Why Linux and Not Windows?
http://www.devx.com/opensource/Article/16969 http://www.computerhope.com/issues/ch000575.htm
Comfort in the Linux Environment
Learning Linux commands – Hands On
Linux Exercise – File Manipulation
Reminder:
Testing your knowledge
Do you have your
Things to Remember
Know: large FASTA formatted nucleotide sequence file stored on your computer?
Keep Challenged –
While U Wait
Try :
Start Cygwin
By double clicking the Cygwin Icon
Cygwin Terminal
You can right click the icon above, select properties, and change the window appearance prior to start up
Know : Linux is case sensitive. Hello is not the same as HELLo
Things you take for granted within a GUI:
GUI:
Graphical
User
Interface
Things that can create bigger challenges
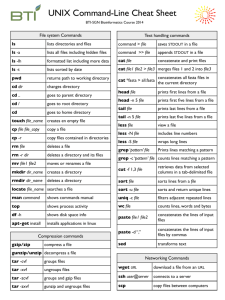
Linux Commands
pwd print working directory: where are you now?
Response: /cydrive/c/Documents and Settings… man manual: see what’s out there man pwd
Know : ‘q’ = quit:
‘b ‘ = back: hit spacebar = forward
Know : The terminal is also called Standard
Output
Linux Commands
Listing, Previewing, Manipulating files in
Windows
Know : GUI: Graphical User Interface (graphical environment for the user)
Linux Commands: ls, cd, mkdir
ls list contents: take a look inside ls –a view everything even hidden files ls –l view ownership of files ls –1 list 1 (one) file per line ls –nt* list all file names starting with nt
Try Now that you know some ls commands view “ man ls ” cd changes directory to a different location cd c:/ go to the c directory ( Try : pwd then ls to see what’s there) cd / go to your root directory ( Try : pwd then ls ) mkdir will make a directory mkdir Bioinfo HCS_Storage create 2 directories ( Try : ls to view) cd /Bioinfo go inside the Bioinfo directory just created above cd .. go backwards 1 level of directory (in this case, back to root /)
Linux Commands: cat
(> & >>)
cat create, join, append, view: You can do it all with cat and text files cat Cygwin.bat display contents of Cygwin.bat on the terminal window
Create 2 files using
‘cat’ in FASTA format as shown below
(CTRL D to save and quit) cat > file1 (> redirects output)
>Seq 1
AATTAATT
>Seq 2
GCGCGC
>Seq 3
CCCCGGGG cat > file2
>Seq 4
TATATATA
>Seq 5
ATATATAT
>Seq 6
TTTTAAAA
Know : >> used to append to an already existing file. No example shown
Linux Commands: cat
(concatenation, sort ) cat file1 display and confirm that file1 has the correct output cat file2 display and confirm that file2 has the correct output cat file1 file2 combine and display both file1 and file2
Now let’s take a look at sort sort file1 sort file1 file2
OUTPUT:
Try
Try
Try
: redirect output of file1 and file2 to file3 display sorted output of file1 display sorted output of file1 and file2
Sequence names first, followed by sequences
: redirect sorted output to a file called mysort
: change a command’s name using ‘alias’: alias dog = “cat” cat file1 file2
>Seq 1
AATTAATT
>Seq 2
GCGCGC
>Seq 3
CCCCGGGG
>Seq 4
TATATATA
>Seq 5
ATATATAT
>Seq 6
TTTTAAAA remove dog command using ‘unalias’: unalias dog
Linux Commands: cat
(| pipe, sort, head) cat file1 file2 | sort (combine then sort)
Output after sort
‘>’ is considered a character before ‘A’
’|’ known as the pipe operator. Takes the output of one command and feeds it into another.
Try : send sorted output to file called mysort
>Seq 1
>Seq 2
>Seq 3
>Seq 4
>Seq 5
>Seq 6
AATTAATT
ATATATAT
CCCCGGGG
GCGCGC
TATATATA
TTTTAAAA
Create a file called name_only and write only the 6 seq names to it.
First take a look at head… head -n 4 mysort : displays the head of a file. -n 4 means: show only 4 lines head is most useful when viewing extremely large files
Linux Commands: tail, head
Can you write only the sequences to a file called seq_only?
tail -n 4 mysort : displays from the end of a file. -n 4 means: show last 4 lines
Try : Visit the man pages, man sort and “reverse” the direction of a sort
Know : Notice above –n 4: That’s adding “parameters” to the tail command.
Know : Want to learn more about cat? http://www.linfo.org/cat.html
Remember that large sequence file, you will need it now.
