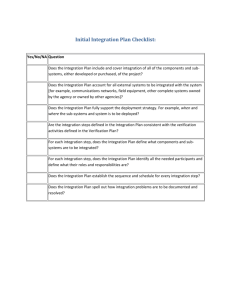
This procedure is not applicable to research activities that precede
advertisement

VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 1 of 11 1. PURPOSE The purpose of this procedure is to ensure that product is developed in a systematic way, ensuring that risk control measures are incorporated in the design, that all design outputs are verified against specifications and validated against user requirements, and that regulatory and standards requirements are fulfilled. 2. SCOPE This procedure covers design and development of new medical devices, including their packaging and labeling, and to modifications and upgrades of existing devices. This procedure applies from the approval of the initial Design Plan. This procedure is not applicable to research activities that precede design and development. This is the primary document meeting the applicable regulatory requirements for Product Development as defined in [Company Name]’s Quality System Manual (POL-001) but approved suppliers providing design services and having procedures for Design Controls that have been approved by [Company Name], may rely on their own procedure(s) instead of this procedure. 3. REFERENCES AND RELATIONSHIPS SYS-025 Technical Documentation Procedure SYS-003 Management Review Procedure SYS-009 Clinical Procedure SYS-040 Usability Procedure SYS-019 Post-Market Surveillance Procedure SYS-011 Supplier Quality Management Procedure SYS-008 Product Development Procedure SYS-010 Risk Management Procedure SYS-013 Servicing Procedure SYS-026 Regulatory Affairs Manual 4. DOCUMENT APPROVAL Changes to this procedure document and associated forms and templates are to be approved by: Product Development Process Owner Quality Manager. 5. REVISION HISTORY Rev Date mm/dd/yy DCN A mm/dd/yy YY-nnn What changed? Why did it change? Author Initial release -- R Packard VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 2 of 11 6. RESPONSIBILITIES AND AUTHORITIES Role Responsibilities and Authorities Senior Management Shall support the resource needs of the project. Project Manager A project manager will be designated by Senior Management and will be responsible for: Project Team Members Reporting team progress to senior management. Ensuring proper documentation is filed in the DHF throughout the design process. Ensuring that a DMR / TF Index is completed during the design transfer Stage of the project or updated for existing products. Ensuring that team members are trained in this procedure as required. Overall responsibility for the design control process. The project team members will include representation from the following functions as necessary, with a minimum of four team members: Research (chemistry, etc.) Engineering (hardware/software/mechanical/etc. – may use a contract service provider to perform this function) Quality/Regulatory (may use external resource for this function) Marketing Operations Manufacturing (may use an external resource such as a contract manufacturer for this function) Clinical Trials (may use external resource for this function such as a Clinical Research Organization) Finance 7. PROCEDURE 0. PRODUCT DEVELOPMENT STAGES As indicated in Section 13 of this procedure, the design control process consists of 5 stages. Typically there is a “gate” at the end of each stage where a design review is conducted and management makes a decision as to whether the project will be funded for the next stage or funding will be redirected to other projects. Stages can be combined, particularly Stage 4 and 5, but this must be reflected in the design plan. In addition, it is not required to have more than one design review—at the end of the project. However, fewer design reviews is only recommended for projects where the product is a revision of an existing design. The preferred approach is to have five design reviews, but to have shorter stage durations between the design reviews when the design project is a revision of an existing design. The Change Control Process, SYS-006, is not required during the design process. It is recommended that technical documentation created during the design process be controlled with a “Date Revision” or a different system that used for the Change Control Process. For example, if the Change Control Process uses alphabetic characters to identify revisions then design controls may use numeric characters to identify revisions. It is also recommended to document revisions to the following documents when significant design changes are made during a design project: Design Plan Design Requirements Trace Matrix (DRTM) VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 3 of 11 1. STAGE 1: CONCEPT STAGE 1 WHEN TO START DOCUMENTATION Design control documentation is essential during product investigations for protection of intellectual property. Therefore, all design activities should be documented from the initial concept through final product launch. Design Team meetings should be documented by the Project Manager or a designee throughout the project and included in the DHF. 2 WHO STARTS THE DESIGN PROJECT When a design project is started, there may be no funding for the project. During the Concept Stage the project definition needs to be documented, a design team needs to be established, user needs should be documented and a design plan should be drafted. Marketing is typically responsible for writing the project definition and identifying preliminary user needs. It is recommended to incorporate the project definition into the project plan to ensure that the project definition is also reviewed, approved and updated as the project evolves. 