Exempt vs. Expedited, Exemption Categories, Educational Research
advertisement

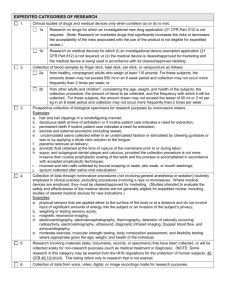
IRB: Scene 2 Elizabeth A. Buchanan, Ph.D. Endowed Chair in Ethics Center for Applied Ethics University of Wisconsin-Stout buchanane@uwstout.edu 7152325184 Follow-Ups and Reminders • One-person IRBs: Not boards, offices; boards must have 5 members per regs • The sessions are recorded; we will need to determine if/how to share. • Ask questions at any time! In the News: • http://www.research.umn.edu/news/docume nts/final_report.pdf Today’s Topics • Processes – Retention – Dating • Reviewing protocols – Exempt – Expedited – Full Board • Consent • Modifications – Significant versus minor modification • Special Topics: SOTL Recall an Important Contextual Point: • Regs • Institutional specificity In order to contextualize today’s session, a few things about us at Stout: • We review all research, student, classroom projects, campus research (we think!), etc • Our process entails: • Researcher submits PDF; documents are checked in by administrative staff; routed to a board member; review conducted and results sent back to admin staff; notification of review status sent via email to researcher; we do not use an IRB management system • 10 working day turnaround for exempt/expedited; • Pilot test this semester: 5 day turn-around for class submissions Retention of Records It is more than keeping records three years. Protocol files need to be kept for three years after completion of the research. Current and previous written procedures required by the regulations need to be retained until all research ever reviewed under under an procedure has been closed for three years. A similar retention requirement applies to current and previous lists of IRB members (rosters), minutes of IRB meetings, and correspondence between the IRB and the investigator. For example, if you had a protocol open in 2000, and close in 2012, then you would have to keep all versions of written procedures, rosters, and minutes back to 2000 until at least 2015. If you retain records longer than required by the regulations, OHRP and FDA can ask for any records in your possesion and you have to hand them over. I have had a number of clients over the years that were unhappy when they found this out by direct experience. If OHRP or FDA ask you for records and you destroyed them as part of a systematic records destruction process, OHRP and FDA will acknowledge that and leave you alone. If you destroy records en masse the day after you get a notification of an audit, you are in a heap of trouble. If you keep an electronic copy and destroy the paper copy, FDA will consider that to be a retained record that they can ask to inspect. If you destroy records, you must destroy all versions, electronic, paper, including backup files and tapes. Retention and Destruction: Jeff Cooper 4.1. Review the study files that can be destroyed. 4.1.1. Omit destruction of records on a legal hold. 4.1.2. Previously approved non-exempt studies: Three years after the date on which all research sites overseen by the [Organization]’s IRB have been completed either through closure, <Termination of IRB Approval>, disapproval, or lapse of approval 4.1.3. Non-exempt studies never approved and exempt studies: Three years after the last IRB action or after withdrawal by the submitter 4.2. Shred paper documents and dispose the shredded materials securely. 4.3. Notify information technology to destroy electronic documents by either deleting the files or replacing the files with stub files documenting the date of deletion. 4.4. Document your name and the date of destruction with the following for each study file destroyed: 4.4.1. Study title 4.4.2. IRB ID 4.4.3. Date of completion 4.4.4. Paper, electronic, or both Dating • No regulatory requirement to put date stamps on consent forms, survey documents, etc. BUT: for good housekeeping: • OHRP recommends that IRBs consider using methods that will allow the IRB to readily recognize the most current version of the IRBapproved informed consent document, for example, using date stamps, version numbers, or initialing and dating documents to indicate when a version was approved. Models of/for Reviewing Protocols • Pre-review by admin/IRB coordinator/IRB analyst • Review by Chair • Sent to a decentral reviewer – Expert review? • Sent to board members • Full board – QUESTION: WHEN DO YOU DETERMINE A PROTOCOL MUST GO TO FULL BOARD? PLEASE DESCRIBE Protocol Review: Always Begin at the Beginning: • Is it research? – Consider if a main goal of the activity is to learn something for the purpose of benefitting people other than the research subjects – Are the study aims clear? – Is there appropriate justification for the project? – What is the primary intent of a project? – What’s NOT research? • Innovative therapy, nonvalidated practices, QI (aimed at improving local systems, not generalizable); public health surveillance, public health program evaluation • Does it involve human subjects? • Are you engaged? • Does it fall under an exempt category? What is Exempt? • Research activities in which the only involvement of human subjects will be in one or more of the exempt categories defined by the federal regulations, will be given an exempt determination, rather than IRB approval. Exempt studies are so named because they are exempt from some of the federal regulations. However, they are not exempt from state laws, institutional policies, or the requirements for ethical research. Who may determine that research is exempt? • The regulations do not specify who at an institution may determine that research is exempt under 45 CFR 46.101(b). However, OHRP recommends that, because of the potential for conflict of interest, investigators not be given the authority to make an independent determination that human subjects research is exempt. Who Decides Exemptions? • Persons making an exemption determination should have access to sufficient information to make a correct determination. Evaluation tools and resources may take a variety of forms, including but not restricted to: checklists, Standard Operating Procedures, or specialized training for individuals authorized by the institution to make an exemption determination. • When an exemption determination is made, the specific exemption category or categories should be included in the record and this information should be available for oversight and audit purposes. • Institutional policies and procedures should identify clearly who is responsible for making exemption decisions. This may be done in a variety of