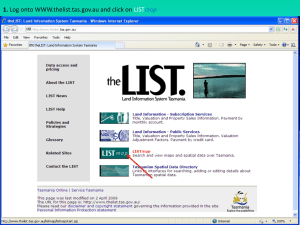
Document 10591551
advertisement

Influence of recent Asian SO 2 and Asian NO. emissions change (2001 to 2010) on particulate matter: shifts in Asian sulfate enhancement over US surface, major production pathway, and lifetime by MASSACHUSE TTS RNM"79E OF TEC HNOLOGY Flora Min 6 B.S., Biological Environmental Engineering Cornell University, 2010 LIBR) ARIES M.Eng, Biological Environmental Engineering Cornell University, 2011 Submitted to the Department of Earth, Atmospheric and Planetary Sciences in Partial Fulfillment of the Requirements for the Degree of Master of'Science in Atmospheric Science at the Massachusetts Institute of Technology September 2014 D 2014 Massachusetts Institute of Technology. All rights reserved Signature redacted .--------------------------Signature of A uthor .................... r --------------------------Department of Earth, Atmospheric and Planetary Sciences Signature redacted ........... Certified by ..... ... Susan Solomon Professor of Earth, Atmospheric and Planetary Sciences Thesis Supervisor Signature redacted Accepted by.......... 20 ........................... Robert D. van der Hilst Schlumberger Professor of Earth Sciences Head, Department of Earth, Atmospheric and Planetary Sciences 2 Influence of recent Asian SO 2 and Asian NO. emissions change (2001 to 2010) on particulate matter: shifts in Asian sulfate enhancement over US surface, major production pathway, and lifetime by Flora Min Submitted to the Department of Earth, Atmospheric and Planetary Sciences on August 27, 2014 in Partial Fulfillment of the Requirements for the Degree of Master of Science in Earth, Atmospheric and Planetary Sciences Abstract A 3-D chemical transport model with coupled oxidant-aerosol chemistry (GEOSChem) is used to analyze the influence of recent (2001 to 2010) growth in Asian NOx and Asian SO 2 emission on transpacific transport of Asian particulate matter, especially sulfate, by quantifying and analyzing the details of chemistry and its sequential influence on transpacific transport during spring and summer. From 2001 to 2010, the spring averaged Asian sulfate enhancement in the western US model surface layer increases by 0.01-0.03 pg M-3 (about 4% of the surface sulfate concentration over the US at 2010 emission level). The net chemical production rate increases in the troposphere over Asia but decreases over the Pacific Ocean. The sulfate production pathway shifts toward gas phase production by OH (7-8% increase-China; 2-3% increase-Upper atmosphere Pacific Ocean) with corresponding reductions in aqueous phase production by H 2 0 2 ; resulting in a switch of the most important production pathway from aqueous production by H 2 0 2 to gas phase production by OH. Almost doubling the Asian NO. emission during the period has a negligible influence on Asian sulfate enhancement over the US; this change in Asian NOx emission causes the net chemical production rate to increase in the Asian boundary layer, but to decrease in the upper Asian atmosphere, and in the atmosphere over the Pacific Ocean and the US. This is because the Asian NOx emission growth strengthens the oxidizing power in the Asian boundary layer to more actively form sulfate (mostly removed by wet scavenging), reducing available SO 2 for further production away from the source. The Asian NOx emission growth is the major driver in changing the sulfate production pathways toward the gas phase. The 20% increase in Asian SO 2 emission during the period is the primary driver in increasing Asian sulfate enhancement over the US; this change accelerates net sulfate production rate, and makes a minor contribution to shifting toward gas phase production by OH over the course of the transpacific transport. The calculated sulfate column burden shows a notable increase as a response to the changes in Asian emissions. However, the shifts in sulfate lifetime over China due to changes in Asian SO 2 and/or Asian NO, emission are almost negligible (generally 1-2%), and lifetimes over the upper level Pacific Ocean are generally reduced by 3-4%. Thesis Supervisor: Susan Solomon Title: Ellen Swallow Richards Professor of Atmospheric Chemistry & Climate Science 3 Acknowledgements First and foremost, I would like to express my sincere gratitude to my advisor Professor Susan Solomon, who is my esteemed promoter, thanks for accepting me as a graduate student, your warm encouragement, thoughtful guidance, critical comments, and correction of the thesis. I would also like to thank the rest of my thesis committee: Professor Noelle Selin and Professor Colette Heald for their encouragements and insightful comments that allowed me the opportunity to develop deeper thoughts and understanding of this research. Lastly, I would like to express gratitude toward my family. My father, Euoo Sung Min, for his wisdom, bottomless support, and warm encouragements, which seeded the motivations for me to dream of being a better person. My mother, Soon Uk Chung, for her warm heart, measureless care and supports, which in all nurture me and give me the strength to take big steps forward. My older sister, Yoonyoung Min, for her ardent enthusiasm as a scholar as well as for being a loving and caring sister, and a lighthouse to shine and guide. 4 Introduction Aerosols influence diverse aspects of the atmosphere as well as human well-being. Some influences on our atmosphere include direct and indirect radiation effects [IPCC, 2013] and stratospheric ozone depletion [Solomon, 1999]. Sub-micro aerosols in the free troposphere are primarily composed of sulfate and organic materials [Murphy et al., 2006], which are major components of particulate matter. Particulate matter is a complex mixture of extremely small aerosols (particles and liquid droplets), which is composed of various chemical species, including acids (nitrates and sulfates), organics, metals, and dust particles [Seinfeld and Pandis, 1998]. In remote national parks, particulate matter may cause visibility impairment while in cities, particles are linked to severe health issues including cardiovascular mortality, inflammatory lung injury, stroke mortality, and pulmonary inflammation [Pope, 2000; Pope and Dockery, 2006]. In the past, the United States faced severe particulate matter pollution. This sparked active mitigation on domestic particulate matter precursor emissions through legislative actions, such as implementation of National Ambient Air Quality Standards (NAAQS) under the Clean Air Act. Since then, emissions of precursors for particulate matter, including sulfur dioxide (SO 2), have been successfully decreased and current particulate matter exceedances of health related National Ambient Air Quality Standards (NAAQS) have decreased dramatically (with remaining problems in regions over California and the eastern US). However, reductions in health related issues didn't necessarily transit into reductions in visibility impairment issues in wilderness areas and national parks. In order to solve these problems, US Environmental Protection Agency (EPA) implemented the Regional Haze Rule, which mandates reductions in domestic anthropogenic pollutant emissions over the period of 2004 through 2018, with a goal of achieving "natural visibility conditions" by 2064 [US EPA, 2003]. In the US, visibility problems are mostly due to aerosols including carbonaceous, sulfate, nitrate, and soil dust [US EPA, 2003]. Domestic emission control will be the primary concern, but prior studies have shown that transboundary influences from foreign countries may hinder the goal of achieving "natural visibility conditions" [Park et al., 2004; Park et al., 2006]. Also, prior studies showed that Asian sulfate affects visibility problems, especially over the western US [Park et al., 2004; Park et al., 2006; Heald et al., 2006]. In contrast to the successful reduction of particulate matter precursor emissions in the US, China experienced a steep growth in particulate matter precursor emissions, including sulfur dioxide (SO 2) and nitrogen oxides (NO, = NO + NO 2) over the last decade [Granier et al., 2011; Zhao et al., 2013a, 2013b; Lu et al., 2011; Zhang et al., 2009; Streets, 2003; Lin et al., 2010,2013; Gu et al., 2013; Mijling et al., 2013], as an undesirable byproduct of rapid economic advancement. This growth in emissions raised concerns in large cities of China, as it could cause visibility impairment issues, as well as severe health problems such as an increase in premature deaths and respiratory illness. Also, growth in Chinese emissions can expand its influences on an intercontinental scale from China to the US and Canada through long-range transport of Asian pollution going across the Pacific. Thus, there have been a number of studies that analyze enhancements of particulate matter levels over North America attributable to Asian influence [Park et al., 2004; Park et al., 2006; Heald et al., 2006; Dunlea et al., 2009; Liu et al., 2008]. The episodic transpacific transport of Asian pollution plumes monitored through satellite data [Heald et al., 2006; Zhang et al., 2008; Lin et al., 2012], showed a signature of Asian plumes that are lofted from the Asian boundary layer and reach the US. Aircraft measurements provided detailed snapshots of transpacific transports of Asian pollution through chemical composition analysis on plumes that were encountered while crossing the Pacific Ocean over to the US, focusing especially on air masses with distinctive 5 characteristics attributable to Asia. Often, plumes with notably high sulfur content are found to originate from Asia [Liang et al., 2007; Lin et al., 2012; Dunlea et al., 2009; Donkelaar et al., 2008; Andreae et al., 1988]. Also, high contents of chemical tracers, such as methyl tertiary butyl ether (MTBE) and Halon H-1211, are used as indicators of Asian origin [Dunlea et al., 2009]. Plumes with large amounts of MTBE, which is used as a gasoline additive in Asia, signal that the air masses are from Asia because North America no longer uses MTBE as gasoline additives [West et al., 2007]. Plumes with high Halon H-1211 level may similarly be identified to be from Asia, since this Halon is largely linked to emissions from developing countries like China [Barletta et al., 2006]. These Asian plumes are most often identified within the free troposphere over the Pacific Ocean, and they can be traced over land, including the western US [Zhang et al., 2008; Heald et al., 2006] and Canada [Donkelaar et al., 2008]. Aerosols in the free troposphere over the Pacific are largely from Asian origin [Andreae et al., 1988; Donkelaar et al., 2008], especially from East Asia [Donkelaar et al., 2008], and are estimated to account for more than 50% of sulfate column burden between 500-900hPa in the free troposphere over British Columbia [Donkelaar et al., 2008]. The lower free troposphere (600-900 hPa) is a main pathway for transpacific transport of aerosols [Heald et al., 2006] including sulfate [Donkelaar et al., 2008], because the marine boundary layer has active wet scavenging [Heald et al., 2006]. Also, during lifting, condensation and wet deposition efficiently removes pollutants in the boundary layer [Brock et al., 2004; Donkelaar et al., 2008]. In the free troposphere, an increasing trend of ozone during the period under higher Asian influence has been recognized [Cooper et al., 2010], which further supports active transport of Asian pollution. The plumes that enter the US boundary layer are less readily identifiable at the surface, presumably due to dilution of plumes while descending into the boundary layer [Jaffe et al., 2005; Price et al., 2004; Zhang et al., 2008; Lin et al., 2012]. Thus, the influence of Asian pollution on the surface is manifested as an increase in background pollution levels [Zhang et al., 2008], rather than as distinguishable plume air masses. This increase may be associated with simultaneous large-scale enhancement of pollutants such as sulfate and ozone over the western US in the surface layer, which has been attributed to increases in Asian emissions [Zhang et al., 2008; Cooper et al., 2011; Zhang et al., 2011]. Modeling studies indicate a significant Asian sulfate enhancement in the surface layer, with Asian sulfate accounting for about 30% of total sulfate background levels over the western US (pollutant emissions held at 2001 level) [Park et al., 2004; Heald et al., 2006], in contrast to Asian nitrate and Asian ammonium, which was found to have almost negligible influence [Park et al., 2004]. A number of prior studies based on observational data give further evidence of Asian sulfate influences over the US. Episodes of elevated sulfate concentrations with signals of Asian origin have been observed [Jaffe et al., 1999, 2001, 2003a, Bertschi et al., 2004], and the sulfate concentration from surface data averaged only for selected days with. maximum simulated Asian influence is found to be higher than the seasonal average [Heald et al., 2006]. A high spatial correlation with no significant time lag on Asian aerosol levels among an ensemble of northwestern US sites with geographic proximity were found in a modeling study [Heald et al., 2006], in concordance with observed large scale influences of Asian sulfate over the northwestern US. Often Asian dust and sulfate have been found together, with high correlations between Asian sulfate contributions predicted from a model and observed dust concentrations from surface measurement sites in the US [Heald et al., 2006]. Studies on other chemical species, like ozone, further support an Asian influence on the US surface layer. Asian ozone is considered to be more efficiently transported than Asian sulfate, because the wet removal efficiency of particulate matter is 6 higher than that of ozone [Park et al., 2004; Dunlea et al., 2009; Brock et al., 2004]. Increases in surface background ozone over the US have also been attributed to rising Asian emissions, [Zhang et al., 2008; Cooper et al., 2011; Zhang et al., 2011; Fiore et al., 2002] which is supported by the finding that 11 widely scattered surface ozone measurement sites in the western US showed similar inter-annual correlations (sometimes with almost simultaneous enhancement in nearly all sites for certain times), which suggest a large scale process operating across those regions when Asian influences are relatively large [Jaffe et al., 2010; Jaffe & Ray, 2007]. Mechanisms for lofting pollution from the Asian boundary layer, and the subsequent transport across the Pacific have been investigated [Liang et al., 2004; Liu et al., 2003; Dickerson et al., 2007; Luan et al., 2012]. Pollution in the Asian planetary boundary layer is typically lofted into the free troposphere through frontal lifting (associated with warm conveyor belts within midlatitude cyclones), deep convection, orographic forcing, and mountain-chimney effects [Cooper et al., 2004; Liang et al., 2004; Liu et al., 2003; Dickerson et al., 2007]. Warm conveyor belt lifting has been identified as the more important transport pathway, and a few transport events associated with warm conveyor belt lifting have been shown to cause notable effects over the Pacific and the US [Cooper et al., 2004; Lin et al., 2012; Dunlea et al., 2009]. These warm conveyor belts are particularly active during spring, and work as the major mechanism throughout the spring season [Liang et al., 2005; Luan et al., 2012]. In general, the maximum Asian pollution outflow occurs during spring [Jacob et al., 2003; Jaffe et al., 2010; Zhang et al., 2008, 2011; Lin et al., 2012]. In addition to warm conveyor belt lifting, convective processes can be also important in summer for a longrange transport [Thompson et al., 1994; Li et al., 2005; Luan et al., 2012]. While ascending to the free troposphere, air masses often experience cloud formation and precipitation processes. Consequently, soluble gas species as well as hygroscopic aerosols are lost through wet scavenging. Once in the free troposphere, lifted Asian pollutants are efficiently transported across the Pacific to the west coast of the US by prevailing free-tropospheric westerlies, while experiencing chemical transformation on a time scale of about 1-2 weeks [Jaffe et al., 2001; Stohl et al., 2002]. A sequence of chemical transformations affecting Asian aerosols and aerosol precursors has been mapped out: from ascending in the Asian boundary layer, crossing the Pacific, to descending over the North America, based on findings from aircraft campaigns, which examined the particle size distribution and composition measurements of plumes with Asian signatures [Brock et al., 2004; Dunlea et al., 2009]. The efficiency of transpacific transport of Asian pollution in relation to chemistry depends on sequential linkages among precursor emissions and corresponding chemistry: in the planetary boundary layer over the source region, wet scavenging removal of soluble gas species and hygroscopic particles during lifting to the free troposphere, and interactions of species available for further chemical transformation in the free troposphere over the Pacific during the transport [Brock et