd, D.F. Hudson, R. Saffery, P. Kalitsis, E. Earle, and K.H. Choo. 1999. Defective chromosome segregation, microtubule bundling and nuclear bridging in inner centromere protein gene (Incenp)-disrupted mice. Hum Mol Genet. 8:1145-55. Dai, W., Q. Wang, T. Liu, M. Swamy, Y. Fang, S. Xie, R. Mahmood, Y.M. Yang, M. Xu, and C.V. Rao. 2004. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. CancerRes. 64:440-5. Dash, B.C., and W.S. El-Deiry. 2004. Cell cycle checkpoint control mechanisms that can be disrupted in cancer. Methods Mol Biol. 280:99-161. De Antoni, A., C.G. Pearson, D. Cimini, J.C. Canman, V. Sala, L. Nezi, M. Mapelli, L. Sironi, M. Faretta, E.D. Salmon, and A. Musacchio. 2005. The Madl/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. CurrBiol. 15:214-25. De Wulf, P., A.D. McAinsh, and P.K. Sorger. 2003. Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev. 17:2902-21. DeLeo, A.B., G. Jay, E. Appella, G.C. Dubois, L.W. Law, and L.J. Old. 1979. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. ProcNatl Acad Sci US A. 76:2420-4. Dimri, G.P. 2005. What has senescence got to do with cancer? Cancer Cell. 7:505-12. Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635-45. Dolle, M.E., R.A. Busuttil, A.M. Garcia, S. Wijnhoven, E. van Drunen, L.J. Niedernhofer, G. van der Horst, J.H. Hoeijmakers, H. van Steeg, and J. Vijg. 2006. Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat Res. 596:22-35. Donehower, L.A., M. Harvey, B.L. Slagle, M.J. McArthur, C.A. Montgomery, Jr., J.S. Butel, and A. Bradley. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356:215-21. Douville, E.M., D.E. Afar, B.W. Howell, K. Letwin, L. Tannock, Y. Ben-David, T. Pawson, and J.C. Bell. 1992. Multiple cDNAs encoding the esk kinase predict transmembrane and intracellular enzyme isoforms. Mol Cell Biol. 12:2681-9. Draviam, V.M., S. Xie, and P.K. Sorger. 2004. Chromosome segregation and genomic stability. Curr Opin Genet Dev. 14:120-5. Duesberg, P., and R. Li. 2003. Multistep carcinogenesis: a chain reaction of aneuploidizations. Cell Cycle. 2:202-10. Ellis, C.A., and G. Clark. 2000. The importance of being K-Ras. Cell Signal. 12:425-34. Ewart-Toland, A., P. Briassouli, J.P. de Koning, J.H. Mao, J. Yuan, F. Chan, L. MacCarthy-Morrogh, B.A. Ponder, H. Nagase, J. Burn, S. Ball, M. Almeida, S. Linardopoulos, and A. Balmain. 2003. Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat Genet. 34:403-12. Fang, G. 2002. Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Biol Cell. 13:755-66. Fischer, M.G., S. Heeger, U. Hacker, and C.F. Lehner. 2004. The mitotic arrest in response to hypoxia and of polar bodies during early embryogenesis requires Drosophila Mpsl. CurrBiol. 14:2019-24. Fisk, H.A., C.P. Mattison, and M. Winey. 2003. Human Mpsl protein kinase is required for centrosome duplication and normal mitotic progression. Proc Natl Acad Sci U SA. 100:14875-80. Fisk, H.A., and M. Winey. 2001. The mouse Mpslp-like kinase regulates centrosome duplication. Cell. 106:95-104. Friday, B.B., and A.A. Adjei. 2005. K-ras as a target for cancer therapy. Biochim Biophys Acta. 1756:127-44. Fujiwara, T., M. Bandi, M. Nitta, E.V. Ivanova, R.T. Bronson, and D. Pellman. 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 437:1043-7. Fukasawa, K., T. Choi, R. Kuriyama, S. Rulong, and G.F. Vande Woude. 1996. Abnormal centrosome amplification in the absence of p53. Science. 271:1744-7. Furukawa, T., S. Azakami, H. Kurosawa, Y. Ono, Y. Ueda, and Y. Konno. 2003. Cystic partially differentiated nephroblastoma, embryonal rhabdomyosarcoma, and multiple congenital anomalies associated with variegated mosaic aneuploidy and premature centromere division: a case report. J PediatrHematol Oncol. 25:896-9. Fuss, J.O., and P.K. Cooper. 2006. DNA repair: dynamic defenders against cancer and aging. PLoS Biol. 4:e203. Futamura, M., H. Arakawa, K. Matsuda, T. Katagiri, S. Saji, Y. Miki, and Y. Nakamura. 2000. Potential role of BRCA2 in a mitotic checkpoint after phosphorylation by hBUBR1. CancerRes. 60:1531-5. Galipeau, P.C., D.S. Cowan, C.A. Sanchez, M.T. Barrett, M.J. Emond, D.S. Levine, P.S. Rabinovitch, and B.J. Reid. 1996. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. ProcNatl Acad Sci USA. 93:7081-4. Gardner, R.D., and D.J. Burke. 2000. The spindle checkpoint: two transitions, two pathways. Trends Cell Biol. 10:154-8. Gergely, F., V.M. Draviam, and J.W. Raff. 2003. The ch-TOG/XMAP215 protein is essential for spindle pole organization in human somatic cells. Genes Dev. 17:336-41. Giet, R., D. McLean, S. Descamps, M.J. Lee, J.W. Raff, C. Prigent, and D.M. Glover. 2002. Drosophila Aurora A kinase is required to localize D-TACC to centrosomes and to regulate astral microtubules. J Cell Biol. 156:437-51. Gilliland, W.D., S.M. Wayson, and R.S. Hawley. 2005. The meiotic defects of mutants in the Drosophila mpsl gene reveal a critical role of Mpsl in the segregation of achiasmate homologs. CurrBiol. 15:672-7. Green, R.A., and K.B. Kaplan. 2003. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J Cell Biol. 163:949-61. Gupta, A., S. Inaba, O.K. Wong, G. Fang, and J. Liu. 2003. Breast cancer-specific gene 1 interacts with the mitotic checkpoint kinase BubR1. Oncogene. 22:7593-9. Habu, T., S.H. Kim, J. Weinstein, and T. Matsumoto. 2002. Identification of a MAD2binding protein, CMT2, and its role in mitosis. Embo J. 21:6419-28. Haigis, K.M., J.G. Caya, M. Reichelderfer, and W.F. Dove. 2002. Intestinal adenomas can develop with a stable karyotype and stable microsatellites. Proc Natl Acad Sci USA. 99:8927-31. Halazonetis, T.D. 2004. Constitutively active DNA damage checkpoint pathways as the driving force for the high frequency of p53 mutations in human cancer. DNA Repair (Amst). 3:1057-62. Hall, A., C.J. Marshall, N.K. Spurr, and R.A. Weiss. 1983. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature. 303:396-400. Hanks, S., K. Coleman, S. Reid, A. Plaja, H. Firth, D. Fitzpatrick, A. Kidd, K. Mehes, R. Nash, N. Robin, N. Shannon, J. Tolmie, J. Swansbury, A. Irrthum, J. Douglas, and N. Rahman. 2004. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB 1B. Nat Genet. 36:1159-61. Hansemann, D. 1890. Ueber asymmetrische Zelltheilung in epithel Krebsen und deren biologische Bedeutung. Virschows Arch. Pathol.Anat. 119:299-326. Hardwick, K.G., E. Weiss, F.C. Luca, M. Winey, and A.W. Murray. 1996. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 273:953-6. Harris, S.L., and A.J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24:2899-908. Hartwell, L., S. Dutcher, J. Woood, and B. Garvik. 1982. The fidelity of mitotic chromosome reproduction in S. cerevisiae.Rec. Adv. Yeast Mol. Biol. 1. Hartwell, L.H., and T.A. Weinert. 1989. Checkpoints: controls that ensure the order of cell cycle events. Science. 246:629-34. Hassold, T., and P. Hunt. 2001. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2:280-91. Hauf, S., R.W. Cole, S. LaTerra, C. Zimmer, G. Schnapp, R. Walter, A. Heckel, J. Van Meel, C.L. Rieder, and J.M. Peters. 2003. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 161:281-94. He, X., M.H. Jones, M. Winey, and S. Sazer. 1998. Mphl, a member of the Mpsl-like family of dual specificity protein kinases, is required for the spindle checkpoint in S. pombe. J Cell Sci. 111:1635-47. Hernando, E., Z. Nahle, G. Juan, E. Diaz-Rodriguez, M. Alaminos, M. Hemann, L. Michel, V. Mittal, W. Gerald, R. Benezra, S.W. Lowe, and C. Cordon-Cardo. 2004. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 430:797-802. Hirota, T., N. Kunitoku, T. Sasayama, T. Marumoto, D. Zhang, M. Nitta, K. Hatakeyama, and H. Saya. 2003. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 114:585-98. Hogg, D., C. Guidos, D. Bailey, A. Amendola, T. Groves, J. Davidson, R. Schmandt, and G. Mills. 1994. Cell cycle dependent regulation of the protein kinase TTK. Oncogene. 9:89-96. Hoyt, M.A., L. Totis, and B.T. Roberts. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 66:507-17. Hwang, L.H., L.F. Lau, D.L. Smith, C.A. Mistrot, K.G. Hardwick, E.S. Hwang, A. Amon, and A.W. Murray. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 279:1041-4. Jablonski, S.A., G.K. Chan, C.A. Cooke, W.C. Earnshaw, and T.J. Yen. 1998. The hBUB 1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma. 107:386-96. Jacks, T., L. Remington, B.O. Williams, E.M. Schmitt, S. Halachmi, R.T. Bronson, and R.A. Weinberg. 1994. Tumor spectrum analysis in p53-mutant mice. CurrBiol. 4:1-7. Jackson, E.L., K.P. Olive, D.A. Tuveson, R. Bronson, D. Crowley, M. Brown, and T. Jacks. 2005. The differential effects of mutant p53 alleles on advanced murine lung cancer. CancerRes. 65:10280-8. Jackson, E.L., N. Willis, K. Mercer, R.T. Bronson, D. Crowley, R. Montoya, T. Jacks, and D.A. Tuveson. 2001. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15:3243-8. Jasin, M. 2002. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 21:8981-93. Jiang, Y., Y. Zhang, E. Lees, and W. Seghezzi. 2003. AuroraA overexpression overrides the mitotic spindle checkpoint triggered by nocodazole, a microtubule destabilizer. Oncogene. 22:8293-301. Jones, M.H., J.B. Bachant, A.R. Castillo, T.H. Giddings, Jr., and M. Winey. 1999. Yeast Damlp is required to maintain spindle integrity during mitosis and interacts with the Mpslp kinase. Mol Biol Cell. 10:2377-91. Jonkers, J., R. Meuwissen, H. van der Gulden, H. Peterse, M. van der Valk, and A. Berns. 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 29:418-25. Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277-82. Kallio, M., J. Weinstein, J.R. Daum, D.J. Burke, and G.J. Gorbsky. 1998. Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events. J Cell Biol. 141:1393-406. Kallio, M.J., M. Nieminen, and J.E. Eriksson. 2001. Human inhibitor of apoptosis protein (IAP) survivin participates in regulation of chromosome segregation and mitotic exit. Faseb J. 15:2721-3. Kamijo, T., S. Bodner, E. van de Kamp, D.H. Randle, and C.J. Sherr. 1999. Tumor spectrum in ARF-deficient mice. CancerRes. 59:2217-22. Kamijo, T., J.D. Weber, G. Zambetti, F. Zindy, M.F. Roussel, and C.J. Sherr. 1998. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. ProcNatl Acad Sci US A. 95:8292-7. Kamijo, T., F. Zindy, M.F. Roussel, D.E. Quelle, J.R. Downing, R.A. Ashmun, G. Grosveld, and C.J. Sherr. 1997. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 91:649-59. Kaplan, K.B., A.A. Burds, J.R. Swedlow, S.S. Bekir, P.K. Sorger, and I.S. Nathke. 2001. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 3:429-32. Kapoor, T.M., T.U. Mayer, M.L. Coughlin, and T.J. Mitchison. 2000. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol. 150:975-88. Kasai, T., Y. Iwanaga, H. Iha, and K.T. Jeang. 2002. Prevalent loss of mitotic spindle checkpoint in adult T-cell leukemia confers resistance to microtubule inhibitors. J Biol Chem. 277:5187-93. Kastan, M.B., and J. Bartek. 2004. Cell-cycle checkpoints and cancer. Nature. 432:31623. Kim, C.F., E.L. Jackson, A.E. Woolfenden, S. Lawrence, I. Babar, S. Vogel, D. Crowley, R.T. Bronson, and T. Jacks. 2005a. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 121:823-35. Kim, H.S., K.H. Park, S.A. Kim, J. Wen, S.W. Park, B. Park, C.W. Gham, W.J. Hyung, S.H. Noh, H.K. Kim, and S.Y. Song. 2005b. Frequent mutations of human Mad2, but not Bub , in gastric cancers cause defective mitotic spindle checkpoint. Mutat Res. 578:187-201. Kops, G.J., B.A. Weaver, and D.W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer.5:773-85. Lanni, J.S., and T. Jacks. 1998. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol. 18:1055-64. Latonen, L., and M. Laiho. 2005. Cellular UV damage responses--functions of tumor suppressor p53. Biochim Biophys Acta. 1755:71-89. Lee, H. 2003. Impaired phosphorylation and mis-localization of Bub 1 and BubR1 are responsible for the defective mitotic checkpoint function in Brca2-mutant thymic lymphomas. Exp Mol Med. 35:448-53. Lee, H., A.H. Trainer, L.S. Friedman, F.C. Thistlethwaite, M.J. Evans, B.A. Ponder, and A.R. Venkitaraman. 1999. Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol Cell. 4:1-10. Lee, M., M.J. Daniels, and A.R. Venkitaraman. 2003. Phosphorylation of BRCA2 by the Polo-like kinase Plkl is regulated by DNA damage and mitotic progression. Oncogene. Leisti, J.T., K.O. Raivio, M.H. Rapola, E.J. Saksela, and P.P. Aula. 1974. The phenotype of human triploidy. Birth Defects Orig Artic Ser. 10:248-53. Leng, M., D.W. Chan, H. Luo, C. Zhu, J. Qin, and Y. Wang. 2006. MPS1-dependent mitotic BLM phosphorylation is important for chromosome stability. ProcNatl Acad Sci USA. 103:11485-90. Lengauer, C., K.W. Kinzler, and B. Vogelstein. 1997. Genetic instability in colorectal cancers. Nature. 386:623-7. Levine, A.J. 1997. p53, the cellular gatekeeper for growth and division. Cell. 88:323-31. Li, R., and A.W. Murray. 1991. Feedback control of mitosis in budding yeast. Cell. 66:519-31. Li, X., and R.B. Nicklas. 1995. Mitotic forces control a cell-cycle checkpoint. Nature. 373:630-2. Li, Y., J. Bachant, A.A. Alcasabas, Y. Wang, J. Qin, and S.J. Elledge. 2002. The mitotic spindle is required for loading of the DASH complex onto the kinetochore. Genes Dev. 16:183-97. Li, Y., and R. Benezra. 1996. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 274:246-8. Liao, M.J., X.X. Zhang, R. Hill, J. Gao, M.B. Qumsiyeh, W. Nichols, and T. Van Dyke. 1998. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol. 18:3495-501. Lin, A.W., M. Barradas, J.C. Stone, L. van Aelst, M. Serrano, and S.W. Lowe. 1998. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 12:3008-19. Lin, H.R., N.S. Ting, J. Qin, and W.H. Lee. 2003. M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2P/CAF complex. JBiol Chem. 278:35979-87. Linzer, D.I., and A.J. Levine. 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 17:43-52. Liu, S.T., G.K. Chan, J.C. Hittle, G. Fujii, E. Lees, and T.J. Yen. 2003. Human MPS 1 Kinase Is Required for Mitotic Arrest Induced by the Loss of CENP-E from Kinetochores. Mol Biol Cell. 14:1638-51. Loeb, L.A. 1991. Mutator phenotype may be required for multistage carcinogenesis. CancerRes. 51:3075-9. Loeb, L.A., and K.C. Cheng. 1990. Errors in DNA synthesis: a source of spontaneous mutations. Mutat Res. 238:297-304. Loeb, L.A., K.R. Loeb, and J.P. Anderson. 2003. Multiple mutations and cancer. Proc NatlAcadSci USA. 100:776-81. Lowe, S.W., and C.J. Sherr. 2003. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 13:77-83. Luo, X., Z. Tang, J. Rizo, and H. Yu. 2002. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Madl or Cdc20. Mol Cell. 9:59-71. Ma, W., D. Zhang, Y. Hou, Y.H. Li, Q.Y. Sun, X.F. Sun, and W.H. Wang. 2005. Reduced expression of MAD2, BCL2, and MAP kinase activity in pig oocytes after in vitro aging are associated with defects in sister chromatid segregation during meiosis II and embryo fragmentation after activation. Biol Reprod.72:37383. Malkinson, A.M. 1998. Molecular comparison of human and mouse pulmonary adenocarcinomas. Exp Lung Res. 24:541-55. Mapelli, M., F.V. Filipp, G. Rancati, L. Massimiliano, L. Nezi, G. Stier, R.S. Hagan, S. Confalonieri, S. Piatti, M. Sattler, and A. Musacchio. 2006. Determinants of conformational dimerization of Mad2 and its inhibition by p31 comet. Embo J. 25:1273-84. Marshall, C.J. 1996. Ras effectors. Curr Opin Cell Biol. 8:197-204. Martin-Lluesma, S., V.M. Stucke, and E.A. Nigg. 2002. Role of Hecl in spindle checkpoint signaling and kinetochore recruitment of Madl/Mad2. Science. 297:2267-70. Martinez-Exposito, M.J., K.B. Kaplan, J. Copeland, and P.K. Sorger. 1999. Retention of the BUB3 checkpoint protein on lagging chromosomes. Proc Natl Acad Sci US A. 96:8493-8. Marx, J. 2001. Cell biology. Do centrosome abnormalities lead to cancer? Science. 292:426-9. Masuda, A., and T. Takahashi. 2002. Chromosome instability in human lung cancers: possible underlying mechanisms and potential consequences in the pathogenesis. Oncogene. 21:6684-97. Matsuura, S., Y. Matsumoto, K. Morishima, H. Izumi, H. Matsumoto, E. Ito, K. Tsutsui, J. Kobayashi, H. Tauchi, Y. Kajiwara, S. Hama, K. Kurisu, H. Tahara, M. Oshimura, K. Komatsu, T. Ikeuchi, and T. Kajii. 2006. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 140:35867. McAinsh, A.D., J.D. Tytell, and P.K. Sorger. 2003. Structure, function, and regulation of budding yeast kinetochores. Annu Rev Cell Dev Biol. 19:519-39. McKeller, R.N., J.L. Fowler, J.J. Cunningham, N. Warner, R.J. Smeyne, F. Zindy, and S.X. Skapek. 2002. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. ProcNatl Acad Sci U S A. 99:384853. Mei, J., X. Huang, and P. Zhang. 2001. Securin is not required for cellular viability, but is required for normal growth of mouse embryonic fibroblasts. Curr Biol. 11:1197201. Meraldi, P., V.M. Draviam, and P.K. Sorger. 2004. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 7:45-60. Meraldi, P., R. Honda, and E.A. Nigg. 2002. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. Embo J. 21:483-92. Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V. Murty, and R. Benezra. 2001. MAD2 haploinsufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 409:355-9. Mills, G.B., R. Schmandt, M. McGill, A. Amendola, M. Hill, K. Jacobs, C. May, A.M. Rodricks, S. Campbell, and D. Hogg. 1992. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. JBiol Chem. 267:16000-6. Miranda, J.J., P. De Wulf, P.K. Sorger, and S.C. Harrison. 2005. The yeast DASH complex forms closed rings on microtubules. Nat Struct Mol Biol. 12:138-43. Miyoshi, Y., K. Iwao, C. Egawa, and S. Noguchi. 2001. Association of centrosomal kinase STK1 5/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer.92:370-3. Mollinedo, F., and C. Gajate. 2003. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 8:413-50. Moore, L., S. Venkatachalam, H. Vogel, J.C. Watt, C.L. Wu, H. Steinman, S.N. Jones, and L.A. Donehower. 2003. Cooperativity ofpl9ARF, Mdm2, and p53 in murine tumorigenesis. Oncogene. 22:7831-7. Morales, J.C., S. Franco, M.M. Murphy, C.H. Bassing, K.D. Mills, M.M. Adams, N.C. Walsh, J.P. Manis, G.Z. Rassidakis, F.W. Alt, and P.B. Carpenter. 2006. 53BP1 and p53 synergize to suppress genomic instability and lymphomagenesis. Proc Natl Acad Sci U S A. 103:3310-5. Morgan, D.O. 1999. Regulation of the APC and the exit from mitosis. Nat Cell Biol. 1:E47-53. Narayan, S., and D. Roy. 2003. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer.2:41. Nezi, L., G. Rancati, A. De Antoni, S. Pasqualato, S. Piatti, and A. Musacchio. 2006. Accumulation of Mad2-Cdc20 complex during spindle checkpoint activation requires binding of open and closed conformers of Mad2 in Saccharomyces cerevisiae. J Cell Biol. 174:39-51. Nicklas, R.B., J.C. Waters, E.D. Salmon, and S.C. Ward. 2001. Checkpoint signals in grasshopper meiosis are sensitive to microtubule attachment, but tension is still essential. J Cell Sci. 114:4173-83. Nigg, E.A. 2002. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer.2:815-25. Nowak, M.A., N.L. Komarova, A. Sengupta, P.V. Jallepalli, M. Shih le, B. Vogelstein, and C. Lengauer. 2002. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci U S A. 99:16226-31. Nowell, P.C. 1976. The clonal evolution of tumor cell populations. Science. 194:23-8. Ota, T., S. Suto, H. Katayama, Z.B. Han, F. Suzuki, M. Maeda, M. Tanino, Y. Terada, and M. Tatsuka. 2002. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability. Cancer Res. 62:5168-77. Palmero, I., C. Pantoja, and M. Serrano. 1998. pl9ARF links the tumour suppressor p53 to Ras. Nature. 395:125-6. Pei, L., and S. Melmed. 1997. Isolation and characterization of a pituitary tumortransforming gene (PTTG). Mol Endocrinol. 11:433-41. Pellman, D. 2001. Cancer. A CINtillating new job for the APC tumor suppressor. Science. 291:2555-6. Perez-Mancera, P.A., and D.A. Tuveson. 2005. Physiological analysis of oncogenic kras. Methods Enzymol. 407:676-90. Pierotti, M.A., and T.A. Dragani. 1992. Genetics and cancer. Curr Opin Oncol. 4:127-33. Polager, S., Y. Kalma, E. Berkovich, and D. Ginsberg. 2002. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene. 21:437-46. Pomerantz, J., N. Schreiber-Agus, N.J. Liegeois, A. Silverman, L. Alland, L. Chin, J. Potes, K. Chen, I. Orlow, H.W. Lee, C. Cordon-Cardo, and R.A. DePinho. 1998. The Ink4a tumor suppressor gene product, pl9Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 92:713-23. Poss, K.D., A. Nechiporuk, K.F. Stringer, C. Lee, and M.T. Keating. 2004. Germ cell aneuploidy in zebrafish with mutations in the mitotic checkpoint gene mps 1. Genes Dev. 18:1527-32. Poss, K.D., L.G. Wilson, and M.T. Keating. 2002. Heart regeneration in zebrafish. Science. 298:2188-90. Purdie, C.A., D.J. Harrison, A. Peter, L. Dobbie, S. White, S.E. Howie, D.M. Salter, C.C. Bird, A.H. Wyllie, M.L. Hooper, and et al. 1994. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 9:603-9. Putkey, F.R., T. Cramer, M.K. Morphew, A.D. Silk, R.S. Johnson, J.R. McIntosh, and D.W. Cleveland. 2002. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell. 3:351-65. Quelle, D.E., F. Zindy, R.A. Ashmun, and C.J. Sherr. 1995. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 83:993-1000. Rao, C.V., Y.M. Yang, M.V. Swamy, T. Liu, Y. Fang, R. Mahmood, M. Jhanwar-Uniyal, and W. Dai. 2005. Colonic tumorigenesis in BubRl+/-ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. ProcNatl Acad Sci USA. 102:4365-70. Rask, K., A. Nilsson, M. Brannstrom, P. Carlsson, P. Hellberg, P.O. Janson, L. Hedin, and K. Sundfeldt. 2003. Wnt-signalling pathway in ovarian epithelial tumours: increased expression of beta-catenin and GSK3beta. Br J Cancer. 89:1298-304. Reimann, J.D., B.E. Gardner, F. Margottin-Goguet, and P.K. Jackson. 2001. Emil regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev. 15:3278-85. Ren, B., H. Cam, Y. Takahashi, T. Volkert, J. Terragni, R.A. Young, and B.D. Dynlacht. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 16:245-56. Rieder, C.L., R.W. Cole, A. Khodjakov, and G. Sluder. 1995. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 130:941-8. Rieder, C.L., and A. Khodjakov. 2003. Mitosis through the microscope: advances in seeing inside live dividing cells. Science. 300:91-6. Rieder, C.L., and H. Maiato. 2004. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 7:637-51. Rieder, C.L., A. Schultz, R. Cole, and G. Sluder. 1994. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 127:1301-10. Saez, C., M.A. Japon, F. Ramos-Morales, F. Romero, D.I. Segura, M. Tortolero, and J.A. Pintor-Toro. 1999. hpttg is over-expressed in pituitary adenomas and other primary epithelial neoplasias. Oncogene. 18:5473-6. Sancar, A., L.A. Lindsey-Boltz, K. Unsal-Kacmaz, and S. Linn. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 73:39-85. Sato, N., K. Mizumoto, M. Nakamura, K. Nakamura, M. Kusumoto, H. Niiyama, T. Ogawa, and M. Tanaka. 1999. Centrosome abnormalities in pancreatic ductal carcinoma. Clin Cancer Res. 5:963-70. Scarbrough, P.R., W.H. Finley, and S.C. Finley. 1982. A review of trisomies 21, 18, and 13. Ala J Med Sci. 19:174-88. Schutz, A.R., and M. Winey. 1998. New alleles of the yeast MPS1 gene reveal multiple requirements in spindle pole body duplication. Mol Biol Cell. 9:759-74. Seeley, T.W., L. Wang, and J.Y. Zhen. 1999. Phosphorylation of human MAD1 by the BUB 1 kinase in vitro. Biochem Biophys Res Commun. 257:589-95. Serrano, M., A.W. Lin, M.E. McCurrach, D. Beach, and S.W. Lowe. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and pl6INK4a. Cell. 88:593-602. Shah, J.V., and D.W. Cleveland. 2000. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell. 103:997-1000. Sherr, C.J. 2001. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2:731-7. Shi, Q., and R.W. King. 2005. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature.437:1038-42. Shibata, D., M.A. Peinado, Y. Ionov, S. Malkhosyan, and M. Perucho. 1994. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet. 6:273-81. Shichiri, M., K. Yoshinaga, H. Hisatomi, K. Sugihara, and Y. Hirata. 2002. Genetic and epigenetic inactivation of mitotic checkpoint genes hBUB1 and hBUBR1 and their relationship to survival. Cancer Res. 62:13-7. Shimizu, K., M. Goldfarb, Y. Suard, M. Perucho, Y. Li, T. Kamata, J. Feramisco, E. Stavnezer, J. Fogh, and M.H. Wigler. 1983. Three human transforming genes are related to the viral ras oncogenes. ProcNatl Acad Sci U S A. 80:2112-6. Sieber, O.M., K. Heinimann, P. Gorman, H. Lamlum, M. Crabtree, C.A. Simpson, D. Davies, K. Neale, S.V. Hodgson, R.R. Roylance, R.K. Phillips, W.F. Bodmer, and I.P. Tomlinson. 2002. Analysis of chromosomal instability in human colorectal adenomas with two mutational hits at APC. ProcNatl Acad Sci U S A. 99:169105. Sironi, L., M. Mapelli, S. Knapp, A. De Antoni, K.T. Jeang, and A. Musacchio. 2002. Crystal structure of the tetrameric Madl-Mad2 core complex: implications of a 'safety belt' binding mechanism for the spindle checkpoint. Embo J. 21:2496-506. Sironi, L., M. Melixetian, M. Faretta, E. Prosperini, K. Helin, and A. Musacchio. 2001. Mad2 binding to Madl and Cdc20, rather than oligomerization, is required for the spindle checkpoint. Embo J. 20:6371-82. Stern, B.M., and A.W. Murray. 2001. Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. CurrBiol. 11:1462-7. Stoler, D.L., N. Chen, M. Basik, M.S. Kahlenberg, M.A. Rodriguez-Bigas, N.J. Petrelli, and G.R. Anderson. 1999. The onset and extent of genomic instability in sporadic colorectal tumor progression. ProcNatl Acad Sci U S A. 96:15121-6. Stucke, V.M., C. Baumann, and E.A. Nigg. 2004. Kinetochore localization and microtubule interaction of the human spindle checkpoint kinase Mps 1. Chromosoma. 113:1-15. Stucke, V.M., H.H. Sillje, L. Arnaud, and E.A. Nigg. 2002. Human Mpsl kinase is required for the spindle assembly checkpoint but not for centrosome duplication. Embo J. 21:1723-32. Su, L.K., K.W. Kinzler, B. Vogelstein, A.C. Preisinger, A.R. Moser, C. Luongo, K.A. Gould, and W.F. Dove. 1992. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 256:668-70. Sudakin, V., G.K. Chan, and T.J. Yen. 2001. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 154:925-36. Sugimoto, M., M.L. Kuo, M.F. Roussel, and C.J. Sherr. 2003. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 11:415-24. Tago, K., S. Chiocca, and C.J. Sherr. 2005. Sumoylation induced by the Arf tumor suppressor: a p53-independent function. ProcNatl Acad Sci U S A. 102:7689-94. Takahashi, T., B. Sano, T. Nagata, H. Kato, Y. Sugiyama, K. Kunieda, M. Kimura, Y. Okano, and S. Saji. 2003. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. CancerSci. 94:148-52. Tanaka, K., J. Nishioka, K. Kato, A. Nakamura, T. Mouri, C. Miki, M. Kusunoki, and T. Nobori. 2001. Mitotic checkpoint protein hsMAD2 as a marker predicting liver metastasis of human gastric cancers. Jpn J CancerRes. 92:952-8. Tanaka, T.U., N. Rachidi, C. Janke, G. Pereira, M. Galova, E. Schiebel, M.J. Stark, and K. Nasmyth. 2002. Evidence that the Ipll-Slil5 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 108:317-29. Tang, Z., R. Bharadwaj, B. Li, and H. Yu. 2001. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubRl. Dev Cell. 1:227-37. Taparowsky, E., K. Shimizu, M. Goldfarb, and M. Wigler. 1983. Structure and activation of the human N-ras gene. Cell. 34:581-6. Tavormina, P.A., and D.J. Burke. 1998. Cell cycle arrest in cdc20 mutants of Saccharomyces cerevisiae is independent of Ndc 10p and kinetochore function but requires a subset of spindle checkpoint genes. Genetics. 148:1701-13. Taylor, S.S., E. Ha, and F. McKeon. 1998. The human homologue of Bub3 is required for kinetochore localization of Bub 1 and a Mad3/Bubl -related protein kinase. J Cell Biol. 142:1-11. Taylor, S.S., and F. McKeon. 1997. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 89:727-35. Temme, A., M. Rieger, F. Reber, D. Lindemann, B. Weigle, P. Diestelkoetter-Bachert, G. Ehninger, M. Tatsuka, Y. Terada, and E.P. Rieber. 2003. Localization, dynamics, and function of survivin revealed by expression of functional survivinDsRed fusion proteins in the living cell. Mol Biol Cell. 14:78-92. Thomas, N.S., S. Ennis, A.J. Sharp, M. Durkie, T.J. Hassold, A.R. Collins, and P.A. Jacobs. 2001. Maternal sex chromosome non-disjunction: evidence for X chromosome-specific risk factors. Hum Mol Genet. 10:243-50. Tighe, A., V.L. Johnson, M. Albertella, and S.S. Taylor. 2001. Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2:609-14. Uhlmann, F. 2003. Chromosome cohesion and separation: from men and molecules. Curr Biol. 13:R104-14. Uren, A.G., L. Wong, M. Pakusch, K.J. Fowler, F.J. Burrows, D.L. Vaux, and K.H. Choo. 2000. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. CurrBiol. 10:1319-28. Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substratespecific activators of APC-dependent proteolysis. Science. 278:460-3. Wang, J.Y., Z.L. Lei, C.L. Nan, S. Yin, J. Liu, Y. Hou, Y.L. Li, D.Y. Chen, and Q.Y. Sun. 2006. RNA Interference as a tool to study the function of MAD2 in mouse oocyte meiotic maturation. Mol Reprod Dev. Wang, Q., T. Liu, Y. Fang, S. Xie, X. Huang, R. Mahmood, G. Ramaswamy, K.M. Sakamoto, Z. Darzynkiewicz, M. Xu, and W. Dai. 2004a. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 103:1278-85. Wang, R.H., H. Yu, and C.X. Deng. 2004b. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. ProcNatlAcadSci USA. 101:1710813. Wang, X., J.R. Babu, J.M. Harden, S.A. Jablonski, M.H. Gazi, W.L. Lingle, P.C. de Groen, T.J. Yen, and J.M. van Deursen. 2001. The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins. JBiol Chem. 276:26559-67. Wang, X., D.Y. Jin, R.W. Ng, H. Feng, Y.C. Wong, A.L. Cheung, and S.W. Tsao. 2002. Significance of MAD2 expression to mitotic checkpoint control in ovarian cancer cells. CancerRes. 62:1662-8. Warren, C.D., D.M. Brady, R.C. Johnston, J.S. Hanna, K.G. Hardwick, and F.A. Spencer. 2002. Distinct chromosome segregation roles for spindle checkpoint proteins. Mol Biol Cell. 13:3029-41. Wassmann, K., and R. Benezra. 2001. Mitotic checkpoints: from yeast to cancer. Curr Opin Genet Dev. 11:83-90. Wassmann, K., V. Liberal, and R. Benezra. 2003a. Mad2 phosphorylation regulates its association with Madl and the APC/C. Embo J. 22:797-806. Wassmann, K., T. Niault, and B. Maro. 2003b. Metaphase I arrest upon activation of the Mad2-dependent spindle checkpoint in mouse oocytes. CurrBiol. 13:1596-608. Waters, J.C., R.H. Chen, A.W. Murray, and E.D. Salmon. 1998. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol. 141:1181-91. Weaver, B.A., Z.Q. Bonday, F.R. Putkey, G.J. Kops, A.D. Silk, and D.W. Cleveland. 2003. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol. 162:551-63. Weber, J.D., J.R. Jeffers, J.E. Rehg, D.H. Randle, G. Lozano, M.F. Roussel, C.J. Sherr, and G.P. Zambetti. 2000. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 14:2358-65. Weber, J.D., L.J. Taylor, M.F. Roussel, C.J. Sherr, and D. Bar-Sagi. 1999. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1:20-6. Wei, J.H., Y.F. Chou, Y.H. Ou, Y.H. Yeh, S.W. Tyan, T.P. Sun, C.Y. Shen, and S.Y. Shieh. 2005. TTK/hMps 1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. JBiol Chem. 280:7748-57. Wei, K., R. Kucherlapati, and W. Edelmann. 2002. Mouse models for human DNA mismatch-repair gene defects. Trends Mol Med. 8:346-53. Weinert, T.A., and L.H. Hartwell. 1988. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 241:317-22. Weiss, E., and M. Winey. 1996. The Saccharomyces cerevisiae spindle pole body duplication gene MPS 1 is part of a mitotic checkpoint. J Cell Biol. 132:111-23. West, S.C. 2003. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 4:435-45. Wilkens, L., P. Flemming, M. Gebel, J. Bleck, C. Terkamp, L. Wingen, H. Kreipe, and B. Schlegelberger. 2004. Induction of aneuploidy by increasing chromosomal instability during dedifferentiation of hepatocellular carcinoma. ProcNatl Acad Sci USA. 101:1309-14. Winey, M., L. Goetsch, P. Baum, and B. Byers. 1991. MPSI and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol. 114:745-54. Winey, M., and B.J. Huneycutt. 2002. Centrosomes and checkpoints: the MPS I family of kinases. Oncogene. 21:6161-9. Yoon, D.S., R.P. Wersto, W. Zhou, F.J. Chrest, E.S. Garrett, T.K. Kwon, and E. Gabrielson. 2002. Variable levels of chromosomal instability and mitotic spindle checkpoint defects in breast cancer. Am JPathol.161:391-7. Yu, H. 2002. Regulation of APC-Cdc20 by the spindle checkpoint. Curr Opin Cell Biol. 14:706-14. Yu, R., W. Lu, J. Chen, C.J. McCabe, and S. Melmed. 2003. Overexpressed pituitary tumor-transforming gene causes aneuploidy in live human cells. Endocrinology. 144:4991-8. Zhou, H., J. Kuang, L. Zhong, W.L. Kuo, J.W. Gray, A. Sahin, B.R. Brinkley, and S. Sen. 1998. Tumour amplified kinase STKl5/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 20:189-93. Zindy, F., C.M. Eischen, D.H. Randle, T. Kamijo, J.L. Cleveland, C.J. Sherr, and M.F. Roussel. 1998. Myc signaling via the ARF tumor suppressor regulates p53dependent apoptosis and immortalization. Genes Dev. 12:2424-33. Chapter 2 A mutation in Mpsl causes chromosomal instability and accelerates tumorigenesis in mice Note: All figures are the author's own work except for supplemental figure 3 where the metaphase spreads and spectral karyotyping was performed by Alexei Protopopov and Elena Ivanova of Ron Depinho's group. Roderick Bronson provided histology analysis. The alignment of the vertebrate Mps 1 sequences was kindly provided by Esther Rheinbay. Abstract Many human tumors exhibit chromosomal instability (CIN). One route to CIN is inaccurate chromosome segregation to daughter cells during mitotis. The spindle checkpoint is a key surveillance system conserved through all eukaryotes that acts to ensure faithful sister chromatid segregation. Since 1914, when Boveri first observed abnormal chromosome numbers in cancer, an age old question has plagued the field of cancer biology: is chromosomal instability (CIN) simply a consequence of the steps leading to cancer or can CIN also function as a causal agent? The argument that CIN causes cancer remains largely circumstantial. Here, I describe the generation of a conditional hypomorphic mouse model of the spindle checkpoint gene Mpsl that induces a CIN phenotype in mice. My aim is to determine if elevating CIN can promote cancer a prioribased on the data from this study. I argue that CIN does function in tumorigenesis as a causal agent but requires the inactivation of the p53 and possibly other pathways. I show that a hypomorphic MpslA allele induces CIN and is sufficient for lymphomagenesis, although tumor incidence is sporadic. However, CIN functions as a robust facilitator of tumorigenesis when p53 is also inactivated. The MpslA mutation induces tumorigenesis in p53 heterozygous knockout thymocytes with full penetrance. MpslA accelerates tumor onset in both p53- heterozygous and p53-null thymocytes suggesting MpslA-CIN are promoting additional events that contribute to tumorigenesis. In summary, CIN caused by a weakened checkpoint is sufficient to facilitate cancer when the p53 pathway is also impaired. Introduction Genomic instability is a hallmark of many human tumors (Jallepalli and Lengauer, 2001). Cell cycle checkpoints exist to protect the genome and prevent events that might promote carcinogenesis (Hartwell and Kastan, 1994; Hartwell and Weinert, 1989). The spindle assembly checkpoint is one such checkpoint that acts during mitosis to ensure accurate chromosome segregation (Draviam et al., 2004; Taylor et al., 2004). The spindle checkpoint delays the onset of anaphase until all sister chromatid pairs have achieved proper bipolar attachment to the mitotic spindle. Checkpoint genes are conserved from yeast to man and include the MAD and BUB genes, and MPS1 (Amon, 1999; Hoyt et al., 1991; Li and Murray, 1991; Weiss and Winey, 1996). Inactivation of the spindle checkpoint is thought to cause chromosomal instability (CIN) and lead to aneuploid cells (Draviam et al., 2004; Kops et al., 2005). CIN has been proposed to act as a mutator, facilitating tumorigenesis by increasing the rate at which growth-promoting mutations accumulate in cells (Duesberg and Li, 2003; Loeb, 1991; Loeb et al., 2003; Nowak et al., 2002). It was Theodore Boveri who initially hypothesized that abnormal chromosome number plays a causal role in cancer (Boveri, 1914). However, a clear demonstration that CIN can be a cause of cancer and not merely the result of events leading to tumor formation is lacking. Will CIN resulting from abrogation of the spindle checkpoint cause cancer? While a disease called mosaic variegated aneuploidy clearly implicates mutations in BUBR1 (Hanks et al., 2004), few other spindle checkpoint mutations have been found in human cancers (Cahill et al., 1998; Hempen et al., 2003; Kim et al., 2005; Langerod et al., 2003; Matsuura et al., 2006; Saeki et al., 2002; Wang et al., 2004b). The spindle checkpoint is essential in metazoans and all checkpoint gene knockouts in mice are embryonic lethal (Dobles et al., 2000; Kalitsis et al., 2000; Wang et al., 2004a). Thus, we sought to create a viable CIN prone mouse model by introducing a conditional partial loss-of-function mutation in the spindle checkpoint gene MPS 1 into mice to test if CIN explicitly can cause cancer in vivo. This mutation in Mps1 acts as an accelerator of T cell tumorigenesis in a p53 heterozygous background and suggests that CIN is likely more involved in tumor progression than tumor initiation. Furthermore, the spindle checkpoint is shown to be absolutely essential for viability and provides additional explanation for the selection against full loss-of function spindle checkpoint mutations in human cancer. Results Mps1 is a serine/threonine kinase whose catalytic domain is essential for checkpoint signaling (Fisk et al., 2004).We found through an alignment of the available MPS 1 vertebrate sequences that the most conserved regions of the coding sequence are located in the N and C terminal regions, with 22% and 44% identity respectively (Fig. 2.1 A, C). The kinase domain is located in the C-terminal region. To generate a conditional hypomorphic mouse model of the spindle checkpoint gene Mps 1 and determine if elevating CIN can promote tumorigenesis a priori,we deleted the 107 amino acid region encoded by exons 2 and 3 of MPS1 in mouse embryonic stem cells using Cre/loxP technology and standard gene targeting methods and introduced this mutation (denoted as MpslA) into mice (Fig 2.1B, D). MpslA maintains the reading frame and results in a shortened mRNA (Fig 2.1D). An analogous A mutation was introduced to a human Mps 1-GFP fusion protein (hMps 1A-GFP) to determine localization of the mutant protein during mitosis. In HeLa cells transfected with hMpslA-GFP or wildtype Mpsl- Figure 2.1 I Kinetochore binding domain Kinase domain I 50 aa m 1 Mps1 2%(58%) 1 2 00 520 856 deleted in MpslA mMpsl Genomic Locus 6 13 5'UTR - S- rna -I 2 5 4 N 15 19 16 20 22 1718 21 3'UTR 8 8 10 12 13 MN n E E . .. FLOX allele Aallele -. - i 3' 665 -p p1rctbe 5' 664 bp p obe ~A ME a MEN IN. Wt ~ i i; A/+ A/+ f/+ f/+ Mpsl Gene Targeting Vector Ncol 5'UT S6 23 SphI - Nptl 11 7,8%, Not -- Noti Nhel HSV-TK NeoR I IoxP FRT - n 1L Mpsl+/+ MpslA/+ Wt A IoxP FRT The alignment of vertebrate Mpsl L8 h~s 745 aa 857 5( aa I A as 53-158 hMps 1-GFP IiI :Y AD hCenDB-mRFP u. - A-I A ' l t Rn ... ... .. ... ... ... . . l R r A POF E 7 ET . Figure 2.1: MpslA is a truncationmutation that retains kinetochore localization. (A) A schematic of mouse Mps protein divided into three regions based on the sequence alignment of nine vertebrate Mpsl proteins showing % similarity (red) and % identity (green). The kinetochore binding domain and the kinase domain are also shown. MpsA is a truncated protein where residues 47-154 are deleted. (B) The MpslA conditional targeting vector is shown. LoxP sites were introduced between exons 2 and 3 of the Mpsl genomic locus. (C) The alignment of the first 280 residues of Mpsl from nine vertebrate species. (D) Southern blotting of tail DNA from founder mice harboring conditional and conventional MpslA alleles to show germline transmission. (E) RT-PCR for Mpsl from Mpsl +/' and Mps l/A thymocytes showing the MpslA allele results in a shortened mRNA. (F) hMps 1A-GFP localizes to kinetochores and responds to nocodazole. (top) A schematic of hMpslA-GFP, a truncation of human Mpsl protein removing residues 53-158 fused to GFP. (middle) Representative images of mitotic HeLa cells transfected with either hMps lA-GFP or hMps 1-GFP (in green) and H2B-RFP (blue). (bottom) A representative HeLa cell transfected with hMps1A-GFP (green) and hCenpB-RFP (red) arrested in mitosis by nocodazole treatment. Figure 2.2 A Genotype % expected % Mpsl+/+ Mpsl +/ A Mpsl A/A 38% 62% 0% 25% 50% 25% Genotype Mpsl+/+ Mpsl1+/ A N.D. reabsorbed embryos n= 237 % 40% 41% 19% n=96 litter# = 33 litter# = 10 = L wtMpsIA -- - Iwe wt Figure 2.2: MpslA causes embryonic lethality in mice before embryonic day 10.5. (A) Live births from Mpsl+/A crosses. (B) el0.5 embryos from Mpsl +/ crosses. (C) Phase-contrast image showing el0.5 embryos from B. Embryo E is Mpsl/A and of normal size. Embryo F MpslA/ and has been mostly reabsorbed. (D) PCR genotyping of yolk sac DNA obtained from e10.5 embryos. DNA from Embryo F was obtained from the absorbed embryo shown in 2.2C. GFP and histone 2B-RFP to visualize DNA, the fusion proteins were found to decorate the chromosomes (Fig 2.1F). hMpslA-GFP co-localizes with the kinetochore marker hCenpB-RFP showing the A mutant protein can specifically bind to kinetochores. Moreover, hMps 1A-GFP maintains kinetochore localization in the presence of the microtubule destabilizing drug nocadozole in HeLa cells which implies that the MpslA allele is checkpoint proficient (Fig 2.1F). To investigate the function of MpslA in vivo, Mpsl /Amice were interbred and their progeny examined. Mps1 +/' and Mps 1+/ mice were born in expected numbers, but no A/A homozgygous mice were born from the MpslA heterozygotes (Fig 2.2A). Thus, the MpslA mutation causes lethality during embryogenesis. MpslA is a recessive mutation since Mps 1l+' mice are born in normal percentages and have fairly normal lifespans (Fig 2.2A and data not shown). Embryos were examined from timed matings, and MpslA induced lethality found to occur before embryonic day 10.5 (Fig. 2.2B, C and D). Mice from three separate founders yielded identical results. Thus, our data suggest that Mps lA is a hypomorph with a partial loss of function mutation in the spindle checkpoint that causes embryonic lethality but retains kinetochore localization and responds to spindle damage. We used the Cre/loxP technique to create a conditional (FLOX or f) allele of the 2Mps 1A mutation that was also introduced into mice (Fig. 2.1B, D). We obtained p53F 10 conditional knockout mice (Jonkers et al., 2001), crossed them to Mps l f/f mice, and then crossed in Lck-Cre to cause T cell specific inactivation (Lee et al., 2001). T cells were chosen for targeting based on the following reasons: 1) Developing T cells are highly mitotic. In fact, human Mpsl/TTK was originally cloned from a T expression cell library and is highly expressed in the thymus (Mills et al., 1992). 2) Mad2 -' embryos die during embryogenesis exhibiting chromosome missegregation and apoptosis, and simulataneous deletion of p53 permits Mad2 -/ blastocyt survival (Burds et al., 2005; Dobles et al., 2000). These data suggest spindle checkpoint defective cells undergo p53-dependent apoptosis and imply that deletion of p53 should rescue potential MpslA cellular lethality as it does for Mad2. 3) The thymus is a non-essential tissue and cellular inviability would still result in live animals. 4) Animals that develop thymomas are often marked by distinctly labored breathing allowing for easy diagnosis of tumor phenotype. To study the effects of simultaneous Mps 1 mutation and p53 inactivation in the thymus, a cohort of Mps 1A; p53 F2-10 compound conditional mice expressing Lck-cre were followed over 10 months and their survival assessed. Hereafter, conditional alleles are referred to as MpslA/A and p53/- in the presence of Lck-cre and as Mpslffand p53f in its absence. Figure 2.3A depicts Kaplan-Meyer survival curves for Mpsl /A; p53 /-and Mpsl /A; p53 / 1'mice. All Mpsl+/A; p53 +/-mice survive past 270 days tumor free (Fig. 2.3A). The MpslA mutation appears to be recessive as Mpsl+/A does not change p53 +/survival (data not shown). Conventional p53 +/-mice typically develop tumors no earlier than 9 months of age and only 50% develop tumors by 18 months of age although precise age of tumor development varies with genetic background (Attardi and Donehower, 2005). Remarkably, Mpsl/ A,p53+/- mice develop thymomas between 2.5 to 5.2 months with 100% penetrance (Fig. 2.3A, B). This result is atypical for spontaneous compound mutant tumor studies with p53+/- mice. For instance, while inactivating one copy of Sds3, a chromatin regulator, is sufficient to decrease the lifespan of conventional p53 -/- mice, it has no effect on p53 ÷/-mice (David et al., 2006). The DNA repair gene, Brcal when Figure 2.3 A weight Thymus Mpsl p53 15 mg A/ A +/+ f/f +/+ 41 mg * AA 942 mg +/- months PCR 1 2 1500 E 1250 3 4 Mps1 p53 S1000D I $ 750D E 500- I U +/+ m +/- I +/- m f/+ I w A f: IMs 1 S250' 0C f 8-9 10-11 12-13 14-16 Time (weeks) 5-6 RT-PCR 1 2 3 Msl/- 4 5 6 p53 I+/+ +/_- +/- -/-. -/-I -/-I Mpsl i +/+ A/A[ Al/ A/ +/+ I+/A-] I 7 I p53 Mpsl -I- -I Gapdh p53 p53 -i 5f - Lkce Figure 2.3: MpslA facilitates tumorigenesis in p53-heterozgyousthymocytes by inducing LOH ofp53. (A) Kaplan-Meyer survival curves for Mpsl+'A; p53+" mice (n=13), Mpsl"/A; p53k+ " mice " -mice (n=9). (n=27) and Mpsl+'/+; p53- (B) MpslA induces lymphomagenesis in p53 heterozygous thymocytes. The pictured thymi with the indicated genotypes were isolated and weighed. (C) Thymus size of Mpsl A/; p53+/- mice increases exponentially with age. Thymi were isolated at the indicated ages and weighed. At least two thymi from each age group were examined, except for at 5-6 weeks, where only one was weighed. (D) PCR analysis of thymi and thymomas for Mpsl and p53. (E) mRNA expression of p53 and Mpsl in thymi and thymomas. GAPDH is used as a loading control. (F) Histological analysis of a normal thymus from a Mpsl+/A; p53 +'/ mouse and a thymoma from a Mpsl"A/; p 5 3 +1" mouse, stained with hematoxylin and eosion showing a mitotic cell at anaphase with lagging chromosomes. inactivated in T cells causes tumors in <10% of p53 +' animals with the time of tumor onset similar to p53-'- animals (McPherson et al., 2006). Furthermore, p53 +' mice normally are not prone to develop thymic lymphoma (Donehower et al., 1992; Jacks et al., 1994; Purdie et al., 1994). In contrast, MpslA/A, p53+/- terminal thymomas are massive (often over 1gram), leading to morbidity in the mouse due to extreme breathing difficulties. Analysis of the weight of MpslA' , p53 +1-thymi from between 1to 4 months of age shows that thymus size in these mice increases exponentially with age with the majority of tumor growth occurring between 8 to 11 weeks of age in the mice analyzed (Fig. 2.3C). These results suggest that when an environment favorable for tumor development is achieved in Mpsl A/, p 5 3 +/-thymocytes, then tumor progression is aggressive and proceeds extremely rapidly. We investigated the molecular basis for this synergy between Mps 1al' and p53' - in tumor development by examining p53 expression in the thymomas. PCR genotyping of MpslA thymocytes express the MpslA allele and are wildtype for p53 (Fig. 2.3D, lane 1). Thymocytes that are p53"'+ express wildtype Mpsl and both the conditional and wildtype alleles for p53 (Fig. 2.3D, lane 4). However, in MpslA/A, p53 +/ tumor cells, PCR genotyping shows they have lost wildtype p53 gene (Fig. 2.3D, lane 2 and 3). RTPCR results confirm LOH of p53 in MpslA/A, p53 /-tumor cells (Fig. 2.3E). Wildtype thymocytes express both p53 and Mpsl mRNA (lane 1), while p53-null tumor cells do not express p53 mRNA (lanes 4-6). Mps A/A, p53 +' tumor cells have experience LOH of p53 (lanes 2 and 3) as no detectable levels of p53 mRNA are observed (Fig. 2.3E). When conventional p53 +/-mice develop tumors, as many as 50% of these p53 +/ tumors exhibit loss of heterozygosity (LOH) of the wildtype allele (Attardi and Donehower, 2005). MpslA seems to be inducing LOH of p53 in an accelerated fashion, causing all Mpsl/ A, p53' - mice to develop thymomas. Are Mps 1AA,p53'- tumors "p53-like"? MpslA/A, p53 ÷' tumor histology show typical lymphoblastic lymphoma with large, immature looking thymocytes exhibiting a "starry sky" pattern and many mitotic and apoptotic bodies (Fig. 2.3F). This histology is also characteristic of thymic lymphomas nullizgyous for p53 (Bassing et al., 2003; Morales et al., 2006; Seo et al., 2005). Typical p53 nullizygous thymomas can be either double positive (DP ) or single positive (SP ÷) for the T cell markers CD4 or CD8 (Bassing et al., 2003). We examined four Mpsl'AA, p 5 3 +1-terminal thymomas and found two to be a mixture of CD8÷/DP÷, one DP+ and one CD8 ÷, again typical of p53-null thymomas (Fig. 2.4C). Mitotic figures were observed showing lagging chromosomes at anaphase inMps 1A/A, p53÷'- suggesting MpslA may be inducing CIN and promoting LOH of p53 (Fig 3.2F). Thus, Mps 1A acts as a powerful tumorigenesis accelerator through inducing CIN in the thymus when one allele of p53 is inactivated inducing formation of thymomas reminiscent of p53 nullizygous tumors. To further dissect the synergy between Mpsl and p53 in tumor suppression, we followed the survival of a cohort of p53 -/- animals with the following Mpsl genotypes: p5 3+Mps 1' +,Mpsl +/,and MpslA / and compared their lifespans to those of Mps1 /A, mice (Fig. 2.4A). As expected, inactivating p53 in T cells causes lymphomagenesis recapitulating conventional p53 -'-knockout mice survival data (Donehower et al., 1992; Jacks et al., 1994; Purdie et al., 1994). Mpsl+/1; p53 -/ mice develop thymic lymphomas with an average time onset (Tso0) of 4.78 months, and Mps 1 +/A; p53-/' mice have a similar Ts0 (Fig. 2.4A, B). However, Mpsl'A/, p53 ÷/-mice develop tumors faster than with T50 = Figure 2.4 Genotype Mps1A/ p53 3.25 Mps /p53 Mps1 + p53 3.15 4.76 2 4 months 6 " p53 Mpsl 0 T50(months) 4.78 8 Mpslr/A p53 +/- Mps1 AA p53 +/ + Mpsl+/+ p53-/- 0.47 98.3 0.42 0.85 CD8 Mpsl p53 /. A/A 1 /-/ O - nocadozole S. nocad - ÷/A 1.... AA • +C/÷ 1c MpslA/A p53+/- Mpsl1 A p53-/- 2n 4n 6n DNA content 55 chromosomes :•. ,,• S =. ..,' .. rll • · .°";f-,e•T,. 800 1000 Mps1 +/+ p53 °/ " Mpsl +/A p53J'- .. 47 chromosomes : "r.0 e-'* ;·P · Figure 2.4: MpslA accelerateslymphomagenesis inp53-null knockout thymocytes (A) Kaplan-Meyer curves of MpslA; p53 +"' mice (n=27), Mpsl A; p53-1- mice (n=l 1), Mpsl+'A; p53' " mice (n=18), Mpsl+/+; p53-/-mice (n=9), and Mpsl+/'; p53 +"' mice (n=13). The survival between Mpsl+/'; p53 --mice and MpslA'6; p53 /+' mice or Mpsl ;p53 / -- mice are statistically significant based on an unpaired t test with P= 0.006 and P=0.0133 respectively. (B) Average time of tumor onset (Tso) for mice in figure 2.4A. (C) FACS profiles for CD4 and CD8 expression in thymocytes from the thymi or thymic lymphoma with the following genotypes: Mpslvf; p53 +' + , Mpsl" /; p53'+/+ , Mpsl"AA; p53 +1- ,and Mpsl+/+; p53'" . One MpslA"/; p53k+ - is CD8 single positive and the other MpslAA; p53 +"- is double positive for CD4 and CD8 expression. (D) DNA FACS profiles using propidium iodide to visualize DNA from passage 3 tumor cells isolated from thymic lymphomas with the indicated genotypes, plus and minus treatment with 100ng/nl nocodazole for 18 hours prior to FACS analysis. (E) DNA FACS profiles using propidium iodide to visualize DNA from thymocytes isolated from thymi with the indicated genotypes at 2.5 months to 4 months of age. (F) Two representative karyotypes of Mpsl"A/; p53 +' thymic lymphoma cells showing the DAPI metaphase spread and the spectral karyotyping showing aneuploid karyotypes. 3.25 months, a decreased time of tumor onset relative to Mpsl++; p53 -' mice with P value= 0.006 (Fig. 2.4A, B). Moreover, all MpslA/A; p53 - - mice develop thymomas and die before 4.5 months, with similar kinetics to MpslIA; p53+/' mice (Fig. 2.4A). Thus, Mps 1A is dramatically decreasing lifespan when p53 is completely inactivated. Therefore, the Mps 1A allele increases tumorigenicity in ways that do not simply involve LOH of p53, but presumably do involve an increase in CIN. Abrogation of spindle checkpoint function is commonly assessed in two ways: 1) inability to respond to a microtubule destabilizer, such as nocoadozole, which normally provokes mitotic arrest and 2) presence of chromosome missegregation events as reflected by a change in the normal chromosome complement. To evaluate the effect of the MpslA mutation on checkpoint function, tumors were excised from mice, placed in culture and passaged 3 times and then treated with 100ng/ml nocodazole for 18 hour and analyzed by flow cytometry. Comparison of the DNA content of the untreated and treated samples shows that nocadazole treatment causes a significant accumulation of cells in mitosis for all 4 tumor samples, regardless of genotype (Fig 2.4D). Live cell microscopy of Mps 1a A; p53'/- tumors treated with nocodazole also arrest in mitosis (data not shown). Thus, the Mps 1A allele is still checkpoint proficient in that it can respond to spindle damage and arrest the tumor cells in mitosis. This result was not unexpected since we had deliberately left the kinase domain intact in the Mps 1Amutation. A functional kinase domain has been shown to be absolutely essential for spindle checkpoint function (Abrieu et al., 2001; Fisk and Winey, 2001). Since Mps 1A leads to embryonic lethality, this mutation presumably is causing CIN in T cells as well. To determine if MpslA had led to chromosome missegregation events in the Mps1 ' ; p53'- tumors, cells from a terminal thymoma were fixed immediately after isolation and its DNA content examined by flow cytometry and compared to controls (Fig. 2.4E). The Mpsl /A; p53' /- tumor sample shows a shift in the G1 peak relative to its littermate control (Mpslf/f; p53+/+; Lck-cre-) demonstrating aneuploidy. An additional four Mpsl A/A; p53+/' tumors were profiled (three are shown from mice which are littermates or age matched) and also shown to be aneuploid. One MpslA/A; p53 ÷/-tumor was found to be polyploid with a broad primary peak at around 6N, but all other MpslA/A; p53 /-thymomas profiled were between 2N and 4N (Fig. 2.4E and data not shown). In contrast, cells isolated from a Mpsl+/+; p53/'-, Cre÷ thymus displayed a normal DNA profile indistinguishable the Cre- control (Fig. 2.4E). However, p53 conventional null tumors are known to be aneuploid as well (Bassing et al., 2003; Liao et al., 1998). An Mps1+/+; p53-/- littermate which did not display an enlarged thymus, nonetheless has abnormal DNA content and displayed an additional population of aneuploid cells other than the typical 2N peak. These mice were all profiled at around 3 months of age. When a Mps1 +/; p53 -/-terminal thymic lymphoma was examined at 5 months of age, the DNA profile was indistinguishable from one for a Mps 1 A/; p53 ÷' terminal thymoma isolated at 3 months of age (data not shown). These data show that MpslA/ A; p53/-tumors, like p53-null tumors, are aneuploid by flow cytometry and have accelerated tumor onset. In another test for Mps lA-induced CIN, we performed karyotype analysis on early passage tumor cells isolated from two Mps1 A/A; p53÷/- terminal thymomas. A total of 18 metaphases were counted with each cell displaying an average of 52 and 53 chromosomes (ranges of 45-57 and 48-68 chromosomes) (Table 2.1). Thymi Table 2.1: Karyotype analysis of Mpsl Tumor Mpsll"A; p53/ - -1 Mps l ; p53 /- -2 Mpsl +*; p53 " -1 Mpsl +*; p53- -2 Mps1*/*; p53- -3 53BP1-3 ; p53 -1 53BP1; p533- -2 53BP1 '; p53 -3 Avg 53 52 49 42 39 50 52 49 ; p53.- thymic lymphomas Chromosome no. Range 47-68 45-58 48-50 39-60 39 39-60 50-54 40-54 Note: Chromosome number data from Mpsl +/;p53' and 53BP14 ; p53' thymic lymphomas were previously published in Liao, et.al 1998 and Morales, et. al, 2006. Figure 2.5 A Mpslf/f, cre+ cre- control, cre+ FII. 100 OMpslf/f; cre- * Mpslf/f; cre+ 80 - nh ArmLI A 0S A A AA .0 AA fIf -•rrn -I. J I- A Q)60 - 0· EU U * U AA 40 - U· 20 - 0- ýn 4n DNA content II 3 A .4,*£ A I UV 0 o - o Mps flf; cre* Mpslf/f; cre+ o Mpslf/f; cre+ 80- 7 20 C 20 o • 0a · E · s A A u C 60 - 0a t- IC. u, 10" 40 20- P%. 0 Lim, I zn 4n DNA content i Figure 2.5: Mpsl A decreases thymus size andincreases heterogeneity in thymocytes. (A) Mpsl f; cre+ (n=17), control, cre+(n=12), and cre- (n=17) thymi ranging from one to six months of age were isolated from age matched littermates and weighed. Statistical analysis by Mann-Whitney tests found the following: for Mps f; cre+ versus control, cre+ thymi, P=0.0007; for Mpsl f/f; cre+ versus control, cre- thymi, P=0.0004; for control, cre+ versus control, cre- thymi, P=0.6261. (B) MpslEf; cre+ (n=4), control, cre+ (n=5), and cre" (n=7) thymocytes were isolated from 6 week old littermates and counted. Mps lA-induced decrease in thymocyte number from mice at 6 weeks of age was found to be statistically significant between Mpsl f/f; cre + and cre- by a Mann-Whitney test with P=0.0424. (C) Representative hematoxylin and eosion images of Mps f; cre+, Mpsl +;cre+, and Mps f/+; cre- thymi sections fixed at 4 months of age. Arrows highlight thymocytes from a Mpsl f; cre+ thymus are more heterogeneous in size and shape than thymocytes from the two control thymi. (D) DNA FACS profiles using propidium iodide to visualize DNA in thymocytes isolated from a Mpsl f; cre- control (in unfilled red) overlayed over two Mpsf/f; cre+ samples (filled and unfilled green). Thymoctes were isolated from two sets of littermates at 2.5 months of age. Figure 2.6 A 1 I control; cre+ - Mpslf/f; cre+ CL 5 Time C 100- 1 2 3 4 5 I Control Thymus: MpslAi+; cre- I Thvmoma: Moslflf: cre+ 80- I Lck-cre I-I-I + + + Mps I f/f 60- f/f f/f f/f f/f np3 I If/f +/+ +/+ +/+ +/+ uvv II 40- Mpsl 200- i p53 ___, 4n DNA content Figure2.6: Mpsl A is a partialloss offunction mutation in the spindle checkpoint that induces CIN and sporadiclymphomagenesis. (A) Kaplan-meyer survival curves of Mpsl f; cre÷ (n=29) and control, cre÷ (n= 13) mice. (B) Representative hematoxylin and eosion images of Mpsl+/'; cre+ control thymus at 5 months and three Mpslf/f; cre÷ lymphomas at 7.3 months (top right), 4.3 months (bottom panels). The histology is consistent with lymphoblastic lymphoma with mitotic figures at anaphase displaying lagging chromosomes (insets, arrowheads). (C) DNA FACS profiles of thymocytes from Mpsl 1+/; cre÷ control thymus (in blue) and tumors cells from a Mpsl f; cre+ thymoma (in green, histology pictured in 2.6B top right). Tumor cells are aneuploid with a DNA profile that is shifted compared to the control thymocytes. (D) PCR genotyping of thymocytes isolated from thymi with the indicated genotypes. Lane 5 is of the thymoma shown in 2.6B, top right and 2.6C. lymphomas nullizygous for p53 have been shown to be aneuploid with no clonal translocations (Liao et al., 1998; Morales et al., 2006). These data show a slight increase in absolute number and a broader distribution of chromosome numbers than those reported for conventional p53 -1terminal thymomas, suggesting MpslA is causing LOH of p53 in MpslA/A; p53 +/' tumor cells by inducing numerical CIN, presumably through chromosome missegregation events (Table 2.1). Spectral karytyping (SKY) was difficult to perform on Mpsl AA; p53 / 1- tumor cells. We found these cells to be quite fragile and few metaphase cells were recovered when prepared for SKY. We are able to conclude that the distribution of chromosomes gained and lost in any two cells from the same tumor can be vastly different, further demonstrating the highly unstable nature of these tumors (Fig. 2.4F). In one tumor from a male mouse, one cell has 6 copies of the Y chromosome while the other cell has none. Mpsl has been reported to phosphorylate the DNA damage checkpoint kinase Chk2 and the RecQ helicase Blm, raising the possibility that MpslA may contribute to lymphomagenesis by dysregulating the function of these substrates (Leng et al., 2006; Wei et al., 2005). However, no apparent translocations were observed in any of the metaphases examined, suggesting MpslA is not contributing to DNA damage and causing DNA breaks. In conclusion, while p53 nullizygous tumors are aneuploid even without the Mps 1Amutation, aneuploidy is at least as severe or more severe with the presence of Mps 1A. To investigate the effect of the MpslA on thymocytes in the presence of p53, the survival of a cohort of Mps IA a mice was followed for 15 months. The majority of Mpsl"A; p53 /' ÷ mice have normal lifespans with no overt phenotypes (Fig. 2.6A). However, when we weighed the thymi from these mice, they were found to be 30%-40% smaller than age matched control Lck-cre ÷ and Lck-cre- mice with P<0.001 by a MannWhitney test (Fig 2.5A). Variation between the Lck-cre + and Lck-cre- control groups were found to be statistically insignificant by a Mann-Whitney test, with P=0.63. There were also fewer cells from the thymi of Mpslff,Lck-cre + mice relative to control mice (Figure 2.5B and Table 2.2). Decreased cell number may result from premature senescence resulting in a proliferation defect. However, Mps 1A/A thymi are positive for the proliferation antigen Ki-67 (data not shown). Thymocyte development was not overly affected by the MpslA mutation as assayed by flow cytometry of CD4, CD8 and TCRp expression in the thymi of mice at 6 weeks of age or by the percentage of peripheral T cells in the blood of mice at 3-4 weeks of age (Table 2.3 and 2.4). These data indicate the decrease in overall size and cell number of MpslA/A thymi is not due to a defect in proliferation or thymocyte development. To investigate the effect of Mps lA on thymus tissue organization, thymi from - 10 animals ranging from 1 to 5.5 months of age of each category in Fig 5E were fixed for histological analysis. We observed irregular cell packing and highly variable nuclear volumes in the cortex of Mps A/A; p53+'+ thymi (n= 11). Histology of a representative Mps A/A; p53 +/+ thymus at 4 months of age show that neighboring thymocytes often exhibit a 2-3 fold difference in nuclear diameter (Fig 2.5F). In contrast, control thymocytes negative for Lck-cre or positive for Lck-cre are fairly uniform in both size and arrangement (Fig. 2.5C). We found the presence of Lck-cre can sometimes slightly alter the morphology of thymocytes and cause a heterogeneous population, either due to the toxic nature of Cre recombinase (Loonstra et al., 2002) or possibly abnormal gene expression due to transgene intergration (seen in 2 of 11 animals), though not as severely C) U) C' Lj o to (CD co () 0) 0 c*%j c) CO CO tO LO N-i (Nl a.L 0) cNI L. -J LO (NI cyi '4- - LOU-u C'4 I,*- N 0O0 0 4- +000 .