3 ESTABLISH A TEAM The Project Manager should be designated by Senior Management once the project definition is completed. The Project Manager will assemble the team which may include additional members to those specified under Responsibilities, e.g. customer service, technical services. The Project Manager, assignment of team members and the project budget should be formally approved when the design plan is approved. 4 INITIATE A DESIGN PLAN Initial planning meeting(s) will be organized by the Project Manager. The intention is to meet the requirements of design planning and bring the team together. A Risk Management Plan (refer to the Risk Management Procedure, SYS-010) should be incorporated in the Design Plan. The best way to create an initial design plan is to start with a previous design plan that was completed for a similar product and revise that plan. Ideally the previous design team conducted a post-mortem on the design project and identified recommended changes for future design plans. Whenever possible, verification and validation activities should be standardized for consistency and to save time and resources. The design plan should identify the following: all the major tasks in the design project (especially verification and validation activities and creating technical documentation), timing of each design review should be documented, responsibilities for implementation of all tasks, and interfaces with different groups or activities that provide, or result in, input to the design and development process. The Design Plan must be reviewed and approved by the Project Manager, the Project Team Members, and Senior Management. After approval the Design Plan may need to be conveyed to additional parties. Updates & subsequent approvals of the Design Plan should occur as the design evolves. 5 RISK MANAGEMENT The Risk management process is defined in SYS-010 and is applied throughout product realization. A risk management plan is required, but it is recommended to incorporate the risk management plan into the design plan at this stage by identifying the risk management activities that shall be performed throughout the design process. Recommended risk management activities are outlined in the flowchart in Section 13 of this procedure. During the design transfer process, a post-market surveillance (PMS) plan should be created. The risk VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 4 of 11 management plan should be incorporated into the PMS plan, because risk management must continue to gather post-production information and the risk management file must be updated for the life of the product. Risk management begins with the development of the design input requirements. 6 REGULATORY PATHWAY DOCUMENTATION The regulatory pathway for a given product defines the verification and validation testing that must be performed, and thereby it has a dramatic effect upon the project design plan and the cost of the project. Therefore, the regulatory pathway should be documented as part of the design plan for all products. The regulatory pathway document should also identify any relevant harmonized standard, guidance documents and any applicable “Special Controls” guidance documents from the FDA. Previous regulatory submissions are useful to make sure that no regulatory requirements have been inadvertently omitted. The regulatory representative on the project team should work with the Project Manager to make sure that all the required technical documentation is included in the plan and that appropriate personnel are identified for each technical document. 7 INITIATING THE DESIGN REQUIREMENTS TRACE MATRIX (DRTM) The DRTM is initiated during Stage 1 of the design process. User Needs are added to the User Needs column of the DRTM (i.e., FRM-019). The remaining columns of the DRTM are populated in subsequent stages of the design process. 