Micelles and Membranes
advertisement

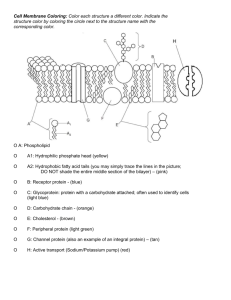
Detergents, Micelles and Membranes. Self-Assembly of Amphiphiles The hydrophobic aspect of lipids and detergents limits their solubility, as monomers, in water. However, above their true solubility limit, the need to minimize the surface area of interaction between water and the hydrophobic parts leads to the formation of small, aggregated structures, or micelles, which remain fully dispersed in solution. The point at which this happens is called the critical micelle concentration, or CMC. Above this the concentration of monomers stays almost constant and further added material forms more micelles. The onset of micelle formation is essentially a phase transition, i.e., it is cooperative, and the sharpness of the transition reflects the cooperativity inherent in the process of micelle formation: N lipid (N monomer) ↔ lipidN (one micelle) Emulsions form when amphiphilic molecules reduce the oil-water tension Salad dressing (vinaigrette) separates into oil and water, despite the superficial increase in order (decrease in entropy) that such separation entails. Water molecules are attracted to oil molecules, but not as much as they are attracted to one another: the oil-water interface disrupts the network of hydrogen bonds, so droplets of water coalesce (get together, unify) to reduce their total surface area. Mayonnaise, too, is mostly a mixture of oil and water; yet it does not separate. What is the difference? One difference is that mayonnaise contains a small quantity of egg. An egg is a complicated system, including many large and small molecules. But even very simple, pure substances can stabilize suspensions of tiny oil droplets in water for long periods. Such substances are generically called emulsifiers or surfactants; a suspension stabilized in this way is called an emulsion. Particularly important are a class of simple molecules called detergents, and the more elaborate phospholipids found in cell membranes. Oil-water interface a) An oil-water interface stabilized by addition of a small amount of surfactant (detergent). Some surfactant molecules are dissolved in the bulk oil or water region, but most migrate to the boundary as shown in the inset. b) An oil-water emulsion stabilized by surfactant: the situation is the same as (a), but for a finite droplet of oil. Two classes of amphiphiles a) Structure of sodium dodecyl sulfate (SDS), a strong detergent. A nonpolar, hydrophobic tail (left) is chemically linked to a polar, hydrophilic head (right). In solution, the Na+ ion dissociates. Molecules from this class form micelles. b) Structure of a generic phosphatidyl-choline, a class of phospholipid molecule. Two hydrophobic tails (left) are chemically linked to a hydrophilic head (right). Molecules from this class form bilayers. The transition is very sharp for some materials, and is similar to the phase separation seen for immiscible liquids, or at the solubility limit for any solute. At low concentrations, the material is soluble and the chemical potential (µi) of the solute (e.g., monomers of detergent) increases as more is added. At some point, however, the chemical potential becomes equal to that of the compound in the pure phase (µi*), whatever that might be (solid, separated liquid, micelle, etc). µ = µio + RT ln ci = µi* Since the chemical potential (free energy per mole) of a pure phase is a constant, the chemical potential in solution must now also remain constant and any further material added creates a separate, pure phase. This represents the solubility limit - or the critical micelle concentration, in the case of amphiphiles. For amphiphiles, reaching the solubility limit is not straightforwardly solved by a separation of two bulk phases, as for a simple hydrocarbon and water. The polar headgroups demand some solvation interactions that are not provided by an apolar environment such as the hydrophobic tails, and dragging water in to the detergent bulk phase would be costly. Thus, the phase that detergents and other amphiphiles withdraw to is a microscopic one, the micelle, that satisfies the needs of the polar heads and the apolar tails, simultaneously. Formation of Micelles (Aggregates) from Monomers (simplified model) Apply the Mass Action rule to the reaction (N monomers) ↔ (one aggregate). N is an unknown parameter, which we will choose to fit the data. It will turn out to be just a few dozen, justifying our picture of micelles as objects intermediate in scale between molecules and the macroscopic world. The concentration c1 of free monomers in solution is related to that of micelles, cN, by cN/(c1)N = Keq, (1) where Keq is a second unknown parameter of the model. The total concentration of all monomers is then ctot = c1 + N·cN = c1·(1 + N·Keq·(c1)N-1). (2) This is the relation between the total number of amphiphilic molecules