-- + 0 Lo W( omc 0 o + 0 >0 S-0 0, m + +00 >~0+ 0>c o+ a0 00 I 0 00 1- 7) CF N (1 COCI T- c C CI r- C as seen in Mps 1A/A; p53+/+ thymi. Analysis of thymocytes isolated from two independent sets of three month old littermates by flow cytometry show slightly broader histograms for the DNA content of two Mps 1ff; cre + samples (solid and outlined green) compare to the Mpsl"f; cre- control (red) (Fig. 2.6D). These data shows MsplA increases heterogeneity in the thymocyte population, but decreases thymocyte number and thymus size when p53 is wildtype. Both the accelerated kinetics of tumor development in compound mutant animals and the aneuploid karyotypes of MpslAA; p53'1 thymomas as indicated by metaphase counts suggest that Mps lA is causing genomic instability, specifically numerical chromosome changes. To determine if inactivation of at least one allele of p53 is required for tumorigenesis of Mps 1A thymocytes, we studied mice carrying only the Mps 1A allele. MpslA/A; p53 ÷'1 mice display a slightly decreased mortality (25%) relative to the control cohort of Mpsl /A; p53' /- mice (<10%) by 11 months of age (Fig 2.5A). Although the majority of Mps1AA mice expressing p53 did not develop tumors, three Mps1A; p53 +/+ mice were found with tumors (between 4 to 7 months), two thymomas and one peripheral lymphoma. Histologically, these tumors are typical of lymphoblastic lymphoma and are indistinguishable from tumors isolated from the Mpsl, p53 compound mutants (Fig 2.5B). Importantly, Mps 1 /; p53÷/+ thymic lymphomas contain mitotic cells with chromosome segregation defects (Fig. 2.6D). Anaphase figures were observed with lagging chromosomes (Fig. 2.6B). Furthermore, one terminal thymoma was assayed by flow cytometry for its DNA content and found to be aneuploid, demonstrating the MpslA allele is causing CIN (Fig. 2.6C). The two animals with thymomas were found alive, but tumor cells that were isolated fail to survive in culture. This is probably reflecting the fact that these tumors still contain p53 as assayed by PCR (Fig 2.6D). Thus, MpslA-induced CIN is sufficient to promote lymphomagenesis in the absence of p53, but tumor incidence is sporadic. Discussion We have shown the MpslA allele is a hypomorph, exhibiting partial loss of spindle checkpoint function which is manifested by embryonic lethality when expressed in all cells and by increased mortality, sporadic lymphoma, and abnormal thymus size and architecture when expressed conditionally in T cells. This hypomorphic Mps 1 allele promotes a modest predisposition to thymic lymphomas with p53 and dramatic predisposition to lymphomas when p53 is inactivated. We propose that the MpslA mutation promotes cancer in thymocytes by inducing CIN. Mps A/A tumors exhibit mitotic figures with lagging chromosomes suggesting the Mps 1Amutation is causing CIN through chromosome missegregation events. The Mps 1Amutant protein localizes to kinetochores upon nocadazole treatment in HeLa cells, and Mpsl A-expressing tumor cells arrest in mitosis in response to nocodazole suggesting Mps lA retains partial checkpoint proficiency. Remarkably, MpslA/A; p53 +/ mice develop thymomas at 100% penetrance suggesting Mps lA-induced CIN can robustly facilitate cancer. In fact, MpslA appears to increase tumor malignancy as tumor growth is rapid in MpslA/A, p53+/thymocytes, with thymi mass increasing 10-fold between 2-3 months of age and the average time of tumor onset accelerated in p53-heterozygous thymocytes when MpslA is expressed. Furthermore, Mps 1Aaccelerates the onset of cancer in conventional p53heterozygous knockout mice. These mice are also prone to thymic lymphoma demonstrating p53 inactivation prior to expressing MpslA does not change tumor phenotype (Fig. 2.7). A consequence of CIN is an enhanced rate of LOH (Nowak et al., 2002). LOH of p53 is exactly what is observed in MpslA/A; p53' - tumors. However, the MpslA mutation might be inducing additional tumor-promoting mutations since Mps1 AA; p53-'- thymocytes form tumors almost two months earlier on average than Mpsl' +; p53/ thymocytes. Thus, Mps lA-induced CIN is proficient at facilitating tumorigenesis in thymocytes when p53 is also inactivated. What is causing MpslA-induced CIN? Mpsl appears to be required for centrosome duplication in budding yeast and possibly in mammals as well although this is controversial, in addition to its role in the spindle checkpoint (Fisk et al., 2003; Fisk and Winey, 2001; He et al., 1998; Liu et al., 2003; Stucke et al., 2004; Stucke et al., 2002; Weiss and Winey, 1996; Winey et al., 1991). Chromosome missegregation can result from both abnormal centrosome numbers and spindle checkpoint defects. However, immunohistochemistry of two control mice and two MpslIA thymi show that centrosomes are not amplified in thymocytes that express MpslA suggesting increased aneuploidy in the presence of Mpsl A is not due to centrosome defects (Table 2.5). No centrosomal abnormality was observed for MpslAA mouse embryonic fibroblasts either (chapter 3). Therefore, MpslA induced CIN is likely due to chromosome missegregation events resulting from a partially defective checkpoint function. The biochemical nature of this phenotype is currently unknown, but Mps 1Adoes not appear to be inducing CIN through dysregulating the DNA damage response or V(D)J recombination. Mpsl has been reported to phosphorylate Chk2 and BLM, the RecQ helicase mutated in Bloom syndrome and thus intersect the spindle checkpoint with the DNA damage response (Leng et al., 2006; Wei et al., 2005). If Mpsl does connect spindle checkpoint function to the DNA damage and repair response, a possible consequence of mutating Mps 1 is increasing the frequency of DNA translocations due to unrepaired DNA double-strand breaks (DSBs). However, inactivation of DNA damage sensing genes tends to result in clonal translocations in tumors, which we have not " tumor cells (Bassing et al., 2003; Gao et al., 2000; Morales et observed in Mpsl A/; p53 +al., 2006). MpslA/A; p53 ÷1 thymic lymphomas developed into and through the DP stage. Moreover, Mps 1 AA thymocytes develop normally and appear to have properly executed V(D)J recombination, demonstrating MpslA does not provoke structural DNA lesions. Unlike Blm-deficient cells, which display a hyper-recombination phenotype and chromsomal structure aberrations, MpslA promotes tumorigenesis through numerical chromosome defects (Chester et al., 2006; Wu and Hickson, 2003) Recent studies of spindle checkpoint knockout mouse models suggest checkpoint inactivation can lead to two outcomes: early aging (for hypomorphic BubR1 and Bub3' - /Rael +'-) or cancer (Mad2÷' -) (Baker et al., 2004; Baker et al., 2006; Michel et al., 2001). Our data suggests Mpsl belongs in the latter category and is involved in tumor suppression. Why are Mps 1al thymi not more susceptible to tumors? One hypothesis is that CIN is insufficient for tumor initiation, and requires another lesion to "prime" tumorigenesis (Nowak, 2002). The majority of MpslA' thymi are smaller than average and have fewer thymocytes. These can be caused by increased apoptosis or decreased proliferation. Van Deursen and colleagues have reported that early aging in Bub3/Rae 1 double heterozygous knockout mice may be due to early onset of cellular senescence (Baker et al., 2006). Mpsl1A thymi were found to be positive for the proliferation antigen Figure 2.7 0 2 1 3 4 5 6 7 Age (Months) RT-PCR 7005 6245 Control 505 bl Mpsl 184bp Sbeta actin 8 Figure 2.7: MpslA acceleratestumorigenesis in p53-heterozygous conventional knockout mice. p53+/'; cre+ mice (A) Survival curves of Mpsl+';p53+'; cre+ mice (n=10), Mpsl f; (n=24), Mpsl f; p53 "-mice; cre- (n=17), Mpsl -; p53+'; cre+ mice (n=18), and Mpslf/f; p53v+; cre+ mice (n=15). The latter group of mice are a subset of what is described in figure 2.3A and 2.4A. The MpslA mutation accelerates tumor onset in p53-heterozygous mice with the conventional knockout allele with similar kinetics as in conditional p53heterozygous mice (solid versus dotted light blue lines). (B) White light and and eosion image of thymic lymphoma from mouse 7005, Mps 1f'; p53k+-; cre+. (C) RT-PCR analysis of Mpsl in tumor cells (7005 and 6245) isolated from Mpslf; p53+-; cre+ thymic lymphomas. Tumor cells express the the truncated MpslA mRNA. 1B6 T cells were used as the control. Beta actin was used as a loading control. 100 Table 2.5: Mpsl A does not induce centrosome duplicationin thymocytes mouse # genotype N cells 1 centrosome 1206 1366 1458 1459 1524 a Mpsl+/+, Lck-cre+ Mpslf/+, Lck-cre+ Mpslf/f, Lck-cre+ Mpslf/f, Lck-cre+ Mpslf/f, Lck-cre+ 90 104 105 114 102 94% 96% 95% 98% 83% The # of gamma-tubulin foci were counted per cell. a This animal developed a thymoma 101 2 centrosomes 6% 4% 5% 2% 17% Ki-67 suggesting premature senescence is unlikely to be inhibiting MpslA lymphomagenesis. Another possibility is that Mps A/A cells are prone to apoptosis and require genetic or epigenetic changes that sustain self-renewal or prevent apoptosis for tumor development. What is the relationship between p53 inactivation and MpslA-induced CIN in lymphomagenesis? While MpslA/A animals are susceptible to tumor formation without loss of p53, tumor onset and rate of formation is greatly increased when this is the case. The most frequently mutated gene in human cancers is p53, emphazing the crucial nature of this tumor suppressor to maintaining genome stability (Levine, 1997). In response to cellular stress such as DNA damage, p53 transactivates genes that causes cell cycle arrest or apoptosis (Harris and Levine, 2005). Genomic instability per se may be insufficient to cause cancer. This scenario has been observed for both DNA damage checkpoint gene -' and H2AX /- and spindle checkpoint mutants like BubR1WH, knockouts like 53BPF1 where these mouse models exhibit genomic instability, but are not prone to cancer (Baker et al., 2006; Bassing et al., 2003; Morales et al., 2006; Ward et al., 2005). Moreover, not all p53 conditional knockout animals examined here developed thymomas further suggesting multiple lesions are necessary for tumorigenesis. Mutating the DNA damage checkpoint or the spindle checkpoint induces genomic instability either through DNA breaks or chromosome missegregation. The majority of genes that synergize with p53 in cancer mouse models are DNA repair or DNA binding proteins such as 53BP1 and H2AX or Sds3 and Cdtl whose inactivation results in DNA breaks and require complete inactivation of p53 for accelerated tumorigenesis (Bassing et al., 2003; David et al., 2006; Morales et al., 2006; Seo et al., 2005). Cells deficient for 53BPI experience genomic 102 stability because of aberrant DSB repair and appear to be targeted for apoptosis by p53 (Morales et al., 2006). When p53 is inactivated in 53BP1-deficient mice, thymic lymphomas form (Morales et al., 2006). Although MpslA induces CIN and not structural DNA defects, p53 deficiency is also required for cancer. We propose that MpslA provokes p53-dependent apoptosis, a hypothesis that would enrich viable cells in MpslA/A; p53' /- tumors for those that have selected for p53 loss. In fact, MpslA/A; p53 ÷/ do experience LOH of p53. In conclusion, we have shown that a CIN prone mouse model has a modest predisposition to cancer, but in the majority of cases, tumors form only in the context of a lesion such as p53 inactivation presumably through destabilizing the genome and preventing apoptosis. Another gene that demonstrates a connection between CIN and spindle checkpoint inactivation in mice is BubR1. BubR1 mutations have been found in mosiac variegated aneuploidy, a rare human disorder prone to cancer. (Matsuura et al., 2006). However, BubR1 mutant mice do not develop tumors despite displaying high rates of aneuploidy (Baker et al., 2004; Wang et al., 2004a). Instead, BubR1 hypomorphic mice have early aging phenotypes and show upregulation of p53 (Baker et al., 2004). However, inactivating p53 in BubR1H/H mice may also accelerate in tumor development much like in compound Mpsl and p53 conditional mice. Like Mpsl, BubR1 has been suggested to function in DNA damage (Fang et al., 2006; Futamura et al., 2000). BubR1 has been shown to phosphorylate the tumor suppressor gene BRCA2, which is critical for DNA replication and repair, in vitro and also to interact with breast cancer specific gene1 (Futamura et al., 2000; Gupta et al., 2003). Furthermore, BubR1 has been shown to be transcriptionally regulated by p53 and has been demonstrated to inhibit centrosome 103 amplification in p53-null cells (Oikawa et al., 2005). These data suggest BubR1H/H mice may also be prone to CIN-driven tumorigenesis if p53 is inactivated. Is CIN resulting from a weak spindle checkpoint also sufficient to cause cancer in humans? Inactivating the spindle checkpoint in mouse models causes embryonic lethality. Here, mutating Mps 1 also results in embryonic lethality further demonstrating the essentiality of the spindle checkpoint to dividing cells. However, at least in an environment where p53 is lost, MpslA is extremely proficient at promoting tumorigenesis in mice. Mutations in spindle checkpoint genes are present in human tumors, but are infrequent (Cahill et al., 1998; Hempen et al., 2003; Kim et al., 2005; Langerod et al., 2003; Matsuura et al., 2006; Saeki et al., 2002; Wang et al., 2004b). This has been interpreted to mean that spindle checkpoint mutations do not play a role in tumor development. The extreme CIN resulting from inactivation of the spindle checkpoint may be too deleterious for even genetically unstable cancer cells. However, impaired spindle checkpoint function is found in many human cancer cell lines (Saeki et al., 2002). Few studies have examined Mpsl for mutations in human tumors, but of the available studies, no mutations of Mps 1 were found in colorectal tumors, breast cancer cell lines or mantle cell lymphoma (Cahill et al., 1999; Camacho et al., 2006; Yuan et al., 2006). However, data presented here suggests additional searches in human tumors may be informative. Since human Mpsl was identified from a T-cell library (Mills et al., 1992), and MpslA alone is sufficient to induce thymomas is a small subset of mice, lymphomas may be an ideal tumor type to begin the search for mutations. The work we present here addresses an age old question in the field of cancer biology: Is CIN simply a consequence of the steps leading to cancer or can CIN also 104 function as a causal agent? We generated a partial loss of function allele in the spindle checkpoint gene Mpsl to induce a CIN phenotype in mice. We argue that CIN does function in tumorigenesis as a causal agent but requires the inactivation of at least the p53 pathway. We have shown that spindle checkpoint dysfunction by a weakened checkpoint allele is a valid route to cancer. Our work underscores the necessity of additional studies based on other cell types for spindle checkpoint dysfunction in human tumor 105 Methods Generation of MpslA conditional and conventional mutant mice. A 129/Sv mouse genomic lambda-phage library was screened for the genomic locus of Mpsl as described (Dobles et al., 2000). A 14 kb portion of the Mpsl genomic locus encoding exons 1-8 was isolated and cloned into pBluescript (Invitrogen) via NotI sites. DNA encoding Diptheria toxin A (DT-A) was cloned into the NotI site as a negative selection marker. A LoxP site was cloned into an NcoI site between exons 1 and 2. A 4 kb selection cassette containing thymidine-kinase and the neomycin-resistance gene flanked by two LoxP and FRT recombination sites (LFNT) was cloned into a Sphl site between exons 3 and 4. The resultant 22.6 kb targeting vector was introduced into embryonic stem (ES) cells and clones isolated as previously described (Dobles et al., 2000). Incorporation of the gene targeting vector by homologous recombination into ES clones was determined by southern blotting using a 664 bp 5' probe and a 665' 3' probe after digestion with Nhe I or Bgl I as indicated in Sup. Fig 2.1A as described (Dobles et al., 2000). The 5' probe was generated by digesting another lambda phage clone with SacI and Xmal which contained sequences 5' of the targeting vector and then subcloned into pBluescript (invitrogen). The 5' probe yields a 11.7 kb wildtype band and a 9 kb targeted band. The 3' probe is homologous to sequences containing exon 9 of Mps 1 and was generated by PCR with 5'GGACAATAGAAAGGGGTCAGGAC and 5' TGACACAATTAAGTAGCATTCCC yields a 13.4 kb wildtype band and a 9.4 kb targeted band. Two properly targeted ES clones were isolated. The LFNT cassette was removed in these ES clones by transfecting cells with vectors expressing Cre recombinase (pCrePac) or Flp recombinase (pFlpe) which resulted in either a conditional 106 (FLOX) allele or the A allele. Southern blotting after restriction digestion with Nco I and probing with the 5' 664 bp probe yields 8kb for wildtype, 12 kb for the targeted locus (containing LFNT), 15.2 kb for the Aallele and 18.6 for the FLOX allele. Two FLOX/+ and three A/+ subclones from the two original clones were injected into blastocyts and successfully generated founder lines for the conditional and conventional A allele. Analysis of Mice Mice used in this study were on a mixed C57BL/6 and 129/Sv genetic background. p53 conditional knockout mice were obtained with permission from Anton Berns through Tyler Jacks' group (Jonkers et al., 2001). Lck-Cre transgenic mice were obtained from Taconic (Lee et al., 2001). DNA was prepared from mouse tails, cells and tissues with the NucleoSpin 96 Tissue Kit (Clontech, cat # 636968). The following genotyping oiligonucleotides were used: For Mps 1: Mpsl-l: 5'CCTGGTAGTCTACCCATCCTCCTGCTC Mpsl-2: 5'GACACAGACATGGTTGGAGAGTCCTGAG Mpsl-3: 5'GAATACCGAATGAGCGAAAAGCCCC For p53 (Jonkers et al., 2001): p53FL A: 5'CACAAAAACAGGTTAAACCCAG p53FL B: 5'AGCACATAGGAGGCAGAGAC For Lck-cre (Lee et al., 2001): Lck-F: CCTTGGTGGAGGAGGGTGGAATGAA Lck-R: TAGAGCCCTGTTCTGGAAGTTACAA CreT2-R: CGCATAACCAGTGAAACAGCATTGC 107 For embryonic lethality experiments, Mps1 +/A mice were intercrossed in timed matings. At embryonic day 10.5, embryos were isolated and the yolk sac taken to determine genotype by PCR as described (Burds et al., 2005). Mpsl+ /Amice obtained from the original founder ES clones had identical results. Thymomas and thymi were isolated from mice and weighed using a Ohaus CS200 scale or Mettler AE50 scale. For survival studies, mice were monitored for tumor development every week starting at 2.5 months of age by observing for difficulty in breathing. Tissues were fixed in 10% formalin for histology. Histology images were taken at 40X with a Zeiss Axiophor microscope. Animal protocols were approved by the Massachusetts Instutute of Technology Committee on Animal Care. Tissue Fractionation and Flow Cytometry Single cells were isolated from thymoma, thymus, spleen and lymph nodes by dissociation between 2 frosted slides (VWR, # 48312-002) and were cleared of debris by passage through a 40 [pM cell strainer (BD Falcon, cat # 5273324). The number of single cells were isolated from various tissues were counted on a hemacytometer. Mice at 3-4 weeks of age were bled by retroorbital bleeding. Red blood cells were lysed with the following lysis buffer: 1 mM KHCO3;0.15M NH4Cl; 0.1mM EDTA. For analysis of T cell markers, 1*106 isolated cells were treated with purified anti-mouse CD16/CD32 (BD Pharmigen) to block the Fcyll/II receptor and then stained with the following fluorescent antibodies (all BD Pharmigen): FITC-conjugated rat anti-mouse CD8a (Ly-2), PEconjugated anti-mouse TCRp; APC-conjugated anti-mouse CD4. Ten-thousand cells per sample were analyzed by using a FACScaliber (Becton Dickinson). 108 Cell Culture, and Metaphase Spreads Tumor cells were cultured in DMEM supplemented with 10% FCS (Hyclone), 1mM LGlutamine (GIBCO) and ImM penicillin (GIBCO). Tumor cells in logarithmic growth phase were arrested in metaphase with 0.1 tg/ml colcemid (GIBCO) and metaphase spreads prepared as described (David et al., 2006). RNA Isolation and RT-PCR RNA was isolated by using the RNAease kit (Qiagen, cat# 74104). Reverse transcription was performed by using OligoDT primers and MMLV_RT from the RETROscript kit (Ambion, 1710) according to the manufacturer's instructions. The following primer sets were used: Mps 1-exon 1-for, (5'GGAGGCTGAAGAGTTAATTGGC); Mpsl-exon4-rev, (5'CCCAGTCTCTACAGCTTTATGAAG); p53ex7, (5'CGCGCCGGCTCTGAGTAA C); p53ex8, (5'CGCTTGCGCTCCCTGGGGGC); and GAPDH primers as described (Sato et al., 1999) [GAPDH - sense, (5'CCCATCACCAT CTTCCAGGAGC); GAPDH - antisense, (5'CCAGTGAGCTTCCCGTTCAGC)]. Nocodazole Arrest and Determination of DNA content by FACS analysis Cells were treated with 100ng/ml nocodazole for 18 hours and cells were prepared for analysis by flow cytometry as described (Burds et al., 2005). Briefly, 1* 106 cells were isolated and washed in PBS, fixed in 90% ethanol, treated with RNAse A (0.5 mg/ml) and stained with propidum iodide (50 [ig/ml). Ten-thousand cells per sample were analyzed by using a FACScaliber (Becton Dickinson). FACS data analyzed with FlowJo. 109 Cloning hMpslA-GFP, cell culture and live cell imaging hMpslA-GFP is a truncated form of hMps I-GFP (kindly provided by Robert Hagan) where amino acids 53-158 were deleted. (mMpslA protein has aa 47-154 deleted.) The following oligos corresponding to sequences in exon 1 of hMpsl were annealed and then digested with HindIII and HpaI: 5'GCCGTAAGCTTGAATAAAATTTCTGCTGATACTACAGATAACTCGGGAACT GTTAACCGTGCAAT and 5'ATTGCACGGTTAACAGTTCCCGAGTTATCTGTAGTATCAGCAGAAATTTTAT TCAAGCTTACGGC. To generate the exon 4 fragment of hMps 1, 5'CACTTGGTTTAGATCCAGGCAC and 5'CGAGGTTACGTAAAAAAAAGTAAACAACTTCT were used to generate a 484 bp fragment by PCR which was then digested by SnaB 1 and BstelIl to yiled a 286 bp fragment. The exonl and exon 4 fragments were then ligated by 3-way ligation into the hMps 1-GFP vector where exons 2 and 3 have been removed by a HindlI/BstEII digest to yield hMpslA-GFP. HeLa cells were grown as described (Martinez-Exposito et al., 1999). HeLa cells stably expressing H2B-GFP were the kind gift of P. Meraldi and V. Draviam (Meraldi et al., 2004). Cells were seeded onto ATO. 15 mm dishes (Bioptechs), transfected as described with hMpslA-GFP or hMpsl-GFP and CenpB-RFP (kindly provided by Markus Prost of the Swedlow group) (Meraldi et al., 2004) with fugene (Invitrogen) and imaged approximately 24 hours after transfection. 50 ng/ml nocodazole was added to cells in a C02-independent medium (GIBCO-BRL) prior to imaging. Images were using a 60x 110 NA0.75 objective on a Zeiss Applied Precision Deltavision microscope equipped with a Mercury 100W lamp, GFP-long pass filter set (Chroma), and a Coolsnap camera. Histology and Immunohistochemistry Tissues were fixed in 10% formalin overnight and then paraffin-embedded. Immunohistochemistry on 0.4[tm thick thymic sections were performed as described (Wong et al., 2005). For centrosome staining, a rabbit anti-mouse gamma tubulin antibody (Abcam catalog # ab 11317, 1:1000) was used overnight at room temperature. A cross-adsorbed, donkey anti-rabbit-rhodamine red secondary antibody (Molecular Probes) was used at a dilution of 1:200. Samples were mounted with Vectashield hard set with Dapi mount (Vector Lab) and analyzed on a on a Zeiss Applied Precision Deltavision microscope equipped with a Mercury 100W lamp with a Coolsnap camera using a 60x NA0.75 objective. The number of centrosomes per cell was quantified by counting 5 random fields. To examine proliferation in thymic tissue, sections were stained with a rabbit antibody to the proliferation antigen Ki-67 (1:1000) overnight. A cross-adsorbed, pig anti-rabbit-biotinylated secondary antibody (Molecular Probes) was used at a dilution of 1:200. Staining was amplified with Vecastain ABC kit (Vector Labs), developed with Vector VIP peroxidase substrate and counterstained with methyl green. 111 Acknowledgements I would like to thank Chris Dillon and Qing Ge for their assistance in immunology techniques. I thank Roderick Bronson for pathology expertise. I thank Michael Brown and the Core Histology facility at the Center for Cancer Research at MIT for preparing tissue sections and H&E slides. I thank Sunny Wong for reagents and helpful discussions. I thank members of Tyler Jacks lab, especially Mandy Tam, for mice and advice. I thank Annagret Schulze-Lutum and the members of the Sorger lab for reagents and helpful discussions. 112 References Abrieu, A., L. Magnaghi-Jaulin, J.A. Kahana, M. Peter, A. Castro, S. Vigneron, T. Lorca, D.W. Cleveland, and J.C. Labbe. 2001. Mpsl is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 106:83-93. Amon, A. 1999. The spindle checkpoint. CurrOpin Genet Dev. 9:69-75. Attardi, L.D., and L.A. Donehower. 2005. Probing p53 biological functions through the use of genetically engineered mouse models. Mutat Res. 576:4-21. Baker, D.J., K.B. Jeganathan, J.D. Cameron, M. Thompson, S. Juneja, A. Kopecka, R. Kumar, R.B. Jenkins, P.C. De Groen, P. Roche, and J.M. Van Deursen. 2004. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. Baker, D.J., K.B. Jeganathan, L. Malureanu, C. Perez-Terzic, A. Terzic, and J.M. van Deursen. 2006. Early aging-associated phenotypes in Bub3/Rael haploinsufficient mice. J Cell Biol. 172:529-40. Bassing, C.H., H. Suh, D.O. Ferguson, K.F. Chua, J. Manis, M. Eckersdorff, M. Gleason, R. Bronson, C. Lee, and F.W. Alt. 2003. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 114:359-70. Boveri, T. 1914. Zur Frage der Entstehung maligner Tumoren. Fischer. Burds, A.A., A.S. Lutum, and P.K. Sorger. 2005. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. ProcNatl Acad Sci US A. 102:11296-301. Cahill, D.P., L.T. da Costa, E.B. Carson-Walter, K.W. Kinzler, B. Vogelstein, and C. Lengauer. 1999. Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics. 58:181-7. Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300-3. Camacho, E., S. Bea, I. Salaverria, A. Lopez-Guillermo, X. Puig, Y. Benavente, S. de Sanjose, E. Campo, and L. Hernandez. 2006. Analysis of Aurora-A and hMPS 1 mitotic kinases in mantle cell lymphoma. Int J Cancer. 118:357-63. Chester, N., H. Babbe, J.Pinkas, C. Manning, and P. Leder. 2006. Mutation of the Murine Bloom's Syndrome Gene Produces Global Genome Destabilization. Mol Cell Biol. 26:6713-26. 113 David, G., J.H. Dannenberg, N. Simpson, P.M. Finnerty, L. Miao, G.M. Turner, Z. Ding, R. Carrasco, and R.A. Depinho. 2006. Haploinsufficiency of the mSds3 chromatin regulator promotes chromosomal instability and cancer only upon complete neutralization of p53. Oncogene. Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635-45. Donehower, L.A., M. Harvey, B.L. Slagle, M.J. McArthur, C.A. Montgomery, Jr., J.S. Butel, and A. Bradley. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356:215-21. Draviam, V.M., S. Xie, and P.K. Sorger. 2004. Chromosome segregation and genomic stability. Curr Opin Genet Dev. 14:120-5. Duesberg, P., and R. Li. 2003. Multistep carcinogenesis: a chain reaction of aneuploidizations. Cell Cycle. 2:202-10. Fang, Y., T. Liu, X. Wang, Y.M. Yang, H. Deng, J. Kunicki, F. Traganos, Z. Darzynkiewicz, L. Lu, and W. Dai. 2006. BubRl is involved in regulation of DNA damage responses. Oncogene. 25:3598-605. Fisk, H.A., C.P. Mattison, and M. Winey. 2003. Human Mpsl protein kinase is required for centrosome duplication and normal mitotic progression. ProcNatl Acad Sci U SA. 100:14875-80. Fisk, H.A., C.P. Mattison, and M. Winey. 2004. A field guide to the Mpsl family of protein kinases. Cell Cycle. 3:439-42. Fisk, H.A., and M. Winey. 2001. The mouse Mpslp-like kinase regulates centrosome duplication. Cell. 106:95-104. Futamura, M., H. Arakawa, K. Matsuda, T. Katagiri, S. Saji, Y. Miki, and Y. Nakamura. 2000. Potential role of BRCA2 in a mitotic checkpoint after phosphorylation by hBUBR1. CancerRes. 60:1531-5. Gao, Y., D.O. Ferguson, W. Xie, J.P. Manis, J. Sekiguchi, K.M. Frank, J. Chaudhuri, J. Homer, R.A. DePinho, and F.W. Alt. 2000. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 404:897-900. Gupta, A., S. Inaba, O.K. Wong, G. Fang, and J. Liu. 2003. Breast cancer-specific gene 1 interacts with the mitotic checkpoint kinase BubR1. Oncogene. 22:7593-9. Hanks, S., K. Coleman, S. Reid, A. Plaja, H. Firth, D. Fitzpatrick, A. Kidd, K. Mehes, R. Nash, N. Robin, N. Shannon, J. Tolmie, J. Swansbury, A. Irrthum, J. Douglas, and 114 N. Rahman. 2004. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 36:1159-61. Harris, S.L., and A.J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24:2899-908. Hartwell, L.H., and M.B. Kastan. 1994. Cell cycle control and cancer. Science. 266:18218. Hartwell, L.H., and T.A. Weinert. 1989. Checkpoints: controls that ensure the order of cell cycle events. Science. 246:629-34. He, X., M.H. Jones, M. Winey, and S. Sazer. 1998. Mphl, a member of the Mpsl-like family of dual specificity protein kinases, is required for the spindle checkpoint in S. pombe. J Cell Sci. 111:1635-47. Hempen, P.M., H. Kurpad, E.S. Calhoun, S. Abraham, and S.E. Kern. 2003. A double missense variation of the BUB 1 gene and a defective mitotic spindle checkpoint in the pancreatic cancer cell line Hs766T. Hum Mutat. 21:445. Hoyt, M.A., L. Totis, and B.T. Roberts. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 66:507-17. Jacks, T., L. Remington, B.O. Williams, E.M. Schmitt, S. Halachmi, R.T. Bronson, and R.A. Weinberg. 1994. Tumor spectrum analysis in p53-mutant mice. CurrBiol. 4:1-7. Jallepalli, P.V., and C. Lengauer. 2001. Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer. 1:109-17. Jonkers, J., R. Meuwissen, H. van der Gulden, H. Peterse, M. van der Valk, and A. Berns. 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 29:418-25. Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277-82. Kim, H.S., K.H. Park, S.A. Kim, J. Wen, S.W. Park, B. Park, C.W. Gham, W.J. Hyung, S.H. Noh, H.K. Kim, and S.Y. Song. 2005. Frequent mutations of human Mad2, but not Bub 1, in gastric cancers cause defective mitotic spindle checkpoint. Mutat Res. 578:187-201. Kops, G.J., B.A. Weaver, and D.W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer.5:773-85. Langerod, A., M. Stromberg, K. Chin, V.N. Kristensen, and A.L. Borresen-Dale. 2003. BUBI infrequently mutated in human breast carcinomas. Hum Mutat. 22:420. 115 Lee, P.P., D.R. Fitzpatrick, C. Beard, H.K. Jessup, S. Lehar, K.W. Makar, M. PerezMelgosa, M.T. Sweetser, M.S. Schlissel, S. Nguyen, S.R. Cherry, J.H. Tsai, S.M. Tucker, W.M. Weaver, A. Kelso, R. Jaenisch, and C.B. Wilson. 2001. A critical role for Dnmtl and DNA methylation in T cell development, function, and survival. Immunity. 15:763-74. Leng, M., D.W. Chan, H. Luo, C. Zhu, J. Qin, and Y. Wang. 2006. MPS 1-dependent mitotic BLM phosphorylation is important for chromosome stability. Proc Natl Acad Sci USA. 103:11485-90. Levine, A.J. 1997. p53, the cellular gatekeeper for growth and division. Cell. 88:323-31. Li, R., and A.W. Murray. 1991. Feedback control of mitosis in budding yeast. Cell. 66:519-31. Liao, M.J., X.X. Zhang, R. Hill, J. Gao, M.B. Qumsiyeh, W. Nichols, and T. Van Dyke. 1998. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol. 18:3495-501. Liu, S.T., G.K. Chan, J.C. Hittle, G. Fujii, E. Lees, and T.J. Yen. 2003. Human MPS I1 Kinase Is Required for Mitotic Arrest Induced by the Loss of CENP-E from Kinetochores. Mol Biol Cell. 14:1638-51. Loeb, L.A. 1991. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 51:3075-9. Loeb, L.A., K.R. Loeb, and J.P. Anderson. 