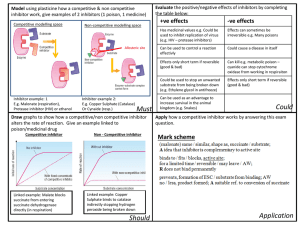
SEP 10 2008 jrUTE LIBRARIES
advertisement

Investigations into the Role of Protein Phosphatases in the EGFR Signaling Network jrUTE MASSACHUSETTS INST OF TEOHNOLOG-N By SEP 10 2008 Kuojung Gordon Lu B.A. Molecular Cell and Biology Universitv of California. Berkeley. 2002 I LIBRARIES Submitted to the Department of Biology in Partial Fulfillment of the Requirements for the Degree of Master of Science in Biology at the Massachusetts Institute of Technology September 2008 © 2008 Massachusetts Institute of Technology All rights reserved Signature of Author: Department of Biology September 9, 2008 Certified by: Visiting Prfessor of Biol Accepted by: -V -- Pete K Sorger ngineering Thesis Supervisor Stephen P Bell Professor of Biology Chairman, Graduate Committee Aic gVE8 S Investigations into the Role of Protein Phosphatases in the EGFR Signaling Network by Kuojung Gordon Lu Submitted to the Department of Biology on September 9, 2008 in Partial Fulfillment of the Requirements for the Degree of Master of Science in Biology ABSTRACT Protein phosphatases regulate the phosphorylation state of intracellular signaling molecules in conjunction with protein kinases. In order to better understand the role of phosphatases in the ErbB signaling network, experimental studies were carried out to validate assays and phosphatase inhibitors for gathering dynamic, system-wide data of phosphatase activity. We verified the use of In-Cell Westerns and phospho-ErbB ELISAs for these studies, as well as the use of different cell lines and both general and specific phosphatase inhibitors in system-wide experiments. The phosphatase inhibitors we used perturbed cellular systems in complex ways that can help elucidate the regulation and activity of specific phosphatases in the ErbB signaling pathway in the future. Thesis Supervisor: Peter Sorger Title: Visiting Professor of Biological Engineering Table of Contents I. Introduction.................................................... ............................................... 4 1. M otivation ................................................................................................ ...... 4 2. Background ............................................................................................. 4........ 3. Assay and Experim ental Design ................................................................ 10 II. Materials and Methods .................................................. 14 1. Reagents ........................................................................................................ 2. In-Cell W esterns ................................................... ............. 14 3. ELISA - Phospho-ErbB fam ily .................................................................. 15 4. Tim e Courses and IC50 Experiments...................................................... 17 5. Cell Culture................................................. III. ............ 14 ............................................ 18 Results ................................................................................................................. 19 1. Optimization of using phosphatase inhibitors..........................19 2. Co-treating cells with phosphatase inhibitors together with protein synthesis or kinase inhibitors ......... ....................................................... 26 3. Phosphatase inhibitors in phospho-ErbB ELISA Assays.....................29 IV. Discussion ............................................................................................................ 31 1. Tools developed for studying phosphatase activity.............................. 31 2. Future work ................................................................................................. 37 V. References .......................................................................................................... 38 VI. Figure captions ................................................................................................... 43 VII. Figures................................................................................................................. 45 I. Introduction 1. Motivation Protein phosphorylation plays an important role in a variety of cellular functions. This is especially true in the ErbB signaling network, where protein phosphorylation is involved in signal transduction leading to various cellular outcomes such as proliferation, survival, motility, and differentiation. The level of phosphorylation is regulated in part by the opposing enzymatic activities of protein kinases and protein phosphatases. Thus, the activity of protein phosphatases is crucial in the regulation of ErbB network signals. In order to advance our understanding of the ErbB network, it is important to obtain quantitative information of the specificity and regulation of various phosphatases at the receptor and downstream levels. This is especially true in the context of aberrant signaling involved in diseases such as cancer. A combination of specific inhibitors, cancer cell lines, and system wide signaling assays can help elucidate the role of phosphatases in the ErbB signaling network. Assays and tools were developed and validated for this purpose. 2. Background System-wide experiments measuring intracellular signals will be used to investigate the effect of perturbing the activities of various phosphatases with small molecule inhibitors. These experiments will be conducted in a panel of tumor cell lines that represent different human cancers, in order to capture similarities and differences in cellular behavior that can then be explored in a computational model. Treatment of the cell lines with different ErbB receptor ligands will stimulate downstream response pathways, and co-treatment with phosphatase inhibitors will inhibit specific phosphatase nodes to help elucidate the role of phosphatases in ErbB signaling pathways. The project will initially focus on the behavior of the Erk and Akt pathways downstream of the ErbB signaling network, and the roles of the protein tyrosine phosphatase (PTP) and protein phosphatase (PP) families of phosphatase proteins. The PTP and PP family of proteins have been shown to be crucial in many types of cellular signaling systems (Hunter 2007). Phosphorylation and dephosphorylation of proteins mediate many signal transduction events involved in ErbB signaling (Tiganis 2002; Tonks 2006). PTPs and PPs are groups of structurally diverse and complex enzymes that act in collaboration with protein tyrosine kinases (PTKs) to modulate a broad range of physiological processes (Alonso et al. 2004). Recent findings suggest that deregulation of protein phosphatases are linked to various human disease states including cancer and diabetes (Easty et al. 2006; Ostman et al. 2006; Zhang and Lee 2003). Thus, it should be interesting to investigate the importance of these phosphatase families in cellular signaling networks. The initial focus of the project will be on the PTP family member PTP1B and PP family member PP2, as they are thought to be intimately involved in growth factor signaling networks and have been to shown to be linked to human disease processes (Lee and Wang 2007; Perrotti and Neviani 2008). Protein Tyrosine Phosphatase 1B (PTP1B) The PTPs are a large family of enzymes, over 100 of which has been characterized thus far. They possess a 230-amino acid common catalytic domain, and a signature active site motif sequence (H/V)C(X)SR(S/T) (Alonso et al. 2004; Mauro et al. 1994). PTPs work together with PTKs to catalyze the addition and removal of phosphate groups on specific substrates to regulate cell signaling, and contain a number of functional subunits that help regulate their subcellular activity and function (Mauro et al. 1994; Yudushkin et al. 2007). Some PTPs, such as phosphatase and tensin homolog (PTEN), are considered products of tumor suppressor genes as they reverse the effects of RTKs and are inactivated by mutation in multiple types of cancers (Li et al. 1997). Other PTPs, however, can also potentiate the actions of PTKs, leading to cellular transformation. Examples of these include SHP2, involved in growth factor-mediated, Ras-dependent cellular transformation, and PTPa, involved in Src activation (Bentires-Alj et al. 2004; Zheng et al. 1992). This demonstrates that PTPs are involved in the complex regulation of cellular networks and are potential drug targets in aberrant signaling (Jiang and Zhang 2008). PTP1B is a widely expressed family member that is a 435 amino acid, 50 kDa protein localized to the endoplasmic reticulum (Chernoff et al. 1990). It has the ability to dephosphorylate the EGFR, PDGFR, and insulin receptor (Bourdeau et al. 2005). PTP1B has been shown to be involved in insulin receptor mediated pathways, and is being investigated as a potential target in treating type 2 diabetes and obesity (Klaman et al. 2000). More recently, genetic studies of PTP1B have linked it to breast and colon cancers (Julien et al. 2007; Bentires-Alj and Neel 2007; Zhu et al. 2007). PTP1B is overexpressed along with HER2 in 90% of all breast tumors (Wiener et al. 1994). It has been found that PTP1B-' mice are protected against HER2-induced breast tumors and have down-regulated pERK, while PTP1B /' fibroblasts show slightly up-regulated pEGFR and pERK following EGF stimulation (Bentires-Alj and Neel 2007; Haj et al. 2003). As there are a number of findings establishing PTP1B as a potential therapeutic target, there has been an effort to develop specific and potent PTPIB small molecule inhibitors. However, this has been difficult due to the conserved nature of the PTP active site motif (Jiang and Zhang 2008). One strategy involves the binding of an allosteric site