Inborn Errors of Metabolism
advertisement

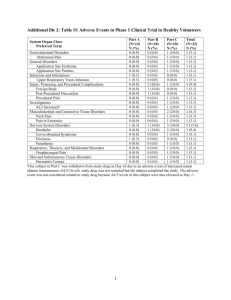
Board Review 11/27/2012 What topic should we do for December Board Review? A. Poisonings and Environmental Exposures B. Substance Abuse Acute Presentation Metabolic crisis Sudden onset of lethargy, vomiting, irritability, respiratory compromise, seizures, encephalopathy Typically due to hyperammonemia, acidosis, ketosis, hypoglycemia Chronic Presentation Indolent course Can affect multiple organ systems CNS Liver Heart Kidney Muscle Eye Dysmorphic features Appear normal at birth Metabolic intermediate responsible for symptoms is removed by maternal placenta Within days-months (rarely years) will develop a septic/shock-like picture Acute onset: lethargy, vomiting, tachypnea (or apnea), irritability, seizures Encephalopathic Workup for infection/sepsis yields normal results An infant who presented with vomiting is now lethargic and progressing to a comatose state. Mom says she just found out the newborn screen was abnormal. Which two tests are most important EMERGENTLY so that interventions may be started? A. Potassium and brain MRI B. WBC and ketones C. Lactate and platelets D. Glucose and ammonia E. Cardiac stress test and glucose tolerance test Acidosis pH <7.3, pCO2<30,bicarb <15 Can suggest: Metabolic disorder Infection Dehydration Intoxication Anoxia Elevated lactate Measured on blood gas Can suggest: hypoxia or poor perfusion (dehydration) Metabolic disorders: Glycogen storage disease Pyruvate defect Fructose 1,6 biphosphonate deficiency Mitochondrial disease Ketosis Ketones are a normal part of physiology, but not when they generate acidosis Organic acidemias Hyperammonemia Urea cycle defects Organic acidemias Fatty acid oxidation defects Hypoglycemia Hyperinsulinism Liver failure Glycogen storage disease, tyrosinemia, galactosemia, NiemannPick Organic acidemias Fatty acid oxidation defect A 3 day old infant was initially vigorous at birth but now has poor feeding, tachypnea, and lethargy. Septic work-up is negative. Serum electrolytes, glucose, and lactate are normal. An ABG shows: pH 7.53, pCO2 20, HCO3 25. Serum ammonia level is 465mcmol/L (elevated). Urine ketones are negative. What is the most likely diagnosis? A. Fatty acid oxidation defect B. Urea cycle defect C. Organic acidemia D. Glycogen Storage Disease Type I E. Renal tubular acidosis Urea Cycle Defects Organic Acidemias Amino Acid Disorders Carbohydrate Disorders Fatty Acid Oxidation Defects Classic presentation: First few days of life: poor feeding, vomiting, tachypnea, lethargy coma Late onset: Partial enzyme deficiencies Recurrent vomiting, developmental delay, learning difficulties, seizures, brittle hair, protein intolerance Failure to thrive Precipitating cause for acute hyperammonemic encephalopathy Infection, trauma, fasting, medications (glucocorticoids) Hyperammonemia WITHOUT acidosis Usually have respiratory ALKALOSIS No ketones No hepatomegaly Types: Ornithine transcarbamylase deficiency (X-linked) Carbamoyl phosphate synthase deficiency (AR) Citrullinemia (AR) Argininosuccinic acidemia (AR) Argininemia (AR) (arginase deficiency) Does not present with hyperammonemia Neurologic manifestations Diagnosis: plasma amino acid concentrations Urine orotic acid value can help distinguish types Enzyme analysis of tissue samples Treatment: Reduce ammonia! (more on this later) Reduce protein intake Avoid catabolism Arginine supplements can be helpful Urea Cycle Defects Organic Acidemias Amino Acid Disorders Carbohydrate Disorders Fatty Acid Oxidation Defects A 2 day old newborn presents with feeding difficulties and lethargy. Septic work-up is negative and urine organic acid values suggest an organic acidemia. The following are lab values which could be seen on initial presentation EXCEPT: A. Low pH B. Hyperammonemia C. Hyperglycemia D. Elevated lactate E. Ketones in the