First Law of Thermodynamics
advertisement

First Law of
Thermodynamics
Doba Jackson, Ph.D.
Dept of Chemistry & Biochemistry
Huntingdon College
Outline of Chapter 2
•
•
•
•
•
•
•
•
Basic Concepts: Heat, Work, Energy
First Law of Thermodynamics
Expansion work
Measurement of Heat
Enthalpy changes
Adiabatic changes (Cooling)
Thermochemistry
Inexact Differntials
System, Surroundings,
Universe
• System- Part of the world of interest
• Surroundings- Region outside the
system
• Open system- Allows matter and energy
to pass
• Closed system- Cannot allow matter to
pass
• Isolated system- Cannot allow matter or
energy to pass
Work
• Work- the amount of energy transferred by
motion against an opposing force
▪
Heat, and Heat Transfer
Heat- is the transfer of energy from one body
to another by thermal contact
Heat can be transferred by
Convection
Conduction
Radiation
Internal Energy: Energy that
exist internal to the system
First Law: The internal energy of an isolated system is a
constant; Energy cannot be created or destroyed.
Utotal = U system U surroundings 0
U system = q + w
Internal energy can have different
forms:
Translational Kinetic Energy
Rotational Kinetic energy
Vibrational Kinetic energy
*Energy within the atom
ΔU= change in internal energy
q = heat (released or absorbed)
w = work done on or by the system
Internal Energy of a monoatomic
gas
RT
Monoatomic gas
3
U m T U m 0 RT
2
RT
Linear molecule
RT
5
U m T U m 0 RT
2
Non-Linear molecule
6
U m T U m 0 RT
2
RT
RT
Expression for Work
Work (W) = force x distance
W = - F Z
Change equation to relate
pressure and volume
Force
F
P=
=
Area
A
F = PA
W = - P A ΔZ
Volume change (ΔV) = A ΔZ
W = - P ΔV
True expression for Work
Vf
dW = - Pex dV W= -V Pex dV
i
Types of Expansion Work
• Free Expansion- work done by
expansion against zero opposing force
or zero pressure
Vf
w Pex dV
Vi
Pex 0
W 0
U q
Types of Expansion Work
• Expansion against a constant external
pressure (irreversible)
Vf
w Pex dV
Vi
If external pressure does, not vary,
this expression can be integrated
Vf
w = -Pex dV
V1
w = -Pex Vf -Vi
w = -Pex ΔV
Types of Expansion
Work
• Expansion is reversible
Vf
w Pex dV
Vi
In this case, the external pressure Pex
exactly equals the internal pressure P
at each stage of the expansion.
Vf
w = - P dV
Pex = P
V1
w = -
Vf
Vi
nRT
dV
V
The integral cannot be evaluated because
temperature is not a constant throughout the
integration.
Types of Expansion
Work
• Expansion is Isothermal, reversible
w
Vf
Vi
Pex dV
Pex = P
The system is kept at a constant temperature.
The external pressure Pex equals the internal
pressure P at each stage of the expansion.
w = -
Vf
Vi
Vf 1
nRT
dV = -nRT
dV
V
i V
V
Vf
w = -nRT Ln
Vi
Moles is always assumed
constant unless otherwise noted.
ΔT = 0
Problems: Calculate the work
needed for a 65 kg person to climb
up 4.0 meters on (a) the earth, (b) the
moon (g=1.60 m/s2)
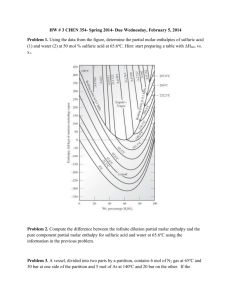
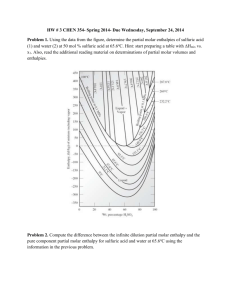
Problem 1: A chemical reaction takes place in a
container of cross sectional area of 50.0 cm2.