away from the common PTP motif, stabilizing an inactive conformation of PTP1B. This resulted in a compound that is highly selective for PTP1B, potent (IC50 = 8 uM), and cell permeable (Wiesmann et al. 2004). This compound is commercially available from Calbiochem (see materials section) and is used to perturb and investigate PTP1B function in the current study. There have also been additional, more recent efforts to develop even more potent PTP1B inhibitors, using a strategy involving bidentate ligands that bind both the active site and a unique adjacent peripheral site. This has resulted in cell permeable compounds with both high PTP1B selectivity and IC50s in the nM range (Boutselis et al. 2007; Combs et al. 2006; Lee and Wang 2007). These compounds could be interesting for future investigations of PTP1B function in ErbB signaling. Protein Phosphatase 2A (PP2A) Serine/threonine protein phosphatases are grouped into two families, the phosphoprotein phosphatases (PPP) and metal-dependent protein phosphatases (PPM). The PPPs and PPMs share a common ancestor gene, and are classified based on their metal-ion dependence, substrate sensitivity, and sensitivity to inhibitory molecules (Lechward et al. 2001). PP2A belongs to the PPP family, and is widely expressed in all eukaryotic organisms. It is a hetero-multimeric holoenzyme consisting of a common catalytic subunit and a large group of regulatory and adaptor proteins (Virshup 2000). Upon association with different subunits, PP2A can dephosphorylate a wide range of substrates and is involved in many cellular processes, including cell cycle regulation, DNA replication and transcription, survival, and the MAPK, Akt, and mTOR pathways (Zolnierowicz 2000). PP2A regulates and mediates processes downstream of growth factors and is involved in the ErbB signaling network. PP2A can dephosphorylate MEKi and ERK, down-regulating mitogenic signals (Gomez and Cohen 1991). It can also activate Raf-1 through dephosphorylation of an autoinhibitory residue and may be involved in ERK-induced feedback on Raf-1 (Dhillon et al. 2007). The PP2A holoenzyme, depending on regulatory unit composition, can also directly dephosphorylate Akt or act downstream of PI3K/Akt by inhibiting the mTOR pathway (Ivaska et al. 2002; Peterson et al. 1999). In addition, mutation or aberrant expression of PP2A subunits have been found in a number of human cancers, including breast cancer and leukemia (Perrotti and Neviani 2008). Thus, PP2A is an important target of study for better understanding the ErbB network in the context of aberrant signaling. For the perturbation of PP2A function, there are also many cell permeable PP2A inhibitors available commercially, such as okadaic acid, microcystin, and endothall (Zolnierowicz 2000). Erk and Akt For initial characterization of the output of the ErbB network following ligand stimulation, we will be measuring the levels of phosphorylated Erk (pERK) and phosphorylated Akt (pAkt). Aberrant signaling in the Erk and Akt pathways have been implicated in the growth and proliferation of many types of tumors (Roberts and Der 2007; Dillon et al. 2007). Erk and Akt pathways have also been used as downstream output readouts in modeling cellular behavior (Schoeberl et al. 2002; Hatakeyama et al. 2003). Mitogen-activated protein kinases (MAPKs) are involved in a wide range of key cellular processes such as differentiation, cellular growth, adaptation to stress, and survival, in response to stimuli such as growth factors, cytokines, adherence, and stress (Roux and Blenis 2004). The p44/42 MAPK ERK(1/2) is the downstream MAPK in the three-kinase pathway consisting of Raf, MEK, and ERK (Rubinfeld and Seger 2005). Growth factors such as EGF and PDGF can activate ERK through the MAPK kinases MEKI and MEK2, and through ERK effect cellular responses such as proliferation and differentiation (Chang and Karin 2001). MEK1 and MEK2 activate ERK through dual phosphorylation of activation loop residues Thr182 and Tyr184, and phosphorylation of these residues can be used as a readout of ERK activity (Widmann et al. 1999; Murphy and Blenis 2006). The phosphatidyl inositol 3-kinase (PI3K) pathway is also involved in a number of cellular processes such as cell growth, proliferation, and survival (Franke et al. 1995). Protein kinase B, or Akt, are a 3-member serine-threonine kinase family that is the primary downstream mediator of the PI3K pathway and affects various downstream processes by phosphorylation of downstream substrates (Franke 2008). The PI3K pathway can be activated by insulin and various growth factors such as IGF and EGF, or the integrin receptor pathway, to promote cell survival and proliferation by inhibiting apoptosis through the activity of Akt (Zhou et al. 2000). Dysregulation of the PI3K pathway, through mutation of its components such as PTEN, is often found in human cancers (Dilton et al. 2007). Akt can be activated by recruitment to the membrane via various scaffolding proteins and phosphorylation of activating residues Thr308 and Ser473 by phosphoinositide dependent kinase 1 (PDK1) and mTOR, and phosphorylation of those residues can be used as a functional readout of Akt activity (Yang et al. 2004; Song et al. 2005). 3. Assay and Experimental Design In the current project, we will focus on using phosphatase inhibitors in parallel with ligand stimulation to perturb the activity of PTP1B and PP2A, to deduce quantitative information about their function in the ErbB network. In order to obtain a large dataset for the purpose of modeling and predicting the role of these phosphatases, we will develop further an existing In-Cell Western (ICW) based assay system that will be able to quantitatively measure signaling dynamics in a high-throughput and reproducible manner. We will also use this assay system to study the signaling dynamics specifically involved in cellular response to phosphatase inhibitors and collect a dataset for future extension and construction of a predictive dynamic model using existing ODE models. The signaling assays used will look at molecules that are known to be important in system behavior from previous studies and computational models, specifically phosphoErbBs 1-4, phospho-Erk, and phospho-Akt. The phosphorylation status of receptors will be measured using total- and phospho-receptor ELISAs, while downstream and intracellular molecules will be measured using ICW, on the LiCor Odyssey infrared imaging system. The ICW assay allows quantitative, dynamic signaling data to be gathered in a high throughput fashion. ICW requires smaller reagent quantities and less biological samples compared to traditional assays. It is also a relatively simple protocol that requires almost no pre- processing of the samples and consists of few wash and staining steps, when compared to kinase activity assays or traditional western blots. The ICW protocol allows cells to be seeded, grown, treated with cue, stained, and visualized in the same 96 well plate and reduces the number of steps required for processing samples. Finally, the use of dyes in the far-red spectrum allows for visualization with low signal overlap and high intensity, and a DNA stain is used to normalize for cell number variation in each well. The phosphorylation status of receptors and downstream molecules will serve as a proxy for their activation state. Since it is not clear what the best metric for measuring receptor activation is, both fractional phosphorylation as a functional of all receptors and absolute phosphorylated receptor numbers will be measured. Intracellular signaling assays will initially use previously worked out protocols in the lab for the measurement of phosphoErk and phospho-Akt using ICW. In order to investigate and capture differential cell signaling behavior, a combination of different ErbB ligands, phosphatase inhibitors, downstream inhibitors, tumor cell lines, and experimental designs will be used. We will perform time course experiments of co-treating available cell lines, such as H1666, H3255, A431, SKBR3, and HeLa with ligand and phosphatase inhibitors to look at dynamic activity of phosphatases. The following types of time course experiments can be informative. A) Treating cells with ligand in parallel with general phosphatase inhibitors, such as vanadate, will demonstrate the overall contribution of phosphatases to immediate signaling following ligand stimulation. B) Using specific phosphatase inhibitors, like okadaic acid, PTP1B inhibitor, endothall, dephostatin, and menadione, will demonstrate the specificity and activity of a particular phosphatase on specific network nodes such as MEK and ERK. This will be more informative in the context of the current ErbB model. C) Cotreatment with phosphatase inhibitors and specific inhibitors of particular nodes, such as tyrosine kinase inhibitors (TKIs), a MEK inhibitor, or a PI3K inhibitor, will stop additional signaling downstream of that node and demonstrate how a particular phosphatase regulate that node. An example of this could be to treat cells with heregulin (HRG), then add a PI3K inhibitor and a PTP1B inhibitor 10 minutes post ligand stimulation, and follow the Akt time course to see the effect of PTP1B on the PI3K/Akt pathway. Adding phosphatase inhibitors at different times (Pre-incubate, co-treat with Ligand, post-ligand stimulation) and in different combinations (phosphatase inhibitor alone, phosphatase inhibitor plus ligand, phosphatase inhibitor plus ligand plus node inhibitor) can help identify the contributions of different phosphatases on particular network nodes. Another type of experiments could be using different concentrations of phosphatase inhibitors to find the IC50s of phosphatases in an intracellular context. Comparing the