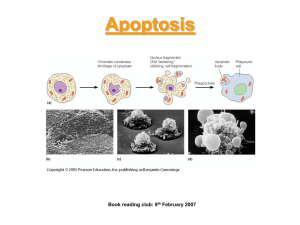
K'; Genetic Control of Death in Chinese Hamster Ovary Cultures
advertisement

K'; Genetic Control of Death in Chinese Hamster Ovary Cultures by Joydeep Goswami M.B.A., MIT-Sloan School of Management, Cambridge, MA, 1998 M.S.C.E.P., Massachusetts Institute of Technology, Cambridge, MA, 1994 B.Tech. Chemical Engineering, Indian Institute of Technology, Bombay, INDIA, 1993 Submitted to the Department of Chemical Engineering in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY September, 1998 ©1998 Massachusetts Institute of Technology. All rights reserved. Signature of Author: __ __ c~t7 Department of Chemical Engineering September 1, 1998 v Certified by: O-Daniel I. C. Wang Institute Professor Thesis Supervisor Certified by: SAnthony J. Sinskey Professor of Biology Thesis Supervisor Accepted by: n c-s-s'-. INSr.TU E OF TECHNOLOGy TEs LIBRARIES Robert E. Cohen St. Laurent Professor of Chemical Engineering Chairman, Committee for Graduate Students 1 Genetic Control of Death in Chinese Hamster Ovary Cultures by Joydeep Goswami Submitted to the Department of Chemical Engineering on September 4th, 1998 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemical Engineering One of the main problems in mammalian cell culture systems, including Chinese Hamster Ovary (CHO) cultures, is the inability to maintain viability of these cultures over extended periods of time. This inability translates into lower final protein titers and higher protein production and recovery costs. This thesis was undertaken to better understand the processes of death in CHO cells and to find ways to extend the viability of CHO cultures. A majority of CHO cells in culture were found to die by apoptosis, a genetically controlled form of cellular suicide. Protein synthesis inhibition in CHO cells led to rapid death, indicating that CHO cells were pre-disposed to death and that survival proteins needed to be continuously synthesized to protect cells from death. Caspases, a class of proteins found to be universally important in inducing apoptosis, were found to be activated in apoptotic CHO cells. Surprisingly, inhibition of caspase activity using z-VAD.fmk, a universal peptide inhibitor of caspases, failed to significantly extend viability in batch culture although it did prevent cleavage of known intracellular caspase substrates. In contrast, expression of bcl2, a well-characterized anti-apoptotic gene, was able to significantly increase the life of CHO batch cultures in response to both glucose limitation and growth factor withdrawal. Using these results, a pathway for apoptosis in CHO cells, focusing on the caspases and bcl-2, was suggested. An experiment was devised to statistically measure the ability of individual cells to replicate. Replication competence was found to correlate well with viability results from the acridine orange / ethidium bromide assay, but not with results from the trypan blue assay. These experiments proved that early apoptotic cells, which have lost membrane integrity but not chromatin integrity, can be considered dead since they lose the ability to replicate. In addition, the experiments proved that bcl-2 expression is able to extend the replication competence of cells under normal culture conditions. Bcl-2 expression was shown to improve both final product titers and integrated viable cell densities in CHO fed-batch cultures. It was also able to maintain insulindeprived fed-batch cultures in a viable and productive state for much longer than insulinsupplemented cultures, thus suggesting an easy way to maintain viability and productivity coupled with slower growth. A slower growth rate has been reported in literature to yield higher product quality and productivity. Concentrations of insulin, a growth and survival factor used in CHO culture, were observed to drop rapidly in fed-batch cultures of CHO cells. The loss of insulin was concurrent with the accumulation of cells in the GO/G1 state and an increase in expression levels ofp53, a well-documented growth-inhibiting and apoptosis-inducing gene. Insulin degrading activity was found to be at least partially caused by release of proteases from cells into the culture medium. Insulin degradation was sharply reduced by adding sodium glycocholate, an amino-peptidase inhibitor, suggesting that amino-peptidases play a major role in insulin degradation in CHO fed-batch cultures. Based on the above results, possible ways to further reduce death and improve productivity in CHO cultures are also suggested. Thesis Supervisor: Dr. Daniel I.C. Wang Title: Institute Professor Thesis Supervisor: Dr. Anthony J. Sinskey Title: Professor of Biology Acknowledgments Although the title of my thesis would suggest that I spent most my time at MIT studying death, my time here was really not that morbid. I feel that my experiences over the last five years, both inside and outside the lab, have made me a better all-round individual. I would thus like to take this opportunity to thank the people who my made my stay at MIT so thoroughly enjoyable. First, I would like to thank my advisors Prof. Tony Sinskey and Prof. Daniel Wang. Thank you for pushing me to look harder for answers and teaching me to focus on the most relevant issues quickly. These are skills I will value a lot in my career. I also appreciate that both of you were so very understanding and encouraging when the going was not easy, especially initially when the transfections refused to work. I also appreciate that you had the confidence to give me the freedom to explore issues that interested me and the patience to listen when I explained these issues to you. I understand that I was not the traditional graduate student, and I want to thank you for your support and recommendations when I applied to the MBA program at Sloan. Without your encouragement and understanding, working simultaneously on both the MBA and Ph.D. would never have been possible. I believe that the biggest support that an advisor can give his students is by expressing confidence in them and their work, and both of you did a splendid job of letting this confidence show. Dr. Wang I enjoyed our tennis games a lot, and I am sorry that we did not get a chance to play more often. Of course, I must confess that your threats of not letting me graduate till you were able to beat me did not influence my game. Tony, no matter how stressed out I was, your good humor always cheered me up. I will always remember the day when I walked into your office and said that I wanted to apply to Sloan, and you said, "Of course, I would be disappointed if you did not". Your confidence in me has meant and will continue to mean a lot to me. Thank you also for the great picnics that you and Prof. Rha had for us. It was nice to have well balanced individuals as advisors. I would also like to thank the other members of my thesis committee - Dr. Mike Glacken, Prof. Doug Lauffenburger, Dr. Morris Rosenberg, Prof. Hermann Steller, Prof. Greg Stephanopoulos. It was great that you spent so much time with me discussing various parts of my research and life in general. Hermann, despite your protests, I still think you are a better tennis player than I am. Thanks for all the help with the apoptosis assays and the p35 experiments. Greg thanks for having those long meetings with me to discuss my research and for helping me put everything in perspective. Doug thanks for all the help with the insulin experiments and for making it to every committee meeting. Mike and Morris, I am grateful to you two for helping me put my work into perspective from an industrial standpoint. I would also like to thank two other members of faculty who were not formally a part of my thesis committee - Prof. Harvey Lodish and Prof. Charlie Cooney. Harvey you amazed me with your insight and knowledge, and the extent to which you were involved in the research in your lab. I went to you several times with problems which were vexing me, and I never came away disappointed. Charlie, I don't think I have ever met anyone goes more out to his/her way to help others. Thank you not only for your insightful comments regarding my research, but also for all your help with getting into and out of Sloan. Your courage, determination and positive attitude toward life have taught me a lot. Congratulations again, on scaling Mt. McKinley. Of course, my studies at MIT would not have been possible without generous financial support. I would like to thank Unilever Ltd. for awarding me the Unilever/ Lever Brothers Practice School Fellowship during my first semester here. In addition, I would like to thank the National Science Foundation which provided financial support for most of my graduate study at MIT. I would like to thank the MIT-Merck Fellowship Foundation which supported me during the last year. Lastly, I would also like to thank Merck and Co. for their fellowship in 1995 which supported my trip to San Diego to attend the Cell Culture Engineering Conference V. I would also like to express my appreciation to the BPEC staff over the years Audrey Childs, Sonia Foster, Lynne Lenker, Joya Gargano, James Leung, John Galvin, Darlene Ray, Lorraine Cable and Sara Puffer. Thank you for promptly attending to all my requests and helping me to work out the various administrative details associated with the life of a graduate student. I would also like to thank the administrative crew in Chemical Engineering graduate student office - Janet Fischer and Elaine Aufiero. Life in ChemE would have been unimaginable without your support - starting with helping me out with lost forms even before I came to MIT, to sorting out late registrations and the tuition-detail headaches which come with being registered in two departments at the same time. Business School types professing quality of service should take a page out of your book. I have had the opportunity to supervise some wonderful undergraduate and high school students over the years - Eve Li, Anya Freedman, Gus Blomquist, Brian Roland, Brock Bienkowski, Sujata Bhatia, Katiuscia Porter and Kevin Sullivan. Working with you was a lot of fun, and I think I learnt a lot more from you than I taught you. Most of my time of course was spent with my fellow grad students at BPEC and Biology, and from you I have learnt the most during my stay at MIT. Along with all the lab techniques and academic knowledge that I gained through our interactions, I will also always cherish our discussions on various life-saving topics (read gossip). Of course, it was fun to feign the part of evil, rampant, heartless capitalist against Populist Steve! Thanks to Robert Balcarcel, David Chang, Jianxin Chen, John Chung, Grace Colon, Dave Schaffer, Brian Follstad, Peter Frier, Sherry Gu, Stephane Guillouet, Bryan Harmon, Bettina Knorr, Araba Lamous-Smith, Dan Lasko, Phil Lessard, Kai-Chee Loh, Margaret McCormick, Steve Meier, Gautam Nayar, Gregg Nyberg, Chandra Papudesu, Cliff Rutt, Anna Sanfeliu, Eric Scharin, Marc Shelikoff, Troy Simpson, Rahul Singhvi, Christi Snell, Margaret Speed, Espe Troyano, Chia-lin Wei, Bruce Woodson, Inn Yuk, Liangzhi Xie and Jifeng Zhang. Thanks also to Mike Hengartner who helped me start off my career in cell death. I would also like to thank Glenn Paradis and Mike Connolly for helping me with flow cytometry experiments. I would also like to thank some of the many friends I made during my tenure at MIT, who kept me sane and prevented me from working too hard. Thanks to Ravi Kane, Arpan Mahorowala, Robert Balcarcel, Suman Banerjee, Aleks Engel, Tim Benish, Fred Colhoun, Kiko Aumond, Chris Dowd, Radha Nayak, John Konz, Ravi Srinivasan, Steffen Ernst, Jeff White and Yoky Matsuoka. It was fun hanging out with you guys. Kiko and Aleks I will always remember your parties as one of the high points of my graduate life. Vivek Mohindra and Gokaraju Raju, thank you for all the advice and guidance that you gave me throughout the years. Thanks also to my friends and teammates from Sloan who helped me juggle both research and classes - Geoff Bloss, Michelle Go, Michelle WilsonClarke, Claudio Ribiero, Eiji Harada, William Dutcher, Paul Evers, Marc Osofsky, Preetish Nijhawan, Joel Serface, Stephanie Tan, Debjani Deb, Chuck Goeringher, Michelle LeBlanc, Frank Martelli, Paul White, Julie Suh and Tina Baumgartner. I also want to thank my tennis partners for getting onto court whenever I requested them to. Thank you Yoky, Joel, Colin Walden, Manish Bhatia and Jonathan Seelig. Thanks also to my squash buddies, Ravi Kane, Ravi Srinivasan, Arup Chakraborty, Preetish, Phillip Hirschon and Ashu Atwal. Lastly, I would like to thank Jennifer Weiloch for making the incredibly difficult last year so incredibly wonderful. Thanks for all your help and support. Finally, I would like to thank my parents. Ever since I was young you have supported me and encouraged me more than any parents I know. I would have never made it here if were not for the time and energy you spent in helping me distinguish right from wrong, and teaching me to have the courage to do what was right. I know it was incredibly tough for you to let your only child leave to go thousands of miles away, but I thank you for making that decision with a smile. Thank you for all the love, caring and dedication throughout the years. I will always be indebted to you and I dedicate this Ph.D. thesis to both of you. I TABLE OF CONTENTS ABSTRACT....................................................................................................................... ........................................................ ACKNOWLEDGMENTS ........................................... ....................................................... TABLE OF CONTENTS........................................... 3 5 9 LIST OF FIGURES................................................................................................................ LIST OF TABLES................................................................................................................. 13 1. INTRODUCTION .............................................................................................................. 21 .............................................. 1.1 Background............................................................... 1.2 M otivation ...................... ....................................................................................... ...................... 1.3 Thesis Objectives ............................................................................. ..................... 1.4 Thesis Organization .......................................................................... 21 23 24 25 ..................... 27 2. LITERATURE REVIEW .......................................................................... 2.1 Cell Death -Necrosis and Apoptosis ..................................... ...... . 20 ...... 27 27 2.......7.................2.... 2.1.1 Necrosis................................................................................... 30 .................................. 2.1.2 Apoptosis........................................................................ 2.2 Genes Regulating Apoptosis........................................................................................34 2.2.1 p53 - a signaling molecule that is involved in both cell cycle control and apoptosis37 .......... .............................. 40 2.2.2 The Bcl-2 family of proteins...................................... 2.2.3 Cysteine Proteases or Caspases.................................................49 2.3 Mitochondria - the meeting ground for the key players in apoptosis ...................... 59 2.4 Are caspases essential for apoptosis?............................................ ............................ .. 60 62 ................... 2.5 Apoptosis in M amm alian Cell Culture ......................................... 3. MATERIALS AND M ETHODS ............................................................................ 65 3.1 Cell Culture .................................................................................................................. 65 3.1.1 Cell Line............................................................................................................. 65 3.1.2 Culture M edium......................................................................................................65 3.1.3 Culture M aintenance.............................................................................................. 66 3.1.4 Caspase-InhibitingPeptide Experiments......................................... 67 67 3.1.5 Fed Batch Culture............................................................................................... 68 ...... ....... ........................... 3.1.6 Insulin binding studies.................................... 3.2 Analytical M ethods ..................................................................................................... 70 .......................... 70 3.2.1 Cell Number and Viability...................................................... 3.2.2 Dry Cell Weight...................................................................................................... 73 73 3.2.3 Sugar and Lactate Assays....................................................... 73 .......................... ...... Concentration 3.2.4 Determination of IFN-y 3.2.5 Determination of insulin concentration........................................ 74 3.2.6 Western Blot........................................................................................................... 74 77 .................................. 3.2.7 Total Protein Assay .................................. 3.2.8 M easurement of Caspase Activity......................................................................... 77 3.2.9 DNA Ladder Technique for Detection of Apoptosis ................................................. 79 ..................... 80 ................................... 3.2.10 Cell cycle assay...................................... 3.3 Transfection and Cloning of cells .................................................... 81 81 3.3.1 Preparationof plasmids .................................................................. 3.3.2 Transfection of Suspension CHO Cells ................................................. 81 3.3.3 M onitoring Transfection Efficiency.........................................................................83 3.3.4 Cloning .......................................................... .................................................. 85 4. APOPTOSIS AND ITS CONTROL IN CHINESE HAMSTER OVARY BATCH CULTURE..........87 4.1 CHO Cells Die by Apoptosis in Serum-free Batch Culture ..................................... 88 4.2 Protein synthesis inhibition causes rapid, dose-dependent apoptosis in CHO cells...93 4.3 Cysteine protease inhibiting peptides are unable to significantly enhance viability of CH O cells in culture ...................................................... ............................................... 95 4.4 Bcl-2 extends viability in batch cultures of CHO cells ..................................... 100 4.5 Bcl-2 protects better than caspase inhibiting peptides in response to growth and survival factor withdrawal............................... 104 4.6 Protective effect of bcl-2 is enhanced in clonal batch cultures ................................ 109 4.7 Caspase-independent death pathways which are blocked by bcl-2 expression appear to exist in CHO cells ........................................ 113 4.8 D iscussion and Conclusions.................................................................................... 118 5. CORRELATING VIABILITY AND REPLICATION COMPETENCE .................................... 125 5.1 Experimental Approach ........................................ 126 5.2 The ability of cells to replicate correlates much better with AO/EB assay results than with TB assay results ................................................... ............................................. 128 5.3 Early apoptotic cells lose the ability to replicate............................... ...... 132 5.4 Bcl-2 is able to prolong the replication competence of cells in culture...................132 5.5 Discussion and Conclusions................................................. ........................ 133 6. IMPROVING CHO FED-BATCH CULTURE PERFORMANCE USING BCL-2 EXPRESSION ................................................................................ .................................................... 135 6.1 Bcl-2 expression significantly extends viability and enhances product titers in normal CHO fed-batch cultures ........................................ 136 6.2 Bcl-2 expression allows insulin-deprived fed-batch cultures to survive longer than norm al fed-batch cultures .......................................... ................................................. 141 6.3 CHO cells progressively arrest in the GO/Gi phase during fed-batch culture........ 147 6.4 p53 expression increases with time in CHO fed-batch culture but the expression of cyclin E does not change......................................................... ................................... 151 6.5 Specific glucose consumption rate drops off sharply just before viable cell density stops increasing .......................................................................... ................................ 155 6.6 Discussion and Conclusions................................................. ........................ 156 7. THE FATE OF INSULIN IN CHO FED-BATCH CULTURES ............................................. 163 7.1 Reduced binding of insulin to its receptor cannot fully explain cessation of growth in CHO fed-batch cultures ........................................ 164 7.2 Insulin rapidly disappears with time in CHO fed-batch cultures ........................... 165 7.3 Insulin degrading activity in CHO fed-batch cultures is concentrated in the supernatant................. ....................................................................................... 168 7.4 Boiling of fed-batch supernatant removes all insulin degrading activity ................. 173 7.5 Aminopeptidase inhibitors are able to substantially reduce the degradation of insulin in the fed-batch supernatant.......................................................................................175 7.6 Adding large quantities of excess insulin also reduces the rate of degradation of insulin in the fed-batch supernatant.......................................................................................178 179 7.7 Discussion and Conclusions.............................. 8. CONCLUSIONS AND RECOMMENDATIONS ................................................................. 185 .......... 185 8.1 C onclusions ................................................................................................ 8.2 Recommendations ................................................................................................... 189 9. REFERENCES ....................................................................................................... 195 LIST OF FIGURES Figure 2-1: Events in necrosis of a typical mammalian cell as characterized by changes in cellular and organelle morphology. The solid arrows indicate the path of progression 29 of the cell in the necrotic process .................................... ................ Figure 2-2: Morphological features identified in the typical apoptotic death of mammalian cells. The thick solid arrows indicate the path of progression of the apoptotic process. All stages of the process need not be observed ..................................... 33 Figure 2-3: A map of bcl-2's (and bcl-xL's) interactions with other proteins which help explain the mechanisms by which it exerts it antiapoptotic effects. See text for details of specific protective effects. Adapted from Reed, 1997 ........................... 45 Figure 2-4: Examples of receptor, adapter and protease complexes in regulation of caspase activity. In this illustration, Fas ligand and TNF- (tumor necrosis factor) receptor are the receptor molecules. FADD (Fas-associated-death-domain protein), TRADD (TNF-receptor-associated-death-domain protein), RIP (receptor-interacting-protein), RAIDD (RIP-associated ICH-1/CED-3 -homologous protein) are the adapter molecules. Pro-caspase-2 and pro-caspase-8 are the end proteases (or caspases) which are activated through this signaling pathway. Adapted from Cohen, 1997....54 Figure 2-5: The mitochondrial caspase cascade. Release of cytochrome c from the mitochondria leads to the activation of caspase-9 in the presence of Apaf-1 and dATP. Caspase-9 subsequently activates other caspase-3 and potentially other downstream caspases. Adapted from Li, et al., 1997 ............................................... 58 Figure 2-6: A map of apoptosis focusing on caspases and the mitochondrial bcl-2 The diagrams representing receptor-adapter-protease activation of proteins. caspases, bcl-2 family regulation and activation of downstream caspases are shown in greater detail in Figure 2-4, Figure 2-3 and Figure 2-5, respectively. The release of AIF (apoptosis inducing factor) is in a positive feedback loop with activation of downstream caspases. Loss of mitochondrial potential and release of reactive oxygen species (ROS) by themselves can lead to death, but there is an active debate in literature as to whether this death is apoptotic or necrotic (see text)................. 61 Figure 4-1: Viable (VCD) and total cell densities (TCD) obtained in normal serum-free ....... 89 batch culture of suspension CHO cells ......................................... Figure 4-2: Apoptosis in batch cultures of CHO cells. Percentage of dead, apoptotic necrotic cells obtained during a normal serum-free batch culture of CHO cells. percentage of dead cells is obtained by adding the percentages of apoptotic necrotic cells. Almost all death is seen to occur via apoptosis. ............................. and The and 90 Figure 4-3: Apoptosis in batch cultures of CHO cells. Agarose gel photograph of genomic DNA from CHO cells shows presence of a DNA ladder which coincides with massive apoptosis on day 4................................... ............... 92 Figure 4-4: Extent of apoptosis induced by varying doses of cycloheximide (CHX). Cycloheximide induces protein synthesis inhibition in cells. Apoptosis accounted for almost all the death observed............................. ....... .................... 94 Figure 4-5: Fluorescence obtained by combining cell-lysate from apoptotic CHO cells with a fluorescent substrate z-YVAD.AFC, indicating the presence of caspases.....96 Figure 4-6: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures CHO cells: Total cell density (TCD) and viable cell density (VCD) as a function tim e..................................................................................... .................................. of of of 98 Figure 4-7: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor of caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures of CHO cells: Total cell density and viability of cultures as a function of time. Viability declines rapidly after 72 hours ................................................ 99 Figure 4-8: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor of caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures of CHO cells: Medium glucose concentration as a function of time. Medium glucose concentration dropped to almost zero after 72 hours in both cultures .................... 101 Figure 4-9: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor of caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures of CHO cells: Poly-ADP-Ribose Polymerase (PARP) cleavage is evident in the control cultures but not the z-VAD.fmk protected cultures ...................................... 102 Figure 4-10: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Total cell density (TCD) and viable cell density (VCD) as a function of tim e .................................................... .............................................. 105 Figure 4-11: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Total cell density (TCD) and viability of cultures as a function of time. Viability of the control, but not the bcl-2 protected cell line, declines rapidly after 72 hours............................................... .......................................... 106 Figure 4-12: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Medium glucose concentration as a function of time. Medium glucose concentration dropped to almost zero after 72 hours in both cultures...... 107 Figure 4-13: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Poly-ADP-Ribose Polymerase (PARP) cleavage is clearly evident in the control cultures but almost all the PARP is uncleaved in the bcl-2 protected cultures..................................................................................................... 108 Figure 4-14: Comparison of the protective effects of bcl-2 expression and z-VAD.fmk, together and separately, in batch cultures of CHO cells that have been deprived of insulin and transferrin. To cultures which did not contain z-VAD.fmk we added a volume of Dimethyl Sulfoxide (DMSO) equal to the volume that was used to deliver z-VAD.fmk to the other cultures (also see text). This graph plots total cell density (TCD) as a function of time ..................................................................................... 10 Figure 4-15: Comparison of the protective effects of bcl-2 expression and z-VAD.fmk, together and separately, in batch cultures of CHO cells which have been deprived of insulin and transferrin. To cultures which did not contain z-VAD.fmk we added a volume of Dimethyl Sulfoxide (DMSO) equal to the volume that was used to deliver z-VAD.fmk to the other cultures (also see text). This graph plots viability of ............................ 111 cultures as a function of time ................................................... Figure 4-16: Comparison of the protective effects of bcl-2 expression and z-VAD.fmk, together and separately, in batch cultures of CHO cells which have been deprived of insulin and transferrin. To cultures which did not contain z-VAD.fmk we added a volume of Dimethyl Sulfoxide (DMSO) equal to the volume that was used to deliver z-VAD.fmk to the other cultures (also see text). Medium glucose concentration is 112 plotted as a function of time. ..................................... Figure 4-17: Bcl-2 CHO clonal populations are able to maintain their viability for a much longer period of time as compared to the control, pcd-CHO cells in a batch culture. The main cause of death is glucose depletion at about 72 hours. The total cell density (TCD) for both cultures is approximately the same. The variation in total cell density at the end of the culture is due to cells sticking to the walls of the flask. 114 Figure 4-18: Clonal populations of CHO cells expressing viability for much longer (as compared to the control to insulin-transferrin deprivation in batch culture. density of the cultures.............................................. bcl-2 are able to maintain their pcd-CHO cell line) in response TCD refers to the total cell ......................................... 115 Figure 4-19: Cleavage of caspase-6 substrate lamin A in CHO cells, in response to insulin and transferrin deprivation. 'p' refers to the control pcd-CHO cell line. 'b' refers to the bcl-2 expressing CHO cell line. '+z' and '-z' indicate whether caspase inhibitor, z-VAD.fmk was or was not added to the culture. The results indicate that addition of z-VAD.fmk is able to prevent cleavage of lamin A, but is not able to protect cells from death. Bcl-2 expression prevents both lamin cleavage and death. 117 Figure 4-20: Conventional pathway of death in the mammalian cells, focusing on the bcl-2 and caspase (C) nodes. S1 and S2 are the same or different external stimuli for apoptosis.................................................................................................................. 12 1 Figure 4-21: Suggested death (including both apoptosis and necrosis) pathway in a CHO cell. The pathway focuses on interaction between caspases and bcl-2 family proteins. FADD, AIF and ROS refer to Fas associated death domain, apoptosis inducing factor and reactive oxygen species, respectively. See text for more details. 123 Figure 5-1: A pictorial representation of the four kinds of cells which can be distinguished by the acridine orange/ ethidium bromide (AO/EB) assay. Cells which have not lost their membrane integrity are penetrated by acridine orange only, appear green under the microscope, and are lightly shaded in the figure. Cells which have lost their membrane integrity also incorporate ethidium bromide and appear orange under the microscope. These cells are hatched in the figure. The dark spots in the early and late apoptotic cells represent condensed and fragmented chromatin material..........129 Figure 5-2: Acridine orange (AO) viability is a good indicator of the ability of a cell to replicate. AO viability is defined as the percentage of viable cells in the population of cells with intact membrane integrity. Normalized replication competence is a normalized measurement of the number of wells in which the cells were able to replicate .................................................................................................................... 13 1 Figure 6-1: Total (TCD) and viable cell densities (VCD) of bcl-2 expressing and control (pcd) cell lines in a normal fed-batch culture seeded at 5x105 cells/mL................ 138 Figure 6-2: Total cell density (TCD) and viability of bcl-2 expressing and control (pcd) cell lines in a normal fed-batch culture seeded at 5x10 5 cells/mL .......................... 139 Figure 6-3: y-interferon production with bcl-2 expressing and control (pcd) cell lines in a normal fed-batch culture seeded at 5x10 5 cells/mL............................ 142 Figure 6-4: Total (TCD) and viable cell densities (VCD) of bcl-2-protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 5x10 5 cells/mL ................................................................................................................... 144 Figure 6-5: Total cell density (TCD) and viability of bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 5x10 5 cells/mL.145 Figure 6-6: y-interferon production with bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 5x10 5 cells/mL............. 146 Figure 6-7: Total (TCD) and viable cell densities (VCD) of bcl-2-protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 8x10 5 cells/mL . ................................................................................................................. 148 Figure 6-8: Total cell density (TCD) and viability of bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 8x1 05 cells/mL. 149 Figure 6-9: y-interferon production with bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 8x1 05 cells/mL ............ 150 Figure 6-10: Cell cycle trends in fed-batch cultures of clonal populations of pcd (a) and bcl-2 (b) transfected CHO cells. It can be seen that the fraction of cells in the GO/G1 phase increases monotonically with culture time, while the proportion in the S-phase drops off. VCD refers to viable cell density of the culture. The viable cell density of the cultures' stops increasing when more than 70% of cells are in the GO/G1 phase. 