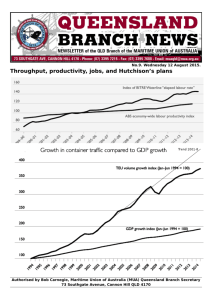
Design framework of the MuA remodeling signal that
advertisement

Design framework of the MuA remodeling signal that confers preferential complex disassembly by the AAA+ unfoldase ClpX by Lorraine Ling B.A. Molecular and Cell Biology, University of California, Berkeley (2007) Submitted to the Department of Biology in partial fulfillment of the requirements for the degree of Doctor of Philosophy at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY September 2014 © 2014 Lorraine Ling. All rights reserved. The author hereby grants to MIT permission to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part in any medium now known or hereafter created. Signature of Author . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Department of Biology July 22, 2014 Certified by . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Tania A. Baker E. C. Whitehead Professor of Biology Thesis Supervisor Accepted by . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Michael Hemann Associate Professor of Biology Co-Chair, Biology Graduate Committee 2 Design framework of the MuA remodeling signal that confers preferential complex disassembly by the AAA+ unfoldase ClpX by Lorraine Ling Submitted to the Department of Biology on July 22, 2014, in partial fulfillment of the requirements for the degree of Doctor of Philosophy Abstract The cell employs many classes of molecular chaperones to facillitate proteins in adopting the proper structure and preventing non-functional and potentially toxic non-native states. The Clp/Hsp100 family of ATPases are unfolding chaperones that remodel macromolecular complexes and facilitate ATP-dependent protein degradation. They are members of the superfamily of AAA+ enzymes (ATPases Associated with various cellular Activities), which is conserved across all kingdoms of life. Efficient selection of multimeric protein complexes over constituent subunits is key to successful remodeling and disassembly reactions. Using E.coli ClpX as a model for AAA+ ATPases, I characterized the mechanism by which ClpX discriminates between two oligomeric states of one of its natural multimeric substrates, phage MuA tranposase. I elucidated many strategies for ClpX’s preference for the assembled Mu transpososome (MuA complex) over unassembled subunits. First, the target substrate makes multiple weak interactions with the AAA+ ATPase via the pore in the conserved ATPase domain and a class-specific auxiliary domain. Second, recognition tags should be at the weaker end of the affinity spectrum to allow effective synergy of multiple tags in the assembled complex. Third, multimeric complexes can "divide the labor" of making these interactions among their subunits. Thus the holistic complex-specific targeting signal is accessible only in the assembled complex. The work of this thesis has provided a framework to understand the design of recognition signals that specify and target macromolecular complexes to unfolding chaperones and remodelers of the AAA+ superfamily. Thesis Supervisor: Tania A. Baker Title: E. C. Whitehead Professor of Biology 3 4 Acknowledgments I am a confidant scientist today thanks in large part my advisor Tania Baker. She is my mentor and role model. Even as an established professor many years away from the bench, Tania answered my nitty-gritty questions and would offer technical help in my MuA assays. Her encyclopedic memory saved me many times from repeating experiments with negative/ uninterpretable results because hardly anyone publishes inconclusive data. Her guidance and enthusiasm help build my confidence as a researcher. She has helped me become a better scientific writer. As a role model, Tania has shown me how to handle stress and life’s complications with grace and perseverance. She is like the CEO of the Tania Baker company. I thank Bob Sauer, my co-advisor, for great advice at our monthly SJ meetings and reminding me to look at the bigger picture in my research. Thank you to my thesis committee members, Amy Keating and Mike Laub, who have been with me since the start of my thesis research. They provided wonderful constructive criticism at all my thesis meetings, helped me pivot my project at a critical time, and ensured that I graduated in a timely manner. Thank you to Jodi Camberg for sitting on my thesis defense committee and suggesting improvements to this thesis. As an undergraduate who majored in Genetics, biochemistry at the graduate level seemed intimidating. I thank all my biochemistry teachers at MIT who taught the subject in an engaging, thought-provoking, and accessible manner. Bob and Frank Solomon taught Graduate Biochemistry. Amy and Bob taught Special Topics in Biochemistry. They all helped me unlock my "biochemistry" power. Many thanks to all past and present members of the Baker Lab. I am so lucky to be colleagues with this group of smart, witty, and compassionate people. Special thanks to fellow graduate student Ben Stein, with whom I shared the lab room. I will miss our ’Party-time’ music-science mash ups. Although I didn’t have the pleasure of overlapping with Aliaa whose research my thesis work has built upon, Aliaa was so generous answering my emails. I thank Anne Meyer who was my rotation mentor. She was uncertain, maybe even skeptical, that I would join the Baker lab due to my wildly different rotations but I did! Lastly, thank you to my family and friends who provide a wonderful counterbalance to graduate school. 5 6 Contents 1 Introduction 11 1.1 Protein homeostasis in the cell . . . . . . . . . . . . . . . . . . . . . . . . 12 1.2 Clp/Hsp100 ATPases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 1.3 Structural features of Clp/Hsp100 family . . . . . . . . . . . . . . . . . . 20 1.3.1 ATPase domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 1.3.2 Auxiliary domain . . . . . . . . . . . . . . . . . . . . . . . . . . . 22 Substrate selection by Clp/Hsp100 ATPases . . . . . . . . . . . . . . . . 22 1.4.1 Direct recognition . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 1.4.2 Assisted recognition . . . . . . . . . . . . . . . . . . . . . . . . . . 25 1.4 1.5 Remodeling enzymes in AAA+ superfamily . . . . . . . . . . . . . . . . 27 1.6 The virus, Bacteriophage Mu . . . . . . . . . . . . . . . . . . . . . . . . 28 1.6.1 MuA transposase . . . . . . . . . . . . . . . . . . . . . . . . . . . 30 1.6.2 Transposition pathway . . . . . . . . . . . . . . . . . . . . . . . . 30 1.6.3 Transpososome remodeling by ClpX . . . . . . . . . . . . . . . . . 33 Motivation for thesis research . . . . . . . . . . . . . . . . . . . . . . . . 34 1.7 2 Design logic of a multivalent recognition signal confers preferential complex disassembly by the AAA+ unfoldase ClpX 37 2.1 Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38 2.2 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39 2.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43 2.3.1 Identification of a region critical for enhanced recognition of transpososomes by ClpX . . . . . . . . . . . . . . . . . . . . . . . . . . 7 43 2.3.2 A peptide encompassing the critical region interacts with the Nterminal zinc-binding domain of ClpX 2.3.3 . . . . . . . . . . . . . . . Step-wise loss of the Enhancement tag trends with ClpX’s weaker affinity for complexes . . . . . . . . . . . . . . . . . . . . . . . . . 2.3.4 49 Mu pore-binding tag is an intrinsically poor ClpX signal without adaptor-like contacts . . . . . . . . . . . . . . . . . . . . . . . . . 2.3.5 47 54 Transpososomes with a strong pore-binding tag do not require Enhancement tags . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54 2.4 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58 2.5 Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64 2.6 Appendix: Geometry experiments on MuA monomer variants . . . . . . 67 2.6.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 2.6.2 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 2.6.3 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69 3 Conclusion & Future Directions 71 3.1 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72 3.2 Future Directions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74 8 List of Figures 1-1 A simplified protein life cycle . . . . . . . . . . . . . . . . . . . . . . . . 13 1-2 Prokaryotic Heat Shock Protein chaperones . . . . . . . . . . . . . . . . . 16 1-3 Model of AAA+ ATPase unfolding and translocation cycles . . . . . . . 18 1-4 Domain structure of bacterial Clp/Hsp100s . . . . . . . . . . . . . . . . . 19 1-5 ClpX structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 1-6 Mechanism of replicative transposition . . . . . . . . . . . . . . . . . . . 29 1-7 Domain structure of MuA transposase . . . . . . . . . . . . . . . . . . . 30 1-8 Phage Mu in vivo replicative transposition . . . . . . . . . . . . . . . . . 33 1-9 Structure of type1 and type 2 Mu transpososomes . . . . . . . . . . . . . 34 2-1 In vitro assays for Mu complex assembly and recognition by ClpX . . . . 41 2-2 Mutation of a sequence region R622-S624 reduces disassembly and degradation rates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 2-3 Comparison of all reaction rates for Mu aspartate variants . . . . . . . . 45 2-4 Residues P623 S624 form a critical interaction between MuA complex and ClpX . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46 2-5 The N-terminal zinc-binding domain of ClpX binds to the enhancement peptide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2-6 Mu transpososome is an asymmetric complex 48 . . . . . . . . . . . . . . . 50 2-7 Tools for making homogeneous mixed mutant complexes . . . . . . . . . 51 2-8 Chimeric complexes disassembly controls . . . . . . . . . . . . . . . . . . 52 2-9 All four subunits can provide the Enhancement tag in MuA complexes . 53 2-10 Mu pore-binding tag is a weak ClpX recognition signal . . . . . . . . . . 55 9 2-11 Mu complexes with a strong pore-binding tag are recognized as well as native Mu complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56 2-12 Mu∆8ssrA monomer degradation by ClpXP . . . . . . . . . . . . . . . . 57 2-13 Permutations of tag engagement in Mu transpososome by ClpX . . . . . 61 2-14 MuSspB monomer degradation by ClpXP 69 10 . . . . . . . . . . . . . . . . . Chapter 1 Introduction 11 Overview The foundational theme of this thesis is understanding the mechanism and regulation of substrate selection by the Hsp100/Clp chaperones. In the cell, chaperones are molecular machines that facilitate a change in a protein’s functions by altering the structure of proteins. Depending at which stage of the protein life cycle that the target protein is in, chaperones aid in both folding and unfolding of the target protein to bring about changes in structure. This introduction will focus on the chaperones found in prokaryotes. The first section situates the many roles of chaperones within the protein life cycle. The next few sections introduce the Hsp100/Clp family of chaperones and chaperone-linked proteases found in E.coli, their structure, some interacting partners which aid in substrate recognition, and a sampling of biological targets. The last section introduces phage MuA transposase, one of many substrates of ClpX, an E.coli Hsp100/Clp unfolding chaperone. The ClpX-MuA transposase interaction is my model system to study mechanisms of specificity and design of recognition signals. 1.1 Protein homeostasis in the cell Inside the densely packed environment of a cell, proteins perform many roles; as structural macromolecules providing support and shape, as enzymes catalyzing chemical reactions, and as signaling molecules and receptors communicating information between the extracellular environment and the cell. For these diverse roles, proteins must adopt the correct structure leading to the biochemists’ axiom “structure equals function.” For ideal cellular function, proteins must be properly folded into their native three-dimensional forms, associated with appropriate partners or oligomerize if required, and disposed of when no longer needed or damaged. Nature has evolved machinery, referred to as molecular chaperones, to guide proteins through these major milestones in the protein life cycle. The life cycle of a protein begins at its birth by the protein-making factory called a ribosome, itself a protein-RNA complex. The newly synthesized polypeptide contains all the information needed within its primary amino acid sequence to define its final folded 12 Clp/Hsp100 proteases Degraded protein Hsp90 Clp/Hsp100 Cl p pr /H ot sp ea 10 se 0 s Protein Complex Clp/Hsp100 proteases Non-native state Folded protein Hsp60 Hsp70 Hsp90 Unfolded protein Clp/Hsp100 Hsp70 Clp/Hsp100 Hsp70 Holdases Aggregated protein Figure 1-1: In this simplified protein life cycle, the upper-left represents on-pathway folding and associations for a functional protein, which in this example forms a multiprotein complex. The lower-right represents off-pathway states due to stress, represented by yellow lightning bolts. Many arrows were omitted for clarity; stress on protein complexes can lead directly to unfolded and non-native states. Actions of various HSP chaperones are indicated by their respective arrows to facilitate proteins adopting proper structure and to prevent or rescue proteins from the aggregated state. The Holdases belong to a diverse group of sHSP, small heat shock proteins, which bind to misfolded proteins and maintain them in a refolding competent state. 13 (native) structure (Anfinsen, 1973). However, in the crowded intracellular environment, nascent polypeptides need the help of molecular chaperones to properly fold and to avoid non-functional non-native and aggregated states (Figure 1-1). Chaperones do not mold proteins into their native structure but simply facilitate the self-directed folding process (Hartl, 2011; Mayer, 2013). In many cases, proteins combine with other proteins to form large macromolecular complexes which are the biologically active states. Individual proteins within a larger complex are referred to as subunits. When these complexes need to perform new functions, the complex may gain, lose, or exchange subunits. These changes in quaternary structure very often reflect changes in biological function. Once again, chaperones facilitate the assembly, alteration, and deactivation of macromolecular complexes. These multi-faceted processes performed by chaperones are encompassed in the term “remodeling.” Several molecular chaperones participating at initial protein folding stages of the protein life cycle come from the highly conserved heat shock protein (Hsp) family, discovered in their roles in heat-shock response (Parsell & Lindquist, 1993). The Hsp60s group includes E.coli GroEL and its co-chaperone GroES (Figure 1-2A). Together they oligomerize into a barrel-shaped structure reminiscent of a basket with a domed lid to provide a defined and isolated environment for the polypeptide to fold and avoid aggregation prior to reaching its native state (Braig et al., 1994; Xu et al., 1997; Ellis, 2003). GroEL has a preference for binding stretches of hydrophobic amino acids, an elegant way to corral unfolded or non-native proteins (Fenton et al., 1994; Mayhew et al., 1996). Using non-specific substrate binding strategies and sequestration mechanisms, GroEL/GroES aids in folding of at least half of the proteins in the cell(Houry et al., 1999; Viitanen et al., 1992). Another group of Hsp, Hsp70s also help proteins reach their native state. In E.coli, the Hsp70 member is DnaK (Figure 1-2B). Working with a co-chaperone,DnaJ, and a nucleotide exchange factor GrpE, the DnaK/ DnaJ/ GrpE chaperone team works at the site of the ribosome, to protect emerging polypeptides and help in folding (Deuerling et al., 1999; Teter et al., 1999). At this stage, these emerging polypeptides may have very 14 little folded structure, so stretches of exposed hydrophobic amino acids are particularly susceptible to aggregation. DnaK chaperone team binds non-specifically to stretches of hydrophobic residues, shielding them from neighbors. Then, powered by ATP, the chaperone releases the chain when it is ready to fold (Fourie et al., 1994; Flynn et al., 1989). Thus, both Hsp60s and Hsp70s use similar non-specific binding mechanisms to prevent inappropriate interactions and facilitate on-pathway folding. Although not required for de novo folding of most proteins, the Hsp90s and Hsp100/Clp chaperones use the energy from ATP to aid the final maturation of selected proteins substrates ("clients") that function as a multicomponent or larger oligomeric complex. In most eukaryotes, Hsp90 is an essential chaperone and its clients include the steroid hormone receptors, protein kinases, and transcription factors (Li & Buchner, 2013). The growing list of Hsp90-associated co-factors enables the chaperone to have a broad substrate repertoire (Eckl & Richter, 2013). Clp/Hsp100 unfolding chaperones facilitate protein complex remodeling and disassembly. Similar to Hsp90 group, Clp/Hsp100 chaperones utilize different co-factors to enlarge substrate range. However, many Clp/Hsp100 unfolding chaperones target just one protein complex; often remodeling these multiprotein complexes for the goal of recycling the subunits. In addition to functions during normal growth, chaperones are key protective agents during times of stress. If proteins adopt non-native conformations due to damage or environmental stress, they may form insoluble aggregates which deactivate protein functions and are often toxic to the cell. Unfolding chaperones in the Hsp100/Clp group working as disaggregases can re-solubilize aggregates (Squires et al., 1991). Then the misfolded polypeptide has a chance to refold into its native conformation, often with help from Hsp60 and Hsp70 folding chaperones, or be degraded(McCarthy et al., 1998; Laskowska et al., 1996). Lastly at the end of a protein’s useful “life”, proteins are degraded into their amino acid building blocks. Whether caused by regulatory responses or quality control mechanisms, proteins which are at the end of their lives are funneled to intracellular chaperone-linked proteases also from the Hsp100/Clp group (Figure 1-2C). Unlike Hsp60s and Hsp70s, the Hsp100/Clp group of unfolding chaperones generally use specific 15 A. Hsp60 group : GroEL/ES C. Hsp100 group : HslU B. Hsp70 group : DnaK ~70kDa ~800kDa ~900kDa top view HslV GroEL GroEL HslU GroES side view David Goodsell & RSCB Protein Data Bank Molecule of the Month Series side view Figure 1-2: Atomic structures of members of the Hsp family of chaperones illustrated by David Goodsell. Figures are not to scale. A. Seven GroEL proteins (Hsp60 chaperone) form a ring. Depending on its nucleotide state, the double stacked GroEL rings are capped by a GroES particle (a 7-mer). B. DnaK, ATP-binding domain on the left, peptide binding domain on the right with a bound peptide colored in red. C. HslUV compartmentalized protease. HslU unfoldase forms a hexameric ring. HslV peptidase is a “double donut” of hexameric rings. 