in solution, ctot and the number that remained unaggregated, c1. It's more meaningful to express the answer not in terms of Keq, but in terms of the critical micelle concentration (CMC), ccmc. By definition, ccmc is the value of ctot at which half the monomers are free and half are assembled into micelles. In other words, c1 = N·cN = ½·ccmc. Substituting into Eq. (1) gives 1 1 ccmc ccmc 2N 2 N K eq We now substitute Keq into Eq. (2), finding 2c ctot c1 1 1 ccmc N 1 (3) Once we have chosen values for the parameters N and ccmc, we can solve Eq. (3) to get c1 in terms of the total amount of surfactant ctot stirred into the solution. Although this equation has no simple analytical solution, we can understand its limiting behavior. At low concentrations, ctot << ccmc, the first term dominates and we get ctot ≈ c1: Essentially all the surfactants are loners. But well above the CMC, the second term dominates and we instead get ctot ≈ N·cN; now essentially all the surfactants are accounted for by the micelles. Osmotic pressure Historical background. As early as 1913, J. McBain had deduced the existence of well-defined micelles from his quantitative study of the physical properties of soap solutions. One of McBain's arguments went as follows. We know the total number of molecules in a solution just by measuring how much soap we put in and checking that none of it precipitates out of solution. But we can independently measure how many independently moving objects the solution contains, by measuring its osmotic pressure and using the van't Hoff relation. For very dilute solutions, McBain and others found that the osmotic pressure faithfully tracked the total number of amphiphilic ions, just as it would for an ordinary salt like KCl. But the similarity ended at a well-defined point, the critical micelle concentration (CMC). Beyond this concentration, the ratio of independently moving objects to all ions dropped sharply. The CMC typically decreases at higher temperature, thus pointing to the role of the hydrophobic interaction in driving the aggregation (self-assemble). McBain's results were not immediately accepted. But eventually, several physical quantities (e.g. electrical conductivity) were all found to undergo sharp changes at the same crtitical concentration as that for the osmotic pressure, and the chemical community agreed that he was right. Calculation of the osmotic pressure. The contribution from the dissociated Na+ ions is simply ctot.kBT, as usual. The contribution from the amphiphiles is, however, more sophisticated with one key difference: each micelle counts as just one object, not as N objects. Thus, the contribution of the amphiphiles to the osmotic pressure consists of the monomers (c1) and the micelles (cN). The osmotic pressure relative to the value 2·ctot·kBT of an ordinary salt like NaCl: N 1 1 1 2c1 N ccmc ctot (c1 c N ) 1 pos (rel ) 1 N 1 2 ctot 2 2c 1 1 c cmc (4) where cN is expressed from Eq. (2) and ctot. from Eq. (3). To use this formula, solve Eq. (3) numerically for c1 as a function of ctot. Then substitute into Eq. (4) to get the relative osmotic activity in terms of the total concentration of amphiphiles. Looking at the experimental data, we see that we must take ccmc to be around 1 mM; the fit shown used ccmc = 1.4 mM. Two curves are shown: the best fit (solid line) used N = 30, whereas the poor fit of the dashed line shows that N is greater than 5. Osmotic pressure vs. concentrations of micelle-forming surfactant (potassium oleate) and ordinary salt (KCl) Conclusion We have obtained a qualitative explanation of the very sudden onset of micelle formation by the hypothesis that geometrical packing considerations select a narrow distribution of "best" micelle size N. Indeed, the sharpness of the micelle transition could not be explained at all if stable aggregates of two, three, four,... monomers could form as intermediates to full micelles. In other words, many monomers must cooperate to create a micelle, and this cooperativity sharpens the transition, mitigating the effects of random thermal motion. Without cooperativity, the curve would fall gradually, not suddenly. Micelle Shape and Bilayer Formation Micelle structures in water (with one exception) have the polar headgroups on the outside and the hydrocarbon tails on the inside. The shapes adopted to achieve this are varied, but fall into the categories of ellipsoids of revolution (prolate and oblate), cylinders or disks, and bilayer sheets. The micelle shape can be understood in terms of an effective “packing shape” of the molecules, which reflects the relative strengths of interaction between the headgroups and between the tails. 