Benzene Structure & Reactions: Aromaticity Explained
advertisement
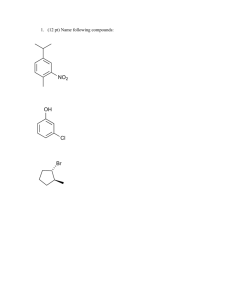
The Structure of Benzene Chemists first isolated benzene from coal tar distillate and found its molecular formula: C6H6. The question of benzene's structure remained for quite some time. The problem lay with the fact that its formula implied substantial unsaturation, yet benzene did not undergo reactions typical of alkenes. There were several proposed structures put forward at the time (1858): Klaus Dewar benzene Now known as Prismane. The Kekulé structure. One problem with the structure of benzene was that there were three known dibromide derivatives. They could be explained by the following three structures: Br BUT: closer inspection suggests that there should be two ortho isomers: Br Br Br Br Br Br Br ortho To resolve this, Kekulé proposed that a rapid interchange of the double and single bonds in benzene. This would make all six carbon atoms of benzene and the bonds between them identical. Br meta Br para In accordance with Kekulé's proposition, all six carbon-carbon bonds are the same length: 1.40Å. This is intermediate between the single C-C and double C=C bond length suggested by the hypothetical molecule 1,3,5-cyclohexatriene. 1,3,5-Cyclohexatriene (drawn to scale) Structure aside, we still need an explanation for the unusual reactivity (or unreactivity) of benzene. Another puzzle is the heat of hydrogenation of benzene. Once again, the reaction of benzene is quite different from that of alkenes. H2 + 28.6 kcal/mol 3H2 The addition of H2 to a double bond is usually exothermic. + 85.8 kcal/mol 3H2 + 49.8 kcal/mol For some reason, benzene is 36 kcal/mol more stable than the hypothetical cyclohexatriene. Cyclohexatriene + 3 H2 Enthalpy 36 kcal Benzene + 3 H2 85.8 kcal 49.8 kcal Conjugated double bonds tend to be more stable than isolated double bonds. Could this be the reason for benzene's unusual stability? CH2 CH3 + 3 H2 CH2 + 80.5 kcal/mol CH3 The Molecular Orbitals of Benzene Ψ6 Ψ4 Ψ5 Ψ2 Ψ3 6 x 2p Ψ2 Ψ1 The combination of the six atomic p-orbitals of the carbons of the ring leads to six π -orbitals. The lowest energy orbital is a very stable, low-energy π -orbital that is bonding between all six carbon atoms . The only node in this orbital is the one in the plane of the ring (which is common to all π -orbitals). With six electrons to put in these orbitals, benzene has just enough to fill the bonding MOs. Therefore, benzene has a filled set of low-energy molecular orbitals, just as the Noble Gases have a filled set of atomic orbitals. Resonance Energy The stability of benzene can be attributed to the set of filled bonding π -type molecular orbitals that result from the atomic p-orbitals around the ring. Molecules that share this kind of stability are often referred to as "aromatic" molecules. Originally, this had to do with their smell, but now the term aromatic is used to refer to compounds which have electrons that are delocalized around the ring. The degree to which benzene is stabilized by its aromatic nature is defined by the difference between its heat of hydrogenation relative to the hypothetical 1,3,5-cyclohexatriene. This energy, 36 kcal/mol, is called the resonance energy. For other aromatic molecules, the resonance energy is defined in an analogous manner. H H H H + H2 H In the first addition of H 2 to benzene, the resonance energy must be sacrificed. This initial hydrogenation is endothermic (by 5.6 kcal/mol)!. No wonder benzene is not inclined to undergo addition reactions. H If benzene is aromatic, then how about the following: After a great deal of difficulty these molecules were finally prepared. Cyclobutadiene is unstable (less stable than expected even for a diene!). We refer to such molecules as anti-aromatic . Cyclooctatetraene is not planar and is considered to be non-aromatic. How about: H Dihydropyrene 14 π-electrons H Cyclobutadiene 4 π-electrons 1,3,5,7-Cyclooctatetraene 8 π-electrons This molecule which has a perifery of 14 π-electrons is aromatic. Clearly, whether or not a molecule is aromatic or not depends on the number of π -electrons. Erich Hückel laid out a rule which accurately predicts which cyclic polyenes will be aromatic and which will not: to be aromatic, a monocyclic compound must contain 4n+2 π -electronss in the cyclic conjugated system, where n is an integer (0, 1, 2 etc.). Criteria for Aromaticity 1.) The molecule must be cyclic. 