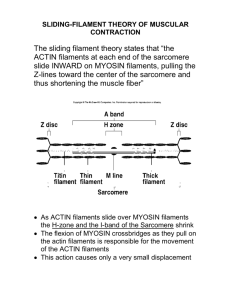
Actin Dynamics in the Cell Cytoplasm and... Associated Proteins
advertisement

Actin Dynamics in the Cell Cytoplasm and the Role of Actin Associated Proteins by James L. McGrath B.S. Mechanical Engineering, Arizona State University, 1991 M.S, Mechanical Engineering, Massachusetts Institute of Technology, 1994 Submitted to the Harvard/MIT Division of Health Sciences and Technology in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biological Engineering at the Massachusetts Institute of Technology June 1998 MIT LIBRARIES © Massachusetts Institute of Technology J l 16 1999 All rights reserved SCHERING ......................................... Signature of A uthor...................... Harvar'/MIT Division of Health Sciences and Technology Certified by........ 7 6/ C. Forbes Dewey, Jr. Professor of Mechanical Engineering, MIT Thesis Supervisor ................................................ ................... John H. Hartwig Associate Pros of Cell Biology, Harvard Medical School Thesis Supervisor Certified by............. A ccep ted b y ....................................................... f J 4 OGY ............... .................................................. Martha Gray J.W. Kieckhefer Ass ciate Professor of Electrical Engineering nces and Technology, Harvard/MIT MOM. Abstract Cytoplasmic actin distributes between monomeric and filamentous phases. In steady crawling, actin polymerization near the expanding cell periphery is balanced by depolymerization elsewhere in the cell.Thus the finite lifetime of actin filaments, the diffusivity of actin monomer, and the distribution of actin between its polymerized and unpolymerized phases are predictably key parameters in cell motility. We have developed approaches for measuring these parameters in living cells and in vitro. In the techniques of photoactivated fluorescence (PAF) or fluorescence recovery after photobleaching (FRAP), fluorescently derivatized actin is microinjected into a cell where it incorporates into the native cytoskeleton. The evolution of fluorescence following local photoactivation or photobleaching reveals the dynamics of cellular actin. A new mathematical model describes the evolution of fluorescence from a photoactivated band. Using this model to interpret PAF and FRAP experiments in bovine aortic endothelial cells (BAECs), simultaneously measures monomer diffusion coefficients, filament turnover rates and the fraction of actin polymerized. The results demonstrate that filament turnover is rapid (- 6 minutes) but not diffusion limited in the bulk actin cytoskeleton of BAECs. BAECs in mechanically wounded monolayers separate into zones with distinct motility and morphology, providing a model system to study the role of actin in determining these characteristics. PAF and FRAP experiments reveal that filament turnover and polymer fraction correlate with motility in these monolayers. This is the first demonstration that cells of a single type can alter actin dynamics to increase their speed. The most motile cells turnover actin filaments quickly (- 6 minute lifetimes) and have most of their actin in a diffusive pool (- 60% of total actin). The slowest cells turnover actin filaments slowly (- 40 minute lifetimes) and have less actin in a diffusive pool (-20% of total actin). A kinetic model of the actin cycle in cells predicts that a simultaneous decrease in filament lifetime and polymerized actin requires accelerating disassembly at 'pointed' filament ends.The loss of gelsolin in fibroblasts, slows motility and filament turnover and increases the fraction of actin polymerized. This result suggests that the regulation of filament capping and/or severing can produce trends similar to those seen in BAECs. The loss of the actin gelation protein ABP-280 from human melanoma cells slows motility without affecting filament turnover, demonstrating that actin dynamics and cytoskeletal architecture can independently control cell motility. A new PAF based system improves over existing techniques for measuring actin dynamics in vitro. With minimal invasion in the same actin preparation, the system provides simultaneous estimates of filament turnover and the fraction of actin polymerized, and indirectly, filament lengths. The system also provides the first visual evidence of actin filament turnover in vitro. Acknowledgments I thank my advisors, Drs. John H. Hartwig and C. Forbes Dewey Jr. for the opportunity to work in an exciting field and in world class laboratories. Thanks to both men for their patience, enthusiasm and encouragement or, when appropriate of course, impatience, skepticism, and criticism. I am indebted to Dr. Yanik Tardy for his friendship and many scientific contributions to this thesis. I thank my committee members Drs. Doug Lauffenburger and Michael Gimbrone, for their help with the thesis, for their examples in science, and for essential help in my pursuit of an independent, interdisciplinary curriculum in Biological Engineering. Thanks to Dr. Kurt Barkalow who made many important suggestions and listened closely to every idea no matter how looney. Thanks to the all the students, faculty, and staff at the Experimental Medicine Division at the Brigham and Women's Hospital and the Fluid Mechanics Laboratory at MIT. Most rewarding is the number of remarkable people I have come to know during my Master's and Doctoral studies. I am grateful to Dr. Roger Mark for making possible my independent doctoral program in Biological Engineering, and to Drs. Lee Gehrke, Joe Bonventre, and Don Ingber for facilitating its completion. I dedicate this thesis to my loving wife Raymonde ("Nicky") McGrath who has worked as hard as I have and sacrificed more so I could have this dream. Our son Adam, born in October of 1996, taught me that sometimes life is perfect, but that I needed to slow down to see it. I hardly know our beautiful daughter Emily, born a few weeks ago and a few days before my defense (whew!), but I'm sure she too has lessons in store for me. Nicky and I did not pull this off without considerable help. Thanks to my mother-in-law, Carmen Violette for coming to Boston at least a halfdozen times to help us through labor or out of jams with baby-sitters, and to my parents, Jim and Gerry McGrath, who's support was always close despite the 3000 miles. Table of Contents Table of Contents Abstract Acknowledgements Table of Contents List of Figures List of Tables CHAPTER 1 Background and Literature Review An Introduction to Actin 1-2 The Importance ofActin in Cells 1-2 Actin Structure 1-3 Actin self-assembly 1-4 The Actin Cycle is Regulated in Cells 1-6 Section Summary 1-6 Monomer sequesteringproteins 1-7 Barbed end capping/filamentsevering proteins 1-8 Profilin 1-10 ADF/cofilin 1-11 Actin DirectedSignalling Pathways 1-12 Actin Dynamics in the Cell Cytoplasm 1-13 Photoactivationoffluorescence andfluorescence recovery after photobleaching1-13 Monomer diffusion 1-14 Filament turnover and retrogradeflow 1-15 The Story of this Thesis Works Cited 1-17 1-19 Actin Dynamics in the Cell Cytoplasm TOC-1 Table of Contents CHAPTER 2 Experimental Set-up and Design Introduction 2-2 Photoactivatedfluorescence andfluorescence recovery after photobleaching 2-2 Propertiesof Derivatizedactin 2-3 Synthesis of caged resorufin-iodoacetamide The Synthesis of Caged-Actin 2-4 Measurement apparatus 2-4 2-6 System characterization 2-11 Lamp stability 2-11 Dynamic response and linearityof the imaging system 2-11 Discussion 2-12 Works Cited 2-13 CHAPTER 3 InterpretingPAF/FRAPMeasurements of Steady-State Actin Dynamics Introduction 3-2 Mathematical model 3-3 Assumptions/idealization/simplifications: 3-3 Derivation of the dimensionalproblem: 3-4 Non-dimensionalproblem 3-6 Solution 3-7 Results 3-9 Concentrationandfluorescence profile 3-9 Center-linedecay 3-9 Long time behaviorwhen fl << 1 3-11 Discussion 3-13 Appendix 3-14 Works Cited 3-17 CHAPTER 4 Baseline and control measurements in subconfluent endothelial cells Introduction 4-2 Materials and Methods Cell Culture 4-5 TOC-2 4-5 Table of Contents Electron Microscoscopy 4-5 Actin preparationand injections 4-6 PAF/FRAP Experiments 4-6 Photobleachingcontrols and corrections 4-7 Analysis 4-8 Geometry Considerations 4-9 Jasplakinolideand cytochalasin D treatments 4-9 Results 4-11 Both PAF and FRAP experiments exhibit biphasic decay consistent with the presence of two phases of actin 4-11 Estimates of D, FF,and t 4-12 PAF and FRAP experiments are sensitive to jas and cyto D 4-14 Discussion Appendix 4-15 4-20 Works Cited CHAPTER 5 4-22 Correlationof actin dynamics with cell motility and with expression of actin associatedproteins Introduction 5-2 Methods 5-3 Cell culture and motility analysis 5-3 PAF and FRAP analysis 5-3 Results 5-4 Actin dynamics correlatewith cell motility in a wounded endothelialmonolayer 5-4 PAF and FRAP experiments agree in subconfluent and confluent endothelialcells 5-5 Gelsolin null and positivefibroblasts andABP+ melanoma cells agree with endothelial cells correlations,but ABP- melanoma cells do not 5-8 ABP+ andABP- cells migrate with different mechanisms 5-10 Discussion 5-10 Works Cited CHAPTER 6 5-12 A MechanisticModel ofActin Dynamics Introduction 6-2 The Mathematical Model 6-5 Methods 6-10 System solution 6-10 Actin Dynamics in the Cell Cytoplasm TOC-3 Table of Contents Pyrene assay 6-11 Results 6-12 Subconfluent cells have more and shorterfilaments than confluent cells 6-12 Accelerating pointed end off rate effects dynamic state while acceleratingnucleotide exchange does not 6-13 Exposingfilament barbed ends counters the accelerationofpointed end off rates 6-14 Filamentsevering shortensfilaments and increasesturnoverbut has a negligible effect on polymerfraction 6-14 Experiments and simulations agree qualitatively but not quantitatively 6-16 Discussion 6-18 Works Cited 6-19 CHAPTER 7 A System for the Assay of Steady-State Actin Dynamics In Vitro Introduction Theory 7-2 7-3 Distinguishingfilament turnoverfromfilament diffusion in PAF experiments 7-3 Adjusting w to visualizefilament turnover 7-5 Methods 7-7 Preparationof caged-actin 7-7 Microcapillarypreparationand actinpolymerization 7-7 PAF experiments 7-8 Results 7-9 The role of an aci-nitro intermediate influorescence rise after uncaging 7-9 Demonstrationoffilament turnover by PAF 7-10 Discussion 7-10 Works Cited 7-14 CHAPTER 8 TOC-4 Conclusions 8-1 List of Figures List of Figures CHAPTER 1 Background and LiteratureReview FIGURE FIGURE FIGURE FIGURE FIGURE FIGURE CHAPTER 2 1.1. The structure of skeletal-muscle actin monomer. 1-2 1.2. Structure of the actin filament. 1-3 1.3. Evidence of actin nucleation. 1-4 1.4. Steady-state actin dynamics in vitro. 1-5 1.5. The actin cycle in cells and the role of actin associated proteins. 1-7 1.6. A recent model of actin dynamics in lamellae. 1-16 ExperimentalSet-up and Design FIGURE 2.1. FIGURE 2.2. The synthesis of caged-actin. 2-5 Absorbance spectra demonstrating the successful synthesis of cagedactin. 2-6 FIGURE 2.3. Schematic of the PAF measurement system. 2-7 FIGURE 2.4. Filter configurations. 2-8 FIGURE 2.5. Computer controlled automation of experiments. 2-9 FIGURE 2.6. Dynamic response of CCD and intensifier following sudden exposure to experimental light levels. 2-10 CHAPTER 3 InterpretingPAF/FRAP Measurements of Steady-State Actin Dynamics FIGURE 3.1 FIGURE 3.2 FIGURE 3.3 Schematic of the model system. 3-4 Evolution of total fluorescence and fluorescence in filaments or monomers. 3-10 Decay of fluorescence in a photoactivated band. 3-11 Actin Dynamics in the Cell Cytoplasm LOF-1 List of Figures CHAPTER 4 Baseline and control measurements in subconfluent endothelialcells FIGURE 4.1 FIGURE 4.2 FIGURE 4.3 FIGURE 4.4 FIGURE 4.5 CHAPTER 5 Photobleaching and background controls in PAF experiments. 4-10 Electron micrographs showing the region of BAECs studied by PAF and FRAP. 4-11 PAF and FRAP experiments in BAECs. 4-12 Corrected PAF and FRAP data. 4-13 Effects ofjasplankinolide. 4-14 Correlation of actin dynamics with cell motility and with expression of actin associated proteins FIGURE 5.1. Relationship between BAEC morphology and average cell speeds.5-5 FIGURE 5.2. PAF images sequence demonstrating different actin dynamics in cells in the a) wound edge and b) confluent regions of a recovering BAEC monolayer. 5-6 FIGURE 5.3. Cytoplasmic actin dynamics correlate with BAEC morphology. 5-7 FIGURE 5.4. Correlations of motility and actin dynamics. 5-8 FIGURE 5.5. Actin dynamics in gelsolin positive and null fibroblasts. 5-9 FIGURE 5.6. ABP+ and ABP- melanoma cells migrate with different mechanisms. 5-11 CHAPTER 6 A MechanisticModel ofActin Dynamics FIGURE 6.1. Schematic of the cell actin cycle. 6-3 FIGURE 6.2. Procedure to identify actin cycle regulatory mechanisms important in endothelial cell motility. 6-4 FIGURE 6.3. Effects of pointed end disassembly and nucleotide exchange on the cell cycle parameters. 6-12 FIGURE 6.4. Levels of ATP and ADP bound monomer with accelerating filament turnover. 6-13 FIGURE 6.5. Effects of increasing filament barbed exposure on the cell cycle parameters. 6-14 FIGURE 6.6. Effects of regulating filament number on the actin cell cycle. 6-15 LOF-2 List of Figures FIGURE 6.7. FIGURE 6.8. CHAPTER 7 Profilin mediated barbed end assembly affects the actin cell cycle. 6-16 Comparison between experimentally determined and kinetic predictions of the actin cycle state in confluent and subconfluent endothelial cells. 6-17 A System for the Assay of Steady-State Actin Dynamics In Vitro FIGURE FIGURE FIGURE FIGURE 7.1 7.2 7.3 7.4 FIGURE 7.5 FIGURE 7.6 System for the assay of actin dynamics using PAF 7-3 PAF experiment in the presence and of physiological levels of salt. 7-4 Photolysis of 2-nitrobenzyl caged compounds. 7-9 The effects of continuous exposure, pH and DTT on fluorescence rise. 7-11 Visualization of filament turnover. 7-11 Example PAF experiment demonstrating filament turnover. 7-12 Actin Dynamics in the Cell Cytoplasm LOF-3 List of Figures LOF-4 List of Tables List of Tables CHAPTER 4 Baseline and control measurements in subconfluent endothelial cells TABLE 4.1: TABLE 4.2: CHAPTER 6 Parameter estimates from PAF and FRAP in BAECs. 4-19 Dynamic parameters in cells before and after treatment with Cyto D. 4-19 A Mechanistic Model ofActin Dynamics TABLE 6.1. TABLE 6.2. Rate constants used in the kinetic model of the actin cycle in cells 6-6 Steady-state rate equations describing the actin cycle in cells 6-9 Actin Dynamics in the Cell Cytoplasm LOT-1 List of Tables LOT-2 CHAPTER 1 Background and Literature Review Monomeric actin spontaneously polymerizes in physiological buffers to form filaments. In cells, actin filaments are important mechanically. They organize into bundles and networks that define cell shape, strength, and adhesiveness. They are also highly dynamic. In most cells, the entire actin cytoskeleton is remodeled through steady-state filament assembly and disassembly at least once in a half hour. The dynamics of actin are important determinants of remodeling events in cell motility and endocytosis. This chapter introduces the protein actin and reviews what is known about its dynamic behavior in vivo and in vitro. The chapter concludes with an overview of the thesis. Actin Dynamics in the Cell Cytoplasm 1-1 Background and Literature Review An Introduction to Actin An Introduction to Actin FIGURE 1.1. The structure of skeletal-muscle actin monomer. A cleft divides the monomer into two domains and binds a nucleotide and cation.(Sheterline, et aL, 1994). The Importance of Actin in Cells Actin is a highly conserved and abundant 42 kD monomeric protein in eukaryotes. Actin spontaneously self-assembles into filaments in the presence of physiological cations and salt. Cells distribute actin filaments into a multifunctional network known as the actin cytoskeleton. As a organized scaffolding, the actin cytoskeleton determines cell shape and strength and participates in the transport and distribution of organelles. Actin polymerization drives the expansion of the cell membrane during cell spreading, crawling, and activation. Actin filament bundles associate with myosin to apply traction to substrates. Both contraction in the cell body 1-2 Background and Literature Review An Introduction to Actin and actin dependent membrane extension are required for cell crawling. barbed end actin monomer Actin filaments continuously assemble and disassemble throughout the cell cytoplasm. Filament turnover is important in cell motility and other actin dependent processes such as endocytosis. Actin Structure The three dimensional structure of the actin molecule has been solved by x-ray diffraction (Kabsch and Vandekerckhove, 1992;McLaughlin, et al., 1993;Schutt, et al., 1993) and is shown in Figure 1.1. The molecule is divided into two domains by a cleft which holds a divalent cation (Ca 2+ or Mg 2+ ) and a nucleotide (ATP or ADP). Association between multiple residues on neighboring monomers stabilize the double helical actin polymer (Figure 1.2) (Holmes, et al., 1990). The ends of the polar filament are termed "pointed" and "barbed" to reflect the arrowhead appearpoinTed end ance of myosin -labeled filaments in electron micrographs. FIGURE1.2. Structure of the acutnflament. Actin monomers Three isoforms of actin (a, 3,y) are found in higher mammals (Herman, into a withble assemble 1993). Most cells express all three isoforms, however a is the primary barbed and pointed ends. (Alberts, et al., isoform in muscle and P and y are the primary isoforms in non-muscle 1994) tissue. The isoforms are very similar in sequence and length indicating that actin has been heavily conserved during mammalian evolution. Small sequence differences concentrated near the N-terminus (Vanderkerchekhove and Weber, 1978;Vanderkerchekhove and Weber, 1979), give the isoforms different activities. The actin iso- Actin Dynamics in the Cell Cytoplasm 1-3 Background and Literature Review An Introductionto Actin time (min) -FIGURE 1.3. Evidence of actin nucleation. A delay in actin polymerization is overcome with the addition of pre-formed nuclei. The result demonstrates that the unfavorable formation of nuclei is the first step in actin polymerization. (Oosawa and Asakura, 1975) forms distribute into different cell structures (DeNofrio, et al., 1989;Herman and D'Amore, 1989;Hoock, et al., 1991;Peng and Fischman, 1991), and have differing affinities for profilin (Larsson and Lindberg, 1988;Ohshima, et al., 1989), 034 thymosin (Weber, et al., 1992), and myosin (Hennessey, et al., 1993). Actin self-assembly Physiological levels of divalent cations and monovalent ions cause actin monomers to spontaneously polymerize. Consistent with the theory of Oosawa (Oosawa and Asakura, 1962), polymerization begins with the unfavorable formation of a trimeric nucleus (Wegner and Engel, 1975). The slow formation of nuclei produces a lag in the polymerization process, which can be overcome by adding pre-formed filaments (Figure 1.3). Once short filaments are 1-4 Background and Literature Review An Introduction to Actin pointed barbed I. net monomer flux I 0 ATP-Actin ADP*Pi-Actin 0 ADP-Actin FIGURE1.4. Steady-state actin dynamics in vitro. In the presence of excess ATP net actin assembly at barbed ends and net disassembly at pointed ends drives monomerflux throughfilaments. The hydrolysis ofATP soon after assembly provides energy driving actin cycle known as treadmilling. formed, monomers add to barbed ends approximately 10 times faster than to pointed ends (Kondo and Ishiwata, 1976; Bonder, et al. 1986; Korn, et al., 1987;Pollard, 1986). In the presence of ATP, actin polymerizes until only 0.1 jgM remains unpolymerized (Drenckhanh and Pollard, 1986;Pollard, 1986;Wegner and Isenberg, 1983). This concentration of monomer, the minimum or 'critical concentration' required for filament assembly, exists in a dynamic steadystate with filaments (Figure 1.4). At steady-state, ATP-actin assembles onto filament barbed ends and rapidly hydrolyzes its bound ATP to the intermediate ADP*P i (Carlier and Pantaloni, 1986). Pi is slowly released from the sides of actin filaments as monomers flux from barbed to pointed ends where they are predominantly bound to ADP (Carlier and Pantaloni, 1986;Korn, et al., 1987). ATP hydrolysis provides the energy required to maintain the biochemical differences between filament ends that permit this monomer flux known as "treadmilling" (Wegner and Isenberg, 1983). Actin Dynamics in the Cell Cytoplasm 1-5 Background and Literature Review The Actin Cycle is Regulated in Cells Actin depolymerization occurs when fully polymerized actin is diluted below the critical concentration. In these experiments, ATP-actin capped barbed ends disassemble monomer faster than ADP-actin capped pointed ends by a factor of 5 -10 (Janmey and Stossel, 1986;Pollard, 1986). Slow rates of disassembly at filament pointed ends (0.03 - 0.27 sec 1 ) predict that actin filaments should be relatively stable in cells where access to the majority of barbed ends is blocked by barbed end capping proteins. The Actin Cycle is Regulated in Cells Section Summary In cells actin-associated proteins regulate the dynamics of the ATP-powered actin cycle (Figure 1.5). Some proteins bind to the barbed ends of actin filaments to block filament assembly, others to the pointed ends to accelerate disassembly, and still others to the sides of actin filaments to sever them. Monomer sequestering proteins maintain unpolymerized actin in cells at concentrations hundreds of times higher than the critical concentration for filament formation. Monomer affinity for sequestering proteins is higher than for pointed ends but lower than for barbed ends. Consequently, a sudden exposure of barbed ends leads to massive actin polymerization. Proteins enhance assembly by catalyzing the exchange of nucleotide on actin monomer and escorting the charged monomer to barbed ends. Many of the proteins regulating the actin cycle are themselves regulated by signaling pathways. 1-6 Background and Literature Review The Actin Cycle is Regulated in Cells filament severing ADP ac"tin S ADP*Pi actin ATP ac tin acceler ated disassernbly * barbed end capping nucleotide exchange ( * aw monomer sequestration 1444 r FIGURE 1.5. The actin cycle in cells and the role of actin associatedproteins.The proteins involved in barbed end capping, severing, accelerateddisassembly,and nucleotide exchange are regulatedand representcontrol points in the actin cycle. Monomer sequestering proteins Profilins, ADF/cofilins, and P-thymosins are ubiquitously expressed proteins that bind actin monomers in one-to-one complexes. Of these, only P4 thymosin is present in quantities large enough to account for the levels of sequestered actin in most cells (Safer, et al., 1990). The others, discussed below, appear to have catalytic roles in the actin cycle. 0-4 thymosin, the most common 3-thymosin isoform, is a 5 kD protein with gLM affinity for ATPactin and a 100 fold lower affinity for ADP-actin (Carlier, et al., 1993). P-4 thymosin hinders Actin Dynamics in the Cell Cytoplasm 1-7 Background and Literature Review The Actin Cycle is Regulated in Cells nucleotide exchange on actin (Carlier, et al., 1993). Because it is unregulated, present in cells at similar concentrations to actin, and sequesters actin in 1:1 complexes in vitro, 0-4 thymosins were originally thought of as a simple monomer sequestering protein. However, overexpression does not decrease polymerized actin in NIH3T3 cells (Safer and Nachmias, 1994;Sun, et al., 1995) and at high concentrations, 3-4 thymosin (> 20 pM) does not sequester actin efficiently in vitro (Carlier, et al., 1996). No function has been attributed to 0-4 thymosin's departure from simple sequestering behavior. Barbed end capping/ filament severing proteins Sequestering proteins have a lower affinity for monomer than actin filament barbed ends. To maintain high levels of sequestered monomer, cells express proteins that bind the majority of barbed ends with nM affinity. The most widely expressed are the gelsolin family of proteins and capping protein (CP). Gelsolin requires tpM calcium to bind barbed ends (Yin, et al., 1981) whereas CP is constitutively active. Both gelsolin (Janmey and Stossel, 1987) and CP (Heiss and Cooper, 1991) can be removed from filament barbed ends by phosphatidylinositol 4,5-bisphosphate (PIP 2 ) and other phosphoinositides. Gelsolin and CP are present in many cells at quantities sufficient to cap all their filaments (Amatruda, et al., 1992;Barkalow, et al., 1996;Hartwig, 1992). The regulatory features of gelsolin and capping protein allow extracellular signals to control actin polymerization by mobilizing intracellular Ca 2+ and regulating phospholipid metabolism (discussed below). The importance of the barbed end capping activity of CP in maintaining and remodeling cellular actin has been demonstrated in a number of model systems. The loss of CP in yeast 1-8 Background and Literature Review The Actin Cycle is Regulated in Cells mutants disrupts the organization and dynamics of the actin cytoskeleton (Amatruda, et al., 1990;Amatruda, et al., 1992). Increased expression of CP in Dictyostelium cell lines decreases the number of free barbed ends, decreases the fraction of actin polymerized, and increases cell motility (Hug, et al., 1995). Regulation of CP activity by PIP 2 is involved in barbed end exposure during platelet (Barkalow, et al., 1996) and Dictyostelium activation (Eddy, et al., 1996). Gelsolin caps, severs and nucleates actin filaments (Burtnick, et al., 1997,Yin et al., 1981, Kwiatkowski et al., 1985). After severing, gelsolin binds to a barbed end to produce one free and one capped filament. Activation of gelsolin is most effective in vitro at > 10-20 PM Ca 2+ (Lamb, et al., 1993), a level -~1000 times higher than resting cell levels and 10 times higher than average cell values during Ca2 + mobilization. Gelsolin may be activated by high Ca 2+ concentrations near stores or the Ca 2+ requirement may be lowered by changes in cellular pH (Lamb, et al., 1993). Regardless of how it occurs, gelsolin is active in cells. Gelsolin-actin form and then dissociate when cells are stimulated to move (Barkalow, et al., 1996;Hartwig, 1992;Howard, et al., 1990; Cunningham, et al., 1991). Cells isolated from gelsolin null mice have phenotypes distinguishable from wild type cells. Most notably, the loss of gelsolin slows motility in fibroblasts (Witke, et al., 1995), neutrophils (Witke, et al., 1995), and neurites (Lu, et al., 1997), enhances stress fiber formation in fibroblasts (Witke, et al., 1995), and hinders actin assembly in platelets after thrombin or glass activation (Witke, et al., 1995). Experiments in gelsolin null fibroblasts demonstrate gelsolin's importance in membrane ruffling as these fail to form ruffles or lammelipodia (Azuma, et al., 1998). Actin Dynamics in the Cell Cytoplasm 1-9 Background and Literature Review The Actin Cycle is Regulated in Cells Profilin Profilins are small 12-15 kD proteins originally identified as sequestering proteins because of their ability to prevent actin polymerization in vitro (Carlson, et al., 1977). Follow-up studies, however, revealed that profilin actually enhances actin polymerization in many circumstances. At steady-state, and in the presence of ATP, profilin decreases the critical concentration of actin by a factor of 10 (Pantioni and Carlier, 1993;Pollard and Cooper, 1984) demonstrating that the profilin promotes assembly at barbed ends at steady-state. Profilin-mediated assembly at steady-state is dramatically demonstrated when profilin is used in combination with a large pool of monomer sequestered by I34 thymosin (Pantioni and Carlier, 1993). Mammalian profi- lin accelerates the dissociation of ADP from actin monomer 1000 fold (Goldschmidt-Clermont, et al., 1991), catalyzing the exchange of ADP for ATP. In the treadmilling model of actin dynamics (Figure 1.4) ADP actin is released from pointed ends into the monomer pool. Very high rates of filament turnover may overwhelm actin's intrinsic ability to exchange its bound nucleotide and deplete ATP-actin for barbed end assembly. Under these conditions profilin can accelerate nucleotide exchange to maintain filament turnover (Goldschmidt-Clermont, et al., 1992;Mitchison, 1992;Theriot and Mitchison, 1993). Plant profilins enhance barbed end assembly but do not promote nucleotide exchange indicating that these functions are uncoupled (Perelroizen, et al., 1996). In vivo data also argue for profilin's role in promoting polymerization. Yeast lacking profilin are unable to repolymerize actin following temperature elevation (Yeh and Haarer, 1996). An expression study in CHO cells demonstrates that enhanced profilin expression correlates with longer filament lifetimes and an increased polymer fraction (Finkel, et al., 1994). 