16 substrate selection mechanisms and actively unfold their substrates for two biological outcomes: remodeling and degradation. Because the Hsp100/Clp group deactivate complexes and often promote irreversible protein degradation, these destructive powers must be tightly regulated at multiple levels, such as target binding, spatial location, and developmental timing. 1.2 Clp/Hsp100 ATPases Clp/Hsp100 ATPase family belong to the larger AAA+ (ATPases associated with various cellular activities) superfamily of proteins. These enzymes use ATP hydrolysis to drive repetitive conformational changes that perform mechanical work in the cell (reviewed in Hanson & Whiteheart, 2005). Many AAA+ enzymes function by translocating protein polypeptides or nucleic-acid polymers. Examples include DNA/RNA helicases, proteinsecretion translocation machinery, and viral packaging motors. The Clp/Hsp100 ATPases actively unfold proteins for two biological outcomes: remodeling and degradation. Protein substrates are targeted to Clp ATPases by short peptide sequences, called tags or degrons (discussed in section 1.4.1). Cycles of ATP binding and hydrolysis drive protein unfolding (Figure 1-3A). For the outcome of degradation, the Clp/Hsp100 ATPase partners with a peptidase to form a compartmentalized protease (Figure 1-3B). The Clp/Hsp100 ATPases are ring hexamers, containing one or two ATPase modules per polypeptide (Figure 1-4). In E.coli, the first Clp family member identified was ClpA (Caseinolytic protease A) named for its role as the ATP-dependent regulatory subunit which partners with ClpP peptidase to form the ClpAP protease that degrades casein(Katayama et al., 1988). ClpP is unrelated to the AAA+ superfamily. Later a paralog of ClpA, ClpX was discovered which can also partner with ClpP to form ClpXP protease. Similarly, the HslUV protease is comprised of the ATPase Heat shock locus HslU (also known as ClpY) and the the peptidase HslV (also known as ClpQ). Two additional proteases, Lon and FtsH, have the ATPase and peptidase components encoded on a single polypeptide instead of existing as separate subunits (reviewed in Sauer & Baker, 2011). Lon was the first bacterial energy-dependent protease discovered. 17 A. Unfolding for remodeling/disassembly tag AAA+ unfoldase ATP ATP B. Unfolding for degradation AAA+ unfoldase ATP ATP peptidase Figure 1-3: A. A recognition signal (tag) in a native substrate is initially recognized by the AAA+ unfoldase. Repetitive cycles of ATP hydrolysis then power unfolding of substrates and translocation through the enzyme’s central channel. This leads to unfolding and/or remodeling of the protein complex. B. When the AAA+ unfoldase is associated with a compartmental peptidase, translocation of the polypeptide into the degradation chamber leads to protein destruction. (Adapted from Sauer and Baker, 2011) Lon’s ability to degrade partially folded proteins has led to its designation as the major protease responsible for protein quality control in the cell (Chung & Goldberg, 1981). Although deletion of other AAA+ chaperones and proteases leads to severe pleiotropic phenotypes, FtsH is the only essential protease in E.coli (Ogura et al., 1991). FtsH is anchored to the inner membrane and degrades membrane and cytoplasmic proteins (Tomoyasu et al., 1993). ClpB cannot partner with ClpP and thus has only chaperone and no coupled protease functions. It is essential for thermotolerance and can solubilize almost any protein that becomes aggregated after severe stress (Squires et al., 1991). ClpB collaborates with the DnaK chaperone system (not members of AAA+ superfamily) to reactive proteins from insoluble aggregates. Studies of these chaperones’ activities in vitro have clarified 18 Class-specific Auxiliary domains ClpB AAA+ module AAA+ module N M ClpA/ClpC N N ClpX ClpP protease ClpP protease HslV HslU protease I LonA protease TM FtsH N1 protease N2 Figure 1-4: The Clp/Hsp100 family of chaperones and compartmentalized proteases contain a conserved ATPase domain, the hallmark feature of the AAA+ superfamily. ClpB, ClpA, and ClpC contain two AAA+ modules. ClpX, HslU, FtsH and Lon contain only one AAA+ module. Each ATPase has class-specific auxiliary domains that are not conserved. Proteases that associate with Clps are shown on the right. ClpP and HslV are separate proteins while FtsH and LonA each contain a protease domain. Adapted from (Sauer and Baker, 2011) their individual mechanisms but a combined mechanism with wide consensus has not yet been established. One model is that ClpB breaks apart large protein aggregates into smaller ones by extracting and unfolding polypeptides from the aggregate (Weibezahn et al., 2004). The released polypeptide can spontaneously refold or interact with the folding chaperones such as DnaK or GroEL/GroES. DnaK may also act earlier in the ClpB-mediated remodeling reaction by helping ClpB bind to aggregates or regulating ATP hydrolysis by ClpB (Doyle & Wickner, 2009). The Gram-positive bacterium, Bascillus subtilis, shares many Clp/Hsp100 orthologs with E.coli. However B.subtilis has a species-specific ClpC ATPase and lacks both ClpA and ClpB. ClpC chaperone can associate with ClpP peptidase to form ClpCP protease (Molière & Turgay, 2009). 19 1.3 1.3.1 Structural features of Clp/Hsp100 family ATPase domain Clp/Hsp100 enzymes are members of the AAA+ superfamily. The hallmark feature of the AAA+ superfamily is a structurally conserved ATPase module, which performs cycles of nucleotide binding, hydrolysis and release to convert chemical energy into mechanical work. The ATPase domain contains a conserved ATP binding pocket. Two conserved sequence motifs which interact with ATP are the Walker A and Walker B motifs, which bind and hydrolyze ATP, respectively (Wendler et al., 2012). Most oligomerize into rings with a central pore. Lining the central pore are flexible sequences referred to as pore loops which bind substrates and translocate the polypeptide through the central channel (Figure 1-5). Figure 1-5: Cutaway view of ClpX with pore loops highlighted. The RKH loops are colored yellow, pore-1 loops red, and pore-2 loops blue in a model of the ClpX hexamer (based on Kim et al. 2003, Bochtler et al. 2000). Three subunits of the hexamer were removed to allow visualization of the pore loops. Figure taken from Martin et al. 2007. 20 Located in the center of the channel, the pore-1 loops are the most conserved, with a nearly invariant Aromatic-hydrophic sequence motif, YVG, among all AAA+ ATPases. Mutagenesis studies and structures of ClpX with different nucleotide states argue that the pore-1 loop is key to translocation (Siddiqui et al., 2004; Martin et al., 2008b; Glynn et al., 2009). The RKH and pore-2 loops are conserved among ClpX orthologs and are located at the mouth of the channel and bottom of the channel, respectively. Both RKH and pore-2 loops play a role in initial binding and translocation of substrates (Farrell et al., 2007; Martin et al., 2007, 2008a). Pore-2 loops also mediate ClpP binding and communication (Martin et al., 2007). Because there are no atomic structures of AAA+ ATPases with bound substrates, the mechanism of coupling ATP hydrolysis, enzyme conformation changes and mechanical work is under active investigation. Studies of ClpX have led to a model mechanism in which cycles of ATP hydrolysis are coupled to ClpX subunit conformational changes and thus the orientation of pore loops leading to a net movement of the substrate polypeptide (Stinson et al., 2013). Crystal structures of HslU and ClpX reveal that the pore-1 loop adopts a dynamic range of confirmations that position the tip of the loop throughout the length of the channel (Bochtler et al., 2000; Sousa et al., 2000; Glynn et al., 2009). Repetitive cycles of ATP hydrolysis by ClpX may be needed to unfold the protein and translocate the polypeptide(Martin et al., 2008c). Translocation of the polypeptide is proposed to pull the attached folded protein against the entrance to the axial pore, thereby generating a denaturing force because the pore is smaller than the folded protein (Baker & Sauer, 2006). The observable result is that Clp/Hsp100 chaperones and chaperone-linked proteases exert a pulling force on the polypeptide which leads to cooperative unfolding of the target protein (Aubin-Tam et al., 2011). There appears to be no obligatory directionality to translocation, as ClpXP can degrade substrates starting either from the N-terminus or from the C-terminus (Gottesman et al., 1998; Gonciarz-Swiatek et al., 1999; Lee et al., 2001; Flynn et al., 2003; Hoskins et al., 2002; Kenniston et al., 2005; Farrell et al., 2007). Once ClpX engages with the protein substrate, the translocation process is processive and remarkably tolerant to the structural identity of the polypeptide. ClpX exhibits very little preference for 21 side group size, charge, or chirality and will translocate peptide bonds spaced with long hydrophobic carbon chains (Barkow et al., 2009). 1.3.2 Auxiliary domain A second feature of Clp/Hsp100 ATPases are auxiliary domains specific to each member. In general these auxiliary domains are not conserved between members, dispensable for ATPase function but play roles in substrate recognition (Sauer & Baker, 2011). Often these member-specific domains provide a docking platform for delivery proteins, called adaptors (discussed in section 1.4.2). ClpA, ClpC, and ClpX all have N-terminal auxiliary domains. However the structure of each auxiliary domain and the connection of each to their respective ATPase module differs among the three Clp ATPases (Zeth et al., 2002; Wang et al., 2011; Park et al., 2007) HslU has an I-domain, which is an insertion of 140 residues in the AAA+ module (Bochtler et al., 2000). The I-domain domain is thought to play a role in substrate binding and allosterically regulates ATPase activity (Sundar et al., 2012). The M-domain (middle domain) of ClpB forms a propeller-shaped coiled-coil and has been proposed to act like a “crowbar” to break apart large aggregates, though the crowbar model has not been experimentally validated (Lee et al., 2003). Interestingly, the M-domain serves as the site for species-specific interaction with the DnaK/DnaJ/GrpE chaperone team (Miot et al., 2011). 1.4 Substrate selection by Clp/Hsp100 ATPases Clp ATPases employ two modes of substrate recognition. The first is direct substrate recognition by binding to short peptide sequences, known as tags, on the substrate. The tag interacts with the pore of the ATPase and subsequent engagement results in unfolding. The second mode termed “assisted recognition” is when another protein aids the ATPase in recognition of the substrate. Additional mechanisms for substrate selection not discussed below are subcellular relocalization of either substrates or Clp/Hsp100 ATPases, phosphorylation of proteins turning them into substrates, and regulating expression levels of substrates or ATPases by developmental timing. 22 1.4.1 Direct recognition Recognition tags are found often at the N- or C-termini of substrates. Tags that bind directly to the unfoldase fall into three classes: intrinsic, latent, and co-translational. intrinsic class Intrinsic signals are encoded in the primary sequence of a substrate protein. The tags are often present at the N or C-termini but may not be accessible to proteases until a conformational change or loss of a shielding binding partner. Examples of substrates with N-terminal tags include Dps (DNA-binding protein from starved cells), a stationary phase nucleoid protein that sequesters iron and protects DNA from damage (Flynn et al., 2003), and bacteriophage 𝜆 O replication initiator protein when not bound to orilambda DNA (Gonciarz-swiatek et al., 1999). Additionally, 𝜆O may have an internal intrinsic signal. Residues Q49-M67 bind strongly to ClpX N-domain and a peptide containing this sequence can compete with degradation of the full-length protein by ClpXP (Thibault et al., 2006). Remarkably the stability of a protein can be attributed to a single amino acid at its N-terminus, known as the N-end rule. Present in both prokaryotes and eukaryotes, the N-end rule tags are often the large hydrophobic residues. In E.coli, an N-terminal leucine, phenylalanine, tryptophan or tyrosine directly targets proteins to intracellular proteases (Varshavsky, 1996). An example of an intrinsic C-terminal tag comes from Supressor of lon (SulA) identified in a screen to suppress the lon - sensitivity to ultraviolet radiation (Gayda et al., 1976). SulA is an inhibitor of cell division and upregulated during SOS response otherwise during normal growth it is degraded by Lon and HslUV proteases (Gottesman et al., 1981; Mizusawa, 1983; Seong et al., 1999). The last eight C-terminal residues are crucial for recognition by Lon as a truncated SulA variant was stabilized both in vivo ans in vitro. (Higashitani et al., 1997) A proteomic based screen for in vivo substrates of ClpXP revealed five classes of recognition signals. The screen utilized a tagged and catalytically inactive variant of ClpP (ClpPtrap ) to receive and contain proteins translocated by ClpX (Flynn et al., 23 2003). Analysis of the >50 captured proteins led to two classes of motifs located at the C-terminus and three classes located at the N-terminus. About one quarter of the trapped proteins contain potential intrinsic ClpX recognition signals at both the Nterminus and C-terminus. Similarly to phage 𝜆 O replication initiator protein, many Nterminal sequences from the pool of trapped proteins directly bound to ClpX’s N-domain on a peptide array. However with so few examples of confirmed N-domain interacting sequences, it was difficult to establish a consensus motif (Flynn et al., 2003). It is unclear whether both signals are engaged by the ATPase domain of ClpX or contribute to additional enzyme-binding interactions via the auxiliary domain similarly to adaptor proteins (see section 1.4.2). latent class Latent signals are also encoded in the primary sequence but require processing of the protein to make the tag accessible. Often an endopeptidic cleavage reveals a new termini containing the tag. Examples include LexA and RseA. LexA is a transcriptional repressor of genes involved in DNA-damage response. During the SOS response to DNA damage, LexA undergoes RecA-stimulated autocleavage between the N-terminal DNA binding domain and the C-terminal dimerization domain. Both fragments are rapidly degraded by Lon and ClpXP (Little, 1983; Neher et al., 2003a). The cleavage reveals a tag at the new C-terminus with residues VAA-CO2 which is similar to the region of the ssrA tag (LAA-CO2 discussed below) recognized by ClpX (Neher et al., 2003a). RseA is the anti-sigma factor to 𝜎 E which activates expression of genes involved in the bacterial extracytoplasmic stress response. RseA is an inner membrane-spanning protein. The N-terminal cytoplasmic portion binds 𝜎 E and sequesters the sigma factor from its target promoters. In response to extracytoplasmic stress, RseA undergoes sequential cleavage steps by proteases in the periplasm and cytoplasm which results in an N-terminal fragment of RseA and frees 𝜎 E (Lima et al., 2013). The released 𝜎 E can activate its regulon, while the N-terminal fragment of RseA, revealing residues VAA at the new C-terminus, is degraded by ClpXP (Flynn et al., 2004). co-translational class 24 In contrast to the two previous classes, the co-translation class is not encoded in the primary sequence of the substrate protein. The only member of this class is the ssrA tag which marks proteins for degradation by multiple AAA+ proteases (ClpAP, ClpXP, FtsH, Lon) (Keiler et al., 1996). When ribosomes get stuck on an mRNA, a rescue system of tmRNA (trans-messenger RNA) displaces the offending mRNA. Translation continues on the tmRNA, which encodes an eleven amino acid sequence (AANDENYALAA) and a stop codon. The ssrA tag is appended onto the truncated polypeptide and marks these incomplete translation products for destruction. It is estimated that 0.5% of translation products receive an ssrA tag (Lies & Maurizi, 2008). Thus the ssrA-tagging system rescues stalled ribosomes and destroys potentially dangerous protein fragments (reviewed in Karzai et al., 2000). 