Alterations in Hematologic Function
advertisement

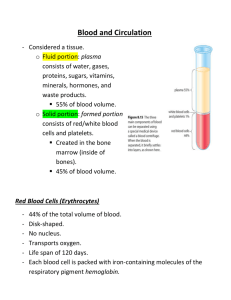
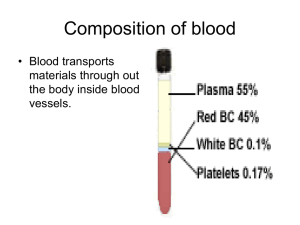
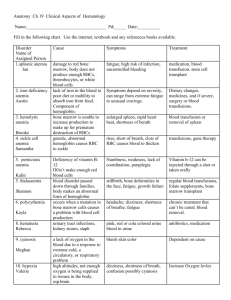
Hematology Jan Bazner-Chandler CPNP, CNS, MSN, RN Blood Blood is the fluid of life Blood is composed of: Plasma RBC WBC Platelets Plasma Plasma consists of: 90% water. 10 % solutes: albumin, electrolytes and proteins. Proteins consist of clotting factors, globulins, circulating antibodies and fibrinogen. Red Blood Cells RBC’s travel through the body delivering oxygen and removing waste. RBC’s are red because they contain a protein chemical called hemoglobin which is bright red in color. Hemoglobin contains iron, making it an excellent vehicle for transporting oxygen and carbon dioxide. RBC’s Average life cycle is 120 days. The bones are continually producing new cells. White Blood Cells The battling blood cells. The white blood cells are continually on the look out for signs of disease. When a germ appears the WBC will: Produce protective antibodies. Surround it and devour the bacteria. WBC’s WBC life span is from a few days to a few weeks. WBC’s will increase when fighting infection. Neutropenia – abnormal percentage of neutrophils compared to total white blood cells; decreases a child’s ability to fight pathologic bacteria Platelets Platelets are irregularlyshaped, colorless bodies that are present in blood. Their sticky surface lets them form clots to stop bleeding. Blood Values CBC with differential and platelet count. Hgb: Normal levels are 11 to 16 g / dl Panic levels are: Less than 5 g / dl More than 20 g / dl Hematocrit Normal hematocrit levels are 35 to 44%. Panic levels: Hmct less than 15 % Hmct greater than 60% Blood Tests Hemoglobin and hematocrit used to screen for anemia. The CBC with differential would be used to help diagnose a specific disorder. A bone marrow aspiration would be the most conclusive in determining cause of anemia – aplastic / leukemia. Coagulation Profile Partial thromboplastin time Prothrombin time Platelet count Fibrinogen Platelet function analysis – clotting analysis Bone Marrow Bone marrow is the spongy substance found in the center of the bones. • It manufactures bone marrow stem cells, which in turn produce blood cells. • • • Red blood cells – carry oxygen to tissue Platelets – help blood to clot White blood cells – fight infection Bone Marrow Aspiration Bone Marrow Transplant Donor is placed under anesthesia. Marrow is aspirated out of the iliac crest. Marrow is filtered and treated to remove bits of bone and other unwanted cells and debris, transferred to a blood bag, and is infused into the patient’s blood just like at transfusion. Treatment Modalities Transfusion: Packed red blood cells – anemia Platelets – platelet dysfunction Fresh frozen plasma – coagulation factors Blood Transfusions 3 types of transfusion reactions Hemolytic Allergic Febrile Hemolytic Reaction Refers to an immune response against transfused blood cells. Antigens, on the surface of red blood cells, are recognized as “foreign proteins” and can stimulate B lymphocytes to produce antibodies to the red blood cell antigens. Hemolytic reaction Flank pain Fever Chills Bloody urine Rash Low blood pressure Dizziness / fainting Nursing Management Stop the blood transfusion. Start normal saline infusion. Take vital signs with blood pressure Call the MD Obtain blood sample and urine specimen. Return blood to blood bank. Document Febrile Reaction Often occurs after multiple blood transfusions. Symptoms:fever, chills, and diaphoresis. Interventions: Slow transfusion and administer antipyretic. Administer antipyretic prior to administration. Allergic Reaction Symptoms: rash, urticaria, respiratory distress, or anaphylaxis. Interventions: administer antihistamine before transfusion Physician may order washed rbc’s Hematologic Conditions Alteration in Hematologic Status Disorders of hemostasis or clotting factors Structural or quantitative abnormalities in the hemoglobin. Anemias Aplastic Anemia Genetic Implications The following have a genetic