153 Figure 6-11: Increase in p53 expression in CHO fed-batch cultures coincides with cessation of an increase in viable cell density and follows the increase in GO/G1 phase population. Cells transfected with bcl-2 are able to maintain their viability for a longer period of time despite p53 expression. '*' indicates a sample from a batch culture in log-phase growth, which was used as a control for p53 expression. 'p' refers to pcd-CHO cells while 'b' refers to bcl2-CHO cells ................................. 154 Figure 6-12: Cyclin E expression remains constant with culture age. The increase in GO/G1 phase population is therefore not related to cyclin E expression. '*' indicates a sample from a batch culture in log-phase growth, which was used as a control for cyclin E expression. 'p' refers to pcd-CHO cells while 'b' refers to bcl2-CHO cells. 154 Figure 6-13: Specific glucose consumption (SGC) and viable cell density (VCD) with time in a normal fed-batch culture of CHO cells. Specific glucose consumption drops off before a fall in viable cell density. ................................................... 157 Figure 6-14: The glucose concentration (glc) of the fed batch media increase sharply after 100 hours due to unavoidable overfeeding and inefficient uptake of glucose. The specific uptake rate of glucose, when measured per mole of glucose in the medium (SGCG) continues to fall after about 90 hours in the culture. The rise in specific glucose uptake rate seen in Figure 6-13 may therefore be explained by the rising 158 concentrations of glucose in the culture (see text)............................... Figure 7-1: Insulin binding to cells taken from different points in a fed-batch culture. Net radioactivity is a proxy for the quantity of insulin bound in a specific manner to its receptor (see text). The higher the net radioactivity value the larger to quantity of insulin bound. 'Old' and 'new' refer to samples taken from day 7 and day 1 of a fed-batch culture, respectively ..................................... ............... 166 Figure 7-2: Insulin concentration as a function of time in a typical fed-batch culture of CHO cells. The initial concentration of insulin in the culture is 5 mg/L. Insulin concentrations drop rapidly to almost non-detectable levels by 100 hours. Culture growth stops soon after. The suffix '-ins' refers to the concentration of insulin in either the bcl2-CHO or pcd-CHO cell culture. VCD refers to the viable cell density in either culture........................................................................................... ........... 167 Figure 7-3: Degradation of insulin in fed-batch supernatant (culture medium with cells spun down and removed) from various days in a CHO fed-batch culture. The degradation of insulin in samples from each day, under cell-culture conditions, was followed for 24 hours. 5 mg/L of insulin was added to the supernatant at '0' hours, and the concentration of insulin measured in the supernatant at this point was denoted as 100%. Insulin in any particular sample was expressed as a fraction of the insulin concentration in the same sample at '0' hours ............................................. 170 Figure 7-4: Degradation of insulin in fed-batch medium (with cells) from various days in a CHO fed-batch culture. The degradation of insulin in samples from each day was followed for 24 hours. 5 mg/L of insulin was added at '0' hours, and the concentration of insulin measured in the supernatant at this point was denoted as 100%. Insulin in any particular sample was expressed as a fraction of the insulin concentration in the same sample at '0' hours ................................................. 171 Figure 7-5: Degradation kinetics of insulin in fresh (cell-free) fed-batch medium under standard cell-culture conditions. 5 mg/L of insulin was added to the medium at 'O' hours. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours ..................................... 172 Figure 7-6: Boiling fed-batch supernatant for three minutes removes its insulin degrading activity. 5 mg/L of insulin was added to the boiled sample and unboiled control at '0' hours. Both samples were incubated under standard cell-culture conditions. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours................................ 174 Figure 7-7: Effect of various protease inhibitors on the degradation of insulin in the supernatant from a fed-batch culture. 'All' indicates that all the protease inhibitors were added to this sample at the concentrations suggested. No protease inhibitors were added to the 'control' culture. 5 mg/L of insulin was added to the medium at '0' hours. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours. All samples were incubated under standard cell-culture conditions for the duration of the experiment............1...77 Figure 7-8: Adding excess insulin reduces the rate of insulin degradation, possibly due to the saturation of proteases or insulin-binding proteins in the fed-batch supernatant. The control indicated has IX or 5gSg/mL of insulin added to it at time zero. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours. All samples were incubated under standard cell-culture conditions for the duration of the experiment ...................... 180 LIST OF TABLES Table 2-I: Changing views in apoptosis .................................................. Table 2-II: Caspases and their target substrates. ....................................... 35 ..... 55 Table 3-I: Antibodies used and the companies from which they were sourced..............78 Table 5-I: Normalized replication competence (norm. repl. comp.), acridine orange viability (AO viab.) and trypan blue viability (TB viab.) data............................130 Table 6-I: A comparison of the results from a normal fed-batch using bcl-2 transfected and control (pcd) clonal cell lines. IVCD represents integrated viable cell density and is the area under the viable cell density curve (plotted against time) ....................... 142 Table 6-II: A comparison of the results from the two insulin-deprived fed-batch runs. All cultures contained clonal population of cells. Run 1 was started with an initial cell density of 5x10 5 cells/mL while Run 2 had an initial cell density of 8x10 5 cells/mL. IVCD represents integrated viable cell density and is the area under the viable cell density curve (plotted against time).........................................................................146 Table 7-I: Concentrations and sources of various protease inhibitors used to study insulin degradation in CHO fed-batch culture supernatants ............................................... 176 1. Introduction 1.1 Background Humans had been unknowingly using bacteria and yeast to produce foods and drinks such as bread, cheese, yogurt, wine, mead and beer long before Pasteur invented the science of microbiology in the mid-1800s. The first recorded use of microorganisms to fight disease was by Edward Jenner in 1796, when he used the material from cowpox lesions as the first effective vaccine against smallpox. Until the mid-1900s, the initial uses of microorganisms on an industrial scale remained confined to the food and beverage industry and the production of simple chemicals such as ethanol, acetone and citric acid. The discovery of the antibiotic penicillin in 1928 by Alexander Fleming gave a huge boost to the biological fermentation industry used for pharmaceutical purposes. The antibiotic industry remains a multi-billion dollar industry. The discovery of the double-helix in 1953 by Watson and Crick, and the subsequent developments in technologies in manipulating DNA led to the development of the biotechnology industry in the 1970s. Once the gene coding for a particular protein was identified, recombinant DNA technology allowed the production of this protein in almost any organism. This meant that large quantities of therapeutic and diagnostic proteins that were difficult to isolate from natural sources, could now be produced industrially in optimally designed host organisms. In 1982, human insulin became the first recombinant pharmaceutical product to be approved by the Food and Drug Administration. The choice of host organism for the production of recombinant product is dictated by several factors, including productivity of host and complexity of the desired protein. Bacterial production is often the most simple, allowing large quantities of product to be manufactured in a short time. However, bacteria are not able to form intramolecular disulfide bonds or glycosylate a protein, both of which are often key to the biological activity of more complex proteins. Nevertheless, bacteria remain important for the production of amino acids, vitamins and simple proteins like insulin. Production in yeast offers the advantages of rapid production and high productivities obtained in bacterial culture. Yeast secrete soluble proteins and are able to form intramolecular disulfide bonds, but are unable to correctly glycosylate most complex proteins. Yeast is still the organism of choice for certain biologics. Animal cell culture, using transformed cells from multicellular animals, is still the only way to manufacture most complex proteins. However, animal cells are slow growing, fragile and offer much lower productivities than bacterial or yeast systems. In addition to production of complex and therapeutic proteins such as erythropoietin, tissue plasminogen activator, monoclonal antibodies and interferons, animal cell culture is also used to produce viruses for vaccines and bioinsectisides. The recent use of transgenic animals to manufacture proteins in their milk has the potential to revolutionize high throughput manufacture of some complex proteins. However, for proteins that may be toxic to the animal in large quantities and for the rapid manufacture of proteins prior to FDA approval (there is not enough time to develop transgenic animal factories), animal cell culture will continue to play an important role. 1.2 Motivation One of the major problems in animal cell culture is the high cost of recovering secreted product from the medium, primarily due to the low concentrations of product in the medium. The main cause of low product concentration is that viability of cells in batch or fed-batch cultures cannot be sustained for sufficiently long periods of time due to their sensitivity to the culture environment and the accumulation of toxic metabolic products. Previous studies have indicated the potential of designing medium and feeding strategies to reduce the production of toxic metabolites such as lactate and ammonia and increase the viability and productivity of cultures. Another way to increase the concentration of the product in the medium would be to improve viability of the cells by increasing the tolerance of the cells to toxic metabolic end products. Recent studies have indicated that a substantial number of the cells that die in vitro during culture, do so by apoptosis, a genetically controlled form of cellular suicide. However, at the time of initiation of this study there were no reported studies of apoptosis in Chinese Hamster Ovary (CHO) cells. Since apoptosis is a genetically controlled process, a cell's commitment to apoptosis, can be altered by recombinant expression of genes controlling the process. To be effective in controlling apoptotic death via genetic means, the genes controlling the apoptotic process in CHO cells first needed to be identified. A clearer understanding of how these genes are ordered in the molecular pathway is also necessary to determine which genes will be the most effective at preventing death. The effect of expression of death controlling genes on the productivity of y-interferon, the model protein expressed by CHO cells studied in our lab, also needed to be studied. Combining the use of feeding strategies and the use of genetic alteration of cells could provide means to further extend viability and improve product concentrations. The use of these genetic techniques could potentially lead to more robust and hardy cell lines and a more costeffective manufacturing process for life-saving drugs and diagnostic proteins. 1.3 Thesis Objectives The central goal of this thesis was to improve viability in CHO cultures, as a means to improving final product concentrations. To achieve this goal, the process of death in the CHO cell was studied at a molecular and genetic level. The specific objectives of these studies were: * To study the process of death in CHO cells and determine whether apoptosis is the main mode of death * If apoptosis is the main mode of death, determine which proteins or genes are the key players in the apoptotic pathway of CHO cells, how they interact, and the molecular ordering of this pathway with respect to these players * Determine whether the viability of CHO cultures can be improved by overexpressing or inhibiting some of the key players in the apoptotic pathway * Determine whether the viability and productivity of CHO fed-batch cultures can be extended beyond that obtained by using just medium design and feeding strategies, by combining these strategies with genetic modulation of the death process In addition to these objectives, the thesis also sought to develop techniques to examine viability at a more fundamental level and studied the interaction of the growth and survival factor insulin with key apoptotic pathway elements in the CHO cell during the culture process. 1.4 Thesis Organization This thesis is organized into eight chapters. The first chapter introduces the research topic and outlines the specific objectives of the thesis. Chapter 2 contains a detailed review of cell death and the key players in apoptosis. Chapter 3 provides a complete description of the techniques and protocols used in the thesis. Chapter 4 describes the results of experiments conducted to determine the mode of death in CHO cells. It also compares the results from attempts to control death in CHO cultures using expression of the antiapoptotic gene bcl-2 and addition of peptide inhibitors of specific apoptotic proteins (caspases). This chapter concludes with an analysis of the apoptotic pathway in CHO cells. The results presented in Chapter 5 confirm that the viabilities measured in Chapter 4 correspond to the ability of cells to replicate (a more stringent test of viability). Chapter 6 presents the results of experiments conducted to investigate the effect of bcl-2 expression on enhancing performance in CHO fed-batch cultures. The chapter also examines some of the underlying causes for cessation in growth of viable cells in CHO fed-batch cultures. Chapter 7 follows the fate of growth and survival factor insulin in fedbatch cultures and suggest methods to reduce its rapid disappearance in these cultures. Finally, Chapter 8 summarizes the results presented in this thesis and presents suggestions for future work in this area. 2. Literature Review 2.1 Cell Death -Necrosis and Apoptosis The study of cell death has not received due recognition until very recently. This may partly have been due to the rather restricted notion of cell death as a degenerative phenomenon caused by injury or age. However, recent studies have clearly shown that this notion is only partially true. eukaryotic organisms. Two distinct forms of cell death exist in most The first subscribes to the previously held 'degenerative phenomenon' idea and is called necrosis. The other process, known as apoptosis, involves the active self destruction of cells (consuming energy and sometimes involving protein synthesis) rather than just passive degeneration. The two processes of cell death can be distinguished by various morphological and biochemical criteria and also by identifying the circumstances of death. The following discussion elucidates some of the characteristic features of necrosis and apoptosis. 2.1.1 Necrosis The occurrence of necrosis is determined, not by factors intrinsic to the cell, but by violent perturbation in the environment (Wyllie, et al., 1980). Examples of such environmental perturbation include severe lack of oxygen (hypoxia) or blood (ischemia), sudden large fluctuations in environmental temperature (hypo- or hyperthermia), disruption of cell membranes by injury or exposure to large doses of toxins. Necrosis is also the mode of cell death by autolysis, in vitro. There is no evidence of necrosis in normal embryonic development and metamorphosis (Wyllie, et al., 1980). Necrosis is seen to affect cells in groups rather than individually. Early morphological changes brought on by necrosis include the marginal clumping of loosely textured chromatin, dilation of the endoplasmic reticulum, mild dispersion of ribosomes and gross swelling of the mitochondrial matrix (see Figure 2-1). The late stages of necrosis are characterized by the rupture of nuclear, organelle and plasma membranes (leading to a loss of organized structure), the dissolution of ribosomes and lysosomes and finally to swelling and rupture of the cell. This burst of cells and spewing out of cell contents causes the familiar exudative inflammation. The debris is eventually ingested and degraded by specialized phagocytic cells. The biochemical basis for this observed rupture of membranes arises from a marked increase in membrane permeability. This increase in permeability is accompanied by potassium loss, sodium entry and a fall in membrane potential. The rise in membrane permeability is also reflected in the failure to exclude vital dyes such as nigrosine, trypan blue, propidium iodide and eosin Y, thus forming a basis for viability tests. The rise in membrane permeability may be a result of toxins directly attacking membrane function. Alternatively, toxins may interfere with the energy supply on which the selective control ER nucleus _ mitochondrion-- * swollen mitochondrial matrix * increase in membrane permeabilityn * cell membrane ruptures * cell contents spewed out * inflammation lysosome o H2( Figure 2-1: Events in necrosis of a typical mammalian cell as characterized by changes in cellular and organelle morphology. The solid arrows indicate the path of progression of the cell in the necrotic process. of fluxes across the plasma membrane depends (Trump and Mergner, 1974). Hence, necrosis does not require the active use of cellular energy. 2.1.2 Apoptosis The word apoptosis is derived from ancient Greek for "falling off of leaves" and was first used to describe the process of physiological cell death by Kerr, et al., 1972. It occurs normally in the steady-state kinetics of healthy adult tissues producing a normal turnover of cells. Salvesen and Dixit, 1997 estimate that as many as 1011 cells die by apoptosis each day in a normal human being. It is interesting to note that this mechanism for 'cell suicide' has not developed in single cell organisms (although there have been some reports to the contrary, see Anderson, 1997), but multicellular organisms have found many uses for this mechanism. It accounts for the deletion of cells during embryonic development and metamorphosis, e.g., death of more than 50% of the neurons during the development of the vertebrate nervous system (Raff, et al., 1993) and the metamorphic degradation of the cells in the tadpole's tail. It may be brought about by a change in concentration of trophic hormones, e.g., removal of the endometrial epithelium by apoptosis after estrogen withdrawal in mice. In addition to morphogenetic and developmental functions, apoptosis is also a major defense mechanism. This is aptly demonstrated in plants where a cell detecting a single bacterium not only kills itself, but also alerts neighboring cells which then follow suit. Apoptosis can also be induced by cell-mediated immunity, e.g., cytotoxic T lymphocytes (CTLs) recognize virus-bearing cells and cause them to undergo apoptosis by transferring enzyme-bearing granules through pores in their surface formed with perforin (see Vaux, et al., 1994 for a review). Failure of cells with defective genomic DNA to die via apoptosis often causes cancer, but the fact that we can induce apoptosis in cells with radiation, cytotoxic cancer-chemotherapeutic agents and hyperthermia provides us with an important tool to fight cancer. In addition, excessive or uncontrolled apoptosis can also lead to pathogenesis. For instance, unregulated apoptosis appears to be the cause of several neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis (ALS) etc. (for a review see Thompson, 1995). As mentioned above, small concentrations of many necrotic agents have been observed to induce apoptosis (Martin and Cotter, 1994). In fact there appear to be crossover concentrations for most drugs under which cells die increasingly by apoptosis than necrosis. This suggests that it is the severity of the insult to cells that determines the type of death the cell will undergo, in addition to the type of insult itself. The morphological features of apoptosis are very distinct (see Wyllie, et al., 1980 and Martin and Cotter, 1994 for reviews). In vivo, the cell initially loses contact with its neighbors. The chromatin condenses into large compact granular masses that lie adjacent to the nuclear membrane. Chromosomal DNA is degraded at nucleosomal intervals (approx. 180 base pairs) leading to the formation of the characteristic 'DNA ladder'. The nuclear outline is abnormally convoluted and later grossly indented, and the nucleolus enlarges with its granules becoming coarse and abnormally scattered. There is usually a marked reduction in cell volume. Most of the cell organelles, however, retain their integrity except for the endoplasmic reticulum, which dilates (see Figure 2-2). There is also some recent evidence that other organelles such as the mitochondria also dilate during apoptosis (Vander Heiden, et al., 1997). In the later phases of apoptosis, cell membrane blebbing leads to cellular fragmentation which in turn leads to the formation of 'apoptotic bodies'. These apoptotic bodies contain nuclear fragments as well as cytoplasmic elements. Finally, neighboring cells and macrophages phagocytose the fragments. Unlike necrosis, apoptosis does not lead to an inflammatory response or the formation of residual scars (Fesus, et al., 1991). In addition, apoptosis is an astonishingly rapid process (Wyllie, et al., 1980) and cells undergoing apoptosis may disappear in vivo within 4 hours. Apoptotic cells do not display increased membrane permeability until well after the characteristic morphological features have appeared (Wyllie, et al., 1980). This may explain the initial insensitivity of apoptotic cells to vital dye exclusion-based viability tests. Furthermore, apoptosis requires the use of energy-rich nucleotides, e.g., death is accelerated in palatal shelf epithelium by artificially raising the level of cyclic adenine monophosphate (cAMP). This energy may be required in the synthesis of enzymes such as endonucleases which serve to cleave chromosomal DNA, or in the various apoptotic signaling cascades (Li, et al., 1997). There is evidence to suggest that the activity of these nucleases is associated with the concentration of divalent cations such as calcium (Cohen, 1991; Wyllie, et al., 1980). Energy is also used in protein synthesis, as it is known that many pathways of apoptosis are genetically coded for and require R nucleus mitochondrion lysosom Chromatin and cytoplasm condense Organelles shrink but retain identity Plasma membrane blebs Chromosomal DNA degraded Convoluted membrane Plasmalemmal sealing - compartments Apoptotic bodies fragmented chromatin Figure 2-2: Morphological features identified in the typical apoptotic death of mammalian cells. The thick solid arrows indicate the path of progression of the apoptotic process. All stages of the process need not be observed. transcription and translation of the genetic code. Some observations that protein inhibitors sometimes induce rather than inhibit apoptosis are explained by the hypothesis that proteins which induce apoptosis have already been synthesized and are countered by active synthesis of proteins which inhibit apoptosis. Protein synthesis inhibition in these cells thus induces apoptosis by preventing the synthesis of apoptosis inhibitor genes (Cuende, et al., 1993; Gottschalk, et al., 1994; Mercille and Massie, 1994; Mosser and Massie, 1994). The field of apoptosis has been in a state of flux, and much of the original concepts are themselves undergoing apoptosis! Table 2-I lists some of the early assumptions in the apoptotic process which are now known to be incorrect (adapted from Kroemer, 1997). This table should serve as a word of caution to the reader that not much in the world of apoptosis can be regarded as absolute truth. 2.2 Genes Regulating Apoptosis Since apoptosis is genetically programmed process, the apoptotic process is controlled both by genes that induce the cell to die and those that protect the cell from death. The process of apoptosis can be subdivided into four different phases - initiation, signaling, effector and degradation. Addition of specific apoptotic stimuli, such as chemotherapeutic drugs, or the loss of survival factors such as IGF-1 and interleukins, can initiate the death process either by turning on the death genes (such as those of the cysteine protease or caspase family) or by suppressing the activity of the survival genes (such as those of the bcl-2 family). The diverse stimuli, in almost all cases (except in the Table 2-I: Changing views in apoptosis Invalidation Assumption Apoptosis involves the de novo synthesis Apoptosis can be induced in all cells in the of 'killer genes' presence of cycloheximide Apoptosis is an abortive cell cycle Apoptosis can be induced during any phase of the cell cycle Apoptosis is a nuclear process Cytoplasts (enucleated cells) can undergo receptor-mediated, bcl-2-regulated cell death Apoptosis is mediated by reactive oxygen Apoptosis can induced in/by the absence of species oxygen, in a bcl-2-regulated fashion Apoptosis involves an elevation of Ca+2 in Ca+2 depletion can induce apoptosis. Nuclear apoptosis can be induced in Ca+2 some subcellular compartment free media Apoptosis is due to cytosolic acidification Acidification inhibits apoptosis in some cases Apoptosis always involves specific Bax and Bak overexpression induce death in proteases (caspases) the presence of caspase inhibitors Apoptosis does not involve mitochondria Cell-free systems of apoptosis require mitochondria or mitochondrial products case of direct DNA damage to the cell), have to transmit the apoptotic signal through a series of signaling molecules and signaling events from the outside of the cell, through the cytosol and to the nucleus to affect gene transcription and translation. The map of signaling pathways, which include FADD (Fas-associated death domain, see Nagata, 1996 for a review) and TRADD (TNF-receptor-associated death domain, see Vaux and Strasser, 1996 for a review), is still incomplete. There is also evidence, that in some instances this signal transduction process is short-circuited. For instance, most caspases already exist in the cytosol as pro-forms of the protein, and are cleaved to the active form via auto-catalyzed reactions, in response to apoptotic stimuli. These pro- and anti- apoptotic genes and their protein products comprise the effector phase of apoptosis. In turn, the effector phase proteins, use common downstream degradation proteins such as nucleases, transglutaminases etc. which actually enable the process of death in the cell. The regulation of apoptosis can occur throughout out the cell - in the nucleus, the cytosol and also, as we shall see later in cell organelles such the mitochondria and endoplasmic reticula. Apoptosis is thought to be beyond regulation once the degradation phase is reached. Much of the original work in identifying the key genetic players in apoptosis was done by studying the process of programmed cell death in the nematode, Caenorhabditis elegans. Three basic genes - ced-3, ced-4 and ced-9 (ced for C. elegans death)- control the process of apoptosis in these animals through mutual interactions (Ellis, et al., 1991; Hengartner, 1996). Subsequently, the mammalian homologues of these basic genes have been discovered, once again pointing to the astonishingly conserved nature of the pathway of apoptosis in evolution. The following sections review some of the signaling pathways and the mammalian homologues of the ced genes. 2.2.1 p5 3 - a signaling molecule that is involved in both cell cycle control and apoptosis p53 codes for a 53-kilodalton nuclear phosphoprotein that has specific DNA-binding capability and can positively and negatively regulate transcription. Normal p53 works as a guardian of the genome (Lane, 1992). It checks for mutations in genomic DNA during cell replication. If it finds mutations or nicks in genomic DNA, it arrests cell division in the Gl phase (Lin, et al., 1992; Marx, 1993; Mercer, et al., 1990; Sherr, 1994). The nicks may be subsequently repaired but it is not clear whether expression of p53 causes it to proceed with its suicide program (Clarke, et al., 1993) or return to its normal cell cycle after damages have been repaired (Lowe, et al., 1993). p 5 3 has also been shown to have 3'-5' exonuclease activity which is believed to be important in DNA replication, recombination and repair (Mummenbrauer, et al., 1996), an observation that further validates its role as the guardian of the genome. Failure to correct damage to or mutation of genomic DNA leads to induction of apoptosis by p53 (Clarke, et al., 1993; Han, et al., 1996; Ko and Prives, 1996; Lowe, et al., 1993; Lowe, et al., 1993; Stasser, et al., 1994). Small quantities of p53 are always present in the cell, but the protein is rapidly turned-over (Ko and Prives, 1996). The activation of p53 is post-transcriptional perhaps through phosphorylation or other post-translational means (Ko and Prives, 1996; Oren and Prives, 1996). These post-transcriptional and post-translational modifications also stabilize the p53 protein. The susceptibility to induction of apoptosis by p53 is linked to the amount of p53 protein that is synthesized. Thus, p53-heterozygous mice (containing only one copy of the p53 gene) displayed an apoptotic response to ionizing radiation and topoisomerase inhibitors (which prevent DNA repair) which was intermediate between that displayed by wild-type (having 2 copies of the p53 gene) and p53 homozygous mutant (a p53 knockout with no copies of the p53 gene) (Clarke, et al., 1993; Lowe, et al., 1993). However, the observation that treatment with apoptotic agents like phorbol ester/calcium ionophore, dexamethasone (Lowe, et al., 1993) and glucocorticoid (Clarke, et al., 1993) lead to similar extents of apoptosis in wild-type, heterozygous mutants and homozygous mutants, indicates that p53-independent apoptotic pathways exist. Some of the above-described experiments suggest that p53 induces apoptosis only when damage to DNA occurs, suggesting that its role is limited to damage control pathways. Another interesting observation made by Clarke, et al., 1993, is that cycloheximide (a protein synthesis inhibitor) blocks apoptosis caused by both p53dependent and p53-independent pathways. This suggests that the two pathways may share common elements that require protein synthesis downstream of p53 and that p53 expression may not be required to activate these downstream proteins. Studies have also indicated that though p53 expression is sometimes necessary for apoptosis, it is not sufficient for cell death (Lowe, et al., 1993). For instance, irradiation of normal cells stabilizes p53, but causes cell cycle arrest without apoptosis (Kastan, et al., 1992). Finally, cells lacking p53 are also known to undergo apoptosis under the influence of chemotherapeutic agents (causing DNA damage), if these are used at sufficiently high concentrations (Sen and D'lncalci, 1992). Hence, even DNA damage related death is possible in the absence of p53, depending on the level of the damage. Several downstream events by whichp53 effects its control on cellular growth and apoptosis have been identified. It was observed that expression of p53 lead to the activation of a gene, alternately called Cipl or WAF1, that produces a 21 kilodalton protein (El-Deiry, et al., 1993; Harper, et al., 1993; Marx, 1993; Sherr, 1994). This protein binds to an enzyme called cyclin-dependent kinase 2 (Cdk2). Cdk2 is responsible for preparing cells to divide by pushing them out of the GI growth phase of the cell cycle into the DNA-synthesizing phase (S). The binding of Cipl to Cdk2 blocks Cdk2's growth-spurring ability. p53 is also thought to control cell cycle progression by activating the GADD45 gene (Kastan, et al., 1992) and the cyclin G gene (Okamoto and Beach, 1994). p53 also induces cells with a damaged genome to undergo apoptosis via several mechanisms, although once again the directory of genes activated by p53 is by no means complete. p53 is known to activate the transcription of an apoptosis inducing member of the bcl-2 family, Bax (Han, et al., 1996; Matsuyama, et al., 1998; Miyashita and Reed, 1995). p 5 3 is also known to transactivate the insulin-like-growth-factor binding protein 3 (IGF-BP3) gene (Buckbinder, et al., 1995). IGF-BP3 is known to inhibit the action of IGF-1 and its secretion can adversely affect mitosis and cellular survival. Lastly, p53 has also been reported to be associated with the Fas apoptotic pathway (Oren and Prives, 1996). Although p53 is an important signaling molecule in the apoptotic pathway, p53 independent apoptotic pathways clearly exist. In addition, the pathways by which p53 induces death are closely linked to those associated with the bcl-2 family of proteins that are discussed below. 2.2.2 The Bcl-2 family of proteins Although the most famous member of this family, bcl-2 (from b-cell lymphoma-2), is one of the strongest anti-apoptotic genes, the bcl-2 gene family includes several other antiapoptotic as well as pro-apoptotic genes. Bcl-2 itself has strong sequence homology to the ced-9 anti-death gene in C. elegans (Hengartner and Horvitz, 1994). The other antiapoptotic members of the family include bcl-xL Mcl-1, Al and BI-1, Bcl-w, Bfl-1, Brag-i (Boise, et al., 1993; Kozopas, et al., 1993; Lin, et al., 1993; Xu and Reed, 1998, while the pro-apoptotic family members include Bax, Bak, Bcl-xs, Bad and Bik (Boise, et al., 1993; Boyd, et al., 1995; Chittenden, et al., 1995; Kiefer, et al., 1995; Oltvai, et al., 1993; Yang, et al., 1995, also see Yang and Korsmeyer, 1996). In addition, genes such as Bag-i, interact with bcl-2 family of proteins to enhance anti-apoptotic activity (Takayama, et al., 1995). Most members of the bcl-2-gene family possess a carboxy-terminal transmembrane region, although Bid and Bad are notable exceptions. This transmembrane region influences the subcellular localization of bcl-2-family proteins. In addition, members of the bcl-2 protein family contain varying amounts of four so-called bcl-2 homology (BH) domains, BH1 to BH4. The BH1 and BH2 domains are thought to allow bcl-2 proteins to form homo- or hetero-dimers with other members of the family. The formation of these dimers allows a level of control in the regulation of death by this family of proteins. The BH3 domain is required for death agonists, such as Bax and Bak, to heterodimerize with death-suppressing members of the family and for their apoptosis promoting function (Zha, et al., 1996). In addition, swapping a 23 aminoacid segment surrounding BH3 from Bax to bcl-2, converted bcl-2 to a death agonist (Hunter and Parslow, 1996). The BH3 domain thus serves as a potent death-domain necessary for the apoptosis inducing activity of the death agonist family members (Kroemer, 1997). The BH4 domain, in contrast, is conserved in all anti-apoptotic bcl-2 family members, and is lacking or ill-conserved in all the proapoptotic members, with the exception of bcl-xs. Deletion of the BH4 domain from death-antagonistic family members, did not inhibit dimerization, but imparted either a loss of function or dominant negative phenotype (yielding mutants that promote rather than suppress apoptosis, see Hunter, et al., 1996). The following sections discuss the activity of anti-apoptotic members of the bcl-2 family in general and some of the emerging mechanisms by which these genes are thought to exert their effects. 