1) Amphiphiles with one tail per headgroup - detergents, soaps and lysolipids (lipids with one fatty acid missing) – are generally “cone shaped”, with large repulsion between headgroups, relative to the tendency for the tails to aggregate, driven by the hydrophobic effect. They form spheroids or ellipsoids, and sometimes cylinders, which can be thought of as prolate ellipses with an infinite axial ratio. Spheres A sodium dodecyl sulfate (SDS) micelle, drawn to scale, with 60 SDS molecules. The hydrocarbon chains pack at liquid hydrocarbon density in the core, where they are almost as disordered as in the bulk liquid state. Each of the 5 spherical shells (dotted lines) contain the correct number of chain segments to ensure even packing density throughout, and all segments spend an appreciable proportion of the time near the micelle surface. Thus, although the core is devoid of water, each segment of each chain samples the hydrophilic environment. (From: Israelachvili, 1992). Cylinders (end caps omitted) 2) Amphiphiles with two tails per headgroup - including natural lipids - generally form bilayer sheets or, occasionally, disks (exaggerated oblate ellipses). Bilayers then close up into vesicles to avoid exposure of the hydrocarbon tails to water at the edges (edge effects). The energy involved in bending the bilayer is the result of several forces acting in the plane of the membrane, including headgroup repulsions and interchain pressure, and the interfacial pressure or surface tension, which tends to minimize the area of the interface between two phases. The curvature of the bilayer requires energy, but this is recovered from the hydrophobic free energy upon sealing up the bilayer edges. Bilayer Lecithin (PC) bilayer in the liquid crystalline state, drawn to scale, showing the forces that determine it and its properties. The lipids diffuse rapidly in the plane of the membrane, covering about 1 µm in 1 s. They also cross the bilayer (flip-flop), and exchange with lipids in solution, but much more slowly – on the order of hours (c.f., the exchange time for a surfactant (detergent) in a micelle is 10-5 - 10-3 s). A rare micelle form - but indicative of the importance of the balance between head and tail influences in natural membranes - is the hexagonal HII phase. This is formed by lipids with small effective headgroup areas, or large relative tail sizes (width and volume), which gives them an effective “inverted cone” shape. The small effective size of the headgroups may be due to small physical size and weak interaction as in the glycolipid, monogalactosyl diglyceride (MGDG) - or it may be due to attractive forces between them, e.g., by hydrogen bonding in phosphatidyl ethanolamine (PE), or cationic cross-linking between negatively charged lipids, as in cardiolipin (diphosphatidyl glycerol, DPG) or phosphatidyl serine (PS). This leads to salt and pH control of the shape factor and, hence, membrane curvature. The HII structure is an inverted micelle, in which the headgroups interact closely on the inside, with the relatively bulky but hydrophobic tails on the outside. The unfavorable aspect of the hydrophobic effect causes aggregation of a large number of the cylindrical, hexagonal micelles, thereby minimizing the surface area. Hexagonal (HII) phase The HII phase is probably not found in biological membranes, but the inverted cone shape of PE and other lipids (see below) is important in allowing and controlling curvature of natural membranes. These lipids more favorably occupy the bilayer leaflet on the inside of a curve. Similarly, MGDG is thought to be especially important in the very tight curvature of chloroplast thylakoids, the functional membranes of plant photosynthetic energy conversion. The Principle of Opposing Forces Quantitation of the opposing forces governing phase separation of amphiphiles requires estimation of the repulsion between headgroups and of the apparent attraction between the hydrocarbon tails. Even though it is a fictitious force, the latter is actually more readily quantified as it is manifested as the surface or interfacial tension between the hydrocarbon phase and water. This can be measured directly, e.g. for monolayers in a Langmuir trough, and can also be estimated from empirical relationships such as between hydrocarbon length or surface area and the free energy of transfer from water to oil, etc. By contrast, the repulsion between headgroups is complex. Although it is essentially electrostatic in nature, it includes large effects of solvation shells and competition between heads for solvent. The problem is approached semi-quantitatively, by recognizing that there is an optimum surface area per headgroup, a molecular volume, v, and an effective length, lC, for the hydrocarbon tails. The optimum area per headgroup, a0, is determined by the maximum separation (to minimize repulsive interactions between polar headgroups) that will not allow water molecules to slip between and enter the hydrocarbon region. It is determined empirically from the results of the analysis, but can be intuitively guessed from the chemical nature of the headgroup. The effective length is usually taken to be close to (and obviously not more than) the maximum length of an extended chain1: lC < lmax ≈ 1.54 + 1.265 nC Å The volume of the chains is taken directly from the molar volumes of liquid hydrocarbons: v = 27.4 + 26.9 nC Å3 We can now categorize different amphiphiles according to their geometric properties, indicated by the dimensionless packing parameter or molecular shape factor, the ratio of the actual molecular volume to the volume of a cylinder with the same area per headgroup and same chain length, v/a0·lC. For a cylinder, v/a0·lC = 1; for a cone, with the fat part at the headgroup, v/a0·lC = 1/3; and for an inverted cone, v/a0·lC > 1. 