IC50s of phosphatase inhibitors on basal versus ligand-stimulated cells demonstrates the basal activity of phosphatases and can help parse out the contribution of each phosphatase to each network node. This is analogous to an EC50 experiment, but instead of ligand stimulation, the stimulus is the inhibition of endogenous phosphatase down-regulation of a particular network molecule. Phosphatases acting on a node reduce activated node species and can lower the in vivo IC50 compared to in vitro measurements (Knight and Shokat 2005). Measuring TKI IC50s in the presence of phosphatase inhibitors can confirm the specificity of a phosphatase for the TKI target. This can be done with combinations of TKIs specific for different ErbB family members to show the regulation of receptor dephosphorylation. A specific example for this type of experiment could be elucidating the functional mechanism for erlotinib inhibition of HRG-stimulated signaling. In Schaefer 2007, it is proposed that erlotinib is binding directly to ErbB2 and inhibiting signaling from ErbB2 homo- and hetero-dimers. This is unexpected given that in vitro experiments suggest erlotinib is 40-3000 fold more specific for ErbB1 than ErbB2. One hypothesis is that ErbB 1 and ErbB2 have differential phosphatase regulation in vivo, resulting in a higher apparent Ki of erlotinib for ErbB2. This can be tested with IC50 experiments looking at the effect of using phosphatase inhibitors in parallel with HRG stimulation on the phosphorylation states of ErbB1 and ErbB2. II. Materials and Methods 1. Reagents Antibodies and Stains, ICW: Phospho-Erk, Thr202/Tyr204, Rabbit monoclonal 197G2, Cell Signaling Technology, #4377, 1:100 Phospho-Akt, Ser473, Rabbit monoclonal 193H12, Cell Signaling Technology, #4058, 1:100 Phospho-EGFR, Tyr1068, Rabbit polyclonal, Cell Signaling Technology, #2234, 1:100 800CW IR-Dye Goat anti-Rabbit, Rockland, 1:800 Topro-3 Iodide DNA stain, Molecular Probes, 1:5000 Ligands: Epidermal growth factor, recombinant human, PeproTech, #100-15 Heregulin, recombinant human HRG1b, PeproTech, #100-03 Amphiregulin, recombinant human, R&D Systems, #262-AR-100 Inhibitors: PTP1B Inhibitor, Calbiochem, cat. no. 539741 Okadaic Acid, Prorocentrumsp., InSolution, Calbiochem, cat. no. 495609 Sodium Orthovanadate, Calbiochem, cat. no. 567540 2. In-Cell Westerns The goal of our work is to quantitatively monitor the activity of proteins that contribute to the ErbB signaling cascade. For this purpose, In-Cell Western (ICW) assays were previously developed to monitor the phosphorylation of phosphor-ERK and phospho-AKT. The ICW assay provides a straightforward means of collecting multiple time or treatment dependent data points. The ICW assay involves the following steps. Cells are seeded in 96 well optical plates at 200 uL/well and grown overnight. The time course or inhibitor experimental treatments are performed on the cells in the same plate. Cells are then fixed with 100 uL/well of 4% formaldehyde at room temperature for 20 minutes. The formaldehyde is then aspirated, and the cells are permeabilized with 4 washes of 0.1 % Triton in PBS, at 5 minutes for each wash, on a rocker. The plates are then blocked with 100ul/well of Odyssey Blocking Buffer, for I hour, with shaking. The Odyssey Blocking Buffer is then aspirated, and 50 uL of the appropriate primary antibody diluted in Odyssey Blocking Buffer is added to each well. The plates are sealed with parafilm and placed at 4oC overnight. The next day, the primary antibodies are aspirated, and the plates are washed with 200 uL/well of 0.1 % Tween in PBS, for 5 washes of 5 minutes each, on a rocker. The final wash is aspirated and replaced with secondary antibody at 1:800 dilution plus Topro-3 DNA stain at 1:5000 dilution in 100 uL of Odyssey Blocking Buffer. The plates are covered with foil to protect from light and are incubated for I hour at room temperature with rocking. The plates are then washed again with 200 uL/well of 0.1 % Tween in PBS, for 5 washes of 5 minutes each, on a rocker. The last wash is aspirated thoroughly to ensure each well has no liquid remaining. The bottoms of the optical plates are then cleaned with 70% ethanol, and the plates are scanned on an Odyssey Infrared Scanner immediately. The resulting images were ana- lyzed using Odyssey software. The phospho-signal is read out as a ratio to the DNA stain intensity to normalize for cell number. 3. ELISA - Phospho-ErbB family For the measurement of phosphorylated receptor levels, we used the Phospho-EGFR, Phospho-ErbB2, Phospho-ErbB3, and Phospho-ErbB4 DuoSet ELISA kits from R&D Systems (DYC1095, DYC1768, DYC1769, and DYC2115). In separate wells, immobilized capture antibodies specific for EGFR, ErbB2, ErbB3, and ErbB4 bind both phosphorylated and unphosphorylated forms of the proteins, and an horseradish peroxidase (HRP) conjugated detection antibody for phosphorylated tyrosine is used to detect the phosphorylated form of the proteins. For the ELISA, cells are plated and grown overnight. Time course or IC50 experiments are performed, and then stopped with PBS at 4VC. The cells are rinsed with PBS and solubilized with lysis buffer #9 (R&D systems), to a concentration of 1x10 7 cells/mL. The cells are then incubated at 4VC on a rocker. The lyrsates are then microcentrifuged at 14,000g for 5 minutes, and the supernatant is transferred and frozen at -20 0 C until use. To prepare the ELISA plates, a 96-well plate was coated with 100 uL/well of capture antibody at 0.8 ug/mL (pEGFR), 4.0 ug/mL (pErbB2), 4.0 ug/mL (pErbB3), or 1.0 ug/mL (pErbB4) in PBS. The plates are sealed and incubated overnight at room temperature. The following day, the wells are aspirated and washed 5 times with 0.05% PBS-Tween. The plates are then blocked with 300 uL/well of 1% BSA in PBS for 1 hour. The wells are then washed again, as before. 100 uL of sample lysate in IC Diluent #12 buffer (R&D Systems, DYC002) was added and incubated for 2 hours at room temperature, with appropriate blanks. The wash step is repeated, and 100uL of HRP-conjugated detection antibody is added and incubated for 2 hours at room temperature. The plates are washed again, and 100uL of a 1:1 mixture of H20 2 + Tetramethylbenzidine (R&D Systems, DY999) is added as substrate and incubated for 20 minutes at room temperature. 50uL of 2N H2SO 4 is then added to stop the reaction, and the ELISA plates are read with a fluorescent plate reader (Spectramax Gemini) at 450 nm, with wavelength correction set at 540 nm. IC Diluent #12 buffer was used as blank and subtracted from the readings from each well. 4. Time Courses and IC50 Experiments Optical 96 well plates (for ICW) or tissue culture treated 12 well plates (for ELISA) were seeded with cells (see tissue culture for numbers) in 200uL total volume of media. Edge wells were not seeded and 200 uL PBs was added to those wells to maintain uniform plate conditions. Semi-permeable membranes were used to seal plates to prevent evaporation, and the cells were incubated overnight at 37 0C. Prior to starting the experiments, the growth media was aspirated and the cells were serum-starved with 100 uL of media containing 0.1% fetal bovine serum for 12-14 hours. For experiments that included inhibitors, the media from the wells was aspirated and replaced with 100 uL of serum-free media containing the correct concentration of the particular inhibitor, and returned to the incubator for the inhibitor pre-treatment period (see results section for pre-treatment times). For time course experiments, plates were removed from the incubator 15 seconds before each time point. 100 uL of serum-free media containing ligand at 2x concentration was added to the corresponding time point wells such that the final concentration of ligand is lx. The plates were then returned to the incubator. The longest time points were treated first, then the next longest time point, and so on until the zero time point, at which time the entire plate was stopped at once. For ELISA experiments the plates were stopped with 40 C PBS and placed on ice while the other plates are being treated, and for ICW experiments the media from all wells was replaced with 150 uL 4% formaldehyde in PBS and incubated for 20 minutes at room temperature. For IC50 experiments, the wells were treated with different concentrations of inhibitor for the same amount of time, and then stopped at the same time. 5. Cell culture HeLa cells were maintained in DMEM supplemented with 10% fetal bovine serum and glutamine. For ICW experiments HeLa cells were plated at 15,000 cells/well in 96 well plates. For ELISA experiments HeLa cells were plated at 50,000 cells/well in 12 well plates. H1666 cells were maintained in ACL-4 medium supplemented with 10% fetal bovine serum. For ICW experiments H1666 cells were plated at 15,000 cells/well in 96 well plates. For ELISA experiments H1666 cells were plated at 50,000 cells/well in 12 well plates. H3255 cells were maintained in ACL-4 medium supplemented with 10% fetal bovine serum. For ICW experiments H3255 cells were plated at 10,000 cells/well in 96 well plates. A431 cells were maintained in DMEM supplemented with 10% fetal bovine serum and glutamine. For ICW experiments A431 cells were plated at 15,000 cells/well in 96 well plates. Cell culture conditions and optimal seeding numbers were determined from previous experiments (data not shown). III. Results 1. Optimization of using phosphatase inhibitors PTP1B Inhibitor, Endothall, Okadaic Acid - Time course In a first-pass experiment, we measured the response of inhibitor pre-treated H1666 cells following EGF and HRG ligand stimulus, over a time course of 60 minutes, with time points at 0, 5, 10, 15, 30, and 60 minutes (see methods section for time course experimental design). The