2.2.2.1 Death-antagonisticfamily members The most well known death antagonistic family members are bcl-2 and bcl-xL. bcl-2 codes for a 26 kDa integral membrane protein, and was first discovered in patients with B-cell malignancies. Bcl-2 and bcl-xL observed to prevent apoptosis in response to a large number of apoptotic stimuli (Lincz, 1998; Reed, 1994; Yang and Korsmeyer, 1996). Expression of bcl-2 inhibits apoptosis brought about by a withdrawal of growth factors, IL-3, NGF etc. In all these cases, bcl-2 alone did not affect the proliferation rate of cells, although there have been some recent reports that bcl-2 delays cell-cycle entry in some cell lines (Huang, et al., 1997). Bcl-2 overexpression allowed cells to survive in the absence of growth factors but without further proliferation. In contrast, reductions in bcl2 levels using antisense-mediated techniques, accelerated cell death, showing a clear function of bcl-2 in reducing apoptosis (Reed, 1994). In all cases examined, expression of bcl-2 prevented the characteristic morphological changes associated with apoptosis such as cell shrinkage, chromatin condensation, nuclear fragmentation and DNA laddering, implying that bcl-2 blocks a relatively early event associated with apoptosis. The extent of protection offered by bcl-2 was proportional to the level of bcl-2 expression in the cells (Hockenberry, et al., 1993). Bcl-2 prevents oxidative injury from resulting in apoptosis (Hockenberry, et al., 1993). Lipid peroxidation, which is a characteristic event observed in apoptosis and induced by a large number of apoptotic stimuli, was also blocked by bcl-2 (Hockenberry, et al., 1993; Kane, et al., 1993). In addition, bcl-2 prevents induction of apoptosis in response to diverse stimuli such as nutrient depletion, accumulation of toxins, viral infection, hypoxic and hyperoxic conditions, and hydrodynamic stresses (Singh, et al., 1996; Levine, et al., 1993; Olsen, et al., 1996; Shimizu, et al., 1996). Bcl-2 and bcl-xL have also been reported to protect against stimuli which normally kill by necrosis (Kane, et al., 1995; Shimizu, et al., 1996; Vander Heiden, et al., 1997). It is thought that many of these necrotic agents induce apoptosis by causing a fall in mitochondrial membrane potential, which bcl-2 by virtue of its sub-cellular localization is able to prevent (see below). Bcl-2 has been observed to counter the ability of p53 to induce apoptosis in response to DNA damage induced by various stimuli (Chiou, et al., 1994; Stasser, et al., 1994). Bcl-2 did not, however, prevent or reduce DNA damage or enhance DNA repair rates in any of these cases, suggesting that it acted downstream of these events and prevented DNA damage from being translated to an apoptotic signal. Bcl-2 was also unable to prevent cell cycle arrest induced by p53. Growth-arrested cells remained viable and when the expression of wild-type p53 was turned off, these cells were able to proliferate even after 10 days in the growth arrested state (Chiou, et al., 1994). Bcl-2 also appears to be very specific in its action of preventing cell death. For instance, it completely inhibits apoptosis induced by c-myc in response to growth factor withdrawal, but does not affect c-myc's mitotic ability (Bissonnette, et al., 1992; Fanidi, et al., 1992; Lotem and Sachs, 1993; Vaux, et al., 1988). Bcl-xL's protective effects closely mimic those of bcl-2 (Boise, et al., 1993; Reed, 1997). 2.2.2.2 Mechanisms of action Although bcl-2-related proteins have been recognized as key players in apoptosis for a while now, their mechanism of action is just being clarified. Subcellular localization, levels of expression, dimerization, post-translational modification and pore-formation sit at the core of these new theories of bcl-2 action, and are discussed below (also see Figure 2-3 for a summary of bcl-2 mechanisms of action). In addition, RNA splicing, as in the case of bcl-xL and bcl-xs, provides another level of control in the manner in which the bcl-2 family regulates death (Boise, et al., 1993). 2.2.2.2.1 Subcellular localization Since most members of the bcl-2 family have transmembrane domains, membrane localization is thought to be necessary for the proteins to perform their proper function in regulating apoptosis. Family members lacking transmembrane domains are thought to exert their function by binding to and affecting the function of the membrane-localized family members. Bcl-2 family members have been detected localized to the nuclear envelope, parts of the endoplasmic reticulum (ER) and the membranes of the mitochondria (Kroemer, 1997; Reed, 1994). Although there has been some speculation that the bcl-2 family may regulate apoptosis by controlling Ca +2 release from the ER (Lam, et al., 1994; Orrenius, et al., 1992) and by being involved in some aspect of nuclear transport or nuclear envelope assembly and maintenance (Krajewski, et al., 1993), not much evidence supporting these hypotheses has been found. In addition, studies in cellfree systems have demonstrated apoptosis in the absence of the ER and nuclei, obviating the need for localization of bcl-2 family proteins to these organelle membranes (Jacobson, et al., 1994; Liu, et al., 1996). Some other studies have suggested that bcl-2 can prevent apoptosis by redistributing calcineurin from the cytosol to intracellular membranes. This prevents calcineurin from dephosphorylating BAD or interacting with substrates such as phosphorylated NF-AT (Reed, 1997; Shibasaki, et al., 1997). Mitochondrial PT (cytochrome c /AIF release) caspases p53 I , I I I I NF-AT Figure 2-3: A map of bcl-2's (and bcl-XL's) interactions with other proteins which help explain the mechanisms by which it exerts it antiapoptotic effects. See text for details of specific protective effects. Adapted from Reed, 1997. However, evidence pointing toward the need for mitochondrial localization is growing steadily (Bossy-Wetzel, et al., 1998; Kroemer, 1997; Kroemer, et al., 1997; Newmeyer, et al., 1994; Petit, et al., 1996; Susin, et al., 1996; Vander Heiden, et al., 1997; Yang, et al., 1997). Preventing members of the bcl-2 family from localizing to the mitochondria severely reduced their ability to control apoptosis. In contrast, once the ability to localize specifically to the mitochondria was added back to the proteins, they regained their function (Zha, et al., 1996). Bcl-2 and Bcl-XL are thought to regulate apoptosis by maintaining mitochondrial membrane potential and preventing the release of cytochrome c and apoptosis-inducing-factor (AIF) (Kluck, et al., 1997; Susin, et al., 1996; Yang, et al., 1997; Zamzami, et al., 1996). The release of these agents from the mitochondria activates caspases, which then push the cell further toward the degradation phase of apoptosis (see below). In addition, it has recently been demonstrated that the mitochondrial FOF 1-ATPase proton pump is necessary for the apoptotic activity of Bax (Matsuyama, et al., 1998), demonstrating that Bax activity is also mitochondria dependent. 2.2.2.2.2 Dimerization and levels of expression The response of a cell to an apoptotic stimulus is determined by the competitive dimerization between death-agonisitic and antagonistic members of the bcl-2 family. These dimerizations, in turn, are affected by the ratios of these proteins to other proteins that they interact with (Yang and Korsmeyer, 1996). Mutant forms of bcl-XL, which are unable to bind to Bax or Bak retain substantial portions of their death repressor activity indicating that family members (at least the death-antagonistic ones) can function without dimerization (Cheng, et al., 1996). In contrast, binding (formation of heteromers) by the death agonistic members of the bcl-2 family seems essential for their apoptotic function (Kroemer, 1997). While it is clear that a dynamic equilibrium between the various homoand heterodimers is instrumental in regulating apoptosis, it is not very clear which of these dimers is the true regulator of apoptosis. 2.2.2.2.3 Post-translational modification Post-translational modification of proteins offers another level of control for apoptosis induction. Serine phosphorylation of bcl-2 related proteins effectively destroys activity of both death agonistic and antagonistic family members (Kroemer, 1997). The disruption of family members often disrupts their ability to bind to other members or the ability to localize to desired organelles. The stimulus for this phosphorylation is sometimes external as with chemotherapeutic agents that act on microtubules. In addition, some bcl-2 family proteins can themselves induce phosphorylation of other family members. For instance, bcl-2 and bcl-XL regulate the phosphorylation of Bad, a death agonist with no transmembrane domain, via Raf-l kinase (Zha, et al., 1996, Kroemer, 1997). Once phosphorylated, Bad loses its ability to bind to bcl-2 and hence its ability to induce apoptosis. Growth factor receptors and kinase cascades can also influence phosphorylation of bcl-2 family members (Kroemer, 1997). For instance, in the presence of growth factors Bad is phosphorylated and bound to the protein 14-3-3, but once growth factors are removed Bad is de-phosphorylated and is free to bind to Bcl-xL and promote apoptosis (Lincz, 1998; Zha, et al., 1996). Bcl-2 and Bcl-XL may also use their mitochondrial localization to interact with and sequester other apoptosis regulating proteins (e.g., Raf-1, BAG-1 etc.) with which they interact. This provides yet another level of apoptosis control (Kroemer, 1997; Reed, 1997). Bcl-2 has also been reported to control the translocation of p53 from the cytosol to the nucleus by binding to a p53 binding protein, p53-BP2 (Naumovski and Cleary, 1996). Interestingly, the death agonistic members of the family are not able to bind with any of the proteins that the death-antagonistic proteins bind to. 2.2.2.2.4 Pore formation Since many of the bcl-2 family of proteins are localized to the mitochondria and regulate mitochondrial membrane potential and release of apoptotic agents from the mitochondria, it has been hypothesized that bcl-2 proteins may have pore-forming ability. Indeed both bcl-2 and bcl-XL contain domains which are similar to the pore-forming domains of bacterial toxins such as colicins and diptheria toxin (Muchmore, et al., 1996). It is suggested that differences in charge and structure (due to formation of homo- or heterodimers, or due to binding with other proteins) could determine ion selectivity and conductance through the pores. Vander Heiden, et al., 1997 suggest that bcl-XL's poreforming ability allows it to regulate volume homeostasis in the mitochondria, and may explain how cytochrome c release can occur without loss in the mitochondrial membrane potential. In addition to mitochondrial pore formation, bcl-2's pore-forming ability and nuclear localization could allow it to catch proteins as they cross over into the nuclear envelope, thus preventing nuclease activity or transmission of the apoptotic signal to the nucleus (McConkey, 1996; Reed, 1997). 2.2.3 Cysteine Proteases or Caspases Though members of the bcl-2 family are important in controlling apoptosis, they are thought to lie fairly upstream in the apoptotic execution pathway. Another family of proteins, which according to some is absolutely essential in apoptosis (Salvesen and Dixit, 1997), is the cysteine protease family of proteins. A cell is thought to commit to death at a point at which the rate of cellular damage outstrips the ability of a cell to repair itself. Cysteine proteases often affect both these parameters determining a cell's commitment to death (Martin and Green, 1995). The cysteine protease family is so called due to the presence of a cysteine residue in the active site(s) of these proteins. In addition, this family displays a highly conserved QACXG pentapeptide active site motif. The fourth residue, 'X', can be R, Q or G. These proteins are also characterized by the absolute requirement of an aspartate residue in the P1 position of their substrates (the P1 position residue is the last residue on the Nterminal side of the cleaved part of the protein, hence substrate cleavage occurs right after the P1 position). Another hallmark of this family of enzymes is that they are all initially produced as inactive zymogens, which are cleaved (often in an autocatalytic fashion) in the presence of apoptotic stimuli to form the active enzyme. The family of cysteine proteases already has at least 10 members, and to reduce the confusion due to multiple names for the same protease (due to simultaneous discovery of the protein by several researchers), a trivial name, caspase, was suggested for all family members. The 'c' in caspase refers to the cysteine protease part and the 'aspase' refers to the ability of these proteases to cleave after an aspartate residue in their substrates. The members are then assigned numbers in the order of their discovery. The first member of the family to be discovered was the interleukin-l-13converting-enzyme or ICE, which is now called caspase-1 (Miura, et al., 1993). Although not now considered a central piece of the apoptotic machinery, a lot of the initial work on the structure, function and mechanism of action of caspases was carried out with this protein. All caspases are now classified as belonging to a family headed by one of the first three caspases discovered, based on sequence homology and specificity of substrates (Alnemri, et al., 1996). The caspase-1 (or ICE) subfamily consists of caspases-1, -4 and 5. Caspases-2 and -9 comprise the caspase-2 (or ICH-1/Nedd2) family, while the caspase 3 (or ced-3/CPP32) subfamily consists of caspases-3, -6, -7, -8, and -10. While caspase-1 and possibly caspases-4 and -5 are primarily involved in procytokine activation, caspases-2, -3, -6, -7, -8, -9 and -10 are considered to promote apoptosis. The caspase family of proteins has been implicated in apoptosis in several cell lines and human diseases (Nicholson, 1996; Takahashi and Earnshaw, 1996). The structure and the specificity of these proteases have provided several clues as to their mechanism of action. In addition, it is being becoming clear that caspases comprise a cascade with upstream members of the family activating downstream members. Thus the caspase family proteins serve both as signaling molecules and as effector molecules. The following section discusses some of the proposed mechanisms by which caspases control the process of apoptosis and their interaction with other families of proteins discussed above. 2.2.3.1 Structureand activationof caspases Most caspases are produced as inactive zymogens (or proenzymes) that are cleaved to the active form. These inactive zymogens consist of an N-terminal peptide prodomain, a large and a small subunit, and sometimes a linker region between the two subunits. Crystal structures of caspases-1 and -3 have shown that the active enzyme is a heterotetramer, consisting of two large and two small subunits (Thomberry and Molineaux, 1995; Wilson, et al., 1994). It is believed that the heterotetramer structure of the active enzyme is maintained in all other caspases. The active site(s) (there are usually at least two active sites per tetramer) span both subunits, making both of them necessary for enzyme activity. The cleavage sites in the inactive proenzyme are aspartate sites that can usually be cleaved by the active form of the enzyme. This supports the hypothesis that these enzymes can be activated via autocatalysis. Activation by autocatalysis thus provides a level of control to the cell in regulating apoptosis through proteolysis via caspases. The structure of active caspase-1 is often considered a model for other caspases (see Cohen, 1997 for a review). In caspase-1, Cys-285 and His-237 form a catalytic dyad in the active site. The conserved QACRG pentapeptide active site is in the larger p20 subunit. However, the Asp pocket (for docking the substrate Asp cleavage site) is formed by Arg-179, Gln-283, Arg-341 and Ser-347, of which only the first two are in the p20 subunit. The two Arg residues form hydrogen bonds with the P1 Asp residue of the substrate, and the presence of these Arg residues is necessary for catalytic activity. In contrast, the P2 and P3 residues in the substrate are solvent exposed, explaining why broad substitution in the P2 position is tolerated in substrates (Thornberry and Molineaux, 1995). The side chain of the P4 tyrosine binds in a hydrophobic channel. Different residues in the P2, P3 and P4 positions are required in substrates for different caspases to ensure that they can bind to the active site and be processed. The structure of the active site thus determines the specificity of the caspase for its substrates. The prodomain in the inactive zymogen might provide an additional level of control and specificity in caspase-regulated apoptosis. Large prodomains function as signal integrators that bind adapter molecules involved in the transduction of the apoptotic signal. The adapter molecules are in turn associated with various cell surface receptors. For instance, the prodomain of caspase-8 binds to the corresponding motif in the adapter molecule FADD (Fas-associated-death-domain) allowing for its recruitment to the CD-95 death receptor signaling complex (Chinnaiyan, et al., 1995; Fraser and Evan, 1996). Figure 2-4 provides an example of some receptor, adapter, protease cascades pertinent to caspase activity. Large prodomains often contain a global homophilic interaction domain referred to as CARD (caspase recruitment domain). Significantly, caspase-2 is recruited to the TNFR-1 signaling complex through an interaction involving the respective CARD domains within the adapter molecule RAIDD and the prodomain of caspase-2 (Duan and Dixit, 1997). CARD domain sequences are highly specific, ensuring that only certain caspases are recruited and activated by particular stimuli/receptors. For instance, only caspase-9, but not other large prodomain caspases, bound to the CARD region of the apoptotic protease activating factor-i (Apaf-1, a mammalian ced-4 homologue) by virtue of a CARD sequence in its prodomain (Pan, et al., 1998). 2.2.3.2 Targetsubstrates of executionercaspases The executioner caspases cleave several key proteins within the cell making the commitment to death an irreversible commitment. A few of these proteins, their significance and the primary caspases responsible for their cleavage are shown in Table 2II (also see Cohen, 1997 for a review). 2.2.3.3 Inhibitorsof caspases There are several genetic and chemical (peptide) inhibitors of caspases. Amongst the genetic inhibitors crmA effectively inhibits caspases-1, -4, -6, and -8 but is a poor inhibitor of caspases-2, -3, -7 and -10. The anti-apoptotic members of the bcl-2 family are able to prevent caspase activation by preventing release of cytochrome c from the mitochondria (see below). However, once caspases (especially downstream caspases) are activated, bcl-2 family proteins are unable to inhibit caspases. It is interesting to note that many caspase-related apoptotic pathways that are resistant to crmA are sensitive to inhibition by bcl-2 family proteins and vice versa (Cohen, 1997). The baculovirus Autographa californica p35 gene-product is a surprisingly wide inhibitor of all known caspases (Bump, et al., 1995; Hershberger, et al., 1994; Rabizadeh, et al., 1993; Xue and Horvitz, 1995). Another family of baculovirus proteins and their human homologues, TNF Fas TNF Ligand RIP - AIDD - FA D D Death domain FADD-like rodomain prodomain DED Pro-caspase-8 .. TRADD Pro-caspase-2 Figure 2-4: Examples of receptor, adapter and protease complexes in regulation of caspase activity. In this illustration, Fas ligand and TNF- (tumor necrosis factor) receptor are the receptor molecules. FADD (Fas-associated-death-domain protein), TRADD (TNFreceptor-associated-death-domain protein), RIP (receptor-interacting-protein), RAIDD (RIP-associated ICH-1/CED-3-homologous protein) are the adapter molecules. Procaspase-2 and pro-caspase-8 are the end proteases (or caspases) which are activated through this signaling pathway. Adapted from Cohen, 1997. Table 2-II: Caspases and their target substrates. Target Protein Biological Significance Cleaving Caspases Poly(ADP- Thought to be responsible for DNA Caspases-3 and -7 ribose) repair polymerase regulation (Lazebnik, et al., 1994). (PARP) However, PARP-null and activity endonuclease mice develop normally (Cohen, 1997). DNA-PK responsible for DNA double-strand- caspase-3 break repair Lamins (A, B responsible for structural integrity of caspase-6 cleaves lamin A (Orth, et al., 1996). Lamin B the nucleus (McKeon, 1991) and C) is cleaved very early in apoptosis and maybe cleaved by a more upstream caspase (Mandal, et al., 1996) U1-70 kD essential for splicing of precursor caspases-3 and -7 mRNA. Cleavage blocks cellular repair pathways dependent on new mRNA synthesis Fodrin responsible for cell membrane integrity. possibly caspase-6 Cleavage leads to plasma membrane blebbing Protein kinase the 5-fragment of PKC is thought to be PKC-8 is a rare protein C (PKC) 8 and responsible for chromatin condensation cleaved by caspase-3 but not Retinoblastoma and nuclear fragmentation. protein (Rb) important mediator of mediation and progression. Rb is an caspase-7. cell cycle Rb is cleaved by caspase-3 called the IAPs (inhibitors of apoptosis proteins), have been reported to broadly inhibit caspase activity (Clem, et al., 1996; Orth and Dixit, 1997). The peptide z-VAD.fmk (benzylocarbonyl-Val-Ala-Asp(OMe).fluoromethyl ketone) is a powerful and irreversible inhibitor of all tested caspases (Fraser and Evan, 1996; Xiang, et al., 1996). 2.2.3.4 The caspase cascade Caspases implicated in apoptosis can be divided into initiators and executioners of apoptosis (Salvesen and Dixit, 1997). Caspases belonging to the caspase-1 family are sometimes thought to play an amplifying role in the signaling process from initiator to executioner caspases, but evidence for this hypothesis is scant (Fraser and Evan, 1996). The initiator caspases (caspases-8, -10 and possibly caspase-2) are located at the apex of the caspase apoptotic cascade, and are distinguished by their ability to directly cleave, and thus activate, zymogens belonging to all other caspases (Cohen, 1997). In contrast, the executioner caspases can only cleave a limited number of the zymogens of other family members. The ordering of some of the executioners of the pathway is controversial and may differ in different cell lines. Cohen, 1997 suggests that since PARP is cleaved before lamins in most cell lines undergoing apoptosis, caspases responsible for cleaving PARP (caspases-3 and -7) may lie upstream of lamin cleaving caspase-6. The best example of a pure caspase cascade can be found in Fas induced death. Here the Fas receptor binds pro-caspase-8 via FADD and activates it. Caspase-8 can then catalytically activate executioner caspases (-9, -3, -6, and -7), either directly or through caspase-10. The executioner caspases then cleave their substrates to initiate the degradation process. This caspase cascade does not require the presence of mitochondria or the release of cytochrome c from the mitochondria (Reed, 1997; Wallach, et al., 1997). This may explain why bcl-2 is not able to protect against Fas induced apoptosis in many cell lines. TNFR mediated death may similarly use caspase-2 as an initiator caspase. Physiologically, proteases such as the cytotoxic cell protease granzyme B can also directly activate the caspase cascade by cleaving caspase zymogens (Pan, et al., 1998). Even more remarkable is the recent demonstration that proteases without aspartate residue specificity can cleave caspase zymogens in the linker regions (regions between subunits), possibly allowing lysosomes or viruses to engage the apoptotic apparatus under pathological conditions (Zhou and Salvesen, 1997). Most of the other caspase cascades however display an absolute requirement for the presence of mitochondria (Kroemer, 1997). Here the primary event in the activation of the caspase pathway is the release of cytochrome c from the mitochondria as a result of as yet incompletely defined upstream events. The release of cytochrome c need not involve upstream caspases since a universal caspase inhibitor failed to block the release of mitochondrial cytochrome c (Bossy-Wetzel, et al., 1998; Yang, et al., 1997). Once cytochrome c (also called Apaf-2; Apaf is an acronym for apoptotic protease activating factor) has been released from the mitochondria it binds to Apaf-1 (see Li, et al., 1997 for a review). The Apaf-1-cytochrome c complex is able, only in the presence of dATP, to bind and cleave the caspase-9 zymogen (see Figure 2-5). Activated caspase-9 then cleaves other downstream executioner caspases such as caspase-3 or caspase-7 (Pan, et al., 1998). These activated caspases can then further act on the mitochondria inducing permeability transition. The permeability transition brings about a reduction in Apoptotic stimuli cytochrome c dATP Im (Apaf-2) mitochondria pro-caspase-9 (Apaf-3) - DOLDA IC DQLDA DQLD Activated caspase-9 pro-caspase-3 activated caspase-3 Q Figure 2-5: The mitochondrial caspase cascade. Release of cytochrome c from the mitochondria leads to the activation of caspase-9 in the presence of Apaf-1 and dATP. Caspase-9 subsequently activates other caspase-3 and potentially other downstream caspases. Adapted from Li, et al., 1997. mitochondrial membrane potential, which in turn triggers the liberation of apoptosisinducing-factor (AIF) from the mitochondria and the generation of reactive oxygen species (Backer, et al., 1990; Bossy-Wetzel, et al., 1998; Susin, et al., 1996; Zamzami, et al., 1996). AIF alone can provoke nuclear apoptosis. In addition to affecting mitochondrial potential, the cytochrome c induced activation of the executioner cascade leaves them free to act on the various biologically critical substrates discussed above and seals the cell's commitment to death. 2.3 Mitochondria - the meeting ground for the key players in apoptosis The release of cytochrome c from the mitochondria, which is a key event in the activation of caspases, is controlled by the bcl-2 family of proteins - bcl-2, bcl-XL and Bax. Pan, et al., 1998 prove that bcl-XL can interact with Apaf-1 (at a binding site different from that required for caspase-9 binding). The authors hypothesize that this interaction also allows bcl-XL another mechanism to prevent caspase-mediated apoptosis. In support of their theory, they demonstrate that other members ofbcl-2 family, namely Bax and Bak, which bind to bcl-XL and disrupt its ability to prevent apoptosis, also prevented its ability to bind Apaf-1. Bcl-XL may be controlling Apaf-l-caspase-9 mediated death by also sequestering Apaf-1 to the mitochondrial matrix and removing it from the cytosol, although this hypothesis remains to be proved. In addition, bcl-2 family members may also prevent death more directly by maintaining mitochondrial membrane potential and preventing the release of AIF and other damaging ROS. The mechanism of cytochrome c release from the mitochondria is not very clear at this point. Since a drop in mitochondrial membrane potential is not essential for cytochrome c release, it is speculated that the release of cytochrome c occurs due to a transitory opening of the permeability transition pore (Bossy-Wetzel, et al., 1998). In addition, it is also believed that bcl-2 proteins are able to mediate the release of cytochrome c via their pore forming domains. Future research will help to elucidate some of the mechanisms of cytochrome c release and the ways in which bcl-2 family proteins regulate this. 2.4 Are caspases essential for apoptosis? Given the central role that caspases play in controlling the executioner phase of apoptosis, there has been a lot of speculation as to whether they are absolutely necessary for apoptosis. In an interesting paper, Xiang, et al., 1996 proved that Bax was able to induce to induce apoptosis-like death in the presence of z-VAD.fmk (considered to be a universal inhibitor of caspases) in response to treatment of cells with Fas. The hallmarks of apoptosis - chromatin condensation, membrane blebbing and cytoplasmic vacuolation were observed to lesser degrees than in cells not treated with z-VAD.fmk. Salvesen and Dixit, 1997, however argue that the observed death was not apoptotic death. Although the apoptotic nature of the caspase-inhibited death process remains in question, the fact remains that rapid death can be induced in the absence of caspases. Bax is thought to mediate this death by altering mitochondrial function, causing a drop in mitochondrial potential and allowing the release of reactive oxygen species. Figure 2-6 shows a map of apoptosis in which both caspase-independent and mitochondria independent forms of death are indicated. Apoptotic Stimuli Direct activators of downstream caspases, e.g. Granzyme B Bcl-2 family regulation of mitochondrial potential and pore-transition Activation of downstream caspases vLoss of mitochondrial potential Release of ROS IMIN1ff activation of degradation proteins I -" 1 Death " Figure 2-6: A map of apoptosis focusing on caspases and the mitochondrial bcl-2 proteins. The diagrams representing receptor-adapter-protease activation of caspases, bcl-2 family regulation and activation of downstream caspases are shown in greater detail in Figure 2-4, Figure 2-3 and Figure 2-5, respectively. The release of AIF (apoptosis inducing factor) is in a positive feedback loop with activation of downstream caspases. Loss of mitochondrial potential and release of reactive oxygen species (ROS) by themselves can lead to death, but there is an active debate in literature as to whether this death is apoptotic or necrotic (see text). 2.5 Apoptosisin Mammalian Cell Culture Several authors have demonstrated apoptosis to be primary cause of death in hybridoma and myeloma cultures (Franek, 1995; Mastrangelo and Betenbaugh, 1998; Mercille and Massie, 1994; Singh, et al., 1994; Vomastek and Franek, 1993). Initial reports by Singh, et al., 1994 suggested that Chinese Hamster Ovary (CHO) cells do not undergo apoptosis in culture. However, later reports demonstrated extensive apoptosis in CHO cells (Moore, et al., 1995; Moore, et al., 1997). Several authors have reported the beneficial effects of bcl-2 expression in delaying death of cells in culture. Bcl-2 suppresses apoptosis in hybridoma cultures under conditions of hyperoxia, hypoxia, glutamine deprivation, glucose deprivation, serum limitation (Simpson, et al., 1997) and insulin withdrawal (Chung, et al., 1998). It has even been reported to prevent apoptosis related to hydrodynamic stress (Singh, et al., 1996). Itoh, et al., 1995 report that bcl-2 is able to protect hybridoma cells at the end of a batch culture from nutrient limitation-induced death, and is also able to enhance productivity of the cells. However, no explanation was offered for the higher productivity observed. Singh, et al., 1997 have also demonstrated that bcl-2 expression reduces specific nutrient consumption, suggesting that bcl-2 expressing cells consume nutrients more efficiently. Mastrangelo, et al., 1996 report that bcl-2 improves recombinant protein production by inhibiting apoptosis in response to viral infection. There have, however, been instances where bcl-2 has failed to prevent apoptosis in the context of mammalian cell culture. Murray, et al., 1996 report that bcl-2 expression failed to prevent the onset of death in an NSO myeloma batch culture. A high indigenous Bax expression level (negating the effects of bcl-2) or the detection of indigenous bcl-XL (leading to an already high base protective level) were offered as possible explanations for these observations. Terada, et al., 1997 also report that bcl-2 is unable to suppress apoptosis induced by the complete absence of serum in hybridoma cultures. An interesting paper by Suzuki, et al., 1997 reports the synergistic effect of expressing both Bag-i and bcl-2 in preventing apoptosis in hybridoma cell lines. In addition to genetic approaches to apoptosis reduction, nutrient and growth factor supplementation approaches have also been suggested (Chung, et al., 1997; Franek, 1995; Franek and Chladkova-Sramkova, 1995; Franek and Dolnikova, 1991). However, the apoptotic pathways involved in cells undergoing apoptosis in mammalian cell cultures remains to be investigated, and could provide more clues to extending the life of mammalian cell cultures. 3. Materials and Methods 3.1 Cell Culture 3.1.1 Cell Line A Chinese hamster ovary cell line producing recombinant human gamma interferon (yCHO) was obtained from Dr. Walter Fiers at the University of Ghent, Belgium (Scahill, et al., 1983). The y-CHO cell line was created from a dihydrofolate reductase deficient (DHFR-) CHO cell line by co-transfecting the cells with genes for DHFR and human gamma interferon (IFN-y). DHFR- cells require added ribonucleosides to survive, while transformed cells are able to produce their own ribonucleosides. The cells were selected for growth in 0.25 gM methotrexate, which is a competitive inhibitor of the DHFR enzyme. Methotrexate selection leads to amplification of the DHFR gene and adjacent DNA (Kaufman and Sharp, 1982), which increases the copy number of genes cotransfected with DHFR (IFN-y in our case). To maintain selection pressure, the cells were grown in ribonucleoside-free medium with 0.25 gM methotrexate. 3.1.2 Culture Medium The cells are routinely grown in suspension in serum-free RPMI medium (Sigma Chemicals) with 2.5 g/L Primatone RL (Quest International), 5 mg/L each of insulin and transferrin (Sigma). Other components added to RPMI to permit serum-free culture include 0.4 g/L 2-hydroxypropyl-(p-cyclodextrin, 1 g/L (0.1%) Pluronic F-68, 5 mg/L insulin, 5 mg/L transferrin, 1 mM sodium pyruvate, 1 jtM putrescine, 11 mg/L choline chloride, 100 jgM ethanolamine, 1.5 gM linoleic acid, 0.25 jtM methotrexate, 10,000 units/L penicillin, 10 mg/L streptomycin, 6.3 mg/L EDTA and trace minerals (10 nM sodium selenite, 1 nM manganese sulfate, 10 nM molybdic acid, 10 nM ammonium metavanadate, 10 nM cupric sulfate, 3 gM zinc sulfate and 5 gM ferric citrate). 