1-10 Background and Literature Review The Actin Cycle is Regulated in Cells Like gelsolin, gelsolin-actin complexes and CP, the profilin-actin complex binds PIP 2 (Lassing and Lindberg, 1985). Once bound to PIP2 , actin disassociates from the complex (Lassing and Lindberg, 1988) and the profilin-PIP 2 complex resists degradation by PLC-y (Goldschmidt-Clermont, et al., 1990). There is also evidence that profilin is a downstream component of cytoskeleton targeted pathways of the Ras family of small GTPases (Tanaka and Takai, 1998). ADF/cofilin The stability of actin filament pointed ends in vitro (Janmey and Stossel, 1986;Pollard, 1986) could not be resolved with the high rates of turnover measured in vivo until a recent demonstration that members of the ADF/cofilin family bind preferentially to ADP-actin near pointed ends to accelerate disassembly by a factor of 25 (Carlier, et al., 1997). The ability of cofilin to accelerate turnover is supported by shortening of Lysteria tails in cell extracts (Carlier, et al., 1997;Rosenblatt, et al., 1997) and by experiments in yeasts demonstrating the importance of ADF/cofilin in supporting the surprisingly high rates of filament turnover in these non-motile cells (Lappalainen and Drubin, 1997). However, the yeast and Lysteria data cannot rule out that filament turnover is accelerated by filament severing, an activity originally proposed to explain coflin's reduction of actin gel viscosity (Cooper, et al., 1986). Carlier and colleagues (Carlier, et al., 1997), however demonstrate exhaustively that filament severing cannot explain their kinetic and structural data. ADF/cofilin is regulated by the reversible phosphorylation of a serine residue near the amino terminus (Nebl, et al., 1996). Dephosphorylation activates cofilin (Moon and Drubin, 1995). The regulation of the phosphatase is complex as phosphatase inhibitors promote the dephosphorylated state (Okada, et al., 1996;Takuma, et al., 1996) but a recent study in neurites identifies Ca 2+ and Actin Dynamics in the Cell Cytoplasm 1-11 Background and Literature Review The Actin Cycle is Regulated in Cells cAMP as second messengers in pathways leading to cofilin deposphorylation (Meberg, et al., 1998). Actin Directed Signalling Pathways The regulation of actin associated proteins by phosphoinositides and their breakdown products in vitro suggests signaling pathways can control phosphoinositide metabolism to control actin assembly. Phosphoinositides recruit CP and gelsolin from barbed ends to promote actin assembly and bind profilin to block PLC-y mediated hydrolysis. The hydrolysis of phosphoinositides liberates gelsolin and capping proteins and produces Ca 2+ to activate gelsolin and (in one example) the phosphatase of cofilin (Meberg, et al., 1998). The in vitro data predict that synthesis and hydrolysis of phosphoinositides in vivo should correlate with filament assembly and filament breakdown respectively. Consistent with this prediction, platelet gelsolin and capping protein are activated during periods of phosphatidylinositol 4, 5 bisphosphate (PIP 2 ) breakdown and lose activity during periods of actin assembly and PIP 2 synthesis (Hartwig, et al., 1995;Hartwig, et al., 1996;Kovacsovics, et al., 1995). PIP2 synthesis also correlates with actin assembly in other cell types (Apgar, 1995;Carson, et al., 1992). Examples where PIP2 synthesis does not correlate with actin assembly demonstrate yet unresolved pathways to actin assembly (Dadabay, et al., 1991;Zigmond, et al., 1997). Members of the Rho family GTPases are essential regulators of signaling pathways directing actin organization (Tapon and Hall, 1997). Constitutively active RhoA promotes filament bundling and adhesion complexes in serum starved cells (Hotchin and Hall, 1995;Ridley and Hall, 1992). Active Rac is essential for generating membrane ruffles found at the leading edge of 1-12 Background and Literature Review Actin Dynamics in the Cell Cytoplasm motile cells (Ridley, et al., 1992). Finally, active Cdc42 elicits narrow membrane extensions known as filopodia (Kozma, et al., 1995). Links between the Rho family GTPases and actin associated proteins are emerging. Rac, Cdc42, and Rho bind phosphoinositide kinases in vitro and in vivo (Carpenter, et al., 1997;Chong, et al., 1994;Hartwig, et al., 1995;Tolias, et al., 1995), and active Rac induces PIP2 synthesis and barbed end uncapping in permeabilized platelets (Hartwig, et al., 1995). Intermediates in RhoA signaling bind profilin in yeast (Imamura, et al., 1997). Gelsolin is essential for Rac mediated membrane ruffling in cultured fibroblasts (Azuma, et al., 1988). Actin Dynamics in the Cell Cytoplasm The term "actin dynamics" includes the kinetics of actin assembly and disassembly, monomer and filament diffusion, and bulk movements of the actin network. Actin is dynamic throughout motile and non-motile cells. The dynamics of actin can vary between cell types and spatially within cells to control cell motility and other actin-dependent processes such as endocytosis. Models and experiments on cell motility focus on the behavior of actin in the protrusive regions of cells. The dynamics of actin in these structures are characterized by net polymerization at the leading membrane, retrograde flow from the membrane, and net depolymerization near the cell body. Photoactivation of fluorescence and fluorescence recovery after photobleaching The dynamics of actin in living cells has been observed using the techniques of fluorescence recovery after photobleaching (FRAP) and photoactivation of fluorescence (PAF) (Cramer, et al., Actin Dynamics in the Cell Cytoplasm 1-13 Background and Literature Review Actin Dynamics in the Cell Cytoplasm 1997;Finkel, et al., 1994;Kreis, 1982;Theriot and Mitchison, 1991;Theriot and Mitchison, 1992;Wang, 1985). In these experiments a fluorophore-labeled actin derivative is microinjected into living cells where it incorporates into the native cytoskeleton. In FRAP, the fluorescence derived from the injected actin is quenched locally under intense laser excitation. In PAF, a focused band of UV excitation converts a non-fluorescent fluorophore derivative (a 'caged' fluorophore) back to its fluorescent parent (Mitchison, 1989). In both techniques the evolution of fluorescence in the illuminated region is monitored to infer information about actin dynamics. Monomer diffusion The diffusion of ficoll and dextran in cells demonstrates that the mobility of inert particles depend more strongly on their size in cytoplasm than in water (Luby-Phelps, 1987). This nonnewtonian behavior is due partly to the sieving effect of cytoskeletal pores and partly to the background of proteins crowding the cytosol (Hou, et al., 1990). The result is that a 28 A actin monomer (Lanni and Ware, 1984) diffusing in cytoplasm experiences a cytoplasmic viscosity which should reduce its mobility 5 fold from aqueous (Luby-Phelps, et al., 1986). However, the consensus values for monomer mobility in cytoplasm are 3-6 x 10-8 cm 2 /sec (Giuliano and Taylor, 1994;Luby-Phelps, 1985), = 15 times lower than values in water (Lanni and Ware, 1984). This disparity has not been resolved but may indicate 1) that the turnover of actin filaments is so rapid that it hinders the ability of monomers to diffuse; 2) that small filaments diffuse through the cytoplasm and contribute to the measured mobility; or 3) that actin monomer binds transiently to sequestering molecules much larger in size than those currently identified (Luby-Phelps, et al., 1988). 1-14 Background and Literature Review Actin Dynamics in the Cell Cytoplasm Filament turnover and retrograde flow The first evidence that actin filaments continuously remodel in living cells came from microinjection studies. These studies demonstrated that fluorescently labeled actin incorporates into all existing actin structures in less than a half hour (Amato, 1986;Kreis, et al., 1979). Local photobleaching of fluorescent actin at the leading edge of motile fibroblasts determined a time scale for complete fluorescence recovery of several minutes (Cramer, et al., 1997;Wang, 1985). Keratocytes, which can move 10 times faster than fibroblasts appear to turn over actin filaments in leading lamella in tens of seconds (Theriot and Mitchison, 1991). Estimates of filament turnover near or in the cell body of fibroblasts and PtK3 cells are on the order of 5-10 minutes (Cramer, et al., 1997;Theriot and Mitchison, 1991). FRAP studies also demonstrated that actin moves rearward in the leading edge of cells (Theriot and Mitchison, 1992;Wang, 1985), consistent with the treadmilling model of filament turnover seen in vitro (Wegner, 1976). Organelles, surface attached beads, and actin rich membrane protrusions resolved by various optical methods move rearward at similar rates (Fisher, et al., 1988;Waterman-Storer and Salmon, 1997), but are faster than the movement of photoactivated and photobleached actin marks (Condeelis, 1993;Theriot and Mitchison, 1992). These results may suggest that PAF and FRAP measure the dynamics of a subset of actin filaments in lamella (Condeelis, 1993;Theriot and Mitchison, 1992). Before the discovery that ADF/cofilin could accelerate pointed end off rates the treadmilling model for actin dynamics in motile cells could not be resolved with in vitro rates of disassembly, and alternative models were proposed in which lamella were comprised primarily of short filaments (Stossel, 1993;Theriot and Mitchison, 1991). Conflicting data on filament lenghts in lamellae contributed to this debate (Hartwig, 1992;Hartwig and Shevlin, 1986;Small, et al., 1995). A Actin Dynamics in the Cell Cytoplasm 1-15 Background and Literature Review Actin Dynamics in the Cell Cytoplasm a) + capping protein very slow turnover accelerated turnover + cofilin c) + capping protein accelerated turnover treadmilling highest turnover funneled assembly FIGURE 1.6. A recentmodel of actin dynamics in lamellae(Carlier,1998). The model describesa synergy between the actin associatedproteins cofilin and cappingprotein. In the absence of cappingprotein or cofilinfilament turnoveris slow andfilaments treadmilla). With the addition of only cappingprotein, disassemblingmonomers arefunneled to a small number offree barbed ends which grow b). With the addition of only cofilin, filament disassembly and barbedend assembly accelerateto give faster treadmillingc) When both cofilin and cappingprotein exist, the funneled growth at barbedends is maximized d). Note thatas filament turnoveraccelerates the pool of actin sequesteredby f4 thymosin grows. current model for lamellar structure and dynamics revises the treadmilling model in light of the ADF/cofilin data (Carlier, 1998;Carlier and Pantaloni, 1997). In this view (Figure 1.6) actin assembly occurs at a small number of free barbed ends beneath the leading edge. The remaining filaments are capped and disassembling quickly due to ADF/cofilin binding, so that on average filament turnover is high. The "funneling" of actin monomer to a small number of 1-16 Background and Literature Review The Story of this Thesis free barbed ends advances the leading edge at rates higher than would be possible if all barbed ends were free as in treadmilling. The new model also predicts that higher levels of filament turnover should correlate with a transfer of actin from filaments to sequestered monomer. The Story of this Thesis The original ambition of this work was to resolve the in vitro/ in vivo turnover paradox. In reviewing the relevant literature, we noticed that FRAP and PAF experiments were interpreted differently. Specifically, FRAP experiments designed to measure monomer diffusion ignored filament turnover, and PAF experiments assaying filament turnover ignored monomer diffusion. Also resorufin was highly photolabile so that fluorescence decay rates could easily be accelerated by inadvertent photobleaching. I investigated if these issues could explain the surprisingly high rates of filament turnover measured in vivo by Theriot and Mitchison, 1991. We developed a mathematical model of actin dynamics designed specifically for the interpretation of PAF and FRAP experiments (Chapter 3), and constructed a system similar to the one developed by Theriot and Mitchison, 1991 but with greater temporal resolution (Chapter 2). Endothelial cells provided a model system because of their capacity to dynamically reorganize their actin cytoskeleton in response to mechanical stimuli. Experiments revealed that filaments turnover in about 7 minutes in the bulk cytoplasm of endothelial cells (Chapter 5). Unlike previous PAF and FRAP studies, our experiments simultaneously produced estimates for the actin monomer diffusion coefficient (3.1 x 10-8 cm 2/sec) and the fraction of actin polymerized (37%) in these cell. This lead to the conclusion that while filament turnover was rapid in the bulk cytoskeleton of endothe- Actin Dynamics in the Cell Cytoplasm 1-17 Background and Literature Review The Story of this Thesis lial cells, it was not diffusion limited. FRAP data obtained in a collaboration demonstrated the general equivalence of the two techniques and that photobleaching had been controlled. The resolution of our imaging system did not allow photoactivation within leading lamellipodia, however the turnover rate in the bulk cytoplasm was still too high to be explained by in vitro turnover rates. In 1996 the in vitro/ in vivo paradox was apparently resolved with the demonstration that ADF/cofilin accelerated pointed end off rates by a factor of 25 (Carlier, et al., 1997). The focus of this work then shifted to elucidating the mechanism of cell motility. While previous work demonstrated that filament turnover was fastest in the fastest cell types (Carlier, et al., 1997;Theriot and Mitchison, 1991;Theriot and Mitchison, 1992;Wang, 1985), there was no evidence that cells of a single type could accelerate turnover as they moved faster. I demonstrated this by applying PAF and FRAP to endothelial cells moving at different rates in wounded monolayers (Chapter 5). Unexpectedly that the fraction of actin polymerized decreased with increasing speed in these cells. This result confirms the predictions of a recent model of cell motility which attributes the trend to accelerated disassembly with ADF/cofilin. However, experiments in gelsolin positive and null fibroblasts exhibited similar trends, raising the prospect that filament severing may be the mechanism involved. In preliminary experiments, subconfluent endothelial cells were found to have three times as many filaments than confluent cells providing further evidence of filament severing (Chapter 6). To elucidate the mechanisms controlling cell motility in endothelial cells I constructed a kinetic model of the actin cycle that includes filament severing and accelerated pointed end disassembly as mechanisms (Chapter 6). Using protein levels and kinetic rate constants from 1-18 Background and Literature Review Works Cited the literature, simulations demonstrate that accelerated filament disassembly is the most effective mechanism producing a simultaneous decrease in the polymer fraction and average lifetime of filaments. In combination with the experimental data, this kinetic model suggests that both filament severing and accelerated disassembly occur as endothelial cells move faster, but that accelerated disassembly has more influence on the dynamic state of the cytoskeleton. I have developed a PAF based in vitro system for examining the effects of actin associated proteins on steady-state actin dynamics (Chapter 7). The system improves over existing techniques in providing a non-invasive assay of filament turnover, polymer fraction, and filament length in a single preparation of actin. The system has provided the first visualization of actin filament turnover. Works Cited 1. Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. Watson. 1994. Molecular Biology of the Cell. Garland Publishing, Inc., New York. 2. Amato, P. A. T., D.L. 1986. Probing the mechanism of incorporation of fluorescently labeled actin into stress fibers. J. Cell Biol. 102:1074-1084. 3. Amatruda, J., J. Cannor, K. Tatchell, C. Hug, and J. Cooper. 1990. Disruption of the actin cytoskeleton in yeast capping protein mutants. Nature. 344:352-354. Actin Dynamics in the Cell Cytoplasm 1-19 Background and Literature Review Works Cited 4. Amatruda, J., D. Gattermeir, T. Karpova, and J. Cooper. 1992. Effects of null mutations and overexpression of capping protein on morhpogenesis, actin distribution and polarized secretion in yeast. J Cell Biol. 119:1151-62. 5. Apgar, J. 1995. Activation of protein kinase C in rat basophilic leukemia cells stimulates increased productionof phosphatidylinositol 4-phosphate and phsphatidylinositol 4,5-bisphosphate: correlation with actin polymerization. Mol Biol Cell. 6:97-108. 6. Azuma, T., W. Witke, T. Stossel, J. Hartwig, and D. Kwiatkowski. 1988. Gelsolin is a downstream effector of rac for fibroblast motility. Embo J. 17:1362-1370. 7. Azuma, T., W. Witke, T. Stossel, J. Hartwig, and D. Kwitkowski. 1998. Gelsolin is a downstream effector of rac for fibroblast motility. Embo J. 17:1362-1370. 8. Barkalow, K., W. Witke, D. Kwiatkowski, and J. Hartwig. 1996. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J Cell Biol. 134:389-399. 9. Burtnick, L., E. Koepf, J. Grimes, E. Jones, D. Stuart, P. McLaughlin, and R. Robinson. 1997. The crystal structure of plasma gelsolin: implications for actin severing, capping, and nucleation. Cell. 90:661-670. 10. Carlier, M. 1998. Control of actin dynamics. CurrOpin Cell Biol. 10:45-51. 11. Carlier, M., D. Didry, I. Erk, J. Lepault, M. Van Troys, J. Vanderkerckhove, I. Perelroizen, H. Yin, Y. Doi, and D. Pantaloni. 1996. Tb4 is not a simple G-actin sequestering protein and interacts with F-actin at high concentration. J Biol Chem. 271:9231-9239. 1-20 Background and Literature Review Works Cited 12. Carlier, M., and D. Pantaloni. 1986. Direct evidence for ADP-Pi-F-actin as the major intermediate in ATP-actin polymerization. Rate of dissociation of Pi from actin filaments. Biochemistry. 25:7789-7792. 13. Carlier, M., and D. Pantaloni. 1997. Control of actin dynamics in cell motility. Mol Biol Cell. 269:459-467. 14. Carlier, M.-F., C. Jean, K. Rieger, M. Lenfant, and D. Pantaloni. 1993. Modulation of the interaction between G-actin and thymosin f34 by the ATP/ADP ratio: possible implication in the regulation of actin dynamics. Proc. Natl. Acad. Sci., U.S.A. 90:5034-5038. 15. Carlier, M.-F., V. Laurent, J. Santolini, R. Melki, D. Didry, G.-X. Xia, Y. Hong, N.-H. Chua, and D. Pantaloni. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin based motility. J. Cell Biol. 136:1307-1322. 16. Carlson, M., P. Babcock, P. Rubenstein, and Y. Wang. 1977. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J Mol Biol. 115:465-483. 17. Carpenter, C., K. Tolias, A. Couvillon, and J. Hartwig. 1997. Signal transduction pathways involving the small G proteins rac and Cdc42 and phosphoinositide kinases. Adv Enzyme Regul. 37:377-390. 18. Carson, M., S. Shasby, S. Lind, and D. Shasby. 1992. Histamine, actin-gelsolin binding, and polyphsphoinositides in human umbilical vein endothelial cells. Am J PHysiol. 263:L664-L669. Actin Dynamics in the Cell Cytoplasm 1-21 Background and Literature Review Works Cited 19. Chong, L., A. Traynor-Kaplan, G. Bokoch, and M. Schwartz. 1994. The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell. 79:507-513. 20. Condeelis, J. 1993. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 9:411-441. 21. Cooper, J., J. Blum, R. Williams, and T. Pollard. 1986. Purification and characterization of Actophorin, a new 15,000 dalton actin -binding protein from Acanthamoeba castellanii. J Biol Chem. 261:477-485. 22. Cramer, L. P., M. Siebert, and T. J. Mitchison. 1997. Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: implications for the generation of motile force. J. Cell Biol. 136:1287-1305. 23. Cunningham, C., T. Stossel, and D. Kwiatkowski. 1991. Enhanced motility in NIH 3T3 fibroblasts that overexpress gelsolin. Science. 251:1233-1236. 24. Dadabay, C., E. Patton, J. Cooper, and L. Pike. 1991. Lack of a correlation between changes in polyphosphoinositide levels and actin/gelsolin complexes in A431 cells treated with epidermal growth factor. J Cell Biol. 112:1151-1156. 25. DeNofrio, D., T. Hoock, and I. Herman. 1989. Functional sorting of actin isoforms in microvascular pericytes. J Cell Biol. 109:191-202. 1-22 Background and Literature Review Works Cited 26. Drenckhanh, D., and T. Pollard. 1986. Elongation of actin filaments is a diffusion-limited reaction at the barbed end and is accelerated by inert macromolecules. J Biol Chem. 261:1275412758. 27. Eddy, R., R. Sauterer, and J. Condeelis. 1996. A major agonist-related capping activity in Dictyostelium is due to the capping protein, cap32/34. Biochim Biophys Acta. 1314:247-259. 28. Finkel, T., J. A. Theriot, K. D. Dise, G. E Tomaselli, and P. J. Goldschmidt-Clermont. 1994. Dynamic actin structures stabilized by profilin. Proc.Natl. Acad. Sci. USA. 91:1510-1514. 29. Fisher, G., P. Conrad, R. DeBiasio, and D. Taylor. 1988. Centripetal transport of cytoplasm, actin and the cell surface in lamellipodia of fibroblasts. Cell Motil Cytoskeleton. 11:235-47. 30. Giuliano, K. A., and D. L. Taylor. 1994. Fluorescent actin analogs with a high affinity for profilin in vitro exhibit an enhanced gradient of assembly in living cell. J. Cell Biol. 124:971-983. 31. Goldschmidt-Clermont, P., M. Furman, D. Wachsstock, D. Safer, V. Nachmias, and T. Pollard. 1992. The control of actin nucleotide exchange by thymosin34 and profilin. A potential regulatory mechanism for actin polymerization in cells. Mol. Biol. Cell. 3:1015-1024. 32. Goldschmidt-Clermont, P., L. Machesky, J. Baldassare, and T. Pollard. 1990. The actin-binding protein profilin binds to PIP2 and inhibits its hydrolysis by phospholipase C. Science. 251:1231-1233. 33. Goldschmidt-Clermont, P., L. Machesky, S. Doberstein, and T. Pollard. 1991. Mechanism of the interaction of human platelet profilin with actin. J Cell Biol. 113:1081-1089. Actin Dynamics in the Cell Cytoplasm 1-23 Background and Literature Review Works Cited 34. Hartwig, J. 1992. Mechanisms of actin rearrangements mediating platelet activation. J. Cell Biol. 1992:1421-1442. 35. Hartwig, J., G. Bokoch, C. Carpenter, P. Janmey, L. Taylor, and T. Stossel. 1995. Thrombin receptor ligation and activated rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 82:643-653. 36. Hartwig, J., S. Kung, T. Kovacsovics, P. Janmey, L. Cantley, T. Stossel, and A. Toker. 1996. D3 phosphoinositides and outside-in signaling by glycoprotein IIb-IIIa mediate platelet actin assembly and filpodial extension induced by phorbos 12-myristate 13-acetate. J Biol Chem. 271:32986-32993. 37. Hartwig, J. H., and P. Shevlin. 1986. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J. Cell. Biol. 103:1007-1020. 38. Heiss, S., and J. Cooper. 1991. Regulation of CapZ, an actin capping protein of chicken muscle, by anionic phspholipids. Biochemistry. 30:8753-8758. 39. Hennessey, E., D. Drummond, and J. Sparrow. 1993. Molecular genetics of actin function. Biochem J. 291:657-671. 40. Herman, I. 1993. Actin Isoforms. Curr Opin Cell Biol. 5:48-55. 41. Herman, I., and P. D'Amore. 1989. Functional sorting of actin isoforms in microvascular pericytes. J Cell Biol. 101:43-52. 1-24 Background and Literature Review Works Cited 42. Holmes, K., D. Popp, W. Gebhard, and W. Kabsch. 1990. Atomic model of the actin filament. Nature. 347:44-49. 43. Hoock, T., P. Newcomb, and I. Herman. 1991. 3 actin and its mRNA are localized at the plasma membrane and the regions of moving cytoplasm during the cellular response to injury. J Cell Biol. 112:653-664. 44. Hotchin, N., and A. Hall. 1995. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular Rho/Rac GTPases. J Cell Biol. 131:1857-1865. 45. Hou, L., E Lanni, and K. Luby-Phelps. 1990. Tracer diffusion in F-actin and Ficoll mixtures towards a model for cytoplasm. Biophys J. 58:31-43. 46. Howard, T., C. Chaponnier, H. Yin, and T. Stossel. 1990. Gelsolin-actin interaction in actin polymerization in human neutrophils. J Cell Biol. 110:1983-1992. 47. Hug, C., P. Jay, I. Reddy, J. McNally, P. Bridgeman, E. Elson, and J. Cooper. 1995. Capping protein levels influnce actin assembly and cell motility in dictyostelium. Cell. 81:591-600. 48. Imamura, H., K. Tanaka, T. Hihara, M. Umikawa, T. Kamei, K. Takahashi, T. Sasaki, and Y. Takai. 1997. Bnilp and Bnrlp: Downstream targets of the Rho family small G-proteins which interact with profilin and regulate actin cytoskeleton in Saccharomyces cerevisiae. EMBO J. 16:2745-2755. 49. Janmey, P. 1994. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu Rev Physiol. 56:169-91. Actin Dynamics in the Cell Cytoplasm 1-25 Background and Literature Review Works Cited 50. Janmey, P., and T. Stossel. 1987. Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature. 325:362-364. 51. Janmey, P. A., and T. P. Stossel. 1986. Kinetics of actin monomer exchange at the slow growing ends of actin filaments and their relation to the elongation of filaments shortened by gelsolin. J. Muscle Res. Cell Motility. 