cells were pre-treated for 5 minutes with the PTP1B Inhibitor at 100uM, Okadaic acid at 100nM, and Endothall at 100uM. The H1666 cells were then stimulated at time zero with saturating ligand concentrations, with EGF at 100ng/mL, and HRG at 50 ng/mL. We sampled the response using intracellular signals for phospho-EGFR, phosphoERK, and phospho-AKT (Figure 1). The control H1666 cells were treated with either EGF or HRG ligand at time zero, without pre-treatment by inhibitor. The phospho-signals for the control H1666 cells exhibited trends similar to previous experiments on these cells. Phospho-EGFR was up-regulated in EGF-stimulated but not HRG-stimulated cells, with a transient response in EGF-stimulated cells that peaked around 15 minutes and then down-regulated over time. Phospho-ERK had a transient response that mostly dropped back to baseline at 30 minutes for both EGF- and HRG-stimulated cells, and phospho-AKT signals had a small transient peak in EGFstimulated cells and a much stronger, more sustained peak in HRG-stimulated cells. No data was available for Okadaic acid treated cells because the treatment seemed to have killed most of the cells and detached them from the well, with very few cells remaining in Okadaic acid treated wells at the end of the time course. This seemed to be a time dependent effect, with more and more of the cells falling off the plate at the later time points. Endothall, a PP2A inhibitor, did not seem to have much of an effect on the intracellular signals when compared with control cells, at the concentration and treatment protocol used. Most of the signals measured for endothall treated cells are very close to those for control cells. The PTP1B inhibitor, however, seemed to have a potent effect on HRG-stimulated H1666 cells. It inhibited phospho-ERK and phospho-AKT signaling when the cells are stimulated with HRG, but not when the cells are stimulated with EGF. The phospho-ERK signal in inhibitor-treated, HRG-stimulated cells was basically flat over the entire time course, while the phospho-AKT response was much reduced compared to control, with a much smaller up-regulation (less than half of the control signal) over the first 30 minutes, with a peak signal at 60 minutes that was still less than the control signal. The PTP1B inhibitor also seemed to inhibit the initial peak of the phospho-EGFR signal in EGFstimulated cells, as can be seen at the 5 and 10 minute time points. It also seemed to slightly up-regulate the phospho-ERK signal compared to control in EGF-stimulated cells, as can be seen at the 5 and 10 minute time points. PTPIB Inhibitor on Additional Cell Lines - Time course In order to compare and confirm the results from the PTP1B inhibitor in the previous experiment, we used the PTP1B inhibitor to pre-treat 3 additional cell lines for a time course experiment that included duplicate data points for each condition. A431, H1666, H3255, and HeLa cells were pre-treated with PTP1B Inhibitor for 1 hour at 250uM, and then the response of these cells following saturating EGF (100ng/mL) or HRG (50ng/mL) treatment was measured at 0, 5, 15, 30, 60, and 120 minutes. Only the phospho-signals for ERK and AKT were measured for these cells, because the phospho-EGFR signal for HRG stimulated cells was too small to be compared using In-cell Westerns. The cells were seeded at an appropriate density for each cell line, with the data normalized by DNA content for each well of cells (see methods section for cell seeding for each line). The phospho-signals for each treatment condition were compared by each cell line for phospho-ERK (Figure 3A) and phospho-AKT (Figure 3B). In H1666 cells, the PTP1B inhibitor results are qualitatively very similar to those of the first experiment they were used in (Figures 1 and 3 - H1666 pERK panels). As in the first experiment, the inhibitor down-regulated phospho-ERK and phospho-AKT signaling when the cells are stimulated with HRG, but not when the cells are stimulated with EGF. The phospho-ERK signal in inhibitor-treated, HRG-stimulated cells was much reduced compared to untreated controls, while the phospho-AKT response was down-regulated compared to control, with a much smaller up-regulation (less than half of the control signal) over the first 30 to 60 minutes. The inhibitor-treated cells also seems to have a reduced baseline for phospho-ERK compared to untreated cells (Figure 3A, H1666 panel), with the inhibitortreated cells having half the signal of untreated cells at time zero. This effect was not observed for phospho-AKT in H 1666 cells. In A431 cells, the PTP1B inhibitor treated cells had much decreased phospho-ERK signals compared to untreated cells over the entire 120 minute time course for HRG stimulation, while inhibitor treated cells had only a slight decrease in signal (about 15%) compared to untreated cells for EGF stimulation (Figure 3A, A431 panel). The inhibitor abolished HRG-stimulated phospho-ERK signal in A431 cells almost completely. Unlike in H1666 cells, the phospho-ERK signal baseline at time zero was the same between inhibitortreated and untreated cells. The phospho-AKT signal in A431 cells was slightly elevated for PTPIB inhibitor treated cells compared to untreated cells for both the EGF and HRG cases (Figure 3B, A431 panel). However, the baseline signal at time zero was slightly elevated for phospho-AKT in inhibitor-treated cells compared to control. In H3255 cells, for phospho-ERK, inhibitor treatment abolished much of the HRGstimulated signal compared to control (Figure 3A, H3255 panel). The activation peak for inhibitor-treated, EGF-stimulated signal was also reduced in duration compared to untreated cells, dropping to a plateau at 15 minutes, while untreated cells had a sustained phosphoERK signal that decreased over the entire 120 minute time course. The drop in the baseline phospho-ERK signal at time zero seen in H1666 cells was also observed for H3255 cells. Like in H1666 cells, the phospho-AKT signal for H3255 cells was down-regulated in inhibitor treated cells compared to untreated cells for the HRG ligand (Figure 3B, H3255 panel). Unlike in H1666 cells, the EGF ligand stimulated signal was increased in inhibitor treated cells compared to untreated cells. However, this could have been due to the elevated baseline signal in inhibitor-treated H3255 cells for phospho-AKT, which was not seen in H1666 cells. For HeLa cells, like in H1666 cells, the phospho-ERK signal was reduced in PTP1B inhibitor-treated cells compared to untreated cells for HRG-stimulated cells but not for EGFstimulated cells (Figure 3A, HeLa panel). The phospho-ERK signal baseline for HeLa cells, however, was increased in inhibitor-treated cells, not decreased like in H1666 cells. Also unlike in H1666 cells, the phospho-AKT signal was increased for inhibitor treated cells compared to untreated cells, not decreased as in other cell lines (Figure 3B, HeLa panel). Both the peak signal and duration were increased for HRG-stimulated, inhibitor-treated cells compared to control, while just the peak signal was increased for EGF-stimulated, inhibitortreated cells compared to control. The baseline signal for phospho-AKT was about the same between inhibitor-treated and untreated HeLa cells. PTP1B Inhibitor, Okadaic Acid, Vanadate - Time course We used additional inhibitors in a first-pass experiment, to increase the panel of available phosphatase inhibitors for perturbing the ErbB system. Okadaic acid and vanadate were the two general phosphatase inhibitors used in this experiment, and the PTP1B inhibitor was included as it had been used in previous experiments and demonstrated an interesting perturbation of cellular signals. The phospho-signals for ERK and AKT were tracked at 0, 5, 15, and 30 minutes post-EGF stimulation, for the A431, H1666, H3255, and HeLa cell lines (Figure 4). We also included amphiregulin (AR), an EGFR ligand that we have had experience using in other signaling experiments in the lab. We used saturating amounts of each ligand for this experiment, with EGF at 100 ng/mL, HRG at 50 ng/mL, and AR at 50 ng/mL. The following protocol was used for those wells that included phosphatase inhibition. Cells were pre-treated with PTP1B Inhibitor for 1 hour at 250uM. Okadaic acid was used at 1 uM. Pervanadate, the activated form of inorganic vanadate, was added at luM. Okadaic acid and pervanadate were added together with the ligand, at time zero, to each well with that experimental condition. As this experiment only had singlet data points for each experimental condition, we used data from previous PTP1B inhibitor treated, EGF and HRG stimulated phospho-ERK and phospho-AKT signaling time courses as a baseline comparison, and only noted large differences in time course trends between inhibitor treated and untreated cells for Okadaic acid and pervanadate. In all four cell lines looked at, the PTP1B inhibitor-treated cells behaved similarly to previously described trends when compared to control untreated cells for EGF and HRG stimulation, with similar differences in signaling profiles and up- or down- regulation of baselines. In all four cell lines, EGF- and HRG- stimulation resulted in phospho-ERK and phospho-AKT signaling profiles similar to those previously described (Figure 4A-D). Amphiregulin, as another EGFR-specific ligand, resulted in similar signaling profiles to EGF stimulation for phospho-ERK signals for all four cell lines. However, AR stimulation resulted in differences in phospho-AKT signal profiles compared to EGF stimulation. H1666 cells had almost no phospho-AKT signals when stimulated with AR compared to a low but detectable phospho-AKT peak when stimulated with EGF (Figure 4B, EGF and AR panels). H3255 cells had a shorter phospho-AKT peak that dropped back