link: implications for genetic screening and fetal diagnosis Sickle cell anemia Thalassemia Hemophilia Bleeding Disorders Three types Hemophilia: males only Type A most common – factor VIII deficiency Type B - lack of factor IX (Christmas Disease) Type C – lack of factor XI Von Willebrand Disease – 1% of population – men or women – prolonged bleeding time Hemophilia Type A Hemophilia type A is the deficiency of clotting factor VIII. A serious blood disorder Affects 1 in 10,000 males in the US Autoimmune disorder with lowered level of clotting factor All races and socio economic groups affected equally Hemophilia Hemophilia is a sex-linked hereditary bleeding disorder Transmitted on the X chromosome Female is the carrier Women do not suffer from the disease itself Historical Perspective First recorded case in Talmud Jewish text by an Arab physician – documentation of two brothers with bleeding after circumcision. Queen Victoria is carrier and spread the disease through the male English royalty. Goals of Care Goals of care: Provide factor VIII (IX) to aid blood in clotting. To decrease transmission of infectious agents in blood products; hepatitis & AIDS. Future: gene therapy to increase production of clotting factor. Assessment Circumcision may produce prolonged bleeding. As child matures and becomes more active the incidence of bleeding due to trauma increases May be mild, moderate or severe Bleeding into joint spaces, hemarthrosis Most dangerous bleed would be intracranial Diagnosis Presenting symptoms Prolonged activated aPTT and decreased levels of factor VIII or IX. Genetic testing to identify carriers Pharmacologic Interventions Products used to treat hemophilia are: Fresh frozen plasma and cryoprecipitate which are from single blood donors and require special freezing Second generation of factor VIII are made with animal or human proteins Fibrin glue – mixture of fibrogen and thrombin can be applied topically on a wound to stop minor bleeding Multidisciplinary Interventions Replace the factor as ordered by physician. Manage pain utilizing analgesics as ordered – no salicylate products. Maintaining joint integrity during acute phase: immobilization, elevation, ice. Physical therapy to prevent flexion contraction and to strengthen muscles and joints. Provide opportunities for normal growth and development. Family Education Medic-Alert bracelet Injury prevention appropriate for age Signs and symptoms of internal bleeding or hemarthrosis Dental checkups Medication administration Long Term Complications 20% develop neutralizing antibodies that make replacement products less effective. Gene therapy providing continuous production of the deficient clotting factor could be the next major advance in hemophilia treatment. Disseminated Intravascular Coagulation or DIC DIC is an acquired coagulopathy that is characterized by both thrombosis and hemorrhage. DIC is not a primary disorder but occurs as a result of a variety of alterations in health. Assessment The most obvious clinical feature of DIC is bleeding. Renal involvement = hematuria Pulmonary involvement = hemoptysis, tachypnea, dyspnea and chest pain. Cutaneous involvement = petechiae, ecchymosis, jaundice, acrocyanosis and gangrene. Multidisciplinary Interventions Treatment of the precipitating disorder. Supportive care with administration of platelet concentration and fresh frozen plasma and coagulation factors. Administration of heparin (controversial in children). Heparin potentates anti-thrombin III which inhibits thrombin and further development of thrombosis. Assessment Rigorous ongoing assessment of all body systems Monitor bleeding No rectal temps Avoid trauma to delicate tissue areas All injections sites and IV sites need to be treated like an arterial stick. Prognosis Depends on the underlying disorder and the severity of the DIC. ITP Idiopathic thrombocytopenic purpura Idiopathic = cause is unknown Thrombocytopenic = blood does not have enough platelets Purpura = excessive bleeding / bruising Immune Thrombocytopenic Purpura Antibodies destroy