2003. Multiple mutations and cancer. Proc NatlAcadSci USA. 100:776-81. Loonstra, A., M. Vooijs, H.B. Beverloo, B.A. Allak, E. van Drunen, R. Kanaar, A. Berns, and J. Jonkers. 2002. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Martinez-Exposito, M.J., K.B. Kaplan, J. Copeland, and P.K. Sorger. 1999. Retention of the BUB3 checkpoint protein on lagging chromosomes. Proc Natl Acad Sci US A. 96:8493-8. Matsuura, S., Y. Matsumoto, K. Morishima, H. Izumi, H. Matsumoto, E. Ito, K. Tsutsui, J. Kobayashi, H. Tauchi, Y. Kajiwara, S. Hama, K. Kurisu, H. Tahara, M. Oshimura, K. Komatsu, T. Ikeuchi, and T. Kajii. 2006. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 140:35867. McPherson, J.P., M.P. Hande, A. Poonepalli, B. Lemmers, E. Zablocki, E. Migon, A. Shehabeldin, A. Porras, J. Karaskova, B. Vukovic, J. Squire, and R. Hakem. 2006. A role for Brcal in chromosome end maintenance. Hum Mol Genet. 15:831-8. 116 Meraldi, P., V.M. Draviam, and P.K. Sorger. 2004. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 7:45-60. Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V. Murty, and R. Benezra. 2001. MAD2 haploinsufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 409:355-9. Mills, G.B., R. Schmandt, M. McGill, A. Amendola, M. Hill, K. Jacobs, C. May, A.M. Rodricks, S. Campbell, and D. Hogg. 1992. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. JBiol Chem. 267:16000-6. Morales, J.C., S. Franco, M.M. Murphy, C.H. Bassing, K.D. Mills, M.M. Adams, N.C. Walsh, J.P. Manis, G.Z. Rassidakis, F.W. Alt, and P.B. Carpenter. 2006. 53BP1 and p53 synergize to suppress genomic instability and lymphomagenesis. Proc NatlAcadSci USA. 103:3310-5. Nowak, M.A., N.L. Komarova, A. Sengupta, P.V. Jallepalli, M. Shih le, B. Vogelstein, and C. Lengauer. 2002. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci US A. 99:16226-3 1. Oikawa, T., M. Okuda, Z. Ma, R. Goorha, H. Tsujimoto, H. Inokuma, and K. Fukasawa. 2005. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol. 25:4046-61. Purdie, C.A., D.J. Harrison, A. Peter, L. Dobbie, S. White, S.E. Howie, D.M. Salter, C.C. Bird, A.H. Wyllie, M.L. Hooper, and et al. 1994. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 9:603-9. Saeki, A., S. Tamura, N. Ito, S. Kiso, Y. Matsuda, I. Yabuuchi, S. Kawata, and Y. Matsuzawa. 2002. Frequent impairment of the spindle assembly checkpoint in hepatocellular carcinoma. Cancer.94:2047-54. Sato, N., K. Mizumoto, M. Nakamura, K. Nakamura, M. Kusumoto, H. Niiyama, T. Ogawa, and M. Tanaka. 1999. Centrosome abnormalities in pancreatic ductal carcinoma. Clin CancerRes. 5:963-70. Seo, J., Y.S. Chung, G.G. Sharma, E. Moon, W.R. Burack, T.K. Pandita, and K. Choi. 2005. Cdtl transgenic mice develop lymphoblastic lymphoma in the absence of p53. Oncogene. 24:8176-86. Stucke, V.M., C. Baumann, and E.A. Nigg. 2004. Kinetochore localization and microtubule interaction of the human spindle checkpoint kinase Mps 1. Chromosoma. 113:1-15. Stucke, V.M., H.H. Sillje, L. Arnaud, and E.A. Nigg. 2002. Human Mpsl kinase is required for the spindle assembly checkpoint but not for centrosome duplication. Embo J. 21:1723-32. 117 Taylor, S.S., M.I. Scott, and A.J. Holland. 2004. The spindle checkpoint: a quality control mechanism which ensures accurate chromosome segregation. Chromosome Res. 12:599-616. Wang, Q., T. Liu, Y. Fang, S. Xie, X. Huang, R. Mahmood, G. Ramaswamy, K.M. Sakamoto, Z. Darzynkiewicz, M. Xu, and W. Dai. 2004a. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 103:1278-85. Wang, Z., J.M. Cummins, D. Shen, D.P. Cahill, P.V. Jallepalli, T.L. Wang, D.W. Parsons, G. Traverso, M. Awad, N. Silliman, J. Ptak, S. Szabo, J.K. Willson, S.D. Markowitz, M.L. Goldberg, R. Karess, K.W. Kinzler, B. Vogelstein, V.E. Velculescu, and C. Lengauer. 2004b. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 64:2998-3001. Ward, I.M., S. Difilippantonio, K. Minn, M.D. Mueller, J.R. Molina, X. Yu, C.S. Frisk, T. Ried, A. Nussenzweig, and J. Chen. 2005. 53BP1 cooperates with p53 and functions as a haploinsufficient tumor suppressor in mice. Mol Cell Biol. 25:10079-86. Wei, J.H., Y.F. Chou, Y.H. Ou, Y.H. Yeh, S.W. Tyan, T.P. Sun, C.Y. Shen, and S.Y. Shieh. 2005. TTK/hMpsl participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. JBiol Chem. 280:7748-57. Weiss, E., and M. Winey. 1996. The Saccharomyces cerevisiae spindle pole body duplication gene MPS 1 is part of a mitotic checkpoint. J Cell Biol. 132:111-23. Winey, M., L. Goetsch, P. Baum, and B. Byers. 1991. MPS I and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol. 114:745-54. Wong, S.Y., H. Haack, D. Crowley, M. Barry, R.T. Bronson, and R.O. Hynes. 2005. Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. CancerRes. 65:9789-98. Wu, L., and I.D. Hickson. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 426:870-4. Yuan, B., Y. Xu, J.H. Woo, Y. Wang, Y.K. Bae, D.S. Yoon, R.P. Wersto, E. Tully, K. Wilsbach, and E. Gabrielson. 2006. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin CancerRes. 12:405-10. 118 Chapter 3 Loss of p53, not p 1 9A RF can rescue MpslA induced lethality in mouse embryonic fibroblasts Note: All figures are the author's own work except Figure 3.1B was provided by Ying Yue. 119 Abstract Chromosomal instability (CIN) is a state where cells frequently gain or lose entire chromosomes during divisions, a common hallmark in human cancer. The spindle checkpoint is one cellular surveillance system that ensures accurate chromosome segregation during mitosis to prevent CIN. Although spindle checkpoint components are infrequently mutated in human cancers, a partial loss of function mutation in the checkpoint kinase Mpsl, called MpslA, has previously been shown to facilitate tumorigenesis in mice in a p53-dependent manner. The tumor suppressor p53 promotes either apoptosis or cellular growth arrest in response to genomic insults. Mouse embryonic fibroblasts (MEFs) are used to gain a better understanding of the synergy between Mpsl and p53. I show here that MpslA MEFs experience chromosome missegregation events, particularly of a few lagging chromosomes. This further implicates MpslA as a CIN inducing mutation which can act as a causal agent for tumor development. CIN decreases viability of Mpsl"• AMEFs, which is rescued by simultaneous deletion of p53. Inactivation of pl 9 ARF does not rescue cellular viability of Mpsl"IA MEFs, although MpslA-induced CIN promotes lymphomagenesis in some pl19 ARF heterozygous mice. In summary, CIN induced by MpslA promotes p53dependent apoptosis, not senescence, and requires downregulation of p53 to facilitate tumor development. 120 Introduction Accurate chromosome segregation, which is essential for maintaining genomic stability, is ensured by the spindle assembly checkpoint (Draviam et al., 2004). The spindle checkpoint delays the onset of anaphase until all sister chromatids have achieved proper bipolar attachment to microtubules from the mitotic spindle (Amon, 1999). Checkpoint proteins are able to sense a "wait anaphase" signal emanating from unattached kinetochores. Canonical checkpoint components, conserved from yeast to man, include Madl, Mad2, Bubl, BubR1, Bub3, and Mpsl (Bharadwaj and Yu, 2004; Fisk and Winey, 2004; Taylor et al., 2004). These proteins along with additional checkpoint proteins such as Aurora B and the Zwl0O/Rod complex localize to kinetochores and act to inhibit mitotic progression by preventing Cdc20 from binding and activating the anaphase promoting complex (APC/C), an E3 ubiquitin ligase (Cohen-Fix et al., 1996; Yu, 2002). Mutation of spindle checkpoint components is one way to cause chromosomal instability (CIN), the continual gain or loss of chromosomes during mitosis, and result in an aneuploid state (Baker et al., 2004; Dobles et al., 2000; Kalitsis et al., 2000; Michel et al., 2001). The unequal segregation of chromosomes to daughter cells during mitosis causes aneuploidy, which since Theodore Boveri, is known to be both detrimental to the daughters and their subsequent progeny (Boveri, 1914). Boveri went on to postulate that a state of aneuploidy may be sufficient to promote tumorigenesis (Boveri, 1914). Although most human solid tumors are aneuploid, and many cancer cell lines exhibit CIN, unequivocal demonstration that CIN can be a causal agent for tumor development and not just a consequence is still minimal in human cancer (Draviam et al., 2004; Kops 121 et al., 2005). Spindle checkpoint mutations have been found in a variety of human cancers, but are not frequent (Cahill et al., 1998; Hempen et al., 2003; Kim et al., 2005; Matsuura et al., 2006; Tighe et al., 2001; Wang et al., 2004b; Yoon et al., 2002). A means to determine if CIN resulting from spindle checkpoint abrogation can promote tumorigenesis in vivo is by genetic inactivation of checkpoint genes in mice. However, complete inactivation - of Mad2, Bub3, and BubRi - causes embryonic lethality, demonstrating the essentiality of the spindle checkpoint to mammalian cell division and provides a basis for infrequent spindle checkpoint mutations in human cancer (Dobles et al., 2000; Kalitsis et al., 2000; Wang et al., 2004a). Therefore, conditional mouse models utilizing such techniques as Cre/LOX are necessary to study the role of the spindle checkpoint in cancer. I previously demonstrated the deletion of 107 residues from the N-terminal region of Mpsl, a spindle checkpoint kinase, creates a hypomorph where the spindle checkpoint can still respond to microtubule insults, but causes embryonic lethality in mice and chromosome missegregation in thymocytes (see chapter 2). This Mps 1Amutation was introduced into mice as a conditional flanked by LoxP (flox or f) allele, crossed to p53 conditional knockout mice and specifically inactivated in the thymus with the Lck-Cre transgene. Intriguely, the combination of this MpslA mutation with just one inactivated allele of p53 is sufficient to cause T cell lymphoma with full penetrance in mice with an average tumor onset of 3.4 months. MpslA appears to promote the loss of heterozygosity / (LOH) of the wildtype p53 allele in these tumors. Time of tumor onset of Mpsl f/f, p53 ", Lckcre ÷ mice are similar to Mpsl f/f, p53"f f, Lckcre÷ mice and are accelerated as compared to Mpsl /+ , p53 f", Lckcre÷ mice. These results suggest the LOH of p53 must be an early 122 event in tumor evolution. Moreover, Mps f/f; Lckcre ÷ thymoma cells were shown to be highly aneuploid and histological tumor sections contained anaphase figures with lagging chromosomes suggesting the mechanism of tumor acceleration is through increased CIN. Therefore, these data suggest that CIN promoted by a weaken checkpoint allele facilitates tumorigenesis in a p53-dependent manner. The tumor suppressor gene, TP53, is the most frequently mutated gene in human cancers, because its protein product, p53 which is often described as a guardian of the genome, is critical for the response to cellular stress, particularly DNA damage (Levine, 1997). Normally, p53 protein levels in a cell are tightly regulated in a number of positive and negative feedback loops primarily through ubiquitin-mediated proteolysis (Harris and Levine, 2005). Upon sensing cellular stress, cells promote the post-translational modification of p53 which increases its stability and allows for transactivation of genes involved in cell cycle arrest, DNA repair or apoptosis. Two key modulators of p53 protein level are Mdm2, an E3 ubiquitin ligase, and pl1 9 ARF, an inihibitor of Mdm2. The primary function of p1 9 ARF is to increase p53 protein stability by antagonizing Mdm2 function in response to cellular stress, by binding and sequestering Mdm2 into the nucleous and thus preventing p53 degradation (Kamijo et al., 1998; Pomerantz et al., 1998; Weber et al., 1999). pl1 9 ARF is normally silenced during embryogenesis as Arf expression results in cellular senescence. Like p53, pl 9 ARF is also a potent tumor suppressor whose locus is frequently mutated in human cancer and whose inactivation in mice promotes tumors, but is primarily dependent on inactivating the p53 pathway (Kamijo et al., 1999; Moore et al., 2003; Weber et al., 2000). 123 What is the cause of organismal and cellular lethality upon spindle checkpoint inactivation when MpslA is expressed? Mad2 null embryos exhibit both chromosome missegregation and apoptosis, which appears to be dependent on p53 (Burds et al., 2005; Dobles et al., 2000). The simultaneous deletion of both Mad2 and p53 rescues the lethality of Mad2 -' -blastocysts and mouse embryonic fibroblasts (MEFs) suggesting p53 is promoting cell death in response to missegregation events (Burds et al., 2005). In contrast, decreased levels of the spindle checkpoint proteins BubR1 causes early onset aging and infertility in mice and cellular senescence in MEFs (Baker et al., 2004). Furthermore, Bub3+/-; Rae 1+' mice also have early aging phenotypes and compound mutant MEFs senesce prematurely and show increased expression of the senescence markers p53, p1 9 , p16 and p21 (Baker et al., 2006). However, Mad2÷/- MEFs do not exhibit premature senescence nor upregulate the p53 and p16 growth inhibition pathways (Baker et al., 2006). Consequently, both p53-dependent apoptosis and cellular senescence responds to decreased levels of spindle checkpoint proteins and suggests inactivation of p53 is required to relieve MpslA lethality. Here, I seek to better understand the synergy of the Mps lA truncation mutation and p53 inactivation in vivo by determining if MpslA causes CIN a prioriin MEFs and if p53 inactivation is required to rescue MpslA lethality ex vivo. I show that the MpslA mutation is lethal to cells in culture in addition to in vivo. Moreover, MpslA dependent cellular lethality appears to be due to chromosome missegregation events as shown through time lapse imaging of MpslA/A; p53 -/-MEFs. The combination of p53 inactivation with the Mpsl truncation mutation rescues the viability of MEFs suggesting MpslA induced cellular lethality is through p53-mediated apoptosis. This rescue occurs 124 when just one allele of p53 is inactivated, albeit with a delay. In contrast, deletion of pl19 ARF is unable to restore viability to Mpsl ' /AMEFs. Furthermore, inactivating one allele of p 1 9ARF in Mpsl conditional mutant mice does not recapitulate the tumor penetrance of Mpslr f, p53f/ +, Lckcre+ mice. Thus, the MpslA mutation activates p53 in cells with lagging chromosomes and suggests there is strong selective pressure for inactivation of the p53 pathway in situations of CIN-facilitated tumorigenesis. Results MpslA decreases viability of MEFs, which is rescued by p53 inactivation To determine if the homozygous expression of the MpslA mutation causes cellular lethality, Mps~fMEFs and Mpsl +/+ MEFs were infected with an adenovirus expressing Cre recombinase fused to green fluorescent protein (AdCre-GFP). Cre-expressing cells were isolated by flow cytometry 48 hours after infection by gating for GFP positive cells. Hence, cells with conditional alleles that have been infected with Cre recombinase are denoted as the recombined allele. Following cell sorting, cells, now at passage 3 (P3) and considered to be at day=0 on growth curves, were placed into culture and their growth analyzed every 3 days (Fig 3.1A). Cre recombinase efficiently converts the Mpsl flox allele from the full-length form (wt) to the truncated Amutation as shown by RT-PCR (Fig 3.1C). Wildtype MEFs (Fig 3.1B shown in blue) initially grew in culture, but stopped growing by day 21 (equivalent to passage 10) and become senescent with positive staining for 13-galactisodase (data not shown) as expected (Todaro and Green, 1963). In contrast, MpslA/A MEFs (Fig 3.1B shown in green) die in culture with cell number decreasing as much as 10 fold after 3 days. In accordance with in vivo 125 Figure 3.1 Figure 3.1 Infect Passage 2MEFs with Adenoviral Cre-GFP (7x106PFU) 1 48 hours post-infection - 40-70% GFP+ Sort by flow cytometry for GFP+ cells 1 Passage cells about every 3 days and determine rate of growth 0 6 12 18 Time (Days) 24 30 RT-PCR RT-PCR 1 2 3 4 1.00E+11 p5% Mp p53 C 1.UUI00Eu 1.00E+05 Mpsl r 1.00E+02 GAPDH 1.00E-01 0 t 6 t 12 18 24 Time (days) 30 36 RT-PCR 1 9 P3 A 5 p Mps p53 I Ito GAPDH I1 126 P6 P15 Figure 3.1: MpslA decreasesMEF viability which is rescued by inactivatingp53. (A) A schematic showing how the MEF growth assay is conducted. MEFs were infected with Ad-CreGFP and placed into culture and their growth followed. (B) Averaged growth curves of Mpsl +/+; p53 /+ (blue), Mps +/+; p53 +/-(orange), Mps +/+; p53'/ - (red), Mpsl'AA; p53' / + (green), Mpsl'A/; p53 + '1(turquoise), and Mpsl/A; p53-'(pink) MEFs. Results are from at least two independent experiments. (C) mRNA expression levels of p53 and Mpsl in GFP + P3 MEFs 48 hours after AdCreGFP infection. GAPDH is used as a control. D) Growth curves of Mpsl+'; p53 -/- (red) and MpslOA; p53 /- (pink) MEFs. (E)mRNA expression levels of p53 in passage 12 MEFs to show decreased expression p53 in Mpsl"A; p53 / -MEFs. GAPDH is used as a control. 127 findings where MpslA causes embryonic lethality and decreased thymus size (Chapter 2), this mutation also decreases viability in MEFs in vitro. The inactivation of p53 in mice where MpslAA is expressed restores thymus size and leads to lymphoma. To determine the effect of deleting p53 and mutating Mps 1 in MEFs, Mpsl+/+; p53 -', Mpsl'AA; p53' -, Mpsl+/+; p53'1- and MpslA/A; p53 ÷1- MEFs were generated and their growth followed (Fig 3.1A). Inactivating p53 has been shown to bypass replicative senescence and immortallize cells in culture (Harvey et al., 1993). As expected, Mpsl +/+; p53 - MEFs (Fig 3.1B shown in red) grow faster than control MEFs and continue to have high growth rates for as long as their growth was followed up to 42 days (Fig 3.1D). Although Mpsl"AA; p53-/' MEFs initially show decreased viability compared to Mpsl+/+; p53 +' + MEFs and Mpsl+/+; p53- MEFs, inactivating p53 restores viability and MpslA/A; p53/ -MEFs also become immortal (Fig 3.1B shown in pink). Inactivating just one allele of p53 also restores growth to MpslA cells, but Mpsl /A; p53 +/ MEFs experience a longer delay of at least 21 days post sorting before having a positive growth rate (Fig 3.1B shown in turquoise). Mps1 +/; p53 /- MEFs also become immortal in culture with growth kinetics similar to Mps1 A; p53-/- MEFs (Fig 3.1B in orange). RT-PCR shows p53 mRNA is lost from Mpslf/f; p5 3f/fMEFs at 48 hours postinfection by Ad-CreGFP (day 0 on growth curves), but does not decrease the levels of p53 mRNA for Mpsl +/; p53 /-MEFs (Fig 3.1C). However, at P12, MpslA/A; p53' /- MEFs show significantly lower levels of p53 mRNA than Mpsl+/+; p53+/' MEFs suggesting there is selective pressure to lose the wildtype allele of p53 (Fig 3.1E). Thus, inactivating one or both alleles of p53 can rescue MpslA induced cell inviability in MEFs. 128 MpslA increases chromosome segregation defects, but not centrosome amplification in p53 null MEFs Two major causes of CIN are defects in the spindle checkpoint and the amplification of centrosomes (Draviam et al., 2004). Histology of the sporadic Mpsl AIA lymphomas showed a few mitotic cells with lagging chromosomes suggesting the spindle checkpoint is partially compromised by the MpslA mutation (Chapter 2). An aberrant spindle checkpoint would result in chromosome missegregation events such as lagging chromosomes during anaphase. Such events are quite dynamic and transitory and as such, live cell microscopy is an appropriate method for observing mitotic cells. To investigate chromosome segregation during mitosis of cells expressing MpslA, Mpsl +/+; p53" /' and Mpsl 'A; p53 -/ -MEFs were infected with retroviral histone 2B-GFP (H2B-GFP) and imaged at passage 16 by time lapse imaging. More than 200 mitotic cells were analyzed for chromosome segregation defects at anaphase by the presence of lagging chromosomes. Cells lacking p53 are known to have centrosome amplification which also can result in chromosome missegregation (Fukasawa et al., 1996). Furthermore, chromosome nondisjunction has been shown to lead to tetraploidy due to lack of cytokinesis in cells imaged by time-lapse imaging (Shi and King, 2005). Therefore, cells undergoing mitosis were observed until they underwent cytokinesis and those mitotic cells that underwent anaphase with lagging chromosomes but did not undergo cytokinesis " / mitotic cells exhibited lagging are categorized separately. While -16% of Mpsl+/+; p53- chromosomes at anaphase but otherwise successfully exited mitosis, more than twice the number of Mpsl /A; p53-/- mitotic cells imaged (-43%) missegregated chromosomes (Fig 3.2A). Unaligned chromosomes at metaphase often led to lagging chromosomes at anaphase which would yield microcnuclei. The percentage of mitotic cells that had 129 Figure 3.2 60% I Mpsl+/+; p53-/- (n=205) I MpslA/A; p53-/- (n=244) 4 50% O 40% E - 30% 4) a 20% 0) a 10% m 0% I,. _L SVandO \ o . \\1Oo ~i~En\"4`m •o 00 aqq. sxeaoe \•o•\•oJ u,- ° • ' - Mpsl x f/f ,,·O p5 3 f/ f at p6 6 4 DNA content 130 0 30 60 90 Time (min) 120 150 Figure 3.2: MpslA is a partialloss offunction mutation that increaseschromosome missegregationin p53-null MEFs. Time lapse imaging of H2B-GFP expressing MEFs were taken every 3 minutes for a 6 hour period to observe chromosome segregation and mitotic timing. (A) Histogram of mitotic defects in Mpsl +/+; p53 -/ (green) and MpslAIA; p53 -'- (blue) MEFs at anaphase. Mitotic cells were placed into 4 catergories: 1) Cells were considered normal if they progressed through mitosis without chromosome segregation errors and successfully completed cytokinesis to yield two daughter cells. 2) Cells with chromosomes at the spindle midzone after anaphase A were characterized as having lagging chromosomes. 3) If cells had lagging chromosomes and did not complete anaphase, they were considered "abnormal with lagging". 4) If cells did not complete cytokinesis, mitosis was otherwise normal, they were characterized as "no lagging, but abnormal". (B) Cumulative frequency graph of mitotic timing in Mpsl+/+; p53 -' - (green) and MpslA"; p53'- - (blue) MEFs. (C) Two representative mitotic cells, one with lagging chromosomes at anaphase (arrow) and one proceeding to anaphase normally are shown from NEBD (T=O) to the onset of anaphase in 3 minute intervals. (D) DNA FACS profiles of P6 Mpsl"A; p53 -' - MEFs with and without nocodazole treatment. 131 Table 3.1: Analysis of mitotic timing in MEFs Genotype Nceii s peak time a Rangec min max Mpsl+/+; p53-/- 205 33.1 10.3 18 75 MpslA/A; p53-/- 244 30.1 7.0 18 57 b a Peak time of the best-fitted frequency distribution. deviation for a best-fitted normal distribution. c Range after exclusion of the top and bottom 3%values. b Standard 132 Table 3.2: Mpsl A does not induce centrosome duplicationin MEFs genotype Mpsl+/+; p53+/+ MpslA/A; p53+/+ MpslA/A; p53+/MpslA/A; p53-/Mpsl+/+; p53-/- N mitotic cells 100 156 197 198 346 % abnormal a 21% 15% 18% 11% 36% Cells are considered to have an abnormal number of centrosomes if # of gamma-tubulin foci per mitotic cell is greater than 2. a 133 lagging chromosomes but did not successfully complete cytokinesis is comparable in Mpsl+/+; p53 -' and MpslA/'; p53-'- MEFs, -10% and -8% respectively (Fig 3.2A). Thus, MpslA causes an extreme CIN phenotype in p53-null MEFs, mostly through improper chromosome segregation. The presence of lagging chromosomes at anaphase can be caused by improper mitotic timing as well as impaired spindle checkpoint function. The checkpoint proteins Mad2 and BubR1 have been shown to regulate mitotic progression, and their depletion by siRNA in human cells causes faster mitotic timing (Meraldi et al., 2004). To determine if there is a difference in mitotic timing between Mpsl+'+; p53 -' and Mpsl"A; p53 -' MEFs, the mitotic cells examined for chromosome segregation defects were analyzed for the time from nuclear envelope breakdown to the onset of anaphase A (Fig 3.2C and Table 3.1). Two representative Mpsl&A; p53 -'- cells are shown in figure 3.2B with NEBD at time=0 (Meraldi et al., 2004; Rieder et al., 1994). There is little difference in mitotic progression in Mpsl+/+; p53 -'- and MpslA/A; p53 -' MEFs as shown in a cumulative frequency plot of mitotic cells (Fig 3.2C). There is only a slight difference in the ranges of mitotic timing for the two cell populations, with Mpsl A/A; p53 -1-MEFs ranging from 18 to 57 minutes and Mpsl+/+; p53 -'- MEFs ranging from 18 to 75 minutes (Table 3.1). MpslA/A; p53 -/-MEFs reach anaphase slightly faster than Mpsl+/'; p53 -/-MEFs with a peak time of 30.1 minutes versus 33.1 minutes (Table 3.1). However, this is within the standard deviation and the timing differential is unlikely to be statistically significant. Hence, MpslA does not function in mitotic progression in MEFs. Mps 1 was initially discovered in Saccharomyces cerevisiaeto be required for both the duplication of the yeast centrosome and for spindle checkpoint function (Weiss 134 and Winey, 1996; Winey et al., 1991). Furthermore, centrosome amplification is a major route to aneuploidy and often observed in tumors (Fisk et al., 2002; Nigg, 2002). Therefore, to ascertain if there is a defect in centrosome duplication in cells with the Mps lA mutation, five MEF lines of various Mpsl and p53 genotypes were infected with adenoviral Cre recombinase, and cells were fixed and stained for the centrosomal protein y-tubulin 5 days post infection. The number of centrosomes was counted for at least 100 mitotic cells per cell line, and those cells with 3 or more centrosomes during mitosis are considered abnormal (Table 3.2). Mpsl+/'; p53 +' ÷ MEFs treated with adenoviral Cre have ~21% of mitotic cells with abnormal centrosomes. Mps 1A does not appear to cause centrosome amplification as the 3 MEF lines with Mpsl mutated - MpslAIA; p53 +/+, MpslV'A; p53 +/ -, and MpslA"A; p53 - - MEFs - have a smaller percentage of cells with abnormal centrosomes than Mpsl +/+; p53 1+ MEFs. In contrast, -36% Mpsl/+; p53 - - MEFs show centrosome amplification which is consistent with the -35% published for early passage p53-null mouse cells (Oikawa et al., 2005). Mpsl AA; p53 -'- MEFs still retain an active spindle checkpoint when challenged by the microtubule destabilizer nocodazole as more cells accumulate at G2/M with 4N DNA content after nocodazole treatment (Fig 3.2D). This is consistent with data from MpslA/A; p53 -/- tumor cells and further suggests the MpslA mutation is a hypomorph (Chapter 2). These data suggest that cells with MpslA truncation protein have a partially impaired spindle checkpoint and not a defect in centrosome duplication as mutant MEFs missegregate chromosomes during mitosis but do not display centrosome amplification.MpslA/A; p53-/ - MEFs exhibit more dynamic CIN than Mpsl+'; p53 /- MEFs 135 In vivo data suggests MpslA is inducing CIN. If this is also the case ex vivo, the ploidy of MpslA-expressing MEFs should show dynamic changes through multiple divisions. Here, I show that MpslA also causes CIN ex vivo through chromosome missegregation in MpslA"A; p53' - MEFs. To investigate the effect of Mps mutation on ploidy in MpslNA; p53/ -' MEFs, over time in culture, MpslAIA; p53 -' and Mpsl'/+; p53/ MEFs were fixed at P3, P6, P15 and P19 and their DNA content analyzed by flow cytometry through propidium iodide staining. P3 MEFs were immediately fixed after sorting for Ad-CreGFP positive cells. The remaining cells were collected from Mps 1 A; p53 -' - and Mps1 /+;p53 '-MEFs whose growth is depicted in figure 3.1D. P3 MEFs show typical cell cycle profiles with distinct GI and G2 peaks and appear mostly euploid regardless of genotype (Fig 3.3A). The percentage of cells in G1 for Mps lNA; p53-'- and Mpsl+/+; p53 -' MEFs are lower than cells with wildtype p53 which has been previously described to occur with inactivation of p53 (Harvey et al., 1993). Cells with inactivated p53 have often been shown to become tetraploid in culture (Andreassen et al., 2001; Cross et al., 1995; Margolis et al., 2003). At P6 (day 9 on the growth curve), Mps1 '+; p53-'1 MEFs display a distinct skewness past the G2/4N peak where cells are no longer euploid with a small population at up to 8N (Fig 3.3B). In contrast, MpslA/A; p53 -/-MEFs appear to exhibit a fairly normal DNA content at P6 with a negligible population of cells at >4N. By P15, Mpsl +';p53 -/-MEFs have all shifted to 4N and 8N in culture and the DNA profile remains largely unchanged 12 days later at P19 (Fig 3.3C). However, Mpsl"A; p53 -/ MEFs are more heterogeneous in DNA content at P15 with distinct peaks at 2N, 4N, and 8N (Fig. 3.3C). By P19, most of the Mpsl"/A; p53 -/-MEFs have genomes that are mostly at 6N, with a negligible 2N population and a small tetraploid population 136 Figure 3.3 A Mpl +/+p53M • Mpal I ff Mpslff P63+ +/+ ps53 M p53 psl+/+ 8n DNA content Mpe1'/' p6if / f/f lfM DNA content C P15 Mpel +/+ p53 P19 53ff DNA content 137 Figure 3.3: MpslA increasesCIN in p53-null MEFs through multiple passages. (A, B, and C) DNA FACS profiles using propidium iodide to visualize DNA of GFP + P3 MEFs 48 hours after Ad-CreGFP infection (A) P6 MEFs (B) and P15 and P19 MEFs (C). P2 wildtype MEFs are used as a control to illustrate the location of the 2N and 4N peaks. 138 " remaining (Fig 3.3C). While Mpsl +';p53 - -MEFs remain mostly tetraploid and octoploid in culture through multiple passages, the MpslA mutation dramatically changes the DNA content in p53-null MEFs presumably due to increased CIN during cell division. Inactivating pl9ARF does not rescue MpsA induced cellular lethality Is the cause of decreased viability in MpsA/A MEFs p53-dependent? Inactivation ofpl 9 ARF , like p53 loss, results in immortal MEFs (Dimri et al., 2000; Weber et al., 1999). Although it is not expressed during embryogenesis, pl 9 ARF expression is induced in cells placed into culture and causes sensescence. To determine if pl 9 ARF inactivation can also rescue the growth defects of MpslA MEFs, I generated Mpsl f; pl9ARFI+/ and Mps 1A; pl9ARF- /- MEFs, infected them with AdCre-GFP and followed their growth in the manner described above. Mpsl /A; p 1 9ARF+ /- and Mpsl1ff; p19 A"- - MEFs Ad-CreGFP) exhibit growth in culture over time, but Mpsl tA; pl9 ARF /- (negative for and Mpsl AA; pl1 9 ARF-/- MEFs do not grow in culture (Fig 3.4A). By 21 days, there were too few Mpsl A; pl9 ARF / " -and Mps 1"; p 1 9 ARF- /- MEFs alive to continue the growth assay. Interestingly, pl9ARF-null MEFs retain expression of p53 (Kamijo et al., 1997). These results show that inactivating pl9ARF does not rescue Mpsl"A MEF inviability, and suggests lethality of MpslA MEFs is p53-dependent. Mps1f'/; pl9ARF+/-; Lckcre+ mice develop tumors with an increase penetrance over Mpslf/f; Lckcre+ mice To further investigate the role ofpl9ARF and the p53 pathway in facilitating Mps lA induced tumorigenesis in T cells, I generated Mps 1 ; pl 9 ARF+/; Lckcre + mice and 139 Figure 3.4 A4 10 C 0 c - 1( 0 N 0. 