to baseline by 15 minutes when stimulated with AR, compared to a peak that was sustained past 15 minutes and only dropped back to baseline by 30 minutes when stimulated with EGF (Figure 4C, EGF and AR panels). HeLa cells also had almost no detectable phospho-AKT signal when stimulated with AR, compared to a definite signal peak for EGF-stimulated cells (Figure 4D, EGF and AR panels). A431 cells, however, had similar EGF and AR activation profiles for phospho-AKT (Figure 4A, EGF and AR panels). Additionally, AR-stimulated cells had similar sensitivities to PTP1B inhibitor pre-treatment with HRG-stimulated cells, with similar differences in signaling profiles and baseline differences between inhibitor-treated and untreated cells. Most of the Okadaic acid pre-treated cells behaved similarly to control untreated cells. The only large differences were a slight decrease in phospho-ERK signals for Okadaic acid treated H1666 cells compared to control H1666 cells, for EGF, HRG and AR stimulation (Figure 4B, ERK panels), and an earlier drop in signaling duration for phosphoERK for all three ligands and an earlier drop in phospho-AKT signal for EGF-stimulation, in HeLa cells (Figure 4D, ERK and AKT panels). The pervanadate, at the concentration used (1 uM), had a large effect on signaling profiles for all four cell lines, all three ligands, and both phospho-ERK and phospho-AKT signals. In all the conditions used in this experiment, it increased both the signal peak and duration for both phospho-ERK and phospho-AKT. The up-regulated signaling profiles are similar between each ligand, for each of the individual cell lines. 2. Co-treating cells with phosphatase inhibitors together with protein synthesis or kinase inhibitors Cycloheximide - Time course In a recent paper from Amit and Yarden et al. (Amit et al. 2007), they demonstrated that cycloheximide co-treatment of HeLa cells resulted in an up-regulation of EGF stimulated phospho-ERK, compared to HeLa cells treated with only EGF, as soon as the first hour post ligand stimulation. Amit et al. showed that this could have been due to a translational upregulation of phosphatases, possibly DUSPs, in response to ligand stimulation. We wanted to confirm this in our cell lines, so we did an experiment where we used EGF to stimulate HeLa and H1666 cells, with or without cycloheximide co-treatment, over a time course sampled at 0, 5, 15, 30, 60, and 120 minutes. We measured phospho-EGFR, phospho-ERK, and phospho-AKT signals, with 2 repeats for each condition [Figure 2]. The lines between each data point are best-fit spline approximations generated using Origin software. The figure from Amit et al. showing their phospho-signal time course was included for comparison. The EGF-stimulated phospho-EGFR, phospho-ERK, and phospho-AKT signals we obtained for this time course are similar to those of previous experiments. H1666 cells showed a low but detectable phospho-EGFR signal that peaked around 5 minutes and dropped over 30 to 60 minutes, a phospho-ERK signal that peaked at 5 minutes and then dropped quickly to a plateau, and a low but detectable phospho-AKT peak. HeLa cells, by comparison, showed phospho-EGFR signals that were almost not detectable by this In-cell Western protocol, a strong phospho-ERK signal that peaked around 10 to 15 minutes, and a low but detectable phospho-AKT signal that peaked around 5 minutes. When Amit et al. co-treated HeLa cells with EGF and cycloheximide, they saw a large and sustained increase in their phospho-ERK signal, as well as a small increase in their phospho-EGFR signal. However, cycloheximide co-treatment, in our experiments, had only a small effect on our HeLa and H1666 cells. Cycloheximide co-treated HeLa cells had a small increase in their phospho-ERK and phospho-AKT signals (less than 15%) past the signal peaks, from the 30 minute time point until the 120 minute time point, and the phospho-EGFR signals were too small to be compared for HeLa cells. On the other hand, cycloheximide cotreated H1666 cells actually had decreased phospho-ERK and phospho-AKT signals compared to H1666 cells only stimulated with EGF ligand, with a decrease in signal of about 10-20% for phospho-ERK at all time points past 5 minutes, and a decrease in signal of about 10% for phospho-AKT between the 5 and 60 minute time points. The phospho-EGFR signal was about the same between cycloheximide co-treated and untreated H1666 cells. Iressa and Vanadate - Time course We performed an Iressa-chase experiment where we initially stimulated H1666 and H3255 cells with EGF at 100 ng/mL, then added either a saturating amount of Iressa (16 uM), 1 uM of pervanadate, or both, at 10 minutes post EGF-stimulation. Phospho-EGFR, phospho-ERK, phospho-SHC, and phospho-AKT signals were sampled at 0 (ligandstimulation), 5, 10 (treatment with or without Iressa and pervanadate), 15, 30, and 60 minutes. Each condition was duplicated in a second identically treated well of cells, and the results for the four phospho-signals are graphed for H1666 (Figure 6A) and H3255 (Figure 6B). Because the phospho-signals from cells treated with pervanadate at 10 minutes are much greater in magnitude than cells untreated with pervanadate, the results for pervanadate treated cells are graphed separately for H1666 (Figure 6C) and H3255 (Figure 6D), with the control cells and Iressa-only cells there as well for comparison. For both H1666 and H3255 cells, addition of Iressa at 10 minutes decreased the amount of phospho-signals almost immediately, at the next time point sampled (15 minutes). With the exception of phospho-ERK and phospho-AKT in H1666 cells, phospho-signals decreased to either baseline levels or below baseline and stayed at that level until 50 minutes post addition of Iressa, at the last time point sampled. For H1666 cells, addition of Iressa corresponded with dropping of the phospho-EGFR and phospho-SHC signals to baseline levels, while the phospho-ERK signal only decreased slightly compared to control cells, and the phospho-AKT signal was very similar in H1666 cells treated or untreated with Iressa at 10 minutes. In both H1666 and H3255 cells, addition of luM pervanadate alone at 10 minutes resulted in a large increase of all phospho-signals, with the exception of phospho-ERK signals in H3255 (Figure 6C and 6D). The phospho-signals either plateaued at a high level (higher than peaks normally seen with saturating amount of ligand), or continued to rise until the last time point sampled, 60 minutes. Co-addition of pervanadate with Iressa resulted in decreased signal up-regulation compared with addition of pervanadate alone for phosphoEGFR and phospho-AKT in H1666s and H3255s, while the phospho-ERK and phospho-SHC signals were similar for pervanadate-treated alone and co-treated cells in both cell lines. 3. Phosphatase inhibitors in phospho-ErbB ELISA assays PTP1B Inhibitor - Time course In this experiment, we used phospho-ErbB ELISA kits to measure phospho-ErbB signals in a time course experiment. We followed identical protocols for ligand-stimulation and phosphatase inhibitor treatment of the cells as the In-cell Western experiments, and measured the response of ErbB1-4 phosphorylation following 100 ng/mL of EGF or 50 ng/mL HRG stimulation of H1666 and HeLa cells, with or without PTP1B inhibitor pretreatment (Figure 5). After incubation for 5 minutes with 100 uM of PTP1B inhibitor, the time course was sampled at 0, 5, 15, 30, 60, and 120 minutes post ligand-stimulation. Each treatment had a single data point; however, this experiment was repeated on a separate day, with similar results (data not shown). For H1666 cells, EGF stimulation resulted in a sustained phospho-EGFR response which only dropped off at the 120 minute time point, and transient peaks for phospho-ErbBs 2, 3, and 4, that peaked at 5 minutes post-stimulation and then decreased over the time course (Figure 5A). HRG stimulation of H1666s resulted in transient peaks for phospho-ErbB2 and phospho-ErbB3 that peaked at 5 minutes and then decreased to a plateau above baseline over a period of approximately 25 minutes, but did not increase phospho-EGFR or phosphoErbB4 levels. Pre-incubation with the PTP1B inhibitor decreased the transient peak for EGFstimulated phospho-ErbBs 2, 3, and 4, and also abolished almost completely the transient peak seen for HRG-stimulated phospho-ErbB2 and phospho-ErbB3. Pre-incubation with the inhibitor also seemed to prolong EGF-stimulated phospho-EGFR signaling, but this was only seen at the 120 minute data point. The PTP1B inhibitor also increased the baseline of phospho-EGFR and phospho-ErbB4 signals in pre-treated cells. For HeLa cells, EGF stimulated time courses showed a large signal peak for phosphoEGFR and phospho-ErbB2 that peaked at 5-15 minutes, and a smaller signal peak for phospho-ErbB4 that peaked at 5 minutes, while phospho-ErbB3 did not seem to be stimulated by EGF (Figure 5B). HRG stimulation of HeLas resulted in a small transient peak for phospho-ErbB2 and phospho-ErbB3 that peaked at 5 minutes and dropped to about baseline over 25 minutes, while HRG stimulation of HeLas did not seem to stimulate phospho-EGFR or phospho-ErbB4. Pre-incubation with 100 uM of PTP1B inhibitor did not seem to affect either EGF or HRG stimulated phospho-ErbB signals in HeLa cells, except for a small decrease in the phospho-ErbB3 signal in HRG-stimulated, PTP1B inhibitor-treated cells. The PTP1B inhibitor also resulted in a slightly elevated baseline like in H1666 cells, but only for phospho-ErbB4 cells. IV. Discussion 1. Tools developed for studying phosphatase activity In this project a set of assays, cell lines, and reagents were tested and verified for the purpose of studying phosphatase activity in the context of EGFR signaling. Specifically, we