1 The value of nC is usually modified (decreased) from the actual number of carbons because it is considered that at least the first one or two, attached to the headgroup, are in substantial contact with the solvent and therefore do not contribute to the difference in free energy between the monomer dispersion and the micelle phase. The rough order of the shape factor can be determined by comparing the experimental aggregation number, N - the number of molecules per micelle - with that expected for certain simple geometric shapes and the values of v and lmax calculated from nC, as above. Most simply we can calculate what we expect for a sphere, with diameter = 2·lmax, and then see how the estimated aggregation number agrees with the experimental value. For maximal packing of the hydrocarbon tails in a micelle, with minimal exposure of the tails to the aqueous phase, at least some of the tails must be fully extended (see Fig. above) and the radius of a spherical micelle is expected to be close to lmax. For other geometries, the constraint of no voids in the interior of the micelle demands that at least one dimension must correspond to this minimum measurement, e.g., in an ellipsoid, a minor axis of 2·lmax. This is consistent with each molecule occupying a fixed volume and with the notion that the headgroups cannot be buried in the hydrocarbon phase. The aggregation number for a sphere (diameter = 2·lmax) is the minimum possible for a given lmax (just as the volume is minimum), and experimental values much greater than the spherical prediction indicate a non-spherical micelle shape. This, in turn, can be interpreted as the result of the balance of forces acting on the headgroups and tails or, equivalently, a specific molecular shape factor. A spherical shape implies strong curvature due to strong headgroup repulsion, or a highly conical molecular shape factor (<0.5); a highly non-spherical shape implies a correspondingly stronger driving force for tail aggregation - a less conical molecular shape factor. Most biological lipids have shape factors close to 1, which favors their packing in-line. However, a single line or layer would expose the ends of the tails to the water, so a bilayer sheet is formed and edge effects are avoided by closing the sheet on itself to form closed vesicles. The precise value of the shape factor for different lipids is determined by second order effects, including (i) specific headgroup interactions such as H-bonding (e.g., PE) or ionic bridging (e.g., DPG), and (ii) specific chain-chain interactions (as in incomplete fluidization.)2. The variability in shape factors is crucial in allowing the flexibility of membrane shapes. Thus, the sharp curvature characteristic of certain (many) natural membranes requires different shape factors for lipids in the inside and outside leaflets of the bilayer. For this reason, it is not surprising that the dominant lipids in animal membranes are PC (v/a0·lC ≈ 1) and PE (v/a0·lC > 1). In chloroplasts and some other plant membranes, the equivalent pair are digalactosyl diglyceride, DGDG, and monogalactosyl diglyceride, MGDG. This can give rise to “lateral phase separations” in which specific lipids aggregate into patches – or “rafts” - in the membrane, sometimes forming gel-like regions in an otherwise liquid membrane - or vice versa. 2 The hydrocarbon interiors of both micelles and bilayers are normally in their fluid state. Repulsive headgroup forces and attractive interfacial forces determine the optimum headgroup area, a0, at which the surface free energy per molecule is at a minimum. The chain volume, v, and chain length, lc, set limits on how the fluid chains pack together, on average, inside an aggregate, depending on the magnitude of the shape factor, v/(a0·lc). Mean (dynamic) packing shapes of lipids and the structures they form Mean (dynamic) packing shapes of lipids and the structures they form (cont.) Self-Assembly in Cells Bilayers self-assemble from two-tailed amphiphiles Puzzle: How can amphiphilic molecules satisfy their hydrophobic tails in a pure water environment? The answer is that they can assemble into sphere. This solution, however, may not always