As a result of the reaction, a piston is pushed
out through 15 cm against an constant external
pressure of 121 kPa. Calculate the work (in
Joules) done by the system
Problem 2: A sample consisting of 2.00 mol
of He is expanded at 22ºC from 22.8 dm3 to
31.7 dm3. Calculate q, w and ΔU (in Joules)
when the expansion occurs under the
following conditions:
(a) Freely
(b) Isothermal, and constant external pressure equal to
the final pressure of the gas
(c) Isothermal, Reversibly
Reversible, Nonspontaneous
Reversible Process: The thermodynamic process in which a system
can be changed from its initial state to its final state then back to its initial
state leaving all thermodynamic variables for the universe (system +
surroundings) unchanged.
A truly reversible change will:
- Occur in an infinite amount of time
- All variables must be in equilibrium with each
other at every stage of the change
Reversible
Irreversible
Irreversible, Spontaneous
Irreversible Process: The thermodynamic process in which a system
that is changed from its initial state to its final state then back to its initial
state will change some thermodynamic variables of the universe.
A truly irreversible change will:
- Occur in an finite amount of time
- All variables will not be in equilibrium with each
other at every stage of the change
Reversible
Irreversible
Heat exchange can be measured
in a Bomb Calorimeter
First Law: The internal
energy of an isolated system
is a constant.
U = q + w
ΔU = q - Pex ΔV
Constant Volume: no work done
ΔU = q v
ΔV = 0
q V = heat at a constant volume
Calorimetry is the
study of heat transfer
during a chemical or
physical process
ΔU = q v
Heat is measured by the
change in temperature of
the water surrounding the
calorimeter
q = C ΔT
C = Calorimeter constant
The calorimeter constant is the
heat capacity of the system
Heat
Heat Capacity at a constant volume
Heat Capacity (Cv) is the
amount of heat needed to
change the temperature of a
system by 1 *C at either a
constant volume.
ΔU = q v CV T
ΔU dU U
CV =
=
=
ΔT dT T V
We need partial derivatives
because the change holds
the volume constant
Heat Capacity at a constant volume
We need partial derivatives because
the change holds the volume constant
ΔU = q v CV T
ΔU U dU
CV =
=
ΔT T V dT
How we can evaluate the Heat
capacity equation
dU
U
CV =
T V dT
ΔU
ΔT
If the heat capacity (Cv) does not vary with
temperature during the range of temperatures
used, then the equation can be broken
down to:
ΔU = CV ΔT
Because at constant volume, change in internal
energy is equal to the heat (qv).
q V = CV ΔT
Problem 2.4: A sample consisting of 1.00 mol of
a perfect gas, for which Cv,m = (3/2)R, initially
at P = 1.00 atm and T = 300 K, is heated reversibly
to 400 K at a constant volume. Calculate the
final pressure, ΔU, q, and w
We cannot measure internal
energy when the volume is not
constant
Typical reactions occur a constant
external pressure
U = q + w
ΔU = q - PΔV
Heat exchange can be measured in a
Differential Scanning Calorimeter at a
constant pressure
First Law: The internal
energy of an isolated system is a
constant.
U = q + w
ΔU = q - PΔV
q = ΔU + PΔV
Lets define Enthalpy as:
ΔH = q P
H = U + PV
ΔP = 0
q P = heat at a constant pressure
Heat Capacity at a constant pressure
Heat Capacity (Cp) is the
amount of heat needed to
change the temperature of a
system by 1 *C at either a
constant pressure.
ΔH = q P CP T
ΔH dH H
CP =
=
=
ΔT dT T P
We need partial derivatives because
the change holds the pressure constant
How we can evaluate the Heat
capacity equation
dH
H
CP =
T P dT
ΔH
ΔT
If the heat capacity (CP) does not vary with
temperature during the range of temperatures
used, then the equation can be broken
down to:
ΔH = CP ΔT
Because at constant volume, change in
internal energy is equal to the heat (qv).
q P = C P ΔT
Problem 2.20: When 2.25 mg of anthracene, C14H10
(s) was burned in a bomb calorimeter, the
temperature rose by 1.35 K. Calculate the
calorimeter constant for the system. By how much
will the temperature rise if 135 mg of phenol
(C6H5OH) is burned in the calorimeter under the
same conditions?