al., 2004; Dunlea et al., 2009]. Even though two prior studies by Brock et al. and Dunlea et al. paint a general picture of the chemistry involved in transpacific transport, much of the topic remains to be studied as was pointed out in Dunlea et al., 2009. Two studies focussed on measurements of individual plume events encountered during aircraft campaigns; thus, they are limited to providing insights on the mean temporal and spatial chemistry as well as detailed chemistry (such as identification of major chemical production pathways, the responses to the recent changes in precursor emissions in Asia, and quantification of the responses in chemistry and physics for transpacific transport); these objectives all can be achieved with simulation studies using global chemistry circulation models. Other studies analyzed overall changes in the Asian sulfate influence over the US [Liu et al., 2008] and shifts in the chemical production pathway over the source region (East 7 Asia/Southeast China) [Manktelow et al., 2007; Barth & Church, 1999], with sensitivity testing on sulfur dioxide emissions in East Asia/Southeast China for the years 1985 and 2000. However, not enough attention has been given to the response to concurrent shifts in oxidizing agents or NO, emissions in the region, despite dramatic changes that have occurred - e.g., almost doubling of the Chinese NO, emissions from 2000 to 2010. The chemical coupling between ozone precursors (including NO) and sulfate [Unger et al., 2006; Meng et al., 1997] has been analyzed for regional chemistry over China, but the chemical relationships also occur during long-range transport, which is further complicated by the coupling between chemistry and physics during the transpacific transport of Asian pollution. The objective of this study is to the broaden the knowledge of the influence of recent (2001 to 2010) Asian emissions changes on transpacific influences on particulate matter, especially on sulfate, by quantifying and analyzing the details of chemistry, and its sequential influence on a transport by (1) comparing the simulation result on Asian particulate matter influences at the US surface against selected prior modeling studies as a model validation step, (2) studying the alterations in Asian sulfate influence over the US with various emission inventory scenarios, (3) identifying the current major chemical production pathway of sulfate and understanding changes in chemistry over China as well as over the Pacific Ocean with various emission scenarios, (4) analyzing the reasons for responses over the Pacific, and (5) considering the possible influence of shifts in major production pathway on sulfate column burdens and lifetimes of sulfate over China and the Pacific Ocean. Background Chemistry & From the planetary boundary layer over China, freshly emitted pollutants experience chemical transformations and form secondary pollutants, such as nitrate, sulfate, and ozone through oxidation. Ozone is mainly formed from oxidation of carbon monoxide (CO), methane (CH 4 ), or nonmethane volatile organic compounds (NMVOCs) in the presence of nitrogen oxides (NOx = NO + NO 2 ). The main source for nitrate aerosol formation is oxidation of NO. to form nitric acid (HNO 3), which is subsequently neutralized with ammonia (NH 3). Sulfate is formed from oxidation of SO 2 to sulfuric acid (H 2SO 4) through three major chemical production pathways, which are gas phase production by hydroxyl radical (OH), aqueous phase production by hydrogen peroxide (H 2 0 2 ), and aqueous production by ozone in a presence of metal catalyst. Sulfuric acid can exist in the gas phase but is extremely soluble (vapor pressure of H 2 SO 4 is much lower against H 2 SO 4 -H 2 0), so sulfuric acid is mostly in aqueous (aerosol) phase [Seinfeld and Pandis, 1998]. Gas phase production by OH and aqueous production by H 20 2 are relatively more significant than aqueous production by 03 globally, as well as over China [Manktelow et al., 2007, Barth Church, 1999; Unger et al., 2006]. Sulfate and nitrate are both neutralized by ammonia to form ammonium sulfate ((NH 4 ) 2 SO 4 ) and ammonium nitrate (NH 4NO 3), respectively. However, neutralization of sulfate proceeds, and then neutralization of nitrate follows next, only if excess ammonia is available after neutralization of sulfate. Ammonium sulfate is predominantly in condensed (aqueous or solid) phase because ammonium sulfate does not exist in equilibrium with the gas phase. In contrast, ammonium nitrate is semi-volatile and exists in equilibrium with the gas phase as nitric acid and ammonia. Therefore, under low ammonia condition, ammonium nitrate can dissociate to release gas phase ammonia, which can subsequently experience irreversible reaction with sulfuric acid to form condensed ammonium sulfate [Kuhns et al., 2003]. This rather simple thermodynamics of H 2SO 4-HNO 3-NH 3 provides an accurate representation [Sienfeld and Pandis, 1988]. 8 , The chemical transformations Asian pollutions experience during a lift from the boundary layer over Asia to the free troposphere are well outlined in Brock et al., 2004 and Dunlea et al., 2009, which are based on aircraft measurements of particle size distributions as well as particles and gas species composition analysis. In summary, while Asian air plumes in the Asian boundary layer are lifted to the free troposphere, highly soluble gas phase species such as HNO 3, H 2SO 4, NH 3, and hygroscopic particle including sulfateammonium-nitrate-organic composites are incorporated into clouds, and then are removed through precipitation. The insoluble species such as 03 and peroxyacetyl nitrates (PANs) remain high in concentration without much influence from the wet removal process. A substantial amount of semi-soluble species such as SO 2 may survive the lifting process; however, efficiency depends on a number of conditions that controls the oxidizing rate of SO 2 , such as PH of the cloud droplets aerosols are being incorporated, and availability of oxidizing agents including hydrogen peroxide, ozone, and metal catalysts. A number of numerical studies as well as observation measurements support that a significant amount of SO 2 survives after a rigorous wet scavenging process in convective clouds [Brock et al., 2004; Dunlea et al., 2009; Barth, 1994; Crutzen and Lawrence, 2000; Kreidenweis et al., 1997]. Thus in the free troposphere over the Pacific, a modest concentration of SO 2 significant amount of PANs [Miyazaki et al., 2003] and ozone [Zhang et al., 2011], and relatively much lower concentrations of soluble gas species (HNO 3, H 2 SO 4 , and NH 3) and hygroscopic particles (including sulfate-ammonium-nitrate-organic composites) are expected to exist [Brock et al., 2004; Dunlea et al., 2009]. Across the Pacific, oxidation of species including SO 2 is expected to continue [Dunlea et al., 2009; Brock et al., 2004], leading to the overall particle growth in these plumes. Subsequently when air masses descend over the Pacific or the US, PAN will thermally decompose to release NO., which will be eventually oxidized to HNO 3 on a timescale of about a day at typical tropospheric OH concentrations [Seinfeld and Pandis, 1998]. Model description and simulation set up The GEOS-Chem (version 9-01-03, http://acmg.seas.harvard.edu/geos/) global chemical transport model (CTM) with coupled oxidant-aerosol chemistry [Park et al., 2004] is used to simulate the sulfate-ammonium-nitrate inorganic aerosol system for springs and summers of 1985 and 2010 with varying emission scenarios. The year 2010 is simulated under fixed meteorological conditions at 2010, but with several emission scenarios in order to analyze a genuine influence shift in Asian emissions may have on chemical relationships. The year 1985 is simulated to consider a condition before the accelerated emission growth in 2000s over China, and to compare with prior studies, which used different models and emission inventories. The GEOS-Chem model is driven by assimilated meteorological data from the Goddard Earth Observing System (GEOS-5) at the NASA Global Modeling and Assimilation Office (GMAO) with 10 latitude by 1' longitude horizontal resolution and with native vertical grid of 72 hybrid eta levels, extending from the surface up to 0.1 hPa. For computational efficiency, horizontal resolution is degraded to 20 x 2.50 and reduced vertical grid is used (total of 47 vertical eta levels). The top 37 vertical levels from -80hPa to 0.01hPa are lumped into 11 levels to reduce the vertical grids. The first 31 vertical levels from the surface are pure sigma levels and levels beyond are at fixed pressure levels. The temporal resolution of the GEOS-5 meteorology is 6 hours with shorter time frame of 3 hours for surface variables and mixing depths. For simulations of year 1985, Modern-Era Retrospective Analysis for Research and Applications (MERRA) from Modeling and Assimilation Data and Information Service Center (MDISC) is used because GEOS-5 is not offered extending back to1985. 9 The coupling between ozone- NO. -hydrocarbon-aerosol chemical mechanism and the H 2SO 4-HNO 3-NH 3 aerosol thermodynamics provided in GEOS-Chem [Park et al., 2004; Park et al., 2006] is an essential modeling tool used in this study. The details of this scheme are provided in Park et al., 2004. In short, tropospheric oxidant chemistry is driven by a detailed ozone- NOx -hydrocarbon chemical mechanism that involves about 80 species with about 300 reactions [Park et al., 2004], and is coupled to the aerosol scheme through aerosol formation, heterogeneous reactions, and aerosol's influence on photolysis rates [Martin et al., 2003]. GEOS-Chem aerosol scheme incorporates the sulfate-ammoniumnitrate system [Park et al., 2004], as well as carbonaceous aerosols [Park et al., 2003; Liao et al., 2007], mineral dust [Duncan Fairlie et al., 2007], and sea-salt [Jaegle' et al., 2011] systems. An external mixture of aerosols with lognormal size distributions is assumed. Thus, the GEOS-Chem aerosol scheme does not provide aerosol micro-physics and it does not model internal mixtures of aerosols. From this aspect, GEOS-Chem may be a less favorable model to be used for studying the radiative influence of aerosols, which is sensitive to internal and external mixtures of aerosols. However, GEOS-Chem offers information on mass concentration of aerosols and it should be sufficient for the purpose of this study. GEOS-Chem uses a standard resistance-in-series dry deposition scheme for gas species and aerosol species including sulfate that depends on surface type and meteorological condition [Wesely, 1989; Wang et al., 1998]. Dust and sea salt dry depositions follow the size dependent scheme by Zhang et al., (2001). Wet deposition is implemented for both soluble gas species [Park et al., 2004] as well as hydrophilic aerosols [Liu et al., 2001]. The aerosol wet deposition scheme implements convective updraft scavenging, rainout and washout from large-scale precipitation, as well as return of aerosols to the atmosphere following evaporation. Wet scavenging of soluble gas species depends on various factors including their effective Henry's law partitioning [Mari et al., 2000; Park et al., 2004], and scavenging of SO 2 is influenced by the local availability of H 20 2 as a fast aqueous-phase oxidant [Park et al., 2004]. Five different sensitivity simulations with linear scaling on Asian NOx and/or Asian SO 2 emissions are conducted for spring and summer of 2010: (1) the default standard simulation with all emissions at 2010 level (hereafter referred to as "2010_base") and four sensitivity simulations with linear scaling on (2) Asian NOx emissions, scaled to correspond to the 2001 emission level ("2010BNOX01"), (3) Asian SO 2 emissions scaled to correspond to the 2001 emission level ("2010B S0201"), (4) both Asian NOx and Asian SO 2 emissions scaled to the 2001 emission level ("2001_base"), and (5) Asian NOx and Asian SO 2 emissions turned off ("2010B zero"). For all five simulations, all pollution precursor emissions (except Asian NOx and/or Asian SO 2) are held at the 2010 emission level, which are automatically obtained from a base year emission inventory to 2010 following a default GEOS-Chem scaling method. The "1985_base" simulation uses the monthly Asian emission at base year 2004 that is scaled down to the emission level at 1985 using the automatic GEOS-Chem scaling method. For every simulation, a spin up time of at least six months are allowed, generally starting the simulation from September of the prior year. The analysis is taken for the following spring and summer. The summary of the simulations and corresponding emission scenarios are outlined in Table 1. The results from these simulations are used to analyze a number of different concepts presented in this study. Please refer to Table 2 for detailed explanations of the calculation method, goals, and abbreviations used for cases that are addressed in the following analysis. The standard global emission inventory for GEOS-Chem is GEIA [Benkovitz et al., 1996], which is replaced by regional inventories such as NE199 (http://www.epa.gov/ttn/chief/net/1999inventory.html) for the US, BRAVO [Kuhns et al., 10 , 2005] for Mexico, EMEP emissions for Europe (http://www.emep.int/), and Zhang et al., (2009) for East Asia. The base year (2006) emission inventory for Asia [Zhang et al., 2009] was developed in order to support the Intercontinental Chemical Transport ExperimentPhase B (INTEX-B) campaign during April and May of 2006. The relative emission trends used for automatic GEOS-Chem emission scaling over Asia are from the Regional Emission inventory in ASia (REAS) [Ohara et al., 2007]. REAS provides gridded emission inventories, which include better spatial detail than national scale emission inventories. The linear scaling on Asian NOx and Asian SO 2 emissions for sensitivity runs is implemented by multiplying the base year emissions (at 2006) with a scalar number as an estimate. The down scaling to 2001 emission level is conducted by dividing the default base year's 2006 emission by China's estimated anthropogenic emissions growth rate during 2001 to 2006 from Zhang et al. (2009). For example, the base year (at 2006) Asian anthropogenic SO 2 emission inventory (47 Tg) used in the model is divided by 1.36 (from the estimated 36% increase of Chinese SO 2 emissions from 2001 to 2006 in Zhang et al., 2009) to represent the Asian SO 2 emission level at 2001. Considering the steep rise in Chinese emissions, scaling of the whole Asian emission inventory by Chinese emission growth rate might give an exaggerated perspective of the shift in Asian emission inventory. However, bearing in mind that the research focuses on transpacific transport of Asian pollution, which is predominantly comprised by East Asian emissions [Donkelaar et al., 2008] with the largest contribution from China, the scaling method used above should be sufficient. Below, the validity of this scaling method is discussed by comparing with prior studies. The percentage shift of Asian NOx and Asian SO 2 emissions from 2006 to 2010 is estimated based on reviews of several emission inventories [Granier et al., 2011; Lu et al., 2011; Zhao et al., 2013a], technology and economy based bottom up inventory analysis [Zhao et al., 2013a, 2013b; Lin et al., 2013; Lu et al., 2011], and satellite observation estimates [Lamsal et al., 2011; Lin et al., 2013; Li et al., 2010; Lin et al., 2010; Gu et al., 2013; Mijling et al., 2013; Lu et al., 2011]. The linear scaling on Asian NO, and Asian SO 2 to the 2010 level is conducted by multiplying the base emission level (2006) by an estimate of about a 10% reduction in SO 2 and about 30% increase in NOx from 2006 to 2010 [Granier et al., 2011; Zhao et al., 2013a, Zhao et al., 2013b; Lu et al., 2011; Lin et al., 2010, 2013; Li et al., 2010; Gu et al., 2013; Mijling et al., 2013; Lamsal et al., 2011]. Monthly average Asian SO 2 and Asian NOx emissions used in the simulations are 1.76 Tg S/month (spring) and 1.16 Tg N/month (summer) for 2010 and 1.43 Tg S/month (spring) and 0.58 Tg N/month (summer) for 2001, respectively. In short, Asian SO 2 emissions increased by about 22% and NOx emissions increased by about 100% from 2001 to 2010. The year 2010 was chosen for this study because meteorological conditions during the spring of 2010 were particularly favorable for Asian inflow into the US. The winter of 2009-2010 was under a strong El-Nino condition, and the transport of Asian pollution during the following spring may be enhanced [Jaffe, 2010] due to elongated eastward extension of the subtropical jets [Koumoutsaris et al., 2008]. Moreover, the amplification of the upper level pattern of a weak ridge over the northeastern Pacific Ocean, a trough over California and a ridge over the central US in 2010 with a surface anticyclone over the midlatitude northeastern Pacific Ocean, enhanced subsidence and northwesterly surface winds over California, allowed more efficient Asian pollution transport into the US boundary layer [Cooper et al., 2011]. The two seasons, spring and summer, are analyzed in this study because spring