be available due to geometrical constraints. Suppose that N amphiphiles pack into a spherical micelle of radius R. Find two relations between ahead, vtail, R and N: 4 R3 N vtail , 4R 2 N ahead 3 vtail R Combine these into a single relation: ahead 3 Suppose instead that amphiphiles pack into a planar bilayer of thicknes 2d. Find relation between ahead, vtail, and d: a d v head tail Although some molecules, like SDS, may be comfortable with the spherical arrangement, it doesn’t work for two-tailed molecules, like the phosphatidylcholines. Schematic figure of a biological membrane. The lipid content of different membranes varies from as little as 25% up to 80% by weight. At highly curved regions the outer convex face contains mainly cone-shaped lipds while the inner concave face has more wedge-shaped lipids. Stressed regions may even adopt a locally non-bilayer structure. Reasons why the phospholipid bilayer membrane is the most ubiquitous architectural component of cells 1) The self-assembly of two-chain phospholipids (like PC) into bilayers is even more avid than that of one-chain surfactants (like SDS) into micelles. The reason is simply that the hydrophobic cost of exposing two chains to water is twice as great as that for one chain. This free energy cost ε enters the equilibrium constant and hence the CMC, a measure of the chemical drive to self-assembly, via its exponential. There’s a big difference between exp(-ε/kBT) and exp(-2·ε/kBT), so the CMC for phospholipid formation is tiny. Membranes resist dissolving even in environments with extremely low phospholipid concentration. 2) Phospholipid membranes automatically form closed bags because any edge to the planar structure would expose the hydrocarbon chains to the surrounding water. Such bags, or bilayer vesicles, can be almost unlimited in extent; it is straightforward to make „giant” vesicles of radius 10 μm, the size of eukaryotic cells. This is many thousands of times larger than the thickness of the membrane; giant vesicles are self-assembled structures composed of tens of millions of individual phospholipid molecules. 3) Phospholipids are not particularly exotic or complex molecules. They are relatively easy for a cell to synthesize, and phospholipid-like molecules could even have arisen abiotically (from nonliving processes) as a step toward the origin of life. In fact, bilayer membranes are even formed by phospholipid-like molecules that fall to Earth in meteorites. 4) The geometry of phospholipids limits the membrane thickness. This thickness in turn dictates the permeability of bilayer membranes, their electrical capacitance, and even their basic mechanical properties. Choosing the chain length that gives a membrane thickness of a few nanometers turns out to give useful values for all these membrane properties; that’s the value Nature has in fact chosen. For example, the permeability to charged solutes (ions) is very low, because the partition coefficient of such molecules in oil is low. Thus, bilayer membranes are thin, tough partitions, scarcely permeable to ions. 5) Bilayer membranes are fluid. No specific chemical bond connects any phospholipid molecule to any other, just the generic dislike of water for the hydrophobic tails. Thus, the molecules are free to diffuse around for membrane-bound cells to change their shape, as, for example, when an amoeba crawls or a red blodd cell squeezes through a capillary. 6) Again because of the nonspecific nature of the hydrophobic interaction, membranes readily accept embedded objects; hence they can serve as the doorway to cells and even as the factory floors inside them. An object intended to poke through the membrane simply needs to be designed with two hydrophilic ends and a hydrophobic waist; entropic forces then automatically take care of inserting it into a nearby membrane. Understanding this principle also immediately gives us a technological bonus: a technique to isolate membrane-bound proteins. Solubilization of integral membrane proteins by detergent Mechanical Properties of Membranes: Bending Stiffness Flat membrane The bilayer membrane prefers to be flat, because this state has the lowest free energy. As the layers are mirror images of each other, there is no tendency to bend to one side or the other. Because each layer is fluid, there is no memory of any previous bent configuration (in contrast to a small patch snipped from a bicycle tire, which remains curved). In short, although it's not impossible to deform a bilayer to a bent shape (indeed, it must so deform in order to close onto itself and form a bag), still bending will entail some free energy cost. Strictly speaking, the argument for minimum free energy state of planar surface applies to artificial, pure lipid bilayers only. Real plasma membranes have significant compositional differences between their two layers, with a corresponding spontaneous