validated In-cell Western and ELISA protocols for the study of intracellular signals resulting from ligands specific for ErbB family members, and tested both specific and general inhibitors for phosphatases that are known to play important roles in EGFR downstream signaling. We also tested and confirmed kinase-specific and translational inhibitors, different ways for using the inhibitors, and various ErbB ligands. These protocols will allow us to distinguish the contributions of phosphatase activity to different layers of the EGFR signaling network. Validation of In-Cell Western and ELISA assays We demonstrated that In-Cell Westerns and commercial phospho-ErbB ELISA kits could gather informative data for the purpose of elucidating the role of phosphatases in the ErbB network. Through these two techniques we were able to gather data on the phosphorylation states of the phospho-ErbB receptors and downstream activation of the Erk and Akt molecules. In-Cell Western assays were used in experiments that showed similar pEGFR, pERK, and pAKT trends to previous experiments for the same cell lines and conditions. In addition, we also gathered informative data using ICW assays for pERK and pAKT. It was seen in previous experiments that it was difficult to compare the response of ligand-stimulated EGFR phosphorylation for some of our cell lines such as HeLa's, due to the low signal of the In-cell Western assay for phospho-EGFR for those lines. Also, we did not have In-cell Western assays for phospho-ErbB2, phospho-ErbB3, or phospho-ErbB4, due to difficulties in finding suitable phospho-antibodies for those receptor moieties. However, we wanted to be able to measure the states for all phospho-ErbB receptors, in order to obtain a more complete signaling dataset for the receptor level following ligand stimulation. This was especially true in experiments involving ligands specific for other ErbB receptors than EGFR, such as heregulin, a ligand with high affinity for ErbB3, or ligands that involved different receptor heterodimerization and trafficking, such as amphiregulin. Therefore, in order to sample receptor-level signals, we used commercially available phospho-ErbB ELISA kits to measure phospho-ErbB signals in time course experiments that can complement and expand the In-cell Western signaling dataset. We demonstrated that it was possible to use phospho-ErbB ELISAs in identical experimental treatments as those of ICW assays, and gather information on the phosphorylation state of the receptors in parallel with phosphatase inhibitor treatment. Validation of Phosphatase Inhibitors In order to determine the combination of phosphatase inhibitors and experimental protocols that will demonstrate measurable effects and allow us to gather reproducible data in our cell lines, we tried available phosphatase inhibitors, both specific (PTP1B Inhibitor and endothall) and broad spectrum (okadaic acid and vanadate) inhibitors. For this purpose, time course experiments were used. Time courses can be used to show the effects of a perturbation on different signaling events occurring over a specific period of time following treatment or cell stimulation. In-cell Western was used for these experiments because of availability of existing verified reagents and protocols, in addition to its reproducibility. In our experiments we saw efficacy of a specific inhibitor for PTP1B, the allosteric inhibitor for PTPIB from Calbiochem. The PTPIB inhibitor we used had an effect on the receptor layer, which was both ligand and cell line dependent (Figure 5, also results PTPIB Inhibitor Time course). In addition, it had a large effect on downstream pERK and pAKT signals, which was again ligand and cell line dependent (Figure 3, also results PTP1B Inhibitor on Additional Cell Lines Time course). It is interesting that PTP1B inhibition resulted in up- or down-regulation of pERK and pAKT, depending on the ligand and cell line used. For example, the effect of the PTP1B inhibitor was much smaller in EGF-stimulated cells compared to HRG-cells. This inhibitor could be used, in combination with additional experimental conditions, to inform the ErbB network model about the differences in phosphatase regulation between different cell lines and receptor configurations. We wanted especially to find a general phosphatase inhibitor that can be used as a comparative baseline against specific phosphatase inhibitors such as the PTP1B inhibitor. It would be interesting to compare inhibition of much of the phosphatase activity in the cell over a short time course versus acute inhibition of just one specific phosphatase. Vanadate is another broad-spectrum inhibitor that has been shown to inhibit general protein tyrosine phosphatase activity through competitive inhibition in many systems (Shisheva and Shechter 1993; Krady et al. 1998). It had not been used in previous experiments because it had not shown effective perturbation of signals when used previously in our lab in kinase experiments with similar cells and assays. However, upon further investigation of literature we discovered that vanadate needs to be activated with peroxide in a manner that allows it to be cell-permeable and active in cell culture experiments (Huyer et al. 1997). We then attempted to use pervanadate, the activated form of inorganic vanadate, in order to see if it provides an interesting comparison to specific PTP1B inhibition. We saw that pervanadate co-treatment with EGF, HRG, or AR increased the pERK and pAKT signal intensity and duration, for all four cell lines. However, as the pervanadate was used to co-treat the cells along with each ligand, it was possible that the similarity between the three ligand-stimulated signaling profiles for each cell line was a result of the pervanadate treatment. Further investigation of the effects of pervanadate along with specific node inhibitors can help elucidate the role of the PTPs in the Erk and Akt pathways downstream of ErbBs 1-4. Another potent broad-spectrum phosphatase inhibitor we tried was okadaic acid. Okadaic acid has been shown to exert an inhibitory effect on many phosphatases of the PPP family, including PP2A (Zolnierowicz 2000; Perrotti and Neviani 2008). The first experiment we tried okadaic acid in, we pre-treated the cells for 5 minutes with it, and it seemed to kill most of the cells and no data was available because most of the cells detached from the plates. The second time we tried okadaic acid, at a lower concentration (100 nM), with no pre-incubation time, we did not see much of an effect on downstream molecules (data not shown). We tried okadaic acid in a third experiment, while it had not shown much efficacy in our hands, due to its demonstrated effects in the literature. We used a new aliquot of this reagent, at a higher concentration, in order to make sure that it does not perturb the signals we are measuring in a significant manner. Although we used ten times the concentration previously used (1 uM in this experiment, versus 100 nM in the previous experiment), it still did not seem to have a significant effect on phospho-ERK and phosphoAKT signaling profiles at the conditions we used in this experiment, for all four cell lines and EGF, HRG, or AR stimulation. This could be due to incorrect pre-incubation times, or that okadaic acid in the form we used was not cell permeable for the lines we used. Reagents and protocols for different ErbB network layers In previous experiments, co-treating with ligand and phosphatase inhibitors resulted in signaling responses that are a combination of the effects of ligand-stimulated receptor phosphorylation and perturbation of steady-state phosphatase activity. In order to distinguish between the contributions of ligand-stimulation and phosphatase inhibition, we used an Iress-chase experiment where we stimulated cells with ligand, then added a saturating amount of Iressa to shut down receptor phosphorylation, with or without pervanadate, to co-inhibit phosphatase function. We saw that adding Iressa decreased phospho-signals almost immediately. In addition, co-addition of pervanadate with Iressa resulted in up-regulation of almost all phospho-signals. This type of experiment is interesting because it could be used to deduce dynamic information about the downregulation of pERK and pAKT by relevant phosphatases, following a pulse of signaling from the receptor layer. We also wanted to sample signals following stimulation for another EGFR ligand that's reported to have different effects on receptor-trafficking following ligand stimulation. For this purpose we used amphiregulin (AR), an EGFR ligand that has been reported to have different heterodimerization and trafficking effects compared to EGF, even though both ligands' favored receptor is EGFR (Salomon et al. 1995; Berasain et al. 2007). We saw that when cell lines are stimulated with AR, it resulted in distinct pERK and pAKT signaling profiles when compared to signaling profiles from cell lines stimulated with either EGF or HRG. Additionally, AR-stimulated cells had similar sensitivities to PTP1B inhibitor pre-treatment with HRG-stimulated cells, with similar differences in signaling profiles and baseline differences between inhibitor-treated and untreated cells. This could be due to the effects of differential receptor trafficking for AR-stimulated cells, and possibly help elucidate the cellular localization of PTP1B in ErbB signaling. In addition, we have previously used cycloheximide in EGF-stimulated signaling experiments, to show that translational responses do not play a big role in EGFR downstream signaling, for the cell lines we are interested in, and at the time scales we are looking at. A recent report (Amit et al. 2007) showed that in HeLa cells, the ligandstimulated translational up-regulation of phosphatases, such as DUSPs, could be significant in the signaling response even as soon as the first 30-60 minutes post ligand addition. We showed that this result is probably not valid in our cell lines and experimental conditions (Figure 2). 