urine Organic acids are the intermediates in the catabolism (break down) of amino acids, lipids and other compounds Products go to Krebs cycle ATP Specific enzyme deficiencies lead to characteristic urine organic acid profiles Organic Acids Defects Isoleucine Valine Methionine Cholesterol Odd chain fatty acids propionic acidemia methylmalonic acidemia biotin Propionyl CoA B12 Methylmalonyl CoA Succinyl CoA Krebs Cycle isovaleric acidemia leucine Isovaleryl CoA 3MCC HMG CoA Even chain fatty acids Lysine Tryptophan glutaric acidemia Glutaryl CoA Crotonyl CoA Acetyl CoA ETS Acetyl CoA ATP Important features High anion gap metabolic acidosis Ketosis Elevated lactate +/- hypoglycemia +/- hyperammonemia Typically presents in first few days of life with introduction of protein into diet Irritable, poor feeding, lethargy coma Diagnosis: urine organic acids Plasma acylcarnitine profile can help distinguish between types Types (multiple!!) Isovaleric acidemia Seizures, high incidence of infection, odor of sweaty feet Propionic acidemia Methlymalonic acidemia May respond to vitamin B12 UCD + + ++ - OA + + +/+ respiratory alkalosis + - urine ketones - + lethargy/coma vomiting hyperammonemia metabolic acidosis Urea Cycle Defects Organic Acidemias Amino Acid Disorders Carbohydrate Disorders Fatty Acid Oxidation Defects Other disorders of amino acid catabolism Similar clinical manifestations to organic acidemias Diagnosed differently Plasma amino acids (vs urine organic acids ) Deficiency of enzyme that converts phenylalanine to tyrosine Not typically seen due to newborn screening Asymptomatic for a few months, then… Severe vomiting, irritability, eczema, mousy odor of urine Blond hair, blue eyes Late signs: profound intellectual disability** Pregnant woman not treated adequately: Fetal risks: miscarriage, SGA, microcephaly, cardiac defects Treatment: low phenylalanine diet (lifelong!**), frequent monitoring, dietary counseling, adequate tyrosine intake Deficiency of enzyme used to breakdown branched-chain amino acids leucine, isoleucine, valine Clinical signs: First week of life: poor feeding, tachypnea with shallow breathing, profound lethargy, hypertonicity Maple syrup smelling urine Hypoglycemia, acidosis Multiple subtypes Type I: liver failure, kidney involvement, nerve problems Type II: oculocutaneous Corneal ulcerations leading to clouding, skin thickening on palms/soles Treatment: low tyrosine and phenylalaine diet Cystathionine synthase deficiency elevated methionine levels 2 subtypes: Pyridoxine (B6) responsive and B6 resistant Better IQ prognosis in responsive type Clinical manifestations: Ectopia lentis (posterior), skeletal abnormalities (marfanoid), cognitive deficits, thromboembolic events Light-colored skin, hair, eyes Diagnosis: homocysteine in urine Treatment: pyridoxine If no response: diet high in cystine and low in methionine Urea Cycle Defects Organic Acidemias Amino Acid Disorders Carbohydrate Disorders Fatty Acid Oxidation Defects Deficiency of galactose-1-phosphate uridyltransferase Appear normal until first meal Develop poor feeding, failure to thrive, vomiting, lethargy, jaundice, abdominal distension, hepatomegaly Labs: Hypoglycemia +Reducing substances in urine (non-glucose) Galactose Metabolism glucose Breast milk, cow’s milk Galactose Lactose (galactose-glucose) (cataracts) galactokinase Gal-1-P galactose-1-P uridyltransferase (classical) Glucose-1-P Glucose-6-P glycolysis pyruvate A 6 day old female who is breast fed is brought to the emergency room due to poor feeding, vomiting, hepatomegaly, and jaundice. You suspect galactosemia. Which of the following would support this diagnosis? A. Blood culture positive for E. coli B. Diabetic mother C. Serum ammonia > 600 mcmol/L D. Cherry red spot on retina E. Microcephaly Diagnosis: enzyme assay on RBCs Complications: Liver disease (cirrhosis with portal