is the season with the strongest transpacific transport of Asian pollution [Jacob et al., 2003; Jaffe et al., 2010; Zhang et al., 2008, 2011; Lin et al., 2012], and summer has stronger convective lifting in the Asian boundary layer [Thompson et al., 1994; Li et al., 2005; Luan et al., 2012], which may alter relative significance amongst transport pathways 11 and also change the upper level chemistry due to the enhancement in wet scavenging of aerosols and soluble gas species. Results Model validity from prior studies Asian enhancements of the aerosol concentration ("2001Asian" and "201OAsian"; Table2) are defined as a contribution of Asian emissions on augmenting pollution levels, which are calculated by subtracting the aerosol concentration simulated with Asian emissions turned off ("201OB_zero") from that of a chosen sensitivity simulation ("2001_base" and "2010_base", respectively). Park et al., (2004) and Heald et al., (2006) provided excellent analyses of the validity of GEOS-Chem (version 5.03 & 7.01.02, respectively) simulation on particulate matter concentration in the US model surface layer by comparing the simulated results with measurements from surface observational networks, such as Interagency Monitoring for Protected Visual Environments (IMPROVE) and Clean Air Status and Trends Network (CASTNET). These studies also examined the validity of GEOS-Chem simulations on Asian sulfate enhancements by comparing the simulation results with aircraft measurements [Park et al., 2004] and IMPROVE data [Heald et al., 2006]. Park et al., 2004 conclude that the model does not overestimate transpacific outflow of sulfate from Asia over the northwest Pacific between February and April of 2001 by comparing the observed mean vertical profiles of non-sea-salt sulfate concentrations from GEOS-Chem to measurements from Transport and Chemical evolution over the Pacific (TRACE-P) aircraft mission. In fact, Park et al., 2004 suggest that the simulated column burden of non-sea-salt sulfate in the lower atmosphere (0-5km), where active Asian outflow is observed from aircraft missions, is underestimated in the model by up to a factor of two. Heald et al., 2006 conclude that GEOS-Chem can simulate the magnitude of Asian sulfate enhancement in the model surface layer over US with reasonable accuracy by taking an average of IMPROVE data selected for specific days for each grid box that are under maximum local Asian influence and comparing this against an average of simulated Asian sulfate enhancement during the corresponding days. The study shows that the seasonal average of surface sulfate concentration during maximum Asian influence (1.04 pg m-3 ) was higher than the seasonal mean (0.69 ptg M- 3 ) over the northwestern US (where Asian influence was highest in the study) by a magnitude that is comparable to the simulated Asian enhancement in the model (0.44 gg M-3 ) averaged during the selected days. Also, the simulated Asian aerosol concentrations among ensembles of IMPROVE sites with spatial proximity in the northwestern US were found to have a high spatial correlation (95%) with no significant time lag. This Asian sulfate influences on the western US surface sites were also found in prior literature based on observational data [Jaffe et al., 2001, 2003a; Bertschi et al., 2004], supporting the large scale plume events over the region. Heald et al., 2006 estimate 50% uncertainty on GEOS-Chem's ability to simulate Asian sulfate enhancement through statistical analysis of bias and correlation with northwestern IMPROVE sites under days with large Asian influence. The study found that when the Asian influence is strengthened (by increasing Asian emissions), the correlation and the bias (compared to IMPROVE) are both increased; and when the Asian influence is zeroed out (by turning off Asian emissions), the correlation and bias are both lowered. An underestimate in Asian SOx (SO 2 + SO 4 -) outflow during the TRACE-P campaign in spring of 2001 was suggested [Park et al., 2004], and an error in the SO 2 oxidation rate was suggested from the conclusion that simulated SO 4 concentrations were higher in comparison to 12 measurements during the ACEAsia campaign [Heald et al., 2005] as well as during the INTEX-B campaign [Donkelaar et al., 2008]. Asian Aerosol influence over the US at 2001 emission level: comparison with prior studies Prior studies [Park et al., 2004; Heald et al., 2006] and this study report similar Asian particulate matter concentrations in the model US surface layer at the 2001 emission level (Figure 1), confirming the validity of the rather simple linear scaling used for estimating Asian emissions in this study. The springtime mean Asian sulfate enhancement in the US model surface layer simulated with emission levels at 2001 and meteorology of year 2010 ("2001Asian" case in Table 2) is highest over the southwestern US, reaching nearly 0.14 pg M-3 and decreasing toward the northwestern US (to 0.06-0.07 pg M- 3 ) and the eastern US (Figure 1-a & Figure 2-a). Relatively prominent northwesterly transport of Asian sulfate (Figure 1-a) towards the southwestern US signals the influence from sustained surface anticyclone over the northeastern Pacific Ocean during the spring of 2010. The mean Asian sulfate enhancement in the model surface layer estimated in this study closely matches the results from prior studies. The estimates of prior studies reported 0.1 Vg m-3 as an annual average [Park et al., 2004] and 0.16 jg M-3 as a spring time average [Heald et al., 2006] over the northwestern US, which is the location of maximum Asian influence driven with the meteorology of 2001. The locations of maximum Asian sulfate influence are probably different between this study (southwestern US) and prior studies (northwestern US), because this study used the meteorology of 2010 while the prior studies used meteorology of 2001 for their simulations. The percentage of Asian sulfate concentration to the total surface sulfate concentration at 2001 emission level is calculated by dividing Asian sulfate enhancement ("2001Asian") by the surface sulfate concentration ("2001_base"), hereafter referred to as "2001Asian/2001" case (Table 2). The percentage is highest over the northwestern US and has a rapid fall toward the central and eastern US (Figure 1-b; Figure 2-b). The high percentage of Asian sulfate concentration (more than 20%) in the northwestern US can be attributed to a large Asian sulfate concentration over the region, as well as a low total surface sulfate concentration over the region. The total surface sulfate concentrations over the northwestern US are low because the domestic emissions of sulfate precursors over the region are relatively small. In comparison, the low percentage of Asian sulfate over the eastern US are primarily attributable to high domestic sulfate precursor emissions that augment the surface sulfate concentration significantly. In contrast to Asian sulfate influences, enhancements from Asian nitrate and Asian ammonium are much less pronounced at the surface layer (Figure 1-c&e), despite observed Asian outflow from flight measurements over Asian marine boundary layer. One of the reasons can be attributed to an active deposition loss of aerosols in the boundary layer, which was discussed as a major difference between CO transport pathway and aerosol transport pathway [Heald et al., 2006]. Despite high NOx emissions in the source region and successful Asian NOx transport resulting in non-negligible Asian contributions in augmenting surface ozone levels over the US [Lin et al., 2012; Cooper et al., 2011; Zhang et al., 2008, 2011; Fiore et al., 2002], Asian nitrate concentration ("2001Asian") is negative in the model US surface layer over most of US (Figure 1-c). This indicates that the total nitrate concentration over the US decreases when Asian NOx and Asian SO 2 emissions are turned on, which is also illustrated in Park et al., 2004. Although the negative contribution could be considered counter-intuitive, this result can be explained from the thermodynamic relationship between sulfate and nitrate that was discussed in the background chemistry section. The probable reason for the negative contribution on nitrate concentration by 13 Asian pollution is that the amount of available ammonium to neutralize nitrate decreases when Asian sulfur dioxide is turned on. The ammonium available for neutralization of nitrate is reduced because the enhancement in sulfate concentration due to augmentation from Asian sources leads to more consumption of ammonium by sulfate for neutralization, which proceeds ahead of neutralization of nitrate. This explanation is further supported with our finding that in contrast to the "2NOX" case (Table 2), which has a large increase in nitrate concentration over most of the atmosphere, with notable enhancements in nitrate concentration reaching the upper atmosphere over the US (more than 30%), the "1.2 S02" case illustrates a significant reduction in nitrate concentration over the whole atmosphere (with more than 40% reduction) and extends beyond the eastern US boundary (Table 2; Supplemental Figure 1). This indicates that the increase in Asian NO, emissions alone from 2001 to 2010 enhances the Asian nitrate concentration over the US, while increases in Asian SO 2 emission alone from 2001 to 2010 leads to a significant reduction in nitrate concentration over the US. Another interesting finding on Asian nitrate is that Asian nitrate represents about 20-50% of the total nitrate concentration over the Arctic region at 2001 emission levels (Supplemental Figure 2). The enhancement in ammonium concentration from Asian sources in the US surface layer is not substantial, but is non-negligible; ranging from approximately 0.05 pg m-3 in the southwestern US and decreasing to 0.02 pg M-3 in the central US (Figure 1-e). However, the contribution in terms of concentration is clearly not as prominent as that from Asian sulfate. Park et al., (2004) stated that Asian ammonium enhancement is negligible over the US by illustrating that Asian ammonium concentration ranges between 0-0.1 jIg M- 3 over the whole US. This range is in broad agreement with the results of our study; however, further analysis in this study indicates that this small contribution (ranging around 0.03-0.05 jg m3) may have significance in terms of the percent of Asian ammonium over the total ammonium concentration. In the western US, the percent of Asian ammonium relative to the total ammonium concentration is about 10-20%. The result should be interpreted with caution, since ammonia emissions are highly uncertain and the significance of this finding is unclear. Thus, for the following sections Asian sulfate is the focus of this study since it plays a dominant role in enhancing the pollution level over the US, in contrast to the negative nitrate enhancement and uncertain Asian ammonium influence. Shifts in Asian sulfate influence over the US from 2001 to 2010 approached with various emission scenarios The response of Asian sulfate enhancement over the US to the rapid growth of Asian NO, and SO 2 emission from 2001 to 2010 is studied by implementing various emission scenarios to modeling simulations, as they are summarized in Table 1. Please refer to Table 2 for the various "cases" used for analysis in the following. These various emission scenarios break down the historical shift in Asian sulfate enhancement from 2001 to 2010 into contributions from individual shifts in Asian NO. or Asian SO 2, and test whether simultaneous enhancement of Asian NO. and Asian SO 2 together has a constructive or destructive relationship on Asian sulfate enhancement. Asian sulfate enhancement in the model surface layer over the US is calculated for various emission scenarios (Figure 2; Table 2). The increase of Asian sulfate concentration from 2001 to 2010 in the US surface layer due to the simultaneous increase in Asian NO. as well as Asian SO 2 is calculated by subtracting surface sulfate concentration of "2001_base" from "2010_base" ("2001to2010 case" in Table 2; Figure 2-c). From 2001 to 2010, Asian sulfate enhancement increases by about 0.03 jg M- 3 in the southwestern US to 0.01 jg M-3 in the intermountain region. The eastern US (more than 0.02 jig M- 3 ) has a greater enhancement than the intermountain region (0.01-0.02 pg M- 3 ). The increases of Asian 14 sulfate concentration corresponds to about 3-4% of the total sulfate concentration at 2010 emission level over a broad region in the western US ("2001to2010/2010" in Table 2; Figure 2-d). The percentage of increase in Asian sulfate concentration from 2001 to 2010 over the total concentration of sulfate at 2001 condition ("2001to2010/2001", not shown) is about 5%. The response of the Asian sulfate enhancement over the US to doubling of Asian NO. emissions ("2NOX" case) is almost negligible (Figure 2-e,f). In contrast, "1.2 S02 case" dominates the shift in Asian sulfate from "2001to2010" case, and is slightly smaller than when both Asian NOx and Asian SO 2 emissions are elevated, especially over the northeastern Pacific Ocean (Figure 2-g,h). Thus, the strengthening in Asian sulfate enhancement is predominantly attributable to about 22% rise in SO 2 emissions over Asia. The enhancement caused by increase of Asian NOx emission increase is much less pronounced and the simultaneous enhancement in Asian NOx and Asian SO 2 together has a small constructive relationship. Recent shift in Asian emissions and responses of chemistry during transpacific transport. & The alterations in chemistry along the path of transpacific transport (from the Chinese boundary layer to the upper levels over the Pacific Ocean) are analyzed to understand possible reasons behind different Asian sulfate enhancements over the US as a response to implementation of various emission scenarios. Prior studies analyzed the sulfate chemistry over the Southeast Chinese/East Asian boundary layer for 1985 [Barth Church, 1999; Manktelow et al., 2007] and 2000 [Manktelow et al., 2007]. By conducting sensitivity analysis on SO 2 emissions over the source region, two studies investigated shifts in sulfate chemical production pathways and the resulting alterations in sulfate burden and lifetime over Southeast China/East Asia. In this section, analysis from the two prior studies are further extended geographically, including Chinese boundary layer and the upper level atmosphere over the Pacific Ocean (major pathway for Asian outflow) to consider the transpacific transport. These findings are also advanced provisionally to consider recent shifts of chemistry from 2000 to 2010, which involve the rapid doubling of NOx emissions during the period and about 22% increase in SO 2 emissions. This analysis involving the influence of doubling of NOx on sulfate chemistry leads to a further investigation on the influence of oxidants on sulfate production pathway, which is a novel approach taken in studies of transpacific transport. The analysis also includes the shifts in chemical production pathways and their sequential influences on sulfate burden, lifetime, and efficiency of transport from China to the Pacific. Major chemical production pathways and the responses to the recent shift in Asian emissions The production rate of secondary pollutants, such as ozone and sulfate, depends on the availability of oxidants, which relies on ozone precursor emissions, such as NOx and Volatile Organic Carbons (VOCs), near the source. A small number of prior studies analyzed chemical couplings between ozone precursors (including NO) and sulfate [Unger et al., 2006; Meng et al., 1997], and found that changing oxidant concentrations can lead to about 20% increases in the sulfate burden in the surface layer over China [Unger et al., 2006]. However, the implications of the relationship have not been fully understood and remain to be explored. Generally, an elevation in sulfate levels may have an influence on ozone concentrations by slowing down the rapid chain production of ozone through enhancing heterogeneous conversion of NOx to nitric acid (HNO 3), which is a reservoir species [Unger et al., 2006]. However, with the recent change in China's steep growth of NOx emissions, it is more important to consider influences that ozone precursors may have on sulfate by 15 changing the total available oxidant budget and by shifting the partitioning of concentrations amongst major oxidizing agents. Nitrogen oxides are important precursors for ozone, and ozone is a source for the production of the hydroxyl (OH) radical. Hydroxyl radical is a precursor for hydrogen peroxide (H 2 0 2 ) indirectly; thus, increases in OH levels from a rise in NO, concentrations may lead to enhancement in H 2 0 2 levels. However, the relationship is more complicated because formation of H 2 0 2 is less favored when the NOx concentration is high. The reason is because the Hydro-peroxy radical (HO 2 ) self-reaction, which forms Hydrogen peroxide (H 20 2) as the termination step of a chain mechanism for ozone production, is suppressed under high NO condition. Under high NOx conditions, Hydro-peroxy radicals react with NO to form