0 C 0.0 s O.0c 0.003 0 6 3 9 12 Days 18 15 21 B D 10 S 8 (A +- 60 4.. = 0r u, I- 40 Mps1f/ p19+ 20 Mps1l/ Mps1 0u 0. Lck-cre + 1 2 3 4 Ow.. - S T pl9ARF WME p53 p19+/ Lck-cre p19+/ Lck-cre + 0 0 T ~~::--- 5 6 months 140 Figure 3.4: Mps1A decreases viability ofpl9ARF knockout MEFs, but increases tumorigenesis inpl9ARF+/- mice. (A) Average growth curves ofMpslI/A; pl19 ARF+/- (purple), Mps 1 A/A; pl 9 ARF-/- (orange), MpslEf; p1 9ARF+ /- (blue) and Mpslf/f; p19 ARF /-(red) MEFs. The MEF growth assay was done as described in the schematic in figure 3.1A, except GFP-negative cells were also collected as controls (where Mpsl is still the wildtype conditional allele denoted as Mps 1 'f). Results are from two independent experiments. (B) Kaplan-Meyer survival curves for Mpsl f;p 1 9 ARF+/-; Lckcre- control mice (n=9), Mpsl If; p 19AR+/-; Lckcre + (n=14) and Mpsl f; p19ARF+/+; Lckcre + (n=30). Data for Mps 1f/f; p 9ARF+/+; Lckcre+ was previously reported in chapter 2. (C) Hematoxylin and eosion stained images of a thymic tumor from Mps 1f/f; pl 9 ARF+/-; Lckcre + mice showing the typical "starry sky" pattern typical of lymphoma and a control for Mpsl/+; p 9ARF+/+; Lckcre ÷ thymus where thymocytes are smaller and more regular. (D) PCR analysis of pl1 9 ARF and p53 of thymic tumor DNA (T) shown in figure 3.4C and spleen DNA (S) from two Mpslf/f; p9ARF+/-; Lckcre + mice. 141 followed the survival of a cohort of 14 mice for at least 6 months. All Mpslrf; pl 9 ARF+/-; Lckcre-control mice are normal at 6 months, but 29% of Mps f/f; pl 9 ARF+/-; Lckcre ÷ were moribund due to lymphoma in the thymus in that time frame (Fig 3.4B). Although pl 9 ARF+- mice can develop tumors, the earliest onset observed was at about 6 months in a previous study and the tumor spectrum leans mostly towards sarcomas, not lymphomas (Kamijo et al., 1999). Histologically, these tumors are typical lymphoblastic lymphoma with large, prominent nuclei and a "starry sky" pattern (Fig 3.5C). Two Mpslf'f; p 1 9ARF+/- ;Lckcre÷ thymomas were examined by PCR and found to still express the wildtype pl 9ARF allele but showed significant under-representation of the p19 knockout allele (Fig 4D). This is contrary to what is seen in Mpslf/f; p53f/+; Lckcre÷ tumors where there is loss of heterozygosity of the wildtype allele of p53 (Chapter 2). Are the thymomas that develop in Mpslf/f; pl9ARF+/-; Lckcre ÷ mice due to the loss of p53 or additional genetic events? Of two tumors analyzed by PCR, one tumor showed loss of p53 while the other tumor did not, suggesting that if decreasing p53 is the cause, then it is not all at the DNA level (Fig 3.4D). These results are consistent with the MpslA; p53 knockout compound tumor study where CIN was shown to have p53-independent effects in tumor progression. Thus, MpslA can facilitate tumor development in the thymi of mice where one allele of pl1 9 ARF is inactivated, providing additional confirmation that CIN can act as a causal agent in carcinogenesis. Discussion The Mpsl A truncation mutation in MEFs is a lethal partial loss of spindle checkpoint function allele that causes chromosome missegregation but does not respond to the microtubule poison nocodazole. These results are consistent with in vivo data from 142 Mpsl /;p53 -1 and Mps A/A; p53' /- T lymphoma cells which also do not respond to nocodazole but have aneuploid karyotypes (Chapter 2). Ifound expression of Mps 1A in MEFs decreases cellular viability, presumably due to chromosome missegregation, which is restored by inactivation of p53. Here, I take advantage of the ability to easily image adherent MEFs by live cell microscopy to show that MpslA does in fact promote chromosome missegregation during mitosis a priori.In p53 -/-MEFs, the expression of Mps 1A caused more than 50% of mitotic cells to missegregate chromosomes. In contrast, only 26% of Mpsl +'+; p53 /-MEFs showed chromosome missegregation. Importantly, 43% of Mpsl"A/; p53 -/-MEFs proceeded through a bipolar mitosis with lagging chromosomes, more than 2-fold higher than Mpsl/+÷; p53 -/' MEFs. These missegregation events are almost as likely to result from multipolar division during mitosis as from a bipolar spindle orientation. Unequal chromosome segregation during mitosis in cells nullizygous for p53 is likely a result of centrosome amplification (Fukasawa et al., 1996). Although 26% of p53-null MEFs also missegregate chromosomes, 10% do not complete cytokinesis after a missegregation event, mostly due to multipolar mitoses. Lagging chromosomes have been shown to prevent proper cytokinesis in cells and lead to tetraploidy (Shi and King, 2005). Mpsl+'/; p53 -/' MEFs become polyploid in culture, but DNA content remain mostly stable over multiple passages. In contrast, the DNA content of MpslA/A; p53 /-MEFs is more dynamic in culture, possibly due to increased CIN, with cells exhibiting distinct 2N, 4N and 8N DNA content at passage 15 becoming primarily 6N by passage 19. Shi and King suggest that nondisjunction does not directly yield aneuploid cells, but rather goes through a tetraploid intermediate state (Shi and King, 2005). The results shown here suggest that MEFs also go through a tetraploid state. 143 Chromosome missegregation defects in MpslAA; p53 -'1MEFs are not a result of centrosome amplification. This is consistent with in vivo data from Mps 1'"thymocytes which also do not show an increase in centrosome numbers. Mpsl;A/A p53 +/+ MEFs have a fewer percentage of mitotic cells with 3 or more centrosomes than wildtype MEFs. In fact, MpslA may suppress centrosome amplification in p53' - MEFs as Mpsl NA; p53 -' MEFs have 3-fold fewer cells with amplified centrosomes than Mpsl+1+; p53 -/-MEFs. This effect could be indirect as increasing the expression level of the spindle checkpoint protein BubR1 in p53-null cells suppresses centrosome amplification (Oikawa et al., 2005). Future work should determine if the expression levels of BubRI are upregulated in Mpsl•A'; p53 -/-MEFs and if that is the cause of the decrease in centrosome amplification. Mps 1A induces cell inviability both in vivo and ex vivo. MpslA causes embryonic lethality in mice and small thymi when expressed in thymocytes. Mpsl conditional MEFs quickly die in culture after expression of the A allele through recombination of the flox allele by Cre recombinase. It is known that expression of Cre recombinase also can inhibit cell growth by activating DNA damage pathways probably due to inappropriate recombination at cryptic LoxP sites (Loonstra et al., 2002). Control MEFs infected with AdCre-GFP do show decreased viability as compared to MEFs without infection presumably due to a growth delay from Cre recombinase expression (data not shown). Importantly, Mpsl +/+ MEFs can recover and exhibit a positive growth rate unlike Mps 1 AA MEFs (Fig 3.1B). In T cells, the inactivation of p53 not only rescues decreased cell number in Mpsl 'Athymi, but results in cancer. Here, I demonstrated that p53 loss rescues MpslA/A 144 MEF inviability and immortalizes Mpsl AA; p53/ -' MEFs. How is inactivating p53 restoring cell viability to Mpsl A " MEFs? Isp53 activating apoptosis or premature senescence in Mps 1 /AMEFs? pl 9 ARF acts in the p53 signaling pathway by stabilizing p53 protein (Pomerantz et al., 1998; Weber et al., 1999). pl19AR F expression is silenced in most embryonic tissues, but its expression is re-activated in MEFs placed into culture and induces cellular senescence (Zindy et al., 2003). P19ARF-null MEFs are highly proliferative and are immortal in culture (Kamijo et al., 1997). Loss of p 1 9A R F does not promote growth of Mpsl 'A; pl9 A R F+ /- MEFs or Mpsl f/f; p19 A R F -/- MEFs, suggesting simply relieving p l9ARF-induced replicative senescence is insufficient to allow growth of Mpsl A MEFs. Interestingly, pl9ARF-null cells not only retain p53 protein expression, but also retain p53-dependent checkpoint responses, particularly to irradiation, and arresting Gl in response to DNA damage (Kamijo et al., 1997). Conceivably, p53 is activated and decreases viability in Mps 1 A/A; pl 9 ARF-/- MEFs. Thus, Mps 1A cell inviability is a result of p53-induced apoptosis or growth arrest that is independent of p 1 9 ARF . MpslA truncation mutation results in a partial loss of spindle checkpoint function. Is CIN resulting from a weakened checkpoint sufficient to induce tumors in mice? I have previously shown that combining Mps 1A with one or both alleles of inactivated p53 is extremely robust at inducing tumors in mouse thymi. Furthermore, in a few cases, MpslA alone was sufficient to induce tumor formation. CIN appeared to be the causal agent as these tumors retain p53 expression, but exhibited mitotic cells with lagging chromosomes. Here, I show that MpslA-induced CIN is able to promote lymphoma in 4 of 14 Mpslf/f; pl9ARF+/-; Lckcre ÷ mice by 6 months of age. Penetrance is significantly lower for Mps l"f/f; pl9ARF+/-; Lckcre ÷ compound mutant mice (29%) than for Mps ff, 145 p53f +, Lckcre + compound mutant mice (100%). However, MpslA is still a potent tumorigenesis facilitator when only one allele of pl 9 ARF is inactivated as Mpsl+/+; p l 9ARF+ /-mice do not develop tumors before 6 months of age and only 16% have been reported to develop spontaneous tumors by one year of age (Kamijo et al., 1999). Furthermore, Mps1 +/+; p19 ARF+/-mice have low susceptibility to lymphomagenesis, /particularly thymomas (Kamijo et al., 1999). The majority of tumors arising in pl9ARF+ mice show LOH of the wildtype allele, classic behavior for a tumor suppressor gene in tumorigenesis (Moore et al., 2003). The limited tumor set is insufficient to determine if LOH of pl9ARF is occurring in Mps 1ff; p19ARF+/-; Lckcre + tumors, but two of the tumors showed decreased expression of the p 9 ARF knockout allele, not the wildtype allele, by PCR analysis. MpslA-induced CIN appears to facilitate tumorigenesis in Mpslf/f; p53f/+; Lckcre+ primarily by increasing the rate of LOH of the wildtype p53 allele. However, CIN is promoting tumorigenesis in other respects than increasing LOH of p53 as the age of tumor onset in Mps l f/f, p53 f f, Lckcre + mice is almost two months faster than in Mps 1+/;p53f/f; Lckcre ÷ mice. Therefore, MpslA may be promoting tumor development in pl 9ARF+/-mice by inducing chromosome missegregation events that reveal tumorigenic gene expression patterns, and not through LOH of pl1 9 ARF . Nonetheless, these data show Mps 1A is extremely potent at increasing the rate of tumor onset in haploinsufficient tumor-prone mouse models. Complete inactivation of p I9ARF has been shown to upregulate aspects of the p53 pathway such as increasing the expression of the cyclin-dependent kinase inhibitor p21CIp (Kamijo et al., 1997). Could complete inactivation of p in Mps lA T cells? Mpslf/f; p9 F ; Lckcre + mice A R -/- 146 9 ARF inhibit tumorigenesis have been generated and their tumor development is currently being followed to ascertain the answer. Inactivation of p19 ARF does not rescue cellular viability of Mps 1 a MEFs. Rather, Mps lA-induced CIN promotes lymphomagenesis in some p19 ARF heterozygous mice suggesting there are different requirements for MpslA cells to proliferate in vivo and ex vivo. What is the lesion sensed by p53 in MpslA cells that decreases cell viability? The tumor suppression abilities of p53 are universally accepted (Levine, 1997; Vousden and Lu, 2002). It can sense signals resulting from a multitude of cellular stress such as DNA damage, oncogenic stress due to increased mitogen signaling and hypoxia (Harris and Levine, 2005; Levine, 1997). p53 has even been implicated to function in the spindle checkpoint, although subsequent investigation has shown this to be unlikely (Cross et al., 1995; Lanni and Jacks, 1998). Spindle assembly insults due to nocodozale treatment have been shown to activate p53 and induce a G1 arrest regardless if cells have a diploid or tetraploid genome (Ciciarello et al., 2001). Cells lacking p53 in culture are known to be genetically unstable, often becoming polyploid which is thought to be the result of inactivating a p53-dependent G1 tetraploidy checkpoint (Andreassen et al., 2001; Fukasawa et al., 1996; Oikawa et al., 2005). However, these experiments were often conducted using cell synchronization drugs such as the mycotoxin cytochalasin. Other reports contradict the claim that mammalian cells have a tetraploidy checkpoint that arrests cells in G 1 in response to improper cell division, and suggest Gl arrest in cells is actually a side effect of drug treatment (Uetake and Sluder, 2004; Wong and Steams, 2005). Nonetheless, nondisjunction events have been shown to inhibit cytokinesis resulting in tetraploidy (Shi and King, 2005). Such failure in cytokinesis resulting in tetraploid cells is proficient at inducing tumorigenesis in nude mice when cells are also 147 p53-null (Fujiwara et al., 2005). Furthermore, tetraploidy inhibits teratoma formation by p53 wildtype embryonic stem cells (Fujiwara et al., 2005) suggesting p53 is somehow able to sense chromosome missegregation events, the lack of cytokinesis, or the tetraploidy state or some combination. I demonstrate here that Mps 1A significantly increases chromosome missegregation in p53-null cells. In p53-wildtype cells, cells with these mitotic defects, presumably have inhibited cytokinesis and are tetraploid, may be sensed and eliminated. These data suggest that one aspect of p53 tumor suppression is to prevent propagation of potentially tumorigenic chromosomal missegregation events. Why is the MpslA mutation causing chromosome missegregation? This mutated protein is a hypomorph that can still localize to kinetochores and cells with MpslA respond to spindle damage caused by nocodazole indicating the spindle checkpoint is at least partially active. Although MpslA can associate with the kinetochore, Mps lA may not interact properly with its targets and result in CIN. Few targets of Mpsl have been characterized, but one possibility is Damlp. Mpslp interacts with the microtubulebinding protein Daml in budding yeast (Jones et al., 1999). Daml is an essential subunit of the DASH complex which is crucial for sister kinetochore biorientation (Janke et al., 2002; Li et al., 2002) Mpsl has been shown to phosphorylate Daml at two sites and Mps 1-mediated phosphorylation is necessary for efficient coupling of kinetochores to the plus ends of spindle microtubules (Shimogawa et al., 2006). The mutation is directed against the N-terminal portion of the protein and should not impact the kinase domain. Conceivably, Mpsl kinase function may be affected and prevent efficient phosphorylation of Daml and result in mono-oriented kinetochores during mitosis which later become lagging. 148 Here, I irrefutably show MpslA is a highly CIN-prone mutation that decreases cell viability through a p53-dependent pathway. MpslA results in a weakened spindle checkpoint that is extremely prone to chromosome missegregation in MEFs, and provides further evidence that this mutation is promoting tumorigenesis in mice through CIN when p53 is also inactivated. Futhermore, MpslA induces thymomas in pl 9 ARF+/ mice before 6 months of age. Thus, Mps 1A induced CIN may act as a universal faciliator of tumorigenesis in mice. 149 Methods Analysis of Mice Mice used in this study are on a mixed C57BL/6 and 129/Sv genetic background. Mpsl conditional A mice were generated as described (chapter 2). p 5 3 conditional and pl 9 ARF knockout mice were obtained with permission from Anton Berns and Charles Sherr through Tyler Jacks' group (Jonkers et al., 2001; Kamijo et al., 1999). Lck-Cre transgenic mice were obtained from Taconic. DNA was prepared from mouse tails, MEFs and tissues with the NucleoSpin 96 Tissue Kit (Clontech, cat # 636968). The following genotyping oiligonucleotides were used: For Mpsl: Mps 1-1: 5'CCTGGTAGTCTACCCATCCTCCTGCTC Mpsl-2: 5'GACACAGACATGGTTGGAGAGTCCTGAG Mpsl-3: 5'GAATACCGAATGAGCGAAAAGCCCC For p53 (Jonkers et al., 2001): p53FL A: 5'CACAAAAACAGGTTAAACCCAG p53FL B: 5'AGCACATAGGAGGCAGAGAC For Lck-cre (Lee et al., 2001): Lck-F: CCTTGGTGGAGGAGGGTGGAATGAA Lck-R: TAGAGCCCTGTTCTGGAAGTTACAA CreT2-R: CGCATAACCAGTGAAACAGCATTGC For p 1l9A RF (Kamijo et al., 1997): ARF 1: 5' AGTACAGCAGCGGGAGCATGG ARF2: 5' TTGAGGAGGACCGTGAAGCCG NEO2: 5' ACCACACTG CTCGACATTGGG For survival studies, mice were monitored for tumor development every week starting at 2.5 months of age by observing for difficulty in breathing. Tissues were fixed in 10% formalin for histology. Histology images were taken at 40X with a Zeiss Axiophor 150 microscope. Animal protocols were approved by the Massachusetts Instutute of Technology Committee on Animal Care. Cell culture Mpsl1+; p53 f/ + mice, MpslE/f; p53'f mice, Mps If/f; pl 9ARF+/-; or Mps If/f; p9A-/- mice were intercrossed and MEFs were prepared from e 13.5 embryos as previous described. Passage 2 MEFs were infected with adenoviral Cre-GFP (Gene Transfer Vector Core, University of. Iowa) at 5*106 PFU/ml and sorted 48 hours post-infection for GFP positive and negative cells by flow cytometry. For growth curves, Cre infected MEFs, now at passage 3, were plated on 6 cm dishes at 2*10 5 to 3*10 5 cells (normalized to 1 at T=0 on growth curves). Cells were trypsinized and the number of cells counted for rate of growth following 3 days and continued to be passaged every 3 days for up to 42 days or until number of cells fell below 5% of starting input. RNA Isolation and RT-PCR RNA was isolated by using the RNAease kit (Qiagen, cat# 74104). Reverse transcription was performed by using OligoDT primers and MMLV_RT from the RETROscript kit (Ambion, 1710) according to the manufacturer's instructions. The following primer sets were used: Mpsl-exonl-for, (5'GGAGGCTGAAGAGTTAATTGGC); Mpsl-exon4-rev, (5'CCCAGTCTCTACAGCTTTATGAAG); p53ex7, (5'CGCGCCGGCTCTGAGTAA C); p53ex8, (5'CGCTTGCGCTCCCTGGGGGC); and GAPDH primers as described (Sato et al., 1999) [GAPDH - sense, (5'CCCATCACCAT CTTCCAGGAGC); GAPDH antisense, (5'CCAGTGAGCTTCCCGTTCAGC)]. 151 Nocodazole Arrest and FACS analysis Cells were treated with 100ng/ml nocodazole for 18 hours and cells were prepared for analysis by flow cytometry as described ((Burds et al., 2005). Briefly, cells were trypsinized, washed in PBS, fixed in 90% ethanol, treated with RNAse A (0.5 mg/ml) and stained with propidum iodide (50 ptg/ml). Ten-thousand cells per sample were analyzed by using a FACScaliber (Becton Dickinson). FACS data analyzed with FlowJo. Live Cell Time-Lapse Imaging and Analysis MEFs that had previously been infected with adenoviral Cre-GFP were infected with a retrovirus expressing H2B-GFP and imaged at passage 16 as described (Meraldi et al., 2004). Briefly, cells were seeded onto ATO.15 mm dishes (Bioptechs) and C02independent medium (GIBCO-BRL) added prior to imaging. Images were acquired every 3 minutes for 6 hours using a 20x NA0.75 objective on a Zeiss Applied Precision Deltavision microscope equipped with a Mercury 100W lamp, GFP-long pass filter set (Chroma), and a Coolsnap camera. Point visiting was used to follow multiple fields of view. Matlab was used to analyze distributions of anaphase A times and determine normal frequency distributions. Immunofluorescence Microscopy Cells were prepared for immunofluorescence as described (Martin-Lluesma et al., 2002; Meraldi et al., 2004). Briefly, cells were prepared for immunofluorescence microscopy at passage 4 with the following antibodies: mouse monoclonal y-tubulin (Sigma-Aldrich, GTU-88; 1:10,000) was used to stain for centrosomes and CREST antiserum (1:100, kind gift of Earnshaw lab) was used as a kinetochore marker. Cross-adsorbed, fluorochrome- 152 labeled secondary antibodies (Molecular Probes) were used at a dilution of 1:200. DNA was visualized with DAPI containing Vectashield (Vector Laboratories). Images were acquired as described (Martinez-Exposito et al., 1999) with a 60X objective on a Nikon Applied precision Deltavision microscope equipped with a Mercury 2 W lamp and Photometrics CH350 camera. Acknowledgements I thank Roderick Bronson for pathology expertise. I thank Michael Brown and the Core Histology facility at the Center for Cancer Research at MIT for preparing tissue sections and H&E slides. I thank Ying Yue for reagents, advice and other help. I thank the rest of the members of the Sorger lab for helpful discussions. 153 References Amon, A. 1999. The spindle checkpoint. Curr Opin Genet Dev. 9:69-75. Andreassen, P.R., O.D. Lohez, F.B. Lacroix, and R.L. Margolis. 2001. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in GI. Mol Biol Cell. 12:1315-28. Baker, D.J., K.B. Jeganathan, J.D. Cameron, M. Thompson, S. Juneja, A. Kopecka, R. Kumar, R.B. Jenkins, P.C. De Groen, P. Roche, and J.M. Van Deursen. 2004. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. Baker, D.J., K.B. Jeganathan, L. Malureanu, C. Perez-Terzic, A. Terzic, and J.M. van Deursen. 2006. Early aging-associated phenotypes in Bub3/Rael haploinsufficient mice. J Cell Biol. 172:529-40. Bharadwaj, R., and H. Yu. 2004. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 23:2016-27. Boveri, T. 1914. Zur Frage der Entstehung maligner Tumoren. Fischer. Burds, A.A., A.S. Lutum, and P.K. Sorger. 2005. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. ProcNatl Acad Sci US A. 102:11296-301. Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300-3. Ciciarello, M., R. Mangiacasale, M. Casenghi, M. Zaira Limongi, M. D'Angelo, S. Soddu, P. Lavia, and E. Cundari. 2001. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem. 276:19205-13. Cohen-Fix, O., J.M. Peters, M.W. Kirschner, and D. Koshland. 1996. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pdslp. Genes Dev. 10:3081-93. Cross, S.M., C.A. Sanchez, C.A. Morgan, M.K. Schimke, S. Ramel, R.L. Idzerda, W.H. Raskind, and B.J. Reid. 1995. A p53-dependent mouse spindle checkpoint. Science. 267:1353-6. Dimri, G.P., K. Itahana, M. Acosta, and J. Campisi. 2000. Regulation of a senescence checkpoint response by the E2F1 transcription factor and pl4(ARF) tumor suppressor. Mol Cell Biol. 20:273-85. 154 Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635-45. Draviam, V.M., S. Xie, and P.K. Sorger. 2004. Chromosome segregation and genomic stability. Curr Opin Genet Dev. 14:120-5. Fisk, H.A., C.P. Mattison, and M. Winey. 2002. Centrosomes and tumour suppressors. Curr Opin Cell Biol. 14:700-5. Fisk, H.A., and M. Winey. 2004. Spindle regulation: Mpsl flies into new areas. Curr Biol. 14:R1058-60. Fujiwara, T., M. Bandi, M. Nitta, E.V. Ivanova, R.T. Bronson, and D. Pellman. 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 437:1043-7. Fukasawa, K., T. Choi, R. Kuriyama, S. Rulong, and G.F. Vande Woude. 1996. Abnormal centrosome amplification in the absence of p53. Science. 271:1744-7. Harris, S.L., and A.J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24:2899-908. Harvey, M., A.T. Sands, R.S. Weiss, M.E. Hegi, R.W. Wiseman, P. Pantazis, B.C. Giovanella, M.A. Tainsky, A. Bradley, and L.A. Donehower. 1993. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene. 8:2457-67. Hempen, P.M., H. Kurpad, E.S. Calhoun, S. Abraham, and S.E. Kern. 2003. A double missense variation of the BUB 1 gene and a defective mitotic spindle checkpoint in the pancreatic cancer cell line Hs766T. Hum Mutat. 21:445. Janke, C., J. Ortiz, T.U. Tanaka, J. Lechner, and E. Schiebel. 2002. Four new subunits of the Daml-Duol complex reveal novel functions in sister kinetochore biorientation. Embo J. 21:181-93. Jones, M.H., J.B. Bachant, A.R. Castillo, T.H. Giddings, Jr., and M. Winey. 1999. Yeast Damlp is required to maintain spindle integrity during mitosis and interacts with the Mpslp kinase. Mol Biol Cell. 10:2377-91. Jonkers, J., R. Meuwissen, H. van der Gulden, H. Peterse, M. van der Valk, and A. Berns. 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 29:418-25. Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277-82. 155 Kamijo, T., S. Bodner, E. van de Kamp, D.H. Randle, and C.J. Sherr. 1999. Tumor spectrum in ARF-deficient mice. CancerRes. 59:2217-22. Kamijo, T., J.D. Weber, G. Zambetti, F. Zindy, M.F. Roussel, and C.J. Sherr. 1998. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. ProcNatl Acad Sci U S A. 95:8292-7. Kamijo, T., F. Zindy, M.F. Roussel, D.E. Quelle, J.R. Downing, R.A. Ashmun, G. Grosveld, and C.J. Sherr. 1997. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product pl 9ARF. Cell. 91:649-59. Kim, H.S., K.H. Park, S.A. Kim, J. Wen, S.W. Park, B. Park, C.W. Gham, W.J. Hyung, S.H. Noh, H.K. Kim, and S.Y. Song. 2005. Frequent mutations of human Mad2, but not Bub 1,in gastric cancers cause defective mitotic spindle checkpoint. Mutat Res. 578:187-201. Kops, G.J., B.A. Weaver, and D.W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer.5:773-85. Lanni, J.S., and T. Jacks. 1998. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol. 18:1055-64. Lee, P.P., D.R. Fitzpatrick, C. Beard, H.K. Jessup, S. Lehar, K.W. Makar, M. PerezMelgosa, M.T. Sweetser, M.S. Schlissel, S. Nguyen, S.R. Cherry, J.H. Tsai, S.M. Tucker, W.M. Weaver, A. Kelso, R. Jaenisch, and C.B. Wilson. 2001. A critical role for Dnmtl and DNA methylation in T cell development, function, and survival. Immunity. 15:763-74. Levine, A.J. 1997. p53, the cellular gatekeeper for growth and division. Cell. 88:323-31. Li, Y., J. Bachant, A.A. Alcasabas, Y. Wang, J. Qin, and S.J. Elledge. 2002. The mitotic spindle is required for loading of the DASH complex onto the kinetochore. Genes Dev. 16:183-97. Loonstra, A., M. Vooijs, H.B. Beverloo, B.A. Allak, E. van Drunen, R. Kanaar, A. Berns, and J. Jonkers. 2002. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Margolis, R.L., O.D. Lohez, and P.R. Andreassen. 2003. G tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem. 88:673-83. Martin-Lluesma, S., V.M. Stucke, and E.A. Nigg. 2002. Role of Hecl in spindle checkpoint signaling and kinetochore recruitment of Madl/Mad2. Science. 297:2267-70. Martinez-Exposito, M.J., K.B. Kaplan, J. Copeland, and P.K. Sorger. 1999. Retention of the BUB3 checkpoint protein on lagging chromosomes. ProcNatl Acad Sci US A. 96:8493-8. 156 Matsuura, S., Y. Matsumoto, K. Morishima, H. Izumi, H. Matsumoto, E. Ito, K. Tsutsui, J. Kobayashi, H. Tauchi, Y. Kajiwara, S. Hama, K. Kurisu, H. Tahara, M. Oshimura, K. Komatsu, T. Ikeuchi, and T. Kajii. 2006. Monoallelic BUBIB mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 140:35867. Meraldi, P., V.M. Draviam, and P.K. Sorger. 2004. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 7:45-60. Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V. Murty, and R. Benezra. 2001. MAD2 haploinsufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 409:355-9. Moore, L., S. Venkatachalam, H. Vogel, J.C. Watt, C.L. Wu, H. Steinman, S.N. Jones, and L.A. Donehower. 2003. Cooperativity ofpl9ARF, Mdm2, and p53 in murine tumorigenesis. Oncogene. 22:7831-7. Nigg, E.A. 2002. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer.2:815-25. Oikawa, T., M. Okuda, Z. Ma, R. Goorha, H. Tsujimoto, H. Inokuma, and K. Fukasawa. 2005. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol. 25:4046-61. Pomerantz, J., N. Schreiber-Agus, N.J. Liegeois, A. Silverman, L. Alland, L. Chin, J. Potes, K. Chen, I. Orlow, H.W. Lee, C. Cordon-Cardo, and R.A. DePinho. 1998. The Ink4a tumor suppressor gene product, pl9Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 92:713-23. Rieder, C.L., A. Schultz, R. Cole, and G. Sluder. 1994. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 127:1301-10. Sato, N., K. Mizumoto, M. Nakamura, K. Nakamura, M. Kusumoto, H. Niiyama, T. Ogawa, and M. Tanaka. 1999. Centrosome abnormalities in pancreatic ductal carcinoma. Clin CancerRes. 5:963-70. Shi, Q., and R.W. King. 2005. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature.437:1038-42. Shimogawa, M.M., B. Graczyk, M.K. Gardner, S.E. Francis, E.A. White, M. Ess, J.N. Molk, C. Ruse, S. Niessen, J.R. Yates, 3rd, E.G. Muller, K. Bloom, D.J. Odde, and T.N. Davis. 2006. Mpsl phosphorylation of daml couples kinetochores to microtubule plus ends at metaphase. Curr Biol. 16:1489-501. 157 Taylor, S.S., M.I. Scott, and A.J. Holland. 2004. The spindle checkpoint: a quality control mechanism which ensures accurate chromosome segregation. Chromosome Res. 12:599-616. Tighe, A., V.L. Johnson, M. Albertella, and S.S. Taylor. 2001. Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2:609-14. Todaro, G.J., and H. Green. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 17:299313. Uetake, Y., and G. Sluder. 2004. Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a "tetraploidy checkpoint". J Cell Biol. 165:609-15. Vousden, K.H., and X. Lu. 2002. Live or let die: the cell's response to p53. Nat Rev Cancer.2:594-604. Wang, Q., T. Liu, Y. Fang, S. Xie, X. Huang, R. Mahmood, G. Ramaswamy, K.M. Sakamoto, Z. Darzynkiewicz, M. Xu, and W. Dai. 2004a. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 103:1278-85. Wang, Z., J.M. Cummins, D. Shen, D.P. Cahill, P.V. Jallepalli, T.L. Wang, D.W. Parsons, G. Traverso, M. Awad, N. Silliman, J. Ptak, S. Szabo, J.K. Willson, S.D. Markowitz, M.L. Goldberg, R. Karess, K.W. Kinzler, B. Vogelstein, V.E. Velculescu, and C. Lengauer. 2004b. Three classes of genes mutated in colorectal cancers with chromosomal instability. CancerRes. 64:2998-3001. Weber, J.D., J.R. Jeffers, J.E. Rehg, D.H. Randle, G. Lozano, M.F. Roussel, C.J. Sherr, and G.P. Zambetti. 2000. p53-independent functions of the pl9(ARF) tumor suppressor. Genes Dev. 14:2358-65. Weber, J.D., L.J. Taylor, M.F. Roussel, C.J. Sherr, and D. Bar-Sagi. 1999. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1:20-6. Weiss, E., and M. Winey. 1996. The Saccharomyces cerevisiae spindle pole body duplication gene MPS 1 is part of a mitotic checkpoint. J Cell Biol. 132:111-23. Winey, M., L. Goetsch, P. Baum, and B. Byers. 1991. MPS I1and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol. 114:745-54. Wong, C., and T. Steams. 2005. Mammalian cells lack checkpoints for tetraploidy, aberrant centrosome number, and cytokinesis failure. BMC Cell Biol. 6:6. Yoon, D.S., R.P. Wersto, W. Zhou, F.J. Chrest, E.S. Garrett, T.K. Kwon, and E. Gabrielson. 2002. Variable levels of chromosomal instability and mitotic spindle checkpoint defects in breast cancer. Am JPathol.161:391-7. 158 Yu, H. 2002. Regulation of APC-Cdc20 by the spindle checkpoint. Curr Opin Cell Biol. 14:706-14. Zindy, F., R.T. Williams, T.A. Baudino, J.E. Rehg, S.X. Skapek, J.L. Cleveland, M.F. Roussel, and C.J. Sherr. 2003. Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. ProcNatl Acad Sci US A. 100:15930-5. 159 Chapter 4 siRNA depletion of Mpsl causes chromosome missegregation and accelerates mitotic timing in HeLa cells Note: All experiments pertaining to Mpsl depletion was conducted by the author. Patrick Meraldi kindly provided the following raw data: the Mad2 nocodazole challenge data in Figure 4.2B, control RNAi and Mad2 RNAi mitotic timing data in Figure 4.3 and control siRNA chromosome segregation data in Figure 4.4 (published in (Meraldi et al., 2004). 160 Abstract To ensure accurate chromosome segregation, a tightly regulated network of proteins control the progression of sister chromatid movement through mitosis. Previous studies have shown that a spindle assembly checkpoint not only monitors chromosomemicrotubule attachment, but two members, Mad2 and BubR1, also control the timing of mitosis. Here, I address the role of Mps 1 in mitotic progression by time lapse imaging of Mps 1-depleted HeLa cells. When Mps 1 is inactivated by RNA interference in HeLa cells, time of anaphase onset is accelerated in addition to loss of spindle checkpoint proficiency. Furthermore, mitotic progression in Mps 1-depleted cells is intermediate between cells undergoing normal mitotic progression and cells depleted of Mad2 possibly through altering the normal cytosolic/kinetochore pools of Mad2. 