2.) Each member of the ring must have an atomic p orbital orthogonal to the plane of the ring (through which the electrons can circulate). 3.) The molecule must be sufficiently planar to allow side-on overlap of all the atomic p-orbitals. 4.) Its gotta fit The Rüle. N N N H O N If the N atom of pyrrole is sp3 hybridized, then pyrrole can't be considered aromatic. N N N H H H N N H H H H N N 1.58 D + 1.81 D + Pyrrolidine Pyrrole H H N H N + H3O + H2O + H3O N N H + H2O H N Pyrrole O Furan S Thiophene H H C C Cyclopentadienyl Anion Cyclopentadienyl Cation Nomenclature of Aromatic Compounds Some benzenes are simply named by the type of substituent followed by the term "benzene". H CH2CH3 NO2 H Br C C - H2 H Vinylbenzene (Styrene) Ethylbenzene Nitrobenzene Bromobenzene Many substituted benzenes have been known for over a century. They have been given trivial names that are firmly entrenched in the language of organic chemistry. These names serve as parent names for their substituted derivatives. H3C CH3 NH2 OH CH OCH3 1.) O2 2.) H3O+ Cumene (Isopropylbenzene) Phenol (not to be confused with phenyl) Anisole Cl Cl Biphenyl (the parent of PCBs) CH3 Aniline (not to be confused with alanine, the amino acid) Toluene O C OH O C H Cl Cl A PCB. Benzoic Acid Benzaldehyde Substituted Benzenes In disubstituted benzenes, the relative positions of the two substituents must be specified. There are two ways in which this is done. IUPAC uses a numbering system. A trivial method also persists. CH3 CH3 CH3 CH3 para-Xylene (intermediate in the preparation of poly(ethylene terephthalate) CH3 meta-Xylene CH3 ortho-Xylene Polysubstituted Benzenes are named as derivatives of the parent benzene (if applicable). Substituent positions are numbered from the dominant functional group, or follow rules akin to those for numbering alkanes. CH3 C Cl NH2 O OH NO2 o-Methylbenzoic acid (2-Methylbenzoic acid) m-Nitroaniline (3-Nitroaniline) Cl p-Dichlorobenzene (1,4-Dichlorobenzene) OH CH3 Cl F F Cl Cl 2,4,5-Trichlorophenol H3C Br 1-Bromo-2,3-difluorobenzene CH3 1,3,5-Trimethylbenzene (Mesitylene) Reactions of Benzene Just like alkenes, benzene has a substantial amount of electron density due to the π-orbitals. As a result, benzene also undergoes reactions with electrophilic species "E+". Alkenes undergo electrophilic addition reactions: + Br2 C C H3C Br H H Benzene, however undergoes electrophilic substitution reactions: Br H HC CH2 H Br FeBr3 + Br2 H3C + HBr The general mechanism for ALL electrophilic aromatic substitution reactions is the following two step process: B H + H—B H E + H E E H σ-complex H The σ-complex is a very important intermediate. It is stabilized by delocalization of the positive charge: H E H H E H H E H Substitution vs Addition Br Energy H H Br H σ-complex σ-complex Reaction Progress Br H Br+ H H Energy H Br+ HBr H H Br H σ-complex Br σ-complex Reaction Progress H Br Br Electrophilic Aromatic Substitution - Electrophiles E+ EAS reactions differ only in the identity of E+ and how it is generated. Nitration + HNO3 H2SO4 heat NO2 O O N O N O + H2SO4 H O H Reduction of nitroaromatics gives anilines. O + HSO4 - N H O O In the case of nitration, E+ is NO2+. Sulfonation + SO3 H2SO4 heat SO3H O O S O S OH + H2SO4 + HSO4 - O O Halogenation Br Fe or FeBr3 + Br2 + HBr Cl + Cl2 Fe or FeCl3 Br Br + FeBr3 + HCl δ+ Br Br δFeBr3 + H2O Alkylation of Aromatic Compounds - The Friedel-Crafts Reaction + RCl H3C CH Cl R AlCl3 + HCl H3C δ+ CH Cl H3C + AlCl3 H3C δAlCl3 In some alkylations, this complex may serve as the alkylating electrophile. H3C H + AlCl4 C H3C When a relatively stable carbocation is possible, then it is likely the electrophile. Again, the mechanism for this reaction is no different than any other Electrophilic Aromatic Substitution. Cl H H3C C CH3 CH3 CH CH CH3 H CH3 + HCl H3C Alternativly, carbocation electrophiles can be generated by the protonation of alkenes by strong acid: H H3PO4 H Full mechanism: p 800. H