3.1.3 Culture Maintenance Cultures were routinely maintained in shake flasks agitated at 70 rpm on an orbital shaker (Bellco, Vineland, NJ) placed in a 370 C incubator with a 5-10% carbon dioxide overlay. Every 2-4 days, cells were resuspended in fresh medium at 2x10 5 cells/mL. Frozen stocks were prepared from cells with viabilities 2 95% by centrifuging the cells at 800 rpm for 7 minutes and resuspending the cells at 7x10 6 cells/mL in freezing medium (7.5% DMSO, 46.3% fresh medium and 46.3% conditioned medium). Vials containing 1 mL of the cell suspension were placed into a cryogenic 1 C/min freezing container (Cole-Parmer, Niles, IL) which was placed into a -70 OC freezer over-night. Cells were later transferred to a liquid nitrogen cell bank for long term storage. New cultures were started by quickly thawing the frozen cells and placing them directly into 20 mL of fresh medium in a shake flask. The following day the cells were resuspended at 2x10 5 cells/mL in fresh medium to remove the DMSO. Cells were re-suspended every 2-4 days until the viability was greater than 95%, at which time experiments were initiated. For all experiments mid-log phase cells were centrifuged and resuspended in fresh medium at an initial density of 4x10 5 cells/mL (unless otherwise noted). All experiments were carried out in duplicate. 3.1.4 Caspase-Inhibiting Peptide Experiments z-VAD.fmk (N-benzylocarbonyl-Val-Ala-Asp-fluoro-methyl-ketone, catalog # FK-009, Enzyme Systems Products) was solubilized in Dimethyl Sulfoxide to yield a 10 mM stock. z-VAD.fmk binds irreversibly to several caspases within the cell. A final concentration of 60 gLM was used for experiments. 3.1.5 Fed Batch Culture Fed batch cultures were performed in 250 mL shake flasks (with a maximum of 80 mL of cell suspension per flask) using the stoichiometric feeding technique developed at MIT (Xie, et al., 1997; Xie and Wang, 1994a; Xie and Wang, 1994b). Agitation was set at 70 rpm, and the flasks were placed in a 370 C incubator with a 5% carbon dioxide atmosphere. Cultures were inoculated at 5x10 5 cells/mL in initial medium containing 3 mM glucose and 0.5 mM glutamine (unless otherwise noted). The initial medium was RPMI-SFM with reduced glucose and glutamine and enhanced buffer capacity. The buffer capacity of RPMI-1640 was enhanced by adding 2.09 g/L of MOPS (Sigma) and 6.50 g/L HEPES sodium salt (Sigma). To maintain proper osmolarity when the extra buffers were added, the sodium chloride concentration was reduced from 6 g/L to 2 g/L. Feeding was performed with a stoichiometrically designed supplemental medium as described in Xie, et al., 1997. Cell counts were performed every 24 hours and feeding was performed every 12 hours. Feeding volume was a function of both cell density and the expected growth rate of cells. Samples were withdrawn periodically for cell counts and to collect supernatant and cells for later analysis. 3.1.5.1 Declumping Protocolfor cells in fed-batch culture CHO cells in fed-batch cultures tended to clump and stick to the walls of the shake flask on reaching high cell densities. The presence of clumps in the cell sample withdrawn for cell number and viability determination introduces high variabilities in these numbers. The following protocol was developed to solve these problems faced due to clumping and wall growth of cells. * Cells are gently scraped off the wall into the medium with a cell scraper (Falcon 3089) * The cell suspension is pipetted up and down repeatedly with a 5 mL pipette to reduce the size of the clumps * After ensuring that the cells and clumps are uniformly suspended, two 0.6 mL samples are withdrawn and subsequently mixed * 1.2 mL of 1X trypsin-EDTA (GIBCO-BRL) is added to sample * The mixture is then gently pipetted up and down for 2 minutes (visible clumps should disappear fairly quickly) * 1.2 mL of 1g/L of soybean trypsin inhibitor (GIBCO-BRL) is added to the suspension followed by brief up and down pipetting * Cells are counted as described below soon after the trypsinization step is completed. 3.1.6 Insulin binding studies Insulin binding was measured using radioactive 1125-insulin (Dupont NEN, catalog # NEX104). Cells from day seven of insulin-free fed-batch culture were trypsinized (to remove clumps) and counted using a Coulter counter. Non-trypsinized cells were then resuspended in the culture medium at 1x10 6 cells/mL. Radioactive insulin was added to the cells at a concentration of 10 ng/mL (0.96 gCi/mL). In this experiment, it was important to measure only the radioactivity due to insulin that is bound specifically to the insulin family receptors on the cell surface, and not the non-specifically bound insulin. To measure the extent of non-specific binding, cold (non-radioactive) insulin was added at a concentration of 10 ptg/mL, to half the population of cells prepared above. This saturating quantity of insulin effectively dislodges radioactive insulin bound specifically to insulin receptors at equilibrium, but does not dislodge non-specifically bound radioactive insulin. Measuring the radioactivity of this population thus permits a measurement of the extent of non-specific binding. Binding measurements were conducted using the Millipore MultiScreen Assay System (catalog # MADP N6550). The cells were then plated directly into a 96-well plate with a 0.65 micron filter base at 1.5x1 0' plates per well in, quadruplicate. As a control, to measure the extent of reduction in insulin binding, cells taken from day one of an insulin-deprived fed-batch culture of CHO cells was similarly treated. The cells were agitated in the 96 well plates for two hours at 40 C. They were then washed with WHIPS (20 mM HEPES, pH 7.4, 130 mM sodium chloride, 5 mM potassium chloride, 0.5 mM magnesium chloride, 1 mg/mL polyvinylpyrolidone) four times, using a vacuum manifold to suck the washing medium through the filter at the bottom of each well. The washing was performed to remove all unbound insulin (radioactive and non-radioactive) from the cells. The filters were dried and the associated radioactivity was measured with a Packard5000 series gamma counter (Packard Instruments). 3.2 AnalyticalMethods 3.2.1 Cell Number and Viability Cell number and viability were determined using a hemacytometer and microscope after staining the sample with trypan blue. Viable cells exclude the dye, while non-viable cells have lost membrane integrity and are stained blue. Total cell number was also determined using a Coulter electronic particle counter (Coulter Electronics, Hialeah, FL) after diluting samples in an isotonic saline solution. Extent of apoptosis was routinely determined by the acridine orange / ethidium bromide (AO/EB) assay (McGahon, et al., 1995; Mercille and Massie, 1994) using a hemacytometer (see details below). In each of the assays at least 200 cells were counted for each sample. 3.2.1.1 Acridine Orange - Ethidium Bromide Apoptosis Assay AO and EB are both DNA intercalating dyes which fluoresce under u.v. light. AO enters all cells regardless of their membrane integrity. EB can only enter cells which have lost their membrane integrity. Using a bluegreen filter and u.v. light, AO stained cells appear green and EB stained cells orange. EB is a stronger dye than AO and overwhelms AO when both dyes are present in a cell. Thus cells which have lost their membrane integrity are stained orange. Materials Stock Solutions: 0.5 g/L Acridine Orange (Sigma catalog # A-6014) and 0.5 g/L of Ethidium Bromide (Sigma catalog # E-8751), prepared in water. Stock solutions should be protected from light (storage in containers wrapped in aluminum foil is recommended). Working Dye Mix: 100 pgg/mL of AO and 100 pg/mL of EB in 1X phosphate buffered saline. Method Working dye mix is added to the cell suspension (5x10 5 - 2x10 6 cells/mL) in the ratio of 1:10-1:30 and mixed well (McGahon, et al., 1995; Mercille and Massie, 1994). For higher cell concentrations a larger proportion of dye may be used. Dye uptake is instantaneous. The mixture can then be counted using a slide or a hemacytometer and a magnification of 20X100X. There are 4 kinds of cells that may be observed (Mercille and Massie, 1994): 1) Viable Cells: Cells are stained bright green with non-fragmented nuclei. Usually the nucleus is very large and the whole cell seems to be stained green. 2) Early Apoptotic Cells: Cells are stained green with several bright green dots or fragments, which correspond to condensed and fragmented chromatin. Condensation and fragmentation of chromatin indicates that the cell is already committed to apoptosis but has not yet lost its membrane integrity. 3) Late Apoptotic Cells: Cells are stained orange with several bright orange dots or fragments, which correspond to condensed and fragmented chromatin. The orange color and the presence of fragmented chromatin indicates that the apoptotic cells have lost their membrane integrity and are in a very late stage of apoptosis. 4) Necrotic Cells: Cells are stained orange (indicating loss of membrane integrity) with randomly degraded chromatin (no condensation of chromatin is observed). Usually the entire cell will be stained orange. Notes 1) AO and EB are both highly mutagenic and hence necessary precautions should be taken while handling them. 2) Some cells will appear to be stained both green and orange. In most cases, these cells will have two distinct bands of chromatin. These cells are undergoing mitosis and hence these should be counted as viable. A good rule of thumb is to count cells as apoptotic only if there are three or more fragments of chromatin. 3) If cells have been dead for a long period of time, they may have begun to lose their chromatin material. These cells were considered apoptotic, since counts performed on the same culture (towards the end of the culture) have indicated that the increase in chromatin-free cells is proportional to the decrease in late-apoptotic cells. It is, however, recommended that this be verified for each cell type used. 4) It is recommended that the cells not be exposed to the dye too long prior to actual counting and at least 200 cells be counted to provide a statistically significant count. 5) Cells where some orange staining seems to lie atop the green chromatin material are not dead cells. The faint orange stain is from single stranded mRNA, which is stained orange by AO. 3.2.2 Dry Cell Weight Dry cell weight was determined by drying samples in a vacuum drying oven. Sample volumes of 60-80 mL were collected from the flask and centrifuged at 250 g for 10 minutes, and the supernatant was removed. The cells were washed twice in phosphate buffered saline and centrifuged. After carefully removing the supernatant, cells were suspended in purified water and transferred to dried, pre-weighed aluminum weigh boats. Samples were dried to constant weights in a vacuum drying oven set at 60 0 C. 3.2.3 Sugar and Lactate Assays Suspension culture samples were centrifuged at 200 g for 8 minutes to remove the cells, and the supernatant was frozen at -20 0 C until ready for analysis. Three hundred and forty pl of thawed sample was deproteinated before performing the lactate and sugar assays by adding 100 pl of 20% rn/v trichloroacetic acid. Two hundred 1 of deproteinated sample was then neutralized with 50 l of 25% m/v potassium bicarbonate. Enzymatic glucose and lactate assays were performed according to the 16-UV and 826UV Sigma assay protocols (Sigma, St. Louis, MO), respectively. Standards were also deproteinated according to the above protocol. 3.2.4 Determination of IFN-y Concentration Gamma interferon (IFN-y) concentrations were measured using a commercially available ELISA kit (Biosource International, Camarillo, CA). Cells were removed from medium by centrifuging at 200 g for 10 minutes, and samples were stored at -20 0 C till further analysis. Just before analysis, samples were thawed at room temperature and diluted to less than 1 gg/L IFN-y (the maximum standard concentration) in a 10 g/L solution of bovine serum albumin in phosphate buffered saline (Pierce, Rockford, IL). It was important to include BSA in the dilution buffer to avoid loss of IFN-,y due to non-specific adsorption to tube walls. The assay was performed according to the manufacturer's protocol. 3.2.5 Determination of insulin concentration Insulin concentration in the medium was determined using a commercially available insulin-ELISA kit (American Laboratory Products Company, Ltd. catalog # 10-1113-01). Cells were removed from medium by centrifuging at 200 g for 10 minutes, and samples were stored at -20 0 C till further analysis. Just before analysis samples thawed at room temperature and were diluted to less than 10 gg/L insulin (the maximum standard concentration) in a 10 g/L solution of bovine serum albumin in phosphate buffered saline (Pierce, Rockford, IL). It was important to include BSA in the dilution buffer to avoid loss of insulin due to non-specific adsorption to tube walls. The assay was performed according to the manufacturer's protocol. 3.2.6 Western Blot 3.2.6.1 Cell Lysis Intracellular protein expression of various proteins were determined by analyzing cell lysates with a western blot. Cells were detergent lysed in a buffer containing 0.5% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl (pH = 8.0), 100 gM EDTA and 0.025% sodium azide . Immediately prior to use, protease inhibitors were added to the following final concentrations: 100 mg/L PMSF, and 10 mg/L aprotinin. Protease inhibitors were added from 100X concentrated stocks prepared in isopropanol (PMSF) or phosphate buffered saline (aprotinin). PMSF is extremely unstable in aqueous solutions and should be added last to the ice-cold lysis buffer, and the buffer should be used within 10 minutes of this addition. All buffer components were from Sigma. Approximately 100 tL of lysis buffer was used per 2x10 6 cells, which ensured efficient extraction. Cells were centrifuged at 200 g for 7 minutes and the supernatant was removed. Cells were washed once in PBS, centrifuged and then suspended in ice-cold lysis buffer. The samples were then vigorously vortexed, after which they were further aliquoted in 10 L samples and stored at -70 0 C until western blot analysis. 3.2.6.2 Electrophoresis Lysates were prepared for western blot analysis by mixing them 1:1 with 2x SDS-PAGE sample buffer (125 mM Tris-HCl pH=6.8, 20% v/v glycerol, 4% w/v SDS, 0.0025% w/v bromophenol blue) before heating the samples in boiling water for 5 minutes. Two hundred mM DTT is added to the buffer just before usage. DTT is stored as a 1 M solution in 0.01 M sodium acetate solution (pH 5.2) at -200 C. DTT is very unstable at a pH different from 5.2. Twenty pl of each sample was loaded per lane onto 12% polyacrylamide gels for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDSPAGE) in the Bio-Rad Mini-Protean II electrophoresis system (Bio-Rad, Hercules, CA). The gels were run at a constant voltage of 100 V for 1.25 hours. 3.2.6.3 Transfer to nitrocellulosemembrane Following electrophoresis, the gels were incubated in transfer buffer (25 mM Tris base and 14.4 g/L glycine in a 20% methanol solution) at 40 C for 15 minutes. Proteins were then transferred from the gels onto nitrocellulose membranes (pre-soaked in transfer buffer for an hour) in the Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad) set at a constant 100 V for 45 minutes. 3.2.6.4 Probingwith antibodiesand development of blot Following the transfer, the membranes were rinsed in TBST, a tris buffered saline solution containing the detergent Tween-20 (10 mM Tris-HC1, 150 mM NaCl, 0.05% Tween-20, pH=8.0). The rinsed membranes were soaked for at least two hours in a blocking buffer which contained 5% non-fat dry milk in TBST to saturate the nitrocellulose membrane's non-specific protein binding capacity. The blocked membranes were then rinsed 3x5 minutes in TBST before adding the primary antibody solution (primary antibody concentrate dissolved in TBST solution containing 5% non-fat milk). After incubation in the primary antibody solution, the membranes were again washed 3x5 minutes in TBST. The next step was an incubation in the secondary antibody solution (horseradish peroxidase conjugated secondary antibody concentrate dissolved in TBST solution containing 5% non-fat milk). In preparation for the development step, the membranes were washed once in TBST and then rinsed 3x5 minutes in TBS (TBST without Tween20), after completion of incubation with the secondary antibody. The membranes were developed using the Renaissance Enhanced Chemiluminescence kit (DuPont NEN, Boston, MA), and the results were recorded by exposing the membranes to X-ray film (X-OMAT AR, Eastman Kodak Co., Rochester, NY) for exposure times ranging from 5 seconds to 2 minutes. Films were developed in a film processor (Eastman Kodak Co.). The antibodies used during the course of the thesis and the companies from which they were sourced are indicated in Table 3-I. 3.2.7 Total Protein Assay Total protein concentrations in cell lysates were determined using the Bio-Rad DC Protein Assay (Bio-Rad, Hercules, CA). The DC Protein Assay is compatible with ionic and non-ionic detergents, and therefore can be used with detergent-lysed cells. Bovine gamma globulin (Bio-Rad) was used as a standard. The standard was prepared in the same buffer used for cell lysis at the following concentrations: 0.1, 0.3, 0.5, 0.7 and 0.9 mg/mL. The assay was performed according to the instructions provided by the manufacturer, and unknown protein concentrations were determined from a linear standard curve. 3.2.8 Measurement of Caspase Activity About 2x10 6 cells were lysed using a single detergent lysis buffer (Sambrook, et al., 1989). Cells from an apoptotic culture were taken for measurement of caspase activity. The substrate used to detect caspases, N-benzylocarbonyl-Tyr-Val-Ala-Asp.AFC (z- YVAD.AFC, catalog # AFC-120, Enzyme Systems Products), has a fluorescent 7amino4trifluromethyl coumarin (AFC) residue. z-YVAD.AFC is a specific substrate for caspases within the cell. Cleavage of this substrate after the aspartate residue, releases Table 3-I: Antibodies used and the companies from which they were sourced protein primary antibody secondary antibody 15131A (Pharmingen) catalog # 107-035-142 (Jackson detected human bcl-2 ImmunoResearch Laboratories) Poly-(ADP- catalog # 1 835 238 (Boehringer catalog # sc-2004 (Santa Cruz Ribose)- Mannheim) Biotechnology) catalog # sc-6215 (Santa Cruz catalog # sc-2020 (Santa Cruz Biotechnology) Biotechnology) catalog # sc-6217 (Santa Cruz catalog # sc-2020 (Santa Cruz Biotechnology) Biotechnology) Polymerase Lamin A Lamin B p53 cyclin E catalog # sc-6243 (Santa Cruz catalog # sc-2004 (Santa Cruz Biotechnology) Biotechnology) catalog # sc-481 (Santa Cruz catalog # sc-2004 (Santa Cruz Biotechnology) Biotechnology) the fluorescent AFC fragment of the protein, which in turn leads to fluorescence. To measure fluorescence, 120 gL of AFC buffer (50 mM HEPES pH 7.5, 1% sucrose, 0.1% CHAPS), 50 gL Dithiothreitol (100 mM DTT stock), and 10 gL of AFC-120 (2.5 mM AFC-120 in Dimethyl Sulfoxide (DMSO)) were added to a cuvette which was then placed in a fluorimeter. The fluorimeter was then zeroed (excitation frequency = 400 nm, emission frequency = 505 nm). 20 gL of the cell lysate was then added to the cuvette and the fluorescence was then monitored with time. Caspase activity was also measured observing cleavage of caspase 3 (CPP32) substrate, Poly-ADP-Ribose Polymerase (PARP). PARP cleavage was determined by western blot analysis of cell lysate using a polyclonal antibody against PARP (Boehringer Mannheim, catalog # 1 835 238). Uncleaved PARP runs as a 113 kDa band, while cleaved PARP is detected either as a 89 kDa fragment or a 24 kDa fragment. 3.2.9 DNA Ladder Technique for Detection of Apoptosis The Apoptotic DNA Ladder Kit from Boehringer Mannheim (catalog # 1 835 246) was used to observe the characteristic ladder obtained due to internucleosomal cleavage of DNA in apoptosis. The protocol for this assay is well described in the kit. DNA from lysed cells bind to microscopic glass fibers in the presence of a chaotropic salt. The salts are then washed away and the DNA is eluted in Tris-EDTA buffer and run on a 2% agarose gel (Sambrook, et al., 1989). 3.2.10 Cell cycle assay Cell cycle analysis of cells from CHO cultures was performed with propidium iodide with the use of a flourescence activated cell sorter (FACS, Becton Dickinson, FACSCAN). Propidium iodide binds to DNA and fluoresces when ultraviolet light is shone on cells (excitation = 488 nm, emission = 640 nm). The extent of fluorescence is dependent on the quantity of DNA in the cell. The assay estimates phase of the cell cycle by estimating the amount of fluorescence in each cell, and hence the amount of DNA in the cell. Sample containing 1x10 6 cells was centrifuged and the supernatant was removed. Cells were then resuspended in 0.5 mL of Stain Solution (1.5% Poly-(Ethylene)-Glycol4000 (PEG-4000), 1.5% PEG-8000, 50 gg/mL propidium iodide (PI), 180 units/mL DNAse-free RNAse, 0.1% Triton-X-100, 4mM sodium citrate pH = 7.8; final pH of solution adjusted to 7.2). The cell suspension is incubated at 370 C for 20 minutes. Onehalf mL of Salt Solution (1.5% Poly-(Ethylene)-Glycol-4000 (PEG-4000), 1.5% PEG8000, 50 gg/mL propidium iodide (PI), 0.1% Triton-X-100, 0.4 M sodium chloride; final pH = 7.2) is added to the suspension at the end of incubation. The suspension must be stored in the dark at 40 C for at least one hour prior to FACS assay, and may be stored for up to four days before the assay is carried out. Storage beyond four days is not recommended as the DNA in the samples starts to degrade. 3.3 Transfectionand Cloning of cells 3.3.1 Preparation of plasmids Human bcl-2 cDNA (graciously donated by Stanley Korsmeyer, University of Washington) was excised using EcoR1 sites, inserted into pcDNA3.1+ vector (Stratagene), and placed under the control of a constitutive CMV promoter. 3.3.2 Transfection of Suspension CHO Cells A protocol was developed for transfecting CHO cells in suspension, rather than transfect anchorage dependent cells and then adapt them to suspension culture. CHO cells grown in suspension were transfected (see transfection protocol below) in standard 6-well plates using Lipofectamine® (GIBCO-BRL). Lipofectamine® uses polycationic liposomes to deliver DNA to the cell. Expression of bcl-2 was verified by western blot analysis using a purified hamster anti-human bcl-2 monoclonal antibody (catalog # 15131A, Pharmingen). The control cell line used was one transfected with the null pcDNA3.1+ vector. 600 pg/mL G-418 (Genetecin, GIBCO-BRL) was used to maintain selective pressure on the mixed culture of cells. 3.3.2.1 Transfection protocol Ensure that there are enough cells at greater than 90% viability, and growing in the log phase. About 2x10 6 cells per well of a 6 well plate are required. PreparationofDNA * For each transfection, dilute 2gg of DNA (Maxipreped DNA) into 100 L of Optimem 1 (Life Technologies). * For each transfection, dilute 5gL of Lipofectamine" into 100p~L of Optimem 1". * The above is most conveniently done in sterile plastic cuvettes, especially if many transfections are done using the same set of DNA. Falcon 2054 12x75 mm tubes were used. Doing at least 3 wells per type of DNA plasmid is recommended. * Combine the DNA and Lipofectamine samples and mix gently (by pipetting up and down). Incubate at room temperature for about 45 minutes to allow the DNA-liposome complexes to form. Preparationof cells * At about 15 minutes before the DNA-lipofectamine mixture is ready: * Spin down the requisite number of cells for n+2 wells, where n is the number of wells that you want to transfect. * Remove supernatant and wash cells once in PBS. * Resuspend cells in Optimem 1" medium at a density of 2.5x10 6 cells/mL. * Pipette 0.8 mL of cell suspension into each well of a 6 well plate. * Add 0.2 mL of the DNA-lipofectamine mixture to each well and mix gently (pipette up and down). * Incubate at 370 C for about 6 hours, and then add 1 mL of Optimem 1" to each well. * After about 20 hours after the start of transfection, harvest cells from plates (this might require a short trypsinization step. Trypsinize for 2-4 minutes, and then quench trypsin by using appropriate amount of trypsin inhibitor). Post TransfectionProcedure * Resuspend cells in normal growth medium and let them grow for 24 hours. * Spin down half the cells, and resuspend in selection medium. We use 600 gg/mL of G-418 (Geneticin®, GIBCO BRL) in RPMI medium. * Split the culture in half again by spinning down the cells, and resuspending the cells in fresh selection medium. * Repeat the above step after 2 days, or sooner if the medium starts turning yellow. * Repeat the above step after 3 days, or sooner if the medium starts turning yellow. * After 1st week, count the number of viable cells and resuspend in fresh selection medium at 0.5x10 6 cells per mL. After this change medium every 3 days dilute cells by 1/2 for the next week or till the culture is about 95% viable. 3.3.3 Monitoring Transfection Efficiency Transfection efficiency was monitored using green fluorescence protein (GFP). Each time the cells were transfected with the desired plasmid, a set of transfections was also carried out using a plasmid containing the GFP gene (pEGFP, catalog # 6077-1, Clontech), using exactly the same transfection conditions as used with the other plasmids. After the last step of the transfection procedure described above, the cells were assayed for GFP expression using a fluorescence activated cell sorter (FACS, Becton Dickinson, FACSCAN). The fraction of cells in the transfected population that displayed green fluorescence (excitation = 488 nm; emission maximum = 507 nm.) was defined as the transfection efficiency, since it was a measure of the fraction of cells transfected which took up the transfecting plasmid. Since about 20,000 cells were counted to arrive at this number, the transfection efficiency measure obtained was statistically very significant. The transfection efficiency obtained with the GFP plasmid was assumed to be indicative of the transfection efficiency obtained with our desired plasmid, since the transfection conditions used for both were identical. The main advantage of using GFP over other techniques, is that it is very rapid and accurate. Cells do not need to be processed at all, before the assay is carried out. Using this technique to monitor transfection efficiency we were able to increase transfection efficiency from under 1% to over 25%, essentially making three changes to the transfection process (all of which are incorporated in the above protocol): a) Optimem 1 (GIBCO-BRL), and not RPMI, was used as the medium during transfection (no insulin, transferrin, antibiotics or primatone was used as it was suspected that small molecules might interfere with transfection) b) Cells were not agitated during transfection (24 hours). Agitation caused many cells to stick to the sides of the 6 well plates and it is possible that these cells did not take up any of the DNA, leading to lower transfection efficiencies. The cells were found to be mostly viable after 24 hours in Optimem 1® without agitation, hence oxygen transport to the cells was not limiting. Some of the cells did tend to stick to the bottom of the plate, but these could easily be trypsinized the following day. c) The ratio of DNA and lipofectamine during transfection was altered. The most optimum ratio amongst the ones tested was 2gg DNA to 5gg lipofectamine. The higher rates of transfection meant shorter times to obtain mixed cultures. This time went down from at least 1.5 months to around two weeks. The monitoring procedure ensures that transfection can rapidly be optimized for any future cell line for which transfection is required. 3.3.4 Cloning Using a fluorescence activated cell sorter (FACS, Becton Dickinson, FACStarPlus), wells of a 384-well plate (NUNC) were seeded with live cells (dead cells were gated out using a propidium iodide stain) from a mixed culture of transfected cells, at one cell per well. Cells were taken from a culture growing in log-phase. Wells were filled with 100gL of 50% conditioned medium (taken from culture where cells were in the exponential growth phase) and 50% fresh GN-RPMI. Selective pressure was constantly maintained during the cloning process. In about 1.5 weeks, wells in which cells had grown were harvested and the cells were transferred to wells of a 24 well-plate. When cells in these wells reached a concentration of about 1x10 6 cells/mL, cells were then transferred to a 6-well plate and then to a T-75 flask. Selective pressure using 600 gg/mL of G-418 was maintained at all times. A sample was taken from each clonal population to check for gene expression using western blotting. The whole process, from the time after harvesting cells from the 384-well plate to the T-75 flask, took about 1.5 weeks. 4. Apoptosis and its Control in Chinese Hamster Ovary Batch Culture The first step in controlling death in cell cultures was to determine the mode of death in serum-free suspension CHO cultures. If the main mode of death in CHO cells under serum-free culture conditions were necrosis, the only way to reduce death would be to make environmental changes in the medium. If on the other hand, cells primarily died due to apoptosis, death could be reduced by both environmental and genetic means. At the time this study was initiated, there were conflicting reports in literature as to the principal mode of death in CHO cells (Moore, et al., 1995; Moore, et al., 1997; Singh, et al., 1994; Singh, et al., 1997). This chapter details the results of several techniques used to ascertain the mode of death in CHO cultures. Since we found that apoptosis was the major mode of death in batch cultures, we were further interested elucidating the apoptotic process in CHO cells. In particular, we were interested in determining whether de novo gene synthesis was required to execute the apoptotic process in our cells. In addition, given the reported predominance of caspases in apoptosis in most mammalian cell lines we wanted to determine whether they played a role in apoptosis in CHO cells. As caspase activity was detected in apoptotic cells, this study attempted to determine whether specifically inhibiting some of the caspases using peptide-caspaseinhibitors could extend the viability of batch cultures. Since the commitment of a cell to apoptosis also depends on expression of survival proteins, we studied the effect of constitutive overexpression of the survival protein, bcl-2, in CHO cells grown in suspension. Bcl-2 has been shown to protect against apoptosis in a large number of cell lines (Kroemer, 1997; Mastrangelo and Betenbaugh, 1998; Reed, 1994). This chapter compares the relative effectiveness of bio-chemical (caspase-inhibiting-peptides) and genetic means (bcl-2 expression) in extending the life of CHO cells in serum-free batch culture. In addition, the chapter compares the relative effectiveness of clonal populations and mixed-culture populations in resisting apoptotic stimuli. The discussion and conclusions section of the chapter summarizes the results and presents additional insights that can be drawn from them. 4.1 CHO Cells Die by Apoptosis in Serum-free Batch Culture The growth kinetics for a typical serum-free batch culture of CHO cells is shown in Figure 4-1. After a period of exponential growth cells die very rapidly. There is a small decline in the number of total cells due to the tendency of dead cells to stick to the walls of the flask. The mode of death involved in these cells, as measured by the Acridine Orange / Ethidium Bromide (AO/EB) assay, is shown in Figure 4-2. The AO/EB assay is measures both membrane integrity and DNA integrity of cells (see Materials and Methods chapter for more assay details). The apoptotic cells included both early (chromatin condensation but no loss in membrane integrity) and late (chromatin condensation and membrane integrity loss) stages of apoptosis. These results indicate 2.OE+6 ..j E 1.5E+6 * -1.OE+6 TCD +-VCD > 5.OE+5 O.OE+O 0 1 2 3 Time (day) 4 Figure 4-1: Viable (VCD) and total cell densities (TCD) obtained in normal serum-free batch culture of suspension CHO cells. 100 80 0 60 cc* % dead -*% apoptotic --- >1 - - -% 40 necrotic 20 0 0 1 2 Time 3 4 (day) Figure 4-2: Apoptosis in batch cultures of CHO cells. Percentage of dead, apoptotic and necrotic cells obtained during a normal serum-free batch culture of CHO cells. The percentage of dead cells is obtained by adding the percentages of apoptotic and necrotic cells. Almost all death is seen to occur via apoptosis. that apoptosis is the main mode of death right from the start of the culture growth process, which is in agreement with the work of Moore, et al., 1995. Furthermore, apoptosis accounts for more than 95% of the death occurring during cultivation. In contrast, necrosis accounts for less than 5% of the final death observed. Apoptosis was also confirmed by the presence of a DNA ladder, when genomic DNA extracted from dead CHO cells was run on an agarose gel (Figure 4-3) (see Materials and Methods chapter for more assay details). The gel-photograph shows that for the first two days of the culture when the viability, as detected by the AO/EB assay, is high the genomic DNA runs as a compact band of high molecular weight. By day 3, some apoptosis has started to occur and this is reflected in the appearance of the beginnings of a DNA ladder - bands of lower molecular DNA, approximately in multiples of 180 bp. This ladder is caused by internucleosomal cleavage of DNA by endonucleases which are activated during the apoptotic process. By day 4 the apoptotic DNA ladder is much more prominent, and further cleavage of DNA is apparent from the appearance of lower molecular bands of DNA. We can conclude that most of the death occurs by apoptosis and not necrosis, due to the presence of an DNA ladder in lane 4 of the gel. Had necrosis been the primary mode of death, a smear caused by random cleavage of DNA would have been observed. Lastly, the complete disappearance of the original band of high molecular weight DNA indicates that most of the cells have perished, confirming the results of the AO/EB assay. viability (%) 99 99 88 day 123 4 Figure 4-3: Apoptosis in batch cultures of CHO cells. Agarose gel photograph of genomic DNA from CHO cells shows presence of a DNA ladder which coincides with massive apoptosis on day 4. 