Molecular Pathology Laboratory
advertisement

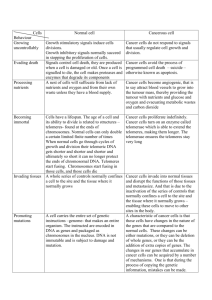
Objectives for this lecture • Understand the mechanism of tumour suppressor inactivation in cancer formation (the two-hit hypothesis) • Gain familiarity with common examples of oncogenes and tumour suppressor genes – their function in normal cells – the effects of aberrations in cancer • Recognise the role of cellular maintenance genes in cancer prevention and grasp the consequences of their inactivation • Understand the concepts of microsatellite instability (MIN) and chromosomal instability (CIN) • Understand the potential use of molecular genetic analysis in cancer diagnosis and treatment selection Retinoblastoma autosomal dominant inheritance • Paediatric tumour of the retina 1/20,000 births – two forms of the disease – familial and sporadic • Due to mutations in the Retinoblastoma (Rb) tumour suppressor gene Knudson’s Two Hit Hypothesis Inherited One normal allele + One mutant allele (1st “hit”) Two mutant alleles (2nd “hit”) Tumorigenesis Sporadic Two normal alleles One normal allele + One mutant allele (1st “hit”) Two mutant alleles (2nd “hit”) Tumorigenesis Genomic instability An abnormal cell state associated with an increased rate of heritable genomic alterations including mutations chromosomal rearrangements deletions inversions Genomic instability Mutator hypothesis: The proposal that genomic instability promotes tumorigenesis by increasing the rate at which mutations in oncogenes and tumour suppressor genes arise during the multistep development of cancer Genomic instability Microsatellite instability (MIN) also known as Replication Error phenotype associated with errors in the DNA mismatch repair system that may lead to an elevated DNA mutation rate genome-wide alterations in repetitive DNA sequences found in tumours of patients with hereditary non-polyposis cancer syndrome and 15% of sporadic (non-inherited) colorectal cancers also found in other types of sporadic cancer Genomic instability Microsatellite instability (MIN) caused by defects in nucleotide mismatch repair machinery HNPCC patients have germline mutations in MMR genes hMSH2, hMLH1, hPMS1, hPMS2 Tumours with MIN have somatic mutations in MMR genes Mismatch repair process G G parental strand newly replicated strand G G Mismatch recognized by hMSH2/GTBP complex G G hMLH1 & hPMS2 join repair complex G C Excision of mismatch and resynthesis of correct base Genomic instability Microsatellite instability (MIN) general increase in mutation rate specifically related to frameshift mutations in genes with repetitive sequences TGFb receptor II has polyA tract mutated in 90% of MIN+ colorectal ca BAX gene involved in apoptosis mutated in 50% of MIN+ colorectal ca Mismatch repair and methylation tolerance Paradox: DNA damaging environment potentiates growth advantage of repair deficiency Methylating agents produce mutations at guanine residues O6-methylguanine-DNA methyltransferase (MGMT) reverses these animals deficient in MGMT hypersensitive to mutagenic and toxic effects of methylating agents MGMT-deficient cells surviving exposure often evolve tolerance due to defects in MMR pathway Mismatch repair and methylation tolerance Paradox: DNA damaging environment potentiates growth advantage of repair deficiency MGMT-/MGMT-, MLH1+/MLH1+ mice are hypersensitive to toxic effects of MNU exposure MGMT-/MGMT-, MLH1-/MLH1- mice are as resistant to toxic effects of MNU exposure as wild-type mice, but develop numerous tumours Explanation: methylating carcinogens produce mutations in growthpromoting genes AND result in general methylation and (MMR) gene silencing Chromosomal Instability and bulky adduct-forming carcinogens Bulky adduct-forming carcinogens UV radiation, free oxygen radicals, many chemicals DNA damage repaired by Nucleotide Excision Repair (NER) involves removal and resynthesis of large DNA fragments promotes chromosomal rearrangments, may distort spindle formation and chromosomal segregation; activates mitotic checkpoint (MCP) Explanation: MCP deficiency may give growth advantage to cells exposed to BAF carcinogens Genomic instability Chromosomal instability (CIN) chromosomal rearrangements, losses and gains measured as abnormal number of chromosomes, shift in nuclear DNA content aneuploidy associated with defect in chromosomal segregation Genomic instability Ways to acquire genomic instability Type Biological process Genes in pathway Associated disorder Mutations: MIN Mismatch repair (MMR) MSH2, PMS1, PMS2, MLH1 HNPCC Nucleotide excision repair (NER) XPA-XPG, CSA, CSB Xeroderma pigmentosum Deletion DNA damage signalling ATM, BRCA1, p53 & Translocation Double-strand break repair Blm, Wrn DNA cross-link repair FANCA-FANCG Ataxia