tendency to bend in one direction. Energetic cost to bend the planar membrane Bending a bilayer involves a combination of stretching the outer layer and squeezing the inner layer. In addition, the bilayer's elasticity also contains contributions from deformation of the tails (not just the heads) of the amphiphilic molecules. These elaborations do not change the general form of the bending elasticity energy established for much more restricted conditions (only the stretching of the heads of the amphiphilic molecules in the membrane is taking into account). With bending one side of the membranes, the polar heads get streched apart, eventually admitting water into the nonpolar core. In other words, each polar head group normally occupies a particular geometrical area ahead; a deviation Δa from this preferred value will incur some free energy cost. For small (Δa << ahead) bends (Hooke relation): elastic energy/phospholipid molecule = ½·k(Δa)2 Bending of bilayer membrane The value of the spring constant, k is an intrinsic property of the membrane. Suppose that we wrap a small patch of membrane around a cylinder of radius R much bigger than the membrane's thickness d. Bending the membrane requires that we stretch the outer layer by ahead d a R Thus, we expect a bending energy cost per head group of the form ½·k(ahead·d/R)2. Because the layer is double, the number of such head groups per area is 2/ahead. The (specific) free energy cost to bend a bilayer membrane into a cylinder of radius R: free energy cost/unit area = ½·κ/R2, where κ is an intrinsic parameter of the membrane, the bend stiffness (its dimension is energy): κ = 2·k·d2·ahead The cost to bend the membrane into a spherical patch of radius R is four times as great as that for cylinder, because each head group gets stretched in two directions, so Δa is twice as great. Thus bending the layer into a spherical shape with radius of curvature R costs free energy per unit area = 2·κ/R2. The total bending energy to wrap a membrane into a spherical vesicle is total free energy cost (flat → sphere) = 8πκ This is already an important result: The total bending energy of a sphere is independent of the sphere’s radius. Estimate of the numerical value of κ. Consider a single layer at an oil-water interface. Bending the layer into a spherical bulge, with radius of curvature R comparable to the length ltail of the hydrocarbon tails, will spread the heads apart and expose the tails to water. Such a large distortion will incur a hydrophobic free energy cost per unit area, Σ, comparable to that at an oil-water interface (surface tension). The corresponding cost for a bilayer in water will be roughly twice this value. We thus have two different expressions for the bending energy of a spherical patch of bilayer. Equating these expressions 2κ/(ltail)2 = 2·Σ. lets us estimate κ. Taking typical values: Σ ≈ 0.05 J/m2 and ltail ≈ 1.3 nm we get κ ≈ 0.8 .10-19 J. Our estimate is crude, but it’s not too far from the measured value κ = 0.6 .10-19 J = 15· kBT for dimyristoyl phosphatidylcholine (DMPC). The total bending energy 8πκ of a spherical vesicle of Lessons from the measured value of κ Suppose that we take a flat membrane of area A and impose on it a corrugated (washboard) shape, alternating cylindrical segments of radius R. The free energy cost of this configuration is ½·κA/R2. Taking A to be 1000 μm2, a value corresponding to a typical 10 μm cell, we find that the bending energy cost greatly exceeds kBT for any value of R under 10 μm. Thus, the stiffness of phospholipid bilayer membranes has been engineered to prevent spontaneous corrugation by thermal fluctuations. At the same time, the bending energy needed for gross, overall shape changes (e.g. those needed for cell crawling) is only a few hundred times kBT, so such changes require the expenditure of only a few dozen ATP molecules. (The free energy change of ATP hydrolysis under normal cell conditions is ΔG = -20 kBT/molecule. The standard free energy change is ΔG’o = -12.4 kBT/molecule, but the cells are far from standard conditions.) Phospholipid bilayer membrane stiffness is thus in just the right range to be biologically useful. Lessons from the measured value of κ Not only are cells themselves surrounded by a bilayer plasma membrane (cell walls). Many of the organelles inside cells are separate compartments, partitioned from the rest by a bilayer (inner membranes). - Products synthesized in one part of the cell (the „factory”) are also shipped to their destinations in special-purpose transport containers, themselves bilayer vesicles. - Incoming complex food molecules awaiting digestion to simpler forms are held in still other vesicles. - The activation of one neuron by another across a synapse involves the release of neurotransmitters, which are stored in bilayer vesicles until needed. Self-assembled bilayers are ubiquitous in cells.