OH and NO 2 rather than reacting with itself to form Hydrogen peroxide. This reaction with NO produces OH and NO 2 back into the system and allows continuation of the chain mechanism for ozone production. Hydro-peroxy radicals are byproducts from reaction between OH and VOCs. Thus, a rapid increase of NOx emissions in China during the past decade may not only have enhanced ozone and total available oxidants levels, but it also may have caused a shift in partitioning between the two major oxidizing agents for sulfate production: OH and H 2 0 2 . Hydroxyl radical concentrations may have increased more rapidly than H 2 0 2 concentrations, since H0 2 has higher possibility to react with NO to form OH, rather than to form H 20 2 by self-reaction. The change in partitioning of concentrations amongst major oxidizing agents (OH, H 2 0 2 , and 03) leads to shifts in partitioning of sulfate production pathways: gas phase production by OH and aqueous production by H 2 0 2 , 03, or sea-salt. The partitioning in each chemical production pathway is defined as a percent of sulfate column burden produced from each chemical production pathway. For example, the gas phase partitioning percentage over China is calculated by dividing sulfate column burden produced from gas phase chemistry (OH) by the total sulfate column burden produced by all four chemical production pathways represented in GEOS-Chem (gas phase (OH), aqueous phases (H 2 0 2 and 03), and sea-salt (03)) over China. Although the enhancement in ozone levels may be large, competition between the gas phase production by OH and aqueous production by H 2 0 2 is the focus of this study, since these two production mechanisms are generally about ten times more important than sulfate production by ozone over China as well as over the globe [Manktelow et al., 2007; Barth & Church, 1999]. Comparison on sulfate production pathways with prior literature on 1985 conditions over Southeast China/East Asia Two prior studies calculated the relative significance among production pathways over Southeast China (Barth & Church, 1999) and East Asia (Manktelow et al., 2007) for 1985 by calculating the percent partitioning of each production rate to the total sulfate production rate (Table 3). There is a large discrepancy between the two prior studies. Manktelow et al., 2007 calculates that about 37.5% production is by gas phase (OH) and 56.9% is by aqueous (H 20 2) production, whereas Barth & Church, 1999 calculate that about 18.8% of the production is by gas phase (OH) and 76.9% of the production is by aqueous phase (H 2 0 2 ) (Table 3). This discrepancy may arise from a number of factors such as differences in models and emission inventories used for simulations or the time frame and spatial extent chosen for their analysis. However, the primary cause of the difference is probably attributable to the use of distinct years on representing the level of oxidizing agents in each simulation. Although both studies had fixed concentration of oxidizing agents for simulation of 1985 conditions (with SO 2 emissions fixed at 1985 level), Barth & Church, 1999 held oxidant concentrations at the 1985 level, whereas Manktelow et al., 2007 held oxidant concentrations at the 2000 level. The significant influence that oxidizing agents may 16 have on determining partitioning between sulfate production pathways is analyzed later in this study. The percentage of contribution made by each production pathway to the total sulfate column contribution over China for year 1985 is calculated and averaged from March to August. The results are in reasonable agreement with prior studies: indicating that about 35.9% of the sulfate production is from gas phase (OH) and that 63.6% is from the aqueous phase (H 20 2) production (Table 3, "1985_base"). The percentage of each production pathway contributing to the sulfate column from Manktelow et al., 2007 is recalculated from Table 1 of Manktelow et al., 2007 and is shown in Table 1 of this study. For 1985, all three studies indicate that the dominant sulfate production pathway is aqueous phase production involving H 2 0 2 chemistry (about 60-70% of total chemical production), and followed by gas phase chemistry involving OH (about 15-40%). Note that the results from this study indicate a relatively much smaller contribution from production through ozone (-1%), in contrast to other two prior studies (-5 to 10%). The sulfate production with sea-salt chemistry makes an insignificant contribution. This negligible contribution from ozone and sea-salt production causes two major chemical pathways, gas phase production (OH) and aqueous phase production (H 2 0 2 ), to balance each other and to total nearly 99% (Table 1). Thus, a growth from one chemistry results in a reduction of the partitioning in the other sulfate production pathway by a similar percentage shift. & Sulfate production pathway partitioning in Historical Years (1985,2001, 2010) Three historical years (1985, 2001, & 2010) are chosen as milestones to examine the partitioning in sulfate production pathway. From 1985 ("1985_base") to 2001("2001_base"), gas phase production (OH) pathway partitioning increases by about 12 percent and aqueous phase (H 20 2) production reduces by about 12% in Chinese atmosphere. However, the comparison between "1985_base" and "2001_base" should be done with caution, since "2001_base" uses emissions held at the 2010 emission level (except Asian NOx and Asian SO 2 held at 2001 condition) whereas "1985_base" uses emissions held at 1985 conditions. Moreover, the meteorological conditions used for simulations are not the same (MERRA vs. GEOS-5). Thus, more precise analysis on shifts in chemistry attributable to change in Asian emissions is conducted by comparing "2001_base" and "2010_base". Two simulations differ only by Asian NO. and Asian SO 2 emissions. Other factors including meteorology and emissions of other precursors are the same and are all fixed at 2010 level. Two regions are specific to the interest of this study: China (25-55N, 100-150E) and the Pacific Ocean (25-55N, 1SOE-120N). These are further divided into four spatial quadrants: lower levels over China (0-4km, 25-55N, 100-150E), upper levels over China (2- 8km, 25-55N, 100-150E), lower levels over the Pacific (0-4 km, 25-55N, 150E - 120 W), and upper levels over the Pacific (2-8km, 25-55N, 150E-120W). These geographic divisions are used for analyzing March to August averaged column burden of sulfate and sulfur dioxide (Table 4) and the sulfate production rate from each production pathways (Table 5). The percent change in sulfate production rate (Tg S/month) between "2010_base" scenario and "2001_base" scenario is calculated (Table 2:"2001to2010 ratio") over the course of transpacific transport and is averaged over the latitude band of 25N to 55N in order to analyze the shifts in chemical production rate in response to simultaneous growth in Asian NOx and Asian SO 2 emissions from 2001 to 2010 (Table 5). In the Chinese boundary layer, spring averaged gas phase (OH) sulfate production rate increases by more than 40% from 2001 to 2010 and the enhancement in production rate extends to the upper atmosphere over China (about 5-15%) and over to the western 17 Pacific (Figure 3-a). The spring averaged aqueous (H 2 0 2 ) sulfate production rate increases by relatively much smaller percentage (less than 10%) in Chinese boundary layer (Figure 3b). An enhancement in sulfate production rate in China during summer (Figure 3, bottom) is similar to spring (Figure 3, top) in spatial pattern as well as the percentage enhancement, but generally enhancement during summer is restricted longitudinally and extends further vertically. This enhancement in vertical extension is probably due to the enhancement in convective processes during summer [Thompson et al., 1994; Li et al., 2005; Luan et al., 2012]. The average taken between March and August is used for the following analysis on partitioning of chemical production pathways. The rapid enhancement in gas phase production rate over most altitudes over China (Figure 3) yields about 7-8% increase in the partitioning of gas phase (OH) and corresponding 7-8% decrease in Aqueous (H 20 2) phase sulfate production (Table 5). The partitioning of gas phase production escalates from 47.7% in 2001 to about 55.3% in 2010 (Table 3), and the partitioning of the H 20 2 Aqueous production pathway decreases from 51.6% in 2001 to 44.0% in 2010. The spring and summer averaged sulfate column burden increases from 2001 to 2010 over China, as a result of concurrent increases in both gas phase (OH) and aqueous phase (H 20 2) chemistry: about 0.011 Tg S (about 23%) increase in the lower atmosphere (0-4km) and 0.004 Tg S (about 17%) enhancement in the upper atmosphere (2-8km) in units of column burden summed for all altitudes over China (Table 4). From these shifts, the relative significance between the two major sulfate production pathways over China is reversed in 2010. The gas phase production (OH) plays a more significant role (55.3%) than aqueous phase (H 20 2) production (44.0%) in 2010 (Table 5). This is in contrast to year 1985 when about 60-70% of total sulfate column over China is produced through aqueous phase production with H 2 0 2 . In summary, the emission change of NOx and S02 from 1985 to 2010 results in about a 20% increase in gas phase chemistry (OH) contribution to the total column sulfate production, which leads to a reduction of partitioning in aqueous phase (H 2 0 2 ) sulfate production by nearly 20%. Due to notable increases in NOx emissions from 2001 to 2010, spring and summer averaged aqueous phase sulfate production rates by ozone also experience about a 30-40% enhancement in the boundary layer over the source region (not shown). However, as discussed, the production by ozone is not further analyzed in this study, since the relative contribution is much smaller than that from gas phase chemistry and aqueous chemistry by . H 202 ) An interesting change evident in Figure 3 is over the upper level Pacific, where the sulfate production rate from all production pathways (ozone and sea-salt production not shown) is reduced during both spring and summer, except for the gas phase production in spring. The gas phase chemistry during spring does not show a significant reduction (less than 5%) and is restricted to the upper level atmosphere over the eastern Pacific and the US; whereas, the reduction in average sulfate production rate from aqueous (H 0 2 2 chemistry is up to 20% in the free troposphere during spring and extends from the western Pacific to the US. The reduction in aqueous phase (H 20 2) production extends over broad regions and altitudes including the upper level Pacific and the US for both spring and summer, but the reduction is more pronounced during spring. Over the Pacific, this significant reduction in aqueous production pathway (H 2 0 2 ) leads to about a 7% reduction in partitioning of aqueous chemistry contributing to the column sulfate production averaged during spring and summer and correspondingly increases the partitioning of gas phase chemistry by about 7% (Table 5). The shift in partitioning of sulfate production pathways for the upper level Pacific (2-8km) is about 2-3%, which increases the partitioning of gas phase chemistry and reduces the aqueous chemistry (H 20 2) partitioning. 18 Despite the reductions in production rates from most of the chemical pathways, the spring and summer averaged sulfate column burden from 2001 to 2010 over the Pacific increases by 0.012Tg S (18%) over the lower atmosphere and about 0.05 Tg S (13%) in the upper atmosphere (Table 2), which is attributable to efficient transpacific transport of sulfate from regions with elevated chemical production, especially from the Chinese boundary layer. For spring, the enhancement in gas phase production in the upper free troposphere over the Pacific is another contributing factor to increasing sulfate column burden. Break down of the response of chemistry to shifts in Asian emissions: Four emission scenarios ("2010_base", "201OBNOX01", "2010BSO201", and "2010_base") Focusing on recent changes during 2000 to 2010, the concurrent emission shift of NO. and SO 2 over Asia is further dissected into individual emission changes of NO. or S02 by analyzing four sensitivity runs ("2010_base", "2010BNOX01", "2010B_SO201", and "2001_base"; please refer to Table 1) to diagnose the shift in chemistry attributable to individual emission precursor changes. The response to -22% increase in Asian SO 2 emissions from 2001 to 2010 can be represented by a comparison between "2010_base" vs. "2010B_S0201" and "2001_base" vs. "2010BNOX01". These two pairs differ in NOx emission levels, but the percentage change in sulfate concentration and chemistry between two pairs are similar (not shown). Thus, only "2010_base" and "2010B_SO201" are used for the following comparison analysis, which gives more weight to the recent NOx condition. The feedback to -100% enhancement in Asian NOx emissions from 2001 to 2010 is considered by comparing the "2010_base" and "2010B_NOX01". Response to about 20% increase in Asian SO 2 emission: "1.2SO2" case The percent ratio of sulfate and SO 2 concentration between 2010 conditions ("2010_base") and the sensitivity test with SO 2 emission scaled down to the 2001 level ("2010BSO201), hereafter referred to as "1.2SO2_ratio" in Table 2, over the transpacific pathway is averaged over the latitude band of 25-55N for spring (Figure 4, top) and summer (Figure 4, bottom). The sulfate concentration (Figure 4-b) increases by more than 15% in spring and 20% in summer in the lower atmosphere (0-4km) over China, indicating a fast oxidation in the boundary layer that allows the increase in sulfate concentration to closely correspond to changes in SO 2 emissions (-22%). The enhancement extends to the upper atmosphere over China, the Pacific, and upper atmosphere (2-10km) over the US (diminishing to about 5-10% in enhancement). Over the Pacific, the enhancement in sulfate concentration is most evident in 2-8km altitudes and the enhancement is suppressed in the boundary layer. This supports the view that an active wet deposition in marine boundary layer causes suppression of transport in the boundary layer, which causes the main transport of Asian aerosol to take place in the lower part of the free troposphere [Heald et al., 2006]. Spring and summer averaged responses of the sulfate column burden over China to about 22% increase in Asian SO 2 emission ("1.2SO2" case) are about 0.08 Tg S (about 17%) increase in the lower atmosphere (0-4km) and about 0.03 Tg S (about 14%) increase in the upper atmosphere (Table 4). Thus, most of the enhancements in sulfate burden over China from 2001 to 2010 can be attributed to the increase of SO 2 emissions (Table 4). The enhancement in SO 2 concentration (Figure 4-a) from the elevation of SO 2 emissions is most pronounced in the Chinese boundary layer, and extends to the upper free troposphere reaching up to about 10km (diminishes to about 8-10%) in spring. During summer, the vertical transport of elevated SO 2 concentration is more evident, and a significant enhancement exceeding 20% is obtained, as high as 10km in altitude over China (Figure 4c), probably due to the enhanced convection during summer. The eastward transport of sulfate and SO 2 is more efficient during spring than summer, probably because upper level 19 ) westerlies are stronger in spring, transporting pollutants eastward more actively. The SO 2 concentration enhancement over the Pacific is higher in the atmosphere (4-10km) than the enhancement in sulfate concentration (2-10km), and is more restricted in longitudinal extent, not showing a significant enhancement beyond the Central Pacific. The enhancement of the chemical production rate (Figure 5) takes place mostly in a region where S02 concentration is elevated (Figure 4-a,c), with the largest enhancement by gas phase production over the Chinese boundary layer (more than 20%), extending to 10 km in altitude over China, diminishing to about 10% in spring and 15% in summer. Most of the enhancement in chemistry is restricted to the atmosphere over China (Figure 5); not showing much of elevated signal in sulfate production over the Pacific nor the US, except for the gas phase production (OH) during spring. The spring enhancement in gas phase production (OH) extends into the Central Pacific, but the enhancement decreases to less than about 10% over the region. Notable enhancements in net chemical production rate (calculated as summation from all sulfate production pathways: Gas phase (OH), Aqueous phase (H 2 0 2 , 03 & sea-salt)) are mostly restricted over China with small signs of increases over the upper atmospheres of the Pacific and negligible change over the US for both spring and summer (Figure 5-c & f). A small reduction in the net production rate in the lower boundary over the US is illustrated. From this rather spatially restricted enhancement in chemical production (Figure 5) and SO 2 concentration (Figure 4 a&c) with small signals of enhancements beyond the Central Pacific, eastward