hypertension/ascites), Gram-negative (E. coli) sepsis, cataracts, MR, speech delay, ovarian failure Cataracts are reversible with diet change Developmental and speech delays are common despite good dietary control** Treatment: galactose free diet, ophthalmology and developmental follow-up A 9 year old male is brought to the emergency room due to vomiting and lethargy shortly after a birthday party. PMHx is significant for FTT in late infancy which resolved without determination of a diagnosis. He had had several bouts of vomiting in the past, usually after consuming candy or soft drinks. Labs reveal elevated AST and ALT. What is the most likely diagnosis? A. Hereditary fructose intolerance B. Glycogen storage disease Type II C. Fatty acid oxidation defect D. Fabry disease E. Zellweger syndrome Deficiency in enzyme to break-down fructose (aldolase B) Exposure to fructose results in vomiting, poor feeding; can see seizures in extreme cases Continued exposure: FTT, hepatomegaly, hypoglycemia, jaundice, renal dysfunction, liver failure, ascites Labs: elevated liver transaminases, elevated direct bilirubin, clotting abnormalities Diagnosis: enzyme assay from liver biopsy Treatment: remove fructose from diet Enzyme converts FDP to F-6-P (part of gluconeogenesis) Deficiency of this enzyme when challenged with fructose, get buildup of FDP which inhibits gluconeogenesis decreased glucose production Labs: hypoglycemia, lactic acidosis, ketosis Hepatomegaly Diagnosis: enzyme assay from liver biopsy Treatment: remove fructose from diet, avoid fasting Urea Cycle Defects Organic Acidemias Amino Acid Disorders Carbohydrate Disorders Fatty Acid Oxidation Defects Multiple enzymatic reactions involved in the degradation of saturated fatty acids Release acetyl-CoAs and ketones which are used for energy production, especially during a FASTING state The enzymes involved in breaking down fatty acids are specific to different fatty acid lengths Short, medium, long, and very-long acyl-CoA dehydrogenases SCAD, MCAD, LCAD, VLCAD Deficiency in any can lead to fatty acid oxidation defect ALL are autosomal recessive Brain Fatty acids VLCAD LCAD MCAD SCAD fasting ketones + acetyl CoA Krebs cycle Medium chain acyl CoA dehydrogenase deficiency(MCAD) is most common defect 25% risk of death with first episode SCAD is typically benign A 2 month old female becomes comatose after an upper respiratory illness. Which of the following lab findings would most suggest a disorder of fatty acid oxidation? A. Hypoglycemia with metabolic alkalosis B. Hypoglycemia with + ketonuria C. Hyperglycemia with - ketonuria D. Hyperglycemia with + ketonuria E. Hypoglycemia with - ketonuria Key feature: Hypoketotic hypoglycemia Reducing substances are also negative +/- hyperammonemia • • Typically presents after recent illness; period of fasting Can be associated with hepatomegaly, liver disease, hypertrophic cardiomyopathy, arrhythmias, adult onset myopathy • Labs: glucose, electrolytes, ammonia level, LFTs, CPK, lactate, uric acid, urinalysis for myoglobinuria Diagnosis: plasma acylcarnitine profile +/- skin biopsy Carnitine transports longer chain fatty acids in to the mitochondria for breakdown Defects in the enzymes involved in this process can lead to similar symptoms of fatty acid oxidation defects Hypoketotic hypoglycemia Hepatomegaly, elevated transaminases, elevated ammonia Seizures (secondary to hypoglycemia) Can typically present in adulthood with muscle weakness, elevated CPK, myoglobinuria Diagnosis: plasma acylcarnitine profile +/- skin biopsy Prompt recognition of possible inborn error of metabolism Appropriate testing Immediate interventions Treat hypoglycemia with glucose infusions Treat encephalopathy due to hyperammonemia Sodium benzoate and sodium phenylacetate to remove ammonia Emergent hemodialysis if level is > 600 Stop all protein intake if urea cycle defect or amino