1.4.2 Assisted recognition Clp ATPases also use a second mode termed “assisted recognition” in which accessory proteins called adaptor proteins modulate substrate choice and often give rise to higheraffinity enzyme-adaptor-substrate complexes. Various mechanisms for adaptors have been observed from acting as delivery vehicles to directly affecting chaperone and protease activity (reviewed in Kirstein et al., 2009). Adaptor proteins are themselves not degradation substrates and thus participate in multiple rounds of delivery or modulation of enzyme activity. A general overview of three E.coli adaptors (SspB, RssB, ClpS) and one B.subtilis adaptor (MecA) represents the diversity of mechanisms to deliver target proteins and modulate chaperone-protease activities yet highlights one common theme of docking to the enzyme’s auxiliary domain. SspB Stringent starvation protein B (SspB) is the best characterized E.coli adaptor protein. SspB enhances degradation of ssrA-tagged substrates by ClpXP via a tethering mechanism. Here, the adaptor binds to both the substrate and the AAA+ unfoldase to increase the effective concentration of the tag near the enzyme’s active center (Wah et al., 2003). SspB binds to ClpX specifically through the unfoldase’s N-domain (Park 25 et al., 2007). In E.coli, ClpX and SspB bind distinct portions of the ssrA tag. ClpX recognizes the last three residues and the carboxyl group whereas SspB recognizes the first four and seventh residue (Levchenko et al., 2000; Flynn et al., 2001; Levchenko et al., 2003; Song & Eck, 2003). As mentioned above, ssrA-tagged substrates are also degraded by ClpAP protease. However, the same adaptor SspB inhibits this reaction because ClpA and SspB bind overlapping residues in the ssrA tag (Flynn et al., 2001). Although the biological consequence of this inhibition remains unclear, a plausible outcome is to turn other substrates into high-priority ClpAP targets, leaving ClpXP to clean up ssrAtagged polypeptides. SspB also recognizes and delivers the N-terminal fragment of RseA to ClpXP for degradation (Flynn et al., 2004). RssB Regulator of sigma-S protein B (RssB) also known as stationary-phase regulator (SprE) is essential for turnover of stationary phase sigma factor, 𝜎 S (Muffler et al., 1996; Pratt & Silhavy, 1996). RssB is a ClpXP-specific adaptor and is activated by phosphorylation. Phospho-RssB protein binds 𝜎 S which exposes a latent tag in the N-terminal region of 𝜎 S . ClpS ClpS adaptor delivers N-end rule substrates to ClpAP protease for degradation (Dougan et al., 2002). Because both ClpS and ClpA recognize the same N-terminal amino acid, ClpS employs a more involved mechanism than simple tethering. ClpA may recognize additional tag features that are not directly bound to ClpS. ClpA, ClpS and the N-end rule substrate form a high-affinity ternary complex (Román-Hernández et al., 2011). Within this complex, ClpA engages the unstructured N-terminal section of ClpS which causes a conformational change and hand-off of the N-end rule residue from the ClpS binding pocket to the pore of ClpA (personal communication Izarys RiveraRivera). ClpS escapes degradation to catalyze another round of delivery because the core substrate binding domain of ClpS carries structural elements that are non-denaturable by ClpA (personal communication Izarys Rivera-Rivera). ClpS interacts with ClpA via the unfoldase’s N-terminal domain. This interaction is necessary for delivery of N-end 26 rule substrates (Zeth et al., 2002). MecA Medium-independent expression of competence (MecA), the best characterized adaptor protein of B.subtilis, was identified in a screen for genes involved in regulation of competence (Dubnau & Roggiani, 1990). MecA binds and inhibits the transcriptional activator of competence, ComK. Furthermore MecA targets ComK for degradation by ClpCP. Unlike the previous adaptor examples, MecA has a unique mechanism to modulate ClpC activity. ClpC is an inactive monomer on it own. MecA triggers the oligomerization of ClpC into the active hexameric chaperone which then can associate with ClpP to form an active protease. This adapter-mediated oligomerization requires MecA to bind to the N-domain of ClpC (Kirstein et al., 2006). 1.5 Remodeling enzymes in AAA+ superfamily In this section, four examples of remodeling enzymes in the AAA+superfamily showcase the breadth of important biological transitions promoted by remodeling. Katanin and Spastin are eukaryotic AAA+ ATPases which remodel microtubules (McNally & Vale, 1993). Microtubules provide support to organelles, shape the cell, and organize into a distinct structure called the spindle that is essential for cell replication and division. They are made up of tubulin subunits, which polymerize into long and branched dynamic polymers. Many factors regulate microtubule assembly and disassembly at the termini of polymers (reviewed in Gardner et al., 2013). However, Katanin and Spastin modulate the dynamics of microtubule from the middle of a polymer. These “microtubule severing enzymes” preferentially unfold and abstract tubulin subunits from the lattice (Roll-Mecak & McNally, 2010). This process of microtubule severing requires the C-terminal tails of tubulin and and the pore loops of Katanin and Spastin (White & Lauring, 2007; RollMecak & Vale, 2008). Another eukaryotic AAA+ family member, N-ethylmalemide sensitive fusion protein (NSF) is an essential factor in intracellular membrane trafficking. NSF in concert with SNAPs (soluble NSF attachment proteins) disassemble SNARE complexes, thus 27 freeing SNARE subunits for additional rounds of membrane fusion (Whiteheart et al., 2001). Although most studied for their functions in intracellular proteolysis, both E.coli ClpA and ClpX have been observed in ClpP-independent remodeling reactions. In vitro, ClpA activates the phage P1 replication initiator protein RepA by remodeling inactive RepA dimers into monomers that are competent to bind DNA (Wickner et al., 1994). ClpX plays a key role in the phage Mu lytic cycle (Mhammedi-Alaoui et al., 1994; Kruklitis et al., 1996). The biochemical steps in phage Mu transposition and the remodeling of the Mu transposase-DNA complex by ClpX have been extensively studied and are summarized in the next section. This well-characterized protein has provided me an ideal model substrate to address questions of target specificity and design principles of ClpX recognition signals. 1.6 The virus, Bacteriophage Mu Bacteriophage Mu is a virus that propagates its genome within a bacterial host using a mechanism of replicative transposition. In replicative transposition, the mobile DNA element cuts, copies itself to a new location, and leaves behind the copy at the previous genomic location. This movement occurs using a branched DNA intermediate called a Shapiro structure (Shapiro, 1979). Extensive study of phage Mu has led to a deeper understanding of the molecular mechanism and regulation of transposition by mobile DNA elements. Phage Mu encodes two proteins necessary for in vivo transposition, MuA and MuB. MuA is the transposase and MuB is a regulatory factor. Together they recombine the correct DNA sequences, transpose at the correct time during the phage lifecycle, and avoid disrupting phage Mu’s own genome (Figure 1-6). Phage Mu regulates this sequence of events by evolving a vectorial process that uses increasingly stable nucleoprotein complexes and co-opting a host chaperone to direct a key transition point. A highly tractable in vitro system was developed early which allowed for biochemical analysis of the transposition reaction. This biochemical system minimally contains the sequences of Mu genomic ends on a supercoiled plasmid, MuA, 28 transposon 3’OH donor DNA cleavage of transferred strand 3’OH donor DNA target DNA strand transfer Cointegrate with 2 copies of transposon 3’OH gap 5’ 3’ gap 3’OH Shapiro Intermediate replication fork assembly (at left gap) lagging strand replication through the transposon Ligation leading strand Figure 1-6: The transpososome introduces a single-strand nick at each end of the ends of the transposon DNA (green). The liberated 3’OH groups then attack the target DNA and become joined to the target by DNA strand transfer. At each end of the transposon, only one strand is transferred into the target at this point, resulting in the formation of a doubly-branched DNA structure, the Shapiro intermediate. The replication apparatus assembles at one of these "forks" (the left one in this figure). Replication continues through the transposon sequence. The resulting product, called a cointegrate, has the two starting circular DNA molecules joined by two copies of the transposon. The ssDNA gaps in the branched intermediate give rise to the target site duplications. These duplications are not shown in the cointegrate for clarity. (Adapted from Figure 11-22 of Watson et al. Molecular Biology of the Gene, 6th ed.) the phage-encoded transposase, host-encoded DNA bending proteins and divalent metal ions (Craigie et al., 1985). Although not required for the core steps of transposition, MuB protein as a key regulator helps ensure transposition into sensible target DNA sites (Reyes et al., 1987). 29 1.6.1 MuA transposase MuA transposase is part of the DDE family of recombinase enzymes which include HIV integrase, Tn5 transposase, and RSV integrase (Rice & Baker, 2001). MuA transposase has a mass of 75,000Da and is monomeric in the absence of phage DNA (Baker & Mizuuchi, 1992). It is organized into three structurally and functionally distinct domains: a DNA binding domain, a catalytic domain, and regulatory domain (Figure 1-7). Figure 1-7: MuA transposase contains three domains. Domain I binds various phage DNA sequences. Domain II contains the catalytic residues, DDE. Domain III contains interaction sites for MuB, a regulator factor, and ClpX, a host unfolding chaperone. Domain I is responsible for site-specific binding to repeat DNA sequences at the ends of the Mu genome. The structure of this domain has a winged helix-turn-helix motif(Clubb et al., 1994). Domain II contains the catalytic DDE motif characteristic of the family (Rice & Mizuuchi, 1995). The active site has dual functions of DNA cleavage and DNA rejoining when the protein is assembled into an active tetrameric enzyme as part of a DNA-protein complex (Lavoie et al., 1991; Mizuuchi et al., 1992). Domain III is the site of interaction with allosteric and regulatory factors such as MuB and the host chaperone, ClpX (Baker et al., 1991; Wu & Chaconas, 1994; Levchenko et al., 1997). 1.6.2 Transposition pathway MuA transposase recombines phage DNA in a reaction pathway characterized by distinct nucleoprotein complexes called transpososomes (Surette et al., 1987; Craigie & Mizuuchi, 1987). MuA binds to DNA attachment sites located at the left and right ends of the phage genome (Craigie et al., 1984). With additional transient binding to an internal enhancer element and help from host histone-like proteins to severely kink the DNA, 30 MuA subunits bring the DNA ends together to form the stable synaptic complex (SSC, Figure 1-8B)(Surette & Chaconas, 1992; Mizuuchi et al., 1992). The SSC consists of four MuA subunits with extensive interprotein contacts. To initiate movement to a new host genomic site (target DNA), MuA transposase cleaves the DNA at the junction of the Mu genome and flanking host DNA to generate 3’ hydroxyl groups at nicked ends. This is the next form of transpososome called the cleaved donor complex (CDC, Figure 1-8C)(Craigie & Mizuuchi, 1987; Surette et al., 1987; Lavoie et al., 1991; Yuan et al., 2005). The freed 3’ hydroxyl groups then attack and join opposite strands of target DNA in a step called DNA strand transfer (Mizuuchi & Adzuma, 1991). This generates recombined DNA synapsed with MuA transposase and the next transpososome called the strand transfer complex (Figure 1-8D) (Surette et al., 1987; Mizuuchi & Adzuma, 1991; Montaño et al., 2012). Structures of the CDC and STC reveal greater interprotein contacts are made as the Mu transpososome progresses through the reaction pathway (Figure 1-9). Selection of the target DNA is regulated by MuB. MuB binds A/T rich target DNA in an ATP-dependent manner and stimulates MuA to catalyze transposition into bound DNA (Baker et al., 1991; Surette et al., 1991; Yamauchi & Baker, 1998). In opposition, MuA stimulates MuB’s ATPase activity. As a result, MuB tends to dissociate near Mu genomic ends where MuA is bound and to associate with DNA far away from the Mu genome (Maxwell et al., 1987; Greene & Mizuuchi, 2002a,b,c). Thus the interactions between MuA and MuB not only promote catalysis but also prevent disruption of its own viral genome, in a phenomenon known as target immunity(Reyes et al., 1987). It is currently unclear what is the molecular mechanism which determines the outcome of the MuA-MuB interaction. In vitro, the minimal system does not require MuB to observe transposition and formation of the STC. After strand transfer, the enzyme has finished its function of recombination but the STC is so stable that the enzyme does not turn over. In vitro, the hyperstable STC resists temperatures up to 75°C and 6M urea (Surette et al., 1987). The stable STC holds onto the recombined DNA products and blocks replication. In fact, continued presence of MuA transposase on the recombined DNA inhibits recruitment of host DNA replication 31 A integrated Mu genome MuA left end right end B Bacterial genome MuB target site DNA Stable Synaptic Complex (SSC) C Cleaved Donor Complex (CDC) D g llin Clp E X e od m re replication machinery remodeled fragile complex G Strand Transfer Complex (STC) Polymerase original Mu genome replication replicated Mu genome 32 F H machinery and thus lytic growth (Nakai & Kruklitis, 1995). At this critical transition, phage Mu switches from its own proteins to ClpX, a host-encoded chaperone, to resolve the replication block (Figure 1-8E). 1.6.3 Transpososome remodeling by ClpX ClpX remodels the stable STC into a fragile complex which then recruits host replication machinery to complete amplification of the phage genome (Figure 1-8F) (Levchenko et al., 1995; Kruklitis et al., 1996; Jones et al., 1998). Deleting ClpX inhibits phage Mu replication in vivo almost completely, but deleting ClpP has almost no effect (MhammediAlaoui et al., 1994). In fact full length ClpX is required for in vivo Mu replication as ClpX lacking its N-terminal zinc-binding domain could not support phage lytic growth (Wojtyra et al., 2003). Thus, it is the chaperone rather than degradation function that is necessary for the Mu lytic cycle. ClpP is irrelevant for transpososome remodeling in vivo. Purification of a host factor that enabled transpososome remodeling led to a single protein fraction with high-specific activity which turned out to be ClpX (Levchenko et al., 1995; Kruklitis et al., 1996). Further in vitro studies of purified ClpX enzyme and transpososomes showed that ClpX was sufficient to disassemble the stable STC and that the last eight C-terminal residues comprised an intrinsic recognition tag (Levchenko et al., 1997). Transpososomes biased to have only one subunit with a tag were sufficient to be destabilized by ClpX (Burton et al., 2001). Highlighting that the unfolding process and not degradation of a Figure 1-8 (preceding page): The in vivo replicative transposition of phage Mu begins with an integrated Mu genome and proceeds through multiple nucleoprotein complexes called transpososomes (A). MuA transposase binds to the left and right ends and brings them together to from the SSC while MuB binds to target site DNA (B). MuA transposase cleaves the DNA to form the CDC while MuB brings the target site closer (C). Recombination into target DNA occurs to form the STC (D). ClpX remodels the hyperstable STC into a fragile complex by unfolding a MuA subunit (E). This remodeled fragile complex then recruits host replication machinery and remaining MuA subunits are released (F). Replication of phage Mu DNA (G) results in two copies of the Mu genome integrated into the bacterial chromosome (H) and the transposition-replication cycle repeats. 33 Cleaved Donor Complex Strand Transfer Complex Figure 1-9: left: Type 1 transpososome, the CDC (cleaved donor complex). An EM structure with MuA subunits colored, DNA in gray. Figure from Yuan et al. 2005. right: Type 2 transpososome, the STC (strand transfer complex). A crystal structure with MuA subunits colored, DNA in gray. PDB: 4FCY MuA subunit was key to remodeling, Burton and coworkers showed that STCs assembled from a MuA variant with an alternative tag (ssrA) at the C-terminus were disassembled by ClpX. Even using an alternative chaperone (ClpA), these alternative ssrA-tagged STCs could be destabilized (Burton & Baker, 2003). Since both free MuA monomers and assembled MuA complexes are in the bacterial cytosol, a pertinent question was how ClpX distinguished between the two oligomeric states of MuA and directed its unfolding activity to the biologically relevant target, the STC. 1.7 Motivation for thesis research AAA+ ATPases use the energy from ATP binding and hydrolysis to drive diverse cellular activities such as DNA replication by helicases and cargo transport along microtubules by dynein. For many of these processes, the ATPase causes key alterations to the structure of the target protein and thus its function. Furthermore, the biologically relevant substrates for some ATPases are large multiprotein structures or a complex in a higherordered oligomeric state. As a consequence, the action of unfolding chaperones must be directed away from subunits or monomers because the cellular process requires a change in structure/function of the macromolecular complex. Thus, it’s important to 34 understand how these enzymes discriminate and prioritize macromolecular complexes over constituent subunits. Using the extensively characterized Mu transpososome remodeling process, I elucidated at the molecular level, the intrinsic recognition signal in transpososomes evolved for disassembly. Then, I uncovered a mechanism used to discriminate between assembled MuA complexes and constituent subunits. Additionally, I articulated the different roles for the multiple intrinsic recognition tags in MuA transposase. Lastly, I engineered MuA variants with different recognition tags to probe the effect of the tags themselves and of the architecture of MuA complex on the holistic “ClpX remodeling” signal. Through this work I present an underlying design framework for how AAA+ enzymes achieve specificity for macromolecular complexes, the biologically relevant targets of remodeling and disassembly reactions. 35 36 Chapter 2 Design logic of a multivalent recognition signal confers preferential complex disassembly by the AAA+ unfoldase ClpX This chapter has been written as a manuscript for publication. A draft is currently being reviewed by collaborators. I performed all experiments for all figures except Figure2-10, which was contributed by A. Abdelhakim. I.Levchenko synthesized the peptides used in Figure 2-5. S.P. Montano and P.A. Rice designed and cloned the initial Sin15Mu chimeric protein and Sin-attachment site DNA oligo, both of which I modified for research in this chapter. 