RED CELL DISORDERS
advertisement

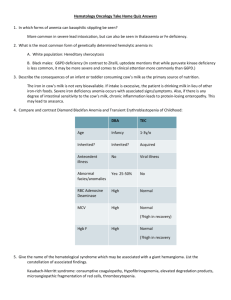
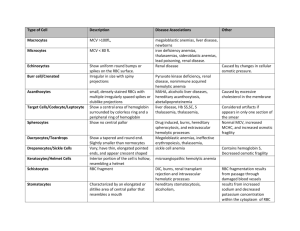
RED CELL DISORDERS. PERIPHERAL BLOOD. ERYTHROCYTES (RED BLOOD CELLS) Peripheral blood smears are important for assessing anemia. Normal erythrocytes are nonnucleated biconcave disks with a uniform diameter of 6-8 μm. Normal red blood cells are round, have a small area of central pallor, and show only a slight variation in size. The erythrocyte cytoplasm contains hemoglobin, a protein complexed to an iron containing porphyrin that gives the cell its characteristic red color. Hemoglobin is vital to oxygen transport in the blood—the main function of the erythrocyte. As the relative amount of hemoglobin in the red cell decreases or increases, the area of central pallor will decrease or increase accordingly. Erythrocytes are extremely pliable cells, able to change shape as they squeeze through the microcirculation. Erythrocytes have a life span in the peripheral blood of approximately 120 days. They are derived from normoblasts in the bone marrow, which lose their nucleus in the final stage of development prior to release into the peripheral blood. The examination of peripheral blood smear is very impotent for identification of disorders hematological system. The peripheral blood is composed of cells suspended in plasma present in table 1. Table 1 Normal Values for Peripheral Blood Elements. Data Units Men Women 14.0Hemoglobin (Hb) g/dL 12.0-16.0 18.0 Hematocrit (HCT, packed cell volume; PCV) % 32-50 37-47 2 3 Mean corpuscular volume (MCV) per mm 76–96 Erythrocyte count (red blood cells [RBCs]) ×106/mm3 4.6–6.0 4.2–5.4 6 Reticulocyte count % 0.5-1.5 Mean cell hemoglobin (MCH)3 pg 27-33 4 Mean cell hemoglobin concentration (MCHC) g/dL (%) 32-35 5 Color index (CI) 0,9-1,0 7 RBC distribution width (RDW) 1 Normal values vary with age; adult values are given here. 2 MCV = PCV / Red cell count (in millions /mm3). It is a measure of the size of individual red cells. 3 MCH = Hemoglobin (in g/dl)x 10/ Red cell count (in millions /mm3) It is content (mass) of Hb per RBC. 4 MCHC = Hemoglobin (in g/dl)/PCV. the average concentration of Hb in a given volume of packed RBCs,. 5 CI= Нв (in g/dL)/RBCs count in millions/mm3) * 0,3 6 1%=10‰ 7 RDW - RBC distribution width is the coefficient of variation of RBC volume. Erythropoiesis is the production of red blood cells (RBCs) in the bone marrow and is dependent on the release of erythropoietin from the kidneys. Stimuli for erythropoietin (EPO) release: hypoxemia, severe anemia, left-shift of oxygen -binding curve, high altitude EPO is synthesized. Increased O2 content suppresses EPO release (e.g., polycythemia vera) in peritubular capillaries. EPO accelerates erythropoiesis in the bone marrow by stimulating the erythroid stem cell to divide. The pathological forms of red blood cells. The smear may show pathological forms of red blood cells. Abnormal forms for peripheral blood erythrocytes found out in peripheral blood smear can be divided into degenerative and regenerative. Degenerative forms: The anisocytosis. It is variation in cell size. In peripheral blood smear may be normocytes, macrocytes, microcytes, megalocytes. The normocytes are normal red blood cells 6-8 µm in diameter. The size of cell may be more then in normal or less then in normal. The macrocytes are large red cells, 8,1-9,35 µm in diameter, with elevated MCV. The microcytes are red cells, 5,7-6,9 µm in diameter. RDW reflects anisocytosis in the peripheral blood. Diametr of megalocytes is 10-15 µm. Significant anisocytosis is observed at severe anemias. The poikilocytosis. It is variation in cell shape. There are Spherocytes, Elliptocytes, Acanthocytosis (Irregular speculation), sickle-shaped, Teardrop cells and so on in peripheral blood smear. Hypochromemia reflects the small maintenance of hemoglobin in erythrocytes. Anisochromemia this various maintenance of hemoglobin in erythrocytes in peripheral blood smear caused by coagulation of hemoglobin, that conducts changes to non-uniform distribution of hemoglobin in erythrocyte and to non-uniform colouring. Hemoglobin Ehrlich's degeneration is presented by more intensively painted