acid disorder suspected Calories supplied with dextrose and intralipids Can give large doses of co-factors while waiting for specific lab tests to come back Vit B12, thiamine, biotin, riboflavin, folic acid, carnitine Once diagnosed Dietary counseling Genetic counseling You are seeing a new 1 year old patient for developmental delay. Based on a positive family history, you are suspecting an inborn error of metabolism as the cause. What are you MOST likely to see on exam?? A. Lack of babbling but normal motor exam B. Inability to sit alone or cruise but says 5-10 words C. Poor babbling, inability to sit alone, no holding toys/objects in her hands D. Normal development E. You are worried about autism but nothing else Difficult to recognize and diagnose Onset from birth to adulthood Neurologic abnormalities Developmental delay that is typically global Seizures, often resistant to medications Movement disorders Dystonias Choreas Abnormal tone Hearing loss Blindness Stroke should suggest homocystinuria or the mitochondrial disorder MELAS You are convinced that a patient is suffering from a chronic inborn error of metabolism. Other than Genetics, who would you MOST likely consult first during your initial work-up. A. Orthopedics B. ENT C. Urology D. Heme/Onc E. Ophthalmology Thorough developmental history and assessment If a metabolic disorder is suspected, initial work-up could include… Glucose, pH, lactate, ammonia level Plasma amino acids and urine organic acids VLCFA and acylcarnitine profile Urinary mucopolysaccharides and oligosaccharides MRI of the brain Skeletal survey May uncover dysostosis multiplex (seen in lysosomal storage diseases) Ophthalmologic examination Results of the initial history and physical exam should guide which tests can be added or eliminated Positive results on the initial screening will typically lead to further, more specific testing DNA analysis For diagnosis For genetic counseling Enzyme assays Liver biopsy Etc…but this would be guided by a friendly Dr. Marble A 3 month old female is found to have hepatomegaly on routine exam. She is asymptomatic. Lab testing shows hypoglycemia, lactic acidosis, hyperuricemia, hyperlipidemia and elevated AST and ALT. Which type of disorder do you suspect? A. B. C. D. E. Glycogen storage disorder Mitochondrial disorder Lysosomal storage disorder Peroxisomal disorder Wilson’s Disease Glycogen Storage Disorders Peroxisomal Disorders and Lipoprotein Disorders Lysosomal Disorders Mitochondrial Disorders Hypoglycemia with hepatomegaly suggests a GSD. Often present when an infant begins sleeping through the night…which results in “prolonged fasting.” Helpful labs include… Glucose Uric acid Lactic acis LFTs Lipids A liver biopsy is often necessary, but DNA testing is becoming more readily available Glycogen Glycogen is a storage form of glucose: •Liver glycogen releases glucose into the circulation •Muscle glycogen is used locally Lactic acidosis pyruvate Glucose – 1- P gluconeogenesis Glucose – 6- P Glucose cytoplasm Glut 2 plasma glucose Acety l CoA Malonyl CoA glycolysis Pentose phosphate shunt (hyperuricemia) Krebs cycle Stimulates fatty acid synthesis and inhibits fatty acid breakdown (Hyperlipidemia) ER Glucose-6phosphatase GSD types 1a and 1b Autosomal recessive deficiency of hepatic glucose-6-phosphatase Clinical manifestations Hypoglycemia with lactic acidosis during fasting Distended abdomen (due to large liver) Doll-like or cherubic face Poor growth Seizures secondary to low glucose Increased TG and cholesterol Treatment Infusion of glucose continuously, especially at night until 2 Cornstarch after 2yo Frequent meals and snacks Allopurinol if uric acid too high Liver transplant (LATER) Long-term complications Sub-optimal growth velocity and short stature in adulthood Delayed puberty in poorly-controlled disease Hepatomegaly, renomegaly Nephrocalcinosis Impaired