7:446-454. 52. Kabsch, W., and J. Vandekerckhove. 1992. Structure and function of actin. Annu. Rev. Biophys. Biomol. Struct. 21:49-76. 53. Korn, E. D., M.-F. Carlier, and D. Pantaloni. 1987. Actin polymerization and ATP hydrolysis. Science. 238:638-644. 54. Kovacsovics, T., C. Bachelot, A. Toker, C. Vlahos, B. Duckworth, L. Cantley, and J. Hartwig. 1995. Phosphoinositide 3-kinase inhibition spares actin assembly in activating platelets but reverses platelet aggregation. The Journalof Biological Chemistry.270:11358-11366. 55. Kozma, R., S. Ahmed, A. Best, and L. Lim. 1995. The Ras-related protein, Cdc42Hs and bradykinin promote formation of peripheral actin micrspikes in filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 15:1942-1952. 56. Kreis, T., Geiger, B. , Schlessinger, J. 1982. Mobility of microinjected rhodamine actin within living chicken gizzard cells determined by fluorescence photobleaching recovery. Cell. 29:835-845. 1-26 Background and Literature Review Works Cited 57. Kreis, T., K. Winterhalter, and W. Birchmeier. 1979. In vivo distribution and turnover of fluorescently labeled actin microinjected into human fibroblasts. Proc.Natl. Acad. Sci. 76:3814-3818. 58. Kwiatkowski, D., P. Janmey, J. Mole, and H. Yin. 1985. Isolation and properties of two actinbinding domains in gelsolin. J Biol Chem. 260:15232-15238. 59. Lamb, J., P. Allen, B. Tuan, and P. Janmey. 1993. Modulation of felsolin function: activation at low pH overrides Ca2 + requirement. J Biol Chem. 268:8999-9004. 60. Lanni, F., and B. Ware. 1984. Detection and characterization of actin monomers, oligomers, and filaments in solution by measurement of fluorescence photobleaching recovery. Biophys J. 46:97-110. 61. Lappalainen, P., and D. Drubin. 1997. Cofilin promotes rapid actin filament turnover in vivo. Nature. 388:1165-1168. 62. Larsson, H., and U. Lindberg. 1988. The effect of divalent cations on the interaction between spleen profilin and different actins. Biochim Biophys Acta. 953:95-105. 63. Lassing, I., and U. Lindberg. 1985. Specific interaction between phosphatidyl-inositol 4,5-bisphosphate and profilactin. Nature. 314:472-474. 64. Lassing, I., and U. Lindberg. 1988. Spcificity of the interaction between phosphatidylinositol 4,5 bisphosphate and the profilin:actin complex. J Cell Biochem. 37:255-267. Actin Dynamics in the Cell Cytoplasm 1-27 Background and Literature Review Works Cited 65. Lu, M., W. Witke, D. Kwiatkowski, and K. Kosik. 1997. Delayed retraction of filopodia in gelsolin null mice. J Cell Biol. 138:1279-1287. 66. Luby-Phelps, K., Lanni, F., Taylor, D.L. 1985. Behavior of a Fluorescent Analogue of Calmodulin in Living 3T3 Cells. J. Cell Biol. 101:1245-1256. 67. Luby-Phelps, K., Castle, P.E., Taylor, D., and Lanni, F. 1987. Hindered diffusion of inert tracer particles in the cytoplasm of mouse 3T3 cells. Proc. Natl. Acad. Sci., USA. 84:49104913. 68. Luby-Phelps, K., F. Lanni, and D. L. Taylor. 1988. The submicroscopic properties of cytoplasm as a determinant of cellular function. Ann. Rev. Biophys. Biophys. Chem. 17:369-396. 69. Luby-Phelps, K., D. Taylor, and F. Lanni. 1986. Probing the structure of cytoplasm. J Cell Biol. 102:2015-2022. 70. McLaughlin, P., J. Gooch, H. Mannherz, and A. Weeds. 1993. Structure of gelsolin segment 1-actin complex and the mechanism of filament severing. Nature. 364:685-692. 71. Meberg, P., S. Ono, L. Minamide, M. Takahashi, and J. Bamburg. 1998. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 39:172-190. 72. Mitchison, T. 1989. Polewards microtubule flux in the mitotic spindle: evidence from photoactivation of fluorescence. J. Cell Biol. 109:637-652. 1-28 Background and Literature Review Works Cited 73. Mitchison, T. 1992. Compare and contract actin filaments and microtubules. Mol Biol Cell. 3:1309-1315. 74. Moon, A., and D. Drubin. 1995. The ADF/cofilin proteins: stimulus responsiveness modluators of actin dynamics. Mol Biol Cell. 6:1423-1431. 75. Nebl, G., S. Meuer, and Y. Samstag. 1996. Dephosphorylation of serine 3 regulates nuclear translocation of cofilin. J Biol Chem. 271:26276-26280. 76. Ohshima, S., H. Abe, and T. Obinata. 1989. Isolation of profilin from embryonic chicken skeletal muscle and evaluation of its interaction with different actin isoforms. J Biochem. 105:855857. 77. Okada, K., H. Takano-Ohmuro, T. Obinata, and H. Abe. 1996. Dephosphorylation of cofilin in polymorphonuclear leukocytes derived from peripheral blood. Exp Cell Res. 227:116-122. 78. Oosawa, F., and S. Asakura. 1962. A theory of linear and helical aggregations of macromolecules. J Mol Biol. 4:10-21. 79. Oosawa, F., and S. Asakura. 1975. Thermodynamics of the Polymerization of Protein. Academic Press, London. 80. Pantioni, D., and M. Carlier. 1993. How profilin promotes actin filament assembly in the presence of thymosin b4. Cell. 75:1007-1014. Actin Dynamics in the Cell Cytoplasm 1-29 Background and Literature Review Works Cited 81. Peng, I., and D. Fischman. 1991. Post-translational incorporation of actin into myofibrils in vitro: evidence for isoform specificity. Cell Motil Cytoskel. 20:151-168. 82. Perelroizen, I., D. Didry, H. Christensen, N. Chua, and M. Carlier. 1996. Role of nucleotide exchange and hydrolysis in the function of profilin in actin assembly. J. Biol. Chem. 271:12302-12309. 83. Pollard, T. 1986. Rate constants for the reactions of ATP-and ADP-actin with the ends of actin filaments. J Cell Biol. 103:2747-2754. 84. Pollard, T., and J. Cooper. 1984. Quantitative analysis of the effect of Acanthameoba profilin on actin filament nucleation and elongation. Biochemistry. 23:6631-6641. 85. Ridley, A., and A. Hall. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70:389-399. 86. Ridley, A., H. Paterson, C. Johnston, D. Diekmann, and A. Hall. 1992. The small GTPbinding protein rac regulates growth factor-induced membrane ruffling. Cell. 70:401-410. 87. Rosenblatt, J., B. Agnew, H. Abe, J. Bamburg, and T. Mitchison. 1997. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the urnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol.: 1323-1332. 88. Safer, D., R. Golla, and V. Nachmias. 1990. Isolation of a 5-kilodalton actin-sequesturing peptide from human blood platelets. Proc Natl Acad Sci USA. 87:2536-2540. 1-30 Background and Literature Review Works Cited 89. Safer, D., and V. Nachmias. 1994. Beta thymosins as actin binding peptides. Bioessays. 16:473-479. 90. Schafer, D., and J. Cooper. 1995. Control of actin assembly at filament ends. Ann Rev Dev Biol. 11:497-518. 91. Schutt, C., J. Myslik, M. Rozycki, N. Goonesekere, and U. Lindberg. 1993. The structure of crystalline profilin-~-actin. Nature. 365:810-816. 92. Sheterline, P., J. Clayton, and J. Sparrow. 1994. Actins. Protein Profile. 1:1-121. 93. Small, J., M. Herzog, and K. Anderson. 1995. Actin filament organization in the fish keratocyte lamellipodium. J Cell Biol. 129:1275-1286. 94. Stossel, T. 1993. On the crawling of animal cells. Science. 260:1086-1094. 95. Sun, H., K. Katarzyna, and H. Yin. 1995. P-thymosins are not simple actin monomer buffering proteins. J Biol Chem. 271:9223-9230. 96. Takuma, T., T. Ichida, N. Yokoyama, S. Tamura, and T. Obinata. 1996. Dephosphorylation of cofilin in paratid acinar cells. J Biochem (Tokyo). 120:35-41. 97. Tanaka, K., and Y. Takai. 1998. Control of reorganization of the actin cytoskeleton by Rho family small GTP-binding proteins in yeast. CurrOpin Cell Biol. 10:112-116. 98. Tapon, N., and A. Hall. 1997. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Cur Opin Cell Biol. 9:86-92. Actin Dynamics in the Cell Cytoplasm 1-31 Background and Literature Review Works Cited 99. Theriot, J., and T. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126-131. 100. Theriot, J., and T. Mitchison. 1992. Comparison of actin and cell surface dynamics in motile fibroblasts. J. Cell Biol. 118:367-377. 101. Theriot, J., and T. Mitchison. 1993. The three faces of profilin. Cell. 75:835-838. 102. Tolias, K., L. Cantley, and C. Carpenter. 1995. Rho family GTPases bind to phosphoinositide kinases. J Biol Chem. 270:17656-17659. 103. Vanderkerchekhove, J., and K. Weber. 1978. At least six different actins are expressed in a higher mammal: an analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J Mol Biol. 122:783-802. 104. Vanderkerchekhove, J., and K. Weber. 1979. The complete amino acid sequence of actins from bovine aorta, bovine heart, bovine fast skeletal muscle and rabbit slow skeletal muscle. Differentiation. 14:123-133. 105. Wang, Y. 1985. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J. Cell. Biol. 101:597-602. 106. Waterman-Storer, C., and E. Salmon. 1997. Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J Cell Biol. 139:417-434. 1-32 Background and Literature Review Works Cited 107. Weber, A., V. Nachmias, C. Pennise, M. Pring, and D. Safer. 1992. Interaction of thymosin 34 with muscle and platelet actin: implications for actin sequestration in resting platelets. Biochemistry. 31:6179-6185. 108. Wegner, A. 1976. Head to tail polymerization of actin. J. Mol. Biol. 108:139-150. 109. Wegner, A., and J. Engel. 1975. Kinetics of cooperative association of actin to actin filaments. Biophys. Chem. 3:215-225. 110. Wegner, A., and G. Isenberg. 1983. 12-fold difference between the critical monomer concentrations of the two ends of actin filaments in physiological salt conditions. Proc.Natl. Acad. Sci. USA. 1983:4922-4925. 111. Witke, W., A. Sharpe, J. Hartwig, T. Azuma, T. Stossel, D. Kwiatkowski. 1995. Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 81:41-51 112. Yeh, J., and B. Haarer. 1996. Profilin is required for the normal timing of actin polymerization in response to thermal-stess. FEBS Lett. 398:303-307. 113. Yin, H., J. Hartwig, K. Marayama, and T. Stossel. 1981. Ca2+ control of actin filament length. Effect of macrophage gelsolin on actin polymerization. J Biol Chem. 256:9693-9697. 114. Zigmond, S., M. Joyce, J. Borleis, G. Bokoch, and P. Devreotes. 1997. Regulation of actin polymerization in cell-free systems by GTPys and Cdc42. J Cell Biol. 138:363-374. Actin Dynamics in the Cell Cytoplasm 1-33 Background and Literature Review Works Cited 1-34 CHAPTER 2 Experimental Set-up and Design This chapter describes a system for measuring actin dynamics in vitro and in vivo using caged resorufin iodoacetamide actin (CRIA). An inverted fluorescence microscope equipped with a microinjection system is fitted with a modified epi-fluorescence port to accept light for excitation and photoactivation from mercury arc lamps. A computer controls sample exposure to excitation and photoactivation light and coordinates exposures with image acquisitions to give high temporal resolution. Macros automate image processing tasks and produce a MATLAB formatted output file that can be readily processed using the interpretive model in Chapter 3. Actin Dynamics in the Cell Cytoplasm 2-1 Experimental Set-up and Design Introduction Introduction Photoactivated fluorescence and fluorescence recovery after photobleaching Actin derivatized with fluorescent groups has been used to study the distribution, mobility, and turnover of actin in vitro and in living cells (Theriot and Mitchison, 1991; Theriot and Mitchison, 1992; Simon, et al., 1988; Wang, 1985; Kreis, 1982; Luby-Phelps, 1985). Early studies established that fluorescently labeled actin rapidly (< 30 minutes) incorporates throughout the cytoskeleton after microinjection (Kreis, et al., 1979; Wang, 1984; Taylor and Wang, 1978). The fluorescence from microinjected actin recovers in two phases after photobleaching (Kreis, 1982), with the earliest phases attributed to the diffusion of actin monomer and the longer phase to the cyclic turnover of filaments. Photoactivation of fluorescence (PAF) was proposed as an alternative to fluorescence recovery after photobleaching (PAF) because of an inherent signal-to-noise-advantage (Mitchison, 1989). In PAF, microinjected actin is coupled to a non-fluorescent "caged" fluorophore that converts to a fluorescent form after exposure to UV light (Theriot and Mitchison, 1991; Theriot and Mitchison, 1992; Krafft, et al., 1986). The decay of a strong signal against a dim background can measure the least dynamic component of a response with greater accuracy than the recovery of a dim signal into a high background (Krafft, et al., 1986). Therefore PAF should be more accurate in measuring filament turnover than FRAP. PAF also requires lower light levels for photoactivation than FRAP experiments require for photobleaching. Intense laser excitation with FRAP can have deleterious effects in cells including photochemical 2-2 Experimental Set-up and Design Introduction cross-linking, localized heating, and oxygen radical induced damage (Vigers, et al., 1988; Leslie, et al., 1984). Properties of Derivatized actin Actin is most commonly labeled at cysteine-374, a reactive sulphydryls targeted by commercially available iodoacetamide or maleimide derivatives (Miki and dos Remedios, 1988). Critical concentrations of actin labeled at cys-374 are near that of unlabeled actin indicating that polymerization is unaffected by cys-374 labeling (Kouyama and Mihashi, 1981; Wang, 1980). However, labeling at cys-374 hinders the interaction of profilin and actin (Malm, 1984). Profilin is a monomer sequestering protein known to catalyze the exchange of ADP for ATP bound to actin monomer (Goldschmidt-Clermont, et al., 1991) and to enhance actin assembly at filament barbed ends in the presence of high levels of I34 thymosin (Pantioni and Carlier, 1993). Lysine labeled actin derivatives retain profilin affinity (Giuliano and Taylor, 1994), and reportedly display incorporation patterns not seen with cys-374 derivatives (Giuliano and Taylor, 1994). Recently, actin fused to green fluorescent protein (GFP) has been expressed in dictyostelium discoideum and yeast (Doyle and Botstein, 1996; Westphal, et al., 1997; Aizawa, et al., 1997). GFPlabeled D. discoideum actin copolymerizes with native actin in vitro and follows the distribution and movement of actin in vivo (Westphal, et al., 1997). The use of GFP-actin as a tracer for actin dynamics has obvious advantages over traditional approaches. While, there are practical limits to the number of cells that can be microinjected, an entire culture of GFP-actin expressing cells can be produced. The viability of GFP-actin cells expressing GFP-actin is not questionable since the cells grow and divide for an extended time while expressing the GFP construct. GFP is linked to Actin Dynamics in the Cell Cytoplasm 2-3 Experimental Set-up and Design Synthesis of caged resorufin-iodoacetamide the N-terminus of actin, whereas chemical modifications of actin target the more functional Cterminus. The primary limitation to the expanded use of GFP-actin is that it has not successfully been expressed in mammalian cells. Because of this limitation, and because of the measurement advantageous of PAF over FRAP, caged-resorufin iodacetimide labeled actin (CRIA), remains the state-of-the-art probe for the study of actin dynamics in living cells. Synthesis of caged resorufin-iodoacetamide The Synthesis of Caged-Actin The two primary steps involved in the synthesis of caged actin are the attachment of a photoliable fluorescence quenching group to a fluorophore and the coupling of the caged compound to actin (Figure 2.1). The need for a fluorophore that performs both of these tasks reduces the choices available. Resorufin (Boehringer Mannheim, Germany) was selected by Theriot and Mitchison (Theriot and Mitchison, 1991) as a fluorophore for PAF experiments involving actin because of its availability as an iodoacetamide derivative for actin labeling and because of the presence of phenolic oxygens for reaction with the caging diazoethane group of Walker, et al., 1989. We synthesized caged-resorufin iodoacetamide (CRI) following the protocols of Theriot and Mitchison, 1991 and Walker, et al., 1989. We then labeled actin according to the method of Kouyama and Mihashi, 1981 to produce CRI-actin (CRIA). The successful synthesis of caged-actin is demonstrated in Figure 2.2, which shows the absorbance spectra for CRIA before and after photoactivation with 365 nm light. The spectrum fea- 2-4 Experimental Set-up and Design Synthesis of caged resorufin-iodoacetamide turing a peak at 460 nm is the Sresorufn -- o 10 N signature of the caged compound, whereas the uncaged compound has a absorption peak at 574 nm -,) UV light H H Ito caged-resorufin /..J - o C . (Figure 2.2a). Figure 2.2b displays the absorbance spectrum of caged-actin. The peak at 290 nm is due to actin, and the 460 nm peak indicates the presence of bound CRIA. Note that the peaks for actin and uncaged resorufin are far separated. Concentrations of resorufin and actin in the final product were therefore determined by completely uncaging the FIGURE 2.1. The synthesis of caged-actin. The first step is to react a resorufin-iodoacetamide with a photoliablefluorescence quenching group. In the second step, the caged-complex is coupled product and measuring abor- bances at 290 nm for actin (extinc- to actin at cys-374. UV light converts the caged dye back to its fluorescent parent. tion coefficient = 26.5 mM-1 cm- 1), and at 575 nm for resorufin (extinction coefficient = 65 mM-1 cm-1). The ratio of these concentrations gave a coupling ratio of 0.2 (1 in 5 actin molecules labeled). This is low compared to the value of 0.65 obtained by Theriot and Mitchison. In preparation for microinjection, CRIA was dialyzed into injection buffer (1 Actin Dynamics in the Cell Cytoplasm 2-5 Experimental Set-up and Design Measurementapparatus 340 520 wavelength [nm] a) 320 460 wavelength [nm] b) FIGURE2.2. Absorbance spectra demonstratingthe successful synthesis of caged-actin.a) Cagedresorufin (peak 405 nm) converts to resorufin (peak577 nm) afterphotolysis with 365 nm light. b) Cagedresorufin is shown coupled to actin (peak270 nm). mM HEPES, pH 7.5, 0.2 mM MgC12 , 0.2 mM ATP), diluted to 23.8 RM, and stored as 5 pl aliquots in 1 ml vials at -80 OC. Measurement apparatus The measurement system centers around a fluorescence microscope (Greyline model; Zeiss, Oberkochen, Germany) (Figure 2.3). A modified epi-fluorescence port accepts an uncaging pathway in addition to the standard excitation illumination pathway. The pathways employ 100W mercury arc lamps and are equipped with electronic shutters. (An excitation shutter is 2-6 Experimental Set-up and Design Measurement apparatus microinjection needle mercury arc for electronic . adjustable slit macintosh computer with Scion LG-3 video board FIGURE 2.3. Schematic of the PAF measurement system. The system centers aroundan epi-fluorescence microscope. Cells microinjectedwith CRIA arephotoactivatedwith UV light extractedfrom an additional mercury arc lamp focused through a narrow slit. A computer controlsthe photoactivationand excitation shutters and acquiresfluorescent imagesfrom an intensified CCD. necessary to minimize sample exposure to excitation light and limit photobleaching artifacts in cells (Chapter 4)). The uncaging lamp is focused upon an adjustable slit (D40,488; Edmund Sci- Actin Dynamics in the Cell Cytoplasm 2-7 Experimental Set-up and Design Measurement apparatus excitation filter 577 nm± 5 dicroic mirror transmits above sample below dicroic mirror transmits above 585 nm reflects below uncaging filtei 350 nm ± 50 barri er filter translmits above 585 r m ---- - - Hg arc broad spectrum 365 nm uncaging light 577 nm excitation light 591 nm fluorescence FIGURE2.4. Filterconfigurations.The broadband emissionfrom mercury arc lamps includes peaks at photoactivationand excitation wavelengths. Resorufin emission at 591 nm is passed to the camera while excluding of all other wavelengths entific; Barrington, NJ), and an external lens images the slit onto the epi-illumination field diaphragm. From the field diaphragm, the slit is imaged by an internal field lens and either a Zeiss 63x or a 16x Ph2 Plan-Neofluar objective onto the sample plane. The Neofluar objectives use non-autofluorescent glass that has high transmission up to near UV. The final image of the slit can be varied from 100 jtm (16x objective) down to 4 gtm (63x objective). The sample plane is imaged by a Hamamatsu C2400-50 CCD camera coupled to a Videoscope VS41845 GEN IV intensifier. Output from the camera is relayed to a video monitor and to a 640 x 480 pixel x 256 grey scale Scion AG-3 image acquisition board. The final spatial resolution of 2-8 Experimental Set-up and Design Measurement apparatus uncaging light e . E .- J -J;- - .- L aXCliation light real-time -n -, image acquisition times and fluorescence values automatically formatted for MATLAB band width measured an inputted manually .4 monomer diffusion - ~ polymer fraction filament turnover FIGURE 2.5. Computer controlled automation of experiments. Macros written for NIH Image 1.61 coordinate photoactivation, excitation, and image acquisition. A digital movie sequence and MATLAB formatted file containing times and fluorescence values are automatically generated. After some manual input, the file can be run in MATLAB with the interpretive model of Chapter 3 to generate estimates of the dynamic parameters. images obtained with the 63x objective was 2.5 pixels/micron and with the 16x objective was 0.7 pixels/micron. Filters in external and internal housing select and direct the wavelengths for excitation, photoactivation, and fluorescence at appropriate points in the light path (Figure 2.4). The Scion AG-3 framegrabber installed in an Apple Macintosh 9500 was equipped with digitalto-analog conversion capabilities to control shutters on the photoactivation and excitation light Actin Dynamics in the Cell Cytoplasm 2-9 Experimental Set-up and Design Measurement apparatus paths. Macros written for the public domain soft" '3: ware NIH Image (v1.61) coordinated the sample exposure to uncaging and excitation light with BL = "+a image acquisition (Figure 2.4). A short real-time Camera Gain max Intensifier gain = 881 movie is created at each measurement time point. Because the imaging system lags behind activation X of the exposure shutter (see System Characteriza- K I - 0 - 3 2 1 x N t' 3 time (sec) . -o 4 5 6 msec. An analysis macro run at the completion of ,Ob), an experiment measures the average fluorescence BL = 7 CW from "-" Camera Gain max .6 tion below), the images within a movie get successively brighter until they stabilize at about 66 in a manually drawn area in each image acquired, Intensifier gain = 948 selects a stabilized image from each movie, and .5 .4 0 0 assembles a new movie composed of only these 0 0 0 0 stabilized images. The analysis macro also records center-line fluorescence values and times in a El I ~-~-- 1 2 m1 - 3 time (sec) - 4 5 6 MATLAB readable output file. The ratio of the band width to total cell width is inputted manually FIGURE2.6. Dynamic response of CCD and intensifierfollowing sudden exposure to experimentallight levels. a) With black level (BL) adjustment off(+) the syste mreaches a steady value in just over 2 videoj rames b) When the BL circuitis active the system overshoots and takes 200 ms to seettle. into the output file. The file can be immediately read into MATLAB and fit in a least-square-fashion to an interpretive model (Chapter 3) to produce simultaneous estimates of the mobility of actin monomer, the fraction of actin polymerized and the turnover time of actin filaments. 2-10 Experimental Set-up and Design System characterization System characterization Lamp stability 100W mercury arc lamps are used on both the excitation and uncaging pathways. Stability of excitation illumination is imperative for quantitative interpretation of fluorescence. The stability of the excitation lamp (an Osram HBO 100 bulb in a Zeiss modified 50W housing), was examined over 5 hour periods and quantifying the fluorescence in a uniform sample of resorufin. The results demonstrate that after a 100 minute start up period the lamp intensity remains stable to within + 4% of its mean value. Dynamic response and linearity of the imaging system A Videoscope VS4-1845 GEN IV intensifier is used in combination with a Hamamatsu C2400-50 CCD camera. This combination was examined for its response to sudden exposure to a bright image by shuttering on the excitation source and measuring fluorescence from a resorufin sample. Figure 2.5 presents the results of these studies. Because the camera and the excitation shutter are not synchronized, the shutter opens during the middle of a camera integration interval, and takes just over 2 video frames (> 0.066ms) to reach a maximum level (Figure 2.5a). The camera controller is equipped with a black level (BL) adjustment. When the knob is turned counter-clockwise from its off position (labeled "+") an analog circuit dims the camera image before it is digitized. The BL knob is used in combination with the gain knob on the intensifier to stretch a subset of image intensities over the dynamic range of the camera. The camera gain is always set to its maximum. The BL circuit creates an overshoot in image intensities that settles after 200 ms or more Actin Dynamics in the Cell Cytoplasm 2-11 Experimental Set-up and Design Discussion depending on the BL setting (Figure 2.5b). The dynamic response of the imaging system is not influenced by the setting of the camera or intensifier gains. Because resorufin is highly photoliable in cells (Chapter 4), sample exposures must be kept to a minimum. The dynamic response of the imaging system requires that the BL circuit be off to obtain a minimum sample exposure of 0.07 seconds. The BL circuit improves images in cell experiments because coverslips produce a strong background intensity. The lack of BL control in these experiments can be compensated for by dimming the excitation light with a neutral density filter and increasing the intensifier gain. Discussion This chapter describes an experimental approach to measuring actin dynamics using caged resorufin iodoacetamide actin (CRIA). A computer controls a modified epi-fluorescence microscope to coordinate photoactivation, excitation, and image acquisition. Macros automate many of the image processing tasks and produce a MATLAB formatted file that can immediately processed to obtain the experiment results. A CCD camera coupled to a Gen IV intensifier provide the high temporal resolution necessary to minimize photobleaching during sample exposure to excitation light. The greatest limitation of the current system is a low spatial resolution (2.6 pixels/micron with a 63x objective compared to 12 pixels/micron on a FRAP system used in collaboration (Chapter 4)), due largely to demagnification by the intensifier. This limitation can be overcome by working with higher powered objectives, however these do not provide the depth of focus necessary to image through the thicker portions of a cell. Another solution is to upgrade the image capture board one with higher spatial resolution. 