37 2.1 Abstract AAA+ enzymes are present in all kingdoms and use the chemical energy of ATP to remodel protein complexes and catalyze substrate protein unfolding. How these powerful enzymes recognize protein complexes and aggregates is poorly understood. Efficient selection of multimeric protein complexes over constituent subunits is key to successful disassembly. Here, we use E.coli ClpX, a AAA+ unfoldase, and the tetrameric MuA transpososome, to investigate how preferential specificity for an assembled complex is achieved. We demonstrate that the MuA tetramer employs a multivalent set of recognition peptides to ensure that the complex has the tightest affinity for ClpX. The critical recognition components are the weak ClpX pore-binding peptide at the Cterminus of MuA and a second peptide ∼ 40 residues away from the pore signal that binds the ClpX N-terminal domain. By constructing chimeric SinMuA proteins with altered DNA-binding specificity we investigated multiple variant complexes carrying different geometries of mutant signals and determined how each subunit contributes to complex-specific recognition. Although individually, the two key recognition peptides bind weakly (70 − 400𝜇M) to ClpX , together within the assembled MuA tetramer they impart an affinity of ∼1𝜇M. All four subunits in the tetramer can donate the ClpX Ndomain-binding peptide and optimal recognition is achieved when all four are present. In contrast, only two specifically located subunits can donate the pore-binding signal. Importantly, the N-domain-binding peptides become unnecessary for complex recognition when the native weak pore-binding signal is replaced with a much stronger compact porebinding tag. Thus, we conclude that the design of signals that are specific for assembled complexes depends on collaboration between multiple weak protein-unfoldase-interaction peptides and that strong-binding signals can prevent multimer-specific recognition. 38 2.2 Introduction Cells are densely packed with proteins performing structural and/or enzymatic roles essential for life. To help respond to environmental changes, manage protein turnover and protein quality control, these cells employ energy-dependent unfoldases/disaggregases and proteases from the AAA+ family (ATPases associated with various cellular activities). Powered by cycles of nucleotide binding, hydrolysis, and release, these ATPases remodel complexes, solubilize aggregates, and degrade proteins (when coupled with partner peptidases). E. coli ClpX is arguably the best-characterized AAA+ unfoldase and is known to disassemble complexes and unfolds proteins (Sauer & Baker, 2011). ClpX acts alone as a protein-remodeling enzyme as well as in complex with ClpP peptidase to make the ClpXP protease. In ClpXP, ClpX recognizes and unfolds many substrate proteins and translocates the unfolded chain to ClpP peptidase where it is degraded. Because of its destructive power, ClpX’s selection of substrates must be exquisite. The protein signals (recognition sequences) and design logic governing recognition of different classes of substrates is being actively investigated. Most bacteria have no ubiquitination system and recognition of protein targets for degradation or disassembly is mediated by a diverse set of short unstructured peptide sequences, or tags. These recognition tags are often located at the termini of otherwise native substrate proteins (Sauer et al., 2004). Examples of substrates with N-terminal recognition tags are 𝜆O, a DNA replication origin-binding protein from phage 𝜆, and UmuD, a subunit of DNA polymerase V, a DNA-repair/tolerance polymerase (Gonciarzswiatek et al., 1999; Gonzalez et al., 2000) and proteins recognized by the N-end rule pathway (Varshavsky, 1996). The best characterized tag is for one class of substrates: truncated polypeptides from stalled ribosomes. These incompletely translated products are marked at their C-termini with an 11 amino acid sequence called the ssrA tag and targeted for degradation principally by ClpXP (Gottesman et al., 1998). However, some substrates may have more complicated, multicomponent recognition signals. A screen of in vivo ClpXP substrates revealed many target proteins carried multiple ClpX-recognition sequences. Furthermore, the screen also indicated that many ClpX substrates ( 60%) are 39 multimeric or subunits in multiprotein complexes (Flynn et al., 2003). For substrates in multiprotein complexes that are remodeled or disassembled by ClpX, how the enzyme distinguishes between assembled complexes versus subunits is not understood. To investigate how preference for an assembled complex is achieved over constituent subunits, we used the MuA transposase, a natural disassembly substrate of ClpX. Phage Mu duplicates its genome by replicative transposition. During transposition, MuA binds DNA sites located at the ends of the Mu genome, forms a tetramer that brings the two ends of the DNA together, and catalyzes the DNA cleavage and joining reactions core to transposition (Craigie et al., 1984; Kuo et al., 1991; Lavoie et al., 1991). This recombination phase of transposition does not require external energy (such as ATP) and is driven forward by proceeding through a series of nucleoprotein complexes (transpososomes) that increase in stability to a final hyperstable DNA product-bound transpososome (Surette et al., 1987). Remodeling converts the hyperstable transpososome (MuA complex) into a fragile complex, which facilitates both disassembly and the recruitment of DNA-replication machinery (Levchenko et al., 1995; Nakai & Kruklitis, 1995; Jones et al., 1998). Completion of a reaction cycle therefore requires that the stable MuA complex be remodeled by ClpX (Mhammedi-Alaoui et al., 1994). MuA transpososome assembly, recombination, and ClpX remodeling have all been reconstituted in vitro. On a native agarose gel, the stable transpososome is observed as a slower migrating band (asterisk, lane 2) as compared to supercoiled substrate plasmid (arrow, lane 1) (Figure 2-1A). In contrast, the fragile complex is unstable to gel electrophoresis and the liberated DNA transposition products are visible as a characteristic series of topoisomerases (Figure 2-1B, bracket). As both monomeric and DNA-bound tetrameric MuA are present in the cytoplasm we sought a molecular understanding to explain how ClpX recognized the transpososome as a high-priority target. Previous analysis revealed that there was information throughout MuA protein that guided ClpX recognition and that information in domain III is most central. MuA contains a C-terminal sequence (RRKKAI) that is necessary for ClpX recognition of both monomeric MuA and assembled transpososomes (Levchenko et al., 1995, 1997; Abdelhakim et al., 2008). It is also established that interaction between the transpososome 40 + pMini-Mu L1 L2 L1 R1 R1 R2 target DNA L1 DNA Ladder 2 1 R1 ≈ supercoiled plasmid (sc) 1 Native agarose gel R2 L2 R2 L2 ≈ MuA ≈ ≈ ≈ ≈ ≈ A. Transpososome assembly ≈ sc 2 Transpososome * Tr sc (Tr) ≈ ≈ R1 ≈ 2 Transpososome electrophoresis R2 L2 ≈ ≈ Native agarose gel time (min) 0.5 1 2 3 +SDS ≈ ≈ L1 ClpX ≈ R2 L2 ≈ ≈ R1 ≈ ≈ ≈ ≈ * ≈ ≈ ≈ ≈ B. ClpX-catalyzed disassembly 3 Fragile Complex 3 recombined DNA products C. ClpX-catalyzed degradation SDS-PAGE time (min) 0 2 4 6 8 10 15 ClpXP MuA MuA Figure 2-1: In vitro assays for Mu complex assembly and recognition by ClpX A. MuA transposase monomers and host protein HU are incubated with a plasmid substrate (“pMini-Mu”) containing “left” and “right” phage Mu attachment sites ( L1, L2, R1, R2). Mu catalyzes DNA cleavage and recombination with target DNA to form the transpososome, a stable complex. When visualized on a native agarose gel, the transpososome appears as a band (“Tr”, asterisk) that migrates slower than supercoiled plasmid alone (“sc”, black arrow) B. ClpX remodels the transpososome (MuA complex) by unfolding a subunit bound to L1 or R1 attachment site to produce the fragile complex. The fragile complex falls apart during gel electrophoresis and produces a stereotypical series of recombined topoisomers (white arrows). Addition of SDS disrupts all inter-protein and protein-DNA interactions within the MuA complex and serves as the “100% disassembly” control. Rates of MuA complex disassembly by ClpX were assayed by measuring the rate of appearance of the lowermost disassembly DNA product on a native agarose gel. C. Schematic of monomeric MuA degradation by ClpXP protease. Rates of protein degradation were assayed by measuring the rate of disappearance of MuA on SDS-PAGE. 41 and the unfoldase is not simply due to avidity contributed by the four C-terminal tags (Mu pore-binding tag). This conclusion is supported by the findings that additional mutations in MuA domain III antagonize only MuA transpososome disassembly by ClpX and not degradation (Abdelhakim et al., 2008). Furthermore, the N-domain of ClpX is exceedingly important for transpososome remodeling but has little role in MuA monomer degradation (Abdelhakim et al., 2008). As previous studies established that ClpX recognizes short peptide-like signals, we characterized whether the remodeling-specific protein-protein contacts were also peptidelike or not. Second, we sought to understand at the molecular level how MuA assembly into a DNA-bound tetramer modulates recognition by ClpX. Because recognition depends on architectural features of the complex, we designed a hybrid Mu protein with novel DNA-binding specificity to assist in specifically placing subunits with altered recognition tags within assembled complexes. Lastly, we modulated the binding affinity of the C-terminal tag to understand the extent of cooperation among the different recognition peptides that together comprise the Mu transpososome remodeling signal “recognon”. Thus, here we establish molecular interactions between MuA and ClpX that enable ClpX to preferentially target the assembled Mu tetramer. This work also elucidates principles attractive to the general problem of designing recognition mechanisms that favor assembled, multimeric protein complexes. 42 2.3 2.3.1 Results Identification of a region critical for enhanced recognition of transpososomes by ClpX MuA consists of three domains and belongs to the DDE family of recombinases (reviewed in Rice & Baker, 2001). Previous analysis revealed that Domain III contributes most of the information recognized by ClpX (Abdelhakim et al., 2008). Although the majority of the structure of MuA is known, including the architecture of the transpososome (Clubb et al., 1994, 1997; Schumacher et al., 1997; Montaño et al., 2012), there is essentially no structural data of Domain III to guide our analysis. Therefore, we tested the primary sequence surrounding three arginines that had been identified previously as participating in transpososome-specific contacts (Figure 2-2A). Selected residues were mutated to aspartic acid as acidic amino acids disrupt ClpX contacts within other recognition tags (Flynn et al., 2003). MuA variants were purified, shown to assemble into stable transpososomes, and assayed for monomer degradation by ClpXP (Figure 2-2B) and for complex disassembly by ClpX (Figure 2-2C) at a substrate concentration significantly below the KM for transpososomes. Most substitution variants had small to modest defects on degradation rates by ClpXP (within 75% of wild-type rate) indicating that the interaction between these altered monomer variants and ClpX was similar to that of wild-type MuA. These same mutations in MuA complexes were also modestly slower (at most 2-fold) in disassembly reactions (Figure 2-3). The exception was that the I620D/F621D variant reduced both degradation of monomers and disassembly of complexes rates to a significant extent. The reason for this dual effect was not analyzed in detail, but it appeared the introduction of negative charges at this position was broadly deleterious. Because the goal of our mutation-based search was to uncover residues specific to the ClpX and transpososome interaction, we did not continue analysis of IF/DD. We categorized these residues as not contributing to ClpX’s distinction between the monomeric and tetrameric states of MuA. However, P623D and S624D displayed similar characteristics as the previously 43 A. MuA domain structure Domain I 1 Ia 77 Domain II 243 Ib 490 IIa DNA binding Domain III IIb 575 615 IIIa IIIb 663 MuA tag ( ClpX recognition ) Catalysis 610 634 PAAPESRIVGIFRPSGNTERVKNQE 635 R D D E Y E T E R D E Y L N H S L D I L E Q N R R K K A I 663 Fraction of start 1.0 time (min) 0.3µM ClpX6 0.8 0 2 4 6 8 +SDS WT * 0.6 0.4 0.2 C. Complex Disassembly WT PS 0 PS DIL GN time (min) 0 2 4 6 8 2 4 6 time (min) WT 8 PS * 0.8 Fraction disassembled B. Monomer degradation 0.6 IVG DIL EQN 0.4 GN 0.2 PS IF 0 0 10 WT 0.1µM ClpX6 2 4 6 8 10 time (min) E. Figure 2-2: Mutation of a sequence region R622-S624 reduces disassembly and degradation rates A. MuA transposase is a 75kDa protein comprised of three domains. Domain III contains the C-terminal Mu pore-binding tag comprised of the last eight residues, which is recognized by ClpX B. Degradation of wild-type MuA and MuA “aspartate” variants by ClpXP protease at sub-saturating enzyme concentrations. All “aspartate” variants are labeled with the endogenous residues that were targeted for aspartate substitution. Substrate concentration was 1uM. Inset shows a representative SDS-PAGE gel of wild-type MuA and MuA(P623D, S624D) monomer. C. Disassembly of complexes assembled from wild-type MuA and MuA “DD” variants by ClpX unfoldase. Transposososmes are marked by asterisks. Initial transpososome concentration was 100nM. DNA disassembly product used for quantification is marked by white arrow. The “+SDS” lane shows the pattern of topoisomer migration upon complete disassembly. Two representative native agarose gels of wild-type MuA complexes and mutant Mu(P623D, S624D) complexes. Quantification of DNA disassembly product appearance. 44 Monomer degradation Complex disassembly Rate relative to WT (%) 100 80 60 40 20 0 WT Δ8 617 620 622 623 PS GN DIL DDD DD A DD DD DDD DDD IVG IF R 625 653 656 EQN Figure 2-3: Comparison of all reaction rates for Mu aspartate variants Quantification of differences in degradation and disassembly rates of MuA variants whose indicated sequences were mutated to alanine or aspartic acid relative to wild-type MuA. Reactions with error bars were performed in triplicate. Error bars are the standard error of the mean. identified transpososome-specific contact residue R622 in which substitution specifically slowed disassembly rates by as much as 10-fold at the sub-saturating protein concentrations (Figure 2-3). We determined the functional interaction of ClpX with double mutant (P623D S624D) variant complexes during disassembly by measuring the concentration of enzyme for half-maximal velocity (KM ). Because it is difficult to obtain transpososomes at high concentration, we started with a fixed substrate concentration, varied the concentration of ClpX, measured the rate of appearance of DNA transposition product released by disassembly, and analyzed these data as previously described to obtain apparent KM values (Pyle & Green, 1994; Abdelhakim et al., 2008). For many ClpX substrates, the KM is nearly equivalent to the KD because catalysis is relatively slower that the binding reactions. Therefore the apparent KM is a measure of the functional affinity of ClpX for transpososomes. The apparent KM for disassembly of PS→DD double-mutant complexes was about 8-fold weaker than that of wild-type MuA complex (Figure 2-4). Additionally 45 WT Reaction Rate (min-1) 2.5 2 1.5 KMapp Vmax app ( µM ) (min −1 ) WT 1.4 ± 0.2 2.7 ± 0.1 PS 0.6 ± 0.1 10.6 ± 2.7 1 PS 0.5 0 0 5 10 15 20 25 ClpX 6 (µΜ) Figure 2-4: Residues P623 S624 form a critical interaction between MuA complex and ClpX Half-maximal velocity determination for ClpX-mediated disassembly of wild-type complexes and MuA(P623D,S624D) mutant complexes. Curves were repeated in triplicate. Error bars are the standard deviation of the average. the Vmax was 5-fold slower compared to that of wild-type MuA complex. These data indicate that the PS/DD mutations impact both recognition and initial post-recognition steps of disassembly (see discussion). Furthermore, these residues are not major contributors to monomer recognition but specific for transpososome recognition by ClpX. A boundary defined by the sharp drop in disassembly rates of the mutant variants spans MuA residues 622-624. This critical region for enhanced recognition of Mu transpososomes behaves in contrast the C-terminal Mu pore-binding tag. Truncations of the C-terminus established that MuA monomers and MuA complexes both rely on the Mu pore-binding tag for ClpX specificity (Figure 2-3D and Levchenko et al. 1995). Point mutations of tag residues also led to a significant decrease of both degradation and disassembly rates (Abdelhakim et al., 2008). Because residues 622-624 appeared to be functionally important to ClpX interaction exclusively in the context of the assembled MuA complex, we hypothesized that this critical region may function as a peptide-signal key for transpososome-specific recognition by ClpX. 46 2.3.2 A peptide encompassing the critical region interacts with the N-terminal zinc-binding domain of ClpX Recognition of Mu transpososomes is strongly influenced by the presence of the Nterminal zinc-binding domain (the N-domain) of ClpX (Abdelhakim et al., 2008). The N-domain binds peptide sequences donated by adaptor proteins, which assist ClpX in recognizing some substrates. For example, the adaptor protein, SspB, enhances degradation of ssrA-tagged substrates by ClpXP (Levchenko et al., 2000). The key interaction for this stimulation is a C-terminal sequence of SspB, known as the ClpX Binding (XB) peptide that binds the N-domain of ClpX (Wah et al., 2003; Song & Eck, 2003). Because Mu transpososome is a multimeric complex and depends on the N-domain for efficient disassembly, we hypothesized that one or