4.2 Protein synthesis inhibitioncauses rapid, dose-dependent apoptosisin CHO cells Since apoptosis is a genetically controlled form of death where the fate of a cell is determined by death suppressing and death inducing proteins, we were interested in determining which class of proteins dominated in CHO cells. One way to determine this was to inhibit protein synthesis using a protein synthesis inhibitor such as cycloheximide, and then follow the fate of the cells. Two scenarios are possible. In the first scenario, if death proteins are present in greater abundance and survival proteins have to be continuously synthesized to keep the cell alive, prevention of protein synthesis would cause rapid apoptosis. In the second scenario, if death proteins have to be synthesized de novo to initiate apoptosis, inhibition of protein synthesis will ultimately lead to death by necrosis. Different concentrations of cycloheximide, ranging from 0 RM to 60 gM, were added to cell suspensions taken from a culture in log-phase growth. Viability of these cultures were monitored at 13, 16 and 19 hours after the addition of cycloheximide. As shown in Figure 4-4, we observed extensive and rapid apoptosis, and almost no necrosis in cultures to which cycloheximide was added. For example, by 19 hours after start of protein synthesis inhibition, more than 85% of the cells in the culture to which 601M cycloheximide was added had undergone apoptosis. By contrast, there was less than 8% apoptosis in the control culture (without cycloheximide). The extent of apoptosis at any 90 80 70 ---- 06 s60 o 50 . < S40 40 0 uM CHX 20 uM CHX 30 uM CHX 40 uM CHX -1- 60 uM CHX ----- 0 30 ~20 w 10 13 16 19 Time (hour) Figure 4-4: Extent of apoptosis induced by varying doses of cycloheximide (CHX). Cycloheximide induces protein synthesis inhibition in cells. Apoptosis accounted for almost all the death observed. point in time was also observed to be dose dependent, since higher levels of cycloheximide at each time point resulted in higher levels of apoptosis. These results suggest that the first scenario in our hypothesis is more accurate in describing apoptosis in CHO cells and survival proteins must be continuously synthesized to protect CHO cells from death. 4.3 Cysteine protease inhibitingpeptides are unable to significantly enhance viabilityof CHO cells in culture. Cysteine proteases (caspases) have been found to be downstream effectors of apoptosis in a large number of cell-lines, including CHO cells. We were interested in determining whether our CHO cell line expressed some of these proteins in response to stresses resulting from normal cultivation conditions. Cell lysate taken from an apoptotic culture of CHO cells was assayed for caspase activity by adding z-YVAD.AFC, a fluorescent substrate for caspase-l (see Materials and Methods for more details of assay). Activated caspase-l in the lysate can cleave z-YVAD.AFC, releasing the fluorescent AFC residue. This event would be detected by the increase in fluorescence. Figure 4-5 indicates that the fluorescence was detected when z-YVAD.AFC was added to the lysate, indicating the presence of activated caspase-1 like caspases in the lysate. Since cysteine protease activity was detected in our cells, we attempted to delay apoptosis in our cells by adding specific caspase-inhibiting peptides to our culture. The peptide we chose was a fluoro-methyl-ketone (fink) with a sequence, benzylocarbonylVal-Ala-Asp(OMe).fluoromethyl ketone (z-VAD.fmk). This is an irreversible inhibitor ICE-like Protease Activity Detected Using YVADFlourescent Substrate control flourescence = 0 10 C MSE 8 6. 4 C 2 0 I 0 --- I I 6 24 30 Time (min) Figure 4-5: Fluorescence obtained by combining cell-lysate from apoptotic CHO cells with a fluorescent substrate z-YVAD.AFC, indicating the presence of caspases. of cysteine proteases (Enari, et al., 1996; Martin, et al., 1996; Takahashi and Earnshaw, 1996; Thornberry and Molineaux, 1995; Wang, et al., 1995; Wang, et al., 1996). The methyl group ensures that the peptide is cell membrane permeable. We added 60gM zVAD.fmk (solubilized in Dimethyl Sulfoxide (DMSO)) to batch cultures to examine whether we could delay the rapid death observed at the end of a batch culture. To the control culture for each of these experiments, we added the same volume of DMSO as was used to deliver the z-VAD.fmk. The results are shown in Figure 4-6 and Figure 4-7. z-VAD.fmk provides only a small improvement in culture viability 96 hours into the culture. Dead cells protected by z-VAD.fmk showed loss of membrane integrity, and condensed but not fragmented chromatin, while those in the control cultures showed a typical apoptotic morphology with condensed and fragmented chromatin. This indicates that z-VAD.fmk is able to inhibit, directly or indirectly, the activity of the endonucleases responsible for fragmenting genomic DNA in the apoptotic process. Figure 4-8 shows that the concentration of glucose in both cultures reaches close to zero levels by 72 hours. We suspect that this depletion of glucose from the medium was the major cause of death in the culture. The results demonstrate that the viabilities and cell densities of the two cultures are almost identical for a period of 72 hours. Death starts to occur almost 24 hours after glucose depletion was detected. We verified that z-VAD.fmk does get into the cells and prevent cleavage of substrates of caspases by monitoring the cleavage of Poly(ADP-Ribose)-Polymerase (PARP) at 96 and 120 hours. PARP is a protein involved in DNA repair and inhibition of nuclease activity, which has been shown to be cleaved by 2.5E+6 2.OE+6 1.5E+6 - -zVAD o-e-m 1.0E+6 C \ - o- 5.OE+5 TCD DMSOTCD -- zVAD VCD --DMSO VCD \ % U) 0.0E+0 I 0 20 I I 40 I 60 I I 80 100 120 Time (hour) Figure 4-6: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor of caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures of CHO cells: Total cell density (TCD) and viable cell density (VCD) as a function of time. 100 2.5E+6 2.OE+6 --80 1.5E+6 -60 _ >% -•--- 40 I\0 S--e--DMSO 1.0E+6 1C I _ 5.OE+5 a) \ 0 I- .0 1 ---- DMSOTCD zVAD viability' viability 20 0 I 0.0E+0 zVAD TCD 20 40 60 80 100 120 Time (hour) Figure 4-7: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor of caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures of CHO cells: Total cell density and viability of cultures as a function of time. Viability declines rapidly after 72 hours. caspase-3 during the apoptotic process in a number of different cell lines (Lazebnik, et al., 1994; Liu, et al., 1996; Mandal, et al., 1996). Figure 4-9 shows that there is no cleavage of PARP in the z-VAD.fmk protected cultures, while a clear cleavage of PARP is seen in the cultures not protected by z-VAD.fmk. The cleavage of PARP in the unprotected culture is further proof of the presence of caspase (caspase-3) activity in apoptotic CHO cells. The presence of intact PARP in cultures to which z-VAD.fmk was added suggests that z-VAD.fmk was able to effectively block caspase activity in these cells. Thus the meager protection in cultures supplemented with z-VAD.fmk cannot be attributed to the inability of z-VAD.fmk to penetrate the cells. In addition, since the control and z- VAD.fmk protected cultures behave almost identically till the cultures start to die, death in the culture with z-VAD.fmk is not due to any toxic side-effect of the peptide. Higher concentrations of z-VAD.fmk (upto 120 gM) did not yield improved culture viabilities. Concentrations higher than 120 jgM could not be used since the DMSO used to solubilize the z-VAD.fmk was toxic to the cells at those levels. 4.4 Bcl-2 extends viabilityin batch cultures of CHO cells A mixed culture of CHO cells transfected with the human bcl-2 antiapoptotic gene (see Kroemer, 1997; Reed, 1997 for a recent review) was used to examine the effect of bcl-2 expression on cell death at the end of a batch culture (see Materials and Methods section for more details on how the mixed culture was obtained). The performance of the bcl-2 expressing culture was compared to that of the pcd-CHO. All cultures were inoculated 100 14 12 E 10 0 C 8 pcd + zVAD - - pcd + DMSO S--o- 6 - E .: 4 0 0 20 40 60 80 100 120 Time (hour) Figure 4-8: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor of caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures of CHO cells: Medium glucose concentration as a function of time. Medium glucose concentration dropped to almost zero after 72 hours in both cultures. 101 cell line pcd + zVAD pcd + DMSO 32 15 viability (%) pcd + DMSO 0 pcd + zVAD 3 uncleaved PARP (113 kDa) hhi cleaved PARP (84 kDa) Figure 4-9: Comparison of the protective effect of z-VAD.fmk (a peptide inhibitor of caspases) on protected (pcd + zVAD) and control (pcd + DMSO) batch cultures of CHO cells: Poly-ADP-Ribose Polymerase (PARP) cleavage is evident in the control cultures but not the z-VAD.fmk protected cultures. 102 with 4x105 cells/mL, and the viabilities and cell densities were measured till the viabilities of both cultures were below 50% (as measured by the AO/EB assay). The results are shown in Figure 4-10, Figure 4-11, Figure 4-12 and Figure 4-13. The fluctuation in total cell densities at the end of the culture results from a tendency of cells to clump and stick to the walls of the flask. The bcl-2 culture is more than 40% viable 72 hours after the control culture is completely dead. As can be seen from Figure 4-10, bcl-2 expression does not affect the growth rate of the culture. Figure 4-12 indicates that the glucose in both cultures had been completely depleted by 72 hours. We suspect that this glucose depletion is the main stimulus for death in the control culture. We checked whether bcl-2 expression is able to block cleavage of caspase-3 substrate PARP. Previous reports have suggested that bcl-2 lies upstream of caspase-3 in the apoptotic pathway, hence if it is effective in preventing apoptosis, it should be able to block caspase-3 activity and hence block cleavage of caspase-3 substrates such as PARP. Bcl-2 prevents activation of downstream caspases such as caspase-3, by preventing the release of cytochrome c from the mitochondrial matrix (Kluck, et al., 1997). Figure 4-13 shows that this is indeed the case. Even after 168 hours the PARP in the bcl-2 protected cultures remains essentially uncleaved, suggesting that bcl-2 prevents caspase-3 activation in CHO cells. We suspect that the small band of cleaved PARP which appears in the bcl-2 lane, may be caused by apoptosis in the mixed population of cells which do not express bcl-2. The protective effect of bcl-2 is similar to that observed by other authors using other cell lines (Itoh, et al., 1995; Simpson, et al., 1997; Singh, et 103 al., 1997; Suzuki, et al., 1997; Terada, et al., 1997). Some hypotheses explaining the surprisingly different results obtained by preventing caspase activation by bcl-2 and zVAD.fmk are presented in the discussion and conclusions section of this chapter. 4.5 Bcl-2 protects better than caspase inhibitingpeptides in response to growth and survival factor withdrawal Growth and survival factor withdrawal is known to be a potent inducer of apoptosis in mammalian cells, especially in the absence of serum (Bottenstein, et al., 1979; Murakami, 1989; Zhou and Hu, 1995). Insulin and transferrin were used as specific growth and survival factors in our serum-free medium. We studied the protective effect of bcl-2 and z-VAD.fmk separately and together in protecting CHO cells in batch culture from the effects of insulin and transferrin withdrawal. The hypothesis underlying these sets of experiments was that if pathways which were bcl-2-dependent and caspase-independent and vice versa, existed, then using both bcl-2 expression and z-VAD.fmk addition would provide better protection than either alone. A volume of DMSO equal to that used to deliver the z-VAD.fmk to the other cultures was added to the control (pcd-CHO) culture and the bcl-2-CHO culture. No insulin or transferrin was added to the medium from the start of the culture. Cells being grown in medium containing insulin and transferrin were spun down and washed once with phosphate buffered saline (PBS), before transferring to medium without insulin and transferrin. All cultures were inoculated at 4x10 5 cells/mL and the experiments were continued till the viability of all the cultures had dropped below 50%. 104 2.OE+6 - m ,-E 1.5E+6 0 c= U - -- 0 O 1.OE+6 ---pcd TCD -o---bcl2 VCD - .-- pcd VCD I C) I 0l I I 5.OE+5 0.0E+0 _: i 50 S I I _ II Time bcl2 TCD _ _ _ 100 a11d .1 I 150 i 1 i I 200 (hours) Figure 4-10: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Total cell density (TCD) and viable cell density (VCD) as a function of time. 105 2.OE+6 100 .. I E , 80 1.5E+6 - o 60 o 1.OE+6 0, U) 0 Q, -40 Ic 0 5.OE+5 - -20 i 0.OE+0 () m | 1 i mii I i I I 50 I pi I. 4 i II 1iW I- 100 I 1 150 I 0 20 0 bcl2 TCD --u--pcd TCD -- o--bcl2 viability --- -- pcd viability Time (hour) Figure 4-11: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Total cell density (TCD) and viability of cultures as a function of time. Viability of the control, but not the bcl-2 protected cell line, declines rapidly after 72 hours. 106 M 10 E 8 O -b- bcl 2: --- pcd 0 0 50 100 Time 150 200 (hour) Figure 4-12: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Medium glucose concentration as a function of time. Medium glucose concentration dropped to almost zero after 72 hours in both cultures. 107 pcd cell line bcl-2 MC time (hour) 96 168 viability 6% 49% uncleaved U PARP (113 kDa) i cleaved PARP (84 kDa) Figure 4-13: Protective effect of bcl-2 expression as compared to a control (pcd) in batch culture of CHO cells: Poly-ADP-Ribose Polymerase (PARP) cleavage is clearly evident in the control cultures but almost all the PARP is uncleaved in the bcl-2 protected cultures. 108 The results of the experiment are shown in Figure 4-14, Figure 4-15 and Figure 416. The potency of insulin and transferrin withdrawal in initiating apoptosis can be judged from the rapid death in the control culture (pcd + DMSO). It is seen that bcl-2 and z-VAD.fmk acting together do indeed provide better protection than either agent alone, confirming our hypothesis presented above. Also, bcl-2 expression provides much better protection than adding z-VAD.fmk to the culture. The sudden decrease in viability for the pcd + z-VAD.fmk culture at 72 hours is probably due to the exhaustion of glucose in the culture (Figure 4-16). Dead cells in the z-VAD.fmk protected cultures showed loss of membrane integrity, and condensed but not fragmented chromatin material. The cells in the control culture showed both loss of membrane integrity coupled with condensed and fragmented chromatin. Again, this indicates that z-VAD.fmk is able to inhibit, directly or indirectly, the activity of the endonucleases responsible for fragmenting genomic DNA in the apoptotic process. The better growth rates of z-VAD.fmk supplemented cultures (Figure 4-14) cannot be explained at this time. 4.6 Protective effect of bcl-2 is enhanced in clonal batch cultures The glucose starvation and insulin-transferrin (growth/survival factor) deprivation experiments performed with bcl-2 mixed cultures were repeated with a clonal population of bcl-2 cells (see Materials and Methods chapter for more details on clone selection procedures). As indicated before, the glucose starvation results naturally at the end of a batch culture when the cells have consumed all the glucose originally present in the medium. In growth factor deprivation experiments, no insulin or transferrin was 109 1.6E+6 E ( 1.2E+6 > 8.-E+5 .. - _ -- ,--pcd ---... ---- -4.E+5 --- bcl-2 + DMSO + DMSO bcl-2 + z-VAD pcd + z-VAD 0.0E+0 0 24 48 Time 72 96 120 144 (hour) Figure 4-14: Comparison of the protective effects of bcl-2 expression and z-VAD.fmk, together and separately, in batch cultures of CHO cells that have been deprived of insulin and transferrin. To cultures which did not contain z-VAD.fmk we added a volume of Dimethyl Sulfoxide (DMSO) equal to the volume that was used to deliver z-VAD.fmk to the other cultures (also see text). This graph plots total cell density (TCD) as a function of time. 110 100 80 60 . - 60t - +- bcl-2 + DMSO 40 - e- - pcd + DMSO .----bcl-2 + z-VAD' S 20 20 -- u pcd + z-VAD 0 0 24 48 Time 72 96 120 144 (hour) Figure 4-15: Comparison of the protective effects of bcl-2 expression and z-VAD.fmk, together and separately, in batch cultures of CHO cells which have been deprived of insulin and transferrin. To cultures which did not contain z-VAD.fmk we added a volume of Dimethyl Sulfoxide (DMSO) equal to the volume that was used to deliver z-VAD.fmk to the other cultures (also see text). This graph plots viability of cultures as a function of time. 111 5.0 4.0 " E i 3.0 %I .- -o---bcl2 + DMSO - --- S2.0 r ---- - -- pcd + DMSO bcI2 b c + zVAD pcd + zVAD 1.0 0.0 24 I 48 I 72 Time I 96 120 (hour) Figure 4-16: Comparison of the protective effects of bcl-2 expression and z-VAD.fmk, together and separately, in batch cultures of CHO cells which have been deprived of insulin and transferrin. To cultures which did not contain z-VAD.fmk we added a volume of Dimethyl Sulfoxide (DMSO) equal to the volume that was used to deliver z-VAD.fmk to the other cultures (also see text). Medium glucose concentration is plotted as a function of time. 112 added to the medium right from the start. The results from the glucose starvation experiment are indicated in Figure 4-17. It can be seen that bcl-2 expressing clones are able to survive more than a 100 hours longer than the control cell line (pcd). The protective effect of a clonal population is much larger than that observed in a mixed culture of bcl-2 CHOs where the additional protective effect over the control was on the order of 48 hours. The much stronger protective effect of bcl-2 in a clonal population is evident in the insulin-transferrin withdrawal experiments as well. Bcl-2 protected cultures were able to maintain viability around 90% 24 hours after the viability of the control culture had fallen below 15% (Figure 4-18). As expected, the cells did not grow very well in the absence of growth factors. The variability in cell density was due to cells sticking to the side of the flask. Again, the protective effect of bcl-2 in a clonal population is enhanced as compared to that of a mixed culture population. For comparison, the mixed culture viability 24 hours after the control culture had substantially perished was about 50%. In summary, these experiments confirm the protective effect of bcl-2 in a clonal population and indicate that the protective effect is stronger in a clonal population than in a mixed culture. 4.7 Caspase-independent death pathways which are blocked by bcl-2 expression appear to exist in CHO cells z-VAD.fmk has been reported to be a powerful and universal inhibitor of cysteine proteases. Yet in our cultures z-VAD.fmk failed to prevent death in response to either 113 2.50E+6 a=~zr 100 ~~ -f -* 90 t A 2.00E+6 80 70 1.50E+6 6o0 1.00E+6 50 . 40 .2 - 30 5.00E+5 -20 - 10 BB 0.OOE+0 50 100 ' 150 0 1 1 200 250 --.-- bcl2-TCD pcd-TCD bcl2-Viability 1----pcd-Viability Time (hour) Figure 4-17: Bcl-2 CHO clonal populations are able to maintain their viability for a much longer period of time as compared to the control, pcd-CHO cells in a batch culture. The main cause of death is glucose depletion at about 72 hours. The total cell density (TCD) for both cultures is approximately the same. The variation in total cell density at the end of the culture is due to cells sticking to the walls of the flask. 114 100 3.5E+5 90 3.OE+5 - 80 E 2.5E+5 - 70 60 o 2.OE+5 - 50 r 1.5E+5 - 40 a a 1.OE+5 30 a o 20 5.OE+4 O.OE+O 10 ----- bcl2-TCD pcd- TCD --- o--bcl2-Viability -- e--pcd-Viability 0 .a 20 . 40 60 80 Time (hour) Figure 4-18: Clonal populations of CHO cells expressing bcl-2 are able to maintain their viability for much longer (as compared to the control pcd-CHO cell line) in response to insulin-transferrin deprivation in batch culture. TCD refers to the total cell density of the cultures. 115 glucose starvation or growth-factor deprivation. To check whether z-VAD.fmk was able to block caspases effectively, we checked for cleavage of substrates of at least two different caspases in lysates from apoptotic CHO cells. Substrate cleavage had to be used as a proxy for caspase activity, since antibodies which could detect native caspases in CHO cells could not be found. The first substrate, which has already been discussed above, is PARP which is cleaved by caspase-3 (CPP32/Yama). The second substrate that we tested for was lamin A, which is cleaved by caspase-6, but not by caspase-3 (Orth, et al., 1996). By selecting these different substrates we could be reasonably sure that zVAD.fmk was indeed universally blocking caspases in CHO cells. As previously shown in Figure 4-9 and Figure 4-13, z-VAD.fmk and bcl-2 were able to block PARP cleavage in apoptotic CHO cells. In contrast, cells not protected by either z-VAD.fmk or bcl-2 suffered complete cleavage of PARP. Figure 4-19 shows the results from the lamin A cleavage study. Cells were exposed to insulin and transferrin withdrawal as a stimulus for apoptosis for this study. The samples shown were taken on day 3 of the culture. Extensive apoptosis was observed in the control pcd-CHO culture to which no z-VAD.fmk was added. The pcd-CHO culture to which 60 gM z-VAD.fmk was added also experienced extensive death. Dead cells lost their membrane integrity and had condensed but not fragmented chromatin. The bcl-2 protected cell-lines remained viable. As shown in Figure 4-19, z-VAD.fmk addition (lane 3) is able to completely block the lamin A cleavage observed in the control (lane 1). Therefore, z-VAD.fmk effectively blocks caspase-6 activity (in addition to blocking caspase-3 activity), but is not able to protect cells from death. No lamin cleavage is observed in bcl-2 protected cell 116 Sample p-z b-z p+z b+z Viability (%) 10 84 11 86 lamin A --- .il 70 kDa Figure 4-19: Cleavage of caspase-6 substrate lamin A in CHO cells, in response to insulin and transferrin deprivation. 'p' refers to the control pcd-CHO cell line. 'b' refers to the bcl-2 expressing CHO cell line. '+z' and '-z' indicate whether caspase inhibitor, zVAD.fmk was or was not added to the culture. The results indicate that addition of zVAD.fink is able to prevent cleavage of lamin A, but is not able to protect cells from death. Bcl-2 expression prevents both lamin cleavage and death. 117 lines (with or without z-VAD.fmk). The results confirm that blocking caspase activity is not sufficient to block death in CHO cells. Secondly, since bcl-2 is able to block death not blocked by addition of caspase inhibitors, bcl-2-dependent but caspase-independent apoptotic pathways exist in CHO cells. 4.8 Discussionand Conclusions There have been conflicting reports in literature as to the mode of death in CHO cells (Moore, et al., 1995; Moore, et al., 1997; Singh, et al., 1994; Singh, et al., 1997). In this chapter we have shown that CHO cells grown in batch culture die primarily via apoptosis, a form of genetically encoded death. In batch culture, this induction of apoptosis followed the depletion of glucose in the medium. In addition, we have also demonstrated, by means of protein synthesis inhibition, that de novo protein synthesis is not necessary for CHO cells to undergo apoptosis. This implies that death inducing and death suppressing proteins are always present within the CHO cell, and that death proteins probably degrade less rapidly than survival proteins (Mercille and Massie, 1994; Mosser and Massie, 1994). Hence, when protein synthesis is inhibited, death proteins are able to exert their effect once the survival proteins have been removed. This hypothesis also implies that in a surviving CHO cell, survival proteins have to be continuously synthesized to enable the cell to escape death. This scenario lends support to our hypothesis that overexpression of the correct survival protein or inhibition of the death proteins in the CHO cell may be able to delay the onset of apoptosis in response to certain stress stimuli. 118 ICE-like cysteine protease (caspase) activity was detected in CHO cells. Hence, our first effort was to determine if countering the effect of cysteine proteases within the cell would be able to extend the life of a batch culture. Experiments were carried out using z-VAD.fmk, a cysteine protease inhibiting peptide. This peptide, though not the most potent inhibitor, was chosen since it was known to have good cell membrane permeability. Our experiments indicate that inhibition of caspases is able to marginally delay the onset of death in batch culture of CHO cells. When apoptotic stimuli such as withdrawal of insulin and transferrin were introduced, cell cultures with z-VAD.fmk initially exhibited a higher viability, with respect to the control. This protective effect was observed right from the start of the culture. However, viability rapidly declined as medium glucose concentrations fell to near zero, suggesting limits to the protective effect of z-VAD.fmk. Other experiments were conducted to determine whether overexpression of bcl-2, a well-studied death-suppressing gene, in CHO cells would be able to extend the life of batch cultures. Bcl-2 has been shown to have a protective effect under cell-culture conditions in several other cell lines (Itoh, et al., 1995; Mastrangelo and Betenbaugh, 1998; Mastrangelo, et al., 1996; Simpson, et al., 1997; Singh, et al., 1996; Singh, et al., 1997; Suzuki, et al., 1997; Terada, et al., 1997). In the case of both normal and insulin and transferrin deprived CHO batch cultures, bcl-2 was able to delay the onset of death substantially. It is interesting to note that both bcl-2 and z-VAD.fmk only start to play a role once apoptotic stimuli are experienced. The growth and viability of the control culture is identical to that of the protected culture before the appearance of apoptotic 119 stimuli. In addition, our experiments clearly indicate that bcl-2 overexpression is a much more effective way of preventing death in CHO cultures than using caspase inhibiting peptides such as z-VAD.fmk. The fact bcl-2 overexpression is a better way of extending the life of CHO cultures than using caspase inhibitors is interesting since the point of action of bcl-2 appears to be upstream of that of the caspases in the apoptotic pathway (Enari, et al., 1996; Wang, et al., 1996) (Figure 4-20). This upstream location would suggest that the protective effect of bcl-2 would at best equal that of caspase inhibitors. We were interested in determining if having both bcl-2 and z-VAD.fmk together provided any additional protection over just having z-VAD.fmk alone in the culture. If certain caspase independent pathways exist, and bcl-2 is also able to block these pathways, then having both bcl-2 and caspase inhibitors should improve culture viability. Figure 4-15 shows that bcl-2 and z-VAD.fmk acting together do indeed provide better protection than either agent alone. This improved survival could not have been due to z-VAD.fmk protection of the non-bcl-2 expressing cells in the mixed culture, since we know that z-VAD.fmk alone is unable to protect cells not expressing bcl-2, once glucose is exhausted (Figure 4-15 and Figure 4-16). Hence, bcl-2-independent and caspase-dependent pathways appear to exist. Our results are bolstered by reports in literature about two separate apoptotic pathway factors that bcl-2 is able to block - cytochrome c (Kluck, et al., 1997; Liu, et al., 1996; Yang, et al., 1997) and Apoptotic Inducing Factor (AIF) (Kroemer, et al., 1997; Susin, et al., 1996). It is suggested that both these factors reside between the inner and outer membranes of the mitochondria. They are released in response to apoptotic 120 S1 S2 bcl-2 I- p35 I death I Figure 4-20: Conventional pathway of death in the mammalian cells, focusing on the bcl-2 and caspase (C) nodes. S and S2 are the same or different external stimuli for apoptosis. 121 stimuli, whereupon they initiate subsequent steps in the apoptotic pathway. Bcl-2, by virtue of its sub-cellular localization to the mitochondrial membrane, is able to block apoptotic death by blocking the release of these agents from the mitochondria. Cytochrome c has been shown to activate cysteine proteases. The pathway by which AIF causes apoptosis can be caspase-independent, since AIF has been shown to directly induce nuclear degradation (Bossy-Wetzel, et al., 1998). In addition, bcl-2 can also protect against death caused by Bax activation, which can kill cells in a caspaseindependent manner (Xiang, et al., 1996). Lastly, bcl-2 can also prevent death by preventing reactive oxygen species (ROS) release and loss of mitochondrial potential (which inhibits oxidative phosphorylation). Given this information and the fact that bcl-2 has been shown to act upstream of certain caspases in the apoptotic pathway, we have attempted to come up with an death pathway (including both apoptotic and necrotic death) in CHO cells which focuses on bcl-2 and caspases (Figure 4-21). Direct activation of downstream caspases, such as caspase-3, by Fas or TNFR activated upstream caspases (caspase-8 and caspase-2) or cytotoxic lymphocyte enzyme, granzyme B, are examples of bcl-2-independent death pathways. Bax mediated death or death due to loss of mitochondrial potential and release of ROSs represent caspase-independent death. The fact that cells still die despite the presence of both z-VAD.fmk and bcl-2, can be explained by one or both of the following hypotheses. First, as the level of insults gets progressively greater, it is possible that the level of bcl-2 and/or z-VAD.fmk in the cell is no longer sufficient to counter the levels of the death protagonist elements of the 122 Direct activators of downstream caspases, e.g. Granzyme B Receptor-Adapter-Protease activation of upstream caspases, e.g. Fas-FADDcaspase-8 Apoptotic Stimuli I bcl-2 nproteins Death Figure 4-21: Suggested death (including both apoptosis and necrosis) pathway in a CHO cell. The pathway focuses on interaction between caspases and bcl-2 family proteins. FADD, AIF and ROS refer to Fas associated death domain, apoptosis inducing factor and reactive oxygen species, respectively. See text for more details. 123 apoptotic pathway. Second, pathways that are both bcl-2- and caspase-independent may exist. These pathways may directly trigger events at an as yet unidentified node in the death pathway, which is downstream of the mitochondrial and caspase checkpoints. At this point we have no evidence to allow us to choose between these two hypotheses. The drawback with studies carried out in the batch mode is that cultures invariably are exposed to a nutrient limitation, which ultimately proves to be too strong a stimulus to counter using either gene expression or peptide induced inhibition of caspases. This observation corroborates batch culture results presented by other authors working with several cell lines (Itoh, et al., 1995; Simpson, et al., 1997; Singh, et al., 1996; Singh, et al., 1997; Suzuki, et al., 1997). Furthermore, this scenario of nutrient limitation is unlikely to occur industrially. Fed-batch studies, described in a later chapter, in which nutrient limitation ceases to be a primary cause of death, will provide an interesting environment in which to further test the protective effect of death suppressing proteins such as bcl-2. In addition, these studies will give us an opportunity to study the effect of bcl-2 expression on total product titers in an environment where nutrient availability is not a limiting factor. 124 5. Correlating Viability and Replication Competence One of the best measures of viability for immortalized mammalian cells is their ability to undergo mitosis when placed in a favorable environment. However, measuring mitosis is time consuming and other assays have been developed to provide a more rapid assessment of viability. One type of assay uses the ability of a viable cell, with its cellular membrane intact, to exclude a vital dye as a measure of viability. Dyes used for these types of assays include trypan blue (TB) (Patterson, 1979) and propidium iodide (PI) (Tanke, et al., 1982; Trost and Lemasters, 1994; Vollenweider and Groscurth, 1992). The underlying assumption in these types of assays is that the commitment of a cell to death and loss of membrane integrity are virtually simultaneous events in the life of a cell. Recently, it has been shown that a large number of mammalian cells in culture undergo death via programmed cell death or apoptosis (Franek and Chladkova-Sramkova, 1995; Goswami, et al., 1998; Itoh, et al., 1995; Mastrangelo and Betenbaugh, 1998; Mastrangelo, et al., 1996; Mercille and Massie, 1994; Moore, et al., 1995; Moore, et al., 1997; Perreault and Lemieux, 1994; Singh, et al., 1996; Singh, et al., 1997; Suzuki, et al., 1997; Terada, et al., 1997; Vomastek and Franek, 1993). However, the loss of cellular membrane integrity is a very late event in the process of apoptotic death, and cells scored as viable by the TB assay may have already committed to death. The condensation and fragmentation of chromatin material has been found to be an event upstream of the loss of membrane integrity, in a cell's commitment to death by apoptosis (Martin and Cotter, 125 1994; Wyllie, et al., 1980). The Acridine Orange/ Ethidium Bromide assay, that is increasingly being used to detect apoptosis in cells (McGahon, et al., 1995; Mercille and Massie, 1994), overcomes the problems of late detection associated with the membrane integrity based viability assays, by using both DNA integrity and membrane integrity to monitor the viability of cells. Our objective was to correlate the viability measurements provided by the aforementioned types of assays to the ability of cells in culture to undergo mitosis. Secondly, we were also interested in determining if early apoptotic cells (cells with intact membrane integrity but fragmented chromatin material) could undergo mitosis. These issues were particularly interesting in the light of the experiments described in the previous chapter where bcl-2 expression was shown to extend viability of CHO cells in batch culture, as measured by the AO/EB assay. We were interested in determining whether this observed extension in 'AO/EB' viability corresponded to an extension in replication competence of the cells. These issues were investigated by following the fate of individual CHO cells. 5.1 Experimental Approach Serum-free batch cultures of four Chinese Hamster Ovary (CHO) cell lines were grown in suspension. For the first data-set, one of these cell lines was a control CHO cell line and the other was a mixed culture of bcl2-CHO cells. Each culture was seeded at 5x10 5 cells/mL and reached a maximum cell density of about 2x10 6 cells/mL on day three. Cultures reached glucose exhaustion on day three (data not shown) and this is most likely 126 the cause of death of the control culture. For the second data set, clonal populations of pcd-CHO and bcl2-CHO were used. These cultures were inoculated at 4x10 5 cells/mL and reached a maximum cell density of about 2x10 6 cells/mL. The control culture started to die on day 4. No additional glucose or any other carbon source was fed to any of the cultures. Cellular viability of a sample taken from each culture was evaluated each day (in duplicate) using a hemacytometer via both the AO/EB and the TB techniques. In each of the assays at least 200 cells were counted for each sample. For replication competence assays, wells of a 384 well plate (NUNC, catalog # 242 765) were filled with 100 gL of 50% conditioned medium and 50% fresh medium. Conditioned medium is medium taken from a culture that is in log phase growth and where the total cell density of the culture is less than 1x10 6 cells/mL. Using a Fluorescence Activated Cell Sorter (FACS; Becton Dickinson, FACStarPlus), the 384 well plates were then seeded at one cell per well with cells from the cultures. Only those cells that excluded propidium iodide, i.e., cells with intact membrane integrity, were used to seed the wells. This was done using the FACS to gate only cells that were not stained by propidium iodide. The 384 well plate was divided into two halves of 192 wells each. One half of these wells were seeded with cells from the pcd-CHO cultures and the other half with cells from the bcl2-CHO cultures. After a week, a light microscope was used to score the number of wells in which the cells had undergone mitosis. The percentage of wells in which cells grew, was defined as the replication competence of that population of cells. Since cells do not grow very well when they are isolated from other cells, for each data-set the percentage of wells in which cells grew in a plate seeded with healthy cells 127 from a log-phase growth culture was used as a base to calculate normalized replication competence (norm. repl. comp.). To summarize: replication competence is defined as the percentage of wells in which cells replicated, and, normalized replication competence is defined as replication competence divided by the percentage of wells in which healthy cells replicated. 