platelet function…bleeding tendency and epistaxis Hepatic adenomas Intrahepatic hemorrhage Malignant transformation Osteoporosis Rickets and anemia PCOS Deficiency in lysosomal breakdown of glycogen Clinical manifestations Infant 1 month or younger Normal at birth but becomes floppy Hypotonia with muscles “hard” on exam Failure to thrive Large liver and macroglossia Progressive cardiomyopathy Eventually, death is due to respiratory failure Glycogen Storage Disorders Peroxisomal Disorders and Lipoprotein Disorders Lysosomal Disorders Mitochondrial Disorders Peroxisomes: organelles involved in the betaoxidation of VLCFA Peroxisomal biogenesis disorders Zellweger Sydrome = most severe High forehead, flat occiput, Large anterior fontanelle Epicanthal folds, Broad nasal bridge, anteverted nostrils Micrognathia Brain defects, seizures, severe intellectual disability Liver disease, adrenal insufficiency, renal abnormalities Death in first year of life Refsum Disease, Adrenoleukodystrophy Mutations of individual enzymes Familial hypercholesterolemia Autosomal dominant Deficiency of LDL receptors Xanthomas after age 10 years NOT associated with obesity Congenital lipodystrophy Adipose tissue resistant to insulin Newborn that is thin AND long Farber’s disease Skin nodules and painful joints noted in 1st week of life Cherry red spot on retina Wolman Disease Due to defective lipoprotein metabolism Triglyceride and cholesterol esters deposited in body tissues Serum levels are normal! FTT, hepatosplenomegaly Calcified and enlarged adrenal gland Autosomal recessive disorder of copper transport Accumulation of copper in tissues, mainly the liver and brain Low serum ceruloplasmin Also affects the eyes and the kidney Visual deficits do NOT occur Clinical manifestations Liver deterioration Jaundice Hepatomegaly Neurologic deterioration Diagnosed by liver biopsy! X-linked recessive disorder of copper transport Copper is poorly distributed to cells in the body Accumulates in some tissues…small intestine and kidney Brain and other tissues have unusually LOW levels Low serum copper and ceruloplasmin Clinical manifestations Usually manifests in infancy Brittle, colorless, twisted hairs Seizures, dev’t delay, MR, hypotonia A. B. C. D. E. Your patient had apparently normal development for the first 6 months of life but begins to slow down. She was able to sit unassisted by 1 year. She was very socially interactive and could grasp objects. She gradually lost her ability to sit and grasp objects, became less interactive, lost interest in eating, and became emaciated. She had splenomegaly. Ophthalmology exam revealed a cherry red spot macula. What family of disorders do you suspect MOST? Glycogen storage disorder Mitochondrial disorder Lysosomal storage disorder Organic acidemia Urea cycle defect Glycogen Storage Disorders Peroxisomal Disorders and Lipoprotein Disorders Lysosomal Disorders Mitochondrial Disorders Lysosomes are organelles that degrade complex molecules to their building blocks. Deficient enzymes result in the accumulation and storage of intermediates. 3 different groups depending on type of glycoprotein involved Mucopolysaccharidoses Oligosaccharidoses Sphingolipidoses • Labs • • • • Urine mucopolysaccharides Urine oligosaccharide Enzyme assay DNA (for genetic counseling and to rule out pseudoalleles) A. B. A 14 month old female presented with developmental delay to your clinic. She was reportedly normal at birth but at 8 months was noted to have mild kyphosis when sitting. Late in infancy, the parents noticed gradual changes in craniofacial features including thickening of the eyebrows, large tongue, prominence of forehead. The patient hand been pulling to stand but lost this ability and seemed to be regressing in overall development. On exam, you notice a scaphocephalic head shape, frontal bossing, relatively thick eyebrows, cloudy