8 STAGE 1 DESIGN REVIEW The primary purpose of the Stage 1 Design Review is to review and approve the initial design plan— including timing and resources required. Resource approval is typically only for the next stage of the design process with tentative approval for the subsequent stages. A design review meeting for Stage 1 should be conducted by the Project Manager, using the Design Review Form FRM-018. An independent reviewer must participate in the design review as well. Attendees of the Stage 1 Design Review must be documented on the Design Review Form and any recommendations for actions to be taken, in addition to execution of the design plan, should be documented on the Design Review Form. 2. STAGE 2: FEASIBILITY STAGE VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: 1 A D8 R. Packard 5 of 11 DEMONSTRATING FEASIBILITY During this stage prototype versions of the product may be constructed virtually or physically in order to evaluate the feasibility of the design concept. Often multiple design concepts will be evaluated in parallel in order to identify the design solutions that are most likely to succeed, but more importantly to identify any design inputs that may not have been identified from previous product versions or competitor products. The most challenging design verification and validation tests should be identified during this stage. As market preference testing or user feedback is required in the form of a Usability Engineering Analysis, simple Prototypes, or mockups may be used. 2 RISK MANAGEMENT Typically the first risk management activity is identification of hazards. These hazards are the risk management outputs used to identify design inputs. As the design evolves, new hazards may become evident and the design inputs may need to be updated. To systematically identify and, when necessary, reduce the risks of each hazard, the risk management process is integrated into the design process and documented in the Design Requirements Matrix. Refer to SYS-010 Risk Management Procedure for hazard identification. 3 DOCUMENTING DESIGN INPUTS Design Inputs are a set of requirements that address the intended use of the product, including the needs of the user and the patient. These inputs should be documented in such a way that the actual requirements are clear and unambiguous. If the design is based on a previous product design that is similar to the device(s) of the current design project, applicable information from the previous similar designs should be used. Design Inputs should be quantitative and/or specific to acceptance criteria as well as identify the testing that is applicable to each user need. Harmonized Standards should be used whenever possible, and where conflicting standards are required for different markets the conflict shall be resolved by the team. If a Harmonized Standard is not available, then internal testing protocols may be referenced instead. 4 DESIGN REQUIREMENTS TRACE MATRIX (DRTM) Each hazard identified shall be added to the appropriate column of the DRTM (FRM-019). Incorporating the hazards into the DRTM facilitates traceability of risk controls to each hazard. The remaining columns of the DRTM are populated in subsequent stages of the design process. 5 STAGE 2 DESIGN REVIEW The primary purpose of the Stage 2 Design Review is to review and approve the design inputs— including all hazards identified. A design review meeting for Stage 2 should be conducted by the Project Manager, using the Design Review Form FRM-018. An independent reviewer must participate in the design review as well. Attendees of the Stage 2 Design Review must be documented on the Design Review Form and any recommendations for actions to be taken, in addition to execution of the design plan, should be documented on the Design Review Form. 3. STAGE 3: DEVELOPMENT STAGE 1 PRODUCT DEVELOPMENT During this stage of the project various design solutions are evaluated—often in parallel. Best practice is to develop a minimum viable product that can be evaluated against a screening test that is a simplified version of the Design Verification or Design Validation protocols that will be used later in Stage 4 and Stage 5. Finite element analysis and bench top simulations should be used whenever possible in lieu of conducting animal testing and prior to conducting fist-in-human testing. After VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 6 of 11 preliminary screening tests have been successful, sometimes cadaver testing may be conducted to verify usability aspects that cannot be verified without human anatomy. 2 Design changes are typically frequent and significant during this stage of the design process. Many of the changes are a direct result of testing failures where new design inputs are identified. Therefore, it is important to update the DRTM and the Design Plan during this Stage. It is also common to begin certain types of design verification early during this stage instead of waiting for Stage 4. For example, cyclic testing for reliability and/or durability may be started at this stage to ensure that it is completed earlier. If the product involves software, software unit validation of various modules may be conducted during this period while final software validation of the complete code may be conducted in Stage 4. If validation of off-the-shelf software is required, that validation may be conducted at this stage. RISK MANAGEMENT A Risk Analysis (refer to SYS-010) shall be performed during this Stage for all known Hazards based upon the Hazard Identification activity performed during Stage 2. Risk Control Option Analysis shall be performed during this stage as well. The option analysis shall be included in the Risk Management File and the final set of risk control options are documented in the appropriate column of the DRTM. The DRTM includes a column for estimation of “Severity of Harm” and “Probability of Occurrence (P1)”. These values are used to compare risk control options and prioritize the development of design risk controls. All activities that verify an aspect of the design must be documented with identification of the design, method(s), date and individual performing the verification task with the individual’s signature or initials. 