more MuA subunit(s) may provide adaptor-like contacts to enhance recognition of MuA complexes by ClpX. In the previous section we provide evidence for a second ClpX-binding sequence in MuA, beyond the C-terminal Mu pore-binding tag, which functions in ClpX’s enhanced recognition of the MuA complex. If the critical region (residues 622-624) behaves as an adaptor-like signal similar to the XB peptide, then we expect this sequence to bind to N-domain of ClpX. We tested for a direct interaction between the critical region of MuA and the Ndomain using both peptide blot and solution binding assays. We designed a MuA peptide blot, wherein each “spot” was a 20 amino acid sequence from MuA, and each neighboring spot was a related peptide, shifted three residues over (C-terminal). In this manner the entire sequence of MuA domain III could be tested for binding in one experiment. The blot was probed with S35 radiolabeled N-domain. By this semi-qualitative measure, we observed binding at spots in three major regions of domain III (Figure 2-5A). One of these regions contained sequences that corresponded to the critical region identified by mutagenesis in section 1 (Figure 2-5A, purple box). To test interaction between this MuA peptide and the ClpX N-domain in solution, an 18-amino acid peptide (Mu-peptide 614-633) containing the critical region 622-624 was synthesized and labeled with fluorescein at its N-terminus. When assayed by fluorescence 47 XB peptide 560 L 563 V 566 N 569 A 572 R 575 R 578 Q 581 L 584 A 587 A 590 K 593 K 596 D 599 E 602 E 605 P 608 A 611 A 620 I 623 P 626 N 629 R 632 N 635 R 638 E 641 T 644 D 614 E X X X X X X X X Peptide Blot Mu Domain III 617 I A 569 A A G R E Y R R R Q K Q L K S A T K A A 614 E S R I V G I F R P S G N T E R V K N Q 644 D E Y L N H S L D I L E Q N R R K K A I B XB peptide RGGRPALRVVK Anisotropy (a.u.) 0.12 KD =13± 3µΜ Tr. enhancement peptide KD = 380 ± 90µΜ ESRIVGIFRPSGNTERVKNQ 0.08 Mutated control peptide KD ≈ 3000µΜ ESRIVGIFDDDGNTERVKNQ 0.04 0 500 1000 N-domain of ClpX (µΜ) Figure 2-5: The N-terminal zinc-binding domain of ClpX binds to the enhancement peptide A. Peptide array of sequences from Domain III of MuA (residues 560-663). Each spot represents a 20-amino-acid-long peptide shifted by three residues. The first amino acid of each peptide is labeled with the position number and single letter. Spots marked with an X have no peptides. The XB peptide serves as a positive control and is derived from a known ClpX-binding sequence in the adapter protein, SspB. Blot was probed with radiolabelled S35 N-domain of ClpX. The sequence of a peptide spot from each of three regions is shown. B. Solution binding of N-terminal Fluorescein-labeled peptides and purified ClpX Ndomain. A fixed amount (200nM) of Fluorescein-labeled peptides with the indicated sequences were incubated with increasing concentrations of purified N-domain. Errors represent standard deviation of the average. 48 anisotropy, the binding of N-domain with peptide, albeit weak (∼400𝜇M), was observed (Figure 2-5B). For comparison, the XB peptide binds to the N-domain with a much tighter affinity, KD ∼13𝜇M. Mutation of the genetically-identified RPS residues (622-624) to aspartates in the Mu-peptide 614-633 essentially abolished binding to the N-domain, verifying that the interaction between the critical region of MuA and the N-domain was specific, although weak. These results establish that the critical region is an N-domainbinding signal in addition to its transpososome-specific signaling attribute. These two features of the critical region suggest that MuA complex makes adapter-like contacts with ClpX. We will refer to the critical region as the N-domain-binding tag or the Enhancement tag for its property of imparting enhanced recognition of MuA complex. The above results, taken together with previous data establish that the natural remodeling signal for transpososomes contains at least two distinct peptide tags recognized by ClpX. These two type of tags are each present on all four subunits in MuA complex. The geometry of this cohort of eight tags in the context of the assembled MuA complex produces the remodeling signal that allows ClpX to discriminate transpososomes from monomers. However, previous work hinted that this cohort of eight tags do not contribute equally to the remodeling signal. Transpososomes with only one active MuA77-663 subunit carrying the pore-binding tag and three tag-truncated subunits are remodeled by ClpX (Burton et al., 2001). Furthermore, one subunit out of the four within the transpososome carrying the pore-binding tag was sufficient to allow ClpX-mediated remodeling (Abdelhakim et al., 2010). To continue to understand transpososome recognition, we addressed how the architecture of MuA complexes affects the Enhancement tag’s contribution to the remodeling signal. 2.3.3 Step-wise loss of the Enhancement tag trends with ClpX’s weaker affinity for complexes A Mu transpososome has rotational symmetry around its vertical axis. However the subunits in the complex adopt two very different conformations and thus form two classes of subunits (Yuan et al., 2005; Montaño et al., 2012). Class 1 are the catalytic subunits; 49 their active-site residues are ordered and positioned at the sites of DNA cleavage. In contrast, Class 2 subunits are distal to the DNA cleavage sites and their active site are disordered (Figure 2-6). With two distinct classes of subunits, we probed the conformations and positions affected their ability to participate in adaptor-like contacts with ClpX. L2 subunit N R2 subunit N Class 2 C Class 1 C L1 subunit R1 subunit Figure 2-6: Crystal structure of Mu transpososome (PDB ID 4FCY) shows residue 77 as the N-terminus and residue 605 as the C-terminus. Dotted ellipses represent an educated guess of where domain III might continue based on EM structure by Yuan et al. Although there is symmetry about the vertical axis, the two lower subunits contain the catalytic residues (Class 1) and two upper subunits play a more structural role with disordered catalytic residues (Class 2). To assay the individual contributions of subunits to donating the N-domain binding tag, we engineered a chimeric variant of MuA transposase that facilitated assembly of homogeneous mixed-mutant complexes. The SinMu chimera is comprised of the DNA binding domain from Sin integrase covalently joined Domains II and III of MuA transposase (Figure 2-7A). The Sin DNA-binding domain recognizes a DNA sequence distinct 50 A. SinMu chimera His6 575 253 Sin (147-200) domain 2 pSinRRSin + MuA ≈ SinMu MuA SinMu Sin R1 663 domain 3 ≈ B. 615 ≈ ≈ R1 Sin C. plasmid Chimeric Complex MuA : SinMu 1:0 0: 1 1:1 +SDS * Figure 2-7: Tools for making homogeneous mixed mutant complexes A. Domain structure of the SinMu chimera. The chimera has the DNA binding domain of Sin recombinase substituting for Mu DNA binding domain. B. Schematic of assembly of chimeric SinMu complexes on the altered- specificity plasmid substrate pSinRRSin. pSinRRSin has the Mu attachment sites, L2 and R2, swapped out for Sin-specific DNA binding sites. C. Assembly of chimeric complexes on pSinRRSin plasmid requires the correct ratio of MuA and SinMu proteins. Lane 1 contains un-reacted pSinRRSin supercoiled plasmid (black arrow). A native agarose gel of the in vitro assembly reaction shows the band associated with assembled complexes (asterisk) in lane 4 with the correct 1:1 protein ratio, but not in lanes 2 and 3 which have incorrect protein ratios. The characteristic pattern of recombined DNA disassembly products (white arrow) can be seen with addition of SDS in lane 5. from that of the MuA DNA binding domain. We derived a set of altered-specificity plasmids from the original pMiniMu by substituting Mu attachment sites individually for the Sin-specific DNA sequences (Figure 2-7B). Assembly of chimeric transpososomes onto a pSinRRSin plasmid depended on the presence of both MuA and SinMu proteins. Transposososmes assembled efficiently on two Mu DNA (or hybrid) right ends and symmetrical complexes have been widely 51 used in biochemical and structural studies (Savilahti et al., 1995; Williams et al., 1999; Yuan et al., 2005). When pSinRRSin was incubated with either protein individually no band corresponding to the assembled complex was observed by electrophoresis (Figure 27C). Hence, we reasoned that intersubunit contacts among MuA Domains II and III are insufficient to drive Mu transpososome assembly in the absence of specific DNA binding sites. Thus,we conclude that the substitutions of Sin DNA sites allow for placement of the SinMu chimeric protein(s) at a specific subunit location(s) within the Mu transpososome. Disassembly Rate (min-1) 3 KMapp Vmaxapp ( µM ) (min-1) 2.5 SinMu 1.3 ±0.2 3.0 ±0.2 2 WT MuA 1.4 ±0.2 2.7 ±0.1 SinMu∆8 n.a. n.a. 1.5 1 0.5 0 0 4 8 12 ClpX6 (µM) 16 Figure 2-8: Half-maximal velocity determination for disassembly of SinMu complexes by ClpX. SinMu complexes, wild-type with respect to the Enhancement tag in black circles. Native wild-type MuA complexes in gray circles. SinMu𝛿8 complexes with mutations in Enhancement tag in both Class 1 subunits, open circles. Errors at each concentration point are standard deviation of average. We assayed the effect of the Sin domain substitution on the functional interaction between ClpX and transpososomes. ClpX disassembled chimeric transpososomes (wildtype with respect to the Enhancement tag) with a KM app of ∼ 1.2𝜇M and Vmax of 3.4 min-1 (Figure 2-8, black). These values are within error of native MuA transpososome, KM app =1.4 ± 0.2𝜇M, suggesting that the Sin DNA binding domain does not significantly alter ClpX interaction with transpososomes. In this background, recognition of SinMu complexes requires the ClpX pore-binding tag in Class 1 subunits as disassembly of SinMu𝛿8 transpososomes by ClpX was barely detectable even at saturating enzyme 52 concentrations (Figure 2-8, dark gray). 3 SinMu “wild type” Disassembly Rate (min-1) PS in Class 1 Vmaxapp 1.3 ±0.2 3.0 ±0.2 4.6 ± 1.6 2.2 ± 0.2 4.4 ± 1.7 1.1 ± 0.1 11.5 ± 1.3 0.5 ± 0.02 ( µM ) 2.5 2 KMapp PS PS (min −1 ) 1.5 PS PS 1 PS in Class 2 PS PS 0.5 0 PS in all four SinMu ∆8 0 10 ClpX6 (µM) PS PS 20 Figure 2-9: All four subunits can provide the Enhancement tag in MuA complexes. Half-maximal velocity determination for disassembly of chimeric complexes with different numbers of subunits mutated at the Enhancement tag (PS/DD). Curves were repeated in triplicate. Error bars at each concentration point are the standard deviation of the average. Errors of the KM are standard deviation of average of three fits. We also mutated the Enhancement tag using the double mutation, P623D/S624D, in Class 1 subunits, Class 2 subunits, and in all four subunits. The mutant subunits in either Class 1 catalytic or Class 2 structural subunits resulted in a weaker KM app , 4.5𝜇M and 4.4𝜇M, respectively (Figure 2-9). The similar magnitude of these KM app values indicated that the two conformations do not differently present the Enhancement tag. Thus, the Enhancement tag is accessible by ClpX (at least in this conformation of the transpososome) from either class of subunits. Only when the Enhancement tag is mutated in all four subunits was the weakest apparent affinity observed, KM app = 11.5 ± 1.5𝜇M (Figure 2-9, black triangles). This value is striking because the KM app of the chimeric complex with all four mutant subunits is within error of the KM app of wild-type Mu monomers (10.5 ± 2.7 𝜇M) (Abdelhakim et al., 2008), revealing that the 10-fold 53 difference in apparent affinity between the wild-type MuA and SinMu-PS4 complexes is the nearly identical to that observed between the wild-type MuA complex and the wild-type Mu monomer. In this altered case, ClpX fails to discriminate assembled MuA complexes from monomeric MuA. Thus, mutations targeting the Enhancement tag erase the adaptor-like function of this sequence and its functional interactions with ClpX. 2.3.4 Mu pore-binding tag is an intrinsically poor ClpX signal without adaptor-like contacts Previous analysis hinted that the Mu pore-binding tag on its own is a weak ClpX recognition signal. A fluorescently-labeled peptide containing the C-terminal pore-binding tag has a KM app of ∼70𝜇M (Barkow, 2009), which is much weaker than the KM app for MuA monomers (∼10𝜇M). To test the strength of the Mu pore-binding tag in a folded protein we constructed a fusion protein (N-𝜆-cI-Mu) in which the Mu pore-binding tag was appended to the C-terminus of the N-domain of 𝜆-cI, a phage repressor protein. N-𝜆-cI-Mu behaved as a folded protein and native N-𝜆-cI protein is not a substrate for ClpXP. Like the peptide, the fusion protein was degraded with a KM app of ∼75𝜇M (Figure 2-10). Because this is roughly 7-fold weaker than ClpXP’s functional affinity for MuA monomers, we infer that ClpX makes contacts with regions of MuA in addition to the pore-binding tag even in MuA monomers, as was hinted previously (Abdelhakim et al., 2008). Thus, the Mu pore-binding tag, as an isolated peptide and as part of a folded protein, is a feeble ClpX recognition signal in the absence of adaptor-like contacts. 2.3.5 Transpososomes with a strong pore-binding tag do not require Enhancement tags The results presented above support a mechanism in which the MuA complex targets itself to ClpX by serving as an auto-adapter. By this model, the N-domain-binding tag acts similar to an adapter for the weak pore-binding tag. We predicted from this model that MuA complexes may not need the N-domain binding tag(s) if MuA is given a stronger pore-binding sequence. To test this model, we constructed a variant of MuA with 54 degradation rate -1 -1 ( µM min enz ) 1 0.8 0.6 0.4 KM 74.7±10.3 µM 0.2 0 Vmax 2.4 ± 0.2 0 10 20 30 min −1 40 N-λcI-Mu (µM) Figure 2-10: Half-maximal velocity determination for degradation of N-𝜆cI-Mu monomers by ClpXP. (A representative experiment.) ClpX6 was 0.3𝜇M, ClpP14 was 0.8𝜇M. its endogenous Mu pore-binding tag substituted with the ssrA tag (called Mu∆8ssrA) (Figure 2-11A). As predicted, disassembly of Mu∆8ssrA transpososomes by ClpX occurred with a similar apparent affinity, KM app =1.6±0.2 𝜇M (Figure 2-11B, filled circle) as wild-type MuA complexes ,KM app =1.4 𝜇M (Figure 2-11B, open circle). Mutating the Enhancement tag in this context of Mu∆8ssrA P623D/S624D resulted in no substantial change in the KM app (2.1± 0.3 𝜇M) (Figure 2-11B, filled square). ClpX recognized both the variants as well as it did wild-type MuA complexes. Thus, we conclude that the N-domain-binding tag is unnecessary for transpososome recognition in the context of the high-affinity ssrA tag. The ssrA-tagged transpososomes were also remodeled with a substantively faster Vmax than native complexes (see discussion). Finally the functional interaction between ClpX and Mu∆8ssrA monomers was determined by measuring the concentration dependence of degradation by ClpXP. These data were fit with the Michealis-Menton equation. As expected for ssrA-tagged proteins, this value for Mu∆8ssrA was strong, KM app ∼0.7 𝜇M (Figure 2-12). Strikingly, KM app for the altered monomers was very similar to the KM app of the complexes. Thus, ClpX loses the ability to discriminate between the two oligomeric states of MuA when transposase 55 A. Mu∆8ssrA Domain IIIβ sequence N-domain binding tag 615 S R I V G I F R P S G N T E R V K N Q E R D D E Y E T E R D 645 E Y L N H S L D I L E A A N D E N Y A L A A 666 Mu∆8ssrA ssrA tag Mu∆8ssrA PS DD 615 666 615 S R I V G I F R D D G N T E R V K N Q E R D D E Y E T E R D 645 E Y L N H S L D I L E A A N D E N Y A L A A 666 B. Complex disassembly by ClpX Disassembly Rate (min-1) 12 8 4 0 KMapp Vmaxapp ( µM ) (min −1 ) Mu∆8ssrA 1.6 ±0.2 12.0 ±0.4 Mu∆8ssrA PS DD 11.4 ±0.5 2.1 ±0.3 MuA - wildtype 1.4 ±0.2 2.7 ±0.1 MuA PS DD 10.6 ±2.7 0.6 ±0.1 0 4 8 12 ClpX 6 (µM) Figure 2-11: Mu complexes with a strong pore-binding tag are recognized as well as native Mu complexes A. Close up of DomainIII𝛽 structure of the variant, Mu∆8ssrA. Enhancement tag is underlined. The ssrA tag is in grey box. Sequence changes are indicated to the right. B. Half-maximal velocity determination for disassembly of wild-type MuA, MuA (PS/DD), Mu∆8ssrA, and Mu∆8ssrA (PS/DD) complexes by ClpX. Reactions were repeated four times. Error bars at each concentration point are standard deviation of the average. Error of the KM are standard deviation of the average of four fits. 56 carries a strong C-terminal pore-binding tag. Degradation Rate (µM min-1 enz-1) 4 3 2 KMapp 0.71 µM 1 Vmaxapp 3.6 min-1 0 0 3 6 Mu∆8ssrA (µM) 9 Figure 2-12: Half-maximal velocity determination for degradation of Mu∆8ssrA monomers by ClpXP. ClpX6 was 0.3𝜇M, ClpP14 was 0.8𝜇M. A representative experiment. 57 2.4 Discussion Here we elucidate a substantial feature of the molecular basis of recognition of the Mu transpososome, a multimeric substrate of the unfoldase ClpX. Our work provides insight into a framework governing the design of recognition signals specialized to favor disassembly/remodeling. Mu transpososome’s disassembly signal is comprised of multiple weak-affinity peptide-like sequences (tags) that are distributed throughout the assembled complex. One class of tag interacts with the central pore whereas the others interact with the N-domain of ClpX. A specific sequence region that interacts with the N-domain of ClpX has a principle role in enhancing recognition specifically of the assembled Mu transpososome. Our results support a strategy of prioritizing selection of an assembled complex over constituent subunits by employing (1) an intrinsically weak pore-binding tag with multiple enhancing signals, and (2) distributing these tags among the subunits of the complex. We propose an auto-adaptor mechanism in which Mu transposase’s disassembly signal is constructed using distinct interactions with the pore and N-domain of ClpX. This multivalent recognition signal is comprised of several short peptide-like sequences (tags). A previously characterized recognition element located at the C-terminus of MuA (Mu pore-binding tag) is accessible to ClpX. This tag is both sufficient and required for unfolding MuA (monomers) by ClpX, thus we infer the Mu pore-binding tag directly interacts with the central pore of ClpX. In this study, we identified and characterized another sequence comprising residues R622-S624 and located in MuA domain III that we term the “Enhancement tag.” Importantly, in contrast to the C-terminal Mu porebinding tag, the Enhancement tag contributes solely (or nearly so) to recognition of MuA complexes by ClpX. Mutations in the Enhancement tag did not appreciably disrupt ClpXMuA monomer interaction (within 75% of wild-type degradation rate) but displayed a serious defect in ClpX-MuA complex interaction (5-8% of wild-type disassembly rate) at sub-saturating concentrations of enzyme (Figure2-3). Peptide interaction blots and solution fluorescence anisotropy binding assays establish that the Enhancement tag binds weakly but specifically to the ClpX N-domain (Figure 2-5). 