2-12 Experimental Set-up and Design Works Cited Works Cited 1. Aizawa, H., M. Sameshima, and I. Yahara. 1997. A green fluorescent protein-actin fusion protein dominatly inhibits cytokinesis, cell spreading, and locomotion in dictyostelium. Cell Structure and Function. 1997:335-345. 2. Doyle, T., and D. Botstein. 1996. Movement of yeast cortical actin cytoskeleton visualized in vivo. ProcNatl Acad Sci USA. 93:3886-3891. 3. Giuliano, K. A., and D. L. Taylor. 1994. Fluorescent actin analogs with a high affinity for profilin in vitro exhibit an enhanced gradient of assembly in living cell. J. Cell Biol. 124:971-983. 4. Goldschmidt-Clermont, P., L. Machesky, S. Doberstein, and T. Pollard. 1991. Mechanism of the interaction of human platelet profilin with actin. J Cell Biol. 113:1081-1089. 5. Kouyama, T., and K. Mihashi. 1981. Fluorimetry study of N-(1-pyrenyl)-iodoacetamidelabelled F-actin. Eur J Biochem. 114:33-38. 6. Krafft, G. A., J. P. Cummings, L. J. Dizig, W. R. Brven, W. R. Sutton, and B. R. Wars. 1986. Fluorescence activation and photodissipation (FDP). In Nucleocytoplasmic Transport. SpringlerVerlag, New York. 35-52. 7. Kreis, T., Geiger, B. , Schlessinger, J. 1982. Mobility of microinjected rhodamine actin within living chicken gizzard cells determined by fluorescence photobleaching recovery. Cell. 29:835845. Actin Dynamics in the Cell Cytoplasm 2-13 Experimental Set-up and Design Works Cited 8. Kreis, T., K. Winterhalter, and W. Birchmeier. 1979. In vivo distribution and turnover of fluorescently labeled actin microinjected into human fibroblasts. Proc. Natl.Acad. Sci. 76:38143818. 9. Leslie, R., W. Saxton, T. Mitchison, B. Neighbors, E. Salmon, and J. McIntosh. 1984. Assembly properties of fluroescein-labeled tubulin in vitro before and after fluorescence bleaching. J. Cell Biol. 99:2146-2156. 10. Luby-Phelps, K., Lanni, F., Taylor, D.L. 1985. Behavior of a Fluorescent Analogue of Calmodulin in Living 3T3 Cells. J. Cell Biol. 101:1245-1256. 11. Malm, B. 1984. Chemical modification of the Cys-374 of actin interferes with the formation of the profilactin complex. FEBS Let. 173:399-402. 12. Miki, M., and C. dos Remedios. 1988. Fluorescence quenching studies of fluorescein attached to Lys-61 or Cys-374 in actin : effects of polymerization, myosin subragment-1 binding, and tropomyosin-troponin binding. J Biol Chem Tokyo. 104:5625-5630. 13. Mitchison, T. 1989. Polewards microtubule flux in the mitotic spindle: evidence from photoactivation of fluorescence. J. Cell Biol. 109:637-652. 14. Pantioni, D., and M. Carlier. 1993. How profilin promotes actin filament assembly in the presence of thymosin b4. Cell. 75:1007-1014. 2-14 Experimental Set-up and Design Works Cited 15. Simon, J., A. Gough, E. Urbanik, F. Wang, E Lanni, B. Ware, and D. Taylor. 1988. Analysis of rhodamine and fluorescein-labeled F-actin diffusion in vitro by fluorescence photobleaching recovery. Biophys J. 54:801-815. 16. Taylor, D., and Y. Wang. 1978. Molecular cytochemisty; Incorporation of fluorescently labeled actin into living cells. Proc.Natl. Acad. Sci. USA. 75:857-861. 17. Theriot, J., and T. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126-131. 18. Theriot, J., and T. Mitchison. 1992. Comparison of actin and cell surface dynamics in motile fibroblasts. J. Cell Biol. 118:367-377. 19. Vigers, G., M. Coue, and J. McIntosh. 1988. Fluorescent microtubules break up under illumination. J Cell Biol. 107:1011-1024. 20. Walker, J., G. Reid, and D. Trentham. 1989. Synthesis and properties of caged nucleotides. Methods Enzymol. 172:289-301. 21. Wang, Y. 1985. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J. Cell. Biol. 101:597-602. 22. Wang, Y. a. T., DL. 1980. Preparation and Characterization of a new molecular cytochemical probe - 5 iodoacetamidofluorescein actin. The JournalofHistochemistryand Cytochemistry. 28. Actin Dynamics in the Cell Cytoplasm 2-15 Experimental Set-up and Design Works Cited 23. Wang, Y. L. 1984. Reorganization of Actin Filament Bundles in Living Fibroblasts. The Journalof Cell Biology. 99: 1478-1485. 24. Westphal, M., A. Jungbluth, M. Heidecker, B. Muhlbauer, C. Heizer, J. Schwartz, G. Marriot, and G. Gerisch. 1997. Microfilament dynamics during cell movement and chemotaxis moitored by a GFP-actin fusion protein. CurrBiol. 7:176-183. 2-16 CHAPTER 3 Interpreting PAF/FRAP Measurements of Steady- State Actin Dynamics This chapter presents a continuum model describing the decay of fluorescence from a photoactivated band of fluorescent actin. The model provides an improved interpretation of photoactivated fluorescence (PAF) and fluorescence recovery after photobleaching (FRAP) experiments in cells. In a simplified cell geometry, the model assumes a uniform concentration of actin and accounts for the exchange of actin between filaments and monomer sequestering proteins. The results demonstrate how fluorescence decay depends on actin monomer diffusion, filament turnover, and the ratio of polymerized to unpolymerized actin. The solution simplifies when monomer diffuses much faster than filaments turnover, a condition predicted to exist both in cells and in purified solutions. Actin Dynamics in the Cell Cytoplasm 3-1 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Introduction Introduction Purified actin at physiological levels of salt and temperature spontaneously assembles into filaments. At steady-state, and in the presence of excess ATP, monomers flux through filaments from barbed to pointed ends (Wegner, 1976) in a process called treadmilling. It is unclear whether treadmilling occurs in cells, however experiments demonstrate that filament turnover does occur. Fluorescent actin monomer microinjected into cells incorporates throughout the cytoskeleton (Taylor and Wang, 1978;Kreis, et al., 1979). Fluorescence from injected actin recovers completely after photobleaching (Wang, 1984;Kreis, 1982) in FRAP experiments. Similarly, the fluorescence from photoactivated bands of actin decays completely in PAF experiments. These experiments demonstrate that filaments steadily exchange subunits with a pool of unpolymerized actin, maintained in cells at hundreds of times the critical concentration for filament formation by monomer sequestering proteins (Sun, et al., 1995). An ideal tracer for cellular actin should distribute between sequestered monomer and filaments. The evolution of fluorescence in a PAF or FRAP experiment will depend on the ratio of polymerized to unpolymerized actin, the diffusion of filaments and monomer, and the turnover of filaments. Previous investigators have applied PAF and FRAP to independently estimate the dynamics of monomer or filaments. In these experiments, the half-times for fluorescence recovery or decay were related to the dynamics of the phase of interest. Because the dynamics of both phases contribute to the evolution of fluorescence in a PAF or FRAP experiment, halftimes may poorly characterize the dynamics of these phases. 3-2 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Mathematicalmodel This chapter presents a mathematical model to guide the interpretation of PAF and FRAP experiments on actin. The model assumes steady-state dynamics and no filament diffusion. The model allows for the exchange of actin between sequestering proteins and filaments. The results describe how fluorescence decay depends on monomer diffusion, filament turnover and the ratio polymerized to unpolymerized actin. Mathematicalmodel Assumptions/idealization/simplifications: * Photoactivation creates a uniform strip of fluorescent actin in a simplified cell geometry (Figure 3.1). This simplifies the problem to one-dimension and permits an analytical solution. A more complex geometry (i.e. the inclusion of a nucleus) would require a computational solution. * There are two uniformly distributed pools of actin: diffusing monomer and non-diffusion filaments. While monomer and filament diffusion and filament turnover should all contribute to fluorescence decay in a PAF experiment, the uniquitious distribution of actin cross-linking proteins such as ABP-280 suggests filament diffusion is limited. * There is no net polymerization of actin, i.e. filaments exchange monomer with sequestering proteins to turnover at steady-state. Actin Dynamics in the Cell Cytoplasm 3-3 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Mathematicalmodel L Xo FIGURE3.1 Schematic of the model system. The cell is modeled with a rectangulargeometry containingtwo actinpools: diffusing monomers and non-diffusingfilaments. Both monomers andfilaments are photoactivatedin a narrowband at the beginningof the experiment (white dots). The fluorescence diffuses out of the band with time. The position of the slit center-line is given by xo and the slit width is 0o. Derivation of the dimensional problem: The equation of species conservation for fluorescent actin sequestered as monomer (denoted by the subscript "um" for uncaged monomer) is JCurm c= t D ac e umx2 + fum aX2 E.3.1 where D is the monomer diffusion coefficient. This expression equates the time rate of change of the concentration of fluorescent actin in the monomer pool, cum to the rate of monomer diffusion and the rate of production or loss of sequestered actin, fum , due to exchange with filaments. 3-4 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Mathematicalmodel The equation of species conservation for fluorescent actin in filaments (denoted with the subscript "uf" for uncaged filaments) is "a = -fum E.3.2 where filaments can't diffuse. The coupling of E.3.1 and E.3.2 conserves total actin, but permits actin exchange between filaments and sequestering proteins. The fraction of actin in filaments that is fluorescent is cf /cf. The fraction of sequestered monomer that is fluorescent is c,,m / Cm, where cm and cf are the concentrations of total actin in sequestered monomer and filaments respectively. The rate of actin exchange between the pools is g , and fum is E.3.3 cu um = gf Eqs. E.3.2 and E.3.3 become ac um at D a 2 c "acu = - +u Uf cum Cf Cm) c cu E.3.4 E.3.5 respectively. These two equations along with the boundary conditions Actin Dynamics in the Cell Cytoplasm 3-5 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Mathematicalmodel acum ax acum ax x =0 x=o =0 E.3.6 x=L and the initial conditions Cur = acm Ix-xo >CO for Cum = Cu! = 0 and cuf = acf for Ix -xo <T2xxI<~ E.3.7 complete the dimensional problem. xo and o are the position and the width of the slit respectively, L is the cell length, and a is the fraction of total actin photoactivated' in the band. Non-dimensional problem Introducing the scaling cA - t*tD 2 SCuf cum acm acf 0o x* =x L E.3.8 Eqs, E.3.5 and E.3.6 become aCA at* 2 - cA 2+ ax*2 f(c7 - c, ) E.3.9 1. With uniform incorporation of injected labelled actin into the endogenous cytoskeleton, and 100% photolysis of caged actin in the band, a is simply the ratio of injected to total actin. 3-6 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Mathematicalmodel 2 C =- at* + ax* 2 (cC - c ) E.3.10 ( where the non-dimensional parameters are P = 02 cD and y = The evolution of fluorescence depends on the parameters y , Cf cm O,/L and xo/L . The geometrical parameters o/L and xo/L are selected prior to an experiment. The parameter y ratios the amount of actin in filaments to that sequestered as monomer, while B ratios the time scales of monomer diffusion and filament turnover. (In the parameter 1, o2/D is the characteristic time for monomer to diffuse from the band and cf/g is residence time of an actin monomer in filaments.) Solution An analytical solution of the non-dimensional system was found with a Fourier-Laplace transform (see appendix 3-A for details). The results are c* = + on cosknx* k= I c = + E.3.11 cosknx* k= with the Fourier coefficients given by Actin Dynamics in the Cell Cytoplasm 3-7 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Mathematicalmodel - On - Cf Here co = c - (s + = CO + P)et*- (s 2 + YP + (s, + J + (nk) 2 + e)S2 * (S- S2) t)est* - (s 2 + ) + (ink) 2 + y )esat* E.3.12 E.3.13 represents the Fourier transform of the initial distribution of fluorescent monomer (or polymer), s, and s 2 are the roots of quadratics in the wave number (nk) 2 (EA.3.6). Experiments follow the sum of the fluorescence from filaments and monomer. Assuming that the fluorescence is proportional to dye concentration and the intensity of excitation light, the fluorescence intensity F can be written as F = IoQ(cuf +Cum) E.3.14 where I0 and Q are the excitation light intensity and the fluorescence quantum efficiency respectively. Using E.3.8 and the definition of y , and normalizing with the maximum fluorescence F0 , the non-dimensional fluorescence is F* 3-8 Fo 1(cA +yc7 ) 1+y E.3.15 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Results Results Concentration and fluorescence profile We programmed E.3.11 to obtain the spatial and temporal evolution of the fluorescence in filaments and in monomer after photoactivation. A maximum summation of 400 terms was used in the calculations. In the example solution shown in Figure 3.2, the fluorescent monomer population quickly diffuses from the photoactivated band while the fluorescence in filament decays slowly. The total fluorescence is a weighted sum of these. The areas under the curves remain equal to w/L , indicating conservation of the fluorescent species. Center-line decay The behavior of center-line decay curves with varying P is shown in Figure 3.3. The decay is clearly bi-phasic. Immediately after photoactivation, fluorescent monomer diffuses from the band and the fluorescence drops rapidly in a 0-independent fashion. At longer times, fluorescence decay depends on P . In the case of P = 0 , actin doesn't exchange between filaments and sequestering proteins. With no filament turnover, the fluorescence from monomer diffuses from the band while the fluorescence from filaments remains indefinitely. When p is large, filament turnover is rapid compared with the time required for monomer to diffuse from the band, and fluorescence decay is diffusion limited (i.e. independent of P ). Actin Dynamics in the Cell Cytoplasm 3-9 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Results 1 Legen d m O. ----- monomer filaments - - 8 c* 0.6 4 - fluorescence _ t*= 0 C* 0.4 oL= 03=20 y= 1 0.2 0 0.2 0.4 0.6 0.8 1 x* 1 t*= 0.04 0.8 0.8 0.6 0.6 0.4 0.4 0.2 0.2 0 . :. 0 0.2 0.4 0.6 1 0.8 x* 0 0.2 . . . . . 0.4 . . . . . . 0.6 0.8 0.6 0.8 . . x* 1 t*= 0.08 0.8 0.6 0.4 0.2 0 . 0 . . . . 0.2 . . . . . . . . 0.6 0.4 x* . . . . 0.8 . 1 0O 0.2 0.4 1 x* FIGURE3.2 Evolution of totalfluorescence andfluorescence in filaments or monomers. In this example fluorescent monomer diffuses quickly while the decay offluorescencefrom filaments lags behind. The totalfluorescence is a weighted sum of these and lies between. 3-10 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Results 1 0.9 monomer diffusion 0.8 Y= 1; xo/L = 0.25; co/L = 0.2 0.7 filament turnover F* 0.6 0.5 = 0.04 0.4~~p 012 0. 0.3 0.2 0 10 5 t* 15 20 25 FIGURE 3.3 Decay offluorescence in a photoactivated band. A rapid initial decay offluorescence is caused by the diffusion offluorescent monomer from the band immediately after uncaging. The long-term fluorescence decay depends on filament turnover. Long time behavior when P << 1 When monomer diffusion is fast compared to filament turnover, P << 1, the long term decay of fluorescence depends strongly on P. Fluorescence decay at long times is examined by restricting the analytical solution to the first wave number, k = 1 .With this, s, - s2 reduces to S 1 -S 2 = 712 + [y + E.3.16 Because Is21> ISI terms with a s2 exponent in E.3.13 and E.3.14 decay faster than terms with a s, exponent. With this result and after renormalizing according to Actin Dynamics in the Cell Cytoplasm 3-11 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Results F*- (0 F2 - E.3.17 1(0 L the long-time center-line fluorescence decay becomes F2* + + + n + s+t* E.3.18 2 Developing s,(see EA.3.6) to first order and assuming P F2* = - < , reduces (Y+ 1) E.3.18 to e-Pt* E.3.19 1+y This convenient expression demonstrates that when filament turnover is slow, long term fluorescence decay is purely exponential with a time constant of -P and a coefficient equal to the fraction of actin polymerized y/(y + 1). The conditions that must be satisfied for E.3.19 to apply at long times are 2 <<1 and j<< n (7y+ 1) Either of these may be limiting. In cells, actin monomer sequestering proteins maintain roughly equal amounts of polymerized and non-polymerized actin (Bray and Thomas, 1976) (i.e. y = 1) and <<1 is the limiting condition. Because monomers will diffuse from an 8 gm photoactivated band in several seconds (employing D = 4 x 10-8 cm 2/s from Chapter 4) while filaments turnover in several minutes, f3 is small in cells and E.3.19 is applicable. This predic- 3-12 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Discussion tion is confirmed for bovine aortic endothelial cells in Chapter 4. PAF experiments may also be conducted in purified solutions (Chapter 7) where the concentration of actin in filaments may be 2 hundreds of times the level of unpolymerized actin. In this case y >>1 and the condition P << (¥ + 1) limits the range of 3 for which E.3.19 applies. Discussion In PAF experiments, microinjected actin coupled to a non-fluorescent "caged" fluorophore converts to a fluorescent form in a region of the cell illuminated with a pulse of UV light (Theriot and Mitchison, 1991;Theriot and Mitchison, 1992). In FRAP, exogeneous fluorescent actin incorporated into a cell is locally bleached with an intense laser. The evolution of fluorescence in photoactivated or photobleached bands will be controlled by the dynamics of actin and therefore PAF or FRAP can be used to assay actin dynamics in living cells. The accuracy of these assays will depend on the accuracy of the physical models used to interpret them. Previously, PAF or FRAP experiments have been interpreted with models that ignore or incorrectly characterize one phase of actin. In FRAP experiments, the short-term recovery of fluorescence was attributed to monomer diffusion and the remaining fluorescence to an "immobile" population (Kreis, 1982;Wang, 1985). In long-term observations, however, complete fluorescence recovery demonstrates that the second population, presumably filaments, was also dynamic. Characterizing actin monomer as 'mobile' and filaments as 'immobile' will be accurate so long as the dynamics of these phases have significantly different time scales. The approach does not describe Actin Dynamics in the Cell Cytoplasm 3-13 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Appendix: Detailed solution filament dynamics and therefore cannot address questions about the influence of filament turnover on monomer diffusion. Ignoring monomer diffusion, the half-life for fluorescence decay in PAF experiments has been used as a measure of filament turnover (Theriot and Mitchison, 1991; Theriot and Mitchison, 1992). With two dynamic phases contributing to fluorescence decay, a half-life may poorly characterize the dynamics of these process. This chapter presents a model that, while highly idealized, captures the physics essential for interpreting actin dynamics in PAF or FRAP experiments. The model assumes that an ideal fluorescent actin tracer distributes between filaments and sequestering proteins and that its transport depends on monomer diffusion and filament turnover, but not filament diffusion. The assumption of negligible filament diffusion is validated by experiments in Chapter 4. The model demonstrates how, in addition to geometric parameters, fluorescence decay depends on the ratio of polymerized to unpolymerized actin, y, and the ratio of the characteristic time for monomer diffusion from a photoactivated band to the time for filament turnover, P. When P >> 1 fluorescence decay is diffusion limited, otherwise it is biphasic with an initial decay due to monomer diffusion and a long term decay that depends on filament turnover. In cells when 3<< 1, the second phase of fluorescence decay will be a pure exponential with a time constant equal to filament turnover time and a coefficient equal to fraction of actin polymerized. Appendix: Detailedsolution In order to solve our coupled system of second order differential equations (E.3.9 and E.3.10), we used the Fourier-Laplace transform defined as1 3-14 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Appendix: Detailed solution f(k, s) = fdtfdc o j(x, t) este-i tkx EA.3.1 o to obtain -1 col s + (L k) 2 + yp) -1P (s +1 [c-cf EA.3.2 1_ where cO is the Fourier transform of the initial conditions. With the initial conditions given by E.3.8, we find cO = 2sin (k(o This system can then be solved for c + L sin(k - L)) EA.3.3 and cm . We obtained 1. The superscript = denotes the double Fourier and Laplace transform. Actin Dynamics in the Cell Cytoplasm 3-15 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Appendix: Detailedsolution ) = c, (s +P+ (s- s)(S - S2 c~ (s + (k) =* 2 + y+ EA.3.4 P) EA.3.5 (s - s)(S - s2) 1 - (itk) 2 S1 with S2 = - 2 1y - 3 + ,I((ik)2 + fy + j) -4(7Ck) -- (k) r 2 - P> < 2 > J.((k) 2 + 13 + 3) 2-4(ck) 2 3 3 -- EA.3.6 Since they are polynomials in the wavenumber, EA.3.4 and EA3.5 can be easily inverse Laplace transformed -, - = coS (s, + YP + P)e-"*- (s2 + YO + 1)e* ((sI 1 -2) - (ink) 2 - Y- + J((k)2 + S1 = - (nk) - ( 7 EA.3.7 y +2-4(k)2 2 2 EA EA.3.8 +- 3y +k)2-4(nk)2 + S2 = Finally, the solution is obtained by taking the inverse Fourier transform of these two expressions. Due to the boundary condition, the inverse transform reduces to a cosine transform and the final solution is 3-16 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Works Cited cA = + Io coskcx* k= I c* = + c EA.3.9 cosk7tx* k= 1 Works Cited 1. Bray, D., and C. Thomas. 1976. Unpolymerized actin in fibroblast and brain. J. Mol. Biol. 16:1055-1069. 2. Kreis, T., Geiger, B. , Schlessinger, J. 1982. Mobility of microinjected rhodamine actin within living chicken gizzard cells determined by fluorescence photobleaching recovery. Cell. 29:835845. 3. Kreis, T., K. Winterhalter, and W. Birchmeier. 1979. In vivo distribution and turnover of fluorescently labeled actin microinjected into human fibroblasts. Proc.Natl. Acad. Sci. 76:3814-3818. 4. Sun, H., K. Kwiatkowska, and H. Yin. 1995. Actin monomer binding proteins. Curr Opin Cell Biol. 7:102-110. 5. Taylor, D., and Y. Wang. 1978. Molecular cytochemisty; Incorporation of fluorescently labeled actin into living cells. Proc.Natl. Acad. Sci. USA. 75:857-861. Actin Dynamics in the Cell Cytoplasm 3-17 Interpreting PAFIFRAP Measurements of Steady-State Actin Dynamics Works Cited 6. Theriot, J., and T. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126-131. 7. Theriot, J., and T. Mitchison. 1992. Comparison of actin and cell surface dynamics in motile fibroblasts. J. Cell Biol. 118:367-377. 8. Wang, Y. 1985. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J. Cell. Biol. 101:597-602. 9. Wang, Y. L. 1984. Reorganization of Actin Filament Bundles in Living Fibroblasts. The Journalof Cell Biology. 99:1478-1485. 10. Wegner, A. 1976. Head to tail polymerization of actin. J. Mol. Biol. 108:139-150. 3-18 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Works Cited Actin Dynamics in the Cell Cytoplasm 3-19 Interpreting PAF/FRAP Measurements of Steady-State Actin Dynamics Works Cited 3-20 CHAPTER 4 Baseline and control measurements in subconfluent endothelial cells Our mathematical description of the dynamics of the actin cytoskeleton (Chapter 3) describes how the evolution of actin fluorecence in photoactivated fluorescence (PAF) and fluorescence recovery after photobleaching (FRAP) experiments depends on the diffusion coefficient of actin monomer, D, the fraction of actin in filaments, FF, and the lifetime of actin filaments, u. Applying this model to interpret PAF and FRAP experiments in subconfluent bovine aortic endothelial cells (BAECs) produces the following parameters PAF: D = 3.1 ± 0.4 x 10-8 cm 2 /s, FF = 0.36 + 0.04, t = 7.5 + 2.0 min. FRAP: D = 5.8 ± 1.2 x 10-8 cm 2/s, FF = 0.5 ± 0.04, I = 4.8 ± 0.97 min. Differences in the parameters are attributed to differences in the actin derivatives employed in the two studies. Control experiments confirm that the evolution of fluorescence is dominated by the diffusion of actin monomer, and the turnover of actin filaments, but not by filament diffusion. The results also demonstrate that filament turnover is rapid, but not diffusion limited in the bulk cytoskeleton of endothelial cells. Actin Dynamics in the Cell Cytoplasm 4-1 Baseline and control measurements in subconfluent endothelial cells Introduction Introduction Actin assembly is the basis for shape changes during cell crawling, spreading and activation. A host of cytoplasmic regulators of actin assembly have been identified. Among these are monomer sequestering proteins (e.g., 034 thymosin, profilin) that prevent assembly at the "pointed" filament end (defined with respect to the stereospecific binding of myosin S1 to actin) and proteins that block monomer access to the fast growing or 'barbed' ends of actin filaments (e.g., capZ, gelsolin). The activities of actin associated proteins and the chemical environment of the cytoplasm results in a steady-state in which roughly half of the cellular actin is maintained in an unpolymerized form, with the remainder in a dynamic polymer phase. Cell crawling is achieved by coupling the polymerization of actin filaments in the protruding regions of the cell, with filament disassembly in regions of the cell being withdrawn. The lifetime of filaments in cytoplasm is therefore a key determinant of the rate at which cells crawl (Theriot and Mitchison, 1991, Theriot and Mitchison, 1992). Estimates of actin filament turnover in living cells (Finkel, et al., 1994, Theriot and Mitchison, 1991, Theriot and Mitchison, 1992, Wang, 1985) are 10 to 100 times faster than for purified actin in vitro (Wegner, 1976). Recently, independent efforts have demonstrated that ADF-cofilin accelerates the depolymerization of actin filaments 5-fold in Xenopus egg extracts infected with lysteria monocytogenes (Carlier, et al., 1997, Rosenblatt, et al., 1997) and by an order of magnitude in purified actin filaments (Carlier, et al., 1997). While these in vitro studies have helped resolve a critical disparity between the dynamics of actin in vivo and in vitro, it remains important to improve upon techniques for the assay of actin dynamics in living cells. 