extension of the enhancement in sulfate concentration (Figure 4 b&d) in the free troposphere over the Pacific and the US can be interpreted as the efficient transport of sulfate across the Pacific, allowing elevation of sulfate concentrations over the regions with suppressed chemical production. In summary, about 22% growth in Asian SO 2 emission increases the SO 2 concentrations, enhances gas phase (OH) and aqueous phase (H 20 2) sulfate production rates over Asia to the Central Pacific, and shifts the chemical production pathway favorable to the gas phase production by about 2-3%. The resulting elevation in sulfate concentration extends further eastward, reaching the free troposphere over the US via the Pacific Ocean. The small enhancement of gas phase production partitioning in response to the elevation in Asian SO 2 emissions from our study is in a good agreement with two prior studies, which conducted sensitivity studies on the shifts in sulfate production pathway partitioning by changing the SO 2 emission over East Asia or Southeast China. Manktelow et al., 2006 studied the influence of increasing East Asian SO 2 emission from 1985 to 2000 compared to oxidizing agent concentrations fixed at 2000 level, and also estimated a 2-3% increase in gas phase production partitioning. The corresponding reduction in H 2 0 2 production pathway was about 1%. Barth & Church, 1999 also reported a sensitivity test of doubling the southeast Chinese SO 2 emission from that of the 1985 level, which also indicated about a 4% increase in partitioning in gas phase sulfate production and a corresponding 5% reduction in partitioning of production by aqueous chemistry (H 2 0 2 over Southeast China. Response to about 100% increase in Asian NO. emission: "2NOX" case The rise in SO 2 emission from 2001 to 2010 does not explain the large shift in percent partitioning among the sulfate production pathways during the period. Next, I present a sensitivity study including doubling in NO, emission to examine the influence that oxidizing agents have in altering the sulfate production pathways as well as the resulting impact on transpacific transport of Asian sulfate. The calculated changes ("2NOXratio" case) of the average concentration of sulfate and SO 2 averaged for the 25-55N latitude band are depicted in Figure 6. Enhancements in sulfate concentration are about 1-10% over the 20 Chinese lower atmosphere (0-4km), but the enhancement is spatially restricted over the Chinese lower atmosphere, and the upper atmosphere over China and the Pacific Ocean does not have much of enhancement (less than 2%). This finding indicates that a doubling of NO, emission does not have a significant influence on the Asian sulfate influence crossing the Pacific Ocean. This is in agreement with Figure 2 e&f, which depicts almost negligible increase in surface sulfate concentration over the Eastern Pacific (less than 1%) and the US (almost negligible). The enhancement in the sulfate column burden averaged from March to August (Table 2) over the Chinese boundary is about 0.03 Tg S (-6%) for the lower atmosphere and about 0.01 Tg S (-3%) over the upper atmosphere. Over the Pacific, the sulfate column burden (Table 2) over the lower atmosphere increases by 0.003 Tg S (-4%) and by about 0.001 Tg S (-1%) for the upper atmosphere. A notable influence that changes in Asian NO, emissions do have is a large reduction of local and tropospheric S02 concentrations as depicted in Figure 6-a&c. The SO 2 concentration over China in the lower atmosphere is reduced by more than 20%, and the influence propagates to broad regions over the Pacific with a secondary maximum reduction of about 15% over the upper level Pacific in spring. The SO 2 concentration reduction extends to the US, but the influence diminishes to about 5-10% with more pronounced reduction in the upper atmosphere (up to more than 10%). The eastward extension of the SO 2 concentration reduction and sulfate concentration increase are more significant during spring than during summer. A strong vertical band of reduction in SO 2 concentration in the upper atmosphere (6-10km) over China during summer overlaps with the narrow vertical band of elevated concentration of sulfate, indicating that the large reduction in sulfate concentration is due to enhanced sulfate production from the elevated NOX concentration, resulting in a reduction in available SO 2 concentration. As is depicted in the figure, the reduction in SO 2 concentration is not strongly pronounced in the boundary layer of the source region, but is more pronounced in the lower free troposphere (1-5km) for both spring and summer; and even in the upper free troposphere (5-9km) over the Pacific during spring. This reduction of SO 2 concentration in the upper atmosphere is also depicted in the column SO 2 mass budget analysis (Table 2). The column of SO 2 mass reduction as a result of enhancement in NOx emission from 2001 to 2010 is about 0.002 Tg S (-10%) in the Chinese lower atmosphere, and about 0.0005 Tg S (-15%) in the Chinese upper atmosphere. Over the Pacific, the reduction is about 0.002 Tg S(-9%) in the lower atmosphere and about 0.0005 Tg S (-12%) in the upper atmosphere. In contrast to the simple congruent increase in sulfate production rate from all sulfate production pathways in the "1.2 S02" case, the feedback of sulfate production pathways to a doubling of Asian NOx emission from 2001 to 2010 is rather more complicated (Figure 7). Despite a small rise (1-2%) in the H 20 2 column budget over all four quadrants along the transpacific transport pathway (Table 4), which supports the idea of a general enhancement in oxidizing power from the increased NOx emission, the aqueous sulfate production rate by H 20 2 chemistry decreases over all four quadrants: from China by more than 20% (15%) in spring (summer) to the US through all altitudes in the troposphere (Figure 7 b&e). Gas phase sulfate production is increased near the Asian boundary layer by 10-25%, extending up to about 4km-Skm for spring and 2-3km for summer. The enhancement stretches to the Pacific in the lower atmosphere in spring (by about 5% for altitudes lower than 2km) and for summer (up to about 3km in altitude). The upper free troposphere over China, Pacific Ocean and the US all experience up to a 10% reduction in gas phase production. The reduction is pronounced in the upper free troposphere over China (3-10km) as well as over the Central and Eastern Pacific (6-10km). The reduction extends all the way to the upper free troposphere over the US. The spring and summer averaged percent shift in sulfate column production pathway partitioning is by about 6 over 21 ) China and is by 5% over the Pacific % toward the gas phase (Table 5). The upper pacific shows a smaller shift in percent partitioning (Table 5) of about 3% shift toward the gas phase. Corresponding percentage reduction in aqueous phase production (H 2 0 2 partitioning exists for all three regions. From these results, we can conclude that the shift in sulfate production pathways over China and the Pacific Ocean from 2001 to 2010 is dominantly driven by the doubling of the Asian NOx emission during the time, with nonnegligible contributions from increase in Asian SO 2 emission. The net chemistry (Figure 7 c&f) reflects the balancing between the two chemistries, i.e., a reduced enhancement (dominant positive influence from gas phase and smaller negative influence from aqueous phase (H 2 0 2 ) chemistry) in the Chinese boundary layer (-10%) that extends to about 23km in altitude, and the general reduction in production through the upper atmosphere with a strong reduction just above the Chinese boundary at about 3-7km for both spring and summer. The notable reduction in sulfate production over the Central/Eastern Pacific at 6-10km is also evident in the net chemistry. & The relationship between sulfate production pathways, column budget lifetime The change in partitioning between gas phase production and aqueous phase production may have an influence on the efficiency of the Asian sulfate transport because the column burden and the lifetime of sulfate may increase due to the shifting of the production pathway toward gas phase production by OH [Barth & Church, 1999; Unger et al., 2006]. Generally, the lifetime of sulfate produced from the aqueous production pathway is shorter than that produced from the gas phase due to the higher risk of removal through wet scavenging [Barth & Church, 1999; Unger et al., 2006] and the subsequent cloud precipitation during the ascent into the free troposphere. This relationship is applicable not only at the boundary layer over the source region, but throughout the transpacific transport pathway. Hence, increase in Asian NO, emissions can lead to an enhancement in total sulfate column burden as well as an increase in the lifetime of sulfate over the source region by increasing the percent of sulfate produced by gas phase production. Increases in NO, emission enhance SO 2 oxidization to form sulfate by strengthening oxidizing power in the boundary layer of the source region (Figure 7 c&f). The consequence of these changes is aggravation of regional air pollution over China and reduction of available sulfur dioxide concentration for the long-range transport (Figure 6 a&c). Transport efficiency to the Pacific Ocean In order to understand the negligible enhancement in Asian sulfate enhancements in the model surface layer over the US as a response to almost doubling of Asian NO. emission, we analyzed chemistry and wet deposition, which are essential processes involved during lifting from the Asian boundary layer to upper atmospheres over the Pacific Ocean. Over the source region, the sulfate burden and lifetime are expected to increase, which is supported by both the anticipated strengthening in oxidizing power from the increase in total available oxidant levels, and by the shift toward gas phase production, which reduces exposures to wet removal processes. Over the Pacific, shifts in column burden and lifetime of sulfate as responses to the Asian NOx emission growth are complicated, because wet deposition is commonly involved during the ascent. The escalated sulfate production s in the boundary layer of the source region from growth in Asian NOx emissions may enhance transport of Asian sulfate over the Pacific Ocean, depending on the wet scavenging efficiency of sulfate during the lift to the free troposphere. If wet scavenging is efficient, enhancement of SO 2 oxidation in the Asian boundary layer can lead to reductions in total available column 22 amount and shorten the lifetime of sulfate over the Pacific, because the active conversion from SO 2 to sulfate is associated with transformation of semi-soluble gas species to hygroscopic species. Thus, active sulfate formation through SO 2 oxidation in conjunction with wet scavenging may result in a general reduction in sulfate concentration as well as sulfate precursor (SO 2) concentrations in the free troposphere over the Pacific. However, if wet scavenging of sulfate is not efficient during the ascent, then the increased oxidizing power could enhance the total sulfate concentration in the Pacific by efficiently transporting the air masses with enhanced sulfate content from the boundary layer to the upper atmosphere over the Pacific Ocean without losses, while continuing the sulfate formation during the course of transport. The net sulfate production rate over the Pacific decreases when Asian NOx emission is doubled (Figure 7 c&f), so the small percentage increase (less than 2%) in sulfate concentration over the Pacific (Figure 6 b&d) cannot be attributed to the enhancement in chemical production in the region. Rather, the elevated sulfate burden over the Pacific can be a result of transport of the excess sulfate produced from the escalated chemical production over the Asian boundary region. Our model results indicate that the enhancement in chemical production over the lower atmosphere in the Asian boundary is largely counter balanced by increases in the two major sink processes, dry and wet depositions, for aerosols. Wet deposition is a continuous sink for sulfate, so that a large amount of sulfate is actively removed during wet processes during the lift to the upper altitudes and toward the Pacific. In response to the shift in NOx emissions, enhancements in sulfate burden and the corresponding increases in wet deposition rates are similar in terms of the spatial pattern as well as the rough magnitude (Figure 8). A budget analysis over a restricted volume (low altitude China defined as 0-4km, 100-150E, 25-55N) indicates that as a response to changes in Asian NOx emissions, enhancements in deposition rates largely balances the growths in sulfate production rates (Table 6). The enhancement in sulfate production rate within a given volume is calculated by subtracting the sulfate production rate in "2010B_NOX01" from that of "2010_base". The enhancement in production rate is hereafter referred to as "chem". Increases in the loss rate from dry and wet deposition are also calculated following the same method, and are referred to as "sink". The summation of "chem" and "sink" within a volume represents how much of the enhanced sulfate production over the lower level Chinese boundary layer escapes without being lost through deposition processes. For spring, about 75% of the increase in sulfate chemical production rate is counter balanced by enhancements of the wet deposition and dry deposition rates ("loss ratio": Table 4). Thus, only 25% of sulfate produced from the enhanced chemistry can survive the loss mechanisms and escape to the upper atmosphere and to the Pacific from the lower atmosphere (0-4km) over the Chinese boundary layer. For summer, about 88% of elevated chemical production is counter balanced by the strengthened wet and dry depositions, allowing only 13% to escape. In summary, for "2NOX" case, a moderate increase in sulfate production rate (0.033 Tg S/month for spring and 0.038 Tg S/month for summer) is largely counter balanced by enhancements in deposition rates, resulting in only a small enhancement in sulfate production rate (0.0085 Tg S/month for spring, 0.0046 Tg S/month for summer) to proliferate to the upper atmosphere over China and to the Pacific Ocean. The budget analysis indicates that for the "1.2SO2" case, about 80% of the increase in sulfate chemical production rate is counter-balanced by increases of wet deposition and dry deposition rates for spring. Thus, about 20% of the increased sulfate production is not counter balanced the enhancements in loss mechanisms. For summer, about 90% of increased chemical production is counter balanced by wet and dry deposition, allowing only 10% to escape without being balanced by loss mechanisms. This indicates that the loss rate 23 for the "1.2SO2" case is higher than that of the "2NOX" case. However, over the Pacific, the enhancement in sulfate concentration from -22% increase in Asian S02 emissions is more significant (Figure 4 b&d vs. Figure 6 b&d) than that from doubling of Asian NO. emission. The difference can be attributed to a significant increase in sulfate production rate (about 0.13 Tg S/month for spring and 0.10 Tg S/month for summer) in the "1.2SO2" case (about 3 to 4 times greater than that of "2NOX" case), which allows the small percentage that escapes the volume (about 20% in spring and about 10 % in summer) to be significant in absolute amount (about 0.024 Tg S/month for spring and 0.01 Tg S/month for summer), proliferating to the free troposphere over the Pacific to increase the average sulfate concentration significantly (Figure 4). Moreover, reasons for stronger enhancements in sulfate concentration over the Pacific in the "1.2SO2" case than that of "2NOX"case can be that the "1.2SO2" case has a positive enhancement in net chemical production rate over the Pacific (Figure 5 c&f); in contrast to "2NOX" case, which has a reduction in net chemical production rate except for the lower atmosphere over Asia (mostly in overlap with the volume defined above: Figure 7 c&o. Column budget, wet and dry deposition loss, and lifetime over China and upper Pacific Lifetime is defined as sulfate column burden divided by sulfate loss rate (wet and dry depositions). Barth & Church, 1999 found that the sulfate burden over Southeast China more than doubles when Southeast Chinese SO 2 emissions were doubled, while emissions of other pollutants (including the precursors for oxidizing agents) are fixed at the 1985 level. This non-linear response - percent of enhancement in sulfate burden is greater than the percent increase of Chinese SO 2 emission-- was explained with the shifts in production pathways. Barth & Church, 1999 stated that due to limited H 20 2 concentration over China, doubling increase in Southeast Chinese SO 2 emissions increases the percent of sulfate produced by gas phase production with OH. Thus, the study found that the lifetime of sulfate increases from 3.5 days to 4.1 days over the region when the Southeast Chinese SO 2 emissions are doubled. In contrast, Manktelow et al., 2007 used oxidant