ways, including delegation by name, role, or position. • Institutions should make policy and procedure information addressing exemption determination readily accessible to investigators and others involved in the conduct and administration of human subjects research. Category 1 • Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as: (i) research on regular and special education instructional strategies, or (ii) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods. • Examples: – Evaluating the use of accepted or revised standardized tests – Testing or comparing a curriculum or lesson – A program evaluation of pharmacy continuing education Category 2 • Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior, unless: (i) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects' responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects' financial standing, employability, or reputation. • Note: The section of this category pertaining to standardized educational tests may be applied to research involving children. This category may also apply to research with children when the investigator observes public behavior but does not participate in that behavior or activity. This section is not applicable to survey or interview research involving children. • Examples: – Surveying teachers, nurses, or doctors about a technique or an outcome – Interviewing managers about a management style or best practice – Conducting a focus group about an experience or an opinion of a community program Can We Use Exempt 2 with Children? • Kelly from UW-O, do you remember?? • IRB Bible: NO • But: • From IRB Forum: “In fact, OHRP notes that a particular research activity may be found to be exempt if it the activity meets the criteria for any one of the exempt categories of research described under HHS regulations at 45 CFR 46.101(b). Therefore, if an activity is found to be meet the criteria for exemption under 45 CFR 46.101(b)(1), it does not need to be evaluated with respect to any other exempt category (such as that category under 45 CFR 46.101(b)(2)). OHRP further notes that some research activities that involve individually identifiable data about student performance reasonably may be found to be exempt under HHS regulations at 45 CFR 46.101(b)(1). Accordingly, in the example given, if an institution determined that the student survey (and the rest of that study) met the requirements for exemption under 45 CFR 46.101(b)(1), then the survey would be exempt, and it would be irrelevant whether or not it met the requirements for exemption under 45 CFR 46.101(b)(2).” Elyse Summers Category 3 • Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior that is not exempt under paragraph (2) of this section, if: (i) the human subjects are elected or appointed public officials or candidates for public office; or (ii) Federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter. • Example: – Interviewing public officials about a local or global issue. Category 4 • Research involving the collection or study of existing* data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects. • *Note: "Existing" means existing before the research is proposed to the institutional review board to determine whether the research is exempt. • Example: – Analyzing existing tissue samples or data set which are recorded by the investigator without identifiers Types of Data • Anonymous – the identity of the respondent cannot be determined; no links exist between the data and the individual about whom the data are recorded; ongoing debates about the reality of anonymity! Is confidentiality where it’s at? • De-identified – identifiers have been removed from the dataset under consideration; links between the data and the individual about whom the data are recorded exist but are not readily accessible to the researcher at CU; • Coded – identifiers have been removed from the dataset under consideration but can readily be replaced through the use of a master list that is accessible to the investigator; • Identifiable or non-coded – the identity of the subject is documented, linked or associated with the data. Coded Data (SO MUCH TO COVER….!) Lots of Discussion and Ambiguity These Days, both SBER and Med/Bio Category 5 • Research and demonstration projects which are conducted by or subject to the approval of department or agency heads, and which are designed to study, evaluate, or otherwise examine: (i) Public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives to those programs or procedures; or (iv) possible changes in methods or levels of payment for benefits or services under those programs. Category 6 • Taste and food quality evaluation and consumer acceptance studies, (i) if wholesome foods without additives are consumed or (ii) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the U.S. Department of Agriculture. – “The sauerkraut protocol” Expedited Review • Expedited reviews are conducted by at least one experienced member of the IRB. In order to qualify for review via expedited procedures, the research must not be greater than minimal risk and fall into at least one of the expedited categories defined by the federal regulations. • The expedited review procedure may not be used where identification of the subjects and/or their responses would reasonably place them at risk of criminal or civil liability or be damaging to the subjects financial standing, employability, insurability, reputation, or be stigmatizing, unless reasonable and appropriate protections will be implemented so that risks related to invasion of privacy and breach of confidentiality are no greater than minimal risk. Full Board Review • Research that is greater than minimal risk and/or does not qualify for exempt or expedited review, as defined by the categories, will be reviewed by the Full Board at a convened meeting. • Expedited review as defined by federal regulations allows the IRB chairperson or one or more experienced reviewers designated by the chairperson from among members of the IRB to evaluate and approve specific types of research. Reviewers conducting an expedited review may exercise all of the authority of the IRB except that they may not disapprove a study. When a subcommittee cannot approve the research under expedited review, the study is referred to the full Committee for review. What Are Our Options? • • • • • Exempt Approve Defer with minor mods Defer with major mods Disapprove • Share your experiences. When is consent required? Research Involving human subjects Conducted or supported by Common Rule agency or covered by applicable FWA Not exempt Not eligible for waiver of