platelets Antibodies see platelets as bacteria and work to eliminate them ITP is preceded by a viral illness URI Varicella Smallpox / measles vaccine Mononucleosis Flu Two Types Acute: occurs in children between 1 and 6 years Chronic (continuous or recurrent): older than 10 years and female Symptoms Random purpura Epistaxis, hematuria, hematemesis, and menorrhagia Petechiae and hemorrhagic bullae in mouth Diagnostic Tests Low platelet count Peripheral blood smear Antiplatelet antibodies Normal platelet count: 150,000 to 400,000 Pharmacologic Interventions For severe cases (platelet count < 20,000) IV gamma globulin for 2-5 days to block antibody production, reduce autoimmune problem and increase platelet count Corticosteroids to enhance vascular stability, increase platelet survival IV anti-D to stimulate platelet production Sickle Cell Disease Autosomal recessive disorder Defect in hemoglobin molecule Cells become sickle shaped and rigid Lose ability to adapt shape to surroundings. Sickling may be triggered by fever and emotional or physical stress Pathophysiology When exposed to diminished levels of oxygen, the hemoglobin in the RBC develops a sickle or crescent shape; the cells are rigid and obstruct capillary blood flow, leading to congestion and tissue hypoxia; clinically, this hypoxia causes additional sickling and extensive infarctions. Whaley & Wong Text Body Systems Affected by SS Brain: CVA – paralysis - death Eyes: retinopathy – blindness Lungs: pneumonia Abdomen: pain, hepatomegaly, splenomegaly (medical emergency due to possible rupture Skeletal: joint pain, bone pain – osteomyelitis Skin: chronic ulcers – poor wound healing Vaso-occlusive Crisis Presents depending on location of vaso-occlusion Bone pain – back, knees, shoulders, elbows Acute chest syndrome – Chest pain / pneumonia Acute abdominal pain Cerebrovascular accident – stoke symptoms Priapism – nonsexual erection of penis Splenic Sequestration Sudden and quickly progressing splenic enlargement Left upper quadrant pain and vomiting Abdominal distension, pallor, dyspnea, tachycardia Hypovolemia and shock Decrease hemoglobin / hematocrit and increased reticulocyte count Aplastic Crisis Diminished production and increased destruction of red blood cells Triggered by viral infection or depletion of folic acid Signs include profound anemia, pallor Diagnostic Screening Newborn screening is mandatory in the United States. Screening done between 24 and 72 hours of age. Verification of results are made by the healthcare provider at first office visit. Newborn Screening – Follow-up Contact family with screening results Refer to specialists if positive Evaluate infant for slenomegaly Initiate daily penicillin VK (125 mg po bid) prophylaxis as recommended Educate regarding risk of sepsis, or signs of splenic sequestration Nursing Management - Hospital Increase tissue perfusion Oxygen / bedrest Blood transfusion / antibiotics if ordered Pain management – meperidine (Demerol) contraindicated Hydration IV fluids as ordered Oral intake of fluids Nursing Management - Home Care • • • Adequate nutrition Emotional Support Discharge instructions • • • • • Information about disease management Daily folic acid Control of triggers Prophylactic antibiotics Immunizations / Pneumococcal Patient Education Necessity of following plan of care Signs and symptoms of impending crisis. Signs and symptoms of infection Preventing hypoxia from physical and emotional stress Proving adequate rest Thalassemia A group of inherited diseases of the blood that affect the ability to produce hemoglobin. Occurs most frequently in people of Italian, Greek, Middle Eastern, Southern Asian and African Ancestry. Two Types of Thalassemia Alpha or beta depending on which part of an oxygencarrying protein the red blood cell is lacking. Child may have mild to severe form of the disease. Cooley’s anemia is severe form of the disease. Tests for Thalassemia Blood tests and family genetics study to show whether an individual has Thalassemia or is a carrier. Prenatal testing at 11th week of pregnancy