granules, clumps, bodies. Heinz bodies are the dark inclusions at the periphery of the red cell. (A special stain is required because the routine Wright stain used on blood smears does not stain Heinz bodies.) Schistocytes are red blood cell fragments that result from membrane damage encountered during passage through vessels. Nucleated red blood cells, (normoblasts, macroblasts, megaloblasts), represent the stages of a red blood cell before it matures. They are not normally found in the peripheral blood. The average size of the normoblast is 7-12 µm in diameter. The cytoplasm is pink. The nucleus is pyknotic (a homogeneous blue-black mass with no structure). Megaloblasts may appear in peripheral blood at megaloblastic type of hemopoiesis. Regenerative forms Regenerative forms it is unmaturation forms which are found out in blood at anemias include Nucleated red blood cells and red blood cells with regenerative structures of nuclear and cytoplasmic nature (Howell-Jolly bodies, Cabo’s rings, basophilic stippling). The polychromatophilic cells The term "polychromasia" refers to the various tinge of these cells. These red cells have been recently released from the bone marrow and still contain significant amounts of RNA, which is the reason for the bluish tinge (remember, RNA stains blue with the Wright's stain). The reticulocytes. Many of polychromatophilic cells prove to be reticulocytes when stained with supravital stains such as brilliant cresyl blue. In this procedure, RNA is precipitated inside the erythrocytes. Because precipitated RNA gives the reticulated appearance shown, the cell is called a reticulocyte. For all practical purposes, reticulocytes and polychromatophilic red cells represent red cells of similar age. These cells remain as reticulocytes in circulation for about 24 hours. At any given time, 0.5-1.5% (5-15‰) of erythrocytes in the peripheral blood are newly released (eg, reticulocytes). Reticulocyte count is a measure of effective erythropoiesis and is corrected for the degree of anemia. Because these cells have been recently released from the marrow, it stands to reason that the number of reticulocytes would reflect the ability of the marrow to respond to the challenges of anemia. In other words, if a person is anemic and has an elevated reticulocyte count, the likely explanation for his anemia is hemolysis. That is to say, the red cells are being destroyed outside the bone marrow. Howell-Jolly bodies are spherical blue-black inclusions of red blood cells seen on Wright-stained smears. They are nuclear fragments of condensed DNA normally removed by the spleen Cabo’s rings the rests of nuclear membrane. The basophilic stippling is basophilic staining inclusions, which is thought to represent aggregates of polyribosomes. ANEMIA Anemia is by far the most common disorder affecting erythrocytes. It is defined as a reduction in the hemoglobin concentration of the blood (with reference to an established normal range for age, sex, and geography) or/and its qualitative disorder, usually associated with a reduction of total circulating red cell mass. It is results in decreasing of the oxygen-carrying capacity of the blood and causes clinical symptoms and signs. Anemias can be classified according to - cell volume (MCV): microcytic, normocytic, or macrocytic) - the ratio of Hb concentration/erythrocyte count (MCH and Сolor index): hypochromic, normochromic, hyperchromic - the type of hemopoiesis – erythroblastic, megaloblastic - the function of bone marrow. The efficiency of the entire erythropoiesis can be measured by determining the number of reticulocytes. Increases in the reticulocyte count provide a reliable measure of the red cell production response to anemia. If the number of reticulocytes is reduced, one must assume an abnormality of cell formation. 