3 DESIGN REQUIREMENTS TRACE MATRIX (DRTM) Each Design Specification or Drawing shall be added to the appropriate column of the DRTM (FRM019). These are the Design Outputs that will be subject to Design Verification in Stage 4. Various aspects of the Design Outputs are risk controls and estimation of risks—which should be documented in the appropriate columns of the DRTM as indicated in the previous step. 4 STAGE 4 DESIGN REVIEW The primary purpose of the Stage 3 Design Review is to review and approve the Design Outputs and to authorize proceeding with Design Verification activities. A design review meeting for Stage 3 should be conducted by the Project Manager, using the Design Review Form FRM-018. An independent reviewer must participate in the design review as well. Attendees of the Stage 3 Design Review must be documented on the Design Review Form and any recommendations for actions to be taken, in addition to execution of the design plan, should be documented on the Design Review Form. 4. STAGE 4: PILOT STAGE VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: 1 A D8 R. Packard 7 of 11 PILOT STAGE During this stage the primary task is to complete Design Verification prior to Design Validation and/or a clinical study. If a clinical study is not required for certain markets, a pilot launch in those markets may be warranted. For example a product may require a clinical study and PMA approval in the USA, but the product may have an abbreviated regulatory approval process in Canada. Therefore, this stage may be used for completing verification testing required prior to a clinical study or for completion of a regulatory submission for markets that do not require a clinical study. 2 RISK MANAGEMENT Verification of effectiveness of risk controls is determined during Design Verification. The Design Verification protocols and reports should be referenced in the DRTM. This is also the Stage during which a Clinical Evaluation via the Literature Search Route should be performed. The purpose of the Clinical Evaluation is to verify that all the potential hazards have been identified and to assess the residual risks against the benefits of the intended use. The Risk/Benefit Analysis is documented in the Clinical Evaluation and the Risk Management File. If a Clinical Study is not required, then the Risk/Benefit Analysis should be performed during the Pilot Stage. If a Clinical Study is required, the Risk/Benefit Analysis is documented in the Clinical Study Summary Report and the Risk Management File. Information from clinical evaluations and clinical studies can also be used to estimate the probability of occurrence of harm resulting from occurrence of possible hazards (P2). Risk is defined as the severity of harm (S) multiplied by the probability of occurrence of harm (P1xP2). Therefore, risk = SxP1xP2 and this value should be used to quantitatively compare relative risks of each hazard. Refer to SYS-010 for the Risk Management Procedure. During this stage of the design project the post-market surveillance (PMS) plan should be drafted; refer to Post-Market Surveillance Procedure SYS-019. This plan should incorporate a risk management plan for collection of post-production information. The PMS plan does not replace the risk management plan in the design plan, because the PMS plan is not implemented until the commercial release of the product. 3 CLINICAL EVALUATION A Clinical Evaluation (Literature Review or sometimes a Formalized Clinical Testing) is required to be done for certain regulatory submissions—such as CE Marking. If formalized Clinical Testing is planned this must be done in accordance with the Investigational Device Exemption procedure. The Investigational Device Exemption Procedure has provisions for Internal Review Board and Informed Consent activities as well as labeling requirements. Refer to SYS-009 Clinical Procedure. 