cornea and stiff elbows. Which type of lysosomal disorder is MOST likely? Mucopolysaccharidoses Sphingolipidoses Normal at birth but progressive in nature. Neurologic involvement Regression and loss of milestones Intellectual disabilities Hepatosplenomegaly Skeletal manifestations Coarsened facial features Joint involvement Short stature Dysostosis multiplex…due to accumulation of glycosaminoglycans in the chondrocytes Diagnosed by enzyme assay LABS Hurler’s (MPS 1) Reduced alpha-Liduronidase in WBC Hunter’s (MPS 2) Reduced iduronate sulfatase activity in WBC Sanfilippo (MPS 3) Increased urine heparin sulfate Morquio (MPS 4) I-cell Disease Coarse Facies + + Cloudy cornea + NO minimal + + + Intellectual difficulties Other features severe Autosomal recessive; hirsutism, HSM, progressive deafness + X-linked recessive, HSM, short stature, joint contractures, “pebbly” skin, progressive deafness + Autosomal recessive; no organomegaly; later onset NO + skeletal involvement severe Neonate, clubfeet, hernias, hip dislocation AKA Lipid Storage Diseases Clinical features that are present in different diseases Hepatosplenomegaly Normal development followed by neurologic regression Demyelination Normal urine glycosaminoglycans and oligosaccharides Cherry red spot on ophthalmologic exam Clinical findings Organomegaly Bone pain Easy bruisability Short stature Thrombocytopenia X-ray Osteosclerosis and lytic lesions Infantile Gaucher Disease Decreased beta glucosidase activity Child in 1st or 2nd year of life with proressive HSM and CNS deterioration Autosomal recessive deficiency of hexosaminidase A, which breaks down GM2 ganglioside High incidence in Ashkenazi Jews (1/30 is carrier) Presentation Normal development through the first 3-6 months Development of nonspecific signs like lethargy and hypotonia, mild weakness, myotonic jerks Continued deterioration including blindness and seizures Death by age 5 Exaggerated startle reflex to loud noise Reduced visual attentiveness and abnormal eye movements Macrocephaly Cherry red spot on retina Management is primarily supportive Adequate nutrition Airway protection Controlling seizures Fabry Disease Orange-colored skin lesions Opacities of the eye Vascular disease of the kidney, heart, and brain Krabbe Disease Demyelination Disorder Progressive neurologic degeneration Death by age 2 years Niemann-Pick Cherry red spot CNS deterioration Hepatosplenomegaly* A. B. C. D. E. Which of the following classes of disorders is mostly MATERNALLY inherited? Glycogen storage diseases Urea cycle defects Amino acid disorders Mitochondrial disorders Sphinogolipidoses Glycogen Storage Disorders Peroxisomal Disorders and Lipoprotein Disorders Lysosomal Disorders Mitochondrial Disorders Mitochondria are involved in Fatty acid oxidation, including carnitine transport, urea cycle, citric acid cycle *Energy production pathway of oxidative phosphorylation Mitochondria have their own DNA that encodes proteins; all are derived from the ovum, so these disorders are MATERNALLY inherited!! Mitochondria harboring mutations are less able to produce energy Clinic symptoms become apparent when energy production is low Brain, liver, and kidney are more susceptible to disease Symptoms of hypoglycemia usually within the first hours/days of life Poor feeding Lethargy Jitteriness Hypotonia Macrosomia Height, weight, HC typically 95% or greater Due to stimulation of IGF1 receptors Persistent hypoglycemia Inappropriately high insulin and C-peptide in the setting of hypoglycemia NO ketonuria Insulin decreases lipolysis and ketogenesis Refractory hypoglycemia is major clue to diagnosis Despite oral feedings glucose remains low IV dextrose required but may NOT always work Glucagon can sometimes correct the low glucose Diazoxide Decreases insulin secretion Stimulates cortisol release Often the “drug of choice” for refractory hypoglycemia*