2. Future work In the future, to help gain a quantitative and mechanistic understanding of the role of phosphatases in the cellular system we can employ a combined approach with computational modeling. A current ordinary differential equations (ODE) model of the ErbB receptor pathway being developed in our lab can be used to help analyze and understand the data from these experiments. Further model characterization and calibration can be done as well with the new experimental results. In addition, model generation and characterization using other methods than the existing ODE model can also be investigated with the phosphatase experimental results, with model generation methods being investigated by other collaborators. To further calibrate the model we can also use quantitative total- and phospho-ErbB ELISAs to measure receptor dynamics, and high-throughput Westerns for absolute quantitation of species in the model. This will enable us to: 1) Validate current model results and 2) Make the model more predictive by encoding behaviours from additional ligandreceptor interactions. The panel of measurements can also be expanded later to include more receptor-proximal signaling molecules such as PIP3K and phospho-MEK, or further downstream readouts such as transcription factor activation, using the Luminex antibodybead based system. We can also expand our panel of phosphatase inhibitors, and look at other interesting phosphatases such DUSPs. Finally, we can also use different concentrations of different phosphatase inhibitors in parallel, to demonstrate dose-dependent effects in IC50 experiments. V. References Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T (2004) Protein tyrosine phosphatases in the human genome. Cell 117(6): 699-711. Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, Tarcic G, Siwak D, Lahad J, Jacob-Hirsch J, Amariglio N, Vaisman N, Segal E, Rechavi G, Alon U, Mills GB, Domany E, Yarden Y (2007) A module of negative feedback regulators defines growth factor signaling. Nat Genet 39(4): 503-512. Bentires-Alj M, Neel BG (2007) Protein-tyrosine phosphatase lB is required for HER2/Neuinduced breast cancer. Cancer Res 67(6): 2420-2424. Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, Richards WG, Du J, Girard L, Minna JD, Loh ML, Fisher DE, Velculescu VE, Vogelstein B, Meyerson M, Sellers WR, Neel BG (2004) Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res 64(24): 8816-8820. Berasain C, Castillo J, Perugorria MJ, Prieto J, Avila MA (2007) Amphiregulin: a new growth factor in hepatocarcinogenesis. Cancer Lett 254(1): 30-41. Bourdeau A, Dub6 N, Tremblay ML (2005) Cytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Curr Opin Cell Biol 17(2): 203-209. Boutselis IG, Yu X, Zhang ZY, Borch RF (2007) Synthesis and cell-based activity of a potent and selective protein tyrosine phosphatase lB inhibitor prodrug. J Med Chem 50(4): 856-864. Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410(6824): 37-40. Chernoff J, Schievella AR, Jost CA, Erikson RL, Neel BG (1990) Cloning of a cDNA for a major human protein-tyrosine-phosphatase. Proc Natl Acad Sci U S A 87(7): 2735-2739. Combs AP, Zhu W, Crawley ML, Glass B, Polam P, Sparks RB, Modi D, Takvorian A, McLaughlin E, Yue EW, Wasserman Z, Bower M, Wei M, Rupar M, Ala PJ, Reid BM, Ellis D, Gonneville L, Emm T, Taylor N, Yeleswaram S, Li Y, Wynn R, Bum TC, Hollis G, Liu PC, Metcalf B (2006) Potent benzimidazole sulfonamide protein tyrosine phosphatase 1B inhibitors containing the heterocyclic (S)-isothiazolidinone phosphotyrosine mimetic. J Med Chem 49(13): 3774-3789. Dhillon AS, von Kriegsheim A, Grindlay J, Kolch W (2007) Phosphatase and feedback regulation of Raf-1 signaling. Cell Cycle 6(1): 3-7. Dillon RL, White DE, Muller WJ (2007) The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene 26(9): 1338-1345. Easty D, Gallagher W, Bennett DC (2006) Protein tyrosine phosphatases, new targets for cancer therapy. Curr Cancer Drug Targets 6(6): 519-532. Franke TF (2008) Intracellular signaling by Akt: bound to be specific. Sci Signal 1(24): pe29 Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81(5): 727-736. Gomez N, Cohen P (1991) Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 353: 170-173. Haj FG, Markova B, Klaman LD, Bohmer FD, Neel BG (2003) Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-iB. J Biol Chem 278(2): 739-744. Hatakeyama M, Kimura S, Naka T, Kawasaki T, Yumoto N, Ichikawa M, Kim JH, Saito K, Saeki M, Shirouzu M, Yokoyama S, Konagaya A (2003) A computational model on the modulation of mitogen-activated protein kinase (MAPK) and Akt pathways in heregulin-induced ErbB signalling. Biochem J 373(Pt 2): 451-63. Hunter T (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 28(5): 730-738. Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C (1997) Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem 272(2): 843-851. Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kiihiiri VM, Heino J (2002) Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol Cell Biol 22(5): 1352-1359. Jiang ZX, Zhang ZY (2008) Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev 27(2): 263-272. Julien SG, Dub6 N, Read M, Penney J, Paquet M, Han Y, Kennedy BP, Muller WJ, Tremblay ML (2007) Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet 39(3): 338-346. Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB (2000) Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol 20(15): 5479-5489. Knight ZA, Shokat KM (2005) Features of selective kinase inhibitors. Chem Biol 12(6):621-637. Krady MM, Malviya AN, Dupont JL (1998) Pervanadate-triggered MAP kinase activation and cell proliferation are not sensitive to PD 98059. Evidence for stimulus-dependent differential PD 98059 inhibition mechanism. FEBS Lett 434(3): 241-244. Lechward K, Awotunde OS, Swiatek W, Muszyi6ska G (2001) Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim Pol 48(4): 921-933. Lee S, Wang Q (2007) Recent development of small molecular specific inhibitor of protein tyrosine phosphatase lB. Med Res Rev 27(4): 553-573. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275(5308): 1943-1947. Mauro LJ, Olmsted EA, Skrobacz BM, Mourey RJ, Davis AR, Dixon JE (1994) Identification of a hormonally regulated protein tyrosine phosphatase associated with bone and testicular differentiation. J Biol Chem 269(48): 30659-30667. Murphy LO, Blenis J (2006) MAPK signal specificity: the right place at the right time. Trends Biochem Sci 31(5): 268-275. Ostman A, Hellberg C, Bbhmer FD (2006) Protein-tyrosine phosphatases and cancer. Nat Rev Cancer 6(4): 307-320. Perrotti D, Neviani P (2008) Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Phl(+) leukemias. Cancer Metastasis Rev 27(2): 159-168. Peterson RT, Desai BN, Hardwick JS, Schreiber SL (1999) Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc Natl Acad Sci U S A 96(8): 4438-4442. Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26(22): 3291-310. Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68(2): 320-344. Rubinfeld H, Seger R (2005) The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol 31(2): 151-174. Salomon DS, Normanno N, Ciardiello F, Brandt R, Shoyab M, Todaro GJ (1995) The role of amphiregulin in breast cancer. Breast Cancer Res Treat 33(2): 103-114. Schaefer G, Shao L, Totpal K, Akita RW (2007) Erlotinib directly inhibits HER2 kinase activation and downstream signaling events in intact cells lacking epidermal growth factor receptor expression. Cancer Res 67(3): 1228-1238. Shisheva A, Shechter Y (1993) Mechanism of pervanadate stimulation and potentiation of insulin-activated glucose transport in rat adipocytes: dissociation from vanadate effect. Endocrinology 133(4): 1562-1568. Schoeberl B, Eichler-Jonsson C, Gilles ED, Muller G (2002) Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat Biotechnol 20(4): 370-375. Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 9(1): 59-71. Tiganis T (2002) Protein tyrosine phosphatases: dephosphorylating the epidermal growth factor receptor. IUBMB Life 53(1): 3-14. Tonks, NK (2006). Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Molecular Cell Biology 7: 833-846. Virshup DM (2000) Protein phosphatase 2A: a panoply of enzymes. rr Opin Cell Biol 12(2): 180-185. Widmann CS, Gibson S, Matthew BJ, Johnson G (1999). Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiology Rev 79(1): 143-180. Wiener JR, Kerns BJ, Harvey EL, Conaway MR, Iglehart JD, Berchuck A, Bast RC Jr (1994) Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression. J Natl Cancer Inst 86(5): 372-378. Yang ZZ, Tschopp O, Baudry A, Dummler D, Hynx D, Hemmings BA (2004) Physiological functions of protein kinase B/Akt. Biochem Soc Trans 32: 350-354. Yudushkin IA, Schleifenbaum A, Kinkhabwala A, Neel BG, Schultz C, Bastiaens PI (2007) Live-cell imaging of enzyme-substrate interaction reveals spatial regulation of PTP1B. Science 315(5808): 115-119 Zhang ZY, Lee SY (2003) PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. Expert Opin Investig Drugs. 