4-2 Baseline and control measurements in subconfluent endothelial cells Introduction The dynamics of actin in living cells has been observed using the techniques of fluorescence recovery after photobleaching (FRAP) and photoactivation of fluorescence (PAF). In these experiments a fluorophore-labeled actin derivative is microinjected into living cells where it incorporates into the native actin cytoskeleton (Amato, 1986, Kreis, et al., 1979). In FRAP, the fluorescence derived from the injected actin is quenched locally under intense laser excitation. In PAF, a focused band of UV excitation converts a non-fluorescent fluorophore derivative (a 'caged' fluorophore) back to its fluorescent parent (Mitchison, 1989). In both techniques the evolution of fluorescence in the illuminated region is monitored to infer information about actin dynamics. While there are theoretical advantages in the signal-to-noise characteristics of PAF compared to FRAP (Krafft, et al., 1986), the techniques are otherwise simple inverses of each other. FRAP experiments on rhodamine and fluorescein labeled actin have revealed two distinct mobilities of actin. The more mobile phase accounts for 20% to 80% of the total fluorescence recovery and has been interpreted as the diffusion of actin monomer, monomer/protein complexes and/or short filaments (Kreis, 1982, Luby-Phelps, 1985, Wang, et al., 1982). The less mobile phase has been attributed to filament remodelling via monomer exchange between filaments and the monomer phase (Wang, 1985). In contrast to FRAP studies, previous PAF experiments exhibit a single decay phase that has been attributed exclusively to filament turnover (Theriot and Mitchison, 1991, Theriot and Mitchison, 1992). The contrast in the behavior of PAF and FRAP experiments may be due to differences in cell types and the location of photoactivated/photobleached regions within cells; however differences in the PAF/FRAP techniques, or in the application and interpretation of these techniques, may also contribute. Actin Dynamics in the Cell Cytoplasm 4-3 Baseline and control measurements in subconfluent endothelial cells Introduction Traditionally, FRAP studies on the dynamics of cellular actin have been interpreted using the model of Axelrod et al., 1976. The Axelrod model considers a single diffusive species and an "immobile fraction" and thus is not strictly applicable to experiments on actin which has two dynamic components. The model in Chapter 3 describes the spatial and temporal evolution of a PAF experiment in which a diffusive monomer pool exchanges subunits with a non-diffusing polymer (Tardy, et al., 1995), providing a more appropriate framework for interpreting PAF and FRAP experiments. The interpretive model produces simultaneous measurements of three important quantities: the diffusion coefficients of actin monomer, D; the fraction of actin in filamentous form, FF; and the turnover time (characteristic lifetime) of actin filaments, t. This paper demonstrates application of our interpretive model (Chapter 3) to PAF and FRAP experiments in the bulk actin cytoskeleton of subconfluent bovine aortic endothelial cells (BAECs). Both PAF and FRAP experiments exhibit biphasic behavior with an early phase consistent with the diffusion of actin monomer and a second phase consistent with the turnover of actin filaments. Filament diffusion does not appear to contribute significantly to the evolution of fluorescence. Results indicate that actin filament turnover, t, is rapid (4-8 minutes) but not diffusion limited in the bulk cytoplasm of endothelial cells. Data are also consistent with previous observations that the mobility, D, of actin monomer differs between cysteine and lysine labeled derivatives (Giuliano and Taylor, 1994), and is hindered beyond expectations for a inert tracer of similar hydrodynamic radius (Luby-Phelps, 1987). 4-4 Baseline and control measurements in subconfluent endothelial cells Materialsand Methods Materialsand Methods Cell Culture The BAEC cell lines (BAEC-77) used in Boston for PAF studies were homogeneous, well charaterized, finite life-span strains of calf aoritc endothelial cells, generously supplied by Dr. M.A. Gimbrone (Vascular Research Division, Brigham and Women's Hospital, Boston). These cells were typically used for experiments at passages 5-15. BAECs used for FRAP studies in Lausanne were obtained from freshly excised calf aortas according to the protocol of Booyse et al., 1975. Cells were grown in Dulbecco's Modified Eagle Medium (DMEM) with 10% bovine calf serum, 50U penicillin/streptomycin (1:1), and 1% L-glutamine. BAECs maintain a highly motile phenotype while subconfluent. To ensure the motile phenotype, cells were plated at low density and examined within 2 days of plating. Time-lapse video microscopy of BAECs confirmed that > 90% of cells are motile under these conditions with root mean square (RMS) velocities of 0.53 ± 0.094 um/min (n = 26). Derivatized actin was microinjected into cells at between 5-15% cell volume, and allowed to incorporate for a minimum of 1 hour. Cells were maintained at 370 C in observation media (DMEM with no phenol red, 10mM HEPES, 10% bovine calf serum, 50U penicillin/streptomycin (1:1), 1% L-glutamine). Injected cells retained the ability to extend and retract lamella. Electron Microscoscopy BAEC monolayers on glass coverslips were permeabilized for 2 minutes at 370 C by adding a large excess of a solution composed of 0.06 M PIPES, 0.025 M HEPES, 0.01 M EGTA, 2 mM Actin Dynamics in the Cell Cytoplasm 4-5 Baseline and control measurements insubconfluent endothelial cells Materialsand Methods MgC12, 0.75% Triton X-100, 1 pgM phallacidin, 5.2 nM leupeptin, 1 nM benzamidine, and 12.3 nM aprotinin in PHEM buffer containing Triton X-100 (Schliwa, et al., 1981). The cytoskeletons were fixed for 10 minutes with PHEM containing 1% glutaraldehyde and 0.1 gM phallacidin. Cytoskeletons were rinsed with PHEM buffer, washed extensively with distilled water, rapidly-frozen on a helium-cooled copper block, freeze-dried at -800 C (Cressington CFE-50 freeze-fracture apparatus, Watford, England) and rotary coated with 1.4 nm of tantalum-tungsten at an application angle of 450 followed by 5 nm of carbon applied at 900 without rotation. The metal replicas were floated from the coverslips, washed in water, and picked up on carbon-formvar-coated copper grids. Replicas were viewed and photographed in a JEOL 1200-EX electron microscope using an accelerating voltage of 100 Kv. Actin preparation and injections Caged-resorufin iodacetamide (CRI) was prepared according to the method of Theriot and Mitchison, 1991 (Theriot and Mitchison, 1991) as described in Chapter 2. For FRAP studies, 5-(and 6) carboxyfluorescein succinimidyl ester labeled actin (CFSA) was purchased from Cytoskeleton (Denver, Colorado). CFSA is labeled at lysine and is >90% polymerization competent. From fluorimetery studies, we estimate that CFSA actin is 11% less fluorescent once polymerized at pH 7.5. This was not taken into account in the interpretation of FRAP data because the result could not be verified in cells. If the fluorescence difference does occur in cells, FRAP estimates of FF would be systematically underestimated by 10%, whereas estimates of D and t would be biased by less than 1%. 4-6 Baseline and control measurements in subconfluent endothelial cells Materials and Methods PAF/FRAP Experiments PAF studies were conducted under a modified epi-fluorescence microscope as described in Chapter 2 using a 63X plan neofluor objective. For FRAP experiments, a computer-controlled laser scanning microscope (LSM410, Zeiss, Germany) was programmed to generate a bleaching/imaging sequence. A 15 mW Argon laser (488 nm) was passed through a 63x Zeiss plan neofluor objective. The laser was set to 50% maximal power for bleaching and 2% maximal power for imaging. In 300 ms the laser bleached a 3 gtm - 7 gtm rectangular band that spanned one dimension of the cell. The time lag between the bleaching period and the first image was 40 ms. Bleaching and imaging were accomplished by raster scanning the laser across the desired areas. A 10% transmission neutral density filter and a 515 nm high pass filter was positioned in front of a photomultiplier that recorded the fluorescence as the laser scan was performed. Images were stored and analyzed with the software controlling the microscope. An average fluorescence was computed in the center-line of photobleached bands (approximately 250 pixels high (= 25 jim) x 6 pixels ( = 0.5 jim) wide). Photobleaching controls and corrections The fluorophore resorufin is highly photolabile under the light levels required to detect it in cells with our intensified CCD. In order to minimize photobleaching contributions to the fluorescence decay in PAF experiments, the excitation light levels were minimized, the number of data points was limited to 10, and the excitation light was precisely shuttered so that the sample was exposed to light for only 80 ms per data point. To estimate contributions from photobleaching, a control experiment was conducted immediately after a PAF experiment. In the control experiments the entire contents of a cell were uncaged to eliminate the local gradients that generate decay due to Actin Dynamics in the Cell Cytoplasm 4-7 Baseline and control measurements insubconfluent endothelial cells Materialsand Methods diffusion and/or turnover. Images were then acquired with the same exposure times and light intensities as in the experiment. The fluorescence in the region defining the original band center-line was monitored to infer the photobleaching contribution. As shown in Figure 4.1a the photobleaching error accumulated with each exposure and was generally not negligible. When the control experiment is processed through the corrective algorithm given in the Appendix the effects of bleaching are removed. When the actual experiment is processed through the control, the correction of the data is slight but is important to a quantitative analysis. The correction is particularly important in estimating the long-term dynamics where the error accumulates. The photobleaching correction assumes that under continuous illumination photobleaching curves are well approximated by an exponential decay. This behavior is demonstrated for resorufin in Figure 4. lb. At the conclusion of a FRAP experiment a sequence of images was acquired in which the entire area of the cell was scanned under imaging conditions. The duration of the scan was equivalent to the scanning time used during the actual experiment. The fluorescence in the region of the recovered band was monitored as a photobleaching control. The photobleaching correction algorithm in the Appendix was applied to FRAP data; however, in most cases photobleaching corrections to FRAP data were negligible. Analysis Fluorescence decay curves were normalized with respect to initial fluorescence levels and fit to the interpretive model in Chapter 3. The extraction of geometric parameters for use in themodel is discussed next. 4-8 Baseline and control measurements insubconfluent endothelial cells Materials and Methods Geometry Considerations A rectangular photoactivated or photobleached band was employed in previous PAF studies (Mitchison, 1989, Theriot and Mitchison, 1991, Theriot and Mitchison, 1992). A 3 jtm - 8 jtm wide strip illuminates 5-20% of the total cellular area of a typical endothelial cell. This leads to a measurable change in the fluorescence throughout the cell as the photoactivated or photobleached band is homogenized over time. The interpretive model assumes a cell of finite dimension and thus accounts for this rising (PAF) or decaying (FRAP) background. For convenience the geometry assumed in the model is that of a rectangular band of fluorescent actin within a bounded rectangular cell. While to a good approximation both the PAF and FRAP experimental set-ups generate rectangular bands within the cell, the assumption of a rectangular cell is not realized. In order to use the one-dimensional model to interpret PAF experiments in arbitrarily shaped cells it is necessary to estimate an equivalent cell length, Leq. Because the ratio A/L is the normalized fluorescence as t - o , we define Leq as Leq = , (4.1) where 0o is the width of the band, Ac is the plan view area of the cell, and As is the plan view area of the uncaged band. Actin Dynamics in the Cell Cytoplasm 4-9 Baseline and control measurements in subconfluent endothelial cells Materialsand Methods * +x 0.8 > A 0.6 Do * 0 0.4 o o 0.2 0 100 200 300 400 Figure 4.1 Photobleachingand background controlsin PAF experiments. After a PAF experiment,microinjected actin throughoutthe cell is uncagedand a second series of exposures is taken. The decay offluorescence in the region of the originalband is measuredas a photobleachingcontrol, a)Raw PAF data (*), correctedPAF data (photobleachingcontrol (A), correctedcontrol (+), and background controlto insurestable excitation (X). The effects ofphotobleachingare completely removedfrom the photobleachingcontroland the experimentaldata using the corrective algorithm of the Appendix. The corrective algorithm assumes an exponential photobleachingdecay during the 80 ms sampling exposures. b) Demonstrationthat under continuous illuminationresorufin photobleachingis well approximatedby a decaying exponential. time (sec) a) 110 100 time constant = 4.99 sec 90 Jasplakinolide and cytochalasin D treatments To demonstrate that PAF and FRAP experiments were sensitive to monomer diffusion and the cyclic turnover of filaments, experiments were conducted in the pres- time (sec) ence of the membrane permeant polymerizing agent jasplakinolide (Jas) (Bubb, et al., 1994) (Molecular Probes, Eugene, Oregon) and the filament barbed end capping agent cytochalasin D (Cyto D) (Sigma Chemical Corp). PAF experiments were conducted in cells incubated with 1 gM Jas for 20 minutes. FRAP experiments were conducted in cells incubated with 2 gLM Cyto D for 20 minutes. In 4-10 Baseline and control measurements in subconfluent endothelial cells Results Figure 4.2 Electron micrographsshowing the region of BAECs studied by PAF and FRAP Stereo view shows uniformity in the cytoskeleton in this region. The height of the bottom panel is the nominal width of photoactivatedand photobleachedregions (7pm). Photoactivatedandphotobleached bands traverse the body of the cell, and an averagefluorescence is measuredalong the centerline. Bar = 1 pm. both cases changes in cell shape occurred and were taken into account in the analysis. The purpose was to strongly perturb the dynamic state of the actin cytoskeleton. The long term viability of the cells was not a concern. Results Both PAF and FRAP experiments exhibit biphasic decay consistent with the presence of two phases of actin Subconfluent endothelial cells display a homogeneous actin cytoskeleton in the cell body (Figure 4.2). PAF and FRAP experiments were conducted in the cell body using 3 Rim - 8 gm wide photoactivated/photobleached bands that spanned one dimension of the cell. Both techniques reveal the presence of two dynamically distinct populations of actin. The initial response Actin Dynamics in the Cell Cytoplasm 4-11 Baseline and control measurements in subconfluent endothelial cells Results a b 0.5 1 b 00 LL 0.5 U 0 0 200 t (sec) 0 Figure 4.3 PAF and FRAP experiments in BAECs. a) PAF data and associated image sequence. The decay of fluorescence from the photoactivated band is accompanied by a rise in fluorescence elsewhere as monomer diffuses throughout the cell. b) FRAP data and associated image sequence. Both PAF and FRAP data are biphasic, confirming the presense of two dynamic phases. Bars = 10 pm. occurs on a time scale of 10 seconds sec while the long-term decay occurs on the order of several minutes. Example PAF and FRAP images sequences are shown in Figure 4.3. Biphasic behavior is seen in 10 sec the center-line fluorescence data shown in Figure 4.3a (upper right inset) and 4.3b = 30 sec 3* (upper inset). The diffusive nature of the - early response is best seen in PAF images of Figure 4.3a where the fluorescence spreads rapidly from the band throughout the cell. 180 sec Estimates of D, FF, and t The evolution of fluorescence in PAF and FRAP experiments was biphasic indicating the presence of two dynamically distinct popula- 4-12 Baseline and control measurements in subconfluent endothelial cells Results Figure4.4 CorrectedPAF and FRAP data. a) CorrectedPAF data and a curve generatedby the averagePAFparameter values in Table 4.1 Also shown is the average evolution offluorescence in 7 cells incubated with Jasfor 20 minutes priorto photoactivation.The error bars indicatemean ± sem at the respective time points. Jas treatment nearly eliminates the diffusive phase, stabilizes filaments, and results in a rise in fluorescence over time due to contraction of the uncaged actin band. b) FRAP data and curve produced by the average parametervalues in Table 4.1 (solid curve). Also is a curve producedfrom the average parameterestimates obtainedin cells exposed to Cyto Dfor 20 minutes (Table 4.2 before a FRAP experiment (dashedcurve). Cytochalasindecreases the fraction of actin in filaments and slows the turnover offilaments. 1.5 a 11 ^ Sshown F/Fo # 0.5 AY# # tions of actin. Corrected PAF and FRAP data are 0 0 100 200 300 time (sec) 400 shown in Figures 4.4a and 4.4b respectively. Each corrected experiment was fit to the interpre- a) tive model to determine D, FF and t. All correla0.8 \ tion coefficients exceeded 0.91. The parameters for PAF are: D = 3.1 ± 0.4 x 10-8 cm 2 /s (n = 20), 0.6 F I/Fo 0.4 -X X x X FF = 0.36 + 0.04 (n = 19), t = 7.5 ± 2.0 min (n = X 17); and for FRAP are: D = 5.8 ± 1.2 10-8 cm 2 /s (n = 25), FF = 0.50 + 0.04 (n = 26), and t= 4.8 + 0.2_ 50 100 time (sec) b) 150 200 1.0 min (n = 26). The differences in the PAF and FRAP estimates of D, FF and T are statistically significant (PD < 0.0005, PFF < 0.0005, P, < 0.025). For each study a simulated experiment was generated using the model in Chapter 3 and Actin Dynamics in the Cell Cytoplasm 4-13 Baseline and control measurements in subconfluent endothelial cells Results average parameter values. These simulations are plotted along with the data in Figure 4.4 to demonstrate the quality of the model fit. PAF and FRAP experiments are sensitive to jas and cyto D To confirm that PAF and FRAP techniques were detecting the diffusion of sequestured monomer and the turnover of filaments studies were conducted in the presence of the polymerizing cytotoxin jas and the Figure 4.5 Effects ofjasplankinolide. a) PAF sequence barbed end capping agent cyto D. Jas the band at short times demonstrates the conversion of induces actin polymerization with an second cell showing the long-term stability of actin filaments with Jas. Jas stabilizes filaments, but induces efficiency and mechanism similar to after treating cells with 1fpM Jas. The increased stability of diffusive monomer to filaments. b) PAF sequence in a bundling and contraction leading to a rise in the fluorescence in the photoactivated band over time. Bars = 1O in. that of phalloidin (Bubb, et al., 1994), but is also cell permeant. After a 20 minute incubation in 1 pM Jas, actin in photoactivated bands is immobile on short time scales (compare Figures 4.5a and 4.3a). The images in Figure 4.5b are typical of the long-term behavior of cells exposed to l1LM Jas. Jas stabilizes filaments and induces a contraction in the actin cytoskeleton. The average trend exhibited by 7 PAF experiments conducted in cells pretreated with Jas is shown in 4-14 Baseline and control measurements insubconfluent endothelial cells Discussion Figure 4.4a. The average decay of fluorescence in the first 30 seconds is less than 12%. A short time after photoactivation the fluorescence in the band begins to rise due to an apparent contraction of the photoactivated band. The rising fluorescence precludes an analysis of these experiments with the interpretive model. FRAP experiments were conducted in cells before and after exposure to 2 gM Cyto D for 20 minutes. Table 4.2 lists the results of fitting the interpretive model to each of the experiments. The average parameters before and after cytochalasin treatment are (n = 6): D + 3.1 ± 1.62 x 10- 8 cm 2/ s, FF = 0.42 ± 0.10, t = 4.2 ± 1.9 min; and D = 3.5 ± 1.7 x 10-8 cm 2 /s, FF = 0.38 ± 0.09, and x = 20.9 ± 18.6 min. A paired t-test indicated that the decrease in FF and increase in t are statistically significant (PFF = 0.059 and P, = 0.043) but the slight increase in D is not. A simulated experiment was generated using the mean parameter values after treatment with Cyto D in the interpretive model. This experiment is shown in Fig. 4b for comparison with the data from untreated cells. Discussion Cellular actin partitions between a monomer phase, maintained by sequestering proteins such as (34 thymosin and profilin, and a dynamic filamentous phase. The dynamics of an ideal actin tracer should be sensitive to the diffusion of actin monomer, the ratio of monomeric to filamentous actin, the dynamic exchange of monomer between filaments and sequestering proteins, and the diffusion of actin filaments. Because of the high degree of cross-linking exhibited by actin networks in cells (Hartwig and Shevlin, 1986), filament diffusion is predicted to be the smallest contributor to tracer dynamics. With these assumptions, the interpretive model (Tardy, et al., 1995) describes the steady-state dynamics of the actin cytoskeleton and predicts the theoretical evolution of fluores- Actin Dynamics in the Cell Cytoplasm 4-15 Baseline and control measurements in subconfluent endothelial cells Discussion cence in a PAF experiment. Here the interpretive model is applied to PAF and FRAP experiments in BAECs to give the first simultaneous measurement of three important parameters in living cells: the diffusion coefficient of actin monomer D; the fraction of actin in filamentous form, FF; and the lifetime, T, of actin filaments. PAF and FRAP experiments in BAECs are biphasic, indicating the presence of two distinct dynamic components. Experiments conducted in the presence of membrane permeant reagents confirm that these phases are due to monomer diffusion and filament turnover, and not to filament diffusion. First, the high mobility phase in PAF studies was nearly eliminated when cells were pretreated for 20 minutes with 1 plM of the actin polymerizing reagent Jas, demonstrating that the bulk of this phase is polymerization competent monomer. Secondly, Cyto D increased the mean estimate of t 5-fold in FRAP experiments, a result expected for filament turnover but not for filament diffusion. Because cytochalasins block monomer access to the growing (barbed ends) of actin filaments while leaving the depolymerizing (pointed) ends of actin filaments free (Cooper, 1987), depolymerization of filaments proceeds until a new equilibrium is achieved between pointed ends and monomer. Filament turnover via monomer exchange at a single filament end is predicted to be much slower than when both ends are free (Wegner, 1976). Thus the increase in r with Cyto D is consistent with the assumption that the dynamics of the low mobility phase is governed by filament turnover. By contrast, filament depolymerization in the presence of cytochalasin should shorten filaments and increasethe mobility of the second phase if significant filament diffusion were present. The following values apply for the bulk actin cytoskeleton in BAECs. PAF: D = 3.1 ± 0.4 x 10-8 cm 2 /s, FF = 0.36 ± 0.04, T = 7.5 + 2.0 min. FRAP: D = 5.8 ± 1.2 x 10-8 cm 2 /s, FF = 0.5 4-16 Baseline and control measurements in subconfluent endothelial cells Discussion ± 0.04, T = 4.8 0.97 min. The differences in the parameters obtained are statistically significant. Cys-374 labeling of actin (as in CRIA for PAF) results in a derivative with a 10-fold lower affinity for profilin than lysine labeled derivatives (as in CSFA for FRAP) (Giuliano and Taylor, 1994), and may interfere with the binding of other actin associated proteins (Kabsch and Vandekerckhove, 1992). Giuliano and Taylor, 1994 directly compared the mobility of lysine and cys-374 labeled actin in FRAP experiments in fibroblasts. The estimates of CFSA and CRIA mobility are in good agreement with their values for lysine and cys-374 labeled actin respectively: Dlysine = 5.6 ± 1.1 x 10-8 cm 2/s (n = 10) and Dcys-374 = 3.8 + 0.85 x 10-8 cm 2/s (n = 10). Also, the filament fractions for CFSA and CRIA agree with the immobile fraction (IF) estimates of Giuliano and Taylor for lysine and cys-374 labeled actin in the peripheral cell body of fibroblasts: IFlysine = 0.48 ± 0.04 (n = 12) and IFcys- 374 = 0.34 ± 0.05 (n = 13). These agreements suggest the discrepancies between PAF and FRAP can be largely attributed to differences in the location of fluorophore on actin and not to inherent differences in the techniques. The results indicate that lysine labeled actin, diffuses faster in the cytoplasm, incorporates more readily into the native cytoskeleton, and cycles through filaments faster than cys-374 labeled actin. Previously, monomer diffusion coefficients were derived from short-term FRAP data (Kreis, 1982, Luby-Phelps, 1985, Taylor and Wang, 1979, Wang, et al., 1982) using the protocol of Axelrod et al., 1976. The theory behind the Axelrod protocol considers a inert diffusive phase and a second "immobile" phase and is not strictly applicable to actin which has two interacting populations. Estimates of D derived from the Axelrod analysis (Giuliano and Taylor, 1994, Luby-Phelps, 1985) are at least 4 times lower than would be expected for an inert particle with the same hydrodynamic radius as monomeric actin (Luby-Phelps, 1987). Because the Axelrod model does not Actin Dynamics in the Cell Cytoplasm 4-17 Baseline and control measurements in subconfluent endothelial cells Discussion consider filament turnover, an analysis by this model would produce underestimates in D if monomer diffusion was hindered by the cyclic incorporation of monomer into filaments. The interpretive model accounts for the influence of filament turnover, yet produces the same estimates for D. This result indicates that filament turnover is too slow to hinder monomer diffusion in the bulk cytoskeleton of BAECs 1 or, equivalently, that filament turnover is not diffusion limited. Filament turnover times in FRAP or PAF experiments have been estimated from half-lives for the total recovery or decay of fluorescence (Theriot and Mitchison, 1991, Theriot and Mitchison, 1992, Wang, 1985). Because actin is equally distributed between monomer and filaments in cells (Bray and Thomas, 1976), the half-life of fluorescence in a PAF or FRAP experiment may not accurately reflect the time scale of either monomer diffusion or filament turnover. For this reason it is not possible to strictly compare the turnover times here to those obtained previously2 . However, estimates of t agree with FRAP measurements of filament turnover in the lamellapodia of motile fibroblasts (Wang, 1985) and with recent PAF estimates in the cell body of fibroblasts (Cramer, et al., 1997) to within a factor of 2. Our measurements are 1 - 7 times slower than PAF estimates of r in the lamellapodia of fibroblasts (Theriot and Mitchi- 1. For hindrance, the characteristic time of monomer diffusion out of the band must be comparable to 2 or slower than the filament turnover time. This condition holds for = ~-D1, where (o is the photoactivated band width, D is the diffusion coefficient of actin monomer, and r is the turnover rate of actin filaments. 