5.2 The abilityof cells to replicate correlates much better with AO/EB assay results than with TB assay results The viability of cells seeded according to the trypan blue assay (TB viability) was 100%, since only cells that were able to exclude vital dye were used to seed wells. The viability of the seeded cells according to the AO/EB assay, defined as acridine orange (AO) viability, is the percentage of cells scored viable (%V) by this assay divided by the sum of the percentages of viable and early apoptotic cells (%EA) (Figure 5-1). Hence, AO viability = %V / (%V + %EA) TB viability = 100% The results of the experiment are shown in Table 5-I. Normalized replication competence was correlated with AO viability and TB viability. The corresponding plot is shown in Figure 5-2. As can be seen from Figure 5-2, normalized replication competence correlates very well with AO viability (R2 = 0.9364 , t-statistic = 25.18, pvalue = 4.46x10"11). Normalized replication competence does not correlate at all with TB viability (R2 = 0; graph not shown). This clearly indicates that the AO/EB assay is a 128 AO EB Viable Late Apoptotic EB AO Early AOE Necrotic EB Figure 5-1: A pictorial representation of the four kinds of cells which can be distinguished by the acridine orange/ ethidium bromide (AO/EB) assay. Cells which have not lost their membrane integrity are penetrated by acridine orange only, appear green under the microscope, and are lightly shaded in the figure. Cells which have lost their membrane integrity also incorporate ethidium bromide and appear orange under the microscope. These cells are hatched in the figure. The dark spots in the early and late apoptotic cells represent condensed and fragmented chromatin material. 129 Table 5-I: Normalized replication competence (norm. repl. comp.), acridine orange viability (AO viab.) and trypan blue viability (TB viab.) data data set cell type day repl. comp.(%) norm. repl. AO viab. (%) comp. (%) TB viab. (%) 1 pcd 3 38.69 100.00 93.75 100 1 pcd 4 1.19 3.08 10.59 100 1 pcd 5 0 0.00 0 100 1 bcl-2 3 36.31 93.85 99.22 100 1 bcl-2 4 27.98 72.31 92.30 100 1 bcl-2 5 19.64 50.77 74.50 100 2 pcd-clone 2 57.22 86.55 99.46 100 2 pcd-clone 3 50.00 75.63 98.02 100 2 pcd-clone 5 0.00 0.00 0.00 100 2 bcl-2-clone 2 66.11 100.00 100.00 100 2 bcl-2-clone 3 60.00 90.76 99.66 100 2 bcl-2-clone 5 50.00 75.63 99.28 100 130 100% o - R 2 =0.94 80% p value = 4.5e-11 cc = 0_ cc Q 20% 0 o 20% E 50% 0% 100% acridine orange viability Figure 5-2: Acridine orange (AO) viability is a good indicator of the ability of a cell to replicate. AO viability is defined as the percentage of viable cells in the population of cells with intact membrane integrity. Normalized replication competence is a normalized measurement of the number of wells in which the cells were able to replicate. 131 better method than the TB assay to determine the ability of CHO cells in a serum-free environment to undergo mitosis. 5.3 Early apoptotic cells lose the abilityto replicate Early apoptotic cells are defined as cells that have condensed or fragmented chromatin material, but which have not lost their membrane integrity. However, it is unclear whether these cells should be classified as viable or dead. From our data, we conclude that early apoptotic cells may be considered dead if the ability to replicate is considered to be a true measure of viability. Table 5-I shows that the pcd-CHO cells from days four and five in data set 1 and day five in data set 2 are mostly in the early apoptotic state and that most of these cells fail to replicate. In fact, on day five all the pcd-CHO cells are in the early apoptotic state (both data sets) and none of the cells in these wells grew, providing strong evidence that early apoptotic cells lose their ability to replicate. 5.4 Bcl-2 is able to prolong the replicationcompetence of cells in culture Previous experiments have indicated that expression of the bcl-2 gene in CHO cells is able to delay apoptosis brought about by glucose limitation in batch culture (Goswami, et al., 1998). Our data proves that these cells, scored as viable the AO/EB assay, also retain the ability to undergo mitosis. As indicated above, both batch cultures ran out of glucose on day three. This glucose limitation is the main cause of the apoptosis observed in the control pcd-CHO cell line. As seen from Table 5-I, the control pcd-CHO cells 132 completely lose their replication competence by day 5 (in both data sets). In contrast, the bcl-2 CHO cells retain their replication competence well into day 5. The clonal population (data set 2) performs even better than the mixed culture population (data set 1), perhaps because a larger fraction of the population expresses the bcl-2 protein. Table 5-I indicates that the cells expressing bcl-2 retain their ability to replicate much longer than cells transfected with the control vector (pcd). Thus transfection with the bcl-2 gene enables cells to extend the period of time during which they retain the ability to undergo mitosis under conditions of stress. More importantly, the viable phenotype that we observe using the AO/EB assay is not merely an artifact related to the assay. 5.5 Discussionand Conclusions From Table 5-I, it is evident that normalized replication competence drops off faster than AO viability. This suggests that some of the cells that are scored viable by the AO/EB assay, are already committed to death by apoptosis. This is entirely plausible since we know that the chromatin condensation and fragmentation that is used as an indicator of death in the AO/EB assay is not the earliest point in the cell's commitment to apoptotic death. For instance, cysteine proteases are activated before chromatin condensation and degradation can begin (Lazebnik, et al., 1994; Lazebnik, et al., 1995; Oberhammer, et al., 1994; Orth, et al., 1996). Hence, even the AO/EB assay, or any other assay that bases its measurement on a later manifestation of apoptosis, is not 100% accurate in determining the viability of cells. More accurate assays would have to detect events further upstream 133 in the apoptotic pathway. However, given that apoptosis is a very rapid process (often taking only a few hours to complete), these assays must be rapid if they are to provide any benefit over the AO/EB assay. In summary, the AO/EB assay is a rapid assay, that is able to assess replication competence of a cell much more accurately than the currently used TB assay. Though we have compared two specific assays in this chapter, the techniques and results used here could easily be extended to other assays. 134 6. Improving CHO Fed-Batch Culture Performance Using Bcl-2 Expression In Chapter 4 we demonstrated the ability of bcl-2 to protect batch cultures from death in response to nutrient starvation and growth factor-depletion. Both these stimuli, however, can easily be avoided by rational feeding strategies. We were, thus, more interested in investigating the ability of bcl-2 expression to extend the life of cultures where nutrient limitation was not a factor. Fed-batch culture allows us to study the protective effect of bcl-2 under conditions in which cells are provided with all the nutrients they require. This chapter studies the fed-batch culture of CHO cells under two conditions. The first uses cells grown in a medium maintained complete with all nutrients and with all growth factors by regular addition of a supplemental medium (Xie, et al., 1997; Xie and Wang, 1994). These cultures are referred to as normal fed-batch cultures. There are several reports in literature which suggest that slowing the growth rate of cells significantly improves the length of time for which cells remain viable and also enhances their productivity (Chung, et al., 1998; Fussenegger, et al., 1997). One way to reduce the growth rate of cells is to remove growth factors from the medium. In the case of CHO cells that are used in this lab, the main growth factor is insulin. Unfortunately, insulin is also a survival factor and cells rapidly die in the absence of insulin. However, as demonstrated in Chapter 4, bcl-2 expression was able to prevent death resulting from insulin deprivation but was not able to spur growth in cells. 135 Thus bcl-2 expression provides us with a tool to reduce growth rate without triggering death. The second kind of fed-batch cultures, referred to in this chapter as insulin-deprived fed-batch cultures, were used to investigate whether bcl-2 expression could significantly increase life and productivity of fed-batch cultures without growth factors, beyond that obtained in normal fed-batch cultures. Cell growth in normal cell cultures was observed to cease after a certain point in time, despite the fact that the cultures were being supplemented with all the necessary nutrients. The second half of this chapter examines some of the underlying phenomena that might provide a clue toward the causes of this growth-arrest. 6.1 Bcl-2 expression significantlyextends viabilityand enhances product titers in normal CHO fed-batch cultures To investigate the beneficial effect of bcl-2 in fed-batch cultures, growth and viability in bcl-2-CHO fed-batch cultures were compared with that in control (pcd-CHO) fed-batch cultures. Both the cultures were clonal populations (see Materials and Methods chapter for clone development protocols) and were seeded at approximately 5x105 cells/mL. G418 was added to both cultures at a concentration of 600 pg/mL, to maintain selective pressure throughout the culture process. The initial medium used was modified RPMI with 3 mM glucose and 0.5 mM L-glutamine (see Materials and Methods section for complete medium composition). The initial medium glucose and glutamine were reduced to minimize production of toxic metabolites lactate and ammonia, respectively (Xie, et al., 136 1997; Xie and Wang, 1994a; Xie and Wang, 1994b). The culture was fed approximately at 12 hour intervals with a calculated volume of supplemental medium (Xie, et al., 1997). The volume of supplemental medium added depended on the cell density of the culture and the expected growth rate of the cells. Cultures were stopped when the viability dropped below 50%. Below this viability, proteases released by lysis of dead cells could seriously affect protein quality. As can be seen from Figure 6-1, the total cell density reached by both cell types was almost the same (about 3.5x10 6 cells/mL). In addition, the growth rates for both cultures were similar throughout the life of the culture. This confirms that bcl-2 expression has no effect on the growth rate of CHO cells in fed-batch culture. A comparison of the viabilities and viable cell densities (Figure 6-1 and Figure 6-2), however, clearly shows the strong protective effect of bcl-2. The bcl-2 expressing cells reach a higher viable cell density and are able to maintain a viability of greater than 50% almost 72 hours longer than the control cultures. The protective effect of bcl-2 starts to become evident after about 72 hours in culture, at which point the control culture begins to die. The stimulus for death in either culture is not yet clear. The dissolved oxygen (D.O.) in the both cultures remained above 40% at all times, which is not considered to be an oxygen limiting condition. However, it is possible that a 40% D.O. is sufficient to trigger apoptosis in unprotected cells. It may also be possible that certain metabolites produced by the cells during the culture have an adverse effect on the viability and growth of cells. In either case, bcl-2 is able to protect against the adverse stimuli and extend viability for almost 72 hours. 137 4.0E+6 3.5E+6 - 3.0E+6 4' 2.5E+6 4' . ---bcl2-TCD ----- pcd-TCD -- 4b -bcl2-VCD S-- -- pcd-VCD 2.OE+6 CO 4' 1.5E+6 4' 4' C. 1.OE+6 5.OE+5 - 0.OE+0 0 50 100 150 Time (hour) 200 250 Figure 6-1: Total (TCD) and viable cell densities (VCD) of bcl-2 expressing and control (pcd) cell lines in a normal fed-batch culture seeded at 5x10 5 cells/mL. 138 4.0E+6 - 100 I1- 9E1 ~ 3.5E+6 - ~ -q T - q \\ S90 -80 3.OE+6 -70 tI 2.5E+6 - -60 1H -40 1.5E+6 - -30 bcl2-TCD S---pcd-TCD - -e- - bcl2-viability -- e--- -pcd-viability -- -50 2.OE+6 - CU 1.0E+6 -20 5.OE+5 - -10 0.0E+0 - - 0 0 50 150 100 Time (hour) 200 250 Figure 6-2: Total cell density (TCD) and viability of bcl-2 expressing and control (pcd) cell lines in a normal fed-batch culture seeded at 5x10 5 cells/mL. 139 The benefit provided by bcl-2 expression can be quantified by comparing the yinterferon produced in the bcl-2 protected cultures to that produced in the control cultures. However, since the y-interferon gene in our CHO cell is controlled by a viral SV40 based S-phase promoter, protein production is growth related and slows down after the cells have stopped growing (Banik, et al., 1996; Fussenegger, et al., 1997; Gu, et al., 1993; Mariani, et al., 1981; Subler, et al., 1992, also see cell cycle studies below). In addition, expression of genes such as p53 (see below) is known to inhibit activity of viral promoters such as SV40 (Subler, et al., 1992). Since most of the protective effect of bcl-2 manifests itself only after this point of cessation of growth, using y-interferon titers as a measure of additional benefit provided by bcl-2 expression would tend to underestimate this protective effect. An alternative way to quantify the protection provided by bcl-2 would be to compare the integrated viable cell densities (IVCD) of the bcl-2 and control cultures over the life of the cultures. The IVCDs calculated represent the sum of the average viable cell densities recorded over the life of the culture, till the culture viability decreased to below 50%. The advantage of using IVCDs to compare performance is that they dissociate the protective effect of bcl-2 from any growth related effects of the promoter used to express the desired protein. If a non-growth associated promoter were used, the final titer of the cultures could easily be calculated by multiplying the IVCD with the specific protein productivity of the cell. In that case of course, the percentage improvements calculated based on IVCD and protein titers would be the same. 140 The final difference in the IVCDs of the bcl-2 protected and control cultures is about 81% of the final IVCD of the control culture. Bcl-2 expression is able to improve final y-interferon titers by about 76% (see Figure 6-3). A comparison of results is shown in Table 6-I. Hence, bcl-2 expression is able to substantially improve fed-batch culture titers by extending culture viabilities and maintaining cells in a productive phase. 6.2 Bcl-2 expression allows insulin-deprivedfed-batch cultures to survive longer than normal fed-batch cultures As described above, the purpose of the insulin-deprived fed-batch cultures was to study whether bcl-2 expression could suppress death in the absence of growth factors in a fedbatch culture mode. Slower growth has been reported to extend viability and enhance productivity of cells. Hence, this set of experiments was performed to investigate whether bcl-2 expression could allow insulin-deprived cultures to survive even longer (and thus produce even higher titers of y-interferon) than normal fed-batch cultures. Two different initial cell densities were investigated in our study. The first set of experiments compared the performance of a bcl-2 transfected and control pcd-culture, when the cultures were seeded at 5x10 5 cells/mL (the same initial cell density as the normal fed-batch cultures). Of course, as expected the performance of the bcl-2 expressing culture was several fold better than that of the control culture, in terms of both IVCD and y-interferon titers (see Figure 6-4, Figure 6-5, Figure 6-6 and Table 6-II). As in batch cultures the difference in viability of the cultures was observed right from the start 141 --- bcl2 --- pcd 50 100 150 200 250 Time (hour) Figure 6-3: y-interferon production with bcl-2 expressing and control (pcd) cell lines in a normal fed-batch culture seeded at 5x10 5 cells/mL. Table 6-I: A comparison of the results from a normal fed-batch using bcl-2 transfected and control (pcd) clonal cell lines. IVCD represents integrated viable cell density and is the area under the viable cell density curve (plotted against time). IVCD (cell-day/mL) y-interferon (mg/L) bcl-2 pcd % improvement due to bcl-2 expression 1.88x10 7 1.04x10 7 81% 9.78 5.55 76% 142 of the cultures, indicating the strong protective effect of bcl-2 expression. More interestingly, the bcl-2 expressing insulin-deprived fed-batch culture survived much longer (-350 hours) than the bcl-2 expressing normal fed-batch culture (-250 hours). However, as expected the culture did not grow as well due to the lack of insulin, and the IVCD and y-interferon titer of the bcl-2 culture was lower than that of the bcl-2 expressing normal fed-batch cultures (Table 6-I and Table 6-II). The y-interferon productivity, which as described above is very growth related, was particularly affected by the poor growth rate. As Figure 6-6 shows, bcl-2 cells continuously synthesized y-interferon, albeit at a slower rate, proving that bcl-2 expression was able to maintain insulin-deprived cells in a viable and productive state. Since cells did not grow very well without insulin, the IVCD of insulin-deprived cultures studied above was low despite a much longer culture life span. To examine whether higher initial cell densities in insulin-deprived cultures could lead to substantially improved IVCDs, another set of fed-batch cultures was initiated at a density of 8x10 5 cells/mL. The bcl-2 transfected culture grew slightly better than the corresponding culture seeded at a lower cell density, perhaps due to the presence of more autocrine factors which were partially able to overcome the absence of insulin. Again, the bcl-2 culture grew more and remained more viable for a much longer time than the control culture (Figure 6-7, Figure 6-8 and Table 6-II). In percentage terms the performance- improvement over the control culture was slightly better than that obtained with the lower initial cell density, although the higher initial cell density culture did not survive as 143 1.6E+6 1.4E+6 1.2E+6 1.0E+6 --- bcl2-TCD _---pcd-TCD -bcl2-VCD ---pcd-VCD --- 8.0E+5 6.0E+5 4.0E+5 2.0E+5 I 0.0E+0 0 50 100 150 200 250 300 350 Time (hour) Figure 6-4: Total (TCD) and viable cell densities (VCD) of bcl-2-protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 5x10 5 cells/mL. 144 100 1.6E+6 1.4E+6 - 90 1 A I -J 1.2E+6 80 E 70 1.0E+6 - 60 8.0E+5 50 . o . 6.OE+5 40 I 30 4.OE+5 4 .' bc2-TCD -----pcd-TCD S-- - bcl2-viability -- s- -pcd-viability 20 2.0E+5 - 10 O.OE+O I i 50 100 150 i_ 0 200 250 300 350 Time (hour) Figure 6-5: Total cell density (TCD) and viability of bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 5x10 5 cells/mL. 145 4.5 4 3.5 3 +bcl-2 -. ---- pcd 2.5 2 1.5 1 0.5 7I 0 0 0 50 50 100 150 200 250 350 300 Time (hour) Figure 6-6: y-interferon production with bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 5x1 05 cells/mL. Table 6-II: A comparison of the results from the two insulin-deprived fed-batch runs. All cultures contained clonal population of cells. Run 1 was started with an initial cell density of 5x10 5 cells/mL while Run 2 had an initial cell density of 8x10 5 cells/mL. IVCD represents integrated viable cell density and is the area under the viable cell density curve (plotted against time). Run 1 Run 2 bcl-2 pcd bcl-2 1.30x10 7 1.74x10 6 1.65x10 7 1.95x10 4.58 1.22 5.51 1.33 Run 1 Run 2 improvement due to pcd CI-Z expression IVCD (cell-day/mL) y-interferon (mg/L) 146 6 646% 746% 274% 315% long (- 300 hours) as the lower initial density one (- 350 hours). Once again, y-interferon was produced throughout the life of the bcl-2 protected culture, indicating that bcl-2 is able to maintain cells in productive state in the absence of insulin (Figure 6-9). More interestingly, the IVCD of the bcl-2 protected culture was much closer to that obtained during normal fed-batch culture (Table 6-I and Table 6-11). This clearly indicates the potential of insulin-deprived bcl-2 expressing slow growing cultures to achieve high IVCDs. Higher initial cell densities could perhaps obtain even higher IVCDs. Cells maintained in the growth arrested state could conceivably have higher productivities (with the right promoter; see Fussenegger, et al., 1997) and produce protein of more uniform quality. There are also added advantages of lower cost and less regulatory issues by not having to add costly growth factors to the culture medium. 6.3 CHO cells progressively arrest in the GO/G1 phase during fed-batch culture The growth arrest observed has interesting implications for the cell cycle in CHO cells. In particular, we were interested in knowing if there was a particular phase in which cells in a fed-batch culture of CHO cells arrested. It is known that hybridomas and other myeloid cell lines arrest in the S-phase of the cell cycle since the c-myc gene in these cell lines is deregulated (Lotem and Sachs, 1993; Miyazaki, et al., 1995; Sherr, 1994). However, cmyc has not been reported to be deregulated in CHO cells. 147 We were also 2.50E+06 --M 2.00E+06 ,_1 - E 1.50E+06 - >cv T\ s bcl2-TCD ----pcd-TCD -- *--bcl2-VCD -- o--pcd a) 0 1.00E+06 - killE O 5.00E+05 - 0.00E+00 0 50 100 150 2 00 250 300 Time (hour) Figure 6-7: Total (TCD) and viable cell densities (VCD) of bcl-2-protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 8x10 5 cells/mL. 148 100 2.5E+6 _ E -80 \ ! 2.0E+6 \ 60 = 1.5E+6 c U) 1.0E+6 O 5.OE+5 - - \ 40 L--I O.OE+O 0 . .0 a bcl2-TCD -.-pcd-TCD - 4 - - bcl2-Viability --- pcd-Viability 20 I I I I I 50 100 150 200 250 0 300 Time (hour) Figure 6-8: Total cell density (TCD) and viability of bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 8x1 05 cells/mL. 149 5 -1 E - 4 -- 0 3 .---bcl2 --- pcd 0 I 0 0 50 I 100 I 150 I 200 I 250 300 Time (hour) Figure 6-9: y-interferon production with bcl-2 protected and control (pcd) cell lines in an insulin-deprived fed-batch with an initial cell density of 8x105 cells/mL. 150 interested in studying how the distribution across the different stages of the cell cycle changed with culture time in fed-batch cultivation of CHO cells. Cell cycle analysis, using the propidium iodide technique (see Materials and Methods section for more details), was performed on CHO cells taken from various time points in a fed-batch culture. Figure 6-10 shows that cells progressively arrest in the GO/G1 phase as the culture progresses. There is no appreciable difference in cell cycle arrest between bcl-2 expressing and control cell lines. At about 60 hours a majority of the cells are still in the S-phase of the culture, suggesting cell division and growth. But the proportion of cells in the S-phase drops monotonically with culture time. By the time the cultures had stopped growing the proportion of cells in the S-phase was below 10%. In contrast, the proportion of cells in the GO/G1 phase had increased from below 40% to above 90% during the same time frame. There was no appreciable change in the fraction of cells in the G2/M phase. Arrest in the GO/G1 phase suggests the expression (or lack thereof) of specific genes involved in both apoptosis and cell cycle regulation, which could further provide insight into the death pathway of CHO cells. Expression of some of these genes is investigated in the following section. 6.4 p53 expression increases with time in CHO fed-batch culture but the expression of cyclin E does not change Since cells in CHO fed-batch cultures were observed to accumulate in the GO/Gi, we were interested in determining whether expression of certain genes could be responsible for this 151 phenomenon. Expression of the p53 gene is known to arrest cells in the GO/G1 phase in several cell lines (Chiou, et al., 1994; Elledge, 1996; King, et al., 1996; Sherr, 1994; Sherr, 1996). p53 has also been reported to mediate death in several cell lines by transactivating genes such as Bax, IGF-BP3 and those involved with the Fas-apoptotic pathway (Oren and Prives, 1996). The observed GO/G1 arrest in CHO fed-batch culture could be mediated by increased p53 expression. A western blot analysis of lysates of cells taken from various points of the fed-batch culture indicated that p53 expression did increase over the life of the culture (Figure 6-11). The expression of p53 in both bcl-2 and control pcd cells increased, indicating that increase in p53 expression was not abrogated by bcl-2 expression. The protective effect of bcl-2 in the presence of overexpressed p53 is confirmed by reports in literature (Chiou, et al., 1994; Stasser, et al., 1994). The stimulus for increased p53 expression however is still unknown. In contrast, the expression of cyclin E, a protein responsible for helping cells move out of the GO/G1 phase (Draetta, 1994; Koff, et al., 1993; Ohtsubo and Roberts, 1993; Ohtsubo, et al., 1995; Sherr, 1994), did not change over the life of the culture (Figure 6-12). This suggests that further overexpression of cyclin E is unlikely to solve the growth problem in CHO cells (also see discussion and conclusion section). 152 3.0E+6 100 90- - 2.5E+6 80 a E 70- 2.0E+6 601.5E+6 50- .> 40 20- - 5.OE+5 GO/G1 G2/M ---- VCD ~-se-VCD 1.OE+6 30- --i- O 100 0 50 100 150 - 0.OE+0 200 Time (hour) 3.0E+6 100 90 2.5E+6 80 " E 70 2.0E+6 60 = a, 0 1.5E+6 50 . C 40 1.0E+6 30 20 5.OE+5 GO/G1 -uG2/M S--VC VCD -- ) 10 0 I 0 0.OE+0 50 100 150 200 Time (hour) (b) Figure 6-10: Cell cycle trends in fed-batch cultures of clonal populations of pcd (a) and bcl-2 (b) transfected CHO cells. It can be seen that the fraction of cells in the GO/G1 phase increases monotonically with culture time, while the proportion in the S-phase drops off. VCD refers to viable cell density of the culture. The viable cell density of the cultures' stops increasing when more than 70% of cells are in the GO/G1 phase. 153 Cell line * culture age (hr.) P b p 88 88 112 112 160 b b 160 208 0 "now- p5 3 .. Figure 6-11: Increase in p53 expression in CHO fed-batch cultures coincides with cessation of an increase in viable cell density and follows the increase in GO/G1 phase population. Cells transfected with bcl-2 are able to maintain their viability for a longer period of time despite p53 expression. '*' indicates a sample from a batch culture in logphase growth, which was used as a control for p53 expression. 'p' refers to pcd-CHO cells while 'b' refers to bcl2-CHO cells. Cell line culture age (hr.) b 88 p rainbow 112 marker 112 -~ cycin E -- :Mi b 160 160 208 - **I i 4=1 W.:qwjm-. anow Figure 6-12: Cyclin E expression remains constant with culture age. The increase in GO/G1 phase population is therefore not related to cyclin E expression. '*' indicates a sample from a batch culture in log-phase growth, which was used as a control for cyclin E expression. 'p' refers to pcd-CHO cells while 'b' refers to bcl2-CHO cells. 154 6.5 Specific glucose consumption rate drops off sharply just before viable cell densitystops increasing To obtain a better understanding of the cause of the observed growth arrest, the consumption of nutrients during the various phases of a fed-batch culture was analyzed. The specific consumption rate of glucose was found to be particularly interesting. Figure 6-13 shows that the specific glucose consumption rate decreases sharply just before the viable cell density in the cultures stops increasing. Interestingly, once the viability of the cultures starts to decrease the glucose consumption rate starts to increase again. This is most probably due to the accumulation of glucose in the culture, which allows for more efficient uptake of glucose by cells through the GLUT-1 transporter, the main glucose transporter in CHO cells (Hausdorff, et al., 1996; Pessin and Bell, 1992). The transport of glucose via the GLUT-1 transporter is concentration gradient dependent (Burant, et al., 1991; Carruthers, 1990). The decrease in glucose consumption indicates that cells may be experiencing a lack of nutrients due to an inability to uptake them, even though we cannot observe any shortage macroscopically in the cellular environment. Possible causes for this inability to uptake glucose are investigated in the next chapter. It is surprising that the specific glucose consumption seems to increase beyond a certain point in both cultures. To understand why this is so, we have to take into consideration that the concentration of glucose rises beyond 100 hours in the cultures due to overfeeding. Since, the transport of glucose via the GLUT-1 transporters is concentration dependent (Burant, et al., 1991; Carruthers, 1990), this increase in concentration in glucose could possibly lead to an increase in glucose uptake rate. To 155 correct for this effect, we measured specific glucose uptake rate on a per cell, per mole of external glucose present per time basis. As shown in Figure 6-14, there is a steady decrease in the specific glucose consumption when measured per mole of glucose present in the medium. The cells therefore are much more inefficient at taking up glucose after about 100 hours into the culture. 6.6 Discussionand Conclusions The results presented in this chapter clearly demonstrate the beneficial effect that bcl-2 expression provides in improving the performance of fed-batch cultures. This improvement in fed-batch performance is brought about primarily by extending the viability of cultures. No difference in growth rates of bcl-2-transfected and control cell cultures was observed. The improvement in the performance of fed-batch cultures was found to be greater than 75% as measured by both integrated viable cell densities (IVCDs) and y-interferon titers. In evaluating these improvements one must remember that performance of the control cultures, against which improvements are measured, has already been optimized by stoichiometric feeding strategies. In addition, the results from insulin-deprived fed-batch cultures suggest that culture viability can be significantly extended beyond that obtained in normal fed-batch cultures by using bcl-2 expression. Confirming results from batch culture experiments, bcl-2 abrogates cell death induced by insulin deprivation, but is unable to induce cell growth. The IVCDs obtained in insulin-deprived cultures increase with increasing initial 156 5.OE-6 3.0E+6 / I I 2.5E+6 - -4.OE-6 ,. E > E ' 2.OE+6 3.0E-6 ,d/ S1.5E+6 - / o 1.OE+6 - dE 2.0E-6 CD - 1.OE-6 5.0E+5 - 0.OE+0 00 0 ! 0.OE+0 I SI 50 100 150 200 250 --. -- bcl2-VCD --- a--pcd-VCD -- -bcl2-SGC .- pcd-SGC Time (hour) Figure 6-13: Specific glucose consumption (SGC) and viable cell density (VCD) with time in a normal fed-batch culture of CHO cells. Specific glucose consumption drops off before a fall in viable cell density. 157 4.0E-7 12 3.5E-7 0 0- - 3.OE-7 Ec 8 6 2.5E-7 2.OE-7 'i o o "^ 4- S1.5E-7 = 1.OE-7 2 5.OE-8 $-0 0 50 100 Time 4) dc i O 0 150 O.OE+O 200 (hour) 250 .0 0 - - bcl2-glc ----pcd-glc - --- bd2-SGCG S.... pcd-SGCG Figure 6-14: The glucose concentration (glc) of the fed batch media increase sharply after 100 hours due to unavoidable overfeeding and inefficient uptake of glucose. The specific uptake rate of glucose, when measured per mole of glucose in the medium (SGCG) continues to fall after about 90 hours in the culture. The rise in specific glucose uptake rate seen in Figure 6-13 may therefore be explained by the rising concentrations of glucose in the culture (see text). 158 cell densities. Interestingly, IVCDs obtained in an insulin-deprived fed-batch culture with an initial cell density of 8x10 5 cells/mL approached those obtained with normal fed-batch cultures. This suggests that higher initial cell densities could perhaps obtain even higher IVCDs. The advantage in using slower growing cultures is that cells waste less of their energy in growth related activities. It has been suggested that they are more productive and produce less waste products in a slow growing state (Fussenegger, et al., 1997; Lao, et al., 1996). In addition, the product produced may be of more uniform quality (since more cells are in the same growth phase). There are also added cost and regulatory advantages of not having to add costly growth factors to the culture medium. Clearly bcl2 expression has the potential to easily permit slow-growing, but longer surviving, fedbatch culture of CHO cells. In almost all of our fed-batch cultures, y-interferon titer improvements did not equal the IVCD improvement. The trend is particularly acute in insulin-deprived cultures. The promoter used to express y-interferon is an SV40 based promoter. The SV40 promoter has been reported by several authors to be a growth-associated promoter, which is most active in the S-phase of the cell cycle. Since CHO cells were shown to arrest in the GO/G1 phase during cell culture, it is not surprising that the productivity of these cultures is lower during the latter part of the cultures. Unfortunately, this is also the part of the culture in which bcl-2 expression provides maximum benefit. Also, it is entirely logical that productivity is most seriously affected in insulin-deprived cultures, where growth is slow throughout the culture. The problem of lower productivity due to 159 slower growth can easily be addressed by using a non-growth-associated promoter. Fussenegger, et al., 1997 have demonstrated this possibility in CHO cells, by placing the product gene and a cytostatic gene under the control of the same non-growth associated promoter. Bcl-2 expression goes a step further than the technique used by Fussenegger, et al., 1997 to enhance productivity, since it also creates a much more hardy and robust cell line in the process. The experiments described in latter part of this chapter focused on identifying the underlying causes behind the cessation in growth observed in fed-batch cultures. This cessation in growth occurs despite ensuring an adequate supply of nutrients. Cell cycle analysis of CHO cells taken from various points in the culture indicated that cells progressively arrested in the GO/G1 phase, suggesting a lack of growth stimulants in the medium. In addition, western blot analysis of cell lysates confirmed that p53 expression was upregulated in cells taken from later phases of the fed-batch culture. This upregulation in p53 expression also indicates loss of growth stimulus or the accumulation of specific death stimuli in the medium (Elledge, 1996; Ko and Prives, 1996; Levine, 1997; Sherr, 1994; Sherr, 1996). Another protein frequently associated with G to S phase transition of cells is cyclin E (Ohtsubo, et al., 1995). Surprisingly, cyclin E expression was found to be unchanged during the entire culture process. Renner, et al., 1995 have reported that cyclin E induces proliferation and reduces surface-attachment requirement in anchorage dependent cell lines. It is possible that during the adaptation of our anchoragedependent CHO cell line to suspension culture, cyclin E expression was deregulated, and hence we see no reduction in cyclin E expression during cessation of growth in a fed160 batch. This also suggests that further overexpressing cyclin E in our cell line, via recombinant techniques, is unlikely to solve the growth problems in CHO fed-batch culture. Another surprising result presented above was that of a sudden decrease in the specific glucose uptake rate just before the viable cell density stopped increasing. This would suggest that cells may be experiencing a lack of nutrients due to an inability to uptake them, even though this nutrient starvation is not apparent through measurement of nutrient concentrations in the external environment. Possible reasons underlying this reduced glucose uptake and stunted growth of cultures are explored in the next chapter. 