1. aregenerative (0 - 10 ‰ reticulocytes) 2. hyporegenerative (10-20 ‰ reticulocytes) 3. regenerative (20-100 ‰ reticulocytes) 4. heperregenerative - more than 100 ‰ reticulocytes 5. disregenerative at megaloblastic type of hemopoiesis Pathogenetic division of the anemias reflects the individual steps of erythropoiesis as well as the life-span of the erythrocytes circulating in blood: decreased production of erythrocytes by the marrow, blood loss too extensive for replacement by the marrow, increased rate of destruction of erythrocytes (more detailed see below) Pathogenetic classification of anemia is based on underlying mechanisms: I. Decreased erythropoiesis II. Increased rate of erythrocyte destruction or hemolytic anemia A. Hereditary intrinsic abnormalities of red cells include 1. Red cell membrane disorder (Hereditary spherocytosis, elliptocytosis) 2. Red cell enzyme deficiencies (Glucose-6-phosphatase deficiency) 3. Disorders of hemoglobin synthesis (sickle-cell anemia, thalassemia) B. Extrinsic abnormalities of red blood cells may be induced by the following: 1. Antibodies (Autoimmune hemolytic anemias associated with warm or cold antibodies, hemolytic blood transfusion reactions, hemolytic disease of the newborn) 2. Mechanical trauma 3. Infections, such as malaria 4. Chemical injury 5. Sequestration in mononuclear phagocyte system III. Blood loss 1. Chronic 2. Acute Clinical signs and symptoms (tachycardia, shortness of breath, and systolic murmurs) may develop secondary to these compensatory processes. If anemia is sufficiently severe (i.e., hemoglobin levels below 7 g/dL), tissue hypoxia is uncompensated. Anemia is characterized by pallor of the skin and mucous membranes and by manifestations of hypoxia (decreased oxygen content of the blood), most commonly weakness, fatigue, lethargy, dizziness, or syncope (fainting). Myocardial hypoxia may result in anginal pain. A hyperdynamic circulation, with an increase in heart rate and stroke volume, occurs in response to hypoxia. Ejection-type flow murmurs may develop, and if the anemia is severe, cardiac failure may ensue. When a patient presents with symptoms of anemia the initial diagnosis may be confirmed by demonstration of a decreased hemoglobin level. Morphologic classification of the anemia is done by examination of blood values (mean corpuscular volume (MCV), mean corpuscular hemoglobin concentration (MCHC)) and a peripheral blood smear. Identification of the cause of anemia may require bone marrow examination, hemoglobin electrophoresis, and determination of serum iron, vitamin B12, and folate levels. The tests required will vary with different patients, eg, bone marrow examination is critical in macrocytic anemia and of limited value in microcytic-hypochromic anemia. I. Anemias Due to Decreased erythropoiesis There are many mechanisms by which production of erythrocytes in the marrow is decreased. Because there is no mechanism to decrease the rate of peripheral destruction of erythrocytes, anemia results. Disorders of erythropoiesis may occur as a result of 1) lack or absence of differentiation of pluripotent, hemopoietic stem cells (aplastic anemia in panmyelopathy or replacement of the bone marrow at leukemia, carcinoma); 2) transient (viral infection) or chronic reduction of only the erythrocytic precursor cells (isolated aplastic anemia) due to autoantibodies against erythropoietin or against membrane proteins of the precursor cells; 3) erythropoietin deficiency in renal failure (renal anemia); 4) chronic inflammation or tumors that can activate, among others, erythropoiesis-inhibiting interleukins (secondary anemia); 5) abnormal cell differentiation result from defective DNA synthesis (megaloblastic anemias), which in addition to gene defectsmay mainly be due to a deficiency in folic acid or vitamin B12; 6) abnormal Hb synthesis (microcytic hypochromic at Iron deficiency, anemia of chronic disease;). Aplastic anemia Aplastic anemia is the result of failure of production, suppression, or destruction of stem cells in the bone marrow, which leads to decreased generation of erythrocytes, leukocytes, and platelets (pancytopenia). The bone marrow shows marked decrease in cellularity. Etiology The cases of aplastic anemia Primary (idiopathic): No known cause Secondary (toxic) Drugs: cytotoxic anticancer drugs Rare idiosyncratic reactions: antiepileptic drugs, oral antidiabetic agents, tranquilizers, antirheumatic drugs, antibacterial agent. hypersensitivity: Immune reaction; requires prior exposure; many drugs (antibodies to drugs often detectable) Industrial and household