2. Furthermore, our turnover times are not half-lives, but are more accurately thought of as l/e times or the time for the fluorescence due to filaments to decay to 37% of its original values. 4-18 Baseline and control measurements in subconfluent endothelial cells Discussion Table 4.1: Parameter estimates from PAF and FRAP in BAECs. Uncertainties shown are the standard error of the means. The differences displayed between the two studies are statistically significant with the following P values obtained from a unpaired t test. PD < 0.0005, PFF < 0.0005, P, < 0.025. These differences are attributable to differences in the actin derivative employed with each technique. Dxl08cm 2/s FF T (min) PAF (CRIA) 3.1 ± 0.4 (n = 20) 0.36 ± 0.04 (n = 17) 7.5 ± 2.0 (n = 17) FRAP (CFSA) 5.8 ± 1.2 (n = 25) 0.5 ± 0.04 (n = 26) 4.8 ± 0.97 (n = 26) Table 4.2: Dynamic parameters in cells before and after treatment with Cyto D. The differences in FF and r are statistically significant while the slight increase in D with treatment cytochalasin treatment is not (PFF = 0.043 and P, = 0.059 determined from a paired t-test). Baseline After 20 minutes in 2uM Cyto D D x 108 cm 2/s FF r (min) Dx 10cm/s FF cell 1 3.3 0.38 4.5 2.4 0.30 13.2 cell 2 3.0 0.32 6.4 6.6 0.38 10.3 cell 3 2.0 0.38 2.2 3.8 0.36 6.0 cell 4 6.2 0.40 3.2 3.9 0.28 57 cell 5 2.1 0.57 2.2 2.1 0.52 26 cell 6 2.0 0.52 6.4 2.0 0.44 13.6 mean ± sem 3.1 ± 1.62 0.42 ± 0.10 4.2± 1.9 3.5 ± 1.7 0.38 ± 0.09 21 108cm2/S Actin Dynamics in the Cell Cytoplasm (min) 9 4-19 Baseline and control measurements insubconfluent endothelial cells Appendix son, 1992) and an order or magnitude slower than PAF estimates in the lamellapodia of highly motile keratocytes (Theriot and Mitchison, 1991). The experiments here establish the general equivalency of the PAF and FRAP techniques when applied to the same cell location and the same cell type. In contrast to FRAP studies in the cell body, however, previous PAF experiments in the lamellapodia of fibroblasts and keratocytes exhibit only a single dynamic component (Theriot and Mitchison, 1991, Theriot and Mitchison, 1992). A single phase decay in a PAF experiment would indicate that only trace amounts of monomer exist or that the exchange of subunits between monomer and filaments occurs so rapidly that the two phases decay simultaneously1 . The = 30 s filament turnover times reported in the lamellapodia of highly motile keratocytes by Theriot and Mitchison (Theriot and Mitchison, 1991) are 2 - 4 times longer than monomer diffusion times from the 2.5 pm photoactivated band used. Even in this case a biphasic decay of fluorescence would be expected if significant sequestered monomer were present. Thus, the disparity between the PAF experiments of Theriot and Mitchison and other studies may indicate a difference in the ratio of sequestured to polymerized actin between the cell body and the lamellapodia. Appendix The photobleaching process has been successfully modeled as a first order reaction with a rate constant proportional to the excitation intensity, I (Axelrod, et al., 1976) 2 1. This is the case of diffusion limited filament turnover. This condition holds for 3, = 4-20 >> 1. Baseline and control measurements insubconfluent endothelial cells Appendix dF dt -lF EA4.1 where F is the fluorescence, and 1Tis a proportionality constant. The final fluorescence of a sample during an exposure to excitation light is given by At Ff = (Fi - F , )e + F. EA4.2 where At is the duration of the exposure, Fi is the sample fluorescence at the beginning of the exposure, and Ff is the sample fluorescence at the end of the exposure. The sample photobleaching time constant t and the unbleachable background fluorescence F. are estimated from the sample photobleaching decay curve under continuous illumination (see Fig. lb). The corrected initial fluorescence is given by F At EA4.3 + F. e During an experiment the effects of photobleaching will accumulate. Defining the drop in fluorescence due to photobleaching during the jth exposure interval as AFj = Fij - Ffj, the fluorescence at the beginning of the nth (n 2 j) exposure interval is corrected for all previous exposures with n F, = f, n e +F + e AF EA4.4 j=1 Actin Dynamics in the Cell Cytoplasm 4-21 Baseline and control measurements in subconfluent endothelial cells Works Cited Equation EA4.4 is the correction algorithm. If the photobleaching is well characterized by t and F*, substituting measured data (a series of Ff's), in EA4.4 produces a series of fluorescence values (a series of Fi's) that are void of photobleaching effects. Works Cited Amato, P. A. T., D.L. 1986. Probing the mechanism of incorporation of fluorescently labeled actin into stress fibers. J. Cell Biol. 102:1074-1084. Bray, D., and C. Thomas. 1976. Unpolymerized actin in fibroblast and brain. J. Mol. Biol. 16:1055-1069. Bubb, M. R., M. Adrian, J. Senderowiez, E. A. Sausville, L. Kimberly, K. Duncant, and E. D. Korn. 1994. Jasplankinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 269:14869-14871. Carlier, M.-F., V. Laurent, J. Santolini, R. Melki, D. Didry, G.-X. Xia, Y. Hong, N.-H. Chua, and D. Pantaloni. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin based motility. J. Cell Biol. 136:1307-1322. Cooper, J. 1987. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 105:1473-1478. Cramer, L. P., M. Siebert, and T. J. Mitchison. 1997. Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: implications for the generation of motile force. J. Cell Biol.:1287-1305. Finkel, T., J. A. Theriot, K. D. Dise, G. F. Tomaselli, and P. J. Goldschmidt-Clermont. 1994. Dynamic actin structures stabilized by profilin. Proc. Natl. Acad. Sci. USA. 91:1510-1514. Giuliano, K. A., and D. L. Taylor. 1994. Fluorescent actin analogs with a high affinity for profilin in vitro exhibit an enhanced gradient of assembly in living cell. J. Cell Biol. 124:971-983. 4-22 Baseline and control measurements insubconfluent endothelial cells Works Cited Hartwig, J. H., and P. Shevlin. 1986. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J. Cell. Biol. 103:10071020. Kabsch, W., and J. Vandekerckhove. 1992. Structure and function of actin. Annu. Rev. Biophys. Biomol. Struct. 21:49-76. Krafft, G. A., J. P. Cummings, L. J. Dizig, W. R. Brven, W. R. Sutton, and B. R. Wars. 1986. Fluorescence activation and photodissipation (FDP). In Nucleocytoplasmic Transport. Springler-Verlag, New York. 35-52. Kreis, T., Geiger, B. , Schlessinger, J. 1982. Mobility of microinjected rhodamine actin within living chicken gizzard cells determined by fluorescence photobleaching recovery. Cell. 29:835-845. Kreis, T., K. Winterhalter, and W. Birchmeier. 1979. In vivo distribution and turnover of fluorescently labeled actin microinjected into human fibroblasts. Proc.Natl. Acad. Sci. 76:3814-3818. Luby-Phelps, K., Lanni, F., Taylor, D.L. 1985. Behavior of a Fluorescent Analogue of Calmodulin in Living 3T3 Cells. J. Cell Biol. 101:1245-1256. Luby-Phelps, K., Castle, P.E., Taylor, D., and Lanni, F. 1987. Hindered diffusion of inert tracer particles in the cytoplasm of mouse 3T3 cells. Proc.Natl. Acad. Sci., USA. 84:4910-4913. Mitchison, T. 1989. Polewards microtubule flux in the mitotic spindle: evidence from photoactivation of fluorescence. J. Cell Biol. 109:637-652. Rosenblatt, J., B. Agnew, H. Abe, J. Bamburg, and T. Mitchison. 1997. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the urnover of actin filaments in Listeriamonocytogenes tails. J. Cell Biol.:1323-1332. Schliwa, M., J. van Blerkom, and K. Porter. 1981. Stabilization of the cytoplasmic ground substance in detergent-opened cells and a structural and biochemical analysis of its composition. Proc.Natl. Acad. Sci., USA. 78: 4329-4333. Actin Dynamics in the Cell Cytoplasm 4-23 Baseline and control measurements in subconfluent endothelial cells Works Cited Spudich, J. A., and S. Watt. 1971. The regulation of rabbit skeletal muscle contraction. J. Biol. Chem. 246:4866-4871. Tardy, Y., J. McGrath, J. Hartwig, and C. Dewey Jr. 1995. Interpreting photoactivated fluorescence microscopy measurements of steady-state actin dynamics. Biophys. J. 69:1674-1682. Taylor, D., and Y. Wang. 1979. Fluorescenctly labelled molecules as probes of the structure and function of living cells. Nature. 284:405-410. Theriot, J., and T. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126-131. Theriot, J., and T. Mitchison. 1992. Comparison of actin and cell surface dynamics in motile fibroblasts. J. Cell Biol. 118:367-377. Wang, Y. 1985. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J. Cell. Biol. 101:597-602. Wang, Y., F. Lanni, P. McNeil, B. Ware, and D. Taylor. 1982. Mobility of cytoplasmic and membrane-associated actin in living cells. Proc.Natl. Acad. Sci., U.S.A. 79:4660-4664. Wegner, A. 1976. Head to tail polymerization of actin. J. Mol. Biol. 108:139-150. 4-24 CHAPTER 5 Correlation of actin dynamics with cell motility and with expression of actin associated proteins The dynamics of actin correlate with cell motility in four distinct bovine aortic endothelial cell (BAEC) phenotypes, demonstrating that cells of a single type can alter the dynamic state of the actin cytoskeleton as they become more motile. The most motile BAECs turnover actin filaments quickly (- 6 minute lifetimes) and have most of their actin in a diffusive pool (- 60% of total actin). The slowest BAECs turnover actin filaments slowly (- 40 minute lifetimes) and have less actin in a diffusive pool (-20% of total actin). The BAEC correlations extend to primary mouse fibroblasts both positive and null for gelsolin, suggesting that the capping and severing activities of gelsolin can be regulated to alter cell speed. The loss of the actin gelation protein ABP-280 from human melanoma cells slows motility without affecting filament turnover, demonstrating that the regulation of actin dynamics and architectural mechanisms independently alter cell motility. Actin Dynamics in the Cell Cytoplasm 5-1 Correlation of actin dynamics with cell motility and protein expression Introduction Introduction Cytoplasmic actin distributes between a monomeric phase maintained by sequestering proteins such as I4 thymosin and profilin, and a dynamic filamentous phase. When cells crawl they recruit actin from the unpolymerized phase for assembly into filaments near the expanding cell periphery. For steady crawling, monomers required for polymerization must derive from depolymerizing filaments elsewhere in the cell. Thus, the finite lifetime of actin filaments, the diffusivity of actin in the sequestered monomer pool, and the distribution of actin between polymerized and unpolymerized phases are predicted to be key parameters in cell motility. These three parameters can be obtained simultaneously in a living cell using fluorescence recovery after photobleaching (FRAP) or photoactivated fluorescence (PAF) (Chapter 4). Previous studies demonstrated that the lifetime of filaments in highly motile fish epithelial keratocytes is 10 times shorter (Theriot and Mitchison, 1991) than in less motile fibroblasts (Theriot and Mitchison, 1992;Wang, 1985). We questioned if this trend applied to cells of the same type moving at different rates. We used PAF to measure the dynamics of actin in endothelial cells migrating at different rates within a wounded endothelial monolayer. Correlating actin dynamics with cell speeds demonstrates that endothelial cells transfer actin from filaments to sequestering proteins and turnover filaments faster as they move faster. The endothelial cell correlation between motility and actin dynamics applies to fibroblasts both positive and null for gelsolin and to melanoma cells. This suggests a common regulation of actin dynamics controls cell motility in disparate cell types and implicates the capping and severing 5-2 Correlation of actin dynamics with cell motility and protein expression Methods activities of gelsolin in this regulation. Data from melanoma cells deficient in the actin gelation protein ABP-280 deviate from the endothelial correlations, demonstrating cytoskeletal architecture and actin dynamics can influence cell motility independently. Methods Cell culture and motility analysis BAECs employed in PAF studies were cultured and prepared for study as described in Chapter 4. Gelsolin null and wild-type fibroblasts were generously provided by Drs. T. Azuma and DJ Kwiatkowski. Cell speeds were determined with time-lapse video microscopy. Wounded monolayers, (BAECs and melanoma cells) were recorded for 10-25 hours and subconfluent cells, (BAECs and fibroblasts) were recorded for 4-8 hours. Melanoma cells at the edge of wounded monolayers were studied. RMS cell speeds were determined by fitting the mean square nuclear displacements of 20-30 cells of each type to a formula describing cell dispersion as a function of time and cell persistence (Othmer, et al., 1988). PAF and FRAP analysis PAF experiments were conducted as described in Chapter 2 and FRAP experiments as described in Chapter 4. FRAP estimates of filament lifetimes in subconfluent and confluent cells agree well with the PAF estimates demonstrating that inadvertent photobleaching has been appropriately controlled in PAF experiments. Intravital staining of plated cells in PAF experiments was done Actin Dynamics in the Cell Cytoplasm 5-3 Correlation of actin dynamics with cell motility and protein expression Results with 5-chloromethylfluorescein diacetate (CMFDA - Molecular Probes, Eugene, OR.) Results Actin dynamics correlate with cell motility in a wounded endothelial monolayer After overnight recovery, wounded bovine aortic endothelial cell (BAEC) monolayers display distinct morphological zones (Gotlieb, et al., 1983) (Figure 5.1a). Far from the wound, cells exhibit the polygonal morphology typical of cells in a confluent endothelial monolayer (Figure (5.1a, zone 1). Cells adjacent to the confluent region are elongated and aligned perpendicular to the wound edge (5. l1a, zone 2). Cells at the edge of the wound have the stereotype fanshape configuration of locomoting cultured cells (5.1 a, zone 3). Root-mean-square (RMS) cell speed monotonically decreases with increasing distance from the wound (Figure 5.1b). All three cell monolayer phenotypes are slower than subconfluent BAECs (i.e. not contacting their neighbors as in zone 4) characterized in Chapter 4. The evolution of fluorescence in photoactivated fluorescent (PAF) bands of actin depends on the mobility of actin monomer, the fraction of total actin in filaments, and the lifetime of actin filaments (Chapter 3). PAF experiments reveal a different dynamic state of actin in BAEC phenotypes (Figure 5.2 and Figure 5.3). The characteristic filament lifetime is 38.9 ± 11.4 (n = 11) minutes in confluent cells and decreases to 7.5 + 2.0 (n = 17) in subconfluent cells. The fraction of actin polymerized is 0.73 ± 0.11 (n = 13) in confluent cells and decreases to 0.36 + 0.04 (n = 19) in subconfluent cells. The mobility of actin monomer increases from 2.3 ± 0.8 x 5-4 Correlation of actin dynamics with cell motility and protein expression Results 10-8 cm 2 /sec in confluent cells to 3.2 ± 0.5 x 10-8 cm 2 /sec in subconfluent cells (the trend including all morphologies is significant, p <0.05). PAF and FRAP experiments agree in subconfluent and confluent endothelial cells FRAP estimates of filament turnover and . 0- ) 6 E 0.4 subconfluent wound edge 0 elongated C.D 0.2 0 confluent polymer fraction in subconfluent and confluent cells agree well with the estimates from PAF experiments. This comparison controls for photobleaching artifacts and for effects due to the fluorescent derivitization of actin (Chapter 4). FRAP experiments V position in monolayer FIGURE 5.1. Relationship between BAEC morphology and average cell speeds. a) Recovering wounded BAEC monolayers exhibit four distinct cell morphologies: subconfluent cells (not contacting neighbors) move in various directions; polarized wound edge cells extend lamellae into the wound; elongated cells align perpendicular to the wound; and the cobblestone morphology typical of confluent BAECs isfoundfarfrom the wound. b) Each BAEC phenotype has a distinct RMS cell speed. Subconfluent BAECs were characterized in Chapter 4. Error bars are SEMs with 20 < n < 50. also measure = 30% reduction in monomer mobility comparing confluent cells to subconfluent cells (p < 0.05). A 2 fold higher magnitude for D in FRAP compared to PAF is likely due to different chemical properties of actin labeled at lysine (in FRAP) vs. cysteine (in PAF) (Giuliano and Taylor, 1994). Actin Dynamics in the Cell Cytoplasm 5-5 Correlation of actin dynamics with cell motility and protein expression Results t 42 ti = 3 l) wound edge confluent FIGURE 5.2. PAF images sequence demonstratingdifferent actin dynamics in cells in the a) wound edge and b) confluent regions of a recoveringBAEC monolayer. Times (in sec) afterphotoactivationare indicated.Intravitalfluorescencestainingof all cells in the lowerpanels shows the overall cell morphology at the wound edge (polarized)and in the wound interior(polygonal). The data yields three physically meaningful correlations (Figure 5.4). First, the turnover rate (the reciprocal of the average filament lifetime) of actin filaments in BAECs correlates positively with RMS cell speeds (Figure 5.4a). While previous studies of filament turnover dem- 5-6 Correlation of actin dynamics with cell motility and protein expression Results onstrate that highly motile keratocytes x 10-8 turnover actin filaments in lamellipodia 10 times faster than less motile fibro- C)_ a1) blasts, the result here is the first exam- E ple of individual cells accelerating filament turnover to increase motility. 0tsj 1 Second, cell speed inversely correlates aC 0.8 with the fraction of actin polymerized E C 0 E 0.6 (Figure 5.4b). This result implicates 0.4 mechanism(s) that accelerate turnover 0 while simultaneously decreasing the a. 0.2 fraction of actin polymerized. Third, 0 the 30% decrease in monomer mobility 50 40 occurs concurrently with a two fold 30 increase in polymer fraction (Figure 20 5.4c). It is not known why actin mono- 10 mer diffuses in cytoplasm slower than 0 inert particles of the same size (Luby- 8 0 Phelps, et al., 1988). The sensitivity of monomer mobility to polymer fraction FIGURE 5.3. Cytoplasmic actin dynamics correlate with BAEC morphology. The mobility of the diffusive actin phase, D, the fraction of actin polymerized, and the characteristic turnover time of actinfilaments, were determined in each BAEC morphology. Subconfluent BAECs were characterized in a previous study 5. Error bars are SEMs with 11 < n < 30. suggests that some fraction of the diffusing population is larger than known sequestered monomer complexes (see Discussion). Actin Dynamics in the Cell Cytoplasm 5-7 Correlation of actin dynamics with cell motility and protein expression Results a) 0.6. 0.5+ 0.4 0.3 C 0.2 a 0.3 melanoma 0.1 0 ABP+ OBAEC 0 0.1 r b) 0.6 0.5 -o 0.4 G_ 0 ABPfibroblast 0.05 0.1 0.15 0.2 turnover rate (min)-1 5 0.2 0 0 Wr G- c) ( E3 ABP+ BAEC 2 ABP- 0 fibroblast 0 0.25 0.5 0.75 polymer fraction 0 -8 4 G+ melanoma x 10 1 1 0 0.5 polymer fraction 1 FIGURE 5.4. Correlationsof motility and actin dynamics. a) The turnoverrate offilaments (the reciprocalof the characteristicturnover time) correlatespositively with the RMS cell speed in BAECs. The BAEC correlationis followed by gelsolin null andpositivefibroblasts,and by ABP-280 positive but not by ABP-280 null, melanoma cells. b) The fraction of actinpolymerized correlatesinversely with RMS cell speed in BAECs. The BAEC correlationisfollowed by all the other cell types. c) The mobility of the diffusive actinpopulation decreaseswith increasingpolymerfraction in BAECs (p < 0.05). Lines are least squaresfit to the endothelial cell data. Gelsolin null and positive fibroblasts and ABP+ melanoma cells agree with endothelial cells correlations, but ABP- melanoma cells do not The relationship between motility and actin dynamics in primary mouse fibroblasts and a human melanoma cell line agree with the BAEC correlations (Figure 5.4a and Figure 5.4b), suggesting a similar regulation of actin dynamics controls the rate of cell motility in these disparate cell types. The primary mouse fibroblasts serve as wild-type cells (G+) for fibroblasts isolated from gelsolin null mice (G-) (Witke, et al., 1995), and the human melanoma cells (ABP+) have been rescued with ABP-280 cDNA from a melanoma line naturally deficient in ABP-280 (ABP-) (Cunningham, et al., 1992). The relationship between motility and actin dynamics in gelsolin null fibroblasts is also in reasonable agreement with the BAEC correla- 5-8 Correlation of actin dynamics with cell motility and protein expression Results FIGURE 5.5. Actin dynamics in gelsolin positive and nullfibroblasts. Photoactivated fluorescent bands persist longer in gelsolin nullfibroblasts a), compared to wild-type b), and indicate a larger fraction of actin is polymerized. Intravitalfluorescence stainingof cells is shown in the lower panels. tions. This result indicates that the capping/or and severing activities of gelsolin are consistent with the mechanisms by which actin dynamics are regulated in BAECs. By contrast, ABP-280, an actin filament cross-linking protein, regulates motility independent of the mechanisms that regulate filament turnover (Figure 4a). Actin Dynamics in the Cell Cytoplasm 5-9 Correlation of actin dynamics with cell motility and protein expression Discussion ABP+ and ABP- cells migrate with different mechanisms Time-lapse video observations of wound recoveries reveal that motile ABP+ melanoma cells extend plasma membrane uniformly to create broad lamellae with ruffles, while ABP- melanoma cells extend their leading edge in tubular projections more akin to filopodia. This observation demonstrates that ABP-280 promotes efficient cell motility by supporting the cytoskeletal architecture necessary for lamellae and ruffles. Surprisingly, the fraction of cellular actin polymerized is a stronger correlate with bulk cell motility than filament turnover, as all the cells studied follow the correlation between polymer fraction and RMS cell speed. Discussion Correlating actin dynamics and motility in wounded BAEC monolayers reveals that these cells increase speed by decreasingthe fraction of actin polymerized and the average lifetime of filaments. Melanoma cells and fibroblasts agree with the BAEC correlation suggesting the mechanisms involved in regulating endothelial motility may apply to other cell types. If the total number of filaments remains the same as a cell accelerates, the transfer of actin from filaments to sequestering proteins will produce shorter actin filaments with shorter lifetimes. One mechanism for transferring actin between its cellular phases is the acceleration of pointed end disassembly, a function proposed as a principal activity cofilin family of proteins (Carlier, et al., 1997;Rosenblatt, et al., 1997). Alternatively, a large increase in filament number could accelerate disassembly and transfer actin from filaments to sequestering proteins. Consistent with this second possibility, the loss of the actin filament severing protein gelsolin from fibro- 5-10 Correlation of actin dynamics with cell motility and protein expression Discussion FIGURE5.6. ABP+andABP- melanoma cells migrate with different mechanisms. ABP+(M2) cells crawl with lamella replete with leading edge ruffles, while ABP- (A7) cells extend repeated tubularprojections. The cells above crawl approximately the same distance in very different times, demonstratingthe inefficiency of motility without the actin gelationprotein ABP-280. Actin Dynamics in the Cell Cytoplasm 5-11 Correlation of actin dynamics with cell motility and protein expression Works Cited blasts slows filament turnover, increases polymer fraction and slows cell motility thereby following the BAEC correlations. However, a kinetic analysis of steady-state actin dynamics (Chapter 6) favors the acceleration of pointed end off rates to filament severing. The analysis shows that if the rate of disassembly at pointed ends remains constant, extensive filament severing transfers only small amounts of actin to sequesturing proteins, far less than the trends seen here in endothelial cells. It is more likely that both pointed end disassembly and filament severing are regulated to control cell motility. The mobility of actin monomer in cytoplasm is about 15 times slower than in water, but only a 5-fold reduction in mobility is due to the enhanced viscosity of cytoplasm (Luby-Phelps, et al., 1988). The small decrease in monomer mobility with increased polymer fraction may indicate that small actin filaments diffuse with sequestered monomer to account for the remaining disparity. As polymerization increases, the number or lengths of short diffusing filaments should increase to slow the measured mobility. Also, cytoskeleton pores should decrease with increasing polymer fraction to retard large molecules but not known monomer sequestering complexes which are small compared to cytoskeleton pores. These arguments suggests that molecules larger than sequestered monomer, perhaps small actin filaments, are components of the diffusive actin population. Works Cited 1. Carlier, M.-F., V. Laurent, J. Santolini, R. Melki, D. Didry, G.-X. Xia, Y. Hong, N.-H. Chua, and D. Pantaloni. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin based motility. J. Cell Biol. 136:1307-1322. 5-12 Correlation of actin dynamics with cell motility and protein expression Works Cited 2. Clark, E., W. King, J. Brugge, M. Symons, and R. Hynes. 1997. Integrin-mediated signals regulated by members of the Rho-family GTPases. unsubmitted manuscript. 3. Cunningham, C., J. Gorlin, D. Kwiatkowski, J. Hartwig, P. Janmey, H. Byers, and T. Stossel. 1992. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 255:325-327. 4. Giuliano, K. A., and D. L. Taylor. 1994. Fluorescent actin analogs with a high affinity for profilin in vitro exhibit an enhanced gradient of assembly in living cell. J. Cell Biol. 124:971-983. 5. Gotlieb, A., W. Spector, M. Wong, and C. Lacey. 1983. In Vitro Reendothelialization, Microfilament Bundle Reorganization in Migrating Porcine Endothelial Cells. Arteriosclerosis.4:91-96. 6. Luby-Phelps, K., F. Lanni, and D. L. Taylor. 1988. The submicroscopic properties of cytoplasm as a determinant of cellular function. Ann. Rev. Biophys. Biophys. Chem. 17:369-396. 7. Mooney, D., R. Langer, and D. Ingber. 1995. Cytoskeletal filament assembly and the control of cell spreading and function by extracellular matrix. J Cell Sci. 108:2311-2320. 8. Othmer, H., S. Dunbar, and W. Alt. 1988. Models of dispersal in biological systems. J Math Biol. 26:263-298. 9. Palecek, S., J. Loftus, M. Ginsberg, D. Lauffenburger, and A. Horwitz. 1997. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 385:537-540. Actin Dynamics in the Cell Cytoplasm 5-13 Correlation of actin dynamics with cell motility and protein expression Works Cited 10. Rosenblatt, J., B. Agnew, H. Abe, J. Bamburg, and T. Mitchison. 1997. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the urnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol.:1323-1332. 11. Southwick, F., G. Dabiri, M. Pashetto, and S. Zigmond. 1989. Polymorphonuclear leukocyte adherence induces actin polymerization by a transduction pathway which differs from that used by chemoattractants. J. Cell Biol. 109:1561-1569. 12. Tardy, Y., J. McGrath, J. Hartwig, and C. Dewey Jr. 1995. Interpreting photoactivated fluorescence microscopy measurements of steady-state actin dynamics. Biophys. J. 69:1674-1682. 13. Theriot, J., and T. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126-131. 14. Theriot, J., and T. Mitchison. 1992. Comparison of actin and cell surface dynamics in motile fibroblasts. J. Cell Biol. 118:367-377. 15. Wang, Y. 1985. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J. Cell. Biol. 101:597-602. 16. Witke, W., A. Sharpe, J. Hartwig, T. Azuma, T. Stossel, and D. Kwiatkowski. 1995. Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 81:41-51. 5-14 CHAPTER 6 A Mechanistic Model of Actin Dynamics A mechanistic model describes the kinetics of the actin cycle in cells. Using rate constants and protein levels from the literature, the model predicts the effects of regulating filament pointed end off rates, profilin levels, barbed end exposure, and filament number, on filament lengths, turnover rate, and the fraction of actin polymerized. Simulations identify the acceleration of pointed end disassembly as the only mechanism that increases filament turnover and the fraction of actin polymerized, trends which correlate with enhanced motility in endothelial cells (Chapter 5). Even at the fastest rates of filament turnover, the 1,000 fold acceleration of nucleotide exchange on actin by profilin has only a small effect on the state of the actin cycle because the intrinsic ability of actin to exchange ADP for ATP is high. In an illustrative comparison with preliminary data, experimental estimates of filament turnover, polymer fraction, filament number, and exposed barbed ends are not quantitatively compatible with the kinetic model although qualitative trends agree. Future comparison with more reliable data will direct revisions in the model. Actin Dynamics in the Cell Cytoplasm 6-1 A Mechanistic Model of Actin Dynamics. Introduction Introduction The interactions of numerous actin regulating proteins contribute to the steady-state actin cycle in cells (Figure 6.1). The monomer sequestering protein 34 thymosin maintains unpolymerized actin at levels hundreds of times higher than in pure actin solutions. 04 thymosin can compete with pointed but not barbed ends for monomer, so to prevent complete polymerization, capping proteins bind the majority of barbed ends. Pointed ends are uncapped and slowly depolymerizing to balance barbed end assembly. Small quantities of profilin bind to actin to accelerate the exchange of ATP for ADP on actin monomer, and shuttle ATP-bound actin to filament barbed ends to promote assembly. The ADF/cofilin family of proteins bind to filament pointed ends to accelerate disassembly. In addition to capping barbed ends, gelsolin regulates filament length and number through severing. The state of the actin cycle is specified by any three of the following parameters: the average length of filaments, the rate of filament turnover, the fraction of actin polymerized, the number of filaments, and the fraction of barbed ends free, all of which are accessible experimentally. Signaling pathways target actin associated proteins to adjust these parameters at points labeled with *'s in Figure 6.1. To elucidate the mechanisms controlling the steady-state actin cycle, the cell cycle parameters may be measured and the data resolved with known activities of actin binding proteins in vitro. This chapter describes a kinetic model of the actin cycle to assist in such analysis. The model employs rate constants and protein concentrations from the literature to predict the state of the actin cycle when various regulatory mechanisms are active. 6-2 I A Mechanistic Model of Actin Dynamics Introduction * profilin • * points of regulation 4 ADP-Actin 34 thymosin I capping proteins * cofilin 1 ATP-Actin KA-D 2* 4Wps 3AD P ~~,b I FIGURE6.1. Schematic of the actin cycle in a cell. Reactions between actin and actin associatedproteins are shown along with associated rate constants. Potentialpoints of regulationare the number offree barbedends (*1), the concentrationof active profilin (*2), the off rate atfilamentpointed ends (*3), and the number offilaments (*4). The state of the actin cycle is specified by the average length offilaments, the rate offilament turnover,and the fraction of actin polymerized. We use the model here to identify candidate mechanisms in the regulation of actin dynamics in endothelial cells (Figure 6.2). Simulations demonstrate that the acceleration of pointed end off rates is the only mechanism that can account for the simultaneous decrease in polymerized actin Actin Dynamics in the Cell Cytoplasm 6-3 A Mechanistic Model of Actin Dynamics Introduction 0.6 E E 0.5 0.4 0.3 i:L 0.2 0 0.1 %NW CL 0 0 I I I a a) ) ( 0.05 0.1 0.6 0.5 E 0.4 0.3 0.2 0.1 0 0.15 0.2 0 rate 1turnover (min turnover rate (min ) 1 , 0.20 - ol 0.8 '-Q) 0.15 0.6 0.10 E a) o o0.05 b) 0.2 0.4 0.6 0.8 1 polymer fraction d) 0.4 0 0.2 -0.0 mechanism - 0 mechanism -- W FIGURE6.2. Procedureto identify actin cycle regulatory mechanisms important in endothelialcell motility. a) and b) As endothelialcells increasespeed in wounded monolayers, the fraction of actin polymerized decreases andfilament turnover rates increase(Chapter5). c) and d) Simulations will identify mechanisms thatsimultaneously decreasethe fraction of actinpolymerized and increasefilament turnover ratesas candidateregulatorsof endothelialcell motility. and filament lifetimes which correlate with increased motility in these cells (Chapter 5). Preliminary experimental data, however, demonstrate higher filament number in subconfluent cells compared to confluent cells, indicating that severing/nucleation occurs in the transformation from confluent to subconfluent. Simulations predict that actin's intrinsic ability to exchange its bound ADP for ATP maintains a large pool of ATP charged monomer even at the 6-4 A Mechanistic Model of Actin Dynamics The MathematicalModel highest rates of filament turnover, and that the acceleration of this exchange by profilin has little effect. Profilin's primary effect is to enhance actin assembly at barbed ends as demonstrated in vitro and in vivo. Experiments measuring the dynamics of the actin cycle do not agree quantitatively with the kinetic predictions. This result is not surprising as some of the experimental data are preliminary and the rate constants employed in the simulations were obtained in varied, often non-physiological, conditions. The Mathematical Model The rate constants and equations describing the cell actin cycle are given in Table 6.1 and Table 6.2 respectively. The model is simplified from an earlier kinetic model (Dufort and Lumsden, 1996) in the following ways: 1)Actin is always bound to a nucleotide. Assuming 0.5 mM ATP and 0.05 mM ADP is maintained in the cell cytoplasm, and employing the rates of Goldschmidt-Clermont et al., 1991 for nucleotide exchange on actin predicts that nucleotide free actin will be short lived and at very low concentrations compared to nucleotide bound actin. 2) We assume that only ATP-actin assembles/disassembles at filament barbed ends and only ADP-bound actin assembles/disassembles from pointed ends. ADP bound actin will interact negligibly with barbed ends so long as sequestered actin monomer is predominately charged with ATP which we demonstrate to be true even at the highest rates of filament turnover. There will be little association of ATP-actin with pointed ends if enough high affinity barbed ends are free. For example at 15% free barbed ends and 5 mM ATP-actin = 7 ATP-monomers will associate at Actin Dynamics in the Cell Cytoplasm 6-5 A Mechanistic Model of Actin Dynamics The MathematicalModel barbed ends for every 1 that associates at a pointed end (Pollard, 1986). This assumption will limit the model's validity at small free barbed end number. Also implicit in this assumption is the equivalence of ATP-actin and ADP*PI-actin disassembly as suggested (Korn, et al., 1987). Disassociated monomer can bind to the sequestering proteins profilin or f4 thymosin. Exchange of ATP for ADP occurs when actin is bound to profilin, or when actin monomer is not bound to sequestering protein. Consistent with experimental data, we assume nucleotide exchange does not occur when actin is sequestered by j4 thymosin (Perelroizen, et al., 1996). TABLE 6.1. Rate constants used in the kinetic model of the actin cycle in cells Constant Kp + Kp + KPAD AD KPAD Reference 0.1 s-1 Janmey and Stossel, 1986 0.09 (pm s)-1 Janmey and Stossel, 1986 profilin association with ATPactin 1 (pm s)-1 Goldschmidt-Clermont et al., 1991b profilin disassociation with ATP- 2.5 s-1 Goldschmidt-Clermont et al., 1991b pointed end disassociation constant pointed end association constant actin 6-6 Value Description A Mechanistic Model of Actin Dynamics The MathematicalModel TABLE 6.1. Rate constants used in the kinetic model of the actin cycle in cells Constant KD-A Description rate of ADP exchange for ATP in profilinactin com- Value Reference 4500 s'l Goldschmidt-Clermont et al., 1992 23.4 s-1 Goldschmidt-Clermont et al., 1992 1 (gms)"1 Goldschmidt-Clermont et al., 1991b plex KA-D rate of ATP exchange for ADP in profilinactin complex + KPAT KPAT rate of profilin association with ATP actin rate of profi- 2.5 s-1 lin disasso- Goldschmidt-Clermont et al, 1991b ciation from ATP actin rate of 04 + KBAD -1 1(LMs) Carlier et al., 1993 600 s-1 Carlier et al., 1993 1 10 (pm s)- Yu et al., 1993 thymosin association with ADP actin rate of 34 thymosin KBAD disassociation from ADP actin rate of f4 + KBAT thymosin association with ATP actin Actin Dynamics in the Cell Cytoplasm 6-7 A Mechanistic Model of Actin Dynamics The MathematicalModel TABLE 6.1. Rate constants used in the kinetic model of the actin cycle in cells Constant Value Description rate of P34 6 s-1 Reference Yu et al., 1993 thymosin KBAT + Kb Kb KADT disassociation from ATP actin rate of ATPactin association with filament barbed ends rate of ATPactin disassociation from filament barbed ends rate of ADP exchange for ATP on free actin monomer rate of ADP KATD 6-8 exchange for ATP on free actin monomer 12.3 (im s)-1 Bonder et. al, 1983 2 s- 1 Bonder et. al, 1983 35 s-1 based on constants from Goldschmidt-Clermont et al., 1992 and 500pM ATP and 50 pM ADP. 0.02 s-1 based on constants from Goldschmidt-Clermont et al., 1992 and 500 pM ATP and 50 pM ADP. A Mechanistic Model of Actin Dynamics The MathematicalModel TABLE 6.2. Steady-state rate equations describing the actin cycle in cells E.6.1 KBAT [AT] [B] steady-state balance of free ATP actin, free 04 thymosin and 04 thymosin/ATP actin complexes [BAT] E.6.2 KBAD = [AD] [B] steady-state balance of free ADPactin, free 04 thymosin and 34 thymosin /ADP-actin complexes [BAD] no net formation of profilin/ATP- E.6.3 actin complex at steady-state. o = KPAT [AT] [P]-KPAT [PAT]-KA-D [PAT]+KD-A [PAD] no net formation of profilin/ADPactin at steady-state. Describes a bal- E.6.4 o= Describes a balance between free ATP- actin, free profilin and profilin/ ATP-actin complexes accounting for nucleotide exchange on complexes KPAD [AD] [P]-KPAD [PAD]+KA-D [PAT]-KD-A [PAD] ance between free ADP- actin, free profilin and profilin/ADP-actin complexes accounting for nucleotide exchange on complexes E.6.5 o= -Kb [barb] [AT] + Kb [barb] - KBAT [B] [AT] + KBAT [BAT] + - KPAT [P] [AT] + KPAT [PAT] - KATD [AT] + KADT [AD] E.6.6 + Kp [point]- KAD [B] [AD] + BAD [BAD] int] [ S=K [point] [AD] + Kp [point] - KBAD [B] [AD] + KBAD[A]and - KPAD [P] [AD] + KPAD [PAD] - KADT [AD] + KATD [AT] E.6.7 ATOT = f +[AT] + [AD] + [PAD] + [PAT] + [BAD] + [BAT] no net formation of free ATP-actin at steady-state. Considers barbed end production and absorption, profilin and thymosin production and absorpand thym osin production and tion, and nucleotide exchange absorpon actin no net formation of free ADP-actin at steady-state. Considering barbed end production and absorption, profilin production and absorp- thymosin and thymosin production and absorption, and nucleotide exchange on actin conservation of actin Actin Dynamics in the Cell Cytoplasm 6-9 A Mechanistic Model of Actin Dynamics Methods TABLE 6.2. Steady-state rate equations describing the actin cycle in cells steady-state balance of free ATP E.6.1 actin, free 34 thymosin and 34 thyKBAT = [AT] [B] mosin/ATP actin complexes [BAT] E.6.8 conservation of profilin PTOT = [PAT] + [PAD] + [P] + [BAD] + [BAT] E.6.9 conservation of 34 thymosin BTOT = [BAT] + [BAD] + [B] E.6.10 0 = -Kb [barb] [AT] + Kb [barb] -Kp [point] [AD] + K [point] no net assembly of filaments at steady-state. This equation is a linear combination of those above and is included as a check. Methods System solution The equations and rate constants in Tables 6.1 and 6.2 were programmed into a least-square optimization routine in MATLAB (Natick, MA). The default parameters for the leastsq function in the Optimization Toolbox were used except that the step size was set to 10 and the algorithm changed to Gauss-Newton. Unless otherwise stated, simulations were initiated by guessing the following solution for the steady-state distribution of actin: ATP-actin unpolymerized = 2 jgM, ADP-actin unpolymerized = 2 gM, profilin/ATP-actin 20 gM, profilin ADPactin 0 gM, P4 thymosin/ATP-actin = 200 LM, P4 thymosin-ADP actin = 0 gM, free profilin = 0 gM, free P4 thymosin = 0 gM, polymerized actin = 200 RM. Protein levels were: total actin = 200 pM, total thymosin P4 = 200 gM, total profilin = 10 gM. Unless otherwise stated 6-10 A Mechanistic Model of Actin Dynamics Methods the filament number was 0.1 gM and exposed barbed ends were 10% of total number of filaments. The off rates at filament pointed ends were accelerated from 0.1 (Janmey and Stossel, 1986) to 25 times this value (Carlier, et al., 1997). Pyrene assay Endothelial cells at 100% or = 40% confluency plated on glass coverslips were transferred to fluorimeter tubes containing phosphate buffered saline (PBS). Cells were washed three times with PBS and transferred to permeabilization buffer (20 mM Hepes, pH 7.5, 150 mM KCl, 4 mM MgC12 , 0.1% Triton, 1 mM ATP, 1%BSA, 2 mM EGTA), containing 1 gM pyrene labeled actin and 42 gM leupeptin, 10 gM benzamidine, and 0.123 .M aprotinin to inhibit proteases, 2 gM cytochalasin B to block barbed ends, and 2 pM phalloidin to stabilize filaments against disassembly. Pyrene-actin polymerization was followed by exciting tubes with 366 nm light and monitoring fluorescence at 388 nm. Initial and final (12 hour) fluorescence levels were measured, and assuming all pyrene-actin eventually polymerized, a fluorescence value per molecule (fvpm) was calculated. The initial rate of polymerization was converted to an assembly rate by dividing by fvpm and the 1.5 molecules per second addition rate at pointed ends (for 1 gM pyrene actin). Experiments were repeated in the absence of cytochalasin to determine the number of barbed ends exposed (Hartwig, 1992). Actin Dynamics in the Cell Cytoplasm 6-11 A Mechanistic Model of Actin Dynamics Results Results 2 1 E 0.9 increasing profilin S0.8 - 0.7 Subconfluent cells have more and shorter filaments than confluent cells S0.6 c 0.5 The rate of addition of pyrene-labeled 0.4 1 profilin values (gM): C0.8.0, 1, 5, 10, 20, 40 b) actin to cytoskeletons in permeabilized subconfluent and confluent endothelial S0.6 cells indicates they contain 7 x 105 and o 0.4 2 x 105 pointed ends respectively. 0 L Assuming 200 pM total actin and 1.4 x 0.2 increasing profilin 0' 0.8 types, and noting that the fraction of 0.7 actin polymerized is = 80% in confluent increasing profilin . 0.6 L 0.5 E o 0.4 0.3 10-12 L cell volume for both pheno- cells and = 40% in subconfluent cells, this result indicates filaments decrease from an average length of 2.34 pgm in confluent cells to 0.66 gm in subconflu- 0 1 2 3 pointed end off rate (sed)1 ent cells. These filament length estimates, along with values for turnover FIGURE 6.3. Effects of pointed end disassembly and nucleotide exchange on the cell cycle parameters. and polymer fraction define the dynamic Accelerating pointed end disassembly affects the dynamic state of the actin cycle consistent with trends correlated state of the actin cycle. Because the with motility. Accelerating nucleotide exchange on has a negligible effect on the unpolymerized actin assumptions in the filament length caldynamic state even at the highest rates offilament turnover. 6-12 A Mechanistic Model of Actin Dynamics Results culation have not been verified, and because the experikadt = 35 s , no profilin ment has not been repeated, this conclusion is very preliminary, but will suffice to illustrate comparison 0.* kadt = 0.35 sl 40 mM profilin S0 between model predictions and experiments. 0. on state while accelerating nucleotide exchange does not The rate of disassembly at pointed ends for purified actin E 0O a) 0.5 factor of 25 (Carlier, et al., 1997). As shown in Figure 1.5 2 2.5 b) 1 at 37 0C is 0.1 molecules/sec (Janmey and Stossel, 1986). The cofilin family of proteins accelerates this rate by a 1 o 0.6 E S0.4 6.3, simulations indicate accelerating pointed end depo- gE lymerization shortens filaments, depolymerizes them, and increases their rate of turnover. Despite profilin's 0 0.5 1 1.5 2 2.5 pointed end off rate (sec)-l demonstrated ability to accelerate the rate of nucleotide FIGURE 6.4. Levels ofATP and ADP exchange on actin by 1000 (Goldschmidt-Clermont, et filamentturnover.Actin exchanges its al., 1991), concentrations from 0 to 40 gM (Figure 6.3) have negligible effect on the state of the actin cycle. The bound monomer with accelerating bound nucleotide fast enough to maintain ATP charge at high turnover rates. Only if this rate is slowed by a factor of 100 does profilin accelerate filament turnover. intrinsic ability of actin to exchange its bound nucleotide is sufficient to maintain nearly all of the actin charged with ATP, primed for assembly at barbed ends (Figure 6.4a). Profilin acceleration of nucleotide exchange on monomer only affects actin dynamics when actin's intrinsic ability for nucleotide exchange is lowered 100 fold (Figure 6.4b). Increasing pointed end disassembly accelerates turnover and depolymerizes actin consistent with Actin Dynamics in the Cell Cytoplasm 6-13 A Mechanistic Model of Actin Dynamics Results the trends correlated with enhanced endothelial cell a) increasing free motility. This result strongly suggests that ADF/cofi- 4barbed ends Sbarbed ends lin is regulated in the confluent/subconfluent transition of endothelial cells. We now examine how the remaining mechanisms modulate the effects of accelerating pointed end off rates. 3 Exposing filament barbed ends counters the acceleration of pointed end off rates Because the concentration of sequestering proteins is high compared to the concentration of filaments, and because exposed barbed ends have a higher affinity 3 increasing free barbed ends 0.8 for actin monomer than sequestering proteins, newly exposing barbed ends causes rapid polymerization, making filaments and their average lifetimes longer (Figure 6.5). 0.60.4- 0 1 2 pointed end off rate (sec)- 13 Filament severing shortens filaments and increases turnover but has a negligible effect on polymer fraction FIGURE 6.5. Effects of increasing filament barbedexposure on the cell cycle As seen in Figure 6.6 increasing filament number parameters.Exposing barbed ends through severing (or nucleation) creates a shorter induces polymerization, lengthens filaments and increasestheir average average filament length, and rapidly accelerates the lifetime. These effects counter the accelerationofpointed end disassemrbly. turnover of filaments, but has negligible effect on 6-14 A Mechanistic Model of Actin Dynamics Results polymer fraction. Net disassembly of filaments will a eventually occur as filament severing produces an 3 increasing number of barbed ends. The results in this 2 • filament number. 1 - ---- --- -- A 1 2.5 - simulation however demonstrate that the effect is negli- increasing 2 -1 gible even when filaments become smaller than 0.2 m. Preliminary experiments indicate that filament 3 severing (or nucleation) does occur in the endothelial 2 transformation from confluence to subconfluence, how- 1.5 ever this simulation suggests that severing alone cannot explain the transformation. S01 (D E 0.5 Profilin-mediated assembly at barbed ends 2 0 1I 1 2 c) filament number = 0.9 Q.1, 0.35, 0.60, 0.85 IgM Profilin mediated assembly at barbed ends is modeled by adding the term -kb[PAT][barb] to E.6.3, and 0.8 equivalently, +kb+[PAT][barb] to E.6.10. Modeled in this way profilin adds ATP-actin onto barbed ends at a 0. 0.61' 0 rate equal to normal ATP-monomer addition (Dufort 2 3 pointed end off rate (sec)-l and Lumsden, 1996;Pantioni and Carlier, 1993;Pollard FIGURE 6.6. Effects of regulatingfilament and Cooper, 1984), and is immediately released after number on the actin cycle. Increasing filament number through severing or nucleation, shortens the average filament the monomer addition. Profilin enhances assembly at length and increases the rate of turnover but has a negligible effect on polymer fraction. barbed ends by competing for sequestered monomer with 34 thymosin and associating with barbed ends where the monomer assembles into filaments (Pantioni Actin Dynamics in the Cell Cytoplasm 6-15 A Mechanistic Model of Actin Dynamics Results and Carlier, 1993). As seen in Figure 6.7 this shuttling U.0 effect enhances polymerization, lengthening filaments S0.7S0.6 cre and increasing their lifetimes. profilin 6 0.5 Experiments and simulations agree qualitatively but not quantitatively 0.4 U 0- The filament turnover rate (lifetime), average filament length, and polymer fraction, and number of filaments have been determined experimentally in subconfluent and confluent endothelial cells, either in a preliminary fashion in this chapter or in Chapter 5. These quantities u specify the dynamic state of the cellular actin cycle in 0.9 o0.8 , 0.7 0~0.6 . I ---- the two phenotypes and are plotted vs. each other in Fig- eas g profilin ure 6.8, where state 1 is associated with confluent cells S0.5 and state 2 with subconfluent cells. Also shown are the 0.4 predictions of the kinetic model at the two cell states. 0.3 0 1 2 3 According to our preliminary measurements, we assume pointed end off rate (sec-1) FIGURE 6.7. Profilin mediated barbed end assembly affects the actin cycle. The equations in Table 6.2 were modified to allow the actin in profilin/ ATP-actin complexes to add to barbed ends, immediately releasing free profilin. This profilin shuttle mechanism invigorates assembly, countering the effects of accelerated disassembly from pointed ends. in state one (confluent) there are 2x10 8 filaments of which 5% are free and in state two (subconfluent) there are 7x10 8 filaments of which 25% are free. 10 gM profilin is active in state 2 and 1 gM in state 1. With these assumptions, the model predicts lengths, turnover rates and polymer fractions for that differ from experimental 6-16 *1 A Mechanistic Model of Actin Dynamics Results 1.5 .0 \ E 0. 0.2 0.3 0.4 0.5 0.6 . 1 0.6 0.41- S0.6 0 c 0.4 50.2 - C 0.2 E 0.1 0.2 0.3 0.4 0.5 0.6 filament number uM 6.1 0.2 0.3 0.4 0.5 0.6 filament number uM FIGURE 6.8. Comparisonbetween experimentally determinedand kinetic predictionsof the actin cycle state in confluent and subconfluent endothelialcells. State 1 is associatedwith confluent cells and state 2 with subconfluentcells. The results agree qualitatively thatthe transitionfrom confluence to subconfluence results in increasedfilament lifetimes, polymerfraction andfilament lengths. The experimentaldata on filament number and barbed end exposure are too preliminaryto permit criticalevaluation of the quantitative differences. estimates. In each case, however, the qualitative results agree that filament lengths, filament lifetimes, and the fraction of actin polymerized are higher in confluent cells than in subconfluent cells. Actin Dynamics in the Cell Cytoplasm 6-17 A Mechanistic Model of Actin Dynamics Discussion Discussion Actin experiences a complex cycle in cells in which filaments continuously turnover by exchanging subunits with a pool of sequestered actin. Regulated proteins act at a variety of points in the cycle to control the limited number of variables that determine the cycle state. These variables are the length, number, and turnover rate of filaments and the fraction of actin in filaments. All of these are experimentally accessible. This chapter compares the experimentally determined state in confluent and subconfluent endothelial cells to a kinetic model of the actin cycle to elucidate the mechanisms regulating this transition. Simulations demonstrate that filament severing and accelerating pointed end off rates both increase filament turnover rates, but that only accelerated disassembly can account for a decrease in polymerized actin. In preliminary experiments, the number of filaments is more than 2 times higher in subconfluent cells than in confluent cells demonstrating that filament severing, or filament nucleation, does occur. Because the increase in filament number is not essential to account for differences in actin dynamics between confluent and subconfluent cells, the role of shorter filaments may be structural. The actin matrix in lamellae, for example, is known to contain short and orthogonally arranged filaments (Hartwig and Shevlin, 1986). The kinetic simulations agree qualitatively but not quantitatively with the experimentally determined state of the actin cytoskeleton in confluent and subconfluent endothelial cells. There are many possible explanations for this disparity, but most importantly the measurement of filament number and barbed end number are very preliminary. Even if these estimates are 6-18 A Mechanistic Model of Actin Dynamics Works Cited improved, it is unlikely that the experimental and numerical results will concur. The chemical environments used to measure rate constants in vitro differ throughout the literature. They also differ from physiological conditions because almost all of the in vitro measurements are made at room temperature. As demonstrated, however, comparisons between in vivo measurements of actin cycle and kinetic models can identify and eliminate mechanisms involved in its regulation. The quantitative convergence of models and measurements should be a goal that refines modeling assumptions and directs experiments both in vitro and in vivo. Works Cited 1. Carlier, M.