161 Introduction The spindle assembly checkpoint ensures accurate chromosome segregation by delaying anaphase onset until all sister chromatids have made proper bipolar chromosome-microtubule attachments for segregation into two equal parts (reviewed in (Amon, 1999)). Spindle checkpoint genes were discovered in S. cerevisiae to be dispensible for viability, but they are essential in higher eukaryotes as shown through a variety of genetic mutants in flies, worms, zebrafish and mice (Dobles et al., 2000; Fischer et al., 2004; Gilliland et al., 2005; Kalitsis et al., 2000; Kitagawa and Rose, 1999; Poss et al., 2004; Wang et al., 2003; Wang et al., 2001; Williams et al., 2003). Spindle checkpoint components include Madl, Mad2, Bub , Bub3, BubR1/Mad3, and Mps 1. These proteins localize to the kinetochore, a multi-protein complex that assembles onto the centromere of each sister chromatid. The kinetochores of each sister chromatid pair must capture microtubules emanating from opposite poles of the mitotic spindle to achieve the tensile forces necessary for separation. Even one unattached kinetochore is sufficient to activate the spindle checkpoint and cause mitotic arrest (Rieder et al, 1994, Nicklas, 1995). The "wait anaphase" signal seems to originate from kinetochores, as perturbation of kinetochores prevents activation of the checkpoint (Rieder, 1995, Gardner 2001). There is evidence for both tension and microtubule occupancy/attachment as the physical basis of checkpoint activation. However, the precise molecular signal sensed by the checkpoint from a maloriented kinetochore remains unknown. . The founding member of the Mpslp family of kinases was discovered in S. cerevisiaeby a genetic screen for spindle pole body mutants (Weiss and Winey, 1996; Winey et al., 1991). It was later found to be required for the spindle checkpoint (Weiss 162 and Winey, 1996). Orthologs have subsequently been identified from fission yeast to man (reviewed in (Winey and Huneycutt, 2002a)). Unlike the Mads and Bubs, Mpslp is essential in budding yeast. However, separation of function mutations in Mps Ip show that it is the defect in SPB (the yeast centrosome) duplication that causes inviability, not its checkpoint function (Hardwick et al., 1996; Schutz and Winey, 1998). Whether Mps has a role in centrosome duplication for mammalian cells has been heavily disputed (Fisk and Winey, 2001; Fisk and Winey, 2004; Liu et al., 2003; Stucke et al., 2002). Interestingly, the fission yeast homolog Mphl does not function in SPB duplication, but can fully complement an Mpsl mutant in S. cerevisiae (He et al., 1998). Nonetheless, the conservation of spindle checkpoint function of Mpsl through evolution has been well documented (reviewed in (Winey and Huneycutt, 2002a)). All family members contain a C-terminal serine/threonine kinase domain, and Mpsl kinase activity is required for checkpoint function (Abrieu et al., 2001; Fisk et al., 2003; Hardwick et al., 1996; Stucke et al., 2002; Winey and Huneycutt, 2002b). Overexpression of Mpslp triggers a checkpoint arrest in budding yeast which was dependent on all other spindle checkpoint genes suggesting that Mpslp acts upstream in the signal transduction pathway (Hardwick et al., 1996). Mpslp has been reported to phosphorylate Madl in vitro. Furthermore, kinetochore localization of Madl and Mad2 requires Mps 1 in human cell lines and Xenopus egg extracts, further suggestive of an upstream function in the checkpoint (Abrieu et al., 2001; Liu et al., 2003; Martin-Lluesma et al., 2002; Wong and Fang, 2006). Mpsl deficiency promotes mitotic defects and chromosomal instability from yeast to man (Straight et al., 2000; Stucke et al., 2002). Recently, human Mpsl has been reported to interact with DNA damage and repair proteins (Leng et al., 2006; Wei et al., 163 2005). This offers the intriguing possibility that Mpsl may link the DNA damage monitoring network to ensuring accurate chromosome segregation and therefore maintain genomic stability. Since chromosome-microtubule capture is stochastic and mitosis a dynamic process, time lapse imaging is a good assay for observing subtle chromosome missegregation defects and temporal disruptions in mitosis. A comprehensive study of 5 spindle checkpoint genes: Madl, Mad2, Bubl, BubR1 and Bub3 using a combination of small interfering RNAs (siRNAs) knockdown and live cell imaging was meticulously done by Meraldi et al in HeLa cells (Meraldi et al., 2004).The authors elegantly show that not only are Mad2 and BubR required for spindle checkpoint activation, they also regulate mitotic timing. Here, I describe the results gathered for hMpsl using similar techniques to further characterize its function in the spindle checkpoint and determine if it also plays a role in mitotic timing. I find that Mpsl is required for spindle checkpoint function in HeLa cells in agreement with previous findings (Stucke, et al, 2002). I confirm that Mps 1 is required for Madl and Mad2 to localize to kinetochores. In addition, I find inactivation of Mps 1 causes acceleration of mitotic timing, but this effect is intermediate between the timing in cells depleted of Mad2 or BubR1 and normal mitotic progression in control cells. I propose that Mpsl also has a dual role in mitotic timing and checkpoint function, which is likely to act through Mad2. 164 Results siRNA depletion of Mpsl abolishes kinetochore localization of Madl and Mad2 RNA interference (RNAi) by siRNAs is a powerful technique to allow for specific inactivation of a gene product in mammalian cells (Elbashir et al., 2001). RNAi is particularly effective in HeLa cells. Inactivation of Mpsl by siRNA depletion had been described previously (Stucke et al., 2002). However, no time lapse imaging of Mps 1 depleted cells had been carried out in that study so the role of Mpsl in mitotic progression was not known. Therefore, I decided to undertake a study to evaluate chromosome dynamics in HeLa cells depleted of Mpsl using a previously described siRNA duplex denoted in this manuscript as Mps l-1. However, since the silencing efficiency of multiple siRNAs can be variable for a single gene and siRNAs can cause off-target effects (Holen et al., 2002; Snove and Holen, 2004), I designed three additional siRNA oligos for Mpsl (Table 1). To determine the level of depletion by the siRNA oligos targeting Mpsl, Mpsl protein level was assessed by western blotting. Mpsl-1 efficiently depletes Mpsl protein from HeLa cells by 24 hours (Fig. 4.1A) as has been described (Stucke et al., 2002). Of the 3 oligos I designed, I found that only Mpsl-4 caused complete depletion of Mps 1 in four independent experiments (Fig 4. 1B and data not shown). Furthermore, similar Mps 1 depletion levels can be sustained for up to 66 hours. To further determine if Mps 1-4 siRNA oligo leads to complete depletion of Mps 1, I used indirect immunofluorescence to show that Mad2 requires Mps 1 to localize to kinetochores in agreement with previous findings using siRNA oligo Mpsl-l (Fig 4. 1Cand (Liu et al., 2003; Martin-Lluesma et al., 2002)). BubR1, whose localization is independent of Mpsl, is used as a control. The kinetochore localization of BubR1 is 165 Table 4.1: Mpsl siRNA sequences Name Sequence Mpsl-1 Mpsl-2 Mpsl-3 Mpsl-4 5'-AAC CCA GAG GAC TGG TTG AGT-3' 5'-AAG ATT CTC AGG TTG GCA CAG-3' 5'-AAA TGC CGA GAT TTG GTT GTG-3' 5'-AAC CAG AAT CCT GCT GCA TCT-3' 166 Figure 4.1 0 0 24 48 66 24 SMpsl 97 kDa Mpsl 97 kDa r ytubulin 48 kDa Mpsl-1 ~ ...... r IIIl- |1"I - --....O ytubulin 48 kDa Mpsl-4 control siRNA MS1 MaS Ovra Mps1-1 siRNA * ub Mps1-4 siRNA BubR Ma6 0S 167 Ovela Figure 4.1: siRNA depletion of Mpsl abolisheskinetochore localizationof Mad2. (A and B) Immunoblots of HeLa cell extracts treated with the following siRNA oligos: Mpsl-1 (A) and Mpsl-4 (B) with anti-Mpsl antibodies to show efficiency of RNAi. Blotting with anti-y-tubulin antibodies is used as a loading control. (C) Immunofluorescence of cells treated for 48 hours with control siRNA and Mps 1-1 siRNA using DAPI for DNA (purple), anti-BubRI antibodies for a kinetochore localization control (red), anti-Mad2 antibodies (blue), and anti-Mpsl antibodies (green). 168 unaffected with either Mpsl-1 or Mpsl-4. In cells treated with a control siRNA duplex against lamin A, Mpsl and Mad2 both localize to kinetochores in a prometaphase cell (Fig 4.1C). However, in a cell transfected with Mpsl-4 siRNA, we see that when Mps 1 is depleted and its kinetochore localization is lost, Mad2 is also lost Fig 4.1C). These data show Mpsl-4 is the most efficient siRNA oligo for Mps 1 depletion and confirm that siRNA oligos directed against the same gene often yield variable levels of depletion. Complete siRNA depletion of Mpsl by Mps1-4 abolishes the checkpoint A functional assay for spindle checkpoint inactivation is to determine the mitotic index after the addition of the microtubule destabilizing drug, nocadazole. When nocodazole triggers the spindle checkpoint in an adherent cell, a characteristic rounded up appearance is evident upon mitotic arrest. In Hela cells with no siRNA treatment (control), an average of 77% of cells are arrested in mitosis after addition of 200ng/ml nocodazole for 16 hours as assessed by cell rounding in two independent experiments (Fig 4.2A and B). In contrast, only about 14% of cells transfected with Mpsl-4 appear rounded up suggesting this siRNA oligo efficiently overrides the spindle checkpoint. This is comparable the level seen for cells depleted of Mad2 of 8%(Fig 4.2B). Surprisingly, Mpsl -1 was found to be quite inefficient at preventing checkpoint arrest by this assay with 63% of cells scored as mitotic despite barely detectable levels of Mpsl protein by western blotting (Fig 4.2B and Fig 4.1A). Cells transfected with Mpsl-2 and Mpsl-3 gave an intermediate phenotype with 38% and 42% of cells arrested in mitotis respectively. Consequently, no additional studies were continued with Mpsl-3. Thus, Mps 1-4 can efficiently override the spindle checkpoint. 169 Figure 4.2 A A B * ~r\n/ IU U/o 80% I o o 60% E S40% 20% no0 I· Hil cono ltvosl2Fol)SA2-s 170 ps2A .ad2 Figure 4.2: Complete siRNA depletion ofMpsl by Mpsl-4 abolishes the checkpoint. (A) Representative phase-contrast images of HeLa cells treated with 200ng/ml nocodazole for 16 hours showing an activated spindle checkpoint with rounded up cells (control) and with the checkpoint abolished by siRNA (Mpsl-4). (B) Histogram of the % of mitotic HeLa cells following transfection with the indicated siRNA oligo or no treatment (control) and then challenged with nocodazole. 171 Table 4.2: Analysis of mitotic timing in cells depleted of Mps RNAi Ncells I skewness peakness a (a, Rangec max 81 Control 263 2.67 25.5 6.5 min 15 Mpsl-1 113 2.13 28.0 6.4 15 52 Mpsl-4 119 1.01 19.5 4.2 15 30 aPeak time of the best-ffited frequency distribution . bStandard deviation for a best-fitted normal distribution. cRange after exclusion of the top and bottom 3%values. 172 Mpsl controls mitotic timing in addition to monitoring chromosome segregation To determine the role of Mpsl in mitotic progression, I turned to time lapse imaging to observe the chromosome dynamics of cells transfected siRNA oligos directed against Mpsl. Cells stably expressing histone 2B-GFP (H2B-GFP) were imaged for 6 hours at 3 minute intervals. The data is summarized in table 2 and compared to the control siRNA data set from Meraldi, et. al. Average time of mitosis is defined as the time a cell progresses from nuclear envelope breakdown (NEBD) to onset of anaphase A when chromosomes begin to move to the poles (Fig 4.3A) (Meraldi et al., 2004; Rieder et al., 1994). NEBD in each mitotic cell is set as T=0. A data set of 263 mitotic cells transfected with a control siRNA gives rise to a skew-normal distribution with a peak value at T=25.5 (Fig 4.3A, 4.3C, Table 4.2). Mitotic timing can range between 15 to 81 minutes for control cells due to spindle checkpoint activation in about 20% of the cells (Table 4.2, Fig 4.3C and (Meraldi et al., 2004; Rieder et al., 1994)). Interestingly, cells transfected with Mpsl-4 not only progresses through mitosis with chromosome missegregation errors, but also faster than control cells with a peak time= 19.5 minutes (Fig 4.3A and Table 4.2). This phenotype is intermediate between Mad2 depleted cells and control cells (Fig 4.3A, 4.3B and (Meraldi et al., 2004)). Timing is also not significantly affected by Mps l-1. Since residual levels of checkpoint proteins from incomplete siRNA depletion have been shown to create hypomorphic phenotypes, it is unsurprising that Mpsl-1 does not display the acceleration in mitotic timing (Table 4.2 and data not shown). Hence, I have demonstrated that efficient depletion ofMpsl protein accelerates mitosis. 173 Figure 4.3 Cotrl N- Control RNAi Mpsl-4 RNAi ?NAi I dat• Control • mmm ConVol fit SContr, dot SContrd fit I IIIIItI1IV I Mlllllm, I 0 Time (min) I 'M rI I I I I I Il I r-I - 40 20 K RlllllllA1 ,- .n 60 80 Time (min) 100 4 dA , I 740 120 Mpsi-1 datal. .Mpa1-1 -\lJ fit Co .0 E 6 1111 II [i- "0 fj. jpý -- I 10io IE ·I ·lI.. _J -- . .. 30 00 50 Time (min) 174 10 I • i - . 20 "30 •i • .. . ' 4030 5 6 ' Time (min) • ... 0 Figure 4.3: Mpsl controls mitotic timing. (A) HeLa Histone 2B-GFP cells were treated with siRNA and followed by time-lapse imaging. Mitotic progression is defined as the time from NEBD (To) to onset of anaphase A. (B) Frequency distribution plots of anaphase times as indicated. (C, D, and E) Matlab analysis depicting anaphase times bin into 3 minute increments as a histogram with a best-fit normal distribution plot for control siRNA (C), Mps1-4 siRNA (D) and Mpsl-l siRNA (E). 175 Figure 4.4 A A R 100% E normal Munaligned, lagging, DNA bridge Omultipolar 03 no cvtokinesis 80% ... . ! .... .. 2- 60% oi Z 40% 20% =L 0% 0% Control Mps1-1 Mps1-2 176 Mpsl-4 Figure 4.4: Mpsl depletion causes chromosome missegregation. (A) Representative Mpsl-depleted mitotic cells showing mitotic defects at metaphase and anaphase. (B) Mitotic phenotype in control, Mpsl-1, Mpsl-2, and Mpsl-4 siRNA transfected cells. Mitotic cells were scored as normal (blue), containing unaligned chromosomes at metaphase and/or progressing to lagging chromosomes or DNA bridges at anaphase (red), undergoing multipolar mitosis (yellow) or failing to undergo any visible anaphase chromosome separation (light blue). 177 To determine the effect of Mpsl1-4 siRNA on the spindle checkpoint, I evaluated the mitotic defects in Mps 1-4 transfected HeLa cells. The checkpoint is efficiently abolished by Mps 1-4 since no skewness is present in the frequency distribution of the 119 mitotic cells imaged with a very tight range of mitosis from 15 to 30 minute (Fig 4.3D, Table 4.2). In contrast, the set of 113 cells transfected with Mpsl-1 have a skew-normal distribution with mitosis ranging between 15 minutes to 52 minutes demonstrating inefficient checkpoint inactivation with this oligo (Fig 4.3E, Table 4.2). When I catalog the complete set of mitotic cells collected by time lapse imaging for mitotic defects, 81% of cells transfected with Mps 1-4 experience chromosome segregation errors in the form of unaligned chromosomes at metaphase that result in lagging chromosomes or DNA bridges at anaphase (Fig 4.4A). An additional 6% of Mps 1-4 transfected cells do not undergo cytokinesis. While 91% of cells transfected with a control siRNA undergo a normal mitosis, less than 13% of Mpsl-4 transfected cells do the same. In contrast, at least 58% of Mps 1-1 treated cells and 34% of Mps 1-2 transfected cells progress through mitosis normally. Thus, Mpsl is a spindle checkpoint protein which monitors chromosome segregation, and its depletion by Mpsl-4 siRNA yields missegregation events. Discussion The complete depletion of Mps 1 by RNAi in HeLa cells abrogates the spindle checkpoint and accelerates mitotic timing. Here, I describe an siRNA oligo, Mpsl1-4, that can efficiently abolish checkpoint activation in cells challenged with nocodazole. Depletion of Mps 1by Mps 1-4 prevents Mad2 from localizing to the kinetochore as expected (Martin-Lluesma et al., 2002). Such depletion induces considerable 178 chromosome missegregation. Furthermore, Mpsl is also required for normal mitotic timing as cells depleted of Mpsl protein by Mps1-4 progresses through mitosis faster than control cells. Thus, Mpsl joins Mad2 and BubR1 in the group of spindle checkpoint proteins that have a dual function in mitotic timing (Meraldi et al., 2004). What is the mechanism for Mpsl regulation of mitotic timing? The depletion of Mps 1 gives an intermediate mitotic timing phenotype in between Mad2-depleted and control cells. The Ndc80 complex is required for the proper kinetochore localization of Mad2 and Madl (Martin-Lluesma et al., 2002). Since the depletion of hNuf2R, one of the components of the human Ndc80 complex did not accelerate mitotic timing, Meraldi et al. propose that it must be the cytosolic pool of Mad2 mediating mitotic timing regulation. Like Mad2, Mps1 also requires the Ndc80 complex for kinetochore localization (Martin-Lluesma et al., 2002). Thus, the cytosolic pool of Mpsl, not the kinetochore pool of Mpsl, is likely regulating mitotic timing (Fig 4.5). Since the localization of Mad2 to the kinetochores requires Mps 1, and the mitotic timing function of Mad2 is monitored by the cytosolic pool of Mad2 ((Liu et al., 2003; Martin-Lluesma et al., 2002; Meraldi et al., 2004), one can imagine that Mpsl is playing a role in mitotic timing by regulating Mad2 pools. Interestingly, the three checkpoint proteins with a role in mitotic timing - Mpsl, Mad2, and BubR1, have been shown to have dynamic kinetics of kinetochore binding by fluorescence recovery after photobleaching (FRAP) experiments in Ptk2 cells (Howell et al., 2004; Meraldi et al., 2004). Potentially, loss of Mpsl is upsetting this rapid dissociation and association of Mad2 onto the kinetochores, and therefore also changing the concentration of the cytosolic pool of Mad2 and slightly 179 Figure 4.5 Cytosolic pool Kinetochore pool Mitotic timing I-1I Mt ' a Metaphase WI % A I n - Anapnase 180 Figure4.5: A modelfor the dualfunctions ofMpsl duringmitosis in the spindle checkpoint and mitotic timing. The spindle checkpoint function and the mitotic timing function of Mpsl are proposed to be mediated by separate pools of Mps 1 protein at the kinetochore and in the cytosol, respectively. Hec I/Nuf2R, members of the Ndc80 complex, are necessary for Mpsl, Mad2 and Madl to localize to unattached kinetochores and along with BubR1, Bubl and Bub3 prevent Cdc20 from activating the APC/C which could cause premature chromosome segregation and segregation defects. Mps 1 may regulate mitotic timing in the cytosol, along with Mad2 and BubR1. 181 altering mitotic timing. Conversely, Mps 1 may have a role in mitotic timing independent of Mad2 through as yet unknown mechanism. Silencing efficieny of multiple siRNA duplexes for a single gene can be quite variable (Holen et al., 2002). One hypothesis for these positional effects is that only a few regions of a particular mRNA may be accessible for binding by the target siRNA. Here, I demonstrate that efficiency of Mpsl knockdown by four siRNA duplexes is also highly variable. Only one siRNA duplex, Mps 1-4, can fully deplete Mpsl to undetectable levels as assayed by western blotting. This oligo also causes the greatest checkpoint impairment as assessed by cell rounding when challenged with nocodazole. Moreover, when cells were treated with three of the oligos (Mps 1-1, Mps 1-2, and Mpsl-4) and imaged by timelapse microscopy, Mpsl-4 caused over 80% of cells to experience chromosome segregation errors. However, only complete depletion of Mpsl by Mps 1-4 abolishes its role in mitotic timing. Results from the nocodazole challenge assay and live cell imaging ranks the efficiency of Mps I depletion as highest for Mps 1-4, Mpsl-2 in the middle, and lowest for Mps 1-1. These data suggest that the mitotic timing function of Mps 1 is less sensitive to Mps 1 protein levels than the spindle checkpoint response of Mps1 as full depletion is required to accelerate mitosis. Knockdown does not equate to a knockout. Residual levels of spindle checkpoint proteins can cause hypomorphic phenotypes and complicate interpretation of protein function. Depletion of the structural kinetochore component Ndc80/Hec1 was reported to arrest human cells in mitosis which is dependent on the spindle checkpoint (MartinLluesma et al., 2002). This is not unexpected, since incomplete inactivation of kinetochores is known to engage the checkpoint in budding yeast (Gardner et al., 2001; 182 Gillett et al., 2004). In contrast, completely inactive kinetochores are unable to recruit checkpoint proteins and abolish checkpoint signaling. This is exactly the case when more efficient Hec depletion was obtained in synchronous HeLa cell populations (Meraldi et al., 2004). Here, I demonstrate that when Mpsl is partially depleted, spindle checkpoint activation is also partially inactivated with a subset of cells experiencing mitotic defects. Mammalian cells appear to be particularly sensitive to concentration changes of mitotic proteins, which is unsurprising since abnormalities in chromosome segregation can be particularly deleterious (Baker et al., 2004). 183 Methods RNA interference, Cell culture and Transfections The sequences of siRNA oligonucleotides targeted against Mps 1 (Dharmacon Research, Inc.) are listed in Table 4.11. Mpsl-1 and the control siRNA against Lamin A were previously described (Elbashir et al., 2001; Stucke et al., 2002) HeLa cells were grown as described (Martinez-Exposito et al., 1999). HeLa cells stably expressing H2B-GFP were the kind gift of P. Meraldi and V. Draviam (Meraldi et al., 2004). Cells were transfected as described (Elbashir et al., 2001) with oligofectamine (Invitrogen) and analysed approximately 48 hours after transfection. Immunofluorescence Microscopy and Immunoblotting Cells were prepared for immunofluorescence as described (Martin-Lluesma et al., 2002; Meraldi et al., 2004). Briefly, following siRNA treatment, cells were prepared for immunofluorescence microscopy with the following antibodies: mouse monoclonal aMpsl (Upstate, NT, product # 05682; 1:1000), rabbit a-Madl (P. Meraldi; (Meraldi et al., 2004); 1:250), rabbit a-Mad2 (A. S. Lutum; (Burds et al., 2005)1:250), sheep aBubR1 (S. Taylor; 1:1000). Cross-adsorbed, fluorochrome-labeled secondary antibodies (Molecular Probes) were used at a dilution of 1:200. DNA was visualized with DAPI containing Vectashield (Vector Laboratories). Images were acquired as described (Martinez-Exposito et al., 1999) with a 60X objective on a Nikon Applied precision Deltavision microscope equipped with a Mercury 2 W lamp and Photometrics CH350 camera. 184 HeLa cell extracts and immunoblots were prepared as described (Martinez-Exposito et al., 1999). Primary antibodies were: mouse monoclonal ot-Mpsl (Upstate, NT, product # 05682; 1:1000) and mouse monoclonal y-tubulin (Sigma-Aldrich, GTU-88; 1:10,000). Immunoblots were developed by using chemiluminescence (SuperSignal West Femto, Pierce) by standard methods and visualized with a FluroChem imaging system (Alpha Innotech Inc.). Nocodazole Arrest Cells were treated with 200ng/ml nocodazole for 16 hours and the fraction of rounded-up cells determined by phase contrast microscopy. Live Cell Time-Lapse Imaging and Analysis Cells were imaged as described (Meraldi et al., 2004). Briefly, cells were seeded onto ATO. 15 mm dishes (Bioptechs) and C02-independent medium (GIBCO-BRL) added prior to imaging. Images were acquired every 3 minutes for 6 hours using a 20x NA0.75 objective on a Zeiss Applied Precision Deltavision microscope equipped with a Mercury 100W lamp, GFP-long pass filter set (Chroma), and a Coolsnap camera. Point visiting was used to follow multiple fields of view. Matlab was used to analyze distributions of anaphase A times and determine normal frequency distributions and skewness. Acknowledgements I thank Patrick Meraldi and Viji Draviam for gifts of reagents, sharing of data and experimental expertise. I thank them and the rest of the members of the Sorger lab for helpful discussions. Anti-BubRi was a kind gift from Steve Taylor. 185 References Abrieu, A., L. Magnaghi-Jaulin, J.A. Kahana, M. Peter, A. Castro, S. Vigneron, T. Lorca, D.W. Cleveland, and J.C. Labbe. 2001. Mpsl is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 106:83-93. Amon, A. 1999. The spindle checkpoint. Curr Opin Genet Dev. 9:69-75. Baker, D.J., K.B. Jeganathan, J.D. Cameron, M. Thompson, S. Juneja, A. Kopecka, R. Kumar, R.B. Jenkins, P.C. De Groen, P. Roche, and J.M. Van Deursen. 2004. BubRl insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. Burds, A.A., A.S. Lutum, and P.K. Sorger. 2005. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl Acad Sci US A. 102:11296-301. Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635-45. Elbashir, S.M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494-8. Fischer, M.G., S. Heeger, U. Hacker, and C.F. Lehner. 2004. The mitotic arrest in response to hypoxia and of polar bodies during early embryogenesis requires Drosophila Mps1. CurrBiol. 14:2019-24. Fisk, H.A., C.P. Mattison, and M. Winey. 2003. Human Mpsl protein kinase is required for centrosome duplication and normal mitotic progression. ProcNatl Acad Sci U SA. 100:14875-80. Fisk, H.A., and M. Winey. 2001. The mouse Mpslp-like kinase regulates centrosome duplication. Cell. 106:95-104. Fisk, H.A., and M. Winey. 2004. Spindle regulation: Mpsl flies into new areas. Curr Biol. 14:R1058-60. Gardner, R.D., A. Poddar, C. Yellman, P.A. Tavormina, M.C. Monteagudo, and D.J. Burke. 2001. The spindle checkpoint of the yeast Saccharomyces cerevisiae requires kinetochore function and maps to the CBF3 domain. Genetics. 157:1493502. Gillett, E.S., C.W. Espelin, and P.K. Sorger. 2004. Spindle checkpoint proteins and chromosome-microtubule attachment in budding yeast. J Cell Biol. 164:535-46. 186 Gilliland, W.D., S.M. Wayson, and R.S. Hawley. 2005. The meiotic defects of mutants in the Drosophila mps 1 gene reveal a critical role of Mpsl in the segregation of achiasmate homologs. CurrBiol. 15:672-7. Hardwick, K.G., E. Weiss, F.C. Luca, M. Winey, and A.W. Murray. 1996. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 273:953-6. He, X., M.H. Jones, M. Winey, and S. Sazer. 1998. Mphl, a member of the Mpsl-like family of dual specificity protein kinases, is required for the spindle checkpoint in S. pombe. J Cell Sci. 111:1635-47. Holen, T., M. Amarzguioui, M.T. Wiiger, E. Babaie, and H. Prydz. 2002. Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Res. 30:1757-66. Howell, B.J., B. Moree, E.M. Farrar, S. Stewart, G. Fang, and E.D. Salmon. 2004. Spindle checkpoint protein dynamics at kinetochores in living cells. CurrBiol. 14:953-64. Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277-82. Kitagawa, R., and A.M. Rose. 1999. Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol. 1:514-21. Leng, M., D.W. Chan, H. Luo, C. Zhu, J. Qin, and Y. Wang. 2006. MPS 1-dependent mitotic BLM phosphorylation is important for chromosome stability. ProcNatl AcadSci USA. 103:11485-90. Liu, S.T., G.K. Chan, J.C. Hittle, G. Fujii, E. Lees, and T.J. Yen. 2003. Human MPS1 Kinase Is Required for Mitotic Arrest Induced by the Loss of CENP-E from Kinetochores. Mol Biol Cell. 14:1638-51. Martin-Lluesma, S., V.M. Stucke, and E.A. Nigg. 2002. Role of Hecl in spindle checkpoint signaling and kinetochore recruitment of Madl/Mad2. Science. 297:2267-70. Martinez-Exposito, M.J., K.B. Kaplan, J. Copeland, and P.K. Sorger. 1999. Retention of the BUB3 checkpoint protein on lagging chromosomes. Proc NatlAcad Sci US A. 96:8493-8. Meraldi, P., V.M. Draviam, and P.K. Sorger. 2004. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 7:45-60. 187 Poss, K.D., A. Nechiporuk, K.F. Stringer, C. Lee, and M.T. Keating. 2004. Germ cell aneuploidy in zebrafish with mutations in the mitotic checkpoint gene mps1. Genes Dev. 18:1527-32. Rieder, C.L., A. Schultz, R. Cole, and G. Sluder. 1994. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 127:1301-10. Schutz, A.R., and M. Winey. 1998. New alleles of the yeast MPS 1 gene reveal multiple requirements in spindle pole body duplication. Mol Biol Cell. 9:759-74. Snove, O., Jr., and T. Holen. 2004. Many commonly used siRNAs risk off-target activity. Biochem Biophys Res Commun. 319:256-63. Straight, P.D., T.H. Giddings, Jr., and M. Winey. 2000. Mpslp regulates meiotic spindle pole body duplication in addition to having novel roles during sporulation. Mol Biol Cell. 11:3525-37. Stucke, V.M., H.H. Sillje, L. Arnaud, and E.A. Nigg. 2002. Human Mps 1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. Embo J. 21:1723-32. Wang, Q., T. Liu, Y. Fang, S. Xie, X. Huang, R. Mahmood, G. Ramasiwamy, K.M. Sakamoto, Z. Darzynkiewicz, M. Xu, and W. Dai. 2003. BUBR1-deficiency results in abnormal megakaryopoiesis. Blood. Wang, X., J.R. Babu, J.M. Harden, S.A. Jablonski, M.H. Gazi, W.L. Lingle, P.C. de Groen, T.J. Yen, and J.M. van Deursen. 2001. The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins. JBiol Chem. 276:26559-67. Wei, J.H., Y.F. Chou, Y.H. Ou, Y.H. Yeh, S.W. Tyan, T.P. Sun, C.Y. Shen, and S.Y. Shieh. 2005. TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. JBiol Chem. 280:7748-57. Weiss, E., and M. Winey. 1996. The Saccharomyces cerevisiae spindle pole body duplication gene MPS 1 is part of a mitotic checkpoint. J Cell Biol. 132:111-23. Williams, B.C., Z. Li, S. Liu, E.V. Williams, G. Leung, T.J. Yen, and M.L. Goldberg. 2003. Zwilch, a New Component of the ZW10/ROD Complex Required for Kinetochore Functions. Mol Biol Cell. 14:1379-91. Winey, M., L. Goetsch, P. Baum, and B. Byers. 1991. MPS I and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol. 114:745-54. 188 Winey, M., and B.J. Huneycutt. 2002a. Centrosomes and checkpoints: the MPSI family of kinases. Oncogene. 21:6161-9. Winey, M., and B.J. Huneycutt. 2002b. Centrosomes and checkpoints: the MPSI family of kinases. Oncogene. 