chemicals: Benzene, trinitrotoluene (TNT), some organic solvents, glue sniffing, certain insecticides (DDT, chlordane, lindane). Radiation: Either external beam or radioactive isotopes Associated diseases: Familial hypoplastic anemia (Fanconi; chromosomal abnormalities and a high incidence of leukemia) With infective hepatitis With pancreatitis With paroxysmal nocturnal hemoglobinuria The pathogenesis of aplastic anemia is not fully understood. Two major mechanisms have been invoked: an immunologically mediated suppression and an intrinsic abnormality of stem cells. In about 70% of cases, marrow failure results from inhibition of stem cell proliferation and differentiation by activated T-cells. It is postulated that at first the stem cells are antigenically altered by exposure to drugs, infectious agents, or other unindentified environmental insults. Cytokines such as interferongamma and TNF-alpha produced by activated T-cells are potent inhibitors of stem cell functions. Pathology and Clinical Features Aplastic anemia shows a markedly hypocellular bone marrow with a reduction of all cell lines. The peripheral blood smear shows the following characteristic features: Erythrocytes are decreased in number, but those that remain are normocytic or macrocytic and normochromic. There is an absence of reticulocytes in the peripheral blood. Granulocytopenia (neutropenia) occurs rapidly because these cells have a very short life span; infections and oral ulcers result. Thrombocytopenia results in a bleeding tendency. Compensatory phenomena include (1) the development of active marrow in sites outside the marrow cavity (extramedullary hematopoiesis or myeloid metaplasia, especially in spleen and liver) and (2) release of cells from the marrow before maturation is complete. The latter results in a shift to the left (immature white cells, including myeloblasts and myelocytes) and the presence of nucleated red cells (normoblasts) in the peripheral blood—hence the designation leukoerythroblastic anemia. The blood picture is characteristic Megaloblastic Anemias Megaloblastic anemias are a subset of macrocytic anemias in which the maturation phase of erythropoiesis in the bone marrow is abnormal, resulting in erythroid precursors that are enlarged and show failure of nuclear maturation (megaloblasts). Etiology Megaloblastic anemias result from conditions in which nucleic acid synthesis is abnormal, as in vitamin B12 and folic acid deficiencies. Vitamin B12 and folic acid play roles as cofactors in the conversion of deoxyuridine to deoxythymidine, an essential step in the synthesis of DNA. Note that vitamin B12 reservoirs in the liver are normally sufficient for several years; following gastrectomy (which removes the source of intrinsic factor, thereby reducing vitamin B12 absorption), a decade may pass before vitamin B12 megaloblastic anemia becomes apparent. Causes of Megaloblastic Anemia. Vitamin B12 deficiency Inadequate dietary intake: very rare; only in strict vegetarians, Failure of absorption due to intrinsic factor deficiency1, Pernicious anemia, Total and subtotal gastrectomy, Terminal ileal disease1, Crohn's disease, Strictures and fistulas that bypass the terminal ileum, Surgical removal of the terminal ileum, Competition for vitamin B12 by intestinal microorganisms, Bacterial overgrowth (blind loop syndromes), Diphyllobothrium latum (fish tapeworm) infection, Drugs: paraaminosalicylic acid (antituberculous agent), Congenital deficiency of transcobalamin II (the vitamin B12 transport protein in blood) Folic acid deficiency - Inadequate intake, Chronic alcoholism, Malnutrition, Failure of absorption2, Tropical sprue, Other malabsorptive states, Increased demand, Pregnancy and infancy, States of increased DNA synthesis (malignant neoplasms with high rate of cell turnover, erythroid hyperplasia in congenital hemolytic anemias), Drugs with folic acid antagonistic activity, Anticancer drugs such as methotrexate, Anticonvulsants such as hydantoins Other causes - Arsenic poisoning, Nitrous oxide inhalation, Some forms of chemotherapy (in addition to folic acid antagonists) Orotic aciduria (a rare condition with abnormal synthesis of purines and pyrimidines) Vitamin