-F., V. Laurent, J. Santolini, R. Melki, D. Didry, G.-X. Xia, Y. Hong, N.-H. Chua, and D. Pantaloni. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin based motility. J. Cell Biol. 136:1307-1322. 2. Dufort, P., and C. Lumsden. 1996. How profilin/barbed-end synergy controls actin polymerization: a kinetic model of the ATP hydrolysis circuit. Cell Motil Cytoskeleton. 35:309-330. 3. Goldschmidt-Clermont, P., L. Machesky, S. Doberstein, and T. Pollard. 1991. Mechanism of the interaction of human platelet profilin with actin. J Cell Biol. 113:1081-1089. 4. Hartwig, J. 1992. Mechanisms of actin rearrangements mediating platelet activation. J. Cell Biol. 1992:1421-1442. Actin Dynamics in the Cell Cytoplasm 6-19 A Mechanistic Model of Actin Dynamics Works Cited 5. Hartwig, J. H., and P. Shevlin. 1986. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J. Cell. Biol. 103:1007-1020. 6. Janmey, P. A., and T. P. Stossel. 1986. Kinetics of actin monomer exchange at the slow growing ends of actin filaments and their relation to the elongation of filaments shortened by gelsolin. J. Muscle Res. Cell Motility. 7:446-454. 7. Korn, E. D., M.-F. Carlier, and D. Pantaloni. 1987. Actin polymerization and ATP hydrolysis. Science. 238:638-644. 8. Pantioni, D., and M. Carlier. 1993. How profilin promotes actin filament assembly in the presence of thymosin b4. Cell. 75:1007-1014. 9. Perelroizen, I., D. Didry, H. Christensen, N. Chua, and M. Carlier. 1996. Role of nucleotide exchange and hydrolysis in the function of profilin in actin assembly. J. Biol. Chem. 271:12302-12309. 10. Pollard, T. 1986. Rate constants for the reactions of ATP-and ADP-actin with the ends of actin filaments. J Cell Biol. 103:2747-2754. 11. Pollard, T., and J. Cooper. 1984. Quantitative analysis of the effect of Acanthameoba profilin on actin filament nucleation and elongation. Biochemistry. 23:6631-6641. 6-20 CHAPTER 7 A System for the Assay of Steady-State Actin Dynamics In Vitro This chapter demonstrates the use of photoactivated fluorescence (PAF) to assay steady-state actin dynamics in vitro. The technique improves over existing approaches because it is minimally invasive and measures filament turnover, filament diffusion and the fraction of actin polymerized within the same preparation of actin at a true steady-state. The experiments are also simple to interpret. When the bulk of actin is polymerized, the decay of fluorescence from broad photoactivated bands is due solely to filament turnover, while the decay of fluorescence from narrow bands is due solely to filament diffusion. Because filament diffusion is a function of filament length, PAF experiments can non-invasively determine the effects of an actin associated protein on filament turnover, polymer fraction, and, indirectly, filament length, in a single preparation of actin. Actin Dynamics in the Cell Cytoplasm 7-1 A System for the Assay of Steady-State Actin Dynamics InVitro Introduction Introduction Understanding the steady-state dynamics of actin in purified systems is essential to unraveling the complexities of actin dynamics in cell crawling. Three in vitro approaches have contributed to our understanding of steady-state actin dynamics. First, the kinetics of actin assembly or disassembly have been measured under transient conditions and extrapolated to steadystate (Bonder, et al., 1983;Pollard and Mooseker, 1981). Also, the incorporation of a trace amount of radiolabeled actin into fully polymerized filaments has been followed to demonstrate that monomers steadily flux through filaments at steady-state in the presence of ATP. Finally, the motility of the pathogen Lysteria Monocytogenes in cell extracts produces complex distributions of actin polymerization and depolymerization in Lysteria's wake that are steady in the Lysteria frame of reference (Carlier, et al., 1997;Rosenblatt, et al., 1997). Immunodepletion of extract components elucidates its role in Lysteria motility and in steady-state actin dynamics. Here I propose the use of photoactivated fluorescence (PAF) to study steady-state actin dynamics in vitro. The PAF approach directly assays steady-state dynamics, is initiated with minimal perturbation of the sample, and can measure filament turnover, actin critical concentration, and filament diffusion in the same preparation. An example in this chapter provides the first direct visualization of filament turnover. 7-2 A System for the Assay of Steady-State Actin Dynamics In Vitro Theory Theory Distinguishing filament turnover from filament diffusion in PAF experiments d = 500 pm The decay of fluorescence length 100 mm from a narrow photoactivated band of actin (Figure 7.1) d = 50 gm IN depends on four factors: the mobility of monomer, the relative amounts of monomer and polymer, the diffusion of filaments, and the turnover of filaments. In the absence of t=0 monomer sequestering pro- t> 0 t- teins the concentration of monomer is the minimum 0" using PAF. UltraFIGURE 7.1 System for the assay of actin dynamics needed to initiate polymeriza- pure cagedfluorescent actin is polymerized in a microcapillary, placed on tion, or the "critical concentraa microscope, and irradiated with a pulse of UV light to create a narrow block offluorescence. The decay offluorescence is governed by the tion" (Oosawa and Asakura, diffusion and turnover offilaments, the mobility of monomer, and the fraction of actin polymerized. 1975). The critical concentra- tion of purified actin is = 0.1 IgM (Bonder, et al., 1983), generally far less the total amount of actin assayed. Assuming a negligible concentration of monomer actin compared to actin in filaments, filament turnover and filament diffusion alone determine the evolution of fluorescence in a PAF experiment. If the time for Actin Dynamics in the Cell Cytoplasm 7-3 A System for the Assay of Steady-State Actin Dynamics InVitro Theory turnover 1 0.8 0.6 1 f/f o 0.4 0.9 0.2 c) o turnover O filament diffusion 0.8 0 f/fo filament diffusion 1 b) 0.8 0.7 0.6 0.5 0.4 f/fo 0.6 0.3 0.2 0.4 DQ13 ! 0.4 0.6 Wo 0.8 0.2 0 - M-+ oo oo FIGURE 7.2 PAF experiment in the presence and ofphysiological levels of salt. Actin is immobile on short time scales in the presence a) but not in the absence b) of physiological salt. (Apparently, an incomplete photolysis of CRIA results in the fluorescence increase with time seen in the band in a) - see text) filament turnover, t, is short compared to the time for filament diffusion, tDf, a photoactivated band of fluorescent actin decays without broadening (Figure 7a). In this case, fluorescence decay is entirely due to filament turnover. If instead filament diffusion is fast compared to filament turnover, (cDf << t) then a photoactivated band broadens as fluorescence decays. In this case fluorescence decay is entirely due to filament diffusion (Figure 7b). A plot of center-line 7-4 A System for the Assay of Steady-State Actin Dynamics In Vitro Theory fluorescence vs. band width (Figure 7c) clearly distinguishes filament diffusion from pure filament turnover. Adjusting o to visualize filament turnover If filament turnover and filament diffusion occur on similar time scales, the width of the photoactivated band in a PAF experiment can be adjusted to distinguish between the processes. The time for filaments to diffuse from a photoactivated band varies according to tDf ~ 2, where o is the band width. The filament turnover time, t, however, is independent of o so long as r << TDm, where rDm is the characteristic time for monomer diffusion'. The decay of fluorescence in a PAF experiment can be attributed to filament turnover alone if monomer is far less concentrated than polymer and o is chosen such that: TDm < < ' << T Df E.7.2 In this case the fluorescence at the center of the photoactivated band, F(t), decays according to t F(t) = e Fo E.7.3 Where, Fo is the initial fluorescence at the band center-line. 1. Because monomer diffusion will also vary according to rDm - co2, it is possible to slow monomer diffusion sufficiently at large band width so that turnover is diffusion limited, i.e. r = tDm. Actin Dynamics in the Cell Cytoplasm 7-5 A System for the Assay of Steady-State Actin Dynamics InVitro Theory If instead a significant fraction of actin is monomeric, yet condition E.7.2 is still satisfied, F(t) is biphasic with an initial decay due to monomer diffusion and a long-term decay due to filament turnover. In this case the second phase of F(t) is governed by I F(t) _ - cc - e Fo E.7.4 co where cc is the critical concentration and co is the total concentration of actin. The decay of fluorescence in a PAF experiment can be attributed to filament diffusion alone if monomer is far less concentrated than polymer and &ois chosen such that: E.7.5 TDf << Dm<< In this case the evolution of fluorescence is given by F(t) F where erf(x) - -1 2 er x+2 2fbft -er x-2 2 2ff t E.7.6 e- dt. If a significant fraction of actin is monomeric, yet condition E.7.5 is still satisfied, F(t) is biphasic with an initial decay due to monomer diffusion and a long-term decay due to filament diffusion. In this case the second phase of F(t)is governed by 7-6 A System for the Assay of Steady-State Actin Dynamics In Vitro Methods F r o2 erf f 2 JDft E.7.7 2 Df t Methods Preparation of caged-actin Rabbit skeletal purified according to the method of Spudich and Watt (Spudich and Watt, 1971) was further isolated from capZ with two passes on a Sephadex-200 column (Casella, et al., 1995). The final preparation did not contain capZ detectable by western blot (< 1 ng). This ultra-pure actin was labeled with caged-resorufin iodacetamide (CRI) to produce CRIA as described in Chapter 2. In the final step, CRIA was dialyzed against buffer A (0.5 mM ATP, 0.5 mM BME, 2 mM TRIS, 0.2 mM CaC12 , pH 7.4). Microcapillary preparation and actin polymerization Hollow glass microcapillaries (Vitro Com Inc., Mountain Lakes, NJ) with a rectangular cross-section (50 pm thick x 500 pm high) were coated with 0.25 mg/ml BSA to minimize actin attachment to glass. To coat a microcapillary one end was placed inside a glass micropipette attached to a positive displacement microcapillary loader. A seal between the microcapillary and the micropipette was made by melting candle wax at their intersection. BSA was loaded into the microcapillary for two minutes and expelled with the loader. 2 p.M CRIA was polymerized with 10X polymerization buffer (1 M KCL, 50 mM MgCl, 1.0 mM EGTA, 5 mM ATP, 250 mM HEPES, 5 Actin Dynamics in the Cell Cytoplasm 7-7 A System for the Assay of Steady-State Actin Dynamics InVitro Methods mM BME, pH 7.5) and immediately loaded into a microcapillary. The seal between the microcapillary and the micropipette was broken with a gentle flame and both ends of the microcapillary sealed with vacuum grease. Actin was allowed to polymerize overnight in the dark at room temperature. PAF experiments Microcapillaries loaded with polymerized CRIA were placed on the stage of a modified epifluorescence microscope equipped with an intensifier and CCD (Chapter 2). Under computer control, a band of UV light focuses on the microcapillaries through a 16x plan-neofluor objective to create a 25-100 pm band of fluorescent actin, and a programmed sequence of image acquisitions executed. Figure 7.3 demonstrates the behavior of a 25 pm photoactivated band with and without the addition of 10X polymerization buffer. In the absence of polymerizing salts, fluorescent actin dissipates from the photoactivated band within 6 minutes. On this time scale, the addition of polymerization buffer creates immobilized filaments (no band broadening), however, the fluorescence in photoactivated bands unexpectedly increases. The rise of fluorescence occurs after uncaging during exposure to 577 nm excitation light (Figure 7.4). Under continuous excitation the fluorescence rises, stabilizes for a short time, and eventually decays due to photobleaching (Figure 7.5). The problem of rising fluorescence is overcome by exposing samples to 4 seconds of continuous excitation after uncaging. Limiting sample exposure to less than 0.5 seconds during the subsequent experiment also prevents photobleaching. The 4 second delay between uncaging and sampling must be small compared to the time scales of the processes being examined. The fluorescence rise apparently indicates an intermediate in the photolysis of caged resorufin-iodacetamide (see Results). 7-8 A System for the Assay of Steady-State Actin Dynamics In Vitro Results 1<0 t=0 t = 4 sec t = 30 sec t = 90 t = 360 t<O t =0 t = 4 sec t = 30 sec t = 360 t = 90 0.15 M KCI 0 M KCI a) b) FIGURE 7.3 Photolysis of 2-nitrobenzyl caged compounds. I hypothesize that an incomplete photolysis produces a non-fluorescent aci-nitro intermediate that is converted to resorufin with exposure to excitation light. Results The role of an aci-nitro intermediate in fluorescence rise after uncaging The rise after photoactivation indicates incomplete photolysis of caged resorufin and conversion of a non-fluorescent intermediate to fluorescent resorufin with exposure to excitation light. Both CRI alone and caged-fluorescein-maleimide-actin demonstrate a transient rise in fluorescence with excitation after uncaging, suggesting this behavior is characteristic of the photolysis of fluorophores caged with 2-nitrobenzyl chemistry. As shown in Figure 7.5, the photolysis of 2Actin Dynamics in the Cell Cytoplasm 7-9 A System for the Assay of Steady-State Actin Dynamics InVitro Discussion nitrobenzyl caged compounds is thought to involve an aci-nitro intermediate. According to the reaction, either a decrease in pH or the presence of a thiol reactive agent eliminating the 2nitro acetophenone product should facilitate the photolysis of caged resorufin. While DTT hastened the transition between fluorescence rise and photobleaching, decreasing pH did not (Figure 7.6). Both DTT and pH affected maximum fluorescence after uncaging. Increasing DTT or glutathione also diminished the capacity of CRIA to uncage (not shown), suggesting DTT reacts directly with CRIA or a non-fluorescent intermediate to block photolysis. These complications preclude implicating the aci-nitro intermediate as the cause of the fluorescence rise. The effects of reducing agents, however, may explain why fluorescence rise does not occur in cells (Chapter 3). Demonstration of filament turnover by PAF Figure 7.7 demonstrates using PAF to measure filament turnover in vitro. The fluorescence in a 75 p.m photoactivated band of 2 pM CRIA decays without significantly broadening (Figure 7.7a). The fluorescence decay is biphasic (Figure 7.7b), and the second phase is well described by equation E.7.4 (R = 0.97). The amount of monomeric actin is 0.2 pM in good agreement with estimates of the critical concentration. The time for filament turnover is 7.5 hours. Discussion Transient assays of actin polymerization and depolymerization predict that at steady-state, and in the presence of excess ATP, monomers flux through filaments from barbed to pointed ends. 7-10 ~-~95ee9~~a~~ai~ -~;i~ti~ A System for the Assay of Steady-State Actin Dynamics In Vitro Discussion CH3 CH 3 CH 3 I I CH I H+ O hv + +® "caged" compound I 0o" aci-nitro intermediate "caged" compound 2-nitro acetophenone FIGURE 7.4 The effects of continuousexposure,pH and DTT on fluorescence rise. a) Under continuous exposure CRIA fluorescence rises transiently but eventually bleaches; increasingpH increasesthe maximum fluorescence reached.b) DTT a thiol scavenger,diminishes the maximum fluorescence and acceleratesthe transitionbetween rise and bleaching. . . b) no DTT 1.5 f/fo 0 1mM DT f/fo 1 10mM DTT 0.5 0 5 10 time (sec) 15 U 0 5 10 time (sec) FIGURE 7.5 Visualization offilament turnover.a) The decay offluorescence in a 75 pm band of 2 pM actin. b) Interpretingthe fluorescence decay accordingto E. 7.4 gives Cc = 0.18 pM and z = 7.5 hours. c) Fluorescencedecay occurs without significant band broadeningindicatingpurefilament turnover. Actin Dynamics in the Cell Cytoplasm 7-11 A System for the Assay of Steady-State Actin Dynamics In Vitro Discussion .U - 0.9 t=0 F Fo O 0.8 92% polymer turnover = 7.5 hours O O 0.7 * background 2uM Actin Oo 0.6 t = 1/2 hours 00 0 0.5 b) b) 0.4 ) 0 OO 2 t (hours) 4 E t = 2 hours 0.8 F(t) Fo t = 6 hours 0.6 0.4 0.2 0.5 1.0 Wo w(t) FIGURE 7.6 Example PAF experiment demonstratingfilament turnover,a) Images of a PAF experiment showing the fluorescence in a photoactivatedbanddecaying over many hours while the band width remains constant b) The fluorescence decay at the centerline of the band. The initialdrop is due to diffusion of the criticalconcentration of monomer out of the band. The bandthen decays as an exponentialas predictedby E7.11. c) The plot offluorescence vs width demonstrates that thedecay is due to turnover and not diffusion. 7-12 A System for the Assay of Steady-State Actin Dynamics In Vitro Discussion This steady-state behavior was demonstrated by Wegner who followed the incorporation of a trace amount of radio labeled monomer into polymerized actin. The Wegner experiment directly assays steady-state actin dynamics. The motility of the pathogen Lysteria monocytogenes in cell extracts provides a second system to study steady-state dynamics. Proteins localized to a region of the Lysteria membrane polymerize actin to propel the bacterium. Cytoplasmic factors break-down filaments polymerized by Lysteria' giving a "comet tail" distribution to actin in Listeria's wake. Depletion of cell extract components identifies factors essential to Lysteria motility, or those important to distributions of filament assembly, break-down, and organization in Lysteria comet-tails. Here we use photoactivation of fluorescence (PAF) in an in vitro assay of steady-state actin dynamics. In these experiments, caged-fluorescent actin is loaded into rectangular tubes and polymerized to steady-state on the stage of a fluorescence microscope. A band of UV light focuses through the sample and creates a locally fluorescent band (Figure 7.1). The evolution of fluorescence is recorded and experiments interpreted according to a two-phase model of actin dynamics. The experiments distinguish between filament diffusion and filament turnover. Filament turnover and the critical concentration of actin can be simultaneously measured. Unlike Wegner's system, the new system does not require a mechanically perturbing pipetting step to initiate experiments. Because measurements are made in a homogeneous environment and with complete knowledge of component proteins, experiments are simpler to interpret than Lysteria studies. The width of photoactivated bands can be adjusted to study filament diffusion or filament turnover exclusively. Controlling filament lengths with gelsolin (Yin, et al., 1981) and using narrow bands, PAF experiments should be done to establish the relationship between filament diffusion and filaActin Dynamics in the Cell Cytoplasm 7-13 A System for the Assay of Steady-State Actin Dynamics In Vitro Works Cited ment length. Then by varying the width of photoactivated bands in successive PAF experiments, the effects of an actin regulatory protein on filament length, filament turnover, and critical concentration could be determined. Works Cited 1. Bonder, E., D. Fishkind, and M. Mooseker. 1983. Direct Measurement of Critical Concentrations and Assembly Rate Constants at the Two Ends of an Actin Filament. Cell. 34:491501. 2. Carlier, M.-F., V. Laurent, J. Santolini, R. Melki, D. Didry, G.-X. Xia, Y. Hong, N.-H. Chua, and D. Pantaloni. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin based motility. J. Cell Biol. 136:1307-1322. 3. Casella, J., E. Barron-Cassela, and M. Torres. 1995. Quantitation of Cap Z in Conventional Actin Preparations and Methods of Furthur Purification of Actin. Cell Motility Cytoskeleton. 30:164-170. 4. Oosawa, E, and S. Asakura. 1975. Thermodynamics of the Polymerization of Protein. Academic Press, London. 5. Pollard, T. D., and M. S. Mooseker. 1981. Direct measurement of actin polymerization rate constants by electron microscopy of actin filaments nucleated by isolated microvillus cores. J. Cell Biol. 88:654-659. 7-14 A System for the Assay of Steady-State Actin Dynamics In Vitro Works Cited 6. Rosenblatt, J., B. Agnew, H. Abe, J. Bamburg, and T. Mitchison. 1997. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the urnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol.:1323-1332. 7. Spudich, J. A., and S. Watt. 1971. The regulation of rabbit skeletal muscle contraction. J. Biol. Chem. 246:4866-4871. 8. Theriot, J., and T. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126-131. 9. Wegner, A. 1976. Head to tail polymerization of actin. J. Mol. Biol. 108:139-150. 10. Yin, H., J. Hartwig, K. Marayama, and T. Stossel. 1981. Ca 2+ control of actin filament length. Effect of macrophage gelsolin on actin polymerization. J Biol Chem. 256:9693-9697. Actin Dynamics in the Cell Cytoplasm 7-15 A System for the Assay of Steady-State Actin Dynamics In Vitro Works Cited 7-16 CHAPTER 8 Conclusions This thesis advances the application of PAF and FRAP in the assay of actin dynamics in cells and in vitro. We demonstrate a new interpretation of the data obtained with these methodologies that provides simultaneous measures of actin monomer mobility, filament turnover rates, and the fraction of actin polymerized. Applying these techniques to endothelial cells in a wounded monolayer, we demonstrate that cells of a single type change the dynamic state of their actin cytoskeleton to become more motile. This cell culture model, experiments in cells with varying expression of key actin binding proteins, and a kinetic model of the actin cycle provide new insight into actin dynamics. A PAF based approach for measuring actin dynamics in vitro, improves upon existing approaches and provides the first visual evidence for actin filament turnover in vitro. Actin Dynamics in the Cell Cytoplasm 8-1 Conclusions The following are the principal conclusions generated in this thesis listed by chapter.: Chapter3: InterpretingPAF/FRAP Measurements of Steady-State Actin Dynamics *A two phase model of actin dynamics improves over previous models interpreting PAF and FRAP measurements providing simultaneous estimates of monomer diffusion coefficients, filament turnover rates, and the fraction of actin polymerized. Chapter4: Baseline and ControlMeasurements in Subconfluent Endothelial Cells * PAF and FRAP experiments in subconfluent BAECs are biphasic and can be interpreted with the model in Chapter 3. * PAF and FRAP experiments give similar estimates for the mobility of sequestered actin monomer, the fraction of actin in filaments, and the lifetimes of actin filaments indicating previous differences between PAF and FRAP experiments were not due to inherent differences in the techniques. * The comparison of PAF and FRAP demonstrate that inadvertent photobleaching in PAF experiments has been controlled. * Control experiments with a membrane permeable barbed end blocking reagent (cytochalasin D) confirm the assumption that filament turnover and not filament diffusion is responsible for the second phase of fluorescence rise in FRAP experiments. 8-2 Conclusions * Control experiments with the membrane permeable polymerizing reagent (jasplankinolide) confirm the assumption that the bulk of actin giving the initial fluorescence decay in PAF experiments is due to polymerization competent monomer. * Small quantitative differences in the PAF and FRAP results agree with previous work showing that cys-374 labeled actin derivatives have cytoplasmic mobilities nearly 2 times lower than lysine labeled derivatives. The results also indicate that cys-374 labeled actin incorporates less readily into filaments and cycles through filaments slower than lysine labeled actin. * The turnover of actin filaments is fast (= 6 minute lifetimes) but not diffusion limited in the bulk actin cytoskeleton of endothelial cells. Chapter5: Correlationof actin dynamics with cell motility and with expression of actin associated proteins *The dynamics of actin correlate with cell motility in four distinct BAEC phenotypes demonstrating for the first time that cells of the same type can alter the dynamic state of their actin cytoskeleton as they become more motile. *A positive correlation between cell speed and filament turnover rate and an inverse correlation between cell speed and actin polymer fraction is demonstrated in the wounded monolayers. * Fibroblasts and melanoma cells agree with the endothelial cell correlations indicating similar factors regulate actin dynamics to control cell motility in these disparate cell types. Actin Dynamics in the Cell Cytoplasm 8-3 Conclusions * The absense of gelsolin in fibroblasts slows motility, increases the fraction of actin polymerized and increases filament lifetimes consistent with the trends in endothelial cells. This result implicates the barbed end capping and/or filament severing activities of gelsolin in the control of endothelial cell migration. * The absense of the actin cross-linking protein ABP-280 slows motility without affecting filament turnover demonstrating that actin dynamics and cytoskeletal architecture can be independently regulated to control cell motility. Chapter6: A Mechanistic Model ofActin Dynamics * Kinetic simulations using rate constants and protein levels from the literature are used to explore the mechanisms involved in confluent to subconfluent transformation in wounded endothelial cell monolayers. Simulations indicate that either increased rates of pointed end disassembly or filament severing increase filament turnover, but that only accelerating disassembly transfers actin from filaments to sequestering proteins. This result strongly suggests the activity of the ADF/cofilin family of proteins in regulation of migration in wounded monolayers. * Simulations uncoupling profilin's barbed end assembly and nucleotide exchange activities demonstrate that the former is important in determining the dynamic state of the actin cytoskeleton. In contrast, profilin's ability to accelerate nucleotide exchange on monomer actin has little effect, even at the highest rates of filament turnover, because actin's intrinsic ability to exchange bound ADP for ATP is sufficiently high. 8-4 Conclusions Chapter 7: A System for the Assay of Steady-State Actin Dynamics In Vitro * A PAF based approach for measuring steady-state actin dynamics in vitro provides a minimally invasive assay to determine the turnover of actin filaments, the fraction of actin polymerized and the average length of actin filaments in a single preparation of actin. The technique improves upon existing approaches which measure these quantities in separate experiments. An example experiment is shown that provides the first visual demonstration of actin filament turnover. Actin Dynamics in the Cell Cytoplasm 8-5