H + H3PO4 H H H + H2PO4 Carbocation Rearrangement during Friedel-Crafts Alkylations + CH3CH2CH2Cl CH2CH2CH3 AlCl3 CH3 CH CH3 40% 35°C 5h 60% PhH PhH H δ+ H3C C C H2 H H3CH2CH2C Cl + AlCl3 Friedel-Crafts Acylation C AlCl3 H3C δAlCl3 C H + AlCl4 H3C O O H3C C Cl Cl Rearrangements like this are always a problem when a more stable carbocation can result from a hydride or alkyl shift. O CH3 H3C C Cl + AlCl3 O H3C C+ Cl δ δAlCl3 An acid chloride. Mechanism O O C H3C H Cl C O H3C C Cl + HCl H3C C O H3C C O The Acylium cation, a resonance stabilized carbocation, is the electrophile. CH3 The Friedel-Crafts acylation can be used to circumvent the problems of carbocation rearrangement. O O H2NNH2, KOH C AlCl3 (Wolff-Kishner reduction) CH2CH3 C Cl H3CH2C or Zn/Hg amalgam, HCl (Clemmenson reduction) CH2CH2CH3 Reactions of Substituted Benzenes CH3 CH3 CH3 CH3 NO2 HNO3, Acetic Acid + + NO2 Toluene 2-Nitrotoluene Statistical Actual 40% 60% 3-Nitrotoluene 40% 3% NO2 4-Nitrotoluene 20% 37% This reaction is faster than the corresponding nitration of benzene. Why? And what is the reason for this particular product distribution? All alkyl groups and several other substituents show this pattern of reactivity. NO2 NO2 NO2 NO2 NO2 HNO3, H2SO4 + NO2 Nitrobenzene NO2 1,3-Dinitrobenzene 93% Yield Minor products This reaction is much slower than the nitration of benzene. Toluene gives mainly ortho and para products while nitrobenzene gives the meta product almost exclusively. What is it about the substituent that directs the position of the incoming electrophile? Cl Cl Cl Cl NO2 HNO3, H2SO4 30% o-Chloronitrobenzene + + NO2 1% m-Chloronitrobenzene NO2 69% p-Chloronitrobenzene This reaction is slower than the nitration of benzene. All of the halogens follow this pattern of reactivity. What's going on? Substituent Directing Effects σ-Complex Intermediates CH3 CH3 CH3 O NO2 NO2 + H N+ CH3 CH3 NO2 H + NO2 H + O CH3 CH3 O O CH3 NO2 NO2 H + NO2 H H CH3 CH3 + + N+ CH3 CH3 CH3 CH3 NO2 CH3 + + O N + O H NO2 + H NO2 H NO2 NO2 The σ -complexes of the ortho- and para-substituted toluenes are stabilized by the methyl group. This is reflected in a lower activation energy leading to these intermediates - they are formed faster. ANY SUBSTITUTENT CAPABLE OF STABILIZING AN ADJACENT POSITIVE CHARGE WILL GIVE PREDOMINANTLY ORTHO- AND PARASUBSTITUTION. More Substituent Directing Effects O N+ O O- σ-Complex Intermediates ON+ O NO2 + N+ O N+ ONO2 H O NO2 N+ O- O O N+ O- O O NO2 ON+ NO2 NO2 NO2 H + O- O NO2 N+ NO2 H NO2 H + O- + + N+ N+ H + O O O- O N+ NO2 H O NO2 O- NO2 NO2 NO2 N+ + + O O N + H NO2 + H NO2 H The σ -complex intermediates for o- and p-substitution are destabilized due to unfavourable charge interactions in their resonance structures. This is reflected in the activation energy for the formation of these complexes (the Eas are higher than that of the m-substitution σ -complex) and they are formed more slowly. ANY SUBSTITUENT WHICH DESTABILIZES AN ADJACENT POSITIVE CHARGE WILL BE A META-DIRECTOR. Substituents with a lone pair of electrons on the atom attached to the ring are a special case. The lone pair can be used very effectively to stabilize the positive charge. An additional resonance structure can be written: : OH : OH : : : : : :O H : OH OH OH + + + H E+ H E H E Examples of this type of substituent: : NR2 : NHR O H : : OR H E E O : NH2 OH N C R : : : : : : : : : : : : : : O C R Halide substituents have three lone pairs of electrons and are capable of using them to stabilize an adjacent carbocation. As such they are also ortho- para- directing groups. But they are different than all other ortho- para- directing groups in one key respect, as we Cl I F Br shall see. : : Z : Z= E Directing Groups and Reaction Rate We have seen how substituents can be placed into two classes: those that are ortho/para-directors and those that are meta-directors. What about the effect of substituents on the rate of reaction? We made the following observations: 1.) Benzenes substituted with ortho/para-directors will react more quickly than benzene itself UNLESS THE SUBSTITUENT IS A HALIDE (F, Cl, Br, I). 