levels held at the 2000 level, and obtained a relatively slower increase in sulfate burden (-28%) as a response to the increase in East Asian SO 2 emissions (-32%). Although lifetime was not calculated in Manktelow et al., 2007, shortening of the lifetime of sulfate over East Asia as a response to the growth in Asian SO 2 emission is expected because the percent of increase in the column burden of sulfate (-28%) is smaller than the enhancement rate of deposition (-40%) as is indicated in Table 1 of Manktelow et al., 2007. Our results indicate that for spring as well as summer, both sulfate column burden and wet deposition rate increase when NO, and/or SO 2 emission shifts from 2001 level. However, the lifetime may increase or decrease, depending on the relative rate of change between the column burden and the wet deposition rate. Our results for the "1.2 S02" case (Table 7; 2010_base vs. 2010B_SO201, and 2010BNOX01 vs. 2001.base) are in closer agreement with Manktelow et al., 2007, indicating a reduction in the lifetime of sulfate over China for both spring and summer; however, the change in lifetime is almost negligible and the shift accounts for about 1% of the lifetime at 2010 emission level. Sulfate burdens over China increases by about 15% as a response to about 20% growth in Asian SO 2 emissions, which is balanced by slightly more rapid growth in deposition rate (about 16%), resulting in about a percent reduction in the lifetime of sulfate. The change in lifetime as a response to the increase in NOx emission is more interesting in that the response differs between the seasons. For spring, the lifetime increases by 1-2% as a result of a balance between about a 5% increase in column burden and a 2-3% increase in wet deposition. In summer, the sulfate lifetime is shortened by about 1-2% as a result of about a 5% enhancement in sulfate 24 column burden and slightly greater enhancement in wet deposition. From these results, it is possible to conclude that for all the scenarios and seasons, the shift in sulfate lifetime over China due to changes in Asian emissions from 2001 to 2010 is almost negligible. Shift in lifetime in the upper level Pacific Ocean (Table 6) is greater than that over China, with maximum reduction of more than 5% in lifetime during summer as a response to shift in Asian NOx and Asian SO 2 emissions from 2001 (4.33 days) to 2010 (4.07 days) (Table 6). The dry deposition is not included in the calculation as a loss mechanism because it is only applicable in the model surface layer. The reduction in lifetime as a response to an increase in NOx emission is almost negligible during spring (much less than 0.1%), but is about 3-4% during summer. Lifetime is reduced over the upper level Pacific by 3-4% in both spring and summer as a response to increase in SO 2 emission. Interestingly, the results indicate that the lifetime over the upper level Pacific decreases from 2001 to 2010, despite the shift in sulfate production pathway toward the gas phase production. In fact the expected increase in lifetime that can be associated with the increase in gas phase production is over China during spring when only NOx emission is increased; all the other possible combinations of shifts in Asian NOx and/or Asian SO 2 emission and seasons indicate reductions in lifetime (although mostly less than 1%) from 2001 to 2010 both over China as well as over the Pacific (mostly about 3-4%). Summary and conclusion In this study, Global CTM (GEOS-Chem) with coupled oxidant-aerosol chemistry was used to broaden the knowledge on the influence of recent (2001 to 2010) growth in Asian NOX and SO 2 emission on transpacific transport of Asian particulate matter, especially sulfate, by quantifying and analyzing the details of chemistry and its sequential influence on the transpacific transport. This work is motivated by prior studies, Park et al., 2004 and Heald et al., 2006, which used GEOS-Chem to conclude that Asian sulfate may make notable enhancements at the US surface layer, which may hinder the efforts to mitigate visibility impairment problems and to achieve an ultimate goal of obtaining "natural visibility conditions" by 2064 set by the Regional Haze Rule. Two other prior studies Barth & Church, 1999 and Manktelow et al., 2007, provided the motivation for analyzing the shifts in chemistry and the sequential influence on lifetime and sulfate column burden along the transpacific pathway. Our model results indicate that Asian pollution has a notable positive influence on sulfate concentrations but negative contribution on nitrate concentrations at the model surface layer over the US. The thermodynamic relationship between sulfate and nitrate explains the negative contributions Asian pollutions have on nitrate, because enhanced sulfate formation from Asian sulfur dioxide emission reduces ammonium concentrations available to neutralize nitrate. From 2001 to 2010, Asian sulfate enhancements in the US model surface layer increase by 0.01-0.03 g M- 3 , which corresponds to about 4% of the sulfate concentration at the 2010 emission level. This increase in Asian sulfate enhancement is mostly attributable to about a 22% increase in Asian SO 2 emission. Almost doubling in Asian NOx emission during the period has a negligible influence on Asian sulfate enhancement over the US, but Asian NOx and SO 2 emissions have a small constructive relationship in augmenting the Asian sulfate enhancement over the US model surface layer. From 2001 to 2010, the net sulfate production rate increases in the Asian boundary layer but is reduced in the upper level Pacific and the US for both spring and summer. The reduction in chemical production over the upper level Pacific is mostly attributable to the increase in NOx emission, which was found to reduce the sulfur dioxide concentration in the upper Pacific and the US. Asian NOx emission increase leads to more active sulfate 25 production in the boundary layer, which reduces Asian SO 2 concentration available for transport from Asian boundary layer to the upper Pacific. Efficient wet scavenging processes are involved during the lift, which remove sulfate aerosols. In contrast, sensitivity test with about a 22% increase of Asian SO 2 emissions show enhancement in sulfate and sulfur dioxide concentrations, and the net chemical production over China as well as Pacific. Gas phase production by OH and aqueous production by H 20 2 are the two major production pathways along the course of transpacific transport. Over China, from 1985 to 2010, the most significant production pathway is changed from aqueous production pathway by H 2 0 2 in 1985 (60-70%) to gas phase production with OH by 2010 (53%). From 2001 to 2010, column burden sulfate production by gas phase processes increases by -6% with corresponding reductions in aqueous phase production by H 2 0 2 (-8%) over China. Over the Pacific Ocean, the shift corresponds to about 7% with increase in gas phase production and reduction in aqueous phase (H 20 2) production. Upper level Pacific has a moderate shift (2-3%) toward the gas phase production. The shift in production pathway is primarily driven by the growth in Asian NOx emission, with minor contributions from increase in Asian SO 2 emission. A budget analysis over a restricted volume of Asian lower boundary explains the reasons for the differences in Asian sulfate enhancement over the Pacific and the US surface layer in response to the shifts in Asian SO 2 or Asian NOx emission. Sensitivity test with Asian NOx emission indicates that the enhancement in sulfate production rate is three to four times smaller than that with Asian SO 2 emission. This modest enhancement in sulfate production from doubling in Asian NOx emission is largely balanced by elevation in deposition rates (75 % is balanced in spring and 88% is balanced in summer) so that significant amounts of enhanced sulfate cannot escape the lower Chinese boundary layer to increase the sulfate concentration over the Pacific and the US. In contrast, despite even more active deposition rates (80 % during spring and 90% during summer), a significant amount of sulfate escapes from the Asian boundary layer as a response to a 20% increase in Asian SO 2 emissions because enhancement in sulfate production rate is much greater (3 to 4 times) in absolute amount than when Asian NOx emissions are increased. Moreover, net chemistry over the upper level Pacific is enhanced when Asian SO 2 emission is elevated, which allows more sulfate production along the path; in contrast to the reduction in net chemical production rate outside of the Chinese boundary layer when Asian NOx emissions are increased. The shift in chemical production pathway toward the gas phase is supposed to increase the column burden and lifetime of sulfate. Our results indicate that the sulfate column burden shows a notable increase as a response to the changes in Asian emissions. However, for all scenarios and seasons, the shifts in sulfate lifetime over China due to changes in Asian SO 2 and/or Asian NOx emission are found to be almost negligible (generally 1-2%). The shift in lifetime over the upper level Pacific is more pronounced, with maximum reductions of more than 5% in lifetime during summer as a response to a simultaneous increase in Asian NO. and SO 2 emissions. Generally, lifetime over the upper level Pacific Ocean is reduced by 3-4% as a response to the imposed growth in Asian NO, and SO 2 emission; except during spring with Asian NO. emission change, which indicated almost negligible reduction in lifetime. Thus in summary, the expected increase in lifetime that can be associated with shift toward the gas phase production is found only when NO, emission is increased during spring over China; all the other possible combinations of shift in Asian NOx and/or Asian SO 2 emissions and seasons indicate reduction in lifetime from 2001 to 2010 both over China as well as over the Pacific. 26 (a) SPR 2001Asian S04(2-) [0.01 ug/m3] (b) SPR 2001 Asian/2001 S04(2-) [%] 0 80 N 80N 70N 70*N 60*N ------ 60 N --- 50*N 50*N 40N 40*N 30*N 30*N 20*N 20*N 1 0*N 1 0*N 0 00 0 120 E 0. -5. 6. 8. 1800 10. 12. 15. 30. 70. 00 0 120*W 60 W 1 ZU-:: 500. 2000.3000. -5. 0. 5. 80*N 0 70 N 70*N 0 60 N 60*N 50*N 50*N 40N 40*N 30*N 30*N 0 20 N 20*N 1 0*N 1 0*N 00 120*E 17 -20. 1800 120*W 0 120 E 0 1800 120 W 60*W n-- -10. 70*N - 00 0 60 W -5. -3. -2. -1. 0. 1. 200. 1500.2000.2500.-250-100.-60. -30. -10. -5. 0. 5. 20. 50. 90. 95. 97. 99. 100. (e) SPR 2001Asian NH4+ [0.01 ug/m3] (f) SPR 2001Asian/2001 NH4+ [%] 0 80*N .---. 60*W 10. 15. 20. 25. 30. 40. 60. 90. 95. 98. 99. (d) SPR 2001Asian/2001 N03- [%J (c) SPR 2001 Asian N03- [0.01 ug/m3] 80*N 120*W 1800 80 N --------- -..-. - - --- - - --- 70*N 60*N 60*N -q .. -.. --- --------- 50N SO*N 40*N 40*N 30*N 30*N--r 20*N 20*N - -- - -- --- -*A-- . .. 10*N 0 00 -5. 0. 0 120 E 1. 3. 4. 1800 6. 120OW 0 60 W 0 0 120 E 1800 rm-MMMMOMM 20. 100. 500.1000.1200.1300.1500. -5. 0. 5. 10. 15. 20. 25. 40. 60. 70. 120*W 60*W 80.- 9 9 ~ 99.~70. Figure 1. Asian inorganic particulate matter (sulfate, nitrate and ammonium) enhancement in the model surface layer: (a,c,e) concentration of Asian enhancement (sulfate, nitrate, ammonium, respectively) [Vg M-3 ] in model surface layer at 2001 emission level; (b,d,f) the percentage of Asian enhancement (sulfate, nitrate, ammonium, respectively) to aerosol concentration (sulfate, nitrate, ammonium, respectively) at 2001 emission level. *Note that contour intervals are different for every figure for the best representation for each figure. Please refer to Table 1 and 2 for abbreviations used. (a) SPR 2001 Asian S04(2-) [ug/m3] (c) SPR 2001 to201 0 S04(2-) [ug/m3] (b) SPR 2001 Asian/2001 S04(2-) [%] (d) 60ON 60*N 60*N 60*N 50*N 50*N 50*N 50*N 40*N 40*N 40*N 40*N 30*N 30*N .30N 20*N 20ON 20*N 11* 1*N 1*N 0 1800 150OW 1200 W 90*W 0 60*W 1800 150*W 1200 W 900 W 60*W 20*N M 1 O"N 00 1800 150*W 120*W 90*W 600 W -0.05 0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 -1. 0. 1 3. 6. 9. 12.15.20.25.30.35.40.45. -0.010.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 (e) SPR 2NOX S04(2-) [ug/m3] (f) SPR 2NOX/2010 S04(2-) [%] (g) SPR 1.2SO2 S04(2-) [ug/m3] 1800 0.0 60*N 60*N 50*N 50*N 50*N 50"N 40*N 40*N 30*N 30*N 30*N 30*N 20*N 20*N 20*N 20*N 0 10 N 00 1800 0 150*W 1200 W 90*W 60W 1800 10 N 150*W 120*W 900 W 600 W -0.010 -0.007 -0.003 0.000 0.003 0.007 0.010 -1.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 0.8 1.0 150*W 1.0 2.0 120*W 90*W 3.0 4.0 (h) SPR 1.2SO2/2010 60*N 40N40*N ..... 00 60*N 10*N 2001 t0201 /201 0 S04(2-) [%] 30*N 0 0 SPR 60OW 5.0 10*N 00 1800 150*W 120*W 90*W 60*W 1800 -0.010.00 0.01 0.02 0.03 0.04 0.05 0.06 0.070.08 0.0 150*W 1.0 2.0 120*W 90*W 3.0 40 60W 5.0 Figure 2. Differences on sulfate enhancements between various emission scenarios in US surface layer during spring: (a) the "2001Asian" sulfate concentration [pg m-]; (b) the percentage of "2001Asian" to sulfate concentration at 2001 emission level; (c,e,g) shifts in Asian enhancement from various emission scenarios; (d,fh) changes in percentage of Asian enhancement over sulfate concentration at 2010 emission level by shifts in emission scenarios (b,d,fh). *Please note the change of scales for (e). "2001to2010" case (c,d); "2NOX" case (e,f); "1.2SO2" case (gh). *Please refer to Table 1 and 2 for abbreviations . O#@t11VAj - ffiIMM~ #MW" - !, , ---- - -- - -- - - ............ i 6.0 S04(2-) [%] 6.0 28 (a) SPR 2001to2010_ratio Gas (OH) [%] - 10- (b) SPR 2001to2010_ratio AQ (H202) [%] - 8 E - 6 0 60. 100 70. 80. 200 Longitude 90. 300 0 100. 110. 120. 130. 140. 60. (c) SUM 2001to2010_ratio Gas (OH) [%] 100 70. 80. 200 Longitude 90. 300 100. 110. 120. 130. 140. (d) SUM 2001to2010_ratio AQ (H202) [%] 10 8 a) V a) *0 0 60. 70. 80. 90. 100. 110. 120. 130. 140. 60. 100 70. 80. 200 Longitude 90. 300 100. 110. 120. 130. 140. . Figure 3. Sulfate production rates: "2001to2010"case. Response of sulfate production rates [kg S/ month] to shifts in Asian emissions from 2001 to 2010 calculated as percent ratios between "2010_base" scenario and "2001_base" scenario averaged for 25-55 N latitude band: (a) spring averaged percent ratio of (a) gas phase sulfate production by OH, (b) aqueous phase sulfate production by H 2 0 2 , and summer averaged percent ratio of (c) gas phase sulfate production by OH, (d) aqueous phase sulfate production by H 2 0 2 *Please refer to Table 1 and 2 for abbreviations used. The color bars are saturated after 140% for (a) and (c). 29 (a) SPR 1.2SO2_ratio S02 [%] 10 - - - -. (b) SPR 1.2SO2ratio S04 [%] . . .10 8- 8- EE _,6 - - 6 a)D 4 2 o 2 0 100 200 Longitude 0 - -300 75. 80. 85. 90. 95. 100.-105.110.115.120.125. (c) SUM 1.2SO2_ratio S02 0 -- - - - 4-4 100 200 Longitude 300 75. 80. 85. 90. 95. 100.105.110.115.120.125. [%J (d) SUM 1.2SO2_ratio S04 [%] 10. 10 8- 8 EE 4- 4 2 2 100 - - - - - - - -0200 300 Longitude 75. 80. 85. 90. 95. 100.105.110.115.120.125. - - - - 0 .. . 0 0 100 200 Longitude 300 75. 80. 85. 90. 95. 100.105.110.115.120.125. Figure 4. Sulfur dioxide and sulfate concentrations: "1.2S02" case. Response of sulfur dioxide and sulfate concentrations to about 20% increase in Asian SO emission calculated 2 as percent ratios between "2010_base" scenario and "2010B_SO201" scenario averaged for 25-55 N latitude band: spring averaged percent ratios on (a) sulfur dioxide concentrations, and (b) sulfate concentrations; and summer averaged percent ratios on (c) sulfur dioxide concentrations, and (d) sulfate concentrations. *Please refer to Table 1 and 2 for abbreviations used. The color bars are saturated after 125% for (a) and (c). 30 (a) SPR 1.2SO2_raWo GAS (OH) [%J 10 (b) SPR 1.2S92_ratIo AQ (H202) [%] 10 (c) SPR 1.2SO2 ratio Net chemistry 1%] 10:1 86-- 6- 4- 4 2- 2 0 200 100 Longitude 60. 70. 80. 90. 300 100. 110. 120. 130. 140. (d) SUM 1.2SO2_ratio Gas (OH) [%j 2 0 60. 100 70, 80. 200 Longitude 90. 300 100.110. 120. 130. 140. 0 60. (a) SUM 1.2SO2_ratio AQ (H202) [%] 100 200 Longitude 300 70. 80. 90. 100. 110. 120. 130. 140. (f)SUM 1.2SO2_ratio Net chemistry [%] 10 E te -C 8- 8 6- 6- 4- 4- 2- 2, 0 60. 100 70. 80. 200 Longitude 90. 300 100. 110. 120. 130. 140. 0 60. 100 70. 80. 200 Longitude 90. 300 100. 110. 120. 130. 140. Figure 5. Sulfate production rates: "1.2S02" case. Response of sulfate production rates [kg S/ month] to about 20% increase in Asian SO 2 emissions calculated as ratios between "2010_base" scenario and "2010B_0S201" scenario averaged for 25-55 N latitude band: spring averaged ratios of (a) gas phase sulfate production by OH, (b) aqueous phase sulfate production by H 2 0 2 , (c) net chemical production (sum of gas phase (OH), aqueous phase (H 2 0 2 , 03, sea-salt), and summer averaged ratio of (d) gas phase sulfate production by OH, (e) aqueous phase sulfate production by H 2 0 2 , (f) net chemical production (sum of gas phase (OH) and aqueous phases (H 2 0 2 , 03, sea-salt)). *Please refer to Table 1 and 2 for abbreviations used. 31 (b) SPR 2NOX ratio 10 -- -- - - - - - - - - 10 8 E -.-.-.-- 8- 6 E 6 4 4- 2 2 0 0 S04 [%] - (a) SPR 2NOXratio S02 [%] 100 200 Longitude 0 300 100 200 Longitude 300 80. 84. 88. 92. 96. 100.104.108.112.116.120. 95. 96. 97. 98. 99. 