B12 is present in high concentration in animal liver and to some degree in most meats but is absent in plants. Folate is widely distributed, especially in leafy green vegetables. Dietary deficiency of vitamin B12 is rare except in strict vegetarians. Dietary deficiency of folic acid is common and occurs in many states of malnutrition. Pathology Red Cell Changes When DNA synthesis is abnormal, erythropoiesis changes from normoblastic to megaloblastic. Megaloblasts differ from normoblasts in that they are larger and show delayed nuclear maturation but normal cytoplasmic hemoglobinization (nuclear-cytoplasmic asynchrony). The late megaloblast, for example, shows a primitive nucleus and fully hemoglobinized cytoplasm—in contrast with the late normoblast, which has a pyknotic nucleus. Delayed maturation leads to accumulation of erythrocyte precursor cells. The bone marrow is hypercellular and contains large numbers of early megaloblasts; prior to the discovery of the beneficial effect of liver extract (contains vitamin B12) on pernicious anemia, this picture was mistaken for that of acute leukemia. As a result of intramedullary hemolysis or ineffective erythropoiesis, many megaloblasts undergo destruction in the bone marrow before maturation, aggravating the anemia and producing mild elevation of serum bilirubin and lactate dehydrogenase (LDH isoenzymes 1 and 2). The peripheral blood smear shows macrocytosis (large red cells with elevated MCV) and marked variation in size (anisocytosis) and shape (poikilocytosis). Oval forms (macro-ovalocytes) are prominent, and Howell-Jolly bodies, consisting of nuclear debris, are occasionally seen. Megaloblastic anemias are therefore macrocytic anemias if morphologic classification is used. Note that not all macrocytic anemias are megaloblastic. Neutrophil Changes The neutrophils are also affected. Neutrophil precursors in the bone marrow show marked enlargement; giant metamyelocytes are characteristic. In the peripheral blood, neutrophils show hypersegmented nuclei, with many cells showing more than 5 nuclear lobes. Changes in Other Cells in the Body 1 1 Vitamin B12 absorption depends both on formation of a complex with intrinsic factor and on absorption of this complex in the terminal ileum. 2 Folate is absorbed throughout the small intestine; it does not require an intrinsic factor. The abnormality in DNA synthesis affects many other cells in the body, notably those that have a high rate of cell turnover. These include the intestinal mucosa and other epithelia, which show cell enlargement and nuclear abnormalities. Recognition of these changes is important contextually if cytologic studies are undertaken—eg, in uterine cervical smears, the nuclear changes of folate deficiency may resemble those of dysplasia. Clinical Features and Diagnosis Patients with megaloblastic anemia present with symptoms of severe anemia. Megaloblastic anemia should be suspected upon finding macrocytic anemia with hypersegmented neutrophils in the peripheral blood. Bone marrow examination is necessary for confirmation and shows megaloblastic erythropoiesis. Establishment of the precise cause of megaloblastic anemia requires further clinical examination and laboratory testing There are two principal forms of megaloblastic anemia: folate deficiency, which has several underlying causes; and vitamin B12 deficiency, again with several causes, including pernicious anemia. (The pernicious anemia is an autoimmune disease, caused by immunologic destruction of the gastric mucosa. This process is associated with failure of secretion of acid and intrinsic factor.) The megaloblastic anemia of folate deficiency is identical to that of vitamin B12 deficiency but anemias of B12 deficiency is characterized by neurologic changes related to involvement of posterior and lateral columns including sensory ataxia and other disorders of sensation. The hematologic manifestations of folic acid deficiency are the same as those of B12 deficiency, but the neurologic changes do not occur. The distinction between folic acid and vitamin B12 deficiency can also be established by measuring serum levels of these compounds. Note that administration of folic acid may thus