58 The IF/DD mutation behaved similarly to MuA∆8 in that it affected both monomer and complex recognition by ClpX (Figure 2-3). These residues may physically bind to ClpX (Figure 2-5) and could be a second dual-function tag. However, more trivially, the aspartate substitutions may disrupt local structure in Domain III, or be severely repulsive to interaction and thus indirectly influence transpososome and monomer contacts with ClpX. The kinetic analysis presented here was fit using Michaelis-Menten equations. For relatively slow reactions, the KM value is dominated by initial binding, and essentially equal to the KD of the interaction. However, MuA-ClpX interactions are not straightforward, and this issue is clearly noticeable by the substantial influence of altering the Enhancement tag on both KM app and Vmax for complexes. The Vmax effect could be explained by the fact that once one ClpX-MuA contact it made, all subsequent proteinprotein interactions are unimolecular, and thus most of the entropic “cost” of subsequent interactions is already paid (McGinness et al., 2007; Sauer & Baker, 2011). Furthermore, the transpososome-specific signals may assist maintaining “grip” on the substrate during initiation of translocation, which could be especially important with very weak pore-interacting tags. Consistent with this model, the MuA-ssrA chimera is processed with a higher Vmax than wild-type MuA. Thus, analysis of the MuA complex remodeling data using Michaelis-Menten formalism is certainly an oversimplification. Multivalent recognition signals and reliance on the N-domain of AAA+ unfoldase is observed with AAA+ enzymes and their substrates. The bacterial cell division protein, FtsZ, a homolog of tubulin, was recently shown to contain two sites important for proteolysis by ClpXP (Camberg et al., 2014). Similarly to MuA, FtsZ contains a C-terminal tag and an internal recognition element located 30 residues from the C-terminus. Another bacterial AAA+ unfoldase, ClpV, disassembles VipA/VipB tubules, components of the type VI secretion system present in many pathogenic proteobacteria. A recent study found multiple interactions between ClpV and VipA/VipB tubules resulting in preference of assembled VipA/VipB complexes over VipB monomers (Pietrosiuk et al., 2011). The eukaryotic AAA+ ATPase p97/ VCP/ Cdc48 is involved in many cellular pathways including membrane fusion, ERAD (endoplasmic reticulum associated degra59 dation), the DNA damage response, autophagy and endosomal sorting. The functional diversity of p97 is mediated through association with distinct, pathway-specific adaptor proteins. These adaptors bind to the auxiliary N-domain or C-terminal tails of the p97/VCP/Cdc48 enzymes. More than three-fourths of the current and growing list of adaptors require the N-domain of their AAA+ ATPase (Baek et al., 2013). Similarly to ClpX, ClpV and p97 may specifically recognize multimeric substrates by binding to complex-specific recognition signals in an N-domain dependent manner. Examination of the canonical ubiquitin tagging system and 26S proteasomal recognition of substrates also reveals a strategy of employing a multivalent signal. The ubiquitin chain is not sufficient to ensure substrate degradation; i.e. association with the proteasome is insufficient to make the protein a degradation substrate. To be a degradation substrate, the protein also must have an unstructured region that the AAA+ ATPases within the proteasome 19S cap can bind and engage (Prakash et al., 2004; Takeuchi et al., 2007). Hence, recognition for ubiquitin-dependent protein degradation is then more accurately described as “two-component” recognition signal (Inobe et al., 2011). This type of multivalent substrate-enzyme interaction for recognition parallels what we have uncovered between Mu transpososome and ClpX. There must be a site of engagement by the AAA+ enzyme pore (via flexible unstructured regions of eukaryotic substrates or the Mu pore-binding tag). Second, there are additional contacts between an auxiliary domain or component on the AAA+ enzyme (i.e. Rpn13, ubiquitin receptor subunit of proteasome or the N-domain of ClpX) and the substrate (Ubiquitin/N-domain-binding tag). In addition to employing multiple tags, the architecture of Mu transpososome restricts which subunits can provide certain tags. MuA complexes contain two classes of subunits: Class 1 (catalytic) and Class 2 (structural). All four subunits can provide the N-domain binding tag (Figure 2-9) whereas only the Class 1 subunits are able to productively provide the C-terminal Mu pore-binding tag (Abdelhakim et al., 2010). Scale representations of the transpososome and ClpX hint at the multiple approaches ClpX may use to bind MuA complexes (Figure 2-13,A). ClpX can span the C-terminal regions of the two class 1 subunits or the C-terminal regions of the Class 1 and Class 2 subunits on the same side of the axis of symmetry (Figure 2-13,B). Also reasonably 60 spaced is ClpX spanning the C-terminal regions of Class 1 and Class 2 subunits on opposite sides of the axis of symmetry. Our data support these orientations of ClpX A. Transpososome ClpX ~113Å ~106Å B. Subunits on the opposite side of symmetry axis E-tag E-tag P-tag E-tag P-tag P-tag E-tag P-tag Subunits on the same side of symmetry axis E-tag E-tag P-tag P-tag Figure 2-13: Permutations of tag engagement in Mu transpososome by ClpX A. Crystal structures of Mu transpososome (4FCY), ClpX ATPase domain hexamer (3HWS), and ClpX N-domain (2DS6) shown at the same scale were modeled to indicate how ClpX may interact simultaneously with multiple subunits in the MuA complex. Transpososome subunits are colored in shades of green and purple. ClpX hexamer is colored in blue. N-domain is colored in light blue. B. For simplicity this diagram considers only two subunits at a time although in principle multiple N-domains of ClpX may interact simultaneously with multiple subunits in the MuA complex. Each subunit contributes either the ClpX-pore-binding tag (P-tag) or the Enhancement a.k.a. N-domain-binding tag (E-tag). 61 as being sufficient to lead to recognition for complex disassembly. By making the Ndomain binding tag accessible on all four subunits, the architecture of the transpososome maximizes the permutations for successful recognition with six possible orientations of ClpX relative to MuA complex. A stricter division of labor in which only Class2 subunits can provide the N-domain-binding tag would result in four possible orientations; likewise if only Class 1 subunits provide both the N-domain-binding tag and the pore-binding tag there would be only two acceptable ClpX “attack” orientations. Multiple N-domaininteracting subunits may also be beneficial as the regulator protein MuB also binds the C-terminal region MuA transposase, and ClpX therefore may be able to make initial contacts with MuA complexes prior to the exit of MuB. Thus the fact that MuA presents the high-affinity multivalent signal only in the context of the assembled complex may have multiple advantages and indicates that the transpososome, by using an intricate set of signals, has evolved to be a high-priority ClpX target. Our work supports a design principle of tuning the strength of recognition signals with modular weakly-binding tags. On its own, the C-terminal Mu pore-binding tag is a poor ClpX recognition signal with an apparent affinity of ∼70𝜇M for ClpX (Figure 2-10; Barkow 2009). In comparison, the ssrA tag has an apparent affinity of ∼1.8𝜇M (Levchenko et al., 2000). However in the context of the assembled MuA complex, the Mu pore-binding tag benefits from multiple “diffuse” ClpX-interacting contacts, one of which is the N-domain binding tag identified in this study. Together, these peptide tags act synergistically to form a high-affinity ClpX recognition signal comparable in affinity to the ssrA tag. According to this model, weak pore-interacting tags favor heavy dependence on accessory recognition elements. In converse, we predicted that accessory recognition elements provide little or no additional benefit to strong, compact ClpX recognition signals. This hypothesis is supported by the MuA-ssrA hybrid proteins, which gained essentially no benefit from the N-domain-binding tag. These observations are consistent with the protein engineering studies of McGuiness et al. who demonstrated that a weakened pore-binding tag gains a much larger magnitude benefit from an adapter (McGinness et al., 2006). This type of “tag tuning” is used during the biologically relevant recognition of MuA by ClpX, as ClpX is no longer able to discriminate between the 62 tetrameric and monomeric states when carrying a strong pore-binding tag. Thus, this analysis of recognition of MuA lays a theoretical framework to understand the design logic of complex-targeting recognition signals employed by AAA+ enzymes. 63 2.5 Methods Buffers Buffer L1, W20, W250 contained 25mM HEPES-KOH pH 7.6, 100mM KCl, 400mM NaCl, 10mM beta-mercaptoethanol, 10% glycerol, and imidazole at concentrations of 10mM, 20mM, and 250mM, respectively. Buffer A contained 25mM HEPES-KOH pH 7.6, 0.1mM EDTA, 1mM DTT, 10% glycerol, 0.3M KCl. Buffer B contained 25mM HEPES-KOH pH 7.6, 0.1mM EDTA, 1mM DTT, 10% glycerol, 1M KCl. PD50 buffer contained 25mM HEPES-KOH ph 7.6, 50mM KCl, 5mM MgCl2, 0.032% NP-40, 10% glycerol. Protein and peptide purification Wild-type and mutant variants of MuA proteins (Baker et al., 1991) , E.coli ClpX (Neher et al., 2003b), ClpX∆N (residue 47-424) (Abdelhakim et al., 2008), HU (Baker et al., 1994), ClpP (Kim et al., 2000), N-domain of ClpX (residue 1-64) with a cleaveable N-terminal His tag (Chowdhury et al., 2010) were purified as previously described. SinMu chimera was cloned based on a plasmid gift, pSin15Mu, from S.P.M and P.A.R. The plasmid, pSin15Mu is residues 147-200 of Sin recombinase followed by a ten-residue SG repeat followed by MuA (residues 253-605). SinMu was generated by appending the remaining MuA transposase sequence such that the construct ends at the natural C-terminus of MuA, (residue 663), cloned into a pET3a vector via NdeI and BamHI restriction sites, and transformed into E. coli strain BL21(DE3). Cells were grown at 37°C to O.D.600nm ≈0.6 in Luria-Bertani broth containing 100𝜇g/mL ampicillin. Protein expression was induced for 3 hours by addition of 0.4mM IPTG. The culture was harvested by centrifugation, resuspended in 10mL of BufferL1 per liter of initial cell culture, and lysed by French press. The lysate was treated with PMSF (phenylmethylsulfonyl fluoride), cleared by centrifugation for 30min at 30,000g 4°C and incubated with Ni-NTA agarose beads equilibrated in BufferL1 for 1 hour at 4°C. The beads were transferred to a column, washed with Buffer W20, and bound protein was 64 eluted using Buffer W250. Fractions containing SinMu variants were identified by SDSPAGE, buffer-exchanged into Buffer A using PD-10 desalting columns. The eluate was further purified by anion exchange chromatography, MonoS equilibrated with Buffer A, and eluted by gradient to Buffer B. Fractions containing SinMu variants were identified by SDS-PAGE, pooled, and concentrated using Amicon (MWCO 5k) filter tubes, and the protein concentration by determined by Bradford reagent. Mu∆8ssrA was generated from pTB1, a pET3d containing MuA tranposase. The last eight C-terminal residues were replaced with the sequence for ssrA tag, generated by PCR with 5’-phosphate primers LLO62: aactacgctttagcagctTAAGGATCCGGCTGCTAACAAAGCC and LLO63: ttcgtcgtttgcggcTTCCAGAATATCCAGCGAATGATTCAGATA. The variant Mu∆8ssrA (P623D S624D) was cloned by PCR using 5’-phosphate primers LLO64: gaTgaCGGTAATACGGAACGGGTGAAG and LLO55: CCGGAAAATACCAACAATTCGTGA. Both Mu∆8ssrA and Mu∆8ssrA (PS/DD) proteins were expressed and purified using the protocol for wild-type MuA. Fluorescein-labeled peptides were synthesized by FMOC technique on an Apex 396 solid-phase synthesizer and purified on a reverse-phage C12 column running a gradient of 0-100% Acetonitrile by HPLC. Peptides were verified by MALDI-TOF mass spectrometry. DNA for transposition pSinRRSin was generated from miniMu plasmid, pMK586. pMK586 was digested with ClaI and EcoN1 to remove the phage left-end attachment sites, treated with Antartic phosphatase, and ligated to 5’-phospshate annealed oligonucleotides: LLO37: CCAAGGAAGCTTGAAGCGGCGCACGAAAAACGCGAAAGCcgtatgattagggtAT LLO38: CGATaccctaatcatacgGCTTTCGCGTTTTTCGTGCGCCGCTTCAAGCTTCCTTG containing the R1-Sin binding sites with appropriate overhangs. The right-end R2 binding site was replaced with Sin attachment sote sequence, generated by PCR with 5’-phosphate primers LLO46: tcatacgGCTTTCGCGTTTTTCGTGCGC and LLO47: ttagggtCTTTAGCTTTCGCGCTTCAAATG. Transpososome Assembly 65 Transpososomes were assembled in vitro in the following buffer: 25mM HEPES pH7.6, 10mM MgCl2, 15% glycerol, 0.1mg/mL BSA, 1mM DTT, 100mM NaCl, 9% DMSO. Transposition reactions contained 16𝜇g/mL supercoiled pMK586, 130nM HU, 100nM MuA and the mixture was incubated at 30°C for 20min. To assemble SinMu chimeric transpososomes, 16𝜇g/mL pSinRRSin, 130nM HU, 50nM MuA variant, 50nM SinMu variant were incubated at 30°C for 60min. Degradation Assay ClpX and ClpP were preincubated with ATP regeneration mix for 1min at 30°C prior to addition of substrate in PD50 buffer. Final concentrations: ClpX6 =0.3𝜇M, ClpP14 =0.8𝜇M, ATP=4mM, creatine phosphate= 5mM, creatine kinase=0.05mg/mL. Samples (5𝜇L) were removed at different times and stopped by addition of 2.5x SDS loading buffer. After SDS-PAGE, products were visualized with Coomassie Blue stain. Disassembly Assay for determination of Steady-State kinetic parameters ClpX was preincubated with ATP regeneration mix for 1min at 30°C prior to addition of substrate in PD50 buffer. Final concentrations: ATP 4mM, Creatine phosphate= 20mM, creatine kinase=0.25mg/mL. For each timepoint, the reaction was stopped by addition of EDTA to 50mM. Samples were electrophoresed on 0.9% High gelling temperature (HGT) Agarose gel (Lonza) containing 10𝜇g/mL BSA and 10𝜇g/mL heparin. Gels were stained with Sybr Green I (Invitrogen) and visualized using a Typhoon imager (GE). Rates of disassembly were quantified using ImageQuant (GE) as previously described. Briefly, for each time point, the DNA product band was calculated as a percent of the total counts in the lane and normalized to the “+SDS lane”, which was used as the “100% disassembly” control (Abdelhakim et al., 2010). Peptide-binding assay Fluorescein-labeled peptides were incubated with increasing amounts of ClpX Ndomain in PD50 buffer at 30°C, and fluorescence was measured using a fluorimeter (Photon Technology International) at 495nm excitation, 520nm emission. The KD values were determined by fitting binding data to a hyperbolic equation. 66 2.6 Appendix: Geometry experiments on MuA monomer variants 2.6.1 Introduction ClpX unfoldase recognizes two oligomeric states of phage MuA transposase. The first state is the biologically-active tetrameric complex synapsed with phage genomic ends and host DNA in the core of the complex (transpososome). The second state is the catalytically-incompetent and unassembled MuA monomeric subunits. However, ClpX has a clear preference to target the transpososome as the substrate for its unfolding activity . In vitro, ClpX has an apparent affinity of ∼1𝜇M for transpososome disassembly while it recognizes MuA monomers with a ten-fold weaker apparent affinity of ∼10𝜇M (Abdelhakim et al., 2008). In my thesis research, I strove to uncover the molecular mechanism for ClpX’s ability to discriminate between the two oligomeric states of MuA transposase and the resulting preference for the assembled MuA complex over MuA monomers. Before the crystal structure of the transpososome was solved, the only available structural reference for an assembled MuA complex was a low-resolution EM (electron microscope) structure of a type 1 transpososome, or CDC (Yuan et al., 2005). In Chapter 2, I identified another ClpX recognition signal, the Enhancement tag, which contributed principally and substantially to transpososome recognition and insignificantly to monomer recognition. With the Mu pore-binding tag, there were now a total of two identified tags that contributed to the holistic remodeling signal. One hypothesis I considered was that these two tags in MuA transposase fit into the "two-component recognition signal" model (Inobe et al., 2011). Studies on the intracellular protease, 26S proteasome, an eukaryotic compartmentalized protease parallel to bacterial ClpXP, and the ubiquitin-tagging system enumerated two minimal components for ubiquitin- tagged proteasome substrates (Prakash et al., 2004). 1) a proteasome-binding site (Ubiquitin or UBL(ubiquitin-like) domains) 2) a flexible unstructured region 67 Futher protein engineering studies by the Matoushek group showed efficient substrate degradation occurred when the initiation region is of a certain minimal length and is appropriately separated in space from the proteasome-binding tag (Inobe & Matouschek, 2014). Inspired by their work on the geometry/ spacing of two-component recognition signals, I probed the spacing of the two tags in MuA transposase. 