100.101.102.103.104.105. (c) SUM 2NOXratio S02 [%] (d) SUM 2NOXratio S04 [%] 80) TIC 0 4- 2 0 100 200 Longitude 300 80. 84. 88. 92. 96. 100.104.108.112.116.120. 0 100 200 Longitude 300 95. 96. 97. 98. 99. 100.101.102.103.104.105. Figure 6. Sulfur dioxide and sulfate concentrations: "2NOX" case. Responses of sulfur dioxide and sulfate concentrations to about 100% increase in Asian NOx emission calculated as ratio between "2010_base" scenario and "2010B_NOX01" scenario averaged for 25-55 N latitude band: spring averaged percent ratio of (a) sulfur dioxide concentrations, and (b) sulfate concentrations, and summer averaged percent ratio of (c) sulfur dioxide concentrations, and (d) sulfate concentrations. *Please refer to Table 1 and 2 for abbreviations used. The color bars are saturated at 80% for (a) and (b) and at 105% for (c) and (d). 32 (b) SPR 2NOX-ratio AO (H202) [%] u 80. 200 Longitude 100 85. 90. 95. 300 0 100 200 300 (c) 0 SPR 2NOX-ratio Net chemistry [%] 100 200 Longitude Longitude 100. 105. 110. 115. 120. 70. (d) SUM 2NOX-ratio Gas (OH) 1%] 78. 85. 92. 100. 108. 115. 122. 130. (e) SUM 2NOX-ratio AQ (H202) [%] 80. 85. 90. 95 300 100. 105. 110. 115. 120. (f) SUM 2NOX-ratio Net chemistry [%] T ZE (D 0 80. 100 85. 90. 200 Longitude 300 95. 100. 105. 110. 115. 120. 0 70. 100 78. 85. 200 Longitude 92. 300 100. 108. 115. 122. 130. Figure 7. Sulfate production rates: "1.2SO2" case. Responses of sulfate production rates [kg S/ month] to about 100% increase in Asian NO, emission calculated as ratio between "2010_base" scenario and "2010B_NOX01" scenario averaged for 25-55 N latitude band: spring averaged ratios of (a) gas phase sulfate production by OH, (b) aqueous phase sulfate production by H 2 0 2 , (c) net chemical production (sum of gas phase (OH), aqueous phase (H 2 0 2 , 03, sea-salt), and summer averaged ratio of (d) gas phase sulfate production by OH, (e) aqueous phase sulfate production by H 2 0 2 , (f) net chemical production (sum of gas phase (OH), aqueous phase (H 2 0 2 , 03, sea-salt)). *Please refer to Table 1 and 2 for abbreviations used. The color bars are saturated at 120% for (a) and (d). 33 (a) SPR [2010_babe-2010- NOXOI 25-55N avg. S04 [ug/n3] (b) SPR [2010_.baae-201B0NOX01 25-55N avg. WETDEP [ug/m3 10 6- R I 42- o 100 200 300 1 IM 200 LongkKdG 0.00 0.01 0.02 0.03 0.05 0.10 0.15 0.20 0.50 1.00 0.00 001 0.02 0.03 0. .oT10 0.15 3W 0.20 0.50 1.Wo Figure 8. Balancing between enhancements in Asian sulfate concentrations and wet deposition rates in spring as a response to about 100% increase in Asian NOx emission: (a) increases in sulfate concentrations [Ig M- 3 ], (b) increases in sulfate wet deposition rates [rIg m-3]. *Please refer to Table 1 and 2 for abbreviations used. *Color bars are saturated after 1 [ptg m-3]. 34 Table 1. Summary of emission scenarios used for simulations. Scenarios 2001_base Asian SO 2 and NO, emissions Both at 2001 level 2010_base Both at 2010 level 2010B_zero Both zero emission 2010B_SO201 Asian SO 2: at 2001 level Asian NO,: at 2010 level 201OB_NOX01 Asian NOx: at 2001 level Asian SO2: at 2010 level 1985_base Both at 1985 level Goal Simulate nitrate, ammonium and sulfate concentrations and sulfate production rates with Asian NO, and SO 2 emissions at 2001 level Simulate nitrate, ammonium and sulfate concentrations and sulfate production rates with Asian NO. and SO 2 emissions at 2010 level Simulate nitrate, ammonium and sulfate concentrations and sulfate production rates without Asian emissions Simulate nitrate, ammonium and sulfate concentrations and sulfate production rates with Asian SO 2 emission at 2001 level and Asian NO. emissions at 2010 level Simulate nitrate, ammonium and sulfate concentrations and sulfate production rates with Asian NOx emission at 2001 level and Asian SO 2 emissions at Meteorology GEOS-5; 2010 condition GEOS-5; 2010 condition GEOS-5; 2010 condition GEOS-5; 2010 condition GEOS-5; 2010 condition 2010 level Simulate nitrate, ammonium MERRA; 1985 and sulfate concentrations and condition sulfate production rates with Asian SO 2 and NOx emissions at 1985 level *Emissions of all other pollutants are held at 2010 emission level. Only Asian NOx and Asian SO 2 are scaled for each scenario (see methods for details). 35 Table 2. Summary of "cases" considered. Goal Calculate concentrations and production rates with Asian emissions at 2001 level Asian aerosol concentrations 2001Asian/2 001 Percent of Asian enhancements to the pollution concentrations in 2001 Percent ratio of concentrations 2001to2010 Calculate change in concentrations or production rates as responses to the growth in Asian NOx and S02 emissions from 2001 to 2010. 2001to2010/ 2010 Calculate change in concentrations or production rates as responses to the growth in Asian NOx and SO2 emissions from 2001 to 2010. Calculate the percentage of change in Asian aeorosol concentration (as a response to Asian NOx and S02 emissions changes from 2001 to 2010) to sulfate concentrations at 2010 condition Calculate the changes in concentrations or production rates as responses to the growth in Asian S02 emissions from 2001 to 2010. Shift in Asian aerosol concentrations or production rates Percent ratios 1.2SO2/2010 2NOX 2NOX/2010 Calculate the changes in concentrations or production rates as responses to the growth in Asian S02 emissions from 2001 to 2010. Calculate the percentage of change in Asian sulfate concentrations (as a response to shift in S02 emissions from 2001 to 2010) to sulfate concentrations at 2010 condition Calculate the changes in concentrations or production rates as responses to the growth in Asian NOx emissions from 2001 to 2010. Calculate the changes in concentrations or production rates as responses to the growth in Asian NOx emissions from 2001 to 2010. Shift in Asian aerosol concentrations or production rates Percent ratios 2001Asian/2001 =(2001_base 201OB-zero) /2001_base*100 2001to2010 = 2010_base 2001_base Figure 1 Figure 2 2001to2010_ratio =2010_base/2001_bas e*100 2001to2010/2010 =(2010_base2001_base) /2010_base*100 Figure3 1.2SO2 =2010_base 2010B_SO201 Figure 2 Table 6 1.2SO2_ratio =2010_base/2010BS Figure 4 Figure 5 Calculate the percentage of change in Asian sulfate concentrations (as a response to shift in NOx emissions from 2001 to 2010) to sulfate 1 concentrations at 2010 condition Figure 2 Figure2 0201*100 Percent ratios 1.2SO2/2010 =(2010_base 2010BSO201) /2010-base*100 Figure 2 Shift in Asian aerosol concentrations or production/los s rates Percent ratios 2NOX =2010_base 2010BNOX01 Figure 2 Figure 8 Table 6 2NOX-ratio =2010_base/2010BN Figure 6 Figure 7 - 1.2SO2 Percent ratios Figures & Tables Figure 1 Figure 2 OX01*100 Percent ratios 2NOX/2010 =(2010_base 2010BNOX01)/2010 base*100 - 2001Asian Abbreviation and Calculation 2001Asian = 2001_base 201OBzero - Cases Figure 2 36 & Table 3. Chemical production Pathways: comparison with prior studies. The percent of sulfate produced through gas phase ("OH %") production pathway and aqueous phase production pathway by H202 ("H202 %") is calculated from this study (2001_base, 1985_base), and two prior studies: Manktetelow et al., 2007 and Barth Church, 1999. 2001 base Manketelow 00 E.Asia 1985 base Maneeo 5EAi Barth &Chad gs Temporal range CHEM Oh OH % [Tg S/ yr] [Tg S/ yr] avg. March-Aug _avg. June-Dec avg. March-Aug avg. Jurne-Dec wk2* 4rldrni 1 H202 H202 % Tg S/ yr] 7.52 3.59 47.7 39.9 3.88 5.38 1.93 35.9 37.51 3.42 2 1 9.0 +H202 % PSO4 China Column 51.6 55.9 63.6 56.91 7, 99.3 95.8 99.5 9. 90.3 China: 100-150 E, 25-55 N. Manktelow 00 E.Asia: Result from [Manktelow et al., 2007] with East Asian emissions at 2000 level. Manktelow 85 E.Asia: Result from [Manktelow et al., 2007] with East Asian emissions at 1985 level. *Please note that [Manktelow et al., 2007] had oxidant levels fixed at 2000 level. Barth & Church 85: Results from [Barth & Church, 1999] with Southeast Chinese emissions at 1985 level. Barth & Church 85_SO2*2: Results from [Barth & Church, 1999] with Southeast Chinese SO 2 emissions doubled from 1985 level; all the other emissions (including oxidants) are fixed at 1985 level. *Please refer to Table 1 and 2 for abbreviations used. 37 Table 4. Column burdens of sulfate and sulfur dioxide concentrations. Left: the column burdens over the four quadrants -- lower level China (LWCHColumn:0- 4km, 25-55N, 100-150E), upper level China (UPCH Column:2-8km, 25-55N, 100-150E), lower level Pacific (LWPCColumn:0-4 km, 25-55N, 150E - 120 W), and upper level Pacific (UPPCColumn:2-8km, 25-55N, 150E-120W )-- are calculated. Right: the ratios of the column burdens calculated (left) against that of 2010_base to calculate the shifts in the column burdens as a response to changes in Asian SO and Asian 2 NOx emissions. *All the results are averaged from March to August. *Please refer to Table 1 for abbreviations used in this table. TgS 2010_base 2010BNOX01 2010_SO201 2001_base TgS 0.059 0.056 0.051 0.048 Tg S 2010_base 2010BNOXO1 20108_SO201 2001_base Tg S 0.024 0.023 0.021 0.020 TgS 2010_base 20108_NOX01 2010BSO201 2001_base TgS 2010-base 1.000 1.000 099 2010B S0202 1.165 1.199 0.995 0.287 0.283 0.287 0.284 2010_base 20108_NOX01 2010BSO201 2001_base 0.670 0.659 0.672 0.662 2010_base 2010BSO201 20&-_6w~e 0.703 0.697 0.704 0.698 2010_base 20WO-ENOX01 201BSO201 2001Obase 2010_base/* 2010_base/* 2010_base/* 1.000 1.000 1.000 1.026 0.861 1.012 1.138 1.155 0.997 1.165 1.004 1.009 Tg 0.027 0.029 0.023 0.025 TgS 0.044 0.043 0.040 0.039 0.306 0.300 0.307 0.302 Tg 0.0028 0.0033 0.0024 0.0028 0.083 0.080 0.073 0.071 TgS 2010_base 20108_NOX01 20108_SO201 2001_base Tg 0.023 0.025 0.019 0.021 1.000 1.000 1.000 L.042 M.913 1.132 1.169 I.V6 1.074 1.0,16 0.996 1.012 1.000 1.000 1.000 1.017 0.882 1.008 1.114 1.110 0.998 1.130 0.990 1.006 Tg 0.0041 0.0046 0.0037 0.0041 38 Table 5. Sulfate production rates column burdens of the two major chemical production pathways. Column burdens of sulfate produced by gas phase production pathway with OH and aqueous phase production pathway with H202 [Tg S/month]. The percentage of sulfate produced from gas phase pathway ("OH %") and aqueous phase production pathway by H202 ("H202 %") is calculated. China: 100-150 E, 25-55 N Pacific Ocean (PC): 150E-120W, 25-55N Upper level Pacific Ocean (UPPC): 150E-12OW, 25-55N, 2-8km *All results are averaged from March to August. *Please refer to Table 1 for abbreviations used in this table. Oh [Tg S/ [Tg S/ QH% H202 [Tg S/ month] month] 2010 base 2010B NOX01 2010B S0201 2001 base 1985 base AVG AVG AVG AVG AVG SPR&SUM SPR&SUM SPR&SUM SPR&SUM SPR&SUM PS04 PC Column 0.77 0.74 0.65 0.63 0.45 CHEM [Tg S/ month] 0.43 0.36 0.35 0.30 0.16 Oh [Tg S/ 55.28 49.35 53.53 47.72 35.90 AVG AVG AVG AVG AVG SPR&SUM SPR&SUM SPR&SUM SPR&SUM SPR&SUM PS04 UPPC [2-8km] Column 0.95 0.92 0.83 0.81 0.6 CHEM [Tg S/ 0.4 0.39 0.38 0.33 0.21 Oh [Tg S/ AVG SPR&SUM AVG SPR&SUM AVG SPR&SUM AVG SPR&SUM AVG SPR&SUM 0.035 0.038 0.031 0.034 0.040 0.017 0.018 0.015 0.016 0.024 43.99 49.96 45.81 51.63 63.63 99.28 99.31 99.35 99.35 99.53 H202 % OH+H202 month] 48.03 42.94 45.67 40.80 3059 0.49 0.52 0.45 0.47 0.4 OH % H202 [Tg S/ month] month] 2010 base 2010B NOX01 2010B S0201 2001 base 1985 base 0.34 0.37 0.30 0.32 0.29 OH % H202 [Tg S/ month] month] 2010 base 2010B NOX01 2010B S0201 2001 base 1985 base H202% OH+H202% % CHEM 51.21 56.29 53.59 58.45 68.61 99.2 99.23 99.26 99.26 99.58 H202 % OH+H202 % PS04 China Column month] 49.15 46.14 49.37 46.52 59.4 0.018 0.021 0.016 0.018 0.016 50.69 53.73 50.4 53.36 40.4 9 99.85 99.86 9986 99.87 99.93 39 Table 6. Budget Analysis: a balance between enhancements in chemical production rates and wet and dry deposition rates. Changes in chemical production rates and the sink rates (dry and wet depositions) over defined volume [0-4km, 100-150 E, 25-55N] to doubling in Asian NOx emissions [top; 2010_base vs. 2010BNOX01] and to increasing Asian SO 2 emissions by about 22% [bottom; 2010 base vs. 2010B_SO201]. The budgets are summed over the defined volume [0-4km, 100-150 E, 25-55N], representing the lower altitudes of China. In units of [Tg S/month]. *Please refer to Table 1 for abbreviations used in this table. 2NOX: 2010_base vs. 2010B_NOX01 SPR 0.034 -0.025 0.0085 0.75 SUM 0.038 -0.034 0.0046 0.88 1.2SO2: 2010_base vs. 201OB_SO201 SPR 0.13 -0.10 0.024 0.81 SUM 0.10 -0.09 0.011 0.90 chem= Difference in chemical production sink= Difference in wet and dry deposition (negative indicates enhancement in sink) chem+sink= net balance between enhancements in chemical production rates and sink rates. (*positive means enhancement in chemical production rate is greater than that of the sink.) Loss ratio = 1- (chem+sink)/chem = the ratio of enhancement in chemistry that is counterbalanced by the enhancement in sink processes. 40 Table 7. Column budgets, wet depositions, dry depositions, sinks and lifetimes over China. Seasonal averages of sulfate column burdens, wet deposition rates, dry deposition rates, sink rates (defined as a sum of dry and wet deposition rates), and lifetimes of sulfate over China (100-150 East, 25-55N) for various emission scenarios. *Please refer to Table 1 for abbreviations used in this table. China Whole Column Column [Ti SI 2010 base SPR 2010B NOX01 2010B S0201 2001 base 0.0782 .0718 . N686 SUM .0819 .0593 0.0564 0.0510 0.0487 Wet dep [Tg S/month] SPR .6655 0.6464 .5730 M.550 0.5968 0.4822 .0994 0.5655 0.0931 0.5071 Dry dep [Tg S/month] SUM SPR 0.0860 .0810 SUM .0740 0.0697 0.0641 Sink [Tg S/monthl 0.0606 SPR 1.7649 0.7395 0.6590 0.6708 3.2830 2.7091 0.6352 Ufetime [days] SUM SPR SUM 3.2416 0.5712 3.34M_ 2.7379 .6390 0.5428 2.7240 3.3009 2.7519 Table 8. Column budgets, wet depositions, dry depositions, sinks and lifetimes in the upper level Pacific Ocean. Seasonal averages of sulfate column burdens, wet deposition rates, dry deposition rates, sink rates (wet deposition rates), and lifetimes of sulfate over upper level Pacific (150E to 120W, 25-55N, 2-8km) for various emission scenarios. *Please refer to Table 1 for abbreviations used in this table. 2-8km Pacific Column [Tg S] SPR SUM Wet dep [Tg S/month] SPR SUM Lifetime [days] SPR SUM 2010_base 0.0573 0.0310 0.3126 0.2331 5.6215 2010B NOX01 2010BS0201 0.0564 0.0517 0.0305 0.0276 0.3076 0.2717 0.2234 0.2003 5.6233 5.8340 4.0798 4.1819 4.2252 2001 base 0.0509 0.0272 0.2676 0.1926 5.8381 4.3259 41 Supplemental Figure 1. Ratio of nitrate concentrations during spring for "1.2SO2_ratio" case (left) and for "2NOXratio" case (right). *Please refer to Table 1 and Table 2 for abbreviations used in this figure. The color bars are saturated at 30% (left) and at 170% (right). SPR 1.2SO2jsio NOS-(%] SR 2 I - -- - 10 XraO W3-1%1 4 2 0 100 __~ngftuds 0[ 0 0 100 100 300 30. 44. 56. 72. 66. 100 114.125. 142Z 156.170 Supplemental Figure 2. The spring nitrate concentration ratio ("2001Asian/2001") focused on Arctic region. *Please refer to Table 1 and Table 2 for abbreviations used in this figure. The color bars are saturated at 0% and at 90%. SPR 2001AsIan/2001 NO3- 1%J S0*N 70*N W O*N .. -... .. 50%N .. .. " 40*N A ......... 30*N ...... -...-..-.. ..- 20ON 10*N - - ........- -- 0 V 120*E 0. 0. 5. 180* 120*W WOW 10. 15. 20. 25. 30. 35. 40. 50. 60. 70. 80. 90. 