Phase Transformations
advertisement

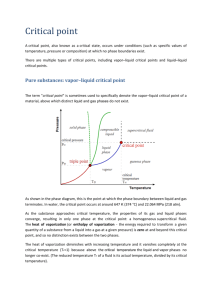
Physical Transformations of Pure Substances Chapter 4 Stabilities of Phase A phase of a substance is a form of matter that is uniform throughout in chemical composition and physical state. A phase transition is the spontaneous conversion of one phase into another. Phase transitions occur at a characteristic temperature and pressure. Stabilities of Phase At 1 atm, < 0 °C, ice is the stable phase of H2O, but > 0 °C, liquid water is the stable phase. The transition temperature, Ttrs, is the temperature at which two phases are in equilibrium. So what happens to Gibbs energy? Stabilities of Phase At 1 atm, < 0 °C, ice is the stable phase of H2O, but > 0 °C, liquid water is the stable phase. The transition temperature, Ttrs, is the temperature at which two phases are in equilibrium. So what happens to Gibbs energy? < 0 °C Gibbs energy decreases as liquid solid. > 0 °C Gibbs energy decreases as solid liquid. Stabilities of Phase Thermodynamics does not provide information regarding the rate of phase change. Diamond graphite Thermodynamically unstable phases that persist due to slow kinetics are called metastable phases. Phase Diagrams Phase boundaries show the values of p and T at which two phases coexist in equilibrium. Vapor Pressure The pressure of a vapor in equilibrium with a liquid is called the vapor pressure. The pressure of a vapor in equilibrium with a solid is called the sublimation vapor pressure. Boiling Point Liquid can vaporize from a liquid surface below it’s boiling point – as we learnt from the Drinking Bird. In an open vessel, the temperature at which the vapor pressure equals the external pressure, is called the boiling temperature. At 1 atm, it’s called the normal boiling temperature, Tb. At 1 bar, it’s called the standard boiling point. Normal point of H2O is 100.0 °C; it’s standard boiling point is 99.6 °C. Critical Point In a closed rigid vessel, boiling does not occur. As the temperature is raised the density of vapor increases and the density of the liquid decreases. When the density of the vapor and liquid phases are equal the surface between the two phases disappears. The temperature at which this occurs is called the critical temperature, Tc. The vapor pressure at the critical temperature is called the critical pressure, pc. Critical Point Melting and Freezing The temperature at which, under a specified pressure, the liquid and solid phases of a substance coexist in equilibrium is called them melting temperature. The freezing temperature is the same as the melting point. At 1 atm, the freezing temperature is called the normal freezing point, Tf. At 1 bar, it’s called the standard freezing point. The difference is negligible in most cases. The normal freezing point is also called the normal melting point. Triple Point There is a set of conditions under which three different phases of a substance (typically solid, liquid and vapor) all simultaneously coexist in equilibrium. This point is called the triple point. For any pure substance the triple point occurs only at single definite pressure and temperature. The triple point of water lies at 273.16 K and 611 Pa. Triple Point The triple point marks the lowest pressure at which a liquid phase can exist. Carbon Dioxide Water Helium Thermodynamics of Phase Transitions The molar Gibbs energy, Gm, is also called chemical potential, m. Phase transitions will be investigated primarily considering the change in m. Thermodynamic definition of equilibrium: At equilibrium the chemical potential of a substance is the same throughout the sample, regardless of how many phases are present. Thermodynamics of Phase Transitions Thermodynamics of Phase Transitions At low temperatures, and provided the pressure is not too low, the solid phase of a substance has the lowest chemical potential and is therefore the most stable. Chemical potentials change with temperature: this explains why different phases exist. Temperature Dependence of Phase Transitions Gm m Sm Sm T p T p As temperature increases, chemical potential decreases. Melting and Applied Pressure Gm m Vm Vm p T p T Molar volume of solid is smaller than that of the liquid. Melting and Applied Pressure Gm m Vm Vm p T p T Molar volume of solid is greater than that of the liquid. Melting and Applied Pressure m Vm p T m Vm p Melting and Applied Pressure Calculate the effect on the chemical potentials of ice and water of increasing pressure from 1.00 to 2.00 bar at 0 °C. The density of ice is 0.917 g cm-3 and that of liquid water is 0.999 g cm-3. Vm M m Mp M 18.02 g mol -1 0.01802 kg mol -1 p 1 bar = 10 5 Pa ice 917 kg m -3 water 999 kg m -3 Melting and Applied Pressure Calculate the effect on the chemical potentials of ice and water of increasing pressure from 1.00 to 2.00 bar at 0 °C. The density of ice is 0.917 g cm-3 and that of liquid water is 0.999 g cm-3. (0.01802 kg mol -1 ) (1.00 Pa) -1 u(ice) = 1.97 J mol 917 kg m -3 (0.01802 kg mol -1 ) (1.00 Pa) -1 u(liquid) = 1.80 J mol 999 kg m -3 Vapor Pressure and Applied Pressure When pressure is applied to a condensed phase, its vapor pressure rises. This is interpreted as molecules get squeezed out of the condensed phase and escape as a gas. p p*e Vm (l )P RT p vapor pressure p* vapor pressure of condensed phase in the absence of an additional pressure P pressure applied Vapor Pressure and Applied Pressure Location of Phase Boundaries Locations of phase boundaries – pressures and temperatures - can be located precisely by making use of the fact that at when two phases are in equilibrium, their chemical potentials must be equal m ( p,T) m ( p,T) Location of Phase Boundaries dG Vdp SdT dm Vm dp Sm dT V ,m dp S ,m dT V ,m dp S ,m dT (S ,m S ,m )dT (V ,m V ,m )dp trsS dp trsV dT Clapeyron equation Location of Phase Boundaries Solid-liquid boundary trsS dp trsV dT trsH trsS T dp fusH dT T trsV Clapeyron equation Solid-liquid boundary Solid-liquid boundary dp fusH dT T fusV fusH dT dp fusV T fusH dT p* dp T * V T fus p T fusH dT p* dp V T * T trs p T fusH T p p ln * fusV T * Solid-liquid boundary fusH T p p ln * fusV T * When T and T * do not differ much p p * fusH T * fusV (T T * ) Liquid-vapor boundary trsS dp trsV dT trsH trsS T dp vap H dT T vapV Clapeyron equation Liquid-vapor boundary dp dT dT dp "small" "large" Liquid-vapor boundary dp vap H dT T vapV vapV Vm (g) vap H dp dT T(RT p) d ln p vap H dT RT 2 - Clausius - Clapeyron equation Liquid-vapor boundary d ln p vap H dT RT 2 vap H d ln p dT 2 RT ln p T vap H ln p* d ln p T * RT 2 dT vap H T dT vap H 1 1 ln p d ln p * ln p* * 2 T R T R T T vap H 1 1 * ln p p * R T T vap H 1 1 p e * * p R T T p p*e Liquid-vapor boundary d ln p vap H dT RT 2 vap H d ln p dT 2 RT ln p T vap H ln p* d ln p T * RT 2 dT vap H T dT vap H 1 1 ln p d ln p * ln p* * 2 T R T R T T vap H 1 1 * ln p p * R T T vap H 1 1 p e * * p R T T p p*e Solid-gas boundary * p p e sub H 1 1 * R T T