2.6.2 Results I hypothesized that spatial constraints prohibited simultaneous engagement of the porebinding tag and the N-domain-binding tag in MuA monomer. Assuming that both tags are accessible in MuA monomer, I tested if increasing the spatial separation between the two tags would improve ClpX’s affinity for MuA monomers. I designed variants of MuA transposase with additional amino acids to increase separation of the C-terminal pore-binding tag from the N-domain-binding tag. The additional amino acids were taken from the sequence in the adaptor protein SspB, a flexible region that connects the folded substrate-binding domain and the XB peptide. I inserted 15 or 25-long sequences just before the Mu pore-binding tag, naming them after the number of inserted amino acids, Mu-SspB15L and Mu-SspB25L. (Figure2-14A). Circular dichroism established that there was no discernible changes in the overall fold of the MuSspB variants as compared to wild-type. I assayed ClpXP-mediated degradation of these Mu-SspB variants. Degradation of Mu-SspB15L and Mu-SspB25L was barely detectable, roughly 3-4% of the wild-type degradation rate at the low enzyme concentration assayed (Figure2-14B). As the Mu-SspB variants seem folded overall, I checked if the inserted SspB linker was somehow evolved to be "undegradable." I then modified the Mu-SspB15L and MuSspB25L genes by removing the Mu pore-binding tag sequence and replacing it with the sequence for the ssrA tag. I was only able to recover clones of Mu-SspB25L-ssrA. Degradation of Mu-SspB25L-ssrA by ClpXP was detectable but slow at ∼25% of the rate for wild-type native MuA monomers. 68 A. Flexible linker of 15 or 25 amino acids pore tag B. Monomer degradation rates by ClpXP Degradation rate %WT (µM min-1 enz-1) 100 80 60 40 20 0 WT MuSspB15L MuSspB25L MuSspB25L-ssrA Figure 2-14: MuSspB monomer degradation by ClpXP A.Diagram illustrating hypothesis of spatial restriction in MuA monomer preventing engagement of pore-binding tag when bound to the N-domain of ClpX. Additional residues from a flexible region of SspB adaptor protein were inserted just before the ClpX porebinding tag.B.Rates of MuSspB monomer variant degradation relative to wild-type MuA by ClpXP 2.6.3 Discussion I conclude that insertion of additional residues from the linker region of SspB into Domain III of MuA disrupted the ClpX recognition signal perhaps by perturbing local structure or contacts. Although all the Mu-SspB variants seem to be properly folded as measured by CD, loss of local structure in domain III may have been masked by the larger signal coming from the properly-folded Domains I and II. The barely detectable degradation rates of the Mu-SspB variants support prior evidence that ClpX recognizes MuA monomers utilizing additional signals other than the C-terminal pore-binding tag. The variant ClpX∆N displays a 3-fold defect in degradation rates of MuA monomer as 69 compared to full-length ClpX (Abdelhakim et al., 2008). Surprisingly, Mu-SspB25L-ssrA monomer variant was degraded slower than wildtype MuA monomers at substrate concentrations well below the KM for degradation. Most likely this difference is reflective of the method used to detect loss of protein; Coomassie Blue staining of SDS-PAGE gels. Coomassie Blue has a smaller dynamic signal range than other protein stains. Thus monitoring fast reactions with Coomassie Blue leads to greater errors and slower-than expected initial rates. For a more reliable comparison between these two variants, I would assay Mu-SspB25L-ssrA degradation by ClpXP over a range of substrate concentrations to get Michaelis-Menten kinetic parameters and would follow degradation using a more sensitive stain like SYPRO Orange or radiolabelled test substrate. 70 Chapter 3 Conclusion & Future Directions 71 3.1 Conclusion E.coli ClpX is a member of the Clp/Hsp100 family of ATPases that remodel multicomponent complexes and facilitate ATP-dependent protein degradation. Previous extensive studies on the interaction of ClpX remodeling Mu transpososomes led to progress toward a molecular understanding of how ClpX recognized MuA complexes. The work of this thesis deepens this understanding by identifying the molecular mechanism for ClpX’s distinction between transpososomes and MuA monomers and thereby the specificity for transpososomes. MuA transposase exists as inactive monomers and as an assembled transpososome, a nucleoprotein complex with four subunits synapsed with recombined DNA. ClpX recognizes the biologically relevant transpososome (MuA complex) with a 10-fold tighter apparent affinity compared to monomeric MuA. I identified a critical region (RPS) in MuA transposase expanding upon a previously identified arginine residue that contributed greatly to the recognition of transpososomes by ClpX and very little to recognition of MuA monomers. This peptide-like signal (residues 622-624) forms a ClpX recognition tag that interacts weakly but specifically to the N-domain of ClpX. Mutation of this Enhancement tag from all four subunits resulted in a 10-fold weaker apparent affinity for disassembly (KM app ∼10𝜇M), which is the same magnitude as ClpX’s affinity for MuA monomers. This newly identified tag (Enhancement tag) act synergistically with the previously characterized C-terminal Mu pore-binding tag (a.k.a "Mu degron" in other studies) to form the holistic remodeling signal imparting ClpX specificity for transpososomes over monomers. I then investigated the geometry of these two recognition tags in the context of the assembled MuA complex. To this end, I engineered a chimeric protein (SinMu) with altered DNA binding specifity by modifying a fusion protein (Sin15Mu) that was initially designed and constructed by Montano and Rice. I also created a library of Mini-mu plasmids with different substitutions in phage attachment sites using the oligo sequence specific for Sin15Mu binding also designed by Montano and Rice. I was able to determine the contributions of Mu subunits to each type of tag. All four subunits in the 72 transpososome can effectively contribute an N-domain-binding tag. Optimal recognition is achieved when all four are present. In contrast, only the catalytic Class 1 subunits productively provide the pore-binding tag. I further showed that for the variant, Mu∆8ssrA, the N-domain-binding tag becomes unnecessary for complex recognition because the native pore-binding tag is replaced by the ssrA tag, a stronger ClpX recognition signal. Complexes of Mu∆8ssrA with and without mutation of the Enhancement tag were recognized by ClpX just as well as native transpososomes. However, ClpX also recognized Mu∆8ssrA monomers with nearly the same apparent affinity, KM app , as assembled complexes. From the above results, I propose the following design framework for recognition signals to target assembled protein complexes to unfolding chaperones and remodelers of the AAA+ superfamily. First, the target substrate makes multiple weak interactions with the AAA ATPase. One type of interaction must be with the pore of the ATPase for engagement and subsequent translocation/unfolding. Another type of interaction occurs with an auxiliary domain of the AAA+ unfoldase. This binding may be direct with the multicomponent substrate such as in the case for Mu transpososome or may be indirect through adaptor proteins. Second, recognition tags should be at the weaker end of the affinity spectrum to allow effective synergy of multiple tags in the assembled complex. As shown in Chapter 2, ClpX failed to discriminate between the tetrameric and monomeric states of a protein that has a strong pore-binding tag. Third, multi-subunit complexes can "divide the labor" of making these interactions among their subunits. The specific architecture of each multimeric complex will determine the extent of this division of labor. Unassembled subunits or monomers are therefore incompetent to provide all the tags available in assembled complexes. Conformational changes within subunits that accompany formation of the assembled complex are a likely mechanism to reveal recognition tags. A reverse-strategy is employed in examples when protein complexes fall apart and a specific subunit is targeted for degradation by a revealed tag. Thus, collaboration between multiple weak substrate-unfoldase signals is an attractive general mechanism for targeting assembled protein complexes to 73 AAA+ enzymes. 3.2 Future Directions The Enhancement tag was described and referred to as a peptide-like signal due to its contiguous nature and ability to interact with purified N-domain when synthesized as a 20-aa peptide. Several secondary structure prediction algorithms (Jpred, CFSSP, NPS@SOPMA) assign these residues to a flexible unstructured linker or random coil immediately following a predicted helix. However, these observations don’t exclude that the residues may be part of a binding surface. Definitive answers would come from a co-crystral structure of ClpX and transpososome assembled using a MuA construct that extends to MuA’s C-terminus as the current crystal structure of the STC transpososome utilized a truncated MuA protein (Montaño et al., 2012). Furthermore, I propose extending the lysine-acetylation footprinting experiments from Abdelhakim et al. 2008 to the SinMu chimeric complexes. This method would likely reveal which residues or surface areas on which subunits are protected in the presence of ClpX and ATP𝛾S compared to SinMu chimeric complexes alone. To further support the insights gained from the transpososome remodeling signal and develop more robust design principles of ClpX recognition signals, I propose that future researchers investigate substrates in a systematic and high-throughput approach. As mentioned in Chapter 1, our lab performed a proteomic screen for in vivo ClpXP substrates and sorted the trapped proteins into five classes of recognition motifs. In light of identifying two distinct classes of recognition tags in Mu transpososome, the results from the 2003 screen may benefit from re-examination with the goal of categorizing the sequences as pore-binding motifs or N-domain-binding motifs. Furthermore, almost 60% of these trapped substrates perform their biological function as subunits within a complex. It would be interesting to elucidate how many of those trapped substrates are recognized by ClpX in an adaptor or N-domain dependent mechanism. Does ClpX recognize these proteins in the context of the assembled complex like Mu transpososome or in the context of a monomeric subunit post-disassembly? To address these questions, 74 I propose a modified version of the original proteomic screen, in which ClpX∆N is used as the partnering unfoldase for the ClpPtrap (Flynn et al., 2003). Secondly, I would reprobe the peptide array from Figure 4 with isolated N-domain of ClpX to observe if these residues/polypeptide regions may be classified as N-domain-binding tags. I would confirm the interaction with solution-based binding assays. Lastly, I would seek to understand if a consensus N-domain-binding motif emerges from these sequences. 75 76 Bibliography Abdelhakim, A. H., Oakes, E. C., Sauer, R. T., & Baker, T. A. (2008). Unique contacts direct high-priority recognition of the tetrameric Mu transposase-DNA complex by the AAA+ unfoldase ClpX.. Mol. Cell 30(1), 39–50. Abdelhakim, A. H., Sauer, R. T., & Baker, T. A. (2010). The AAA+ ClpX machine unfolds a keystone subunit to remodel the Mu transpososome.. Proc. Natl. Acad. Sci. U. S. A. 107(6), 2437–42. Anfinsen, C. B. (1973). Principles that govern the folding of protein chains.. Science 181(4096), 223–30. Aubin-Tam, M.-E., Olivares, A. O., Sauer, R. T., Baker, T. A., & Lang, M. J. (2011). Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine.. Cell 145(2), 257–67. Baek, G. H., Cheng, H., Choe, V., Bao, X., Shao, J., Luo, S., & Rao, H. (2013). Cdc48: A Swiss Army Knife of Cell Biology.. J. Amino Acids 2013, 183421. Baker, T. A., Kremenstova, E., & Luo, L. (1994). Complete transposition requires four active monomers in the mu transposase tetramer.. Genes Dev. 8(20), 2416–2428. Baker, T. A. & Mizuuchi, K. (1992). DNA-promoted assembly of the active tetramer of the Mu transposase.. Genes Dev. 6(11), 2221–32. Baker, T. A., Mizuuchi, M., & Mizuuchi, K. (1991). MuB protein allosterically activates strand transfer by the transposase of phage Mu.. Cell 65(6), 1003–13. Baker, T. A. & Sauer, R. T. (2006). ATP-dependent proteases of bacteria: recognition logic and operating principles.. Trends Biochem. Sci. 31(12), 647–53. Barkow, S. R. (2009). Mechanistic Studies of the AAA + Molecular Motor ClpXP PhD thesis Massachusetts Institute of Technology. Barkow, S. R., Levchenko, I., Baker, T. A., & Sauer, R. T. (2009). Polypeptide translocation by the AAA+ ClpXP protease machine.. Chem. Biol. 16(6), 605–12. Bochtler, M., Hartmann, C., Song, H. K., Bourenkov, G. P., Bartunik, H. D., & Huber, R. (2000). The structures of HsIU and the ATP-dependent protease HsIU-HsIV.. Nature 403(6771), 800–5. 77 Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D. C., Joachimiak, A., Horwich, A. L., & Sigler, P. B. (1994). The crystal structure of the bacterial chaperonin GroEL at 2.8 A.. Nature 371(6498), 578–86. Burton, B. M. & Baker, T. A. (2003). Mu Transpososome Architecture Ensures that Unfolding by ClpX or Proteolysis by ClpXP Remodels but Does Not Destroy the Complex. Chem. Biol. 10, 463–472. Burton, R. E., Siddiqui, S. M., Kim, Y. I., Baker, T. A., & Sauer, R. T. (2001). Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine.. EMBO J. 20(12), 3092–100. Camberg, J. L., Viola, M. G., Rea, L., Hoskins, J. R., & Wickner, S. (2014). Location of Dual Sites in E. coli FtsZ Important for Degradation by ClpXP; One at the C-Terminus and One in the Disordered Linker.. PLoS One 9(4), e94964. Chowdhury, T., Chien, P., Ebrahim, S., Sauer, R. T., & Baker, T. A. (2010). Versatile modes of peptide recognition by the ClpX N domain mediate alternative adaptorbinding specificities in different bacterial species.. Protein Sci. 19(2), 242–54. Chung, C. H. & Goldberg, A. L. (1981). The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La.. Proc. Natl. Acad. Sci. U. S. A. 78(8), 4931–5. Clubb, R. T., Omichinski, J. G., Savilahti, H., Mizuuchi, K., Gronenborn, A. M., & Clore, G. M. (1994). A novel class of winged helix-turn-helix protein: the DNA-binding domain of Mu transposase.. Structure 2(11), 1041–8. Clubb, R. T., Schumacher, S., Mizuuchi, K., Gronenborn, a. M., & Clore, G. M. (1997). Solution structure of the I gamma subdomain of the Mu end DNA-binding domain of phage Mu transposase.. J. Mol. Biol. 273(1), 19–25. Craigie, R., Arndt-Jovin, D. J., & Mizuuchi, K. (1985). A defined system for the DNA strand-transfer reaction at the initiation of bacteriophage Mu transposition: protein and DNA substrate requirements.. Proc. Natl. Acad. Sci. U. S. A. 82(22), 7570–4. Craigie, R. & Mizuuchi, K. (1987). Transposition of Mu DNA: joining of Mu to target DNA can be uncoupled from cleavage at the ends of Mu.. Cell 51(3), 493–501. Craigie, R., Mizuuchi, M., & Mizuuchi, K. (1984). Site-specific recognition of the bacteriophage Mu ends by the Mu A protein.. Cell 39(2 Pt 1), 387–94. Deuerling, E., Schulze-Specking, A., Tomoyasu, T., Mogk, A., & Bukau, B. (1999). Trigger factor and DnaK cooperate in folding of newly synthesized proteins.. Nature 400(6745), 693–6. Dougan, D. a., Reid, B. G., Horwich, A. L., & Bukau, B. (2002). ClpS, a substrate modulator of the ClpAP machine.. Mol. Cell 9(3), 673–83. 78 Doyle, S. M. & Wickner, S. (2009). Hsp104 and ClpB: protein disaggregating machines.. Trends Biochem. Sci. 34(1), 40–8. Dubnau, D. & Roggiani, M. (1990). Growth medium-independent genetic competence mutants of Bacillus subtilis.. J. Bacteriol. 172(7), 4048–55. Eckl, J. M. & Richter, K. (2013). Functions of the Hsp90 chaperone system: lifting client proteins to new heights.. Int. J. Biochem. Mol. Biol. 4(4), 157–65. Ellis, R. (2003). Protein Folding: Importance of the Anfinsen Cage. Curr. Biol. 13(22), R881–R883. Farrell, C. M., Baker, T. A., & Sauer, R. T. (2007). Altered specificity of a AAA+ protease.. Mol. Cell 25(1), 161–6. Fenton, W. A., Kashi, Y., Furtak, K., & Horwich, A. L. (1994). Residues in chaperonin GroEL required for polypeptide binding and release.. Nature 371(6498), 614–9. Flynn, G. C., Chappell, T. G., & Rothman, J. E. (1989). Peptide binding and release by proteins implicated as catalysts of protein assembly.. Science 245(4916), 385–90. Flynn, J. M., Levchenko, I., Sauer, R. T., & Baker, T. A. (2004). Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation.. Genes Dev. 18(18), 2292–301. Flynn, J. M., Levchenko, I., Seidel, M., Wickner, S. H., Sauer, R. T., & Baker, T. A. (2001). Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis.. Proc. Natl. Acad. Sci. U. S. A. 98(19), 10584–9. Flynn, J. M., Neher, S. B., Kim, Y. I., Sauer, R. T., & Baker, T. A. (2003). Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpXrecognition signals.. Mol. Cell 11(3), 671–83. Fourie, a. M., Sambrook, J. F., & Gething, M. J. (1994). Common and divergent peptide binding specificities of hsp70 molecular chaperones.. J. Biol. Chem. 269(48), 30470–8. Gardner, M. K., Zanic, M., & Howard, J. (2013). Microtubule catastrophe and rescue.. Curr. Opin. Cell Biol. 25(1), 14–22. Gayda, R. C., Yamamoto, L. T., & Markovitz, a. (1976). Second-site mutations in capR (lon) strains of Escherichia coli K-12 that prevent radiation sensitivity and allow bacteriophage lambda to lysogenize.. J. Bacteriol. 127(3), 1208–16. Glynn, S. E., Martin, A., Nager, A. R., Baker, T. A., & Sauer, R. T. (2009). Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ proteinunfolding machine.. Cell 139(4), 744–56. 79 Gonciarz-swiatek, M., Um, S.-j., Learn, B. A., Mcmacken, R., Kelley, W. L., Chem, J. B., Wawrzynow, A., Georgopoulos, C., Sliekers, O., & Zylicz, M. (1999). Recognition , Targeting , and Hydrolysis of the 𝜆 O Replication Protein by the Recognition , Targeting , and Hydrolysis of the O Replication Protein by the ClpP / ClpX Protease *. J. Biol. Chem. 274, 13999–14005. Gonzalez, M., Rasulova, F., Maurizi, M. R., & Woodgate, R. (2000). Subunit-specific degradation of the UmuD/D’ heterodimer by the ClpXP protease: the role of trans recognition in UmuD’ stability.. EMBO J. 19(19), 5251–8. Gottesman, S., Halpern, E., & Trisler, P. (1981). Role of sulA and sulB in filamentation by lon mutants of Escherichia coli K-12. J. Bacteriol. 