(ΔcHΘ(C14H10) = -7061 kJ/mol)
Adiabatic Expansion: work is done
but no heat enters the system
• When a gas expands adiabatically,
work is done but no heat enters or
leaves the system.
• The internal energy falls and the
temperature also falls.
U = q + w
q=0
ΔU = w = CV ΔT
wad = U = CV ΔT
Adiabatic changes to an Ideal
Gas can be related by the
following equations
Vi
T f Ti
Vf
Vi
Pf Pi
Vf
1
1
c
C = CV/nR
γ = Cp/CV
The derivation of these equations is
quite long and follows the next slide
Adiabatic Expansion of an Ideal
Gas (Derivation)
U = q + w
Assume the heat capacity does not
vary during the temperature interval
For an adiabatic change: q = 0
CV
dU = w ad
CV dT = -P dV
nRT
CV dT = dV
V
Ti
Ti
Vf 1
1
dT = -nR
dV
Vi V
T
Tf
V
= - nR Ln f
Ti
Vi
CV
Tf
Vf
Ln
= - Ln
nR
Ti
Vi
CV
Tf
Vi
c
=
c Ln
= Ln
nR
Ti
Vf
CV Ln
P=
1
nR
CV dT = - dV
T
V
Tf
Tf
Vf nR
1
CV dT = -
dV
Vi V
T
nRT
V
c
Tf Vi
=
Ti Vf
Problem 2.1- A 3.75 mole sample of an ideal gas with
Cv,m = 3R/2 initially at a temperature Ti=298 K, and Pi= 1.00
bar is enclosed in an adiabatic piston and cylinder
assembly. The gas is compressed by placing a 725 kg
mass on the piston of diameter 25.4 cm. Calculate the work
done in this process and the distance the piston travels.
Assume the mass of the piston is negligible.
Thermochemistry
The study of energy transferred as heat during the
course of chemical reactions.
In thermochemistry, we rely on calorimetry to measure
the internal energy (∆U) enthalpy (∆H).
Changes in matter are:
Physical Changes
Solid Liquid
Melting (Fusion)
Liquid Gas
Boiling (Vaporization)
Solid Gas
Sublimation
Chemical Changes
Formation reactions
Combustion reactions
Other reactions
The Thermodynamic Standard
State
The standard state of a material (pure substance, mixture or solution)
is a reference point used to calculate its properties under different
conditions. The International Union of Pure and Applied Chemistry
(IUPAC) recommends using a standard pressure po = 1 bar
(100 kilopascals).
C3H8(g) + 5O2(g)
3CO2(g) + 4H2O(g)
∆H = -2044 kJ
C3H8(g) + 5O2(g)
3CO2(g) + 4H2O(l)
∆H = -2220 kJ
Thermodynamic Standard State: Most stable form of a substance at 1 bar
and at a specified temperature, usually 25 °C (for non-solutions).
C3H8(g) + 5O2(g)
3CO2(g) + 4H2O(g)
∆H° = -2044 kJ
Physical and Chemical Transitions
State Functions
Internal energy and enthalpy
are is a state function.
ΔU=
ΔH=
f
i
f
i
dU
dH
Work and Heat are path
function. This means we can
never write Δq or Δw
Enthalpies of Physical Change
Hess’s Law
Example of Hess’s Law
Hess’s Law: The overall enthalpy change
for a reaction is equal to the sum of the
enthalpy changes for the individual steps in
the reaction.
Germain Hess
Example 1:
The two reactions below are known methods to produce ammonia by the
Haber process. Use the information to solve for the enthalpy change in the
reaction below.
3H2(g) + N2(g)
N2H4(g) + H2(g)
2NH3(g)
2NH3(g)
∆H°1 = -92.2 kJ
∆H°2 = -187.6 kJ
Solve for the reaction below
2H2(g) + N2(g)
N2H4(g)
∆H°3 = ?