informed consent (Note: not available for FDA-regulated research except for emergency research, and other life-threatening or military situations outlined in FDA regulations at 21 CFR 50.23) Consent Includes Subject Recruitment! • OHRP considers subject recruitment part of informed consent: o Recruitment plan must receive IRB review/approval prior to initiation of research o This often poses challenges in classroom research: o Dealing with “pre-recruitment” o Convenience subjects/sampling Required Elements of Consent • • • • • • • • • • • • • §46.116 Informed Consent Checklist - Basic and Additional Elements A statement that the study involves research An explanation of the purposes of the research The expected duration of the subject's participation A description of the procedures to be followed Identification of any procedures which are experimental A description of any reasonably foreseeable risks or discomforts to the subject A description of any benefits to the subject or to others which may reasonably be expected from the research A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained For research involving more than minimal risk, an explanation as to whether any compensation, and an explanation as to whether any medical treatments are available, if injury occurs and, if so, what they consist of, or where further information may be obtained Research, Rights or Injury: An explanation of whom to contact for answers to pertinent questions about the research and research subjects' rights, and whom to contact in the event of a research-related injury to the subject A statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and the subject may discontinue participation at any time without penalty or loss of benefits, to which the subject is otherwise entitled Waiver of informed consent 45 CFR 46.116(d)[not for FDA-regulated research] • Waiver may be of some or all of the required elements, or of requirement for consent in toto • IRB must find and document: – Minimal risk research; – Waiver will not adversely affect subjects’ rights and welfare; – Research could not practicably be carried out without waiver; and – When appropriate, subjects provided with additional pertinent information after participation Documentation required unless waived [waiver not permitted in FDA-regulated studies] If documentation required, “long form” or “short form” procedure (45 CFR 46.117) For waiver, IRB must find: o Consent document would be only record linking subject to the research and principal risk is breach of confidentiality, and each subject must be asked if documentation wanted; or o Minimal risk research; no procedures requiring consent in a non-research context IRB may require investigator to give subjects written statement regarding the research Continuing Review • http://www.hhs.gov/ohrp/policy/continuingre view2010.html Electronic Signatures Electronic signatures Electronic signatures encompass a broad gamut of technologies and methodologies, ranging from an “I agree” button in a click-thru agreement… to an electronic tablet which accepts a handwritten signature (oftentimes referred to as an eSignature)… to a digital signature cryptographically tied to a digital ID or certificate John B. Harris, Adobe Security blog entry "So what is an electronic signature anyway?", http://blogs.adobe.com/security/2008/02/ Electronic signatures OHRP FAQ: Does not require specific method of electronic signature Provides points to consider for the IRB to evaluate method of obtaining electronic signature Q: In what jurisdiction is the research being conducted? Electronic Signatures • Jurisdictionally-based • Wisconsin laws at http://docs.legis.wisconsin.gov/statutes/statut es/137/II/15 • Discuss with your legal counsel SOTL • With eternal thanks to and sadness at the loss of DR. RENEE MEYER, UWM • In 2007, Renee spearheaded a group to discuss SOTL and IRB concerns • That document remains relevant today. – SOTL researchers seek to advance the practice of teaching and learning through scholarly inquiry into student learning. • SOTL asks: – How can an instructor help students learn to be more critical or reflective thinkers? – How can an instructor help students learn how to use feedback on their assignments so as to make the subsequent assignments better? – How do students view group participation, and how does that impact their learning? – How can instructors help students learn to think creatively? – How do students draw on their prior knowledge to learn about new information or ideas? Particular Concerns with SOTL • Student Coercion/Power differentials • Waste of time? (not just SOTL research…) Case Studies A student PI contacted the IRB regarding a proposed study in which they planned to interview gang members about the meaning behind their tattoos [a very valuable data point]. The IRB and the PI were concerned that a gang member could disclose a past crime or a future intent to harm, and wondered what legal obligations the student would have should such information be disclosed. 10-12 orphanages in Cairo, Egypt (near Tahir Square) and interviews with government officials were undertaken by a student researcher who had previously been told by his faculty advisor that he did not need IRB review to conduct research for his dissertation. Questions were personal and sensitive in nature, and ranged in topic from perceptions of the orphanage system and solutions for improvement, to how staff could be better trained, to the kinds of psychological distress experienced by the children living there. Student collected data about children using their experiences in directed art therapy. Data was also collected by way of observation in order to describe day to day life. Surveys are commonly used in social behavioral research. To reach large numbers of people the field has moved to vendors like mechanicalTurk, and Qualtrics to reach a wide audience and provide an intermediary between subject and researcher. MechanicalTurk is also a venue for posting small jobs and processing the compensation paid to individual account holders who perform the work. A regular mechanicTurk user became disgruntled after a 75₵ payment was denied for completion of a survey. He took his complaint to the president of USC. As in all big institutions, the action to respond went through many administrative layers, all of which passed the issue down, but all of which needed to be informed of the resolution. In other words, a major ruckus over 75₵ Next Session • April 3, 2015, 2-4 • Topics: Internet/social media research, data security, and emerging issues • Send questions/cases!