using chorionic villi sampling. Amniocentesis around the 16th week of pregnancy Assisted reproductive therapy Clinical Manifestations The child may be: pale and listless have poor appetite enlarged spleen, liver and heart bones become thin and brittle Heart failure and infection are the major cause of death. Interdisciplinary Interventions Frequent blood transfusions to keep hemoglobin levels near normal “iron chelators” to rid body of excess iron from numerous blood transfusions Bone marrow transplant: only possible for a small minority of patients who have a suitable bone marrow donor Iron Deficiency Anemia Most common nutritional deficiency Depletion of iron stores due to Inadequate dietary intake Impaired iron absorption American Academy of Pediatrics Recommends screening for iron deficiency at 1 year of age Screening of high risk children (low socio-economic status or poor dietary intake) What a problem? IDA can have long lasting effects on motor development, mental development and auditory and visual function. IDA Occurs in children experiencing: Rapid physical growth Low iron intake Inadequate iron absorption Loss of blood Assessment Associated with low oxygenation of tissue: Pallor Fatigue Shortness or breath Irritability Intolerance of physical work / exercise Diagnostic Tests Initial screen is hemoglobin Hemoglobin levels less than 8 g/dL Decreased levels of Serum Iron or Total Iron Binding or Serum Ferritin Microcytic and hypochromic red blood cells Pharmacologic Treatment Iron supplementation Given in a.m. on an empty stomach To avoid staining of teeth, give using a syringe, dropper or straw Instruct caretaker that child may have dark-colored stools Management Infants younger than 12 months should be on formula with iron Solid foods – introduced about 6 months of age Infants 12 months or older Rice cereal fortified with iron Pureed Vegetables, meats Decrease intake of milk Increase solid foods Children: iron fortified cereals, meat, green leafy vegetables, yogurt, cheeses, low-fat or non-fat milk Teenagers: reduce junk food Aplastic Anemia Acquired or inherited Normal production of blood cells in the bone marrow is absent or decreased. A marked decrease in RBC’s, WBC’s and platelets. Causes Exposure to drugs Exposure to chemicals Exposure to toxins Infection Idiopathic in nature Inherited: Fanconi’s anemia Blood Characteristics Neutophil less than 500 Platelet less than 20,000 Hemoglobin less than 7 Reticulocytes 1% Nursing Diagnosis? Bone marrow reveals hypo-cellular and fatty marrow. Management Immunosuppressive therapy Antithymocyte globulin Administered IV over 4 days Response seen within 3 months Bone Marrow Transplant Neonatal Hyperbilirubinemia Hyperbilirubinemia is defined as excessive levels of bilirubin in the blood. Alert: Jaundice that occurs within the first 24 hours of life or after 2 weeks of life signifies an abnormal physiologic process. Kernicterus: bilirubin encephalopathy Jaundice Yellowing of the skin – occurs when bilirubin is deposited into the subcutaneous tissue and it becomes visible when the serum bilirubin levels exceed 7.0 mg/dL. Normal Physiologic Jaundice Occurs in 45% to 65% of all healthy newborns in the first 3 to 5 days of life Occurs more frequently in infants less than 38 weeks gestation Breast fed infants have a higher incidence of jaundice (due to dehydration and caloric deprivation) Assessment Very yellow or orange skin tones (beginning at the head and spreading to the toes) Increased sleepiness, so much that it is hard to wake the baby High-pitched cry Poor sucking or nursing Weakness, limpness, or floppiness Photo Therapy Fiberoptic Blanket Multidisciplinary Interventions Monitor bilirubin levels Assess activity level – muscle tone – infant reflexes Encourage po intake: May need to supplement with formula if inadequate breastfeeding Weight daily to assess hydration status Monitor stools – amount and number Cover eyes while under bili-lights Facilitate parent - infant bonding Loss of moro or startle reflex can indicate possible brain damage due to Kernicterus