Levels of Protein Structure

Determination of Protein Structure
Methods for Determining Structures
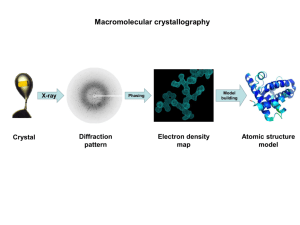
X-ray crystallography – uses an X-ray diffraction pattern and electron density maps
NMR spectroscopy – structure is based on local conformation and distance between atoms that are close to one another
Electron microscopy – relies on an image of the overall shape of the molecule
In most cases, the experimental information is not sufficient to build an atomic model from scratch. Additional knowledge about the molecular structure must be added. For instance, we often already know the sequence of amino acids in a protein, and we know the preferred geometry of atoms in a typical protein (for example, the bond lengths and bond angles). This information allows the scientist to build a model that is consistent with both the experimental data and the expected composition and geometry of the molecule.
NMR Spectroscopy
The protein is purified, placed in a strong magnetic field, and then probed with radio waves.
A distinctive set of observed resonances may be analyzed to give a list of atomic nuclei that are close to one another, and to characterize the local conformation of atoms that are bonded together.
This list of restraints is then used to build a model of the protein that shows the location of each atom.
The technique is currently limited to small or medium proteins, since large proteins present problems with overlapping peaks in the NMR spectra.
1D NMR spectrum from a globin protein
Interaction 1 is between two side chain hydrogens in the protein.
Interaction 2 is between a methyl group and a hydrogen on the heme.
Use of 2D NMR to determine the 3D structure of a globin protein
NMR Spectroscopy
A major advantage of NMR spectroscopy is that it provides information on proteins in solution, as opposed to those locked in a crystal or bound to a microscope grid, and thus, NMR spectroscopy is the premier method for studying the atomic structures of flexible proteins.
A typical NMR structure will include an ensemble of protein structures, all of which are consistent with the observed list of experimental restraints. The structures in this ensemble will be very similar to each other in regions with strong restraints, and very different in less constrained portions of the chain. Presumably, these areas with fewer restraints are the flexible parts of the molecule, and thus do not give a strong signal in the experiment.
Electron Microscopy
A beam of electrons is used to image the molecule directly.
If the proteins can be coaxed into forming small crystals or if they pack symmetrically in a membrane, electron diffraction can be used to generate a 3D density map, using methods similar to X-ray diffraction.
For a few particularly well-behaved systems, electron diffraction produces atomiclevel data, but typically, electron micrographic experiments do not allow the researcher to see each atom.
Electron micrographic studies often combine information from X-ray crystallography or NMR spectroscopy to sort out the atomic details.
This has proven very useful for multimolecular structures such as complexes of ribosomes, tRNA and protein factors, and muscle actomyosin structures.
Electron Microscopy
The tail of the T4 bacteriophage has been examined by combining electron microscopy and atomic structures. The image shows a surface rendering of the EM data ( emd-
1048 ) with atomic coordinates from PDB entries
X-ray Crystallography
Chapter 18
X-ray Crystallography
Chapter 18
Used for most of the structures in the PDB
The protein is purified and crystallized
Crystal is subjected to an intense beam of X-rays.
The proteins in the crystal diffract the X-ray beam into one or another characteristic pattern of spots, which are then analyzed to determine the distribution of electrons in the protein.
The resulting map of the electron density is interpreted to determine the location of each atom.
Unit Cells
Page 373
X-ray diffraction pattern
Page 374
Protein crystals have large solvent filled spaces
Page 375
Page 376
Hanging Drop Method of
Protein Crystallization
A diffraction experiment
Page 377
Structure Factors and Electron Density
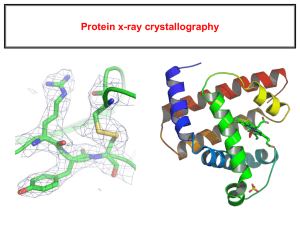
A crystal is subjected to a narrow beam of intense X-rays, and the diffraction pattern is observed with a detector or a sheet of film. This pattern forms a characteristic array of spots, commonly referred to as reflections. Crystallographers measure the intensity of these reflections and use the information to determine the distribution of electrons in the crystal. The result is a map of the crystal that shows the distribution of electrons at each point, which may then be interpreted to find coordinates for each atom in the crystallized molecules.
Diffraction of x-rays by a crystal
Page 378
Amplitude, Phase, and Wavelength
Page 379
Reflection angle of a diffracted beam
Page 379
Heavy Atoms
Page 380
Heavy Metal Diffraction
Page 380 negative positive
Electron density maps
Page 382
Fitting into an electron density map
Page 382
X-ray Crystallography
The experimental electron density from a structure of DNA is shown here (PDB 196d), along with the atomic model that was generated based on the data.
The contours surround regions with high densities of electrons, which correspond to the atoms in the molecule.
Where are the Hydrogen Atoms?
Most crystallographic experiments do not resolve hydrogen atoms.
NMR-determined structures most often include hydrogen atoms since much of the experimental information consists of the distance between hydrogen atoms.
Since crystallographic experiments typically do not see hydrogen atoms, and since oxygen and nitrogen atoms have similar numbers of electrons and thus look similar in crystallographic electron density maps, it is often difficult to determine the exact identity of atoms in sidechains such as asparagines and glutamines.
In some cases, if you look carefully at the hydrogen-bonding pattern with neighboring amino acids, you may find a better match by switching the nitrogen and oxygen in an amide sidechain.
Structure of insulin solved by X-ray crystallography
(top 2ins) and NMR spectroscopy (bottom 2hiu) .