148(1). Gottesman, S., Roche, E., Zhou, Y., & Sauer, R. T. (1998). The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrAtagging system. Genes Dev. 12(9), 1338–1347. Greene, E. C. & Mizuuchi, K. (2002a). Direct observation of single MuB polymers: evidence for a DNA-dependent conformational change for generating an active target complex.. Mol. Cell 9(5), 1079–89. Greene, E. C. & Mizuuchi, K. (2002b). Dynamics of a protein polymer: the assembly and disassembly pathways of the MuB transposition target complex.. EMBO J. 21(6), 1477–86. Greene, E. C. & Mizuuchi, K. (2002c). Target immunity during Mu DNA transposition. Transpososome assembly and DNA looping enhance MuA-mediated disassembly of the MuB target complex.. Mol. Cell 10(6), 1367–78. Hanson, P. I. & Whiteheart, S. W. (2005). AAA+ proteins: have engine, will work.. Nat. Rev. Mol. Cell Biol. 6(7), 519–29. Hartl, F. U. (2011). Chaperone-assisted protein folding: the path to discovery from a personal perspective.. Nat. Med. 17(10), 1206–10. Higashitani, A., Ishii, Y., Kato, Y., & Koriuchi, K. (1997). Functional dissection of a cell-division inhibitor, SulA, of Escherichia coli and its negative regulation by Lon.. Mol. Gen. Genet. 254(4), 351–7. Houry, W. A., Frishman, D., Eckerskorn, C., Lottspeich, F., & Hartl, F. U. (1999). Identification of in vivo substrates of the chaperonin GroEL.. Nature 402(6758), 147– 54. Inobe, T., Fishbain, S., Prakash, S., & Matouschek, A. (2011). Defining the geometry of the two-component proteasome degron.. Nat. Chem. Biol. 7(3), 161–7. Inobe, T. & Matouschek, A. (2014). Paradigms of protein degradation by the proteasome.. Curr. Opin. Struct. Biol. 24, 156–64. 80 Jones, J. M., Welty, D. J., Chem, J. B., & Nakai, H. (1998). Versatile Action of Escherichia coli ClpXP as Protease or Molecular Chaperone for Bacteriophage Mu Transposition Versatile Action of Escherichia coli ClpXP as Protease or Molecular Chaperone for Bacteriophage Mu Transposition *. J. Bacteriol. 273, 459–465. Karzai, A. W., Roche, E. D., & Sauer, R. T. (2000). The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue.. Nat. Struct. Biol. 7(6), 449–55. Katayama, Y., Gottesman, S., Pumphreyll, J., Rudikoffl, S., Clark, W. P., & Maurizi, M. R. (1988). The Two-component , ATP-dependent Clp Protease of Escherichia coli. J. Biol. Chem. 263(29), 15226–15236. Keiler, K. C., Waller, P. R., & Sauer, R. T. (1996). Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA.. Science 271(5251), 990–3. Kim, Y. I., Burton, R. E., Burton, B. M., Sauer, R. T., & Baker, T. A. (2000). Dynamics of substrate denaturation and translocation by the ClpXP degradation machine.. Mol. Cell 5(4), 639–48. Kirstein, J., Molière, N., Dougan, D. a., & Turgay, K. (2009). Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases.. Nat. Rev. Microbiol. 7(8), 589–99. Kirstein, J., Schlothauer, T., Dougan, D. a., Lilie, H., Tischendorf, G., Mogk, A., Bukau, B., & Turgay, K. (2006). Adaptor protein controlled oligomerization activates the AAA+ protein ClpC.. EMBO J. 25(7), 1481–91. Kruklitis, R., Welty, D. J., & Nakai, H. (1996). ClpX protein of Escherichia coli activates bacteriophage Mu transposase in the strand transfer complex for initiation of Mu DNA synthesis.. EMBO J. 15(4), 935–44. Kuo, C. F., Zou, a. H., Jayaram, M., Getzoff, E., & Harshey, R. (1991). DNA-protein complexes during attachment-site synapsis in Mu DNA transposition.. EMBO J. 10(6), 1585–91. Laskowska, E., Kuczyńska-Wiśnik, D., Skórko-Glonek, J., & Taylor, A. (1996). Degradation by proteases Lon, Clp and HtrA, of Escherichia coli proteins aggregated in vivo by heat shock; HtrA protease action in vivo and in vitro.. Mol. Microbiol. 22(3), 555–71. Lavoie, B. D., Chan, B. S., Allison, R. G., & Chaconas, G. (1991). Structural aspects of a higher order nucleoprotein complex: induction of an altered DNA structure at the Mu - host junction of the Mu Type 1 transpososome. EMBO 10(10), 3051–3059. Lee, S., Sowa, M. E., Watanabe, Y.-h., Sigler, P. B., Chiu, W., Yoshida, M., & Tsai, F. T. (2003). The Structure of ClpB. Cell 115(2), 229–240. 81 Levchenko, I., Grant, R. a., Wah, D. a., Sauer, R. T., & Baker, T. A. (2003). Structure of a delivery protein for an AAA+ protease in complex with a peptide degradation tag.. Mol. Cell 12(2), 365–72. Levchenko, I., Luo, L., & Baker, T. A. (1995). Disassembly of the Mu transposase tetramer by the ClpX chaperone.. Genes Dev. 9(19), 2399–2408. Levchenko, I., Seidel, M., Sauer, R. T., & Baker, T. A. (2000). A Specificity-Enhancing Factor for the ClpXP Degradation Machine. Science 289(5488), 2354–2356. Levchenko, I., Yamauchi, M., & Baker, T. A. (1997). ClpX and MuB interact with overlapping regions of Mu transposase: implications for control of the transposition pathway.. Genes Dev. 11(12), 1561–1572. Li, J. & Buchner, J. (2013). Structure, function and regulation of the hsp90 machinery.. Biomed. J. 36(3), 106–17. Lies, M. & Maurizi, M. R. (2008). Turnover of endogenous SsrA-tagged proteins mediated by ATP-dependent proteases in Escherichia coli.. J. Biol. Chem. 283(34), 22918–29. Lima, S., Guo, M. S., Chaba, R., Gross, C. a., & Sauer, R. T. (2013). Dual molecular signals mediate the bacterial response to outer-membrane stress.. Science 340(6134), 837–41. Little, J. W. (1983). The SOS Regulatory System: Control of its State by the Level of RecA Protease. J. Mol. Biol. 167, 791–808. Martin, A., Baker, T. A., & Sauer, R. T. (2007). Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease.. Mol. Cell 27(1), 41– 52. Martin, A., Baker, T. A., & Sauer, R. T. (2008a). Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates.. Mol. Cell 29(4), 441–50. Martin, A., Baker, T. A., & Sauer, R. T. (2008b). Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding.. Nat. Struct. Mol. Biol. 15(11), 1147–51. Martin, A., Baker, T. A., & Sauer, R. T. (2008c). Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes.. Nat. Struct. Mol. Biol. 15(2), 139–45. Maxwell, A., Craigie, R., & Mizuuchi, K. (1987). B protein of bacteriophage mu is an ATPase that preferentially stimulates intermolecular DNA strand transfer.. Proc. Natl. Acad. Sci. U. S. A. 84(3), 699–703. Mayer, M. P. (2013). Hsp70 chaperone dynamics and molecular mechanism.. Trends Biochem. Sci. 38(10), 507–14. 82 Mayhew, M., da Silva, A. C., Martin, J., Erdjument-Bromage, H., Tempst, P., & Hartl, F. U. (1996). Protein folding in the central cavity of the GroEL-GroES chaperonin complex.. Nature 379(6564), 420–6. McCarthy, D., Kramer, G., & Hardesty, B. (1998). Reactivation of thermally inactivated pre-beta-lactamase by DnaK, DnaJ, and GrpE.. Protein Sci. 7(5), 1164–71. McGinness, K. E., Baker, T. A., & Sauer, R. T. (2006). Engineering controllable protein degradation.. Mol. Cell 22(5), 701–7. McGinness, K. E., Bolon, D. N., Kaganovich, M., Baker, T. a., & Sauer, R. T. (2007). Altered tethering of the SspB adaptor to the ClpXP protease causes changes in substrate delivery.. J. Biol. Chem. 282(15), 11465–73. McNally, F. J. & Vale, R. D. (1993). Identification of katanin, an ATPase that severs and disassembles stable microtubules.. Cell 75(3), 419–29. Mhammedi-Alaoui, A., Pato, M., Gama, M.-J., & Toussaint, A. (1994). A new component of bacteriophage Mu replicative transposition machinery: the Escherichia coli CipX protein. Mol. Microbiol. 11, 1109–1116. Miot, M., Reidy, M., Doyle, S. M., Hoskins, J. R., Johnston, D. M., Genest, O., Vitery, M.-C., Masison, D. C., & Wickner, S. (2011). Species-specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation.. Proc. Natl. Acad. Sci. U. S. A. 108(17), 6915–20. Mizusawa, S. (1983). Protein degradation in Escherichia coli : The Lon gene controls the stability of sulA protein. Proc. Natl. Acad. Sci. U. S. A. 80(January), 358–362. Mizuuchi, K. & Adzuma, K. (1991). Inversion of the phosphate chirality at the target site of Mu DNA strand transfer: evidence for a one-step transesterification mechanism.. Cell 66(1), 129–40. Mizuuchi, M., Baker, T. a., & Mizuuchi, K. (1992). Assembly of the active form of the transposase-Mu DNA complex: a critical control point in Mu transposition.. Cell 70(2), 303–11. Molière, N. & Turgay, K. (2009). Chaperone-protease systems in regulation and protein quality control in Bacillus subtilis.. Res. Microbiol. 160(9), 637–44. Montaño, S. P., Pigli, Y. Z., & Rice, P. a. (2012). The 𝜇 transpososome structure sheds light on DDE recombinase evolution.. Nature 491(7424), 413–7. Muffler, a., Fischer, D., Altuvia, S., Storz, G., & Hengge-Aronis, R. (1996). The response regulator RssB controls stability of the sigma(S) subunit of RNA polymerase in Escherichia coli.. EMBO J. 15(6), 1333–9. Nakai, H. & Kruklitis, R. (1995). Disassembly of the bacteriophage Mu transposase for the initiation of Mu DNA replication. J. Biol. Chem. 270(33), 19591–19598. 83 Neher, S. B., Flynn, J. M., Sauer, R. T., & Baker, T. a. (2003a). Latent ClpX-recognition signals ensure LexA destruction after DNA damage.. Genes Dev. 17(9), 1084–9. Neher, S. B., Sauer, R. T., & Baker, T. a. (2003b). Distinct peptide signals in the UmuD and UmuD’ subunits of UmuD/D’ mediate tethering and substrate processing by the ClpXP protease.. Proc. Natl. Acad. Sci. U. S. A. 100(23), 13219–24. Ogura, T., Tomoyasu, T., Yuki, T., Morimura, S., Begg, K. J., Donachie, W. D., Mori, H., Niki, H., & Hiraga, S. (1991). Structure and function of the ftsH gene in Escherichia coli.. Res. Microbiol. 142(2-3), 279–82. Park, E. Y., Lee, B.-G., Hong, S.-B., Kim, H.-W., Jeon, H., & Song, H. K. (2007). Structural basis of SspB-tail recognition by the zinc binding domain of ClpX.. J. Mol. Biol. 367(2), 514–26. Parsell, D. A. & Lindquist, S. (1993). The Function of Heat-shock Proteins in Stress Tolerance: Degradation and Reactivation of Damaged Proteins. Annu. Rev. Genet. 27, 437–496. Pietrosiuk, A., Lenherr, E. D., Falk, S., Bönemann, G., Kopp, J., Zentgraf, H., Sinning, I., & Mogk, A. (2011). Molecular basis for the unique role of the AAA+ chaperone ClpV in type VI protein secretion.. J. Biol. Chem. 286(34), 30010–21. Prakash, S., Tian, L., Ratliff, K. S., Lehotzky, R. E., & Matouschek, A. (2004). An unstructured initiation site is required for efficient proteasome-mediated degradation.. Nat. Struct. Mol. Biol. 11(9), 830–7. Pratt, L. A. & Silhavy, T. J. (1996). regulator SprE controls the stability of RpoS. Proc. Natl. Acad. Sci. U. S. A. 93(March), 2488–2492. Pyle, A. M. & Green, J. B. (1994). Building a kinetic framework for group II intron ribozyme activity: quantitation of interdomain binding and reaction rate.. Biochemistry 33(9), 2716–25. Reyes, O., Beyou, A., Mignotte-Vieux, C., & Richaud, F. (1987). Mini-Mu transduction: cis-inhibition of the insertion of Mud transposons.. Plasmid 18(3), 183–92. Rice, P. A. & Baker, T. A. (2001). Comparative architecture of transposase and integrase complexes.. Nat. Struct. Biol. 8(5), 302–7. Rice, P. A. & Mizuuchi, K. (1995). Structure of the bacteriophage Mu transposase core: a common structural motif for DNA transposition and retroviral integration.. Cell 82(2), 209–20. Roll-Mecak, A. & McNally, F. J. (2010). Microtubule-severing enzymes.. Curr. Opin. Cell Biol. 22(1), 96–103. Roll-Mecak, A. & Vale, R. D. (2008). Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin.. Nature 451(7176), 363–7. 84 Román-Hernández, G., Hou, J. Y., Grant, R. A., Sauer, R. T., & Baker, T. A. (2011). The ClpS adaptor mediates staged delivery of N-end rule substrates to the AAA+ ClpAP protease.. Mol. Cell 43(2), 217–28. Sauer, R. T. & Baker, T. a. (2011). AAA+ proteases: ATP-fueled machines of protein destruction.. Annu. Rev. Biochem. 80, 587–612. Sauer, R. T., Bolon, D. N., Burton, B. M., Burton, R. E., Flynn, J. M., Grant, R. A., Hersch, G. L., Joshi, S. A., Kenniston, J. A., Levchenko, I., Neher, S. B., Oakes, E. S., Siddiqui, S. M., Wah, D. A., & Baker, T. A. (2004). Sculpting the Proteome with AAA+ Proteases and Disassembly Machines. Cell 119(1), 9–18. Savilahti, H., Rice, P. A., & Mizuuchi, K. (1995). The phage Mu transpososome core: DNA requirements for assembly and function.. EMBO J. 14(19), 4893–903. Schumacher, S., Clubb, R. T., Cai, M., Mizuuchi, K., Clore, G. M., & Gronenborn, a. M. (1997). Solution structure of the Mu end DNA-binding ibeta subdomain of phage Mu transposase: modular DNA recognition by two tethered domains.. EMBO J. 16(24), 7532–41. Seong, I. S., Oh, J. Y., Yoo, S. J., Seol, J. H., & Chung, C. H. (1999). ATP-dependent degradation of SulA, a cell division inhibitor, by the HslVU protease in Escherichia coli.. FEBS Lett. 456(1), 211–4. Shapiro, J. A. (1979). Molecular Model for the transposition and replication of bacteriophage Mu and other transposable elements. Proc. Natl. Acad. Sci. U. S. A. 76, 1933–1937. Siddiqui, S. M., Sauer, R. T., & Baker, T. A. (2004). Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates.. Genes Dev. 18(4), 369–74. Song, H. K. & Eck, M. J. (2003). Structural basis of degradation signal recognition by SspB, a specificity-enhancing factor for the ClpXP proteolytic machine.. Mol. Cell 12(1), 75–86. Sousa, M. C., Trame, C. B., Tsuruta, H., Wilbanks, S. M., Reddy, V. S., & McKay, D. B. (2000). Crystal and Solution Structures of an HslUV Protease–Chaperone Complex. Cell 103(4), 633–643. Squires, C. L., Pedersen, S., Ross, B. M., & Squires, C. (1991). ClpB is the Escherichia coli heat shock protein F84.1.. J. Bacteriol. 173(14), 4254–62. Stinson, B. M., Nager, A. R., Glynn, S. E., Schmitz, K. R., Baker, T. A., & Sauer, R. T. (2013). Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine.. Cell 153(3), 628–39. 85 Sundar, S., Baker, T. A., & Sauer, R. T. (2012). The I domain of the AAA+ HslUV protease coordinates substrate binding, ATP hydrolysis, and protein degradation.. Protein Sci. 21(2), 188–98. Surette, M. G., Buch, S. J., & Chaconas, G. (1987). Transpososomes: stable proteinDNA complexes involved in the in vitro transposition of bacteriophage Mu DNA. Cell 49, 253–262. Surette, M. G. & Chaconas, G. (1992). The Mu transpositional enhancer can function in trans: requirement of the enhancer for synapsis but not strand cleavage.. Cell 68(6), 1101–8. Surette, M. G., Harkness, T., & Chaconas, G. (1991). Stimulation of the Mu A Proteinmediated Strand Cleavage Reaction by the Mu B Protein, and the Requirement of DNA Nicking for Stable Type 1 Transpososome Formation. J. Biol. Chem. 266(15), 3118–3124. Takeuchi, J., Chen, H., & Coffino, P. (2007). Proteasome substrate degradation requires association plus extended peptide.. EMBO J. 26(1), 123–31. Teter, S. A., Houry, W. A., Ang, D., Tradler, T., Rockabrand, D., Fischer, G., Blum, P., Georgopoulos, C., & Hartl, F. U. (1999). Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains.. Cell 97(6), 755–65. Thibault, G., Yudin, J., Wong, P., Tsitrin, V., Sprangers, R., Zhao, R., & Houry, W. a. (2006). Specificity in substrate and cofactor recognition by the N-terminal domain of the chaperone ClpX.. Proc. Natl. Acad. Sci. U. S. A. 103(47), 17724–9. Tomoyasu, T., Kunitoshi, Y., Murata, K., Suzaki, T., Bouloc, P., Kato, A., Niki, H., Hiraga, S., & Ogura, T. (1993). Topology and Subcellular Localization of FtsH Protein in Escherichia coli. J. Bacteriol. 175(5), 1352–1357. Varshavsky, A. (1996). The N-end rule: functions, mysteries, uses.. Proc. Natl. Acad. Sci. U. S. A. 93(22), 12142–9. Viitanen, P. V., Gatenby, A. A., & Lorimer, G. H. (1992). Purified chaperonin 60 (groEL) interacts with the nonnative states of a multitude of Escherichia coli proteins.. Protein Sci. 1(3), 363–9. Wah, D. A., Levchenko, I., Rieckhof, G. E., Bolon, D. N., Baker, T. a., & Sauer, R. T. (2003). Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease.. Mol. Cell 12(2), 355–63. Wang, F., Mei, Z., Qi, Y., Yan, C., Hu, Q., Wang, J., & Shi, Y. (2011). Structure and mechanism of the hexameric MecA-ClpC molecular machine.. Nature 471(7338), 331–5. 86 Weibezahn, J., Tessarz, P., Schlieker, C., Zahn, R., Maglica, Z., Lee, S., Zentgraf, H., Weber-ban, E. U., Dougan, D. A., Tsai, F. T. F., Mogk, A., & Bukau, B. (2004). Thermotolerance Requires Refolding of Aggregated Proteins by Substrate Translocation through the Central Pore of ClpB. Cell 119(5), 653–665. Wendler, P., Ciniawsky, S., Kock, M., & Kube, S. (2012). Structure and function of the AAA+ nucleotide binding pocket.. Biochim. Biophys. Acta 1823(1), 2–14. White, S. R. & Lauring, B. (2007). AAA+ ATPases: achieving diversity of function with conserved machinery.. Traffic 8(12), 1657–67. Whiteheart, S. W., Schraw, T., & Matveeva, E. A. (2001). N-ethylmaleimide sensitive factor (NSF) structure and function.. Int. Rev. Cytol. 207, 71–112. Wickner, S., Gottesman, S., Skowyra, D., Hoskins, J., McKenney, K., & Maurizi, M. R. (1994). A molecular chaperone, ClpA, functions like DnaK and DnaJ.. Proc. Natl. Acad. Sci. U. S. A. 91(25), 12218–22. Williams, T. L., Jackson, E. L., Carritte, A., & Baker, T. A. (1999). Organization and dynamics of the Mu transpososome : recombination by communication between two active sites. Genes Dev. 13(20), 2725–2737. Wojtyra, U. a., Thibault, G., Tuite, A., & Houry, W. a. (2003). The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function.. J. Biol. Chem. 278(49), 48981–90. Wu, Z. & Chaconas, G. (1994). Characterization of a region in phage Mu transposase that is involved in interaction with the Mu B protein.. J. Biol. Chem. 269(46), 28829–33. Xu, Z., Horwich, A. L., & Sigler, P. B. (1997). The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex.. Nature 388(6644), 741–50. Yamauchi, M. & Baker, T. A. (1998). An ATP-ADP switch in MuB controls progression of the Mu transposition pathway.. EMBO J. 17(18), 5509–18. Yuan, J. F., Beniac, D. R., Chaconas, G., & Ottensmeyer, F. P. (2005). 3D reconstruction of the Mu transposase and the Type 1 transpososome: a structural framework for Mu DNA transposition.. Genes Dev. 19(7), 840–52. Zeth, K., Ravelli, R. B., Paal, K., Cusack, S., Bukau, B., & Dougan, D. A. (2002). Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA.. Nat. Struct. Biol. 9(12), 906–11. 87