4 DESIGN REQUIREMENTS TRACE MATRIX (DRTM) Each Design Verification protocol and report shall be added to the appropriate column of the DRTM (FRM-018). The verification reports provide objective evidence that the design specifications (i.e., Design Outputs) meet the design inputs, and the reports also verify effectiveness of the risk control options implemented. VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: 5 A D8 R. Packard 8 of 11 DESIGN TRANSFER TO PRODUCTION The completeness and accuracy of the manufacturing documentation is verified in the transfer to manufacturing activities. Transfer to manufacturing is the process that ensures the product design specifications have been correctly translated into manufacturing specifications. This is accomplished at a minimum by releasing documentation, training personnel and conducting trial production runs. Process validation protocols and reports created during this process. Design Transfer activities should begin with initiation of a draft Device Master Record (DMR) / Technical File (TF) Index. As technical documentation, procedures and work instructions are completed these documents are added to the DMR/TF Index. The final DMR/TF Index is approved at the final design review and the product is released commercially. Process Validation, Design Verification and Design Validation require the use of components and finished devices that are representative of what will be used in commercial distribution or these verification and validation activities may need to be repeated. Therefore, it is recommended to coordinate closely with supply-chain management in order to ensure that final component specifications are released early enough to begin verification and validation on-time. The Design Transfer process is not completed until the final design review approving the product for commercial distribution, and the Design Transfer process may begin in Stage 3 for design specifications that are unlikely to change. 6 STAGE 4 DESIGN REVIEW The primary purpose of the Stage 4 Design Review is to review and approve the product design for first in human testing based upon the results of the design verification testing. If no human testing or clinical studies are required for product launch, the Stage 4 and Stage 5 design reviews may be combined. However, if the cost of design validation testing is high the Stage 4 Design Review may be used to authorize initiation of design validation testing. A design review meeting for Stage 4 should be conducted by the Project Manager, using the Design Review Form FRM-018. An independent reviewer must participate in the design review as well. Attendees of the Stage 4 Design Review must be documented on the Design Review Form and any recommendations for actions to be taken, in addition to execution of the design plan, should be documented on the Design Review Form. 5. STAGE 5: RELEASE STAGE 1 DESIGN VALIDATION Design Validation will be performed upon completion of successful Design Verification (i.e., Stage 4) to ensure that product conforms to the User Needs. The final Design Validation will be performed using production (or equivalent) units, representative of the final product. Design Validation will follow a prospective protocol, and where Clinical Studies are involved the Design Validation shall conform to ISO 14155 and shall meet the national or regional regulatory requirements for a Clinical Study. Design Validation should include clinical verification with human subjects as well as validation of customer documentation (including user manuals and product labeling). If there are different intended uses, multiple validations may be necessary. 2 RISK MANAGEMENT If the Risk/Benefit Analysis was not conducted during a Clinical Evaluation using the literature route, then a Risk/Benefit Analysis shall be performed based upon the clinical data and summarized in the Clinical Study Summary Report. VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 9 of 11 The residual risks existing after a clinical evaluation has been conducted shall be documented in the risk management report, the Clinical Evaluation Report (CER) and the DRTM. Any residual risks must also be disclosed to the user/patient in the IFU. The Risk Management File is completed by writing a Summary Technical Document (STED) that references all of the risk management documentation that was generated during the design project. The Risk Management File shall be updated and maintained in accordance with the risk management plan contained within the PMS Plan that is included in the DMR/TF Index. If a trend of adverse events or a new indication for use is identified, the risk management documentation should be reviewed and updated. Refer to SYS-010 for the Risk Management Procedure. 3 CLINICAL STUDY SUMMARY REPORT If the literature route was not used to create a Clinical Evaluation during Stage 4, then a Clinical Evaluation Report (CER) shall be generated using Clinical Study data. Formalized Clinical Testing must be done in accordance with the Investigational Device Exemption procedure. The Investigational Device Exemption Procedure has provisions for Internal Review Board and Informed Consent activities as well as labeling requirements. Refer to SYS-009 Clinical Procedure. 