42 References Andreae, Mo 0., et al. "Vertical distribution of dimethylsulfide, sulfur dioxide, aerosol ions, and radon over the northeast Pacific Ocean." Journal of Atmospheric Chemistry 6.1-2 (1988): 149-173. Barletta, Barbara, et al. "Ambient halocarbon mixing ratios in 45 Chinese cities." Atmospheric Environment 40.40 (2006): 7706-7719. Barth, Mary C. "Numerical modeling of sulfur and nitrogen chemistry in a narrow coldfrontal rainband: The impact of meteorological and chemical parameters." Journal of applied meteorology 33.7 (1994): 855-868. Benkovitz, Carmen M., et al. "Global gridded inventories of anthropogenic emissions of sulfur and nitrogen." Journal of Geophysical Research: Atmospheres (1984- 2012) 101.D22 (1996): 29239-29253. Bertschi, I. T., et al. "PHOBEA/ITCT 2002 airborne observations of transpacific transport of ozone, CO, volatile organic compounds, and aerosols to the northeast Pacific: Impacts of Asian anthropogenic and Siberian boreal fire emissions." Journal of Geophysical Research: Atmospheres (1984-2012)109.D23 (2004). Brock, Charles A., et al. "Particle characteristics following cloud-modified transport from Asia to North America." Journal of Geophysical Research: Atmospheres (1984- 2012) 109.D23 (2004). Brown-Steiner, Benjamin, and Peter Hess. "Asian influence on surface ozone in the United States: A comparison of chemistry, seasonality, and transport mechanisms." Journal of Geophysical Research: Atmospheres (1984-2012)116.D17 (2011). Cooper, 0. R., et al. "A case study of transpacific warm conveyor belt transport: Influence of merging airstreams on trace gas import to North America." Journal of Geophysical Research: Atmospheres (1984-2012)109.D23 (2004). Cooper, 0. R., et al. "Increasing springtime ozone mixing ratios in the free troposphere over western North America." Nature 463.7279 (2010): 344-348. Cooper, 0. R., et al. "Measurement of western US baseline ozone from the surface to the tropopause and assessment of downwind impact regions."Journal of Geophysical Research: Atmospheres (1984-2012) 116.D21 (2011). Crawford, J., et al. "An assessment of ozone photochemistry in the extratropical western North Pacific: Impact of continental outflow during the late winter/early spring." Journal of Geophysical Research: Atmospheres (1984-2012) 102.D23 (1997): 28469-28487. Crutzen, Paul J., and Mark G. Lawrence. "The impact of precipitation scavenging on the transport of trace gases: A 3-dimensional model sensitivity study." Journal of Atmospheric Chemistry 37.1 (2000): 81-112. Dickerson, R. R., et al. "Aircraft observations of dust and pollutants over northeast China: 43 Insight into the meteorological mechanisms of transport."Journal of Geophysical Research: Atmospheres (1984-2012) 112.D24 (2007). Donkelaar, A. van, et al. "Analysis of aircraft and satellite measurements from the Intercontinental Chemical Transport Experiment (INTEX-B) to quantify long-range transport of East Asian sulfur to Canada." Atmospheric Chemistry and Physics 8.11 (2008): 2999-3014. Duncan Fairlie, T., Daniel J. Jacob, and Rokjin J. Park. "The impact of transpacific transport of mineral dust in the United States." Atmospheric Environment 41.6 (2007): 1251-1266. Dunlea, E. J., et al. "Evolution of Asian aerosols during transpacific transport in INTEX- B." Atmospheric Chemistry and Physics 9.19 (2009): 7257-7287. Fairlie, T. D., et al. "Impact of mineral dust on nitrate, sulfate, and ozone in transpacific Asian pollution plumes." Atmospheric Chemistry and Physics 10.8 (2010): 3999-4012. Fiore, Arlene M., et al. "Background ozone over the United States in summer: Origin, trend, and contribution to pollution episodes." Journal of Geophysical Research 107.D15 (2002): 4275. Granier, Claire, et al. "Evolution of anthropogenic and biomass burning emissions of air pollutants at global and regional scales during the 1980-2010 period." Climatic Change 109.1-2 (2011): 163-190. Gu, Dasa, et al. "Reduction in NO x Emission Trends over China: Regional and Seasonal Variations." Environmental science & technology 47.22 (2013): 12912-12919. Hack, James J., et al. "Simulation of the global hydrological cycle in the CCSM Community Atmosphere Model version 3 (CAM3): Mean features." Journal of climate 19.11 (2006): 2199-2221. Heald, Colette L., et al. "A large organic aerosol source in the free troposphere missing from current models." Geophysical Research Letters 32.18 (2005). Heald, Colette L., et al. "Transpacific transport of Asian anthropogenic aerosols and its impact on surface air quality in the United States." Journal of Geophysical Research: Atmospheres (1984-2012) 111.D14 (2006). IPCC, 2013: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change [Stocker, T.F., D. Qin, G.-K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P.M. Midgley (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 1535 pp, 2013. Jacob D. J., Heikes, B. G., Fan, S.-M., Logan, J. A., Mauzerall,D. L., Bradshaw, J. D., Singh, H. B., Gregory, G. L., Talbot,R. W., Blake, D. R., and Sachse, G. W.: Origin of ozone and NOx in the tropical troposphere: A photochemical analysis of aircraft observations over the South Atlantic basin, J. Geophys. Res., 101, 24 235-24 250, 1996. 44 Jacob, Daniel J., et al. "Transport and Chemical Evolution over the Pacific (TRACE-P) aircraft mission: Design, execution, and first results." Journal of Geophysical Research: Atmospheres (1984-2012) 108.D20 (2003). Jaegl6, L., et al. "Global distribution of sea salt aerosols: new constraints from in situ and remote sensing observations." Atmospheric Chemistry and Physics11.7 (2011): 3137- 3157. Jaffe, D. A., Anderson, T., Covert, D., Trost, B., Danielson, J., Simpson, W., Blake, D., Harris, J., and Streets, D.: Observations of ozone and related species in the northeast Pacific during the PHOBEA campaigns: 1. Ground-based observations as Cheeka Peak, J. Geophys. Res., 106, 7449-7461, 2001. Jaffe, Dan, and John Ray. "Increase in surface ozone at rural sites in the western US." Atmospheric Environment41.26 (2007): 5452-5463. Jaffe, Dan, et al. "Transport of Asian air pollution to North America." Geophysical Research Letters 26.6 (1999): 711-714. Jaffe, Dan. "Relationship between surface and free tropospheric ozone in the western US." Environmental science & technology 45.2 (2010): 432-438. Jaffe, Daniel, et al. "Export of atmospheric mercury from Asia." Atmospheric Environment 39.17 (2005): 3029-3038. Jaffe, Daniel, et al. "Increasing background ozone during spring on the west coast of North America." Geophysical Research Letters 30.12 (2003a). Jaffe, Daniel, et al. "Six 'new'episodes of trans-Pacific transport of air pollutants."Atmospheric Environment 37.3 (2003b): 391-404. Koumoutsaris, S., et al. "Influence of El Nifio-Southern Oscillation on the interannual variability of tropospheric ozone in the northern midlatitudes."Journal of Geophysical Research: Atmospheres (1984-2012) 113.D19 (2008). Kreidenweis, Sonia M., Yiping Zhang, and Gregory R. Taylor. "The effects of clouds on aerosol and chemical species production and distribution: 2. Chemistry model description and sensitivity analysis." Journal of Geophysical Research: Atmospheres (1984- 2012) 102.D20 (1997): 23867-23882. Kuhns, Hampden, Eladio M. Knipping, and Jeffrey M. Vukovich. "Development of a United States-Mexico emissions inventory for the big bend regional aerosol and visibility observational (BRAVO) study." Journal of the Air & Waste Management Association 55.5 (2005): 677-692. Kuhns, Hampden, et al. "The Treasure Valley secondary aerosol study I: Measurements and equilibrium modeling of inorganic secondary aerosols and precursors for southwestern Idaho." Atmospheric Environment 37.4 (2003): 511-524. 45 Lamsal, L. N., et al. "Application of satellite observations for timely updates to global anthropogenic NOx emission inventories." Geophysical Research Letters38.5 (2011). Li, Can, et al. "Recent large reduction in sulfur dioxide emissions from Chinese power plants observed by the Ozone Monitoring Instrument." Geophysical Research Letters 37.8 (2010). Li, Qinbin, et al. "North American pollution outflow and the trapping of convectively lifted pollution by upper-level anticyclone." Journal of Geophysical Research: Atmospheres (1984-2012) 110.D10 (2005). Liang, Q., et al. "Summertime influence of Asian pollution in the free troposphere over North America." Journal of Geophysical Research: Atmospheres (1984-2012) 112.D12 (2007). Liang, Qing, et al. "Long-range transport of Asian pollution to the northeast Pacific: Seasonal variations and transport pathways of carbon monoxide."Journal of Geophysical Research: Atmospheres (1984-2012) 109.D23 (2004). Liang, Qing, Lyatt Jaegl6, and John M. Wallace. "Meteorological indices for Asian outflow and transpacific transport on daily to interannual timescales."Journal of Geophysical Research: Atmospheres (1984-2012) 110.D18 (2005). Liao, Hong, et al. "Biogenic secondary organic aerosol over the United States: Comparison of climatological simulations with observations." Journal of Geophysical Research: Atmospheres (1984-2012) 112.D6 (2007). Lin, Jin-Tai, Da Pan, and Rui-Xiong Zhang. "Trend and interannual variability of Chinese air pollution since 2000 in association with socioeconomic development: A brief overview." Atmos. Oceanic Sci. Lett 6 (2013): 84-89. & Lin, Jintai, et al. "Recent changes in particulate air pollution over China observed from space and the ground: effectiveness of emission control."Environmental science technology 44.20 (2010): 7771-7776. Lin, Meiyun, et al. "Transport of Asian ozone pollution into surface air over the western United States in spring." Journal of Geophysical Research: Atmospheres (1984- 2012) 117.D21 (2012). Liu, Hongyu, et al. "Constraints from 21OPb and 7Be on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields." Journal of Geophysical Research: Atmospheres (1984-2012) 106.D11 (2001): 12109-12128. Liu, Hongyu, et al. "Transport pathways for Asian pollution outflow over the Pacific: Interannual and seasonal variations." Journal of Geophysical Research108.D20 (2003): 8786. Liu, Junfeng, Denise L. Mauzerall, and Larry W. Horowitz. "Source-receptor relationships between East Asian sulfur dioxide emissions and Northern Hemisphere sulfate 46 concentrations." Atmospheric Chemistry and Physics Discussions 8.2 (2008): 5537-5561. Lu, Zifeng, Qiang Zhang, and David G. Streets. "Sulfur dioxide and primary carbonaceous aerosol emissions in China and India, 1996-2010." Atmospheric Chemistry and Physics 11.18 (2011): 9839-9864. Luan, Y., and L. Jaegl6. "Composite study of aerosol export events from East Asia and North America." Atmospheric Chemistry and Physics Discussions12.8 (2012): 21977- 22022. Manktelow, P. T., et al. "Regional and global trends in sulfate aerosol since the 1980s." Geophysical Research Letters 34.14 (2007). Mari, C6line, Daniel J. Jacob, and Peter Bechtold. "Transport and scavenging of soluble gases in a deep convective cloud." Journal of Geophysical Research: Atmospheres (1984- 2012) 105.D17 (2000): 22255-22267. Martin, Randall V., et al. "Global and regional decreases in tropospheric oxidants from photochemical effects of aerosols." Journal of GeophysicalResearch:Atmospheres (1984- 2012) 108.D3 (2003). Meng, Z., D. Dabdub, and John H. Seinfeld. "Chemical coupling between atmospheric ozone and particulate matter." Science 277.5322 (1997): 116-119. Mijling, B., R. J. van der A, and Q. Zhang. "Regional nitrogen oxides emission trends in East Asia observed from space." Atmospheric Chemistry and Physics 13.23 (2013): 12003- 12012. Miyazaki, Y., et al. "Synoptic-scale transport of reactive nitrogen over the western Pacific in spring." Journal of Geophysical Research: Atmospheres (1984-2012) 108.D20 (2003). Murphy, D. M., et al. "Single-particle mass spectrometry of tropospheric aerosol particles." Journal of Geophysical Research: Atmospheres (1984-2012)111.D23 (2006). Ohara, T. A. H. K., et al. "An Asian emission inventory of anthropogenic emission sources for the period 1980-2020." Atmospheric Chemistry and Physics 7.16 (2007): 4419-4444. Park, Rokjin J., et al. "Export efficiency of black carbon aerosol in continental outflow: Global implications." Journal of Geophysical Research: Atmospheres (1984- 2012) 110.D11 (2005). Park, Rokjin J., et al. "Natural and transboundary pollution influences on sulfate-nitrateammonium aerosols in the United States: Implications for policy." Journal of Geophysical Research: Atmospheres (1984-2012) 109.D15 (2004). Park, Rokjin J., et al. "Regional visibility statistics in the United States: Natural and transboundary pollution influences, and implications for the Regional Haze Rule." Atmospheric Environment 40.28 (2006): 5405-5423. Park, Rokjin J., et al. "Sources of carbonaceous aerosols over the United States and 47 implications for natural visibility." Journal of Geophysical Research: Atmospheres (1984- 2012) 108.D12 (2003). Pfister, G. G., et al. "Characterizing summertime chemical boundary conditions for airmasses entering the US West Coast." Atmospheric Chemistry and Physics 11.4 (2011): 1769-1790. Pfister, G. G., et al. (2006), Ozone production from the 2004 North American boreal fires, J. Geophys. Res., 111, D24S07, doi:10.1029/2006JD007695 Pope III, C. Arden, and Douglas W. Dockery. "Health effects of fine particulate air pollution: lines that connect." Journal of the Air & Waste Management Association 56.6 (2006): 709742. Pope III, C. Arden. "Review: epidemiological basis for particulate air pollution health standards." Aerosol Science & Technology 32.1 (2000): 4-14. Price, Colin, and David Rind. "A simple lightning parameterization for calculating global lightning distributions." Journal of Geophysical Research: Atmospheres (1984- 2012) 97.D9 (1992): 9919-9933. Price, Colin, Joyce Penner, and Michael Prather. "NOx from lightning: 1. Global distribution based on lightning physics." journal of Geophysical Research: Atmospheres (1984- 2012) 102.D5 (1997): 5929-5941. Price, Heather U., et al. "Photochemistry, ozone production, and dilution during longrange transport episodes from Eurasia to the northwest United States."Journal of Geophysical Research: Atmospheres (1984-2012) 109.D23 (2004). Sanderson, M. G., et al. "A multi-model study of the hemispheric transport and deposition of oxidised nitrogen." Geophysical Research Letters 35.17 (2008). Seinfleld J. H. and S.N. Pandis (1988), Atmospheric chemistry and physics: From Air Pollution to Climate Change, 1326 pp., Wiley-Interscience, New York. Shindell, D. T., et al. "A multi-model assessment of pollution transport to the Arctic." Atmospheric Chemistry and Physics 8.17 (2008): 5353-5372. Solomon, Susan. "Stratospheric ozone depletion: A review of concepts and history." Reviews of Geophysics 37.3 (1999): 275-316. Stohl, A., Eckhardt, S., Forster, C., James, P., and Spichtinger, N.: On the pathways and timescales of intercontinental air pollution transport, J. Geophys. Res., 107(D23), 4684, doi:10.1029/2001JD001396, 2002. Streets, D. G., et al. "An inventory of gaseous and primary aerosol emissions in Asia in the year 2000." Journal of Geophysical Research: Atmospheres (1984-2012) 108.D21 (2003). Thompson, Anne M., et al. "Convective transport over the central United States and its role in regional CO and ozone budgets." Journal of Geophysical Research: Atmospheres (1984- 48 2012) 99.D9 (1994): 18703-18711. Unger, Nadine, et al. "Cross influences of ozone and sulfate precursor emissions changes on air quality and climate." Proceedings of the National Academy of Sciences of the United States of America 103.12 (2006): 4377-4380. US Environmental Protection Agency, US EPA, 2006.Air quality criteria for ozone and related photochemical oxidants; Report Nos. EPA/600/R-05/004af; Office of Research and Development, Research Triangle Park: NC, February 2006. US EPA, 2003. Guidance for Estimating Natural Visibility Conditions Under the Regional Haze Rule, US EPA OAQPS report, EPA 454/B-03-005, Research Triangle Park, NC. van der A, R. J., et al. "Trends, seasonal variability and dominant NOx source derived from a ten year record of N02 measured from space." Journal of Geophysical Research: Atmospheres (1984-2012) 113.D4 (2008). van der Werf, Guido R., et al. "Interannual variability in global biomass burning emissions from 1997 to 2004." Atmospheric Chemistry and Physics 6.11 (2006): 3423-3441. Walcek, C. J., et al. "S02 sulfate and HNO3 deposition velocities computed using regional landuse and meteorological data." Atmospheric Environment (1967) 20.5 (1986): 949- 964. Wang, Y., Jacob, D. J., and Logan, J. A.: Global simulation of tropospheric 03-NOxhydrocarbon chemistry, 3. Origin of tropospheric ozone and effects of non-methane hydrocarbons, J. Geophys. Res., 103/D9, 10 757-10 768, 1998. Wang, Yuhang, Jennifer A. Logan, and Daniel J. Jacob. "Global simulation of tropospheric 03-NO x-hydrocarbon chemistry: 1. Model formulation." Journal of Geophysical Research: Atmospheres (1984-2012)103 1.D9 (1998): 10713-10726. Wesely, M. L. "Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models." Atmospheric Environment (1967) 23.6 (1989): 1293- 1304. Wesely, M. L., and B. B. Hicks. "A review of the current status of knowledge on dry deposition." Atmospheric environment 34.12 (2000): 2261-2282. West, Benjamin, et al. "MTBE as a Tracer for Asian and Mexican Megacity Emissions." AGU Spring Meeting Abstracts. Vol. 1. 2007. Wiedinmyer, C., et al. "The Fire INventory from NCAR (FINN): A high resolution global model to estimate the emissions from open burning." Geoscientific Model Development 4 (2011): 625. Zhang, Guang J., and Norman A. McFarlane. "Sensitivity of climate simulations to the parameterization of cumulus convection in the Canadian Climate Centre general circulation model." Atmosphere-Ocean 33.3 (1995): 407-446. Zhang, Leiming, et al. "A size-segregated particle dry deposition scheme for an 49 atmospheric aerosol module." Atmospheric Environment 35.3 (2001): 549-560. Zhang, Lin, et al. "Improved estimate of the policy-relevant background ozone in the United States using the GEOS-Chem global model with 1/2x 2/3 horizontal resolution over North America." Atmospheric Environment 45.37 (2011): 6769-6776. Zhang, Lin, et al. "Transpacific transport of ozone pollution and the effect of recent Asian emission increases on air quality in North America: an integrated analysis using satellite, aircraft, ozonesonde, and surface observations."Atmospheric Chemistry and Physics Discussions 8.2 (2008): 8143-8191. Zhang, Qiang, et al. "Asian emissions in 2006 for the NASA INTEX-B mission."Atmospheric Chemistry and Physics 9.14 (2009): 5131-5153. Zhang, Qiang, et al. "NOx emission trends for China, 1995-2004: The view from the ground and the view from space." Journal of Geophysical Research: Atmospheres (1984- 2012) 112.D22 (2007). Zhao, B., et al. "NO x emissions in China: historical trends and future perspectives." Atmospheric Chemistry and Physics 13.19 (2013a): 9869-9897. Zhao, Bin, et al. "Environmental effects of the recent emission changes in China: Implications for particulate matter pollution and soil acidification."Environmental Research Letters 8.2 (2013b): 024031.