21:6161-9. Wong, O.K., and G. Fang. 2006. Loading of the 3F3/2 Antigen onto Kinetochores Is Dependent on the Ordered Assembly of the Spindle Checkpoint Proteins. Mol Biol Cell. 189 Chapter 5 Conclusions, Discussion and Future Directions 190 Aneuploidy is a hallmark of cancer cells (Boveri, 1914; Lengauer et al., 1997). Many aneuploid tumors exhibit chromosomal instability (CIN) - the continual gain and loss of entire chromosomes during cell divisions (Draviam et al., 2004). The mitotic spindle checkpoint prevents CIN by ensuring accurate chromosome segregation when it delays anaphase onset until all kinetochores have achieved bipolar attachment to the spindle apparatus (Kops et al., 2005; Taylor et al., 2004). When I began the research described in this dissertation, there was little evidence for a connection between the spindle checkpoint and cancer in mammals. Although abrogation of the mammalian mitotic checkpoint causes various mitotic errors including CIN, few mutations in checkpoint components had been found in human cancers (reviewed in (Draviam et al., 2004)). Moreover, the argument that CIN causes cancer remained largely circumstantial. Of the six core checkpoint components (Bubl, BubRI, Bub3, Madl, Mad2, and Mpsl) only Mad2 and Bub3 had been genetically inactivated in mice and neither displayed an aggressive tumor phenotype (Dobles et al., 2000; Kalitsis et al., 2000 ; Michel et al., 2001). However, chromosome missegregation had long been postulated to play a role in tumorigenesis and an increased incidence of lung cancer in aged Mad2 ÷' - mice made pursuing the study of the spindle checkpoint and cancer extremely attractive (Hartwell and Kastan, 1994; Hernando et al., 2004; Nowell, 1976). The Mad2 and Bub3 knockout mice proved to be embryonic lethal demonstrating that the spindle checkpoint has additional functions in metazoans beyond the classic checkpoint role. One explanation may be that the spindle checkpoint is essential for proper mitosis, and that complete loss of function may be too severe for tumor development. No conditional mouse model of a spindle checkpoint gene had yet been described. However, it was clear that further 191 exploration of a role for the spindle checkpoint in tumorigenesis required conditional inactivation of spindle checkpoint components in adult somatic tissues. Given these postulates, I set out to generate a conditional hypomorphic mouse model of the spindle checkpoint gene Mpsl and determine if elevating the rate of CIN can promote tumorigenesis apriori. The Mps 1 gene, studied here, encodes a serine/threonine protein kinase essential for proper chromosome segregation and in some organisms, also essential for centrosome duplication (Fisk et al., 2004). Mps 1 protein is conserved mostly in the N-terminal region and the C-terminal kinase domain (Stucke et al., 2004). I generated a truncated version called Mps lA that deletes exons 2 and 3 in the genomic locus, thereby removing a highly conserved region of 107 amino acids. The conditional MpslA allele was introduced into mice to investigate the interaction of the spindle checkpoint and CIN in tumor development. Conclusions and Discussion MpslA is a partial loss of function mutation in the spindle checkpoint MpsldA inhibits viability, but responds to nocodazole MpslA induces cell inviability both in vivo and ex vivo. MpslA causes embryonic lethality in mice before embryonic day 13.5. Mpsl conditional mice that harbor the Lckcre transgene to specifically express MpslA in thymocytes have smaller thymi and fewer thymocytes than control mice. Furthermore, I found expression of MpslA in MEFs also decreases cellular viability. However, the analogous MpslA truncation protein localizes properly to kinetochores. Moreover, cells expressing Mps lA responds to the microtubule 192 destabilizing drug nocodazole by arresting cell cycle progression suggesting the checkpoint is at least partially active. Thus, the MpslA truncation mutation is a lethal partial loss of spindle checkpoint function allele that retains kinetochore localization, responds to the microtubule poison nocodazole, but causes cellular lethality. Mps 1A induces CIN through chromosome segregationdefects Why is MpslA inducing cellular and embryonic lethality? I propose that MpslA causes chromosome missegregation during mitosis which leads to cell inviability. Simultaneous deletion of Mad2 and p53 rescued the viability of Mad knockout blastocyts and MEFs suggesting that p53 decreases the viability of cells with an aberrant spindle checkpoint (Burds et al., 2005). Elimination of p53 also rescued Mpsl"AA MEFs, allowing for in vitro study of these cells. Live cell microscopy of p53-null MEFs expressing MpslA show that MpslA does in fact promote chromosome missegregation during mitosis. Though p53 -' MEFs have been shown to exhibit elevated rates of mitotic errors, the additional expression of Mps 1A doubled the number of cells with chromosome segregation defects. Analysis of MpslA/A; p53 -' MEFs showed that chromosome missegregation in these cells is not a result of centrosome amplification. This is consistent with in vivo data from Mps 1 AA thymocytes which also do not show an increase in centrosome numbers. A few Mps 1A/ T cell lymphomas were successfully isolated from mice. CIN appeared to be the causal agent as these tumors exhibited mitotic cells with lagging chromosomes at anaphase by histology, but retain p53 expression as assayed by PCR genotyping. Thus, MpslA is a loss of function checkpoint allele that induces CIN in vivo and ex vivo due to chromosome segregation defects. 193 p53 inhibits CIN Inactivationofp53 cooperates with MpslA-induced CIN to promote tumorigenesis Mps 1A was specifically expressed in thymocytes to determine if CIN can promote tumorigenesis in mice. MpslA appears to cause apoptosis, since Mpsl"WA mice are embryonic lethal and MpslA/A thymi expressing the tissue-specific Lck-Cre are significantly smaller in size. Moreover, MpslA/A MEFs exhibit decreased cellular viability. Once rescue of Mpsl"A MEFs was observe through deletion of p53, the MpslA tumor study was also conducted on a p53-heterozygous and p53-null background. I show in chapter 2 and 3 that an Mpsl hypomorphic mutation causes sporadic spontaneous lymphomas, and in combination with p53 deficiency robustly facilitates tumorigenesis in thymocytes. Strikingly, MpslA induces tumorigenesis in p53-heterozygous knockout thymocytes with full penetrance and accelerates tumor onset for both p53-heterozygous and p53-null thymocytes. These results suggest MpslA-CIN is promoting additional events that contribute to carcinogenesis. In summary, CIN caused by a weakened checkpoint is sufficient to facilitate tumorigenesis when the p53 pathway is also impaired. Inactivation ofp53, but not ofpl 94RF rescues MpsldA cellularlethality The tumor suppressor p53 responds to cellular stress by inducing apoptosis or growth arrest (Harris and Levine, 2005). The tumor suppressor pl 9 ARF antagonizes Mdm2 to stabilize p53 protein and thereby promotes p53-dependent functions (Lowe and Sherr, 2003). I found that inactivation of p53 rescues Mpsl AA MEF inviability. In contrast, although P19ARF-null MEFs are highly proliferative and are immortal in culture, Mpsl VA; p9ARF-/- MEFs do not survive in culture, suggesting simply relieving pl 194 9 ARF_ induced replicative senescence is insufficient to allow growth of Mpsl A/ MEFs (Kamijo et al., 1997). This may be due to the fact that pl9ARF-null cells not only retain p53 protein expression, but also retain p53-dependent checkpoint responses, particularly to irradiation, and experience Gl arrest in response to DNA damage (Kamijo et al., 1997). Conceivably, p53 is activated by MpslA induced CIN and decreases viability of Mpsl A/A; p9ARF-/-MEFs. Nonetheless, MpslA cell inviability is a result of p53-induced apoptosis or growth arrest that is independent ofpl 9ARF . p53 may inhibit MpslA from facilitatingtumorigenesis by inducing apoptosis Why does inactivation of p53 allow MpslA to facilitate tumorigenesis? MpslA/A ; p53 -/-MEFs can grow in culture demonstrating loss of p53 rescues MpslA cellular lethality. Furthermore, all thymi where Mps 1 and p53 are simultaneously mutated develop tumors. MpslA does not appear to be inducing senescence as MpslA/A thymi show proliferation (by Ki-67) and retain p53 DNA. These data suggest that the Mps mutation is activating p53 and causing the majority of MpslA expressing cells to die. In MpslA/A; p53 +/ thymocytes, a subset of cells from each mouse can successfully undergo LOH of p53, survive and develop thymomas. These results suggest that LOH of p53 must be an early event in tumor evolution. Presumably, once cells lose p53 function to prevent apoptosis, Mps lA-induced CIN is causing stochastic loss and gain of chromosomes that result in a favorable environment for a few cells to become tumorigenic. What is the lesion generated by MpslA that activates p53? Mps expression has been shown to be suppressed by p53 after DNA damage (Bhonde et al., 2006). Microarray analysis of human colon carcinoma cell lines treated with the topoisomerase 195 I inhibitor SN-38 showed Mpsl expression was 2-fold higher when cells harbor p53 mutations (Bhonde et al., 2006). When wildtype p53 was introduced into the carcinoma cells, both Mpsl mRNA and protein expression was reduced. Although there is little evidence for chromosome missegregation events directly causing DNA damage (Burds et al., 2005), one possibility is that mutating Mps 1 does increase DNA damage and this is activating p53. Another possibility is that MpslA is causing damage to the mitotic spindle. Since generating the Mps lA allele requires expression of Cre recombinase which is known to cause promiscuous recombination at cryptic LoxP sites in cells and induce DNA damage, it is also possible that expression of Cre recombinase is activating p53 and killing MpslA MEFs. A fourth possibility is that p53 does not only respond to DNA damage, but to changes in DNA content because of MpslA -induced CIN. Aneuploidy could produce global expression changes that causes cellular stress and activates p53. It remains an open question whether any of these hypotheses are in fact correct or if other lesions are produced by expression of Mps lA to activate p53. Integratingthe DNA damage and spindle checkpoints Human Mps 1 has been reported to interact with and phosphorylate the DNA damage checkpoint kinase Chk2 and the RecQ helicase Blm (Ellis et al., 1995; Leng et al., 2006; Wei et al., 2005). BLM is the gene product mutated in Bloom syndrome, a rare human autosomal recessive disorder characterized by genomic instability where patients have a predisposition to diabetes, lung disease and cancer (German, 1995). The Blm helicase maintains genomic stability by suppressing excessive crossing over during homologous recombination, and Blm deficiency elevates the rates of sister chromatid exchange during mitotis (Wu and Hickson, 2003). The Blm helicase is also required in 196 DNA replication, recombination as well as DNA repair. (Hickson, 2003). Phosphorylation of Blm by Mps 1 appears to be necessary for accurate chromosome segregation (Leng et al., 2006). Chk2 is activated in response to DNA damage and prevents genomic instability partly by directly phosphorylating p53 (Hirao et al., 2002; Sancar et al., 2004). Blm also interacts with p53 and Blm-null cells are deficient for apoptosis (Wang et al., 2001). These data supports a model where Mpsl integrates mitotis and accurate chromosome segregation to the DNA damage checkpoint through Chk2 and Blm. These Mpsl substrates may provide a framework for determining the relationship between Mps lA and p53 activation. The spindle checkpoint kinase Mpsl has multiple functions Mpsl functions in mitotic and meiotic timing Mammalian spindle checkpoint genes have multiple functions. Cell biology experiments, particularly from time lapse imaging of siRNA depleted HeLa cells, have demonstrated that the canonical spindle checkpoint genes can be partitioned into three functional groups in mammals: 1) "pure" checkpoint genes (Madl), 2) mitotic timing regulators (Mad2 and BubRI), and 3) chromosome-microtubule attachment genes (Bub and Bub3) (Meraldi et al., 2004). The work I present in chapter 4 of this thesis suggests Mpsl joins Mad2 and BubR1 as a regulator of mitotic timing. Depletion of hMpsl by siRNA results in faster mitotic timing in HeLa cells. Moreover, Mps 1 also appears to be required for meiotic timing (Katja Wassmann, personal communication). In a collaboration to study the effect of MpslA on meiosis, Mpsl conditional mice were crossed to Zp3-Cre transgenic mice to specifically express Mps lA before the first meiotic division. Preliminary data from Mpslf/f, Zp3-Cre + oocytes show that some oocytes can 197 complete nuclear envelope breakdown to anaphase onset in as little as four hours during meiosis I. Wildtype oocytes normally take 8 hours, showing a partial timing defect. MpslA does not appear to significantly affect mitotic timing in MpslA/ A; p53 -1-MEFs. This may reflect different mechanisms of timing for meiosis and mitosis or some adaptation in the Mps 1A ;p53 /-MEFs. Nonetheless, regulation of mitotic and meiotic timing should be added to the list of Mps 1 functions. CIN as a facilitator of tumorigenesis Spindle checkpoint dysfunction in mice universally induces CIN, but rarely causes tumor formation. Spindle checkpoint genes appear to segregate further into two phenotypic groups when mutated in mice: those that induce early aging (BubR1 and Bub3 when combined with Rae 1) and those that induce tumorigenesis (Mad2). Chapters 2 and 3 show that the Mps 1 Amutation is a potent tumorigenesis facilitator and suggest Mps 1 should join Mad2 in the category of spindle checkpoint genes that induce cancer in mice when mutated. Thus, mutating Mps 1 may contribute to a CIN phenotype through dysregulation of various functions. Bub3 +' mice are haploinsufficient for spindle checkpoint function, but are not predisposed to spontaneous tumor formation (Kalitsis et al., 2000). BubR1 hypomorphic mice exhibit increased aneuploidy, but develop early onset aging and infertility without an increase in cancer incidence (Baker et al., 2006). This data would argue that aneuploidy is not sufficient to drive cancer formation. However, a recent study identified missense or truncating mutations in BUBR1 from individuals suffering from a rare human condition called mosaic variegated aneuploidy characterized by growth 198 retardation, microcephaly, childhood cancer and CIN (Hanks et al., 2004). Hence, mutation of spindle checkpoint components can induce CIN and result in cancer. Here, I show that a mutation in Mpsl induces CIN and is sufficient for lymphomagenesis, although tumor incidence is sporadic. However, CIN functions as a robust facilitator of tumorigenesis when p53 is also inactivated. Moreover, MpslA-induced CIN also functions as a facilitator of lymphomagenesis on a p 19-heterozygous background - a mouse model that is not normally prone to T-cell lymphoma. A model of specific gene mutations causing the step by step process of carcinogenesis is not incompatible with a CIN environment as a causal agent of tumor formation (Loeb, 1991; Loeb et al., 2003; Nowak et al., 2002). In fact, the clonal expansion theory of tumor progression which hypothesizes advantageous mutations are accumulated in a tumor cell by successive waves of clonal selection would suggest that a CIN environment should increase the mutation rate and accelerate tumor malignancy (Nowell, 1976). I have shown that CIN does accelerate tumor onset in p53-null animals possibly by favoring aneuploid states that confer a selective advantage. FutureDirections MpslA as a universal facilitator of tumorigenesis? I have shown that MpslA can cooperate with the haploinsufficient tumor suppressor genes p53 and pl 9 ARF to induce maglinant lymphoma in thymocytes. Can this mutation act as a universal facilitator of tumorigenesis? Adenomatous polyposis coli (Apc) is a well known tumor suppressor gene that interacts with the spindle checkpoint (Kaplan et al., 2001). The APC Min allele causes mice to develop multiple tumors in the small intestine and an occasional polyp in the colon (Su et al., 1992). I have begun to 199 intercross the Mpsl+/A mice to APCMinI+ mice, on a pure C57BL/6 background to ascertain if MpslA increases tumor spectrum and/or onset in the compound mutant mice. This study also would test the interaction of microsatellite instability (MIN) and chromosomal instability (CIN). In human colorectal tumors, mutations causing MIN and CIN phenotypes are often mutually exclusive (Lengauer et al., 1997). A study of APCMin/+; BubR1 +/ mice showed these mice had an increase in both colonic tumor number and tumor grade over APCu "in /+ mice, providing validation that spindle checkpoint dysfunction and Apemin does cooperate in tumorigenesis (Rao et al., 2005). Additional mouse models that are attractive candidates to interbreed with the Mps 1A conditional mice are the two recently reported targets for human Mpsl: Chk2 and Blm (Chester et al., 2006; Hirao et al., 2002; Leng et al., 2006; Wei et al., 2005). Although, Chk2 -/ mice are not susceptible to spontaneous tumors (Hirao et al., 2002), and MpslA has a sporadic incidence of spontaneous tumors (chapter 2), combining both mutations may reveal a tumor phenotype in highly proliferating cells given the reported interaction between Chk2 and Mpsl in human cells (Wei et al., 2005). Another informative compound mouse model to study would be Mps 1A; Blm compound conditional mice (Chester et al., 2006). Since Bloom syndrome patients are predisposed to multiple types of cancers (Hickson, 2003), this mouse model could be studied in multiple tissues for cancer disposition using an inducible Cre line such as the tamoxifeninducible Cre mouse (Hayashi and McMahon, 2002). What is the mechanism of MpslA induced chromosome missegregation? The truncated Mps lA protein retains kinetochore localization in HeLa cells, and both tumor cells and MEFs expressing MpslA respond to spindle damage caused by 200 nocodazole indicating the spindle checkpoint is at least partially active. Residues 53 to 158 are deleted in hMpslA protein. Kinetochore localization of human Mpsl requires the N-terminal portion of the protein (Liu et al., 2003; Stucke et al., 2004). Mutational analysis of hMps seeking its kinetochore localization domain showed that a mutant protein encoding residues 1 through 305 was sufficient to localize to the kinetochore (Stucke et al., 2004). Intriguingly, mutant proteins that only encoded the first 128 amino acids of hMps 1 or residues 96 to 318 were unable to localization to the kinetochore (Stucke et al., 2004). The kinetochore localization of the hMpslA protein is consistent with data from previous Mpsl mutants and these results together suggest the two ends of the kinetochore binding domain are necessary for localization while about 100 amino acids (residues 53 to 158) are dispensible. Why this highly conserved deleted region would be dispensible is unclear, but the structure of Mpsl should be quite revealing if biophysical studies are conducted. Presumably, mMpsl A also properly localizes to the kinetochore since the deleted region is virtually identical between mMpsl A (residues 47 to 154) and hMpslA. Thus, I hypothesize that although MpslA protein can associate with the kinetochore, it may not interact properly with its targets and result in CIN. Few targets of Mpsl have been characterized, but possibilities for abnormal association are with Damlp, Chk2 or Blm. Mpslp interacts with the microtubule-binding protein Daml in S. cerevisiae (Jones et al., 1999). Daml is an essential subunit of the DASH complex which is required for sister kinetochore biorientation (Janke et al., 2002; Li et al., 2002) Mpslp has been shown to phosphorylate Daml at two sites and Mpslmediated phosphorylation is necessary for efficient coupling of kinetochores to the plus ends of spindle microtubules (Shimogawa et al., 2006). The MpslA mutation is directed 201 against the N-terminal portion of the protein and should not impact the function of the kinase domain. Conceivably, Mpsl kinase function may be affected and prevent efficient phosphorylation of Daml and result in mono-oriented kinetochores during mitosis which later become lagging. A human homologue of Damlp has been recently identified (Meraldi et al., 2006). Moreover, I have already generated the analogous MpslA mutation for hMpsl. It would be advisable to ascertain if the Mpslp-phosphorylated residues in Damlp are conserved in the human homologue, before undertaking the biochemical analysis of human Mps 1 and Dam1. Nonetheless, the characterization of Mps lA kinase activity in vitro could be extremely informative to mechanism, particularly if MpslA does not recapitulate the phosphorylation results of wildtype Mps 1 on the known human substrates like Blm and Chk2. The intersection of p53 and the spindle checkpoint in tumorigenesis Why does inactivating p53 rescue MpslA MEF inviability, but inactivating pl 9 ARF does not? I propose that mutation of Mpsl is inducing chromosomal instability which activates a p53-dependent apoptotic pathway. Synergy between the spindle checkpoint and p53 has been previously described for Mad2 in vivo and in vitro (Burds et al., 2005). Moreover, recent experiments with compound conditional Mad2, p53 knockout mice that display an increased incidence of liver tumors further supports a connection between the p53 dependent apoptosis pathway and spindle checkpoint inactivation (Ying Yue, personal communication). Intriguingly, Blm interacts with p53 to mediate apoptosis, and Blm-deficient cells are also deficient for apoptosis (Wang et al., 2001). Therefore, the Mpsl; Blm conditional mice proposed above is one way to 202 investigate the hypothesis that lack of apoptosis is allowing Mps 1A to induce lymphomagenesis in vivo. Recent mouse models of the spindle checkpoint genes - BubR1, Bub3 and Rae 1suggest that CIN in these knockout mice are more likely to cause cellular senescence and early aging than tumorigenesis which appears to be dependent on the p53 pathway (Baker et al., 2005; Baker et al., 2004; Baker et al., 2006). BubR1 ÷'- mice exhibit aneuploidy, but have little elevated predisposition to cancer (Wang et al., 2004). Instead, decreased expression of BubR1 causes early onset aging phenotypes and decreased lifespan in mice as demonstrated with a series of BubR1 genetic mutants combining hypomorphic and knockout alleles (Baker et al., 2004). A link between early onset aging and checkpoint induced aneuploidy is further strengthened from a study of Bub3÷/-; Rae +'-compound mice showing early aging (Baker et al., 2006). Like BubR1 haploinsufficient mice, Bub3'/; Rae' / mice have a low propensity for spontaneous tumor formation (Babu et al., 2003). Spindle checkpoint dysregulation appears to promote aging due to premature senescence. Bub3 +-; Rae+'- mouse embryonic fibroblasts (MEFs) undergo premature senescence which appears to be dependent on the p53-regulated growth arrest pathway since several proteins in the pathway including pl 9 ARF , p53, p21, and p16 are upregulated. This begs the question what would be the result of inactivating p53 in the senescence-inducing checkpoint (BubR1, Bub3 and Rael) mouse models? Would these compound mice reveal a tumor phenotype? I propose that inactivation of these spindle checkpoint genes in combination with p53-deficiency will promote tumorigenesis in mice. A recent study has shown that preventing p53-dependent senescence does promote 203 favorable conditions for carcinogenesis in otherwise senescent cells. Inactivation of the tumor suppressor gene Pten induces cellular senescence in a mouse model of prostate cancer and in MEFs (Chen et al., 2005). Intriguingly, this growth arrest is dependent on the p53 pathway and inactivation of p53 prevents senescence in Pten /- cells. Combined inactivation of p53 and Pten in mouse prostate cells intensifies tumorigenicity, decreasing time of tumor onset and increasing metastatic potential. The outcome from inactivating p53 or other members of the p53-dependent growth arrest pathway such as pl 9 ARF in the early aging spindle checkpoint mouse models would significantly add to our understanding of the intersection of spindle checkpoint defects and p53 activation. Tissue specific effects of spindle checkpoint inactivation and genetic modifiers There are clearly tissue-specific effects of various tumor-promoting mutations in cancer. For example, Brcal and Brca2 mutations mostly occur cancers of the breast and ovary (Jasin, 2002; Venkitaraman, 2002). Mutations in APC are often limited to colorectal cancers (Narayan and Roy, 2003). Although, many human cancers contain p53 mutations, genetic inactivation of p53 in mice primarily cause lymphomas and sarcomas (Donehower et al., 1992; Jacks et al., 1994; Purdie et al., 1994). Tumor formation in other p53-inactivated tissues, e.g. the liver, require additional mutations (Farazi et al., 2006). The MpslA truncation mutation exhibits tissue-preference in tumor promotion. In chapter 2, Ipresented survival data for specific expression of Mps 1A in thymocytes. In the appendix, I discuss the effect of Mps 1A in two primarily post-mitotic tissues - the lung and the liver. MpslA thymi are smaller than wildtype thymi. Futhermore, MpslA thymocytes are extremely prone to tumor development when p53 is also inactivated. Unlike in the thymus, expressing the truncated form of Mpsl in the lung or liver has 204 minimal effect on tissue architecture. Combining MpslA either with inactivation of p53 in the liver (with albumin-cre) or an activating allele of K-ras in the lung (with intranasal infection of adenoviral-cre) also does not significantly increase tumor incidence. Expressing KrasG12D in the mouse lung typically causes adenomas and some papillomas (Jackson et al., 2001). Although in the lung, MpslA may be altering tumor type, by increasing the propensity for papillomas. Moreover, mice expressing Mps 1NA in the liver do not exhibit any significant developmental problems or show a propensity for tumor formation. In contrast, conditional knockout of Mad2 in the liver causes neoplasia and increased tumor incidence (Ying Yue, personal communication). Interestingly, while Mps f/, p5 3f+, Lck-cre ÷ mice are fully penetrant with respect to thymoma development, preliminary data from the Mad2; p53 compound conditional knockout study suggests penetrance is significantly lower (Ying Yue, personal communication). What is the basis of these different tissue-specific effects? This discrepancy in tumor incidence may reflect a genetic modifier effect or cell cycle effects such as the presence of mitosis. It is well known that modifiers on the various mouse genetic backgrounds can dramatically influence tumorigenesis including tumor grade, tumor incidence and tissue specificity (Dragani, 2003). For instance, both tumor spectrum and tumor onset varies in p53 knockout mice on different genetic backgrounds (Donehower et al., 1995; Harvey et al., 1993). I carried out the tumor studies described in this dissertation on a mixed genetic background of 129/Sv and C57BL/6. Are the tissue specific effects in tumor development for Mps 1 due to genetic modifiers? I have backcrossed both the Mps 1 Aand flox alleles onto the C57BL/6 background for at least 5 generations. Therefore, it would be feasible to study the effects genetic modifiers may have on tumor phenotypes for MpslA. MpslA 205 accelerates tumorigenesis in both p53 conditional knockout and p53 conventional knockout mice with similar kinetics of tumor onset. I have begun to intercross Mps1 conditional and conventional Mpsl mutant mice to p53 conventional knockout mice on the C57BL/6 background to compare tumor spectrum and tumor onset to those results from chapter 2. Would the tumor incidence of MpslA and p53 knockout compound mutant mice be different on a pure C57BL/6 genetic background from a mixed 129/Sv and C57BL/6 background? This is quite likely as tumor onset in p53 knockout mice is earlier on the 129/Sv background than the mixed background suggesting modifiers in the C57BL/6 background may be inhibiting tumor progression (Harvey et al., 1993). Furthermore, these C57BL/6 mice could be crossed to 129/Sv for loss of heterozygosity studies by comparative genome hybridization (CGH) like Representational Oligonucleotide Microarray Analysis (ROMA) (Hicks et al., 2005). Such experiments would be quite enlightening and could answer, for example, if there are specific regions being selected for or against in the lymphomas. Thus, we would gain a better mechanistic understanding of tumor development in situations of CIN. Another variable impacting the tumor studies presented here is the difference in proliferative capability among tissues. Human Mpsl/TTK was originally discovered to be a dual-specificity kinase that is highly expressed in all human proliferative tissues and cells, particularly the thymus (Hogg et al., 1994; Mills et al., 1992). Hence, a reason for the robustness of Mps 1A-induced tumorigenesis in p53 inactivated thymocytes may be the increased mitosis and/or high expression level of Mpsl in the thymus. In contrast, MpslA does not appear to promote hepatocellular carcinogenesis in p53 knockout mice. Mps 1 mRNA is detected in the liver, but it is much lower than in the thymus. However, 206 the liver is a tissue where mitosis can be reactivated in hepatocytes in response to liver damage (Sell, 2001). The characterization of the polo-like kinase Plk4 heterozygous knockout mouse in hepatoceullular carcinogenesis was through a model utilizing partial hepatectomy, a surgical procedure where a portion of the liver is removed (Ko et al., 2005). Thus, a tumor phenotype may be revealed in the MpslA, p53-knockout compound mice if hepatocytes are induced to re-enter the cell cycle after a partial hepatectomy. Mpsl and human cancer Few spindle checkpoint mutations have been identified in human tumors, but they do exist (Cahill et al., 1998). Fewer studies have examined Mpsl in human tumors for sequence variations or changes in expression levels than for other canonical spindle checkpoint genes like Bubl, BubR1 and Mad2 (Cahill et al., 1999; Cahill et al., 1998; Camacho et al., 2006; Hanks et al., 2004; Hempen et al., 2003; Kim et al., 2005; Langerod et al., 2003; Matsuura et al., 2006; Saeki et al., 2002; Seike et al., 2002; Shichiri et al., 2002; Shin et al., 2003; Takahashi et al., 1999; Tanaka et al., 2001; Wang et al., 2002; Yuan et al., 2006). In three studies investigating sequence variation of Mpsl in human tumors, no mutations of Mpsl were found in colorectal tumors, breast cancer cell lines or mantle cell lymphoma (Cahill et al., 1999; Camacho et al., 2006; Yuan et al., 2006). However, high Mpsl expression levels have been found in human gastric cancer and breast cancer cell lines (Iwase et al., 1993; Yuan et al., 2006). Although upregulation of Mps 1 in these tumors may have some functional significance in tumorigenesis, it is equally likely this is a consequence of the increased proliferative status of tumor cells. Since Mpsl does not appear to be targeted for mutation in human tumors, does my research on mouse Mpsl have any significance in human cancer? Certainly it is possible 207 that Mpsl does not operate as a tumor suppressor gene. Because its mutation is not relevant in human cancers, mutations are not selected for and have not been found. However, so few studies have been published that have examined the human Mpsl locus for mutations that I would reserve my response until more tumors are sequenced. Since human Mpsl was identified from a T-cell library (Mills et al., 1992), and MpslA alone is sufficient to induce thymomas is a small subset of mice, lymphomas may be an ideal tumor type to begin the search for mutations. Summary The work I present here addresses an age old question in the field of cancer biology: Is chromosomal instability (CIN) simply a consequence of the steps leading to cancer or can CIN also function as a causal agent? I generated a partial loss of function allele in the spindle checkpoint gene Mpsl to induce a CIN phenotype in mice. I argue that CIN does function in tumorigenesis as a causal agent but requires the inactivation of at least the p53 pathway. My work shows that spindle checkpoint dysfunction by a weakened checkpoint allele is a valid route to cancer and underscores the necessity of additional studies for spindle checkpoint dysfunction in human tumors. However, the work here also points to problems that afflict the spindle checkpoint field. Only a few spindle checkpoint components have been genetically inactivated or mutated in mice. Mutations in checkpoint components such as Zw 10 have been identified in human cancers, but have not been studied for a causal role in tumor development. Checkpoint proteins, particularly the kinases, appear to have multiple functions and dissecting the mechanistic details of their function has proven difficult. Progress in understanding the spindle checkpoint and its role in tumorigenesis will require additional genetic and 208 biochemical analysis through the generation of additional conditional checkpoint mouse mutants, the identification of checkpoint kinase substrates, a better understanding of how the spindle checkpoint synergizes with DNA damage checkpoint and repair components, and a better characterization of the signals that activate the checkpoint. 