Use … cat my_large_seq_file (see what happens) head my_large_seq_file (how many lines do you see?) tail my_large_seq_file (how many lines do you see?)
Perhaps you need to see more of the file
HINT: Use head and the –n parameter. Choose how many lines you need to see
Linux Commands: less
Try : Send 200 lines of my large sequence file to a file called lines_200 less: the ability to see more less my_large_seq_file best way to controls viewing of large files
Navigation after the less command is similar to the man pages:
‘q’ = quit:
‘b ‘ = back:
Page Up = up 1 page
Page Dn = down 1 page hit spacebar = forward Home = top & End = bottom
Know : Linux/Unix provides powerful commands that allow you to manipulate files of any size much easier than in the Windows environment. I can’t believe I said that
Linux Commands: cp
Make sure you are NOT in the Bioinfo directory cp file1 cp_file1 : make a copy of file1 and call it cp_file1
WARNING! cp_file1 will be created and overwritten without warning
Let’s visit the man pages, man cp and look at the “interactive” option cp file1 /Bioinfo : make a copy of file1 and place it in the Bioinfo directory
Try : list ( ls ) the contents of the Bioinfo directory without using cd cp file1 /Bioinfo/cp_test : copy file1 into the Bioinfo directory & call it cp_test
Linux Commands: mv, rm, rmdir
mv /Bioinfo/cp_test /HCS_Storage : move cp_test from Bioinfo to HCS_Storage
Try : Move cp_test to HCS_Storage and give the file a new name: cp_test2
Know : Moving does NOT retain a copy of the file in its original location
Know : The PATH refers to the location of the file, for example:
/Bioinfo/cp_test means that cp_test is located in the Bioinfo directory
Go to the HCS_Storage directory ( cd ???
). Remove the file cp_test rm cp_test remove the file cp_test from the hard drive
Check to make sure the file cp_test is gone ( ls )
BEFORE YOU LEAVE THE HCS_Storage DIRECTORY MAKE SURE THERE IS A FILE
IN IT. CREATE A FILE IF NEEDED.
Linux Commands: rm, rmdir
1: Go to your root directory ( cd ???
).
2: Use pwd to make sure you are at the root level.
Note: Response to pwd if at root level: /
3: Make a duplicate of HCS_Storage ( cp –r ??? ???
). Call it HCS_Storage2 rm HCS_Storage remove the HCS_Storage directory
(Error: HCS_Storage is a directory) rmdir HCS_Storage remove the HCS_Storage directory
(Error: Failed to remove HCS_Storage) rm –r HCS_Storage remove directory and its contents rm –ri HCS_Storage2 remove directory and its contents interactively
Know : ‘/’ indicates the root or “top” level of the Linux file system
Know : rmdir only removes directories that are empty. HCS_Storage has 1 file
Know : rm only removes empty directories, unless the –r option is used
Linux Commands: grep
grep finds text within a file
You will need: file1 , file2 , and your large
FASTA formatted sequence file . Let’s
1: IMPORTANT: Copy of your seq file ( cp ) and add “cp_” to the beginning of the file name
2: Confirm the new file has some content ( head ) see what’s in them without looking
Using file1 and file2: Does the sequence TATA appear in file1?
grep “TATA” file1
How about in file1 or file2?
grep “TATA” file1 file2
What if you do not know whether the bases are upper or lower case grep -i “TatA” file1 file2
How many matches I have but I am still concerned about the bases grep -ic “TatA” file1 file2
Try : Can you find out how many sequences (entries) are in your file?
Linux Commands:
Putting it all together
1: Create 2 directories called Temp1 and Temp2. ( mkdir )
2: In Temp2 directory and create a file called tempFile1 ( cd, cat, ls )
3: Make a duplicate of tempFile1 and call it tempFile2 ( cat or cp )
4: Move tempFile1 into directory Temp1 ( mv )
5: Move tempFile2 into directory Temp1 but rename to new_tempfile2 ( mv )
6: Confirm that Temp1 has 2 files and Temp2 has 0 files ( cd, ls )
7: Remove the Temp2 directory ( rmdir )
8: Remove the remove Temp1 directory ( rm )
9: Write the first 30 lines of your large sequence file to sampleSeq ( head, > )
10: Append the last 60 lines to sampleSeq ( tail, >> )
11: In sampleSeq, find a match any sequence string of your choice ( grep )
12: Find the same sequence in both your large seq file and sampleSeq ( grep )
13: Display only the total number of sequences you found in #12 ( grep )
 0
0