161 7. The Fate of Insulin in CHO Fed-Batch Cultures The results from the previous chapter indicated that although the expression of bcl-2 is able to extend the life of fed-batch cultures of CHO cells, it is unable to affect the growth of the cultures. It was unclear, however, why the cultures stopped growing after a certain point in time. In addition, it was unclear what was causing the cells to die even when they were supplied with the requisite quantities of nutrients. Analysis of the medium for glucose and the other amino acids indicated that none of them were close to being depleted during the culture, and hence their depletion could be ruled out as a cause of death. Moreover, cells from the stationary phase of the fed-batch that were transferred to fresh RPMI grew normally, indicating that the cell's intrinsic ability to divide and grow was not in any way impaired. Results presented in the previous chapter also indicated a progressive arrest of cells in the GO/G1 phase. This suggested the loss of growth factors from the medium. In addition, p53, a well known growth-suppressing and apoptosis-inducing gene, was expressed in larger quantities as the culture progressed. More interestingly, specific glucose uptake rate dropped off 24 hours prior to the cessation of growth. This dip in glucose uptake rate could be a cause of both cessation of growth and of cell death, since we know from batch experiments that insufficient supply of glucose to cells can trigger death. To understand why the cellular glucose uptake reduces, elements in the glucose uptake pathway to the cell were investigated. One obvious step was the binding of 163 insulin to its receptor, which helps transport glucose into the cell. In CHO cells, insulin binding to its receptor triggers glucose transport mainly by activating the GLUT-i transporter gene (Hausdorff, et al., 1996; Pessin and Bell, 1992). The GLUT-i transporter helps the cell uptake glucose from the surroundings in a concentrationdependent manner (Burant, et al., 1991; Carruthers, 1990). Moreover insulin is a well documented growth factor in many mammalian cell cultures (Bottenstein, et al., 1979; Pimentel, 1994), and any disruption in the insulin signal transduction pathway could hamper growth of cells. Possible events that could hamper the ability of insulin to facilitate glucose uptake and spur growth in a CHO fed-batch culture were investigated, and the results are presented in the following sections. 7.1 Reduced bindingof insulin to its receptor cannot fully explain cessation of growth in CHO fed-batch cultures An event that could explain the observed phenomena of growth reduction and reduced glucose uptake was the reduced ability of insulin to bind to its receptor. This could be caused by a change in the integrity of expressed insulin receptors as the culture progressed. We compared the insulin-receptor binding efficiency in cells taken from day 1 and day 7 of a fed-batch culture. Samples were incubated with radioactive 1125 insulin and then washed to remove unbound insulin. The amount of radioactivity measured in a washed sample provided a measure of the quantity of insulin bound to the sample. The specifically bound insulin was calculated as the difference between the mean values of 164 radioactivity in samples where only radioactive insulin was added to those in which both radioactive insulin and a saturating amount of non-radioactive insulin was added (see Materials and Methods chapter for more assay details). It was found that the amount of insulin specifically bound to the cells taken from day one of a fed-batch culture was about 1.5 times that bound specifically to cells from day seven of a fed-batch culture (see Figure 7-1). Although this represents a significant reduction in insulin binding, insulin is still able to bind to cells in the stationary phase of the fed-batch. This would tend to disprove the hypothesis of failure of insulin binding as the major cause of the stationary phase observed in CHO fed-batch cultures. 7.2 Insulin rapidly disappears with time in CHO fed-batch cultures Since the binding of insulin to receptors on the cell surface was not substantially affected by fed-batch culture, it could be that insulin itself was being affected in the fed-batch culture. To determine if this was the case, we first decided to measure the concentration of insulin in the medium supernatant as a function of time. This was done using a commercially available insulin-ELISA kit (American Laboratory Products Company, Ltd. catalog # 10-1113-01). Figure 7-2 shows the concentration of insulin in a typical (normal) fed-batch as the culture proceeds. The secondary y-axis also shows the corresponding viable cell densities of the bcl-2- and pcd-CHO cultures. Figure 7-2 indicates that the concentration of insulin in the supernatant falls rapidly from an initial value of 5 mg/L to undetectable levels within 96 hours of the start of the culture. The viable densities stop 165 5000 4500 E" 4000o 3500 S3000 0 2500 o 2000 S1500 Z 1000 500 0 old new Figure 7-1: Insulin binding to cells taken from different points in a fed-batch culture. Net radioactivity is a proxy for the quantity of insulin bound in a specific manner to its receptor (see text). The higher the net radioactivity value the larger to quantity of insulin bound. 'Old' and 'new' refer to samples taken from day 7 and day 1 of a fed-batch culture, respectively. 166 3.0 3.0E+6 2.5 2.5E+6 2.0 2.0E+6 u0 1.5 1.5E+6 > 1.0 1.OE+6 Q 0.5 5.OE+5 0 -j -- 4-- bc2-ins 0.0 ! 0 50 100 ,, --- 150 200 -a- -pcd-ins bcl2-VCD1 ----- pcd-VCD 0.OE+0 250 Time (hour) Figure 7-2: Insulin concentration as a function of time in a typical fed-batch culture of CHO cells. The initial concentration of insulin in the culture is 5 mg/L. Insulin concentrations drop rapidly to almost non-detectable levels by 100 hours. Culture growth stops soon after. The suffix '-ins' refers to the concentration of insulin in either the bcl2-CHO or pcd-CHO cell culture. VCD refers to the viable cell density in either culture. 167 increasing approximately 24 hours after the insulin disappeared from the medium. Interestingly, there is no significant difference between the disappearance of the insulin from the bcl-2 transfected and control (pcd) cultures, indicating that the disappearance of insulin from the medium is a phenomenon intrinsic to CHOs that is not affected by transfection with bcl-2. 7.3 Insulin degrading activityin CHO fed-batch cultures is concentrated in the supernatant Although the measurement of insulin in the fed-culture medium indicated that it was disappearing rapidly as the culture progressed, it was not clear what was causing this disappearance. One possibility was that insulin was binding (perhaps non-specifically) to the surface of the cells and thus being removed from the medium. Another possibility was that the cells were using insulin much more inefficiently as the culture progressed, causing a rapid consumption of insulin. Lastly, it was also possible that the cells were secreting either a protease or a binding protein that was either degrading the insulin or preventing it from being used by cells or being detected by ELISA. To differentiate between these alternatives we added fresh doses of 5 mg/L of insulin to a 2 mL sample of supernatant (medium from which cells have been removed) and to cell containing medium on various days of a normal fed-batch culture. The samples were then incubated under culture conditions, and insulin degradation was then quantified over a period of 24 hours by measuring the quantity of insulin in the supernatant or the 168 cell-containing medium at 0, 2, 6, 10 and 24 hours. For ease of comparison, the concentrations of insulin in the samples were expressed as a fraction of the concentration at '0' hours. Figure 7-3 and Figure 7-4 show the results from the experiment. It is evident that the degradative activity in both the supernatant and the cell-containing medium increase as a function of culture time. Also, on any particular day, the concentration of insulin in the sample undergoes a monotonic decrease with time. This is more evident in the samples taken earlier in the culture where the insulin does not disappear too rapidly. In addition, comparing the results from days 6 and 7, the degradative activity of the supernatant alone is almost identical to that of the cell containing medium. Thus the degradative activity is primarily in the supernatant, favoring the hypothesis of a protease or binding protein being responsible for the disappearance of insulin. It should also be noted that almost all the insulin added disappears within 24 hours, especially towards the later part of the culture. This explains why earlier experiments, in which insulin was added to the culture in an effort to spur increases in viable cell density, failed to achieve any results. To guard against the possibility that the conditions of incubation (culture conditions) are responsible for the disappearance of insulin, a control experiment was performed to study the degradation kinetics of insulin under these conditions in fresh cellfree fed-batch culture medium. Figure 7-5 indicates that insulin does not degrade significantly in a fresh cell-free medium under culture conditions even after almost 200 hours, which is far longer than the time scale of 24 hours in which most of the insulin disappears from the supernatant of a fed-batch culture. Hence, the insulin degradation 169 100% '- 90% 80% *" 70% 60o r day 3i 0% Iaday 4 day 6 7 S40% --__ 30% - _ --_ Dday _ 20% LL 10% - 0 2 6 10 24 Time (hour) Figure 7-3: Degradation of insulin in fed-batch supernatant (culture medium with cells spun down and removed) from various days in a CHO fed-batch culture. The degradation of insulin in samples from each day, under cell-culture conditions, was followed for 24 hours. 5 mg/L of insulin was added to the supernatant at '0' hours, and the concentration of insulin measured in the supernatant at this point was denoted as 100%. Insulin in any particular sample was expressed as a fraction of the insulin concentration in the same sample at '0' hours. 170 100% _ 80% w 60% - o day 5 13 day 6 O day 7 40% - .0 20% L U- 0 6 2 10 24 Time (hour) Figure 7-4: Degradation of insulin in fed-batch medium (with cells) from various days in a CHO fed-batch culture. The degradation of insulin in samples from each day was followed for 24 hours. 5 mg/L of insulin was added at '0' hours, and the concentration of insulin measured in the supernatant at this point was denoted as 100%. Insulin in any particular sample was expressed as a fraction of the insulin concentration in the same sample at '0' hours. 171 120% 100% C " 80% E S60% S40% C L_ 0% 0 50 100 150 200 Time (hour) Figure 7-5: Degradation kinetics of insulin in fresh (cell-free) fed-batch medium under standard cell-culture conditions. 5 mg/L of insulin was added to the medium at '0' hours. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours. 172 activity that we observe in the supernatant of a fed-batch culture is due to a substance present in the supernatant that is not present in the original medium. In addition, insulin degradation due to incubator conditions is negligible. 7.4 Boiling of fed-batch supernatant removes all insulin degrading activity To asses the nature of the component in the fed-batch medium which was causing the insulin to disappear, the supernatant was boiled for three minutes and fresh insulin was added to it after cooling. supernatant. Boiling destroys any protease activity present in the As Figure 7-6 indicates the boiled supernatant showed almost no degradation of insulin after 24 hours, while in the unboiled control almost all the insulin had disappeared after the same period of time. In fact, the boiled control retained almost complete insulin activity even after 120 hours. Therefore the component responsible for the disappearance of insulin could be a protease. However, a binding protein, secreted by the cells during the culture process, which binds to insulin and prevents its detection by ELISA could also cause the disappearance of insulin. There is evidence that p53 expression in cells (p53 expression increases during CHO fed-batch cultivation, see results from previous chapter) leads to secretion of an IGF-1 binding protein (Buckbinder, et al., 1995). It is possible that CHO cells secrete a similar binding protein, which directly affects insulin. Boiling of the supernatant would also destroy any such binding protein activity. The boiling experiment confirmed the presence of degradative 173 100% 0 90% E 80% E * . E S70% 60% 50% 40% 30% - ---- boiled unboiled control 20% 10% u.0% o o 0 10 20 Time (hour) 30 Figure 7-6: Boiling fed-batch supernatant for three minutes removes its insulin degrading activity. 5 mg/L of insulin was added to the boiled sample and unboiled control at '0' hours. Both samples were incubated under standard cell-culture conditions. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours. 174 activity in the supernatant, but, further experiments were needed to distinguish between protease and binding protein activity. 7.5 Aminopeptidase inhibitors are able to substantially reduce the degradation of insulinin the fed-batch supernatant If the disappearance of insulin in the supematant were due to a protease, addition of protease inhibitors to the supernatant should prevent its insulin degrading activity. A literature search on insulin degrading proteases that are normally produced by cells (Fukuda, et al., 1995; Morishita, et al., 1992; Tozaki, et al., 1997; Yamamoto, et al., 1994) suggested several protease inhibitors that prevented degradation of insulin in vivo. The concentrations of various protease inhibitors added to supematant samples are indicated in Table 7-1. At the start of the experiment (zero hours) 5 mg/L of insulin was added to each supernatant sample. The concentrations of insulin in the supernatant were measured at 0, 0.5, 1.5, 3 and 13.5 hours, respectively. Degradation activity in these samples was compared to the degradation in a control supematant to which no protease inhibitors were added. The results from the study are shown in Figure 7-7. For ease of comparison, the concentrations of insulin in the samples were expressed as a fraction of the concentration at '0' hours. The bacitracin obtained from Sigma contained some intrinsic protease activity, and this explains the rapid disappearance of insulin in the bacitracin and 'All' samples. Figure 7-7 indicates that sodium glycocholate was able to reduce insulin 175 Table 7-I: Concentrations and sources of various protease inhibitors used to study insulin degradation in CHO fed-batch culture supernatants. Inhibitor Used Final Concentration Source Soybean Trypsin Inhibitor 10 mg/mL Sigma-Aldrich, T-9128 Bacitracin 20 mM Sigma-Aldrich, B-0125 Aprotinin 100 gg/mL Sigma-Aldrich, A-6279 Sodium Glycocholate 10 mM Sigma-Aldrich, G-7132 All above concentrations of all inhibitors added to sample 176 120% "E 100% E a 80% All r---- Trypsin Inhibitor --&--Bacitracin -=o60%0 40% - '-v-Aprotinin 20% - ---- Na Glycocholate ---- Control o _ 0% 0 5 10 15 Time (hour) Figure 7-7: Effect of various protease inhibitors on the degradation of insulin in the supernatant from a fed-batch culture. 'All' indicates that all the protease inhibitors were added to this sample at the concentrations suggested. No protease inhibitors were added to the 'control' culture. 5 mg/L of insulin was added to the medium at '0' hours. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours. All samples were incubated under standard cell-culture conditions for the duration of the experiment. 177 degradation in the supernatant to the largest extent, with almost 90% of the insulin remaining after 13.5 hours. It was confirmed that sodium glycocholate did not cause a false positive with the insulin assay used. Aprotinin and Soybean Trypsin Inhibitor (STI) displayed the next best insulin-protease-inhibitor activity. Supematants treated with these inhibitors managed to retain 50% of original insulin activity after 13.5 hours. In contrast, the control, which had no added protease inhibitors, only retained 33% insulin activity after 13.5 hours. Sodium Glycocholate is an aminopeptidase inhibitor (Fukuda, et al., 1995). Hence, the results indicate that the protease(s) in the supernatant of a fed-batch culture degrades insulin from the N-terminal position. Furthermore, since serine protease inhibitors such as aprotinin and STI also reduced the degradation of insulin, it is possible that there are also some trypsin- and chymotrypsin-like proteases in the supernatant. 7.6 Adding large quantities of excess insulin also reduces the rate of degradation of insulinin the fed-batch supernatant If the degradation of insulin in the fed-batch supernatant was caused by binding of insulin to a binding protein secreted by the cells, the addition of large excesses of insulin should saturate the binding protein available and prevent disappearance of insulin. In order to determine the effect of excess insulin on the rate of insulin disappearance from the supernatant, we added 500 gg/mL and 50 gg/mL insulin to the supernatant, and compared insulin disappearance in these samples to that in the control (5 gg/mL insulin). Insulin 178 concentrations were measured at 0, 0.5, 1.5, 3 and 13.5 hours after the initial addition of insulin. For ease of comparison, the concentrations of insulin in the samples were expressed as a fraction of the concentration at '0' hours. Figure 7-8 shows that increasing the concentration of insulin to 100X (500 gg/mL) and to 10X (50 gg/mL), caused only 73% and 66% of the insulin, respectively, to degrade after 13.5 hours. In contrast, only 33% of the insulin at time zero was remaining in the control (IX insulin) after 13.5 hours. The reduction in the rate of insulin degradation due to the addition of excess insulin could be due to saturation of binding protein present in the supernatant.. However, if non- catalytic insulin proteases were present in the medium, the observed reduction in protease activity could also be due to saturation of the activity of these proteases. 7.7 Discussion and Conclusions The experiments described in this chapter focused on identifying the underlying causes of the cessation in growth observed in CHO fed-batch cultures. This cessation in growth occurs despite ensuring an adequate supply of nutrients and adding extra doses of the growth factor insulin. The results presented in the previous chapter also indicated that specific glucose consumption dips just before cessation in viable cell density growth. Cells in fed-batch cultures progressively arrested in the GO/G1 phase and upregulated p53 expression. This chapter explored some events that might explain these observations. The ability of insulin to bind its receptor was found to be reduced in later phases of a fed-batch culture, but the magnitude of this decrease was not sufficient to explain the 179 120% 100% E 80% S -, 100X insulin I--e-1OX insulin -- control 60% - --- ) 40%0 o 20% 0% 0 5 10 15 Time (hour) Figure 7-8: Adding excess insulin reduces the rate of insulin degradation, possibly due to the saturation of proteases or insulin-binding proteins in the fed-batch supernatant. The control indicated has 1X or 5gg/mL of insulin added to it at time zero. The concentration of insulin in the medium at various time points was expressed as a fraction of the concentration at '0' hours. All samples were incubated under standard cell-culture conditions for the duration of the experiment. 180 observed phenomena. However, the increase in p53 expression and the cessation of growth both appear correlated to the rapid disappearance of insulin from fed-batch cultures. Insulin is a known growth and survival factor for the culture of several mammalian cell lines (Bottenstein, et al., 1979; Murakami, 1989; Pimentel, 1994; Zhou and Hu, 1995). Insulin concentrations in the culture were observed to fall below detectable levels at least 24 hours before the viable cell density stopped increasing. The lag in cessation of growth could be due to insulin receptors remaining phosphorylated for some time after the insulin has disappeared from the medium. Interestingly, these results corroborate the work of Drapeau, et al., 1994 and Lao, et al., 1996, who demonstrated insulin degrading activity in batch-refeed processes and perfusion cultures, respectively. The authors did not, however, explore the cause of the insulin degrading activity. Insulin degrading activity in CHO fed-batch culture was shown to be concentrated in the supernatant, since there was no observed difference in degradation rates of insulin between culture medium in which cells were still present and culture medium from which cells were removed. Since the rate of insulin degradation is almost completely removed by boiling and significantly reduced by specific protease inhibitors, insulin degradation is definitely protease related. Since the best reduction in protease activity was provided by sodium glycocholate, an amino-peptidase inhibitor, amino-peptidases appear to play a major role in insulin degradation in CHO fed-batch cultures. Furthermore, trypsin- and chymotrypsin-like proteases may also play a role in insulin degradation since serine protease inhibitors such as aprotinin and soybean trypsin inhibitor were able to reduce insulin degradation. Interestingly, adding large excesses of insulin to the supernatant also 181 reduces the rate of insulin degradation. This may be due to the saturation of non-catalytic protease activity or due to the quenching of any insulin-binding-protein activity that may be present. Binding-protein activity, while not specifically confirmed, cannot be ruled out at this stage. It may be noted that p53 expression has been shown to cause secretion of a IGF binding protein, IGF-BP3, in some cell lines. It is not unreasonable, therefore, to expect that the observed p53 expression in CHO fed-batch cultures can trigger secretion of similar binding proteins that affect insulin. No insulin degrading activity was observed in fresh medium. In addition, the insulin degrading activity in a fed-batch culture progressively increases with time, as evidenced by increasingly rapid rates of disappearance of added insulin. Insulin degrading proteases and binding proteins (if any) must therefore be secreted by the cells. Also the degradative activity starts to accumulate before extensive death and cell lysis occur, indicating that the insulin degrading agents are released as a part of the normal metabolic process. Since the insulin degrading activity increases with culture time, the degrading agents must accumulate with time indicating that they have a fairly long half-life. The above experimental outcomes suggest that the degradation of insulin in fedbatch cultures can be reduced in one of two ways. First, very high doses of insulin could be added to the culture at regular intervals. Of course, cost of adding such large quantities of insulin is a drawback. In addition, large quantities of insulin are known to downregulate the insulin receptor, necessitating even higher quantities of insulin in the culture. A second strategy to reduce the disappearance of insulin would be to add a cocktail of protease inhibitors. The above results suggest that amino-peptidase inhibitors 182 should be an essential part of this cocktail. The cost of these protease inhibitors and the optimum concentrations at which they are most effective without being toxic to cells are issues that will need to be addressed in future studies. 183 8. Conclusions and Recommendations 8.1 Conclusions The results presented in this thesis showed that CHO cells in serum free culture die primarily by apoptosis. A new assay, the acridine orange / ethidium bromide (AO/EB) assay, was optimized to allow rapid analysis of apoptosis in CHO cells. In addition, apoptosis was confirmed by the degradation of genomic DNA into multiples of 180 bp fragments at the onset of apoptosis. Protein synthesis inhibition demonstrated that death proteins dominate in CHO cells and survival proteins must be continuously synthesized in order to protect cells from death. Activation of caspases was observed in apoptotic CHO cells and confirmed by the cleavage of substrates of two different caspases - Poly(ADP-Ribose)-Polymerase (PARP), which is a substrate of caspase-3, and lamin A, which is a substrate of caspase-6. Caspases are perhaps the most important apoptosis agonists in eukaryotes and their activation is considered by some researchers to be essential in the apoptotic process. Surprisingly, inhibition of caspase activity (as confirmed by non-cleavage of substrates) by universal and irreversible caspase inhibitor z-VAD.fmk failed to prevent death in CHO cells. In contrast, expression of the anti-apoptotic gene bcl-2 was able to significantly extend the viability of CHO cells in batch culture, in response to glucose starvation at the end of a batch. Bcl-2 expression was able to completely abrogate death in batch culture response to removal of growth and survival factor, insulin. z-VAD.fmk again failed to have a significant protective effect. In both these cases, bcl-2 expression completely prevented caspase activity confirming previous reports that it lay upstream 185 of caspases in the apoptotic pathway. The failure of caspases to protect cells from death was surprising since previous reports had indicated that caspases were universally required in apoptosis, and that inhibition of their activity would be a more potent agent for prevention of death than overexpressing bcl-2. Our results show this not to be the case, suggesting that bcl-2-dependent and caspase-independent death pathways do exist, at least in CHO cells. These results have since been confirmed in other reports in literature (Xiang, et al., 1996). In conclusion, bcl-2 expression was found to be a much better way of extending batch culture viability than using peptide inhibition of caspases. This better protection afforded by bcl-2 is attributed to its ability to not only prevent activation of downstream caspases but also maintain mitochondrial potential, thus protecting a cell's ability to carry out oxidative phosphorylation and preventing the release of reactive oxygen species. Before claiming significant improvement in cellular viability using bcl-2 expression, it was important to confirm the increased viability with another assay which went beyond measuring membrane permeability and chromatin integrity. The more stringent benchmark chosen for viability was the ability of a cell to undergo mitosis when placed in a more favorable environment. An experiment that allowed us to follow the fate of individual cells in a favorable environment was designed. The results from this experiment (discussed in Chapter 5) proved that the viabilities observed with bcl-2 correlated with the continuing ability of a cell to undergo mitosis. It also showed that the AO/EB assay results correlated much better with replication competence than did the results of the trypan blue assay. Lastly, this set of experiments proved that early 186 apoptotic cells could be considered dead, if replication competence is considered as a stringent measure of viability. Bcl-2 expression combined with stoichiometric medium design and feeding strategies was able to significantly improve the performance of fed-batch cultures beyond that obtained using just stoichiometric medium design and feeding strategies without bcl-2 expression. Final gamma interferon (IFN-y) concentrations (the model protein secreted by the CHO cells used) and integrated viable cell densities (IVCDs) were both improved by a factor of at least 75% as compared to control cultures, by using bcl-2 clonal cell lines. Bcl-2 clonal cultures had at least a 600% higher IVCD as compared to the control culture, in an insulin-deprived environment. Bcl-2 expression also extended culture lifespans in these slower growing insulin-deprived fed-batch cultures beyond that obtained with bcl-2 expressing cells in insulin-supplemented fed-batch medium by as much as 40%. For insulin-deprived cultures seeded at higher densities, the IVCDs obtained with bcl-2 expressing clones approached those obtained with bcl-2 expressing clones in insulin-supplemented cultures. However, although bcl-2 expression allowed cells to remain viable and productive in the absence of insulin, the productivities were lower than those obtained in an insulin-supplemented medium. This was most likely due to the fact that the SV40-based promoter used to express IFN-y is not as active in the absence of growth. Nevertheless, these experiments demonstrate the potential of bcl-2 expression to permit slow growing cultures to achieve much higher lifespans and productivites (provided a non-growth associated promoter is used) than the faster 187 growing cultures. In fact some reports have suggested that slower growing cultures are more productive than faster growing ones (Fussenegger, et al., 1997). Bcl-2 expression provides these benefits in addition to advantages of a more robust cell line, lower cost (no use of costly growth factors) and fewer regulatory issues (related to the use of undefined growth factors). The cessation of growth in fed-batch cultures was observed to be concurrent with accumulation of cells in the GO/G1 phase. The expression of p53, a well-characterized growth-inhibiting and apoptotic protein, which arrests cells in the GO/G1 phase, was also observed to increase throughout the culture. The increase in p53 expression could also be a factor in the increased death observed soon after viable cell density stops increasing. p53 expression has been shown to activate Bax expression. Bax is an apoptotic member of the bcl-2 family, which dimerizes with bcl-2. Increasing quantities of Bax can negate the protective effect of bcl-2 and cause death of cells. This hypothesis may explain why even bcl-2 expressing fed-batch cultures ultimately perish. The expression of cyclin E, a protein known to drive G to S phase transition in cells, was observed not to change during the fed-batch process, indicating that this gene might have been deregulated during the process of adaptation to suspension culture from anchorage-dependent growth. Further overexpression of cyclin E is therefore unlikely to increase growth rates. The specific glucose uptake rate was observed to drop right before the cessation of viable-density growth in fed-batch cultures. This suggests that cells might be experiencing glucose starvation, which has already been shown to be a potent death stimulus, due to a failure to uptake glucose. A closer look at insulin, known to be a 188 critical factor in cellular glucose uptake and growth, provided some surprising results. The ability of insulin to bind to its receptor was found to be significantly reduced, but not enough to explain the complete cessation of growth. More interestingly, insulin was found to disappear at increasingly rapid rates from the fed-batch medium as the culture progressed, falling to undetectable levels about 24 hours before the cessation of growth. The insulin degrading activity was isolated to the medium (and not the cells) and was shown to be blocked by amino-peptidase inhibitors such as sodium glychocholate and to a much lesser extent by serine protease inhibitors. Surprisingly, insulin degradation rates were also reduced by using high concentrations of insulin, suggesting the presence of irreversible protease activity or insulin binding proteins. 8.2 Recommendations The fact that bcl-2 is able to significantly extend viabilities in batch and fed-batch culture of CHO cells does not imply that further improvements cannot be made. There are some reports to suggest that other homologues of bcl-2, such as bcl-XL, prevent apoptosis in cases where bcl-2 does not (Boise, et al., 1993; Cheng, et al., 1996; Gottschalk, et al., 1994). In addition, proteins that interact with bcl-2 to increase its protective effect have been reported. One such protein, Bag-l, enhances bcl-2 activity presumably by helping it interact with Raf-1 and inducing phosphorylation and deactivation of apoptotic protein Bad (Reed, 1997). Co-expression of bcl-xL and/or Bag-1 with bcl-2 in CHO cells could further improve viabilities. 