2.) Benzenes substituted with meta-directors ALWAYS REACT MORE SLOWLY THAN BENZENE ITSELF. Why? Recall that these substitutions are ELECTROPHILIC, i.e. the attacking species are attracted by the large amount of electron density circulating in the aromatic ring. Alkyl groups are "electron releasing" or "electron donating". This is how they are able to stabilize adjacent positive CH + 3 charges in EAS reactions and in other carbocation species. (Recall that 3° carbocations are more stable than 2° carbocations because they bear more alkyl substituents.) Substituents with lone pairs on them can delocalize an electron pair into the aromatic ring thus dramatically increasing the electron density. This is reflected in the resonance structures for phenol. : : : : + : OH + OH + OH _ + OH _ _ In both these cases, electron density is donated by the substituent into the aromatic ring. Substituents that are capable of doing this are called ELECTRON DONATING GROUPS. By donating electron density into the aromatic ring they increase the charge density of the aromatic cloud making it more attractive to attacking electrophiles. These groups are therefore said to be ACTIVATING GROUPS as well as being ortho/para directors. Meta-Directing Groups These are groups which destabilize an adjacent positive charge, usually because the atom attached to the aromatic ring itself carries a formal positive charge, or is positively polarized by virtue of being attached to more electronegative atom(s). Examples (only the destabilized resonance structure of the σ-complex is shown): _ O H O _ O O N+ N+ + + E H H H : NH2 H N+ H H H 3O + + E+ E H N+ H E Protonation of the amino group ties up the lone pair that could stabilize the positive charge. The N atom now bears a +ve charge making the NH3+ group a meta-director. In the the nitro group there is always a +ve formal charge on N. _ + H O H C+ C + + E H :O H : : : :O H C E H E : : ANY KIND of carbonyl attached to an aromatic ring will be a meta-directing group because of the positive polarization of the carbon (as seen in the left-hand resonance structure. Delocalizing the +ve charge (onto the O atom) does not help in this case. O is very electronegative and abhores to be electron deficient (it has only six electrons in the right-hand resonance structure). _ :N N All of the following are meta-directors: O C+ C O Amides Esters C O C + NR2 + OR C O OH O Carboxylic Acids C C R H E H E Cl Aldehydes Acid For similar reasons, nitrile substituents Ketones Halides are also meta-directors. What about meta-directors? These groups destabilize adjacent positive charges because they themselves are electron deficient and/or bear formal positive charges. _ O + O Meta-directors are electron withdrawing groups. They suck N electron density away from the aromatic electron cloud making it less attractive to electrophiles. This slows the reaction between the electrophile and the aromatic ring as a result. Thus, meta-directors are said to be DEACTIVATING. + What about the halides? Well, the halides strike a unique balance. They are ortho/para-directors because their lone pairs are capable of stabilizing an adjacent positive charge in a π -type of interaction. At the same time, the halogens are electronegative enough to diminish the electron density of the ring through the σ-bond.. : : Cl : + Directing influence o,p o,p o,p o,p m m Halides are considered to be electron-withdrawing groups. They are unique among substituent types in that they are ORTHO/PARA DIRECTING AND DEACTIVATING. Summary Substituent -NH2 , -NHR, -NR2 (amino and aminoalkyl), -OH (hydroxy) Effect on Rate Very Strongly Activating -NHCOR (amide), -OR (ether), -OCOR (ester - O end) Strongly Activating R (alkyl), Aryl, Vinyl Activating X = F, Cl, Br, I (Halides) -CHO (aldehyde), -COR (ketone), -CO2H (carboxylic acid), -CO2R (ester), -COCl (acid halide), -CN (nitrile), -SO3H (sulfonic acid) -NO2 (nitro) Deactivating Strongly Deactivating Very Strongly Deactivating
 0
0
advertisement
Related documents







Download
advertisement
Add this document to collection(s)
You can add this document to your study collection(s)
Sign in Available only to authorized usersAdd this document to saved
You can add this document to your saved list
Sign in Available only to authorized users