telangiectasia Bloom’s & Werner’s syndromes Fanconi’s anaemia Ways to acquire genomic instability Type Chromosomal instability Biological process Sister chromatid cohesion Genes in pathway Associated disorder PTTG Pituitary tumours and condensation Loss/Gain of chromosomes Spindle checkpoint BUB1 Colorectal cancers Altered Ploidy Centrosome cytokinesis Aurora A Colorectal cancers Cell death following p53, Bcl-2 Breast cancers prolonged mitotic arrest TP53 is inactivated in many forms of human cancer • one of the most commonly deleted mutated genes in human cancer • Complete loss of functional P53 occurs in over 50% of all human tumours • Loss of function is generally due to point mutation for one allele and loss of the other • Majority of mutations occur in central region of coding sequence TP53 function • Encodes p53 – 393 a.a. nuclear phosphoprotein • DNA-binding protein: role in transcriptional regulation • Controls cell’s decision to replicate DNA at G1/S checkpoint • Causes cells with DNA damage to arrest at G1 • Exerts control over cell’s decision to undergo apoptosis P53 binding DNA BRCA1 • Accounts for 1/2 of the autosomal dominant familial breast cancers • Confers high risk for ovarian cancer as well • May also predispose to prostate and colon cancer • Encodes 1863 a.a. nuclear protein • Most identified mutations result in a truncated protein BRCA2 • Accounts for 1/3 of the autosomal dominant familial breast cancers • Confers high risk for ovarian cancer as well (but not as high as BRCA1) • Confers high risk for male breast cancer (10-20% of all cases have BRCA2 mutations) • May also predispose to malignant melanoma, prostate, pancreatic, gall bladder, bile duct and stomach cancer • Encodes 3418 a.a. nuclear protein Potential roles of BRCA1 & BRCA2 in DNA repair • BRCA1 and BRCA2 function in the same multiprotein complex • May help maintain genomic integrity by promoting repair of DNA double strand breaks that result from damage • Evidence also suggests the complex may play a role in transcriptional regulation BRCA1 & BRCA2 mutations and cancer predisposition What is the lifetime risk for developing breast cancer for women carrying mutations in BRCA1 and BRCA2? Originally thought to be 80%, however, when risk was estimated from pop.studies, 45-60% High penetrance families have additional genetic and/or environmental factors present – many women in the family are affected BRCA1 & BRCA2 mutations in sporadic breast cancer • Initially, very few sporadic tumours were found to have detectable BRCA1 or BRCA2 mutations • It now appears that promoter hypermethylation may represent an important mechanism for BRCA1 inactivation – leads to closed chromatin conformation Telomerase represents a novel proto-oncogene • Full length telomeres are approximately 15 kb long • In germline cells, telomerase, a reverse transcriptase, adds a hexameric DNA repeat to the end to maintain full telomere length after DNA replication Telomerase represents a novel proto-oncogene • As cells differentiate during fetal development, telomerase function declines and the telomeres shorten - with each successive round of DNA replication, the telomere shortens by about 35 bases • Ultimately, as telomeres shorten, chromosome ends become damaged and the cells stop dividing-may be the cause of normal cellular senescence • In transformed cells and many tumors, telomerase activity reappears, enhancing the ability of tumor cells to divide without limit – Telomerase activity detected in more than 30 cancer types – Telomerase activity detected in over 80% of cancer samples Fusion Genes in Solid Cancers Gene A Promoter Breakpoints Gene B Promoter Fusion Gene A/B Promoter Fusion protein Domain from A Domain from B Oncogenes activated by chromosome translocations • Breakpoint can occur within introns of two genes: chimeric protein with novel properties: Chronic Myelogenous Leukaemia • Alternately, translocation may place protooncogene downstream of a strong constitutive promoter from another gene – proto-oncogene is now expressed at inappropriate time/place – Burkitt Lymphoma Chronic Myelogenous Leukemia (CML) • Proto-oncogene ABL (tyrosine kinase) moves from 9q to the “breakpoint cluster region (BCR) on 22q • Chimeric protein has increased tyrosine kinase activity but altered structure and function • Requires secondary mutation to move into crisis phase • Effective drug therapy developed to target novel protein Burkitt Lymphoma • B-cell tumour • C-MYC proto-oncogene (transcription factor) translocated from 8q24 to 14q32, distal to the Ig heavy chain locus • Ig enhancers or activating sequences act on C-MYC – allowing unregulated expression and uncontrolled cell growth http://tooldoc.wncc.edu/Infections/lymphoma.JPG • Solid