209 References Babu, J.R., K.B. Jeganathan, D.J. Baker, X. Wu, N. Kang-Decker, and J.M. van Deursen. 2003. Rael is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 160:341-53. Baker, D.J., J. Chen, and J.M. van Deursen. 2005. The mitotic checkpoint in cancer and aging: what have mice taught us? Curr Opin Cell Biol. 17:583-9. Baker, D.J., K.B. Jeganathan, J.D. Cameron, M. Thompson, S. Juneja, A. Kopecka, R. Kumar, R.B. Jenkins, P.C. De Groen, P. Roche, and J.M. Van Deursen. 2004. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. Baker, D.J., K.B. Jeganathan, L. Malureanu, C. Perez-Terzic, A. Terzic, and J.M. van Deursen. 2006. Early aging-associated phenotypes in Bub3/Rael haploinsufficient mice. J Cell Biol. 172:529-40. Bhonde, M.R., M.L. Hanski, J. Budczies, M. Cao, B. Gillissen, D. Moorthy, F. Simonetta, H. Scherubl, M. Truss, C. Hagemeier, H.W. Mewes, P.T. Daniel, M. Zeitz, and C. Hanski. 2006. DNA damage-induced expression of p53 suppresses mitotic checkpoint kinase hMps 1: the lack of this suppression in p53MUT cells contributes to apoptosis. JBiol Chem. 281:8675-85. Boveri, T. 1914. Zur Frage der Entstehung maligner Tumoren. Fischer. Burds, A.A., A.S. Lutum, and P.K. Sorger. 2005. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl Acad Sci U S A. 102:11296-301. Cahill, D.P., L.T. da Costa, E.B. Carson-Walter, K.W. Kinzler, B. Vogelstein, and C. Lengauer. 1999. Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics. 58:181-7. Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300-3. Camacho, E., S. Bea, I. Salaverria, A. Lopez-Guillermo, X. Puig, Y. Benavente, S. de Sanjose, E. Campo, and L. Hernandez. 2006. Analysis of Aurora-A and hMPS 1 mitotic kinases in mantle cell lymphoma. Int J Cancer.118:357-63. Chen, Z., L.C. Trotman, D. Shaffer, H.K. Lin, Z.A. Dotan, M. Niki, J.A. Koutcher, H.I. Scher, T. Ludwig, W. Gerald, C. Cordon-Cardo, and P.P. Pandolfi. 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 436:725-30. 210 Chester, N., H. Babbe, J. Pinkas, C. Manning, and P. Leder. 2006. Mutation of the Murine Bloom's Syndrome Gene Produces Global Genome Destabilization. Mol Cell Biol. 26:6713-26. Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635-45. Donehower, L.A., M. Harvey, B.L. Slagle, M.J. McArthur, C.A. Montgomery, Jr., J.S. Butel, and A. Bradley. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356:215-21. Donehower, L.A., M. Harvey, H. Vogel, M.J. McArthur, C.A. Montgomery, Jr., S.H. Park, T. Thompson, R.J. Ford, and A. Bradley. 1995. Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog. 14:16-22. Dragani, T.A. 2003. 10 years of mouse cancer modifier loci: human relevance. Cancer Res. 63:3011-8. Draviam, V.M., S. Xie, and P.K. Sorger. 2004. Chromosome segregation and genomic stability. Curr Opin Genet Dev. 14:120-5. Ellis, N.A., J. Groden, T.Z. Ye, J. Straughen, D.J. Lennon, S. Ciocci, M. Proytcheva, and J. German. 1995. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 83:655-66. Farazi, P.A., J. Glickman, J. Homer, and R.A. Depinho. 2006. Cooperative interactions of p53 mutation, telomere dysfunction, and chronic liver damage in hepatocellular carcinoma progression. CancerRes. 66:4766-73. Fisk, H.A., C.P. Mattison, and M. Winey. 2004. A field guide to the Mpsl family of protein kinases. Cell Cycle. 3:439-42. German, J. 1995. Bloom's syndrome. Dermatol Clin. 13:7-18. Hanks, S., K. Coleman, S. Reid, A. Plaja, H. Firth, D. Fitzpatrick, A. Kidd, K. Mehes, R. Nash, N. Robin, N. Shannon, J. Tolmie, J. Swansbury, A. Irrthum, J. Douglas, and N. Rahman. 2004. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB IB. Nat Genet. 36:1159-61. Harris, S.L., and A.J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24:2899-908. Hartwell, L.H., and M.B. Kastan. 1994. Cell cycle control and cancer. Science. 266:18218. 211 Harvey, M., M.J. McArthur, C.A. Montgomery, Jr., A. Bradley, and L.A. Donehower. 1993. Genetic background alters the spectrum of tumors that develop in p53deficient mice. Faseb J. 7:938-43. Hayashi, S., and A.P. McMahon. 2002. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 244:305-18. Hempen, P.M., H. Kurpad, E.S. Calhoun, S. Abraham, and S.E. Kern. 2003. A double missense variation of the BUB 1 gene and a defective mitotic spindle checkpoint in the pancreatic cancer cell line Hs766T. Hum Mutat. 21:445. Hernando, E., Z. Nahle, G. Juan, E. Diaz-Rodriguez, M. Alaminos, M. Hemann, L. Michel, V. Mittal, W. Gerald, R. Benezra, S.W. Lowe, and C. Cordon-Cardo. 2004. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 430:797-802. Hicks, J., L. Muthuswamy, A. Krasnitz, N. Navin, M. Riggs, V. Grubor, D. Esposito, J. Alexander, J. Troge, M. Wigler, S. Maner, P. Lundin, and A. Zetterberg. 2005. High-resolution ROMA CGH and FISH analysis of aneuploid and diploid breast tumors. Cold Spring Harb Symp Quant Biol. 70:51-63. Hickson, I.D. 2003. RecQ helicases: caretakers of the genome. Nat Rev Cancer.3:16978. Hirao, A., A. Cheung, G. Duncan, P.M. Girard, A.J. Elia, A. Wakeham, H. Okada, T. Sarkissian, J.A. Wong, T. Sakai, E. De Stanchina, R.G. Bristow, T. Suda, S.W. Lowe, P.A. Jeggo, S.J. Elledge, and T.W. Mak. 2002. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)dependent and an ATM-independent manner. Mol Cell Biol. 22:6521-32. Hogg, D., C. Guidos, D. Bailey, A. Amendola, T. Groves, J. Davidson, R. Schmandt, and G. Mills. 1994. Cell cycle dependent regulation of the protein kinase TTK. Oncogene. 9:89-96. Iwase, T., M. Tanaka, M. Suzuki, Y. Naito, H. Sugimura, and I. Kino. 1993. Identification of protein-tyrosine kinase genes preferentially expressed in embryo stomach and gastric cancer. Biochem Biophys Res Commun. 194:698-705. Jacks, T., L. Remington, B.O. Williams, E.M. Schmitt, S. Halachmi, R.T. Bronson, and R.A. Weinberg. 1994. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 4:1-7. Jackson, E.L., N. Willis, K. Mercer, R.T. Bronson, D. Crowley, R. Montoya, T. Jacks, and D.A. Tuveson. 2001. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15:3243-8. 212 Janke, C., J. Ortiz, T.U. Tanaka, J. Lechner, and E. Schiebel. 2002. Four new subunits of the Daml-Duol complex reveal novel functions in sister kinetochore biorientation. Embo J. 21:181-93. Jasin, M. 2002. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 21:8981-93. Jones, M.H., J.B. Bachant, A.R. Castillo, T.H. Giddings, Jr., and M. Winey. 1999. Yeast Damlp is required to maintain spindle integrity during mitosis and interacts with the Mpslp kinase. Mol Biol Cell. 10:2377-91. Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277-82. Kamijo, T., F. Zindy, M.F. Roussel, D.E. Quelle, J.R. Downing, R.A. Ashmun, G. Grosveld, and C.J. Sherr. 1997. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product pl 9ARF. Cell. 91:649-59. Kaplan, K.B., A.A. Burds, J.R. Swedlow, S.S. Bekir, P.K. Sorger, and I.S. Nathke. 2001. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 3:429-32. Kim, H.S., K.H. Park, S.A. Kim, J. Wen, S.W. Park, B. Park, C.W. Gham, W.J. Hyung, S.H. Noh, H.K. Kim, and S.Y. Song. 2005. Frequent mutations of human Mad2, but not Bub 1, in gastric cancers cause defective mitotic spindle checkpoint. Mutat Res. 578:187-201. Ko, M.A., C.O. Rosario, J.W. Hudson, S. Kulkarni, A. Pollett, J.W. Dennis, and C.J. Swallow. 2005. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat Genet. 37:883-8. Kops, G.J., B.A. Weaver, and D.W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer.5:773-85. Langerod, A., M. Stromberg, K. Chin, V.N. Kristensen, and A.L. Borresen-Dale. 2003. BUB 1 infrequently mutated in human breast carcinomas. Hum Mutat. 22:420. Leng, M., D.W. Chan, H. Luo, C. Zhu, J. Qin, and Y. Wang. 2006. MPS 1-dependent mitotic BLM phosphorylation is important for chromosome stability. ProcNatl Acad Sci USA. 103:11485-90. Lengauer, C., K.W. Kinzler, and B. Vogelstein. 1997. Genetic instability in colorectal cancers. Nature. 386:623-7. Li, Y., J. Bachant, A.A. Alcasabas, Y. Wang, J. Qin, and S.J. Elledge. 2002. The mitotic spindle is required for loading of the DASH complex onto the kinetochore. Genes Dev. 16:183-97. 213 Liu, S.T., G.K. Chan, J.C. Hittle, G. Fujii, E. Lees, and T.J. Yen. 2003. Human MPS1 Kinase Is Required for Mitotic Arrest Induced by the Loss of CENP-E from Kinetochores. Mol Biol Cell. 14:1638-51. Loeb, L.A. 1991. Mutator phenotype may be required for multistage carcinogenesis. CancerRes. 51:3075-9. Loeb, L.A., K.R. Loeb, and J.P. Anderson. 2003. Multiple mutations and cancer. Proc Natl Acad Sci US A. 100:776-8 1. Lowe, S.W., and C.J. Sherr. 2003. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 13:77-83. Matsuura, S., Y. Matsumoto, K. Morishima, H. Izumi, H. Matsumoto, E. Ito, K. Tsutsui, J. Kobayashi, H. Tauchi, Y. Kajiwara, S. Hama, K. Kurisu, H. Tahara, M. Oshimura, K. Komatsu, T. Ikeuchi, and T. Kajii. 2006. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 140:35867. Meraldi, P., V.M. Draviam, and P.K. Sorger. 2004. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 7:45-60. Meraldi, P., A.D. McAinsh, E. Rheinbay, and P.K. Sorger. 2006. Phylogenetic and structural analysis of centromeric DNA and kinetochore proteins. Genome Biol. 7:R23. Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V. Murty, and R. Benezra. 2001. MAD2 haploinsufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature.409:355-9. Mills, G.B., R. Schmandt, M. McGill, A. Amendola, M. Hill, K. Jacobs, C. May, A.M. Rodricks, S. Campbell, and D. Hogg. 1992. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. JBiol Chem. 267:16000-6. Narayan, S., and D. Roy. 2003. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer. 2:41. Nowak, M.A., N.L. Komarova, A. Sengupta, P.V. Jallepalli, M. Shih le, B. Vogelstein, and C. Lengauer. 2002. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci U S A. 99:16226-31. Nowell, P.C. 1976. The clonal evolution of tumor cell populations. Science. 194:23-8. Purdie, C.A., D.J. Harrison, A. Peter, L. Dobbie, S. White, S.E. Howie, D.M. Salter, C.C. Bird, A.H. Wyllie, M.L. Hooper, and et al. 1994. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 9:603-9. 214 Rao, C.V., Y.M. Yang, M.V. Swamy, T. Liu, Y. Fang, R. Mahmood, M. Jhanwar-Uniyal, and W. Dai. 2005. Colonic tumorigenesis in BubR1+/-ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 102:4365-70. Saeki, A., S. Tamura, N. Ito, S. Kiso, Y. Matsuda, I. Yabuuchi, S. Kawata, and Y. Matsuzawa. 2002. Frequent impairment of the spindle assembly checkpoint in hepatocellular carcinoma. Cancer.94:2047-54. Sancar, A., L.A. Lindsey-Boltz, K. Unsal-Kacmaz, and S. Linn. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 73:39-85. Seike, M., A. Gemma, Y. Hosoya, Y. Hosomi, T. Okano, F. Kurimoto, K. Uematsu, K. Takenaka, A. Yoshimura, M. Shibuya, K. Ui-Tei, and S. Kudoh. 2002. The promoter region of the human BUBRI gene and its expression analysis in lung cancer. Lung Cancer.38:229-34. Sell, S. 2001. The role of progenitor cells in repair of liver injury and in liver transplantation. Wound RepairRegen. 9:467-82. Shichiri, M., K. Yoshinaga, H. Hisatomi, K. Sugihara, and Y. Hirata. 2002. Genetic and epigenetic inactivation of mitotic checkpoint genes hBUB 1 and hBUBR1 and their relationship to survival. CancerRes. 62:13-7. Shimogawa, M.M., B. Graczyk, M.K. Gardner, S.E. Francis, E.A. White, M. Ess, J.N. Molk, C. Ruse, S. Niessen, J.R. Yates, 3rd, E.G. Muller, K. Bloom, D.J. Odde, and T.N. Davis. 2006. Mpsl phosphorylation of daml couples kinetochores to microtubule plus ends at metaphase. CurrBiol. 16:1489-501. Shin, H.J., K.H. Baek, A.H. Jeon, M.T. Park, S.J. Lee, C.M. Kang, H.S. Lee, S.H. Yoo, D.H. Chung, Y.C. Sung, F. McKeon, and C.W. Lee. 2003. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 4:483-97. Stucke, V.M., C. Baumann, and E.A. Nigg. 2004. Kinetochore localization and microtubule interaction of the human spindle checkpoint kinase Mps 1. Chromosoma. 113:1-15. Su, L.K., K.W. Kinzler, B. Vogelstein, A.C. Preisinger, A.R. Moser, C. Luongo, K.A. Gould, and W.F. Dove. 1992. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 256:668-70. Takahashi, T., N. Haruki, S. Nomoto, A. Masuda, S. Saji, and H. Osada. 1999. Identification of frequent impairment of the mitotic checkpoint and molecular analysis of the mitotic checkpoint genes, hsMAD2 and p55CDC, in human lung cancers. Oncogene. 18:4295-300. 215 Tanaka, K., J. Nishioka, K. Kato, A. Nakamura, T. Mouri, C. Miki, M. Kusunoki, and T. Nobori. 2001. Mitotic checkpoint protein hsMAD2 as a marker predicting liver metastasis of human gastric cancers. Jpn J CancerRes. 92:952-8. Taylor, S.S., M.I. Scott, and A.J. Holland. 2004. The spindle checkpoint: a quality control mechanism which ensures accurate chromosome segregation. Chromosome Res. 12:599-616. Venkitaraman, A.R. 2002. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 108:171-82. Wang, Q., T. Liu, Y. Fang, S. Xie, X. Huang, R. Mahmood, G. Ramaswamy, K.M. Sakamoto, Z. Darzynkiewicz, M. Xu, and W. Dai. 2004. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 103:1278-85. Wang, X., D.Y. Jin, R.W. Ng, H. Feng, Y.C. Wong, A.L. Cheung, and S.W. Tsao. 2002. Significance of MAD2 expression to mitotic checkpoint control in ovarian cancer cells. CancerRes. 62:1662-8. Wang, X.W., A. Tseng, N.A. Ellis, E.A. Spillare, S.P. Linke, A.I. Robles, H. Seker, Q. Yang, P. Hu, S. Beresten, N.A. Bemmels, S. Garfield, and C.C. Harris. 2001. Functional interaction of p53 and BLM DNA helicase in apoptosis. JBiol Chem. 276:32948-55. Wei, J.H., Y.F. Chou, Y.H. Ou, Y.H. Yeh, S.W. Tyan, T.P. Sun, C.Y. Shen, and S.Y. Shieh. 2005. TTK/hMpsl participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. JBiol Chem. 280:7748-57. Wu, L., and I.D. Hickson. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 426:870-4. Yuan, B., Y. Xu, J.H. Woo, Y. Wang, Y.K. Bae, D.S. Yoon, R.P. Wersto, E. Tully, K. Wilsbach, and E. Gabrielson. 2006. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin CancerRes. 12:405-10. 216 Appendix Determining the effect of MpslA in two post mitotic tissues: a conditional K-ras lung cancer model and liver-specific deletion by Albumin-Cre 217 Introduction Adult somatic organs have different proliferative capabilities. The thymus is the site of developing T cells where thymocytes can be highly mitotic. In contrast, the liver and lung are mostly composed of differentiated cells and the mitotic index is normally quite low. Cancer can develop in any organ of the body, but many oncogenes and tumor suppressor genes exhibit preference for tumor formation in specific tissues. Although the reasons are uncertain, one possibility is the underlying rate of mitosis in a particular organ. Cancer is thought to require multiple "hits" (Duesberg and Li, 2003; Knudson, 2001). For a normal cell to have acquired a sufficient number of deleterious mutations to allow transformation into a cancerous cell, it has to have the capacity to self-renew or obtain key transforming events early during tumorigenesis. If a tissue is highly mitotic, then pre-cancerous cells can proliferate and increase their potential to acquire the necessary alterations for tumor formation. In a mostly post-mitotic tissue, a different set of initial events may need to occur to initiate tumorigenesis. Chromosomal instability (CIN) is an underlying feature of many human tumors. While it is uncertain whether CIN has a direct role in human cancer progression, I have shown in Chapter 2 that a CIN-causing mutation in the spindle checkpoint gene Mps 1 facilitates tumorigenesis in mouse thymocytes, particularly in the absence of at least one allele of p53. The thymus was chosen for the conditional expression of the hypomorphic Mps 1Aallele through Lck-Cre expression, precisely because it is a highly mitotic tissue where strong selection would presumably occur for those CIN causing events that would promote tumorigenesis. Remarkably, 100% of Mpslf1f; p 53f/+; Lck-cre ÷ mice develop thymomas with an average tumor onset of 3.3 months. These tumors appear to initiate 218 growth as thymocytes enter periods of rapid and extensive proliferation from the double negative to the double positive stages of thymocyte development. Thus, extensive mitosis seems to promote CIN and allow for the selection of cells that have accumulated the necessary events for transformation including loss of heterozygosity of p53. K-ras is a proto-oncogene encoding a monomeric GTPase that transduces mitogenic signals from growth factor receptors to multiple intracellular signaling cascades. K-ras is the most frequently mutated member of the Ras family in human cancer. Oncogenic K-ras mutations are found in as many as one half of human lung adenocarcinomas (Friday and Adjei, 2005). This frequency increases to >90% in mouse models of lung cancer (Malkinson, 1998). A mouse lung cancer model, where the oncogenic allele K-ras Gl 2D is conditionally activated by intranasal delivery of Cre recombinase to the lungs of mice, has been used successfully to study many aspects of lung tumorigenesis (Jackson et al., 2005; Jackson et al., 2001; Kim et al., 2005; Meuwissen et al., 2001). K-ras appears to stimulate proliferation of only a small portion of lung epithelial cells (Guerra et al., 2003). Analysis of the early stages of lung adenocarcinoma induced by oncogenic K-ras suggested the existence of bronchioalveolar stem cells (BASCs) within the lung that may be the cancer precursor cells (Jackson et al., 2001). Subsequent work by Kim et al have isolated these BASCs at the broinchioalveolar duct junction and shown this population of cells responds to activated K-ras by proliferating. Intriguely, the Ras pathway may intersect with the spindle checkpoint through the mitogen activated protein kinase (MAPK) pathway (Eves et al., 2006). One of the main effectors of Ras proteins is Raf kinase, a member of the MAPK pathway (Marshall, 1996). Raf kinase inhibitory protein (RKIP) inhibits this pathway by 219 regulating the kinetochore localization and kinase activity of Aurora B, which is required for proper chromosome segregation (Eves et al., 2006). These recent findings suggest an alternative mechanism for ras mutations to promote tumorigenesis is through creating aberrant mitoses. Here, I describe two mouse aging studies where I seek to determine the effect of MpslA in the lung and the liver. I found MpslA-induced CIN has only a modest effect on survival when expressed in the liver and does not predispose mice to either lung or liver cancer. Results and Discussion MpslA causes a modest decrease in mouse mortality when expressed in the liver To determine the effect MpslA has on mouse survival when it is mutated in the liver, Mpslf"mice were crossed to Albumin-cre (Alb-cre) transgenic mice to specifically express MpslA in hepatocytes (Postic and Magnuson, 2000). A cohort of 30 Mpslf/f; Alb-cre÷ mice and 17 Mps f/f; Alb-cre- mice were followed for up to 14 months and their survival assessed (Fig. Al). While four (13%) Mpsl'/f; Alb-cre ÷ mice died during that period, no Mpslf/f; Alb-cre- did so (Table Al). Two of those Mpslf/f; Alb-cre ÷ animals were recovered and were found to have very pale livers and bleeding in the intestinal area, suggesting liver function may have been compromised. Is death due to MpslA inducing liver tumor formation in these mice that impinges on the proper function of the intestinal organs? Unfortunately, decomposition had set in so histology was uninformative. When I analyzed the remaining 26 Mps l'f; Alb-cre ÷ mice, two animals were found with liver tumors before 14 months of age. However, one Mpsl"ff; Alb-cre- 220 Figure Al 100 90. 80- 70- Mpsl f/f, Alb-cre+ (n=30) _L Mpsl1 f/f, Alb-cre- (n=17) 60II 50I Time (months) IAric If/f 1r-2f/f Alk 12 12 rm-- MDs f/f 0 53f/+ Alb-cre + MDsIf/f 0 53f/+ Alb-cre + Moslf/+ D53f/+ Alb-cre + 221 Figure Al: MpslA causes a modest increase in mortality when expressedin the liver. (A)Kaplan-Meyer survival curve for Mpsl f Alb-cre + (red) and Mps1' f f Alb-cre (blue) mice. (B) Hematoxylin and eosion stained images of liver sections from 3 month old mice with the following genotypes: Mpsl fp53 f f Alb-cre ' , Mps U+ p53f/+ Alb-cre + and Mpslf/fp533 " Alb-cre +. By histology, the liver morphology is comparatively normal between all three groups of animals. 222 ~ I I mouse was also found to have develop hepatocellular carcinoma. Therefore, MpslA does not predispose the liver to hepatocellular carcinoma. Would expressing MpslA on a p53-hetrozygous background in hepatocytes facilitate tumorigenesis in the liver? Tumor development is fully penetrant when MpslA is expressed in a p53-heterozygous background in thymocytes (Chapter 2). Thymomas develop with an average tumor onset of 3.3 months in these mice. Preliminary data suggest no such effect occurs in the liver. The livers of two Mps l'; p53f/+; Alb-cre ÷ and three Mpslf/+; p53/+; Alb-cre÷ mice were isolated at 3 months of age and no neoplasia was found with homozygous expression of MpslA. In fact, the livers of Mpsl /";p53f/+; Alb-cre ÷ mice appear to be normal by histology showing no overall variation from either an albumin-cre negative liver or the liver from a Mps 1f/+; p53f/+; Alb-cre ÷ mouse. Thus, MpslA does not appear to facilitate tumorigenesis in the liver either on a p53-wildtype or a p53-heterozygous background. G 2 Dlungs MpslA has a modest effect on the tumor spectrum of K-rasC To determine if expression of MpslA would exacerbate the lung tumor phenotype in the oncogenic K-ras mouse model, MpslA conditional mice were interbred with conditional K-rasG12D mice. Mice of all six possible genotypes were infected with adenoviral cre by intranasal inhalation at six weeks of age to activate the mutant alleles of K-ras and Mpsl (Fig. A2A). Mice were sacrificed 16 weeks post-infection and their lungs prepared for histological analysis. In the presence of the K-rasGI 2Dmutation, all lungs suffered from some form of lung dysplasia depending on the amount of viral particles successfully inhaled by each mouse (Table Al). MpslA does not affect lung tissue architecture (Fig. A2A, B).When MpslA is combined with oncogenic K-ras, tumor 224 Figure A2 A Mps1 +/+ KrasG12D/+ 12 (12 Kras+/+ 4 (0) Mps if/+ 15 (15) 8 (0) Mps lf/f 16 (16) 24 (1) # animals (# animals with lung tumors) B Mpslf/f, K-ras+/+ h:i "i"~ *; I .·. i,. Q i"i-?J9 t-·:-·, ;i~ ·· ,;i hlS I:~ 3;i·4e c ii:t..i · ·i" : ot~,:~ -.-- ~ MDsl+/+. K-rasG12D/+ MDslf/f. K-rasG12D/+ 225 Mpslf/f, K-rasG12D/+ FigureA2: MpslA causes a modest shift in K-rasGl 2D lungsfrom adenomas to papillomas. (A) The number of animals whose lungs were examined following 16 weeks postinfection with Adenoviral cre bred from crosses between the Mpsl conditional and KrasGl 2Dconditional animals. The numbers in parentheses indicates the number of animals that exhibited lung tumors. In the presence of the oncogenic K-rasG 12D mutation, all animals showed evidence of tumor formation by histological analysis. (B) Hematoxylin and eosion stained images of lungs from a subset of animals described in figure A2A. In a Mpsl f/f K- ras+/' lung, the tissue appears to be normal with no indication of dysplasia. In Mpsl / + K- rasG 12Dlungs, we see both types of tumors, with more of propensity for adenomas. In some Mps l f K- rasGl 2Dlungs, there is no overt change in tumor incidence from Mpsl +/' K- raSGl 2D lungs. In other Mps Vf K- rasGJ 2D lungs (here we show three different set of lungs), there is more of a propensity for papillomas - dysplasia in the bronchial tubes. Black arrows point to adenomas and green arrows to papillomas. 226 incidence is not overtly changed. Neither tumor grade nor the type of lung dysplasia appears to change in K-ras G2D mice when Mpsl A is expressed. Mice expressing KrasGl 2D develop both adenomas and paillomas (bronchial dysplasia), with more of a tendency for adenomas (Fig. A2B and (Jackson et al., 2001)). These tumors typically are categorized as grade 1 and 2 by histological analysis, where cells still remain fairly uniform in size and shape. As in K-ras G 12D/+ lungs, we see both adenomas and some papillomas in the double mutant lungs (Fig. A2B). However, in -30% of double mutant mice, the tumor spectrum is shifted towards more papillomas (Fig. A2B). Why there may be a predilection for bronchial dysplasia in some lung cells when Mps lA is combined with oncogenic Kras is unknown. These data show that MpslA exerts only a moderate effect on tumor incidence in the K-ras G2D lung tumor model. Why does MpslA only exert a minimal effect in the lung and liver? Human Mps I/TTK was originally discovered to be a dual-specificity kinase that is highly expressed in all human proliferative tissues and cells, particularly the thymus (Hogg et al., 1994; Mills et al., 1992). Neither the lung nor the liver is normally proliferative, while the thymus is highly proliferative. Mps 1 mRNA is detected in the liver, but it is much lower than in the thymus. Hence, the difference in proliferative capability among these tissues could reflect the differences in the ability of Mps 1A to facilitate tumorigenesis in these tissues. The robustness of MpslA-induced tumorigenesis in p53 inactivated thymocytes may be due to the increased mitosis and/or high expression level of Mpsl in the thymus. In contrast, MpslA does not appear to promote hepatocellular carcinogenesis in p 5 3 knockout mice or lung tumorigenesis in K-rasCG 2 Dmice. However, upon damage, both tissues have been shown to regenerate. The liver is known for its regenerative 227 abilities where mitosis can be reactivated in hepatocytes in response to liver damage, while the lung has at least one regional stem cell niche that is prone to tumor formation (Kim et al., 2005; Sell, 2001). Moreover, p53-deficiency has been shown to strongly increase the malignancy of K-rasGl 2D-induced lung adenocarcinomas (Jackson et al., 2005). Since, p53-deficiency is also required for MpslA-induced lymphomagenesis, the inactivation of p53 in the lung may be necessary to reveal any MpslA-induced effects on lung tumors. Thus, a tumor phenotype may be revealed in the liver and lung if MpslA is expressed in cycling cells of the lung and liver in the absence of p53. 228 Methods Analysis of Mice Mice used in this study are on a mixed C57BL/6 and 129/Sv genetic background. Mpsl conditional A mice were generated as described (chapter 2). p 5 3 conditional knockout mice were obtained with permission from Anton Berns through Tyler Jacks's group (Jonkers et al., 2001). K-rasGl2D conditional knockin mice were obtained from Tyler Jack's group (Jackson et al., 2001). Alb-Cre transgenic mice were obtained from Jackson labs (Postic and Magnuson, 2000). DNA was prepared from mouse tails, MEFs and tissues with the NucleoSpin 96 Tissue Kit (Clontech, cat # 636968). The following genotyping oiligonucleotides were used: For Mpsl: Mps 1-1: 5'CCTGGTAGTCTACCCATCCTCCTGCTC Mpsl-2: 5'GACACAGACATGGTTGGAGAGTCCTGAG Mpsl-3: 5'GAATACCGAATGAGCGAAAAGCCCC For p53 (Jonkers et al., 2001): p53FL A: 5'CACAAAAACAGGTTAAACCCAG p53FL B: 5'AGCACATAGGAGGCAGAGAC For Alb-cre (oligo sequences courtesy of Aurora Burds Conners): AlbF 1: 5'GTTAATGATCTACAGTTATTGG CreT2-R: 5'CGCATAACCAGTGAAACAGCATTGC For K-ras (Jackson et al., 2001): SD5' (5' mutant): 5'AGCTAGCCACCATGGCTTGAGTAAGTCTGCA LJ3' (3' universal): 5'CCTTTACAAGCGCACGCAGACTGTAGA For Alb-cre survival studies, mice were monitored for tumor development every week starting at 3 months of age. Tissues were fixed in 10% formalin for histology. Histology 229 images were taken at 40X (for liver) and 5X (for lung) with a Zeiss Axiophor microscope. Animal protocols were approved by the Massachusetts Instutute of Technology Committee on Animal Care. Intranasal lung infections Mice were infected with adenoviral Cre (Adcre, obtained from Gene Transfer Vector Core, University of Iowa) at six weeks of age as described (Jackson et al., 2001). Briefly, Mice were anesthetized with avertin. AdCre:CaPi coprecipitates were prepared by mixing 2.5 pLl Adcre (5 * 106 PFU) with 121.9 pl MEM, then 0.6 tplCaC12 added. AdCre:CaPi coprecipitates was allowed to sit for - 20 minutes before intranasal infection of mice in two 62.5-tL instillations. The second instillation was administered about 5-10 minutes after the first instillation, when breathing rates had returned to normal. Mice were sacrificed at 16 weeks post-infection, lungs isolated and infused with 4% paraformaldehyde for histological analysis. Acknowledgements I thank Roderick Bronson for histological analysis. I thank Carla Kim and Amber Woolfenden in Tyler Jack's group for teaching me the techniques for the lung study. I thank Michael Brown of the CCR histology core facility. 230 References Duesberg, P., and R. Li. 2003. Multistep carcinogenesis: a chain reaction of aneuploidizations. Cell Cycle. 2:202-10. Eves, E.M., P. Shapiro, K. Naik, U.R. Klein, N. Trakul, and M.R. Rosner. 2006. Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol Cell. 23:561-74. Friday, B.B., and A.A. Adjei. 2005. K-ras as a target for cancer therapy. Biochim Biophys Acta. 1756:127-44. Guerra, C., N. Mijimolle, A. Dhawahir, P. Dubus, M. Barradas, M. Serrano, V. Campuzano, and M. Barbacid. 2003. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. CancerCell. 4:111-20. Hogg, D., C. Guidos, D. Bailey, A. Amendola, T. Groves, J. Davidson, R. Schmandt, and G. Mills. 1994. Cell cycle dependent regulation of the protein kinase TTK. Oncogene. 9:89-96. Jackson, E.L., K.P. Olive, D.A. Tuveson, R. Bronson, D. Crowley, M. Brown, and T. Jacks. 2005. The differential effects of mutant p53 alleles on advanced murine lung cancer. CancerRes. 65:10280-8. Jackson, E.L., N. Willis, K. Mercer, R.T. Bronson, D. Crowley, R. Montoya, T. Jacks, and D.A. Tuveson. 2001. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15:3243-8. Jonkers, J., R. Meuwissen, H. van der Gulden, H. Peterse, M. van der Valk, and A. Berns. 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 29:418-25. Kim, C.F., E.L. Jackson, A.E. Woolfenden, S. Lawrence, I. Babar, S. Vogel, D. Crowley, R.T. Bronson, and T. Jacks. 2005. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 121:823-35. Knudson, A.G. 2001. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 1:15762. Malkinson, A.M. 1998. Molecular comparison of human and mouse pulmonary adenocarcinomas. Exp Lung Res. 24:541-55. Marshall, C.J. 1996. Ras effectors. CurrOpin Cell Biol. 8:197-204. Meuwissen, R., S.C. Linn, M. van der Valk, W.J. Mooi, and A. Berns. 2001. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 20:6551-8. 231 Mills, G.B., R. Schmandt, M. McGill, A. Amendola, M. Hill, K. Jacobs, C. May, A.M. Rodricks, S. Campbell, and D. Hogg. 1992. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. JBiol Chem. 267:16000-6. Postic, C., and M.A. Magnuson. 2000. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 26:149-50. Sell, S. 2001. The role of progenitor cells in repair of liver injury and in liver transplantation. Wound Repair Regen. 9:467-82. 232