189 Since caspases are important effectors of apoptosis in CHO cells, further research needs to be performed at trying to inhibit caspases using genetic expression of caspase inhibiting proteins. One such protein, the baculovirus protein p35, has been demonstrated to be a strong and universal inhibitor of caspases in the nematode C. elegans, and some insect and mammalian cell lines (Bump, et al., 1995; Clem, et al., 1991; Rabizadeh, et al., 1993). However, preliminary attempts to transfect cells stably with p35 failed in this lab, and reportedly at some other labs (H. Steller, personal communication). The second set of proteins that have been shown to inhibit caspase activity are the IAPs (for inhibitor of apoptosis proteins; Bump, et al., 1995; Clem, et al., 1996; Hawkins, et al., 1996; Liston, et al., 1996; Orth and Dixit, 1997). Several human homologues of these proteins have recently been identified. Since results presented in Chapter 4 (and independent reports in literature, see Cuende, et al., 1993; Lincz, 1998; Reed, 1997) suggest that bcl-2 independent but caspase-dependent death pathways exist in mammalian cells, co-expression of bcl-2 and specific caspase-inhibiting proteins in CHO cells has the potential to significantly improve the resistance of cells to death stimuli. There have recently been significant developments in mammalian expression systems that can help us implement some of the above recommendations more efficiently. Promoters which can provide higher levels of bcl-2 expression than the CMV promoter are now available commercially. One such promoter is tetracycline-inducible (Clontech), which would permit the study of different expression levels of bcl-2 and other proteins in the same cell line. Plasmids with an internal ribosomal entry site (IRES), which can allow 190 the simultaneous expression of two or more genes using the same promoter (translation of two genes of a single mRNA transcript), are also now commercially available (Clontech; Fussenegger, et al., 1998). Bicistrionic plasmid expression systems in which ones of the genes is that responsible for green fluorescence protein expression are also available (Clontech). These expression systems permit the easy and rapid selection of high expression clones using flow-cytometry. The results presented in Chapter 6 suggest that slower growing fed-batch cultures survive much longer than faster growing cultures. In addition, previous work in this lab has suggested that slower growth in cultures leads to higher glycosylation efficiencies (Nyberg, 1998). Further, reports in literature suggest that cells are more productive in the stationary phase (Fussenegger, et al., 1997). However, these advantages have not been translated into higher product titers in our cell line due to the use of a growth phase dependent promoter (Banik, et al., 1996; Gu, et al., 1993; Gu, et al., 1994; Mariani, et al., 1981). Expressing IFN-y with a promoter that is active in the S-phase (see Fussenegger, et al., 1997 for a review) is strongly recommended to be able to better study productivity and quality issues in a slow-growing cultures. Insulin disappearance was shown to be significantly reduced in the supernatant from fed-batch media by using amino-peptidase inhibitor sodium glycocholate and by adding larger doses of insulin. Further studies need to be performed to investigate the effects of directly adding protease inhibitors to fed-batch cultures of CHO cells on insulin degradation, cell growth and cell viability. Protease inhibitors should be chosen based on 191 both effectiveness of protease inhibition and the absence of adverse effects on cells. Adding large doses of insulin to cultures is not recommended due to cost issues and also the possibility of downregulation of insulin receptors on the cell surface. Another more exotic approach would be to design (or select) a clone that can grow in the absence of insulin (Lao, et al., 1996; Renner, et al., 1995). It is interesting to observe that even bcl-2 expressing cultures ultimately die rapidly after a point in fed-batch culture. The level of protection afforded by bcl-2 depends on its level of expression in the cell. Further increases in levels of bcl-2 expression using a different promoter may increase the life of bcl-2 fed-batch cultures. In addition, the level of protection offered by bcl-2 expression also depends on the expression levels of other dimerizing members of the bcl-2 family, most importantly that of Bax. p53 expression, which was shown to increase during the course of a fed-batch culture, has been reported to stimulate Bax expression (Han, et al., 1996; Matsuyama, et al., 1998; Miyashita and Reed, 1995; Oren and Prives, 1996). It would be interesting to see if Bax expression increases with time in CHO fed-batch cultures. p53 expression has also been reported to induce the expression of transforming growth factor beta (TGF-3), another apoptotic and growth inhibiting protein (Fussenegger, et al., 1997). Very preliminary studies in our cell line indicated an accumulation of TGF-P in fed-batch culture of CHOs. Further research into expression of these and other apoptotic stimuli secreted as a part of the normal metabolic process can provide a better idea of the 192 apoptotic pathway in CHO cells and suggest mechanisms to further extend viability and productivity in CHO cell cultures. 193 9. References Alnemri, E. S., Livingston, D. J., Nicholson, D. W., Salvesen, G., Thornberry, N. A., Wong, W. W. and Yuan, J. 1996. Human ICE/CED-3 protease nomenclature (letter). Cell. 87: 171. Anderson, P. 1997. Kinase Cascades Regulating Entry into Apoptosis. Microbiol. Mol. Biol. Rev. 61: 33-46. Backer, J. M., Kahn, C. R. and White, M. F. 1990. The Dissociation and Degradation of Internalized Insulin Occur in the Endosomes of Rat Hepatoma Cells. J. Biol. Chem. 265: 14828-35. Banik, G. G., Todd, P. W. and Kompala, D. S. 1996. Foreign protein expression from S phase specific promoters in continuous cultures of recombinant CHO cells. Cytotechnology. 22: 179-184. Bissonnette, R. P., Echeverri, F., Mahboubi, A. and Green, D. R. 1992. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 359: 552-554. Boise, L. H., Gonzalez-GarcIa, M., Postema, C. E., Ding, L., Lindsten, T., Turka, L. A., Mao, X., NuOez, G. and Thompson, C. B. 1993. Bcl-x, a bcl-2-Related Gene That Functions as a Dominant Regulator of Apoptotic Cell Death. Cell. 74: 597-608. Bossy-Wetzel, E., Newmeyer, D. D. and Green, D. R. 1998. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 17: 37-49. Bottenstein, J., Hayashi, I., Huchings, S., Masui, H., Mather, J., McClure, D., Ohasa, S., Rizzino, A., Sato, G., Serrero, G., Wolfe, R. and Wu, R. 1979. The growth of cells in serum-free hormone supplemented media. In: W. B. Jakoby and I. H. Pastan, Methods in Enzymology, Academic, New York. Boyd, J. M., Gallo, G. J., Elangovan, B., Houghton, A. B., Malstrom, S., Avery, B. J., Ebb, R. G., Subramanian, T., Chittenden, T., Lutz, R. J. and Chinnadurai, G. 1995. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene. 11: 1921-8. 195 Buckbinder, L., Talbott, R., Velasco-Miguel, S., Takenaka, I., Faha, B., Seizinger, B. R. and Kley, N. 1995. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 377: 646-649. Bump, N. J., Hackett, M., Hugunin, M., Seshagiri, S., Brady, K., Chen, P., Ferenz, C., Franklin, S., Ghayur, T., Li, P., Licari, P., Mankovich, J., Shi, L., Greenberg, A. H., Miller, L. K. and Wong, W. W. 1995. Inhibition of ICE Family Proteases by Baculovirus Antiapoptotic Protein p35. Science. 269: 1885-88. Burant, C. F., Sivitz, W. I., Fukumoto, H., Kayano, T., Nagamatsu, S., Seino, S., Pessin, J. E. and Bell, G. I. 1991. Mammalian Glucose Transporter: Structure and Molecular Regulation. Recent Prog. Hormone Res. 47: 349-89. Carruthers, A. 1990. Facilitated Diffusion of Glucose. Physio. Rev. 70: 1135-76. Cheng, E. H., Levine, B., Boise , L. H., Thompson, C. B. and Hardwick, J. M. 1996. Baxindependent inhibition of apoptosis by Bcl-xL. Nature. 379: 554-556. Chinnaiyan, A. M., O'Rourke, K., Tewari, M. and Dixit, V. M. 1995. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 81: 505-12. Chiou, S. K., Rao, L. and White, E. 1994. Bcl-2 blocks p53-Dependent Apoptosis. Mol. Cell. Biol. 14: 2256-2263. Chittenden, T., Harrington, E. A., O'Connor, R., Flemington, C., Lutz, R. J., Evan, G. I. and Guild, B. C. 1995. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 374: 733-736. Chung, J. D., Sinskey, A. J. and Stephanopoulos, G. N. 1998. Growth Factor and Bcl-2 Mediated Survival During Abortive Proliferation of Hybridoma Cell Line. Biotech. Bioeng. 57: 164-171. Chung, J. D., Zabel, C., Sinskey, A. J. and Stephanopoulos, G. N. 1997. Extension of Sp2/0 Hybridoma Cell Viability Through Interleukin-6 Supplementation. Biotech. Bioeng. 55: 439-446. Clarke, A. R., Purdie, C. A., Harrison, D. J., Morris, R. G., Bird, C. C., Hooper, M. L. and Wyllie, A. H. 1993. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 362: 849-852. Clem, R., Fechheimer, M. and Miller, L. K. 1991. Prevention of Apoptosis by a Baculovirus Gene During Infection of Insect Cells. Science. 254: 1388-90. 196 Clem, R. J., Hardwick, J. M. and Miller, L. K. 1996. Anti-apoptotic genes of baculoviruses. Cell Death and Differentiaion. 3: 9-16. Cohen, G. M. 1997. Caspases: the executioners of apoptosis. Biochem. J. 326: 1-16. Cohen, J. J. 1991. Programmed Cell Death in the Immune System. Adv. Immunol. 50: 55-85. Cuende, E., AlEs-Martinez, J. E., Ding, L., GUnzalez-GarcIa, M., MartInez, A. C. and Nunez, G. 1993. Programmed Cell Death by bcl-2-dependent and independent mechanisms in B Lymphoma Cells. EMBO J. 12: 1555-1560. Draetta, G. F. 1994. Mammalian GI cyclins. Curr. Op. Cell Bio. 6: 842-46. Drapeau, D., Luan, Y.-T., Popoloski, J. A., Richards, D. T., Cohen, D. C., Sinacore, M. S. and Adamson, S. R. 1994. Extracellular insulin degrading activity creates instability in a CHO-based batch-refeed continuous process. Cytotechnology. 15: 103-109. Duan, H. and Dixit, V. M. 1997. RAIDD is a new 'death' adaptor molecule. Nature. 385: 86-9. El-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B. and Parsons, R. 1993. WAF1, a Potential Mediator of p53 Tumor Suppression. Cell. 75: 817. Elledge, S. J. 1996. Cell Cycle Checkpoints: Preventing an Identity Crisis. Science. 274: 1664-72. Ellis, R. E., Yuan, J. Y. and Horvitz, H. R. 1991. Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 7: 663-698. Enari, M., Talanian, R. V., Wong, W. W. and Nagata, S. 1996. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 380: 723726. Fanidi, A., Harrington, E. A. and I., E. G. 1992. Cooperative interaction between c-Myc and bcl-2 oncogenes. Nature. 359: 554-556. Fesus, L., Davies, P. J. and Piacentini, M. 1991. Apoptosis: molecular mechanisms in programmed cell death. Eur. J. Cell Biol. 56: 170-177. Franek, F. 1995. Starvation-Induced Programmed Death of Hybridoma Cells: Prevention by Amino Acid Mixtures. Biotech. Bioeng. 45: 86-90. Franek, F. and Chladkova-Sramkova, K. 1995. Apoptosis and nutrition: Involvement of amino acid transport system in repression of hybridoma cell death. Cytotechnology. 18: 113-117. 197 Franek, F. and Dolnikova, J. 1991. Hybridoma growth and monoclonal antibody production in iron-rich protein-free medium: Effect of nutrient concentration. Cytotechnology. 7: 33-38. Fraser, A. and Evan, G. 1996. A license to Kill. Cell. 85: 781-. Fukuda, Y., Tsuji, T., Fujita, T., Yamamoto, A. and Muranishi, S. 1995. Susceptibility of Insulin to Proteolysis in Rat Lung Homogenate and Its Protection from Proteolysis by Various Protease Inhibitors. Biol. Pharma. Bull. 18: 891-94. Fussenegger, M., Mazur, X. and Bailey, J. E. 1997. A Novel Cytostatic Process Enhances the Productivity of Chinese Hamster Ovary Cells. Biotechnol. Bioeng. 55: 927-39. Fussenegger, M., Mazur, X. and Bailey, J. E. 1998. pTRIDENT, a Novel Vector Family for Tricistrionic Gene Expression in Mammalian Cells. Biotechnol. Bioeng. 57: 1-10. Goswami, J., Sinskey, A. J., Steller, H., Stephanopoulos, G. N. and Wang, D. I. C. 1998. Apoptosis in Batch Cultures of Chinese Hamster Ovary Cells. Biotech. and Bioeng. in press: Gottschalk, A. R., Boise, L. H., Thompson, C. B. and Quintens, J. 1994. Identification of Immunosuppressant-induced apoptosis in a murine B-cell line and its prevention by bcl-x but not bcl-2. Proc. Natl. Acad. Sci. USA. 91: 7350-7354. Gu, M. B., Todd, P. and Kompala, D. S. 1993. Foreign gene expression (betagalactosidase) during the cell-cycle phases in recombinant CHO cells. Biotech. Bioeng. 42: 1113-1123. Gu, M. B., Todd, P. and Kompala, D. S. 1994. Analysis of Foreign Protein Overproduction in Recombinant CHO Cells. Annals of N.Y. Acad. Sci. 721: 194-207. Han, J., Sabbatini, P., Perez, D., Rao, L., Modha, D. and White, E. 1996. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes and Development. 10: 461-477. Harper, J. W., Adami, G. R., Wei, N., Keyomarsi, K. and Elledge, S. J. 1993. The p21 CDK-Interacting Protein Cipl Is a Potent Inhibitor of Gl Cyclin-Dependent Kinases. Cell. 75: 805. Hausdorff, S. F., Fingar, D. C. and Birnbaum, M. J. 1996. Signalling Pathways Mediating Insulin-Activated Glucose Transport. Cell & Dev. Biol. 7: 239-47. 198 Hawkins, C. J., Uren, A. G., Hacker, G., Medcalf, R. L. and Vaux, D. L. 1996. Inhibition of Interleukin 1 beta-converting enzyme-mediated apoptosis of mammalian cells by baculovirus IAP. Proc. Natl. Acad. Sci. USA. 93: 13786-90. Hengartner, M. O. 1996. Programmed cell death in invertebrates. Curr. Opin. Genet. Dev. 6: 34-38. Hengartner, M. 0. and Horvitz, H. R. 1994. Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature. 369: 318-320. Hershberger, P. A., LaCount, D. J. and Friesen, P. D. 1994. The Apoptotic Suppressor P35 is Required Early During Baculovirus Replication and is Targeted to the Cytosol of Infected Cells. J. Virol. 68: 3467-77. Hockenberry, D. M., Oltvai, Z. N., Yin, X., Milliman, C. L. and Korsmeyer, S. J. 1993. Bcl-2 Functions in an Antioxidant Pathway to Prevent Apoptosis. Cell. 75: 241-251. Huang, D. C. S., O'Reilly, L. A., Strasser, A. and Cory, S. 1997. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 16: 4628-4638. Hunter, J. J., Bond, B. L. and Parslow, T. G. 1996. Functional dissection of the human Bcl2 protein: Sequence requirements for the inhibition of apoptosis. Mol. Cell . Biol. 16: 877-883. Hunter, J. J. and Parslow, T. G. 1996. A peptide sequence from Bax that converts Bcl-2 into an activator of apoptosis. J. Biol. Chem. 271: 8521-8524. Itoh, Y., Ueda, H. and Suzuki, E. 1995. Overexpression of bcl-2, Apoptosis Suppressing Gene: Prolonged Viable Culture Period of Hybridoma and Enhanced Antibody Production. Biotech. and Bioeng. 48: Jacobson, M. D., Burne, J. F. and Raff, M. C. 1994. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J. 13: 1899-1910. Kane, D. H., Sarafian, T. A., Anton, R., Hahn, H., Gralla, E. B., Valentine, J. S., Ord, T. and Bredesen, D. E. 1993. Bcl-2 Inhibition of Neural Death: Decreased Generation of Reactive Oxygen Species. Science. 262: 1274-1276. Kane, D. J., Ord, T., Anton, R. and Bredesen, D. E. 1995. Expression of bcl-2 Inhibits Necrotic Neural Cell Death. J. Neurosci. Res. 40: 269-275. Kastan, M. B., Zhan, Q., El-Deiry, W. S., Carrier, F., Jacks, T., Walsh, W. V., Plunkett, B. S., Vogelstein, B. and Fornace, A. J. J. 1992. A mammalian cell cycle 199 checkpoint utilizing p53 and GADD45 is defective in atazia-telangiectasia. Cell. 71: 587-597. Kaufman, R. J. and Sharp, P. A. 1982. Amplification and Expression of Sequences Cotransfected with a Modular Dihydrofolate Reductase Complementary DNA Gene. Journal of Molecular Biology. 159: 601-621. Kerr, J. F. R., Wyllie, A. H. and Currie, A. H. 1972. Apoptosis, a basic biological phenomenon with wider implications in tissue kinetics. Br. J. Cancer. 26: 239-45. Kiefer, M. C., Brauer, M. J., Powers, V. C., Wu, J. J., Umansky, S. R., Tomei, L. D. and Barr, P. J. 1995. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature. 374: 736-739. King, R. W., Deshaies, R. J., Peters, J. M. and Kirschner, M. W. 1996. How Proteolysis Drives the Cell Cycle. Science. 274: 1652-59. Kluck, R. M., Bossy-Wetzel, E., Green, D. R. and Newmeyer, D. D. 1997. The Release of Cytochrome c from Mitochondria: A Primary Site for Bcl-2 Regulation of Apoptosis. Science. 275: 1132-36. Ko, L. J. and Prives, C. 1996. p53: puzzle and paradigm. Genes Dev. 10: 1054-1072. Koff, A., Ohtsuki, M., Polyak, K., Roberts, J. M. and Massagu6, J. 1993. Negative Regulation of G1 in Mammalian Cells: Inhibition of Cyclin E-Dependent Kinase by TGF-B. Science. 260: 536-9. Kozopas, K. M., Yang, T., Buchan, H. L., Zhou, P. and Craig, R. W. 1993. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. 90: Krajewski, S., Tanaka, S., Takayama, S., Schibler, M. J., Fenton, W. and Reed, J. C. 1993. Investigations of the subcellular distribution of bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 53: 4701-4704. Kroemer, G. 1997. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 3: 614-620. Kroemer, G., Zamzami, N. and Susin, S. A. 1997. Mitochondrial control of apoptosis. Immunol. Today. 18: 44-51. Lam, M., Dubyak, G., Chen, L., Nufiez, G., Miesfeld, R. L. and Distelhorst, C. W. 1994. Evidence that bcl-2 represses apoptosis by regulating endoplasmic reticulumassociated Ca + + fluxes. Proc. Natl. Acad. Sci. USA. 91: 6569-6573. 200 Lane, D. P. 1992. p53, guardian of the genome. Nature. 358: 15-16. Lao, M.-S., Toth, D., Dannel, G. and Schalla, C. 1996. Degradative activities in a recombinant chinese hamster ovary cell culture. Cytotechnology. 22: 43-52. Lazebnik, Y. A., Kaufmann, S. H., Desnoyers, S., Poirier, G. G. and Earnshaw, W. C. 1994. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 371: 346-347. Lazebnik, Y. A., Takahashi, A., Moir, R. D., Goldman, R. D. and Poirier, G. G. 1995. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc. Natl. Acad. Sci. USA. 92: 9042-46. Levine, A. J. 1997. p53, the Cellular Gatekeeper for Growth and Division. Cell. 88: 323331. Levine, B., Huang, Q., Isaacs, J. T., Reed, J. C., Griffin, D. E. and Hardwick, J. M. 1993. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature. 361: 739-42. Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S. and Wang, X. 1997. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell. 91: 479-489. Lin, D., Shields, M. T., Ullrich, S. J., Appella, E. and Mercer, W. E. 1992. Growth arrest induced by wild-type p53 protein blocks cells prior to or near the restriction point in late G1 phase. Proc. Natl. Acad. Sci. USA. 89: 9210-4. Lin, E. Y., Orlofsky, A., Berger, M. S. and Prystowsky, M. B. 1993. Characterization of Al, a novel hemopoietic-specific early-response gene with sequence similarity to bcl2. J. Immunol. (IFB). 151: 1979-88. Lincz, L. F. 1998. Deciphering the apoptotic pathway: All roads lead to death. Immunol. Cell Biol. 76: 1-19. Liston, P., Roy, N., Tamai, K., Lefebvre, C., Baird, S., Cherton-Horvat, G., Farahani, R., McLean, M., Ikeda, J. E., MacKenzie, A. and Korneluk, R. G. 1996. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature. 379: 349-353. Liu, X., Kim, C. N., Yang, J., Jemmerson, R. and Wang, X. 1996. Induction of Apoptotic Program in Cell-Free Extracts: Requriement for dATP and Cytochrome c. Cell. 86: 147-157. 201 Lotem, J. and Sachs, L. 1993. Regulation by bcl-2, c-Myc, and p-53 of Susceptibility to Induction of Apoptosis by Heat Shock and Cancer Chemotherapy Compounds in Differentiation-component and -defective Myeloid Leukemic Cells. Cell Growth Diff. 4: 41-47. Lowe, S. W., Ruley, H. E., Jacks, T. and Housman, D. E. 1993. p53-Dependent Apoptosis Modulates the Cytotoxicity of Anticancer Agents. Cell. 74: 957-967. Lowe, S. W., Schmitt, E. M., Smith, S. W., Osborne, B. A. and Jacks, T. 1993. p53 is required for the radiation induced apoptosis in mouse thymocytes. Nature. 362: 847849. Mandal, M., Maggirwar, S. B., Sharma, N., Kaufmann, S. H., Sun, S. C. and Kumar, R. 1996. Bcl-2 prevents CD95 (Fas/APO-l)-induced degradation of lamin B and poly(ADP-ribose) polymerase and restores the NF-kappaB signaling pathway. J. Biol. Chem. 271: 30354-9. Mariani, B. D., Slate, D. L. and Schimke, R. T. 1981. S phase -specific synthesis of dihydrofolate reductase in Chinese Hamster Ovary cells. Proc. Natl. Acad. Sci. USA. 78: 4985-4989. Martin, S. J., Amarante-Mendes, G. P., Shi, L., Chuang, T., Casiano, C. A., O'Brien, G. A., Fitzgerald, P., Tan, E. M., Bokoch, G. M., Greenberg, A. H. and Green, D. R. 1996. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO J. 15: 2407-16. Martin, S. J. and Cotter, T. G. 1994. Apoptosis of Human Leukemia: Induction, Morphology and Molecular Mechanisms. In: Apoptosis II: The Molecular Basis of Apoptosis in Disease, Cold Spring Harbor Laboratory Press, Martin, S. J. and Green, D. R. 1995. Protease Activation during Apoptosis: Death by a Thousand Cuts? Cell. 82: 349-52. Marx, J. 1993. Howp53 Suppresses Cell Growth. Science. 262: 1644-1645. Mastrangelo, A. J. and Betenbaugh, M. J. 1998. Overcoming apoptosis: new methods for improving protein-expression systems. TIBTECH. 16: 88-95. Mastrangelo, A. J., Hardwick, J. M. and Betenbaugh, M. J. 1996. Bcl-2 inhibits apoptosis and extends recombinant protein production in cells infected with Sindbis viral vectors. Cytotechnology. 22: 169-178. 202 Matsuyama, S., Xu, Q., Velours, J. and Reed, J. C. 1998. The Mitochondrial FoF 1ATPase Proton Pump Is Required for Function of the Proapoptotic Protein Bax in Yeast and Mammalian cells. Mol. Cell. 1: 327-336. McConkey, D. J. 1996. Calcium-dependent, interleukin 1-beta-converting enzyme inhibitor-insensitive degradation of lamin B 1 and DNA fragmentation in isolated thymocyte nuclei. J. Biol. Chem. 271: 22398-406. McGahon, A. J., Martin, S. J., Bisonette, R. P., Mahboubi, A., Yufang, S., Mogil, R. J., Nishioka, W. K. and Green, D. R. 1995. The End of the (Cell) Line: Methods for the Study of Apoptosis in Vitro. In: L. M. Schwartz and B. Osborne, Methods in Cell Biology, Academic Press, Inc., San Diego. McKeon, F. 1991. Nuclear lamin proteins: Domains required for nuclear targeting, assembly, and cell-cycle-regulated dynamics. Curr. Opin. Cell Biol. 3: 82-86. Mercer, W. E., Shields, M. T., Amin, M., Sauve, G. J., Appella, E., Romano, J. W. and Ullrich, S. J. 1990. Negative growth regulation in a glioblastoma tumor cell line that conditionally expresses human wild-type p53. Proc. Natl. Acad. Sci. USA. 87: 616670. Mercille, S. and Massie, B. 1994. Induction of Apoptosis in Nutrient-Deprived Cultures of Hybridoma and Myeloma Cells. Biotech. and Bioeng. 44: 11401154. Miura, M., Zhu, H., Rotello, R., Hartwieg, E. A. and Yuan, J. 1993. Induction of Apoptosis in Fibroblasts by IL- lb-Converting Enzyme, a Mammalian Homolog of the C. elegans Cell Death Gene ced-3. Cell. 75: 653-660. Miyashita, T. and Reed, J. C. 1995. Tumor Suppressor p53 Is a Direct Transcriptional Activator of the Human bax Gene. Cell. 80: 293-299. Miyazaki, T., Liu, Z. J., Kawahara, A., Minami, Y., Yamada, K., Tsujimoto, Y., Barsoumian, E. L., Perlmutter, R. M. and Taniguchi, T. 1995. Three Distinct IL-2 Signaling Pathways Mediated by bcl-2, c-myc, and Ick Cooperate in Hematopoietic Cell Proliferation. Cell. 81: 223-31. Moore, A., Donahue, C. J., Hooley, J., Stocks, D. L., Bauer, K. D. and Mather, J. P. 1995. Apoptosis in CHO cell batch cultures: examination by flow cytometry. Cytotechnology. 17: 1-11. Moore, A., Mercer, J., Dutina, G., Donahue, C. J., Bauer, K. D., Mather, J. P., Etcheverry, T. and Ryll, T. 1997. Effects of temperature shift on cell cycle, apoptosis and nucleotide pools in CHO cell batch cultures. Cytotechnology. 23: 47-54. 203 Morishita, M., Morishita, I., Takayama, K., Machida, Y. and Nagai, T. 1992. Novel oral microspheres of insulin with protease inhibitor protecting from enzymatic degradation. Int. J. of Pharmaceutics. 78: 1-7. Mosser, D. D. and Massie, B. 1994. Genetically Engineering Mammalian Cell Lines for Increased Viability and Productivity. Biotech. Adv. 12: 253-277. Muchmore, S. W., Sattler, M., Liang, H., Meadows, R. P., Harlan, J. E., Yoon, H. S., Nettesheim, D., Chang, B. S., Thompson, C. B., Wong, S. L., Ng, S. L. and Fesik, S. W. 1996. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 381: 335-41. Mummenbrauer, T., Janus, F., Muller, B., Wiesmuller, L., Deppert, W. and Grosse, F. 1996. p53 Protein Exhibits 3'-to-5' Exonuclease Activity. Cell. 85: 1089-1099. Murakami, H. 1989. Serum-free media used for cultivation of hybridomas. In: Monoclonal antibodies: Production and applications, Alan R. Liss, New York. Murray, K., Ang, C., Gull, K., Hickman, J. A. and Dickson, A. J. 1996. NSO Myeloma Cell Death: Influence ofbcl-2 Overexpression. Biotech. Bioeng. 51: 298-304. Nagata, S. 1996. Apoptosis: Telling cells their time is up. Curr. Biol. 6: 1241-43. Naumovski, L. and Cleary, M. L. 1996. The p53-binding protein 53BP2 also interacts with Bcl2 and impedes cell cycle progression at G2/M. Mol. Cell. Biol. 16: 3884-92. Newmeyer, D. D., Farschon, D. M. and Reed, J. C. 1994. Cell-Free Apoptosis in Xenopus Egg Extracts: Inhibition by Bcl-2 and Requirement for an Organelle Fraction Enriched in Mitochondria. Cell. 79: 353-364. Nicholson, D. 1996. ICE/CED3-like Proteases as Therapeutic Targets for the Control of Inappropriate Apoptosis. Nature Biotech. 14: 297-301. Nyberg, G. B. 1998. Glycosylation Site Occupancy Heterogeneity in Chinese Hamster Ovary Cell Culture. Massachusetts Institute of Technology. Oberhammer, F. A., Hochegger, K., Fr6shcl, G., Tiefenbacher, R. and Pavelka, M. 1994. Chromatin Condensation during Apoptosis Is Accompanied by Degradation of Lamin A+B, without Enhanced Activation of cdc2 Kinase. J. Biol. Chem. 126: 827-37. Ohtsubo, M. and Roberts, J. M. 1993. Cyclin-Dependent Regulation of G 1 in Mammalian Fibroblasts. Science. 259: 1908-11. 204 Ohtsubo, M., Theodoras, A. M., Schumacher, J., Roberts, J. M. and Pagano, M. 1995. Human Cyclin E, a Nuclear Protein Essential for the G 1 -to-S Phase Transition. Mol. Cell. Biol. 15: 2612-24. Okamoto, K. and Beach, D. 1994. Cyclin G is a transcriptional target of the p53 tumor suppressor protein. EMBO J. 13: 4816-4822. Olsen, C. W., Kehren, J. C., Dybdahl-Sissoko, N. R. and Hinshaw, V. S. 1996. bcl-2 alters influenza virus yield, spread, and hemagglutinin glycosylation. J. Virol. 70: 663666. Oltvai, Z. N., Milliman, C. L. and Korsmeyer, S. J. 1993. Bcl-2 Heterodimerizes In Vivo with a Conserved Homolog, Bax, That Accelerates Programmed Cell Death. Cell. 74: 609-619. Oren, M. and Prives, C. 1996. p53: upstream, downstream and off stream. Biochim. Biophys. Acta. 1288: R13-R19. Orrenius, S., Burkitt, M. J., Kass, G. E. N., Dypbukt, J. M. and Nicotera, P. 1992. Calcium ions and oxidative cell injury. Ann. Neurol. 32: S33-S42. Orth, K., Chinnaiyan, A. M., Garg, M., Froelich, C. J. and Dixit, V. M. 1996. The CED3/ICE-like Protease Mch2 Is Activated during Apoptosis and Cleaves the Death Substrate Lamin A. J. Biol. Chem. 271: 16443-46. Orth, K. and Dixit, V. M. 1997. Bik and Bak Induce Apoptosis Downstream of CrmA but Upstream of Inhibitor of Apoptosis. J. Biol. Chem. 272: 8841-8844. Pan, G., Humke, E. W. and Dixit, V. M. 1998. Activation of caspases triggered by cytochrome c in vitro. FEBS Lett. 426: 151-154. Pan, G., O'Rourke, K. and Dixit, V. M. 1998. Caspase-9, Bcl-XL, and Apaf-1 Form a Ternary Complex. J. Biol. Chem. 273: 5841-5845. Patterson, M. K. 1979. Measurement of growth and viability of cells in culture. Methods Enzymol. 58: 141-152. Perreault, J. and Lemieux, R. 1994. Essential role of optimal protein synthesis in preventing the apoptotic death of B cell hybridomas. Cytotechnology. 13: 99-105. Pessin, J. E. and Bell, G. I. 1992. Mammalian Facilitative Glucose Transporter Family: Structure and Molecular Regulation. Annu. Rev. Physiol. 54: 911-30. 205 Petit, P. X., Susin, S.-A., Zamzami, N., Mignotte, B. and Kroemer, G. 1996. Mitochondria and programmed cell death: back to the future. FEBS Letters. 396: 713. Pimentel, E. 1994. Insulin. In: Handbook of Growth Factors, 1. CRC Press, Rabizadeh, S., LaCount, D. J., Friesen, P. D. and Bredesen, D. E. 1993. Expression of the Baculovirus p35 Gene Inhibits Mammalian Neural Cell Death. J. Neurochem. 61: 2318-21. Raff, M. C., Barres, B. A., Burne, J. F., Coles, H. S., Ishizaki, Y. and Jacobson, M. D. 1993. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 262: 695-700. Reed, J. C. 1994. Bcl-2 and the Regulation of Programmed Cell Death. The Journal of Cell Biology. 124: 1-6. Reed, J. C. 1997. Cytochrome c: Can't Live with It - Can't Live without It. Cell. 91: 559562. Reed, J. C. 1997. Double Identity for proteins of the Bcl-2 family. Nature. 387: 773-776. Renner, W. A., Lee, K. H., Hatzimanikatis, V., Bailey, J. E. and Eppenberger, H. M. 1995. Recombinant Cyclin E Expression Activates Proliferation and Obviates Surface Attachment of Chinese Hamster Ovary (CHO) Cells in Protein-Free Medium. Biotech. Bioeng. 47: 476-. Salvesen, G. S. and Dixit, V. M. 1997. Caspases: Intracellular Signaling by Proteolysis. Cell. 91: 443-446. Sambrook, J., Fritsch, E. F. and Maniatis, T. 1989. Molecular Cloning. 2nd. Cold Spring Harbor Laboratory Press, Cold Spring Harbor. Scahill, S. J., Devos, R., van der Heyden, J. and Fiers, W. 1983. Expression and characterization of the product of a human immune interferon cDNA gene in Chines hamster ovary cells. Proc. Natl. Acad. Sci. USA. 80: 4654-58. Sen, S. and D'lncalci, M. 1992. Apoptosis. Biochemical events and relevance to cancer chemotherapy. FEBS Lett. 307: 122-127. Sherr, C. J. 1994. G1 Phase Progression: Cycling on Cue. Cell. 79: 551-555. Sherr, C. J. 1996. Cancer Cell Cycles. Science. 274: 1672-1677. Shibasaki, F., Kondo, E., Akagi, T. and McKeon, F. 1997. Suppression of signalling through NF-AT by interactions between calcineurin and Bcl-2. Nature. 386: 728-731. 206 Shimizu, S., Eguchi, Y., Kamiike, W., Itoh, Y., Hasegawa, J., Yamabe, K., Otsuki, Y., Matsuda, H. and Tsujimoto, Y. 1996. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-XL. Cancer Res. 56: 2161-6. Simpson, N. J., Milner, A. E. and Al-Rubeai, M. 1997. Prevention of Hybridoma Cell Death by bcl-2 During Suboptimal Culture Conditions. Biotech. Bioeng. 54: 1-16. Singh, R. P., Al-Rubeai, M., Gregory, C. D. and Emery, A. N. 1994. Cell Death in Bioreactors: A role for Apoptosis. Biotech. and Bioeng. 44: 720-726. Singh, R. P., Emery, A. N. and Al-Rubeai, M. 1996. Enhancement of Survivability of Mammalian Cells by Overexpression of the Apoptosis-Suppressor Gene bcl-2. Biotech. Bioeng. 52: 166-175. Singh, R. P., Finka, G., Emery, A. N. and Al-Rubeai, M. 1997. Apoptosis and its control in cell culture systems. Cytotechnology. 23: 87-93. Stasser, A., Harris, A. W., Jacks, T. and Cory, S. 1994. DNA Damage Can Induce Apoptosis in Proliferating Lymphoid Cells via p53-Independent Mechanisms Inhibitable by Bcl-2. Cell. 79: 329-339. Subler, M. A., Martin, D. W. and Deb, S. 1992. Inhibition of viral and cellular promoters by human wild-type p53. J. Virol. 66: 4757-4762. Susin, S. A., Zamzami, N., Castedo, M., Hirsch, T., Marchetti, P., Macho, A., Daugas, E., Geuskens, M. and Kroemer, G. 1996. Bcl-2 Inhibits the Mitochondrial Release of an Apoptogenic Protease. J. Exp. Med. 184: 1331-41. Suzuki, E., Terada, S., Ueda, H., Fujita, T., Komatsu, T., Takayama, S. and Reed, J. C. 1997. Establishing apoptosis resistant cell lines for improving protein productivity of cell culture. Cytotechnology. 23: 55-59. Takahashi, A. and Earnshaw, W. C. 1996. ICE-related proteases in apoptosis. Curr. Op. Gen. Dev. 6: 50-55. Takayama, S., Sato, T., Krajewski, S., Kochel, K., Irie, S., Millan, J. A. and Reed, J. C. 1995. Cloning and Functional Analysis of BAG-1: A Novel Bcl-2-Binding Protein with Anti-Cell Death Activity. Cell. 80: 279-284. Tanke, J. J., Van der Linden, P. W. G. and Langerak, J. 1982. Alternative Fluorochromes to Ethidium Bromide for Automated Read Out of Cytotoxicity Tests. J. Immunol. Methods. 52: 91-96. 207 Terada, S., Itoh, Y., Ueda, H. and Suzuki, E. 1997. Characterization and fed-batch culture of hybridoma overexpressing apoptosis suppressing gene bcl-2. Cytotechnology. 24: 135-141. Thompson, C. B. 1995. Apoptosis in the Pathogenesis and Treatment of Disease. Science. 267: 1456-1462. Thomberry, N. A. and Molineaux, S. M. 1995. Interleukin-1-0 converting enzyme: A novel cysteine protease required for IL-1 3 production and implicated in programmed cell death. Protein Sci. 4: 3-12. Tozaki, H., Emi, Y., Horisaka, E., Fujita, T., Yamamoto, A. and Muranishi, S. 1997. Degradation of Insulin and Calcitonin and Their Protection by Various Protease Inhibitors in Rat Caecal Contents: Implications in Peptide Delivery to the Colon. J. Pharm. Pharmacol. 49: 164-168. Trost, L. C. and Lemasters, J. J. 1994. A Cytotoxicity Assay for Tumor Necrosis Factor Emplying a Multiwell Flourescence Scanner. Anal. Biochem. 220: 149-153. Trump, B. F. and Mergner, W. J. 1974. Cell Injury. Second. Academic Press, New York. Vander Heiden, M. G., Chandel, N. S., Williamson, E. K., Schumacker, P. T. and Thompson, C. B. 1997. Bcl-xL Regulates the Membrane Potential and Volume Homeostasis of Mitochondria. Cell. 91: 627-637. Vaux, D. L., Cory, S. and Adams, J. M. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 335: 440-442. Vaux, D. L., Haecker, G. and Strasser, A. S. 1994. An Evolutionary Perspective on Apoptosis. Cell. 76: 777-779. Vaux, D. L. and Strasser, A. 1996. The molecular biology of apoptosis. Proc. Natl. Acad. Sci USA. 93: 2239-2244. Vollenweider, I. and Groscurth, P. 1992. Comparison of four DNA staining fluorescence dyes for measuring cell proliferation of lymphokine-activated killer (LAK) cells. J. Immunol. Methods. 149: 133-135. Vomastek, T. and Franek, F. 1993. Kinetic of development of spontaneous apoptosis in B cell hybridoma cultures. Immunol. Letters. 35: 19-24. Wallach, D., Boldin, M., Varfolomeev, E., Beyaert, R., Vandenabeele, P. and Fiers, W. 1997. Cell death induction by receptors of the TNF family: towards a molecular understanding. FEBS Lett. 410: 96-106. 208 Wang, X., Pai, J., Wiedenfeld, E. A., Medina, J. C., Slaughter, C. A., Glodstein, J. L. and Brown, M. S. 1995. Purification of an Interleukin-l-3 Converting enzyme-related Cysteine Protease that Cleaves Sterol Regulatory Element-binding Proteins between the Leucine Zipper and Transmembrane Domains. J. Biol. Chem. 270: 18044-50. Wang, X., Zelenski, N. G., Yang, J., Sakai, J., Brown, M. S. and Goldstein, J. L. 1996. Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J. 15: 1012-20. Wilson, K. P., Black, J. A., Thomson, J. A., Kim, E. E., Griffith, J. P., Navia, M. A., Murcko, M. A., Chambers, S. P., Aldape, R. A., Raybuck, S. A. and Livingston, D. J. 1994. Structure and mechanism of interleukin-1 beta converting enzyme. Nature. 370: 270-5. Wyllie, A. H., Kerr, J. F. R. and Currie, A. R. 1980. Cell Death: The Significance of Apoptosis. Int. Rev. Cytol. 68: 251-306. Xiang, J., Chao, D. T. and Korsmeyer, S. J. 1996. BAX-induced cell death may not require interleukin 1-b-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA. 93: 14559-14563. Xie, L., Nyberg, G., Gu, X., Li, H., M611born, F. and Wang, D. I. C. 1997. GammaInterferon Production and Quality in Stoichiometric Fed-Batch Cultures of Chinese Hamster Ovary (CHO) Cells under Serum-Free Conditions. Biotech. and Bioeng. 56: 577-82. Xie, L. and Wang, D. I. C. 1994a. Fed-batch Cultivation of Animal Cells Using Different Medium Design and Feeding Strategies. Biotech. Bioeng. 43: 1175. Xie, L. and Wang, D. I. C. 1994b. Stoichiometric Analysis of Animal Cell Growth and its Application in Medium Design. Biotech. and Bioeng. 43: Xu, Q. and Reed, J. C. 1998. Bax Inhibitor-1, a Mammalian Apoptosis Suppressor Identified by Functional Screening in Yeast. Mol. Cell. 1: 337-346. Xue, D. and Horvitz, H. R. 1995. Inhibition of Caenorhabditis elegans cell death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature. 377: 248-251. Yamamoto, A., Taniguchi, T., Rikyuu, K., Tsuji, T., Fujita, T., Murakami, M. and Muranishi, S. 1994. Effects of Varioius Protease Inhibitors on the Intestinal Absorption and Degradation of Insulin in Rats. Pharma. Res. 11: 1496-1500. Yang, E. and Korsmeyer, S. J. 1996. Molecular Thanatopsis: A discourse on the Bcl-2 family and cell death. Blood. 88: 386-401. 209 Yang, E., Zha, J., Jockel, J., Boise, L. H., Thompson, C. B. and Korsmeyer, S. J. 1995. Bad, a Heterodimeric Partner for Bcl-xL and Bcl-2, Displaces and Promoter Cell Death. Cell. 80: 285-291. Yang, J., Xuesong, L., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J., Peng, T., Jones, D. P. and Wang, X. 1997. Prevention of Apoptosis by Bcl-2: Release of Cytochrome c from Mitochondria Blocked. Science. 275: 1129-32. Zamzami, N., Susin, S. A., Marchetti, P., Hirsch, T., Gomez-Monterrey, I., Castedo, M. and Kroemer, G. 1996. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 183: 1533-44. Zha, H., Aime-Sempre, C., Sato, T. and Reed, J. C. 1996. Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J. Biol. Chem. 271: 7440-7444. Zha, H., Fisk, H. A., Yaffe, M. P., Mahajan, N., Herman, B. and Reed, J. C. 1996. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell. Biol. 16: 6494-6508. Zha, J. P., Harada, H., Yang, E., Jockel, J. and Korsmeyer, S. J. 1996. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not Bcl-xL. Cell. 87: 619-628. Zhou, Q. and Salvesen, G. S. 1997. Activation of pro-caspase-7 by serine proteases includes a non-canonical specificity. Biochem. J. 324: 361-364. Zhou, W. and Hu, W.-S. 1995. Effect of Insulin on a Serum-Free Hybridoma Culture. Biotech. Bioeng. 47: 181-185. 210