tumour of B-lymphocytes • Predominantly affecting young children in Africa • one of the fastest growing malignancies in humans • manifested most often as a large jaw lesion expanding rapidly over a period of a few weeks to invade the orbit • Visceral involvement, usually an abdominal mass • Treatment of the jaw and eye areas is by radiotherapy,while visceral involvement requires systemic chemotherapy. In all cases, translocation of C-MYC is the cause Fusion Genes in Solid Cancers CHOP Myxoid liposarcoma ERG Myeloid leukaemia TLS/FUS FEV FLI1 EWS Ewing’s sarcoma ETV1 E1AF WT1 Desmoplastic small round cell tumour ATF1 Clear cell sarcoma TEC Extraskeletal myxoid chondrosarcoma Oncogene amplifications in solid tumours How do oncogenes amplify? Intrachromosomal tandem duplication during recombination, further unequal chromatid exchange double chromatid breaks at fragile site, subsequent telomere fusion, breakage-fusion bridge cycles Extrachromosomal repair replication at fragile site Oncogenes activated by locus amplification • Amplified sequences can be seen in karyotypes as: – double minute (DM) chromosomes - very small accessory chromosomes – additional banding regions called homogeneously staining regions (HSR) • Both contain 20-100s of copies of a DNA region of several hundred thousand bases-extra copies of proto-oncogenes NMYC, HER2 DM Oncogene amplifications in solid tumours N-MYC: originally identified as HSRs or DMs in 20% neuroblastoma less frequent in small cell lung cancer retinoblastoma malignant gliomas peripheral neuroectodermal tumours typically present as 50-100-fold amplification co-amplification of DDX1 in 50% of N- MYC+ retinoblastomas & neuroblastomas Oncogene amplifications in solid tumours MDM2: amplified in neuroblastomas, sarcomas and gliomas in neuroblastomas, only amplified in MYCN+ cases (never p53 mutant) MDM2 protein complexes with p53 overexpression causes p53 sequestration sarcomas with MDM2 amplification plus p53 mutation: worse prognosis HER2 is amplified in many breast cancers • Encodes transmembrane receptor tyrosine kinase, overexpression leads to homodimer formation-> constitutively active expression • HER2 amplification is found in 20-25% of breast cancers • leads to increased gene expression and an increase in cell proliferation • amplification correlated with – More likely lymph node metastasis – Shortened time to relapse – Reduced overall survival Antibodies to HER2 may become part of clinical treatment • Antibodies to erbB2 – are able to convert rapidly dividing breast cancer cells into growth-arrested cells – Remove the receptor from the cell surface – Attract natural killer cells to the cell, targeting it for destruction – Commercially available as Trastuzumab (HerceptinTM) from Genentech and used in conjunction with chemotherapy Molecular Genetics of Breast Cancer Chromosomal location 1p 1q 3p 6q 7q 8p 8q 9p 10q 11q 13q 16q 17p 17q 18q 20q 22q Abnormality % of tumours deletion 45% deletion/amplification 60% deletion 40% deletion 40% deletion 0-80% deletion 50% amplification 15% deletion 45% deletion rare amplification 40% deletion 50% deletion 65% deletion 50% deletion/amplification 30-50% deletion 40% amplification 15% deletion 40% Oncogene Suppressor gene FHIT MYC PTEN CCND1 HER2 BRCA2, RB1 ECDH TP53 BRCA1 Molecular Detection and Analysis of Cancer • Expression of a gene – its transcription from DNA to RNA • All genes are not expressed equally in every cell • Altered gene expression is part of the cancer transformation process • Better monitoring of gene expression in tumour cells vs. normal cells can: – Provide better classification system – Serve as predictors of outcome and response to treatment options Van’t Veer. L.J. et al. Nature, 415, 530-536 (2002) patients Expression patterns of different tumours can be compared Red-upregulated Green-downregulated Identity of the genes is not important-- predictive profile is Conclusions • The three major classes of genes involved in cancer development are – Oncogenes – Tumour suppressors – Genes involved in cellular and genomic maintenance Conclusions • Oncogenes can be activated in several ways: – Point mutations • RAS – Chromosomal translocation • BCR/ABL - CML • MYC/Ig - Burkitt’s Lymphoma – Amplification • HER2 – Breast, ovarian cancers • Telomerase can serve as an oncogene by postponing cell senescence Conclusions • Molecular analysis is used to refine the classification of various forms of cancer molecular profiling • Patient prognosis can be predicted based on the profile of their tumour • Response to various types of treatment can be predicted by the profiles of the tumour • • MIN+ colorectal cancers may have better response to chemotherapy HER2+ tumours are candidates for Herceptin therapy