Example 1:
The two reactions below are known methods to produce ammonia by
the Haber process. Use the information to solve for the enthalpy change
in the reaction below.
3H2(g) + N2(g)
2NH3(g)
N2H4(g) + H2(g)
∆H°1 = -92.2 kJ
∆H°2 = -187.6 kJ
2NH3(g)
Steps to solve the problem
- Find known reactions that will add up to the desired reaction
ΔH Θ A
B = -ΔH Θ B
A
- Add up the reactions and add up the ΔH, ΔU, ΔG or other state functions
2
3H2(g) + N2(g)
2NH3(g)
2NH3
N 2 H 4 (g) + H 2 (g)
Solve for the reaction below
2H2(g) + N2(g)
N2H4(g)
∆H°1 = -92.2 kJ
∆H°2 = 187.6 kJ
∆H°3 = -92.2 kJ + (187.6 kJ)
= 95.4 kJ
Hess’s Law Example
Standard Heats of Formation
Standard Heat of Formation (∆fH° ): The enthalpy change for the formation
of 1 mol of a substance in its standard state from its constituent elements
in their standard states.
Standard states
C(s) + 2H2(g)
CH4(g)
1 mol of 1 substance
∆H°f = -74.8 kJ
Standard Heats of Formation are used to
determine reaction enthalpies (ΔrHº)
Reaction enthalpies (ΔrHº) can be
determined by the difference of the
product enthalpies of formation and
the reactant enthalpies of formation.
Δ r H0 =
v Δf H0 -
Products
Example:
v Δf H0
Reactants
aA + bB
cC + dD
∆rH° = {c ∆fH°(C) + d ∆fH°(D)} – {a ∆fHº(A) + b ∆fH°(B)}
Products
Reactants
Standard Heats of Formation
Using standard heats of formation, calculate the
standard enthalpy of reaction for the photosynthesis of
glucose (C6H12O6) and O2 from CO2 and liquid H2O.
6*CO2(g) + 6*H2O(l)
C6H12O6(s) + 6*O2(g)
∆rH° = ?
Standard State
(ΔfHº)= 0
Answer
ΔrH° = [∆H°f (C6H12O6)] - [6*∆H°f (CO2) + 6*∆H°f (H2O)]
Products
Reactants
∆rH° = [(1 mol)(-1273.3 kJ/mol)] [(6 mol)(-393.5 kJ/mol) + (6 mol)(-285.8 kJ/mol)] = 2802.5 kJ
Heats of Combustion (ΔcHº) is a
special type of reaction enthalpy
Calculate the standard enthalpy of combustion for
methane (CH4(g)).
Answer
CH4(g) + 2O2(g)
CO2(g) + 2H2O(l)
∆cH°= [∆fH°(CO2) + 2 ∆fH°(H2O)] - [∆fH°(CH4)]
= [(1 mol)(-393.5 kJ/mol) + (2 mol)(-285.8 kJ/mol)] ΔfHº (CH4) = -74.8 kJ/mol
ΔfHº (CO2) = -393 kJ/mol
[(1 mol)(-74.8 kJ/mol)] = -890.3 kJ
ΔfHº (H2O) = -285 kJ/mol
Combustion reactions are always balanced with 1 as the coefficient
of the hydrocarbon
Temperature dependence of
reaction enthalpy (ΔrHº)
• Often standard reaction enthalpies
need to be corrected for temperature
differences.
H T1 = H T2 + Cp dT
T2
T1
Δr H
o
T1 = Δ r H T2
Δ r Cop =
o
Products
v Cop,m -
+
T2
T1
r Cop dT
v Cop,m
Reactants
If Cp is independent of temperature during the
temperature range, the integral can be evaluated.
The equation doesn’t work if there is a phase transition
through the temperature range.
Problem: Balance the reactions
and determine ΔrHΘ and ΔrUΘ
NH3 g NO g
N2 g H 2O g
NO: 91.3 kJ/mol
NH3: -45.9 kJ/mol
H2O: -241.8 kJ/mol