4 DESIGN REQUIREMENTS TRACE MATRIX (DRTM) Each Design Validation protocol and report shall be added to the appropriate column of the DRTM (FRM-019). The validation reports provide objective evidence that the device meets the user needs, and the reports also verify completeness of the risk controls implemented. Any residual risks shall be documented in the DRTM, and a reference to the information provided in the IFU about residual risks shall be referenced in the DRTM. 5 STAGE 5 DESIGN REVIEW The primary purpose of the Stage 5 Design Review is to review and approve the product design for commercial release. All Design Validation must be complete and any required regulatory approvals must be received. The DMR/TF Index must be complete and approved during this Design Review—if not prior to the Design Review. The DMR/TF Index also includes approval of the PMS Plan that shall be implemented upon completion of the Stage 5 Design Review. If no human testing or clinical studies are required for product launch, the Stage 4 and Stage 5 design reviews may be combined, but this should be identified with a justification in the Design Plan. A design review meeting for Stage 5 should be conducted by the Project Manager, using the Design Review Form FRM-018. An independent reviewer must participate in the design review as well. Attendees of the Stage 4 Design Review must be documented on the Design Review Form and any recommendations for actions to be taken should be documented on the Design Review Form. 8. MONITORING AND MEASUREMENT In order to identify the greatest opportunities for improvement the process owner responsible for design controls should review design tasks to identify any sub-tasks that can performed externally to the process to prevent those tasks from becoming critical path items. When sub-tasks cannot be performed externally to the design process, resource buffers should be established to prevent delays caused by a lack of resources. A post-mortem should be performed after completion of each design stage and at the end of each design project to identify corrective and preventive actions. Top management should consider scheduling an internal audit of the DHF at the conclusion of each project to identify any nonconformities and to provide additional suggestions for improvement to the design process. The following variables are recommended for monitoring and measurement for the design control process: • • • measure task duration, measure delays (i.e., waiting) between tasks, and measure task variability. VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 10 of 11 9. TRAINING/RETRAINING Role Training or Retraining Required Project Manager Shall be trained and be competent in all aspects of this procedure and Risk Management (SYS-010). Project Team Members Any person required to be a member of the project team shall have a general understanding of the procedure and be trained in those parts of the procedure that they are involved in at the time that they first need to apply them, by the Project Manager. Team members should also be trained on Risk Management (SYS-010). Senior Management Shall have a general understanding of this procedure and the Risk Management (SYS-010) procedure. 10. RISK MANAGEMENT Hazard Risk control measures 1. Design proceeds without inputs being fully defined. Define inputs cross-functionally with the use of Hazard Identification and previous and competitor product designs as inputs. 2. Regulatory requirements are not met. Regulatory pathway is specifically incorporated into Stage 1 of the design process. 3. Design inputs do not meet outputs. Verification, validation and traceability is achieved via a Design Requirements Trace Matrix (DRTM). 4. Flaws in design are not detected till late in the process. Cross-functional design reviews are conducted with an independent reviewer, a stage-gate design process is used with “hard” gates, “Design Freeze” occurs prior to verification and validation in the Stage 3 Design Review and finally the use of preliminary screening tests is incorporated into Stage 3 prior to “Design Freeze.” 5. Design Control Process is not compliant. Perform internal audit of DHF at the end of each design project. 11. RECORDS The DHF is a record that must be retained for the life of the product—which must be defined in the DRM/TF Index. The DHF shall consist of all design team records generated and the DHF shall be closed when the product is commercially released or the project is cancelled. Critical documents that will be maintained as technical documentation after the design project is completed include: 1) PMS Plan, refer to SYS-019; 2) Risk Management File, refer to SYS-010; 3) Clinical Evaluation, refer to SYS-009; and 4) Design Outputs in the form of the DRTM and the DMR/TF Index, refer to SYS-025 Technical Documentation Procedure. VERSION: PRODUCT DEVELOPMENT PROCEDURE EFFECTIVE: SYS-008 AUTHOR: PAGE: A D8 R. Packard 11 of 11 12. FLOWCHARTS Risk Control Option Analysis Risk Control Hazard Effectiveness Risk Management Identification Risk Report Verification Assessment Risk Risk / Benefit Management Analysis Plan Product Launch 510(k) 1. Concept 2. Feasibility 3. Development 4. Pilot 5. Release D R Stage DHF Begins Stage Stage Stage Stage Design Transfer