12(2): 223-233. Zheng XM, Wang Y, Pallen CJ (1992) Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 359(6393): 336-339. Zhou H, Li XM, Meinkoth J, Pittman RN (2000) Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol 151(3): 483-494. Zhu S, Bjorge JD, Fujita DJ (2007) PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res 67(21): 10129-10137. Zolnierowicz S (2000) Type 2A protein phosphatase, the complex regulator of numerous signaling pathways. Biochem Pharmacol 60(8): 1225-1235. VI. Figure captions For all time course In-Cell Western experiments, phospho-intensity as a ratio of the 800 nm (phospho-signal) channel versus the 700 nm (DNA stain) channel is plotted on the y-axis, and time in minutes on the x-axis. Figure 1. PTP1B Inhibitor Time course - ICW Left panels: pEGFR (top), pERK (middle), pAKT (bottom) time courses following 100 ng/ mL EGF treatment. Each colored line is for a different inhibitor treatment. Right panels: pEGFR (top), pERK (middle), pAKT (bottom) time courses following 50 ng/ mL HRG treatment. Each colored line is for a different inhibitor treatment. Figure 2. Cycloheximide comparison Time course - ICW (A) H1666 cells, pEGFR, pERK, and pAKT signals time course, following 100 ng/mL EGF treatment, with or without cycloheximide (CHX) pre-treatment. Each colored line is for a different phospho-signal, with or without CHX. (B) HeLa cells, pEGFR, pERK, and pAKT signals time course, following 100 ng/mL EGF treatment, with or without cycloheximide (CHX) pre-treatment. Each colored line is for a different phospho-signal, with or without CHX. (C) HeLa cells signal time course from Amit et al. 2007 for comparison. Figure 3. PTP1B Inhibitor Time course for Erk and Akt - ICW (A) Phospho-Erk signal profiles for A431 (upper left), H1666 (upper right), H3255 (lower left), and HeLa (lower right), following 100 ng/mL EGF (black and red lines) or 50 ng/mL HRG (blue and green lines) treatment. The red and green lines represent data from wells pre-treated with 250 uM of PTP1B inhibitor for 1 hour. (B) Phospho-Akt signal profiles for A431 (upper left), H1666 (upper right), H3255 (lower left), and HeLa (lower right), following 100 ng/mL EGF (black and red lines) or 50 ng/mL HRG (blue and green lines) treatment. The red and green lines represent data from wells pre-treated with 250 uM of PTP1B inhibitor for 1 hour. 44 Figure 4. PTP1B Inhibitor, Okadaic Acid, and Vanadate - Time course - ICW Phospho-ERK (top panels) and phospho-AKT (bottom panels) signal profiles. The cells were treated with 100 ng/mL EGF (left panels), 50 ng/mL HRG (middle panels), or 50 ng/mL AR (right panels). Each colored line represent a different inhibitor treatment: untreated (black), 250 uM PTP1B inhibitor (red), Okadaic acid (blue), and pervanadate (green). (A) A431 cells (B) H1666 cells (C) H3255 cells (D) HeLa cells Figure 5. PTP1B ELISA for Phospho-ErbB1-4, Time course - ELISA Phospho-ErbB1-4 signals were plotted as the intensity reading, corrected for background, on the y-axis, and time in minutes on the x-axis. (A) H1666 cells, phospho-signals for EGFR (upper left), ErbB2 (upper right), ErbB3 (lower left), and ErbB4 (lower right), following 100 ng/mL EGF (black and red lines) or 50 ng/mL HRG (blue and green lines) treatment. The red and green lines represent data from wells pre-treated with 250 uM of PTP1B inhibitor for 1 hour. (B) HeLa cells, phospho-signals for EGFR (upper left), ErbB2 (upper right), ErbB3 (lower left), and ErbB4 (lower right), following 100 ng/mL EGF (black and red lines) or 50 ng/mL HRG (blue and green lines) treatment. The red and green lines represent data from wells pre-treated with 250 uM of PTP1B inhibitor for 1 hour. Figure 6. Iressa then Vanadate-chase Time course - ICW Cells were treated with 100 ng/mL of EGF at time = 0, and then with or without 16 uM Iressa at 10 minutes. The error bars represent standard error from duplicate experimental wells. Phospho-signals were measured for EGFR (upper left), ERK (upper right), AKT (lower left), and SHC (lower right). Each colored line represent a different inhibitor treatment: untreated (black), 16 uM Iressa at 10 minutes (red), pre-treated with pervanadate + 16 uM Iressa at 10 minutes (blue), and pervanadate pre-treatment alone (green). Since the pervanadate treated cells resulted in much higher signals, each cell line is plotted twice, with or without the pervanadate pre-treated signal profiles, to better show the difference between untreated and Iressa at 10 minute treated signals. (A) H1666 (B) H3255 (C) H1666 replot, with the pervanadate treated signals (D) H3255 replot, with the pervanadate treated sig als VII. Figures Fig. 1 - PTP1B Inhibitor Time course HRG 0.55- 0.550.50EGF 0.45- 0.400.450.50-. OAS. 0640- 0.200.30025- 0.20- 0.20. 0 1o 2o 3• 40 50 60 0 1o 20 lime (mini.) 3o 40 5o 60 rime (min.) EGF I--PTpSlki1 1.e. 1.0- HRG Enota 1.6. 1.6- 1A. 1A- 12- 12- 1.0. 1.0- 0.60.6- o.,- 0.4U.,. 0 1o 20 0i r 0 Time (mmin.) 0 60 Mz 0 1o 2o io ai0 rime (min.) 50 0 EGF . ..... * .. 1.6. PT~~I~u HRG 16. 1.4 1.4- 12. 1.2- 1.0 1.0 I- I IS0.8. 0.6 0.6. 0.4OA 02 I 0.2. 0 10 20 30 40 50 40 0 Time (min.) 10 20 30 Time(min.) 48 40 50 60 TP1 Elnowalw Fig. 2 - Cycloheximide comparison Time course A. B. HeLa H1666 --- EGFR -v- EGFR +CHX --- Erk HX "1) "I, 2.2- 2.0. 2.0- 1.8- 1.8- 1.6. 1.6- 1.4. 1.4- ~1.2- C 1.2- 1.0. I -X 1.0- 0.8- 0.8- 0.6- 0.6- 0.4- 0.4 0.2- 027 0 20 40 60 80 lime (mins) 100a 120 0 20 SEGF fIm 40 60 80 100 120 LcbhedmKide r. 6.n0 A4Da pErk pJNK pp3 8 pEGFR pSIATS pMTOR Jun 0.1 Amit, Yarden et al 2007 Fig. 3 - PTP1B Inhibitor Time course for Erk and Akt A. phospho-Erk pErk A431 -u-EGF -- EGF.Inh Inh = w/PTP1B Inhibitor, 250uM, 1hr pre-treat H1666 E -- HRG HRG.InhI 3.0- EGF.Inh HRG - G In.h .......... I 2.5 2.0- 1.5. l 1.5 1. 1.0. 0.5. O.S 0 20 40 60 80 100 120 0 20 Time (mins) 40 60 80 100 120 Time (mins) H3255 HeLa EGF EGF EGF.Inh EGF.Inh -- HRG HRG 6.0- IRG.Inh 5.5- RG.Inh 5.0- 2.0 454.0- 1.5 S1 ,"3.5 a' 3.0. 2) 0 1.0 0.5. 1.5. 1.00 20 40 60 Time (mins) 0• 100 120 0 20 40 60 Time (mins) 80 100 120 B. phospho-Akt pAkt A431 EGF Inh = w/PTP1B Inhibitor, 250uM, 1hr pre-treat H1666 EGF.Inh 2.0- -OEGF.Inh ý RGý RG.In_ HRG -q- HRG.Inh 2.0- | 1.5 1.0- 1.0. 0. 0.5- · 0 · 20 _·_·····_·· 40 60 w0 100 0 120 20 40 60 80 100 120 Time (mins) Time (mins) H3255 EGF EGF.Inh HRG IRG.Inhl 5.5. 5.0. HeLa 1.2. -- EGF EGF.Inh HRG IRG.Inh 1.0- 4.5. 4.0. 0.8- 3.53.0. a 0.6- C.2.5- 2.0- 0.4- 1.5. 1.0. 0.2- U.320 40T Time (mins) k 1;0 120 0 2 40T0 0 Time (mins) 100 120 Fig. 4 - PTP1B Inhibitor, Okadaic Acid, and Vanadate - Time course A. A431 A431 ERK EGF S-4- EGF.PTP1B.Inh SHRG S-4- HRG.PTP1Binh -HRG.OA 5.0- -v- HRG.Vanadate 5.5- 5.0 4.5 45. 4.0 4.0. 3.5 3.5. 3.0 3.0- ~2-3 2.0 7-0- 1.50 15- 1.0. 1.0. 0.5 0.0 03. , ' 0 5 10 15 ' 20 ~ ' 25 ' ' 30 0 5 Time (mins) 10 15 20 25 30 Time (mins) Time (mins) AKT -*-EGF 25 EGFFTPTBInh is5 -9-- EGF.Vadate EGF.Vanadate - -- HRG HRG.PTP1B.Inh -- AR --- AR.PTP1B.Inh 5 - HRGOA .- AR.OA -v- AR.Vanadate I 0 5 0 15i 20 25 3 rime(mins) 0 s '~'~'~'' 10 I Time (mins) 1 20 25 30 0 5 10 I5 Time (mins) 20 25 3 B. H1666 H1666 ERK - ARPT B. -~AR PTP1B.Inh 0 1 ,- 0 5 10 i (mins) 3 S rume(mins) 5 Time (mins) 10 15Tim ins)20 rime (mins) 25 AKT -0-AR -,- AR.PTPIB.Inh -- AR.OA -,- AR.Vanadate 0 Time (mins) 5 10 15 Trme (mins) 20 25 30 0 5 10 15 20 Time (mins) 25 30 C. H3255 H3255 ERK - -EGF EGF.PTP1B.Inh 12. --I- EGF.OA --- EGF.Vanadate -U-HRG --I nn h.T - HRG.OA -rHRG Vanadate 12- -v- 10. 10. 10- 8. 8- 8- 6. 6. S6- 4. 4. 4- 2. 5 10 15 20 25 3 0 5 10 15 20 lime (mins) Time (mins) AR AR.PTP1B.Inh AR.OA AR.Vanadate 2- 2- d 0 ----- 30 o me10 15 rime (mins) AKT 0- -0- EGF EGF.PTP1B.Inh -- EGF.Vanadate I--EGF.OA 7- 0 5 10 15 Time (mins) 20 25 30 Time (mins) Time (mins) 20 2 3 D. HeLa HeLa ERK 5.04.5- ----9- EGF EGF.PTP1B.Inh EGF.OA EGF.Vanadate ------ -- HRG HRG.PTPIB.Inh HRG.OA HRG.Vanadate 5.0. --- 4.5 4.0- 4.0- 4.0- 3.5- 3.5- 3.5. 3.0- 3.0- 3.0 w 2.5- wL 2.5- w 2.5 10 2.0- 2.0- 2.0 1.5- 1.5- 1.5- 1.0- 1.0- 1.0- V..'ý 0 5 10 15 20 25 0.5 30 0 5 Time (mins) 10 --- 15 20 25 0 30 AR AR.PTPIB.Inh AR.OA AR.Vanadate 5 Time (mins) 1o 15 20 25 30 Time (mins) AKT 3.0. -e-- -V- EGF EGF.PTP1B.Inh EGF.OA EGF.Vanadate -* -&- -,- HRG HRG.PTP1B.Inh HRG OA HRG.Vanadate -U-AR -- *-4.AR.PTP1Binh --- AR.OA --- 2.5 AR.Vanadate 25- 2.0. 2.0 4 1.5. < 1.5 1.0- 1.0* O.S 05" 0 5 10 15 Time (mins) 20 25 30 2.0- 0 5 10 15 Time (mins) 20 25 30 0 5 10 15 20 25 30 Time (mins) Fig. 5 - PTP1B ELISA for Phospho-ErbB1-4, Time course A. H1666 08-29-07 PTP1B Inhibitor ELISA H1666 EGFR ErbB2 -,ar.•- 5 2.02.0- - 0 EGF --EGFInh -A- HRG v- HRGInh 1.5- 1.5- -- EGFF .-..- EGFInh HRG HRGInh W 1.0- 01.0- 05 0.5. U.U 0 10 20 30 40 50 60 120 0 0 Time (mins) 20 30 40 50 60 120 Time (mins) ErbB3 ErbB4 1.4. 1.2-*----- 1.0- S0.6 EGF EGFInh HRG HRGInh -e- EGF --EGFInh 0.3- - HRG -w- .0.2- 0.40.1 0.2an 0 10 20 30 Time (mins) 40 50 60 120 0 10 20 30 Time (mins) 40 50 60 120 HRGInh B. HeLa 08-29-07 PTP1B Inhibitor ELISA HeLa EGFR ErbB2 2.0 U- EGF 4- EGFInh - HRG Pw-HRGInh -- EGF EGFInh -A-HRG -v- HRGInh m W 1.0. 0. 0 10 20 30 40 50 60 Y·V_ 120 0 10 Time (mins) , ,• ,• ., - , • , • , / ----- r 20 30 40 50 60 120 Time (mins) ErbB3 ErbB4 - EGF - EGFInh 6- HRG P- HRGInh m CL 0. --.--v- 0.3- .0.2- ·· UU 0 Time (mins) 0 10 20 30 Time (mins) 40 50 60 m 120 EGF EGFInh HRG HRGInh Fig. 6 - Iressa then Vanadate-chase Time course A. H1666 EGFR - 0.40- ERK EGF - ar-C- 0.350.30 - S0.251.0- 0.20 - 0 5 10 15 20 25 30 60 0 Time (mins) 5 10 15 20 AKT SHC rr-r -a 0.6. S0.5 U x V. CL 0.40.3. L•L__ 0 5 10 15 20 Time (mins) 30 25 30 -,- -- L 0.7- n • .I 25 60 Time (mins) 60 Time (mins) EGF I----- ~nl H3255 EGFR SEGF -- 1.2. 1-' .. ERK -GF 15 25 inc 1.0- US0.8 w 0.6- 0.4- U.Lz "` 0 5 10 15 20 25 30 60 0 5 10 Time (mins) 30 60 Time (mins) AKT SHC -- a FCF: U ) 3.02.52.00 5 10 15 20 Time (mins) 25 30 60 1•231 0 5 10 15 20 Time (mins) 25 30 60 fl" H1666 61 h.I ERK EGFR -EGF 1.4- 3.5- 1.2- 3.0- 1.0- 2.5- a 8- 2.02_O- • 0.6- 1.5- 0.4 0.2 o 1.0 . 0.0 . on b O1 . 5 . 10 Time (mins) . 1'5 , 20 Time (mins) SHC AKT u x I- in CL 0 Time (mins) 5 10 1 Time (mins) 60 - , 23 " i jz m 06 cz___• H3255 EGFR ERK . U. CL Time (mins) Time (mins) AKT SHC -0-EGF 1 -*-&-9- 1 Iressa.10' Iressa.Vi.10' Vi.lO I) M. U- 0 Time (mins) 5 10 15 20 Time (mins) 25 30 60