mask (partially correct) the anemia of vitamin B12 deficiency, and vice versa; however, folic acid administration does not correct the neurologic effects of B12 deficiency. For this reason, it is important to exclude vitamin B12 deficiency before treating megaloblastic anemia with folate. Iron Deficiency Anemia Deficiency of iron is probably the most common nutritional disorder in the world. An iron deficiency may result from dietary lack, impaired absorption, increased requirement, or chronic blood loss. Iron-deficient anemia with microcytic, hypochromic red blood cell indicates a prolonged period of negative iron balance and anemia severe enough to stimulate the production of poorly hemoglobinized cells. Depletion of iron-containing enzymes may cause koilonychia, alopecia, atrophic changes in the tongue and gastric mucosa, and intestinal malabsorption. These changes are seen in patients with severe and long-standing iron deficiency. The clinical manifestations related to the anemia are nonspecific. Both hemoglobin and hematocrit are depressed, usually to moderate levels, and are associated with hypochromia, microcytosis and some poikilocytosis. The serum iron and serum ferritin are low, while the transferrin concentration is high. With iron deficiency, the levels of cell-bound transferrin receptors and their soluble forms that circulate in the blood are elevated. II Hemolytic Anemias Anemias that are associated with a decreased RBC life span are termed hemolytic anemias. Shortened survival may be caused by either inherent (intracorpuscular) RBC defects, which are usually inherited, or external (extracorpuscular) factors, which are usually acquired. Before discussing the various disorders individually, we will describe certain general features of hemolytic anemias. All are characterized by (1) an increased rate of RBC destruction; (2) a compensatory increase in erythropoiesis that results in reticulocytosis; and (3) the retention by the body of the products of RBC destruction, including iron. Because the iron is conserved and recycled readily, RBC regeneration can keep pace with the hemolysis. Consequently, these anemias are almost invariably associated with a marked erythroid hyperplasia within the marrow and an increased reticulocyte count in peripheral blood. If the anemia is severe, extramedullary hematopoiesis may develop in the spleen, liver, and lymph nodes. Destruction of RBCs may occur within the vascular compartment (intravascular hemolysis) or within the cells of the mononuclear phagocyte, or reticuloendothelial (RE), system (extravascular hemolysis). Intravascular hemolysis occurs when RBCs are subjected to mechanical trauma or damaged by a variety of biochemical or physical agents (e.g., fixation of complement, exposure to clostridial toxins, or heat). Regardless of cause, intravascular hemolysis results in hemoglobinemia, hemoglobinuria, and hemosiderinuria. Conversion of the heme pigment to bilirubin may give rise to unconjugated hyperbilirubinemia and jaundice. Massive intravascular hemolysis sometimes leads to acute tubular necrosis. Levels of serum haptoglobin, a protein that binds free Hb, are characteristically low. Extravascular hemolysis, the more common mode of RBC destruction, takes place largely within the phagocytic cells of the spleen and liver. The mononuclear phagocyte system removes erythrocytes from the circulation whenever RBCs are injured or immunologically altered. Because extreme alterations of shape are necessary for RBCs to successfully navigate the splenic sinusoids, reduced deformability makes this passage difficult and leads to splenic sequestration, followed by phagocytosis. This is believed to be an important factor in the pathogenesis of RBC destruction in a variety of hemolytic anemias. Extravascular hemolysis is not associated with hemoglobinemia and hemoglobinuria, but it may result in jaundice and, if of long standing, in the formation of bilirubin-rich gallstones (so-called pigment stones). Serum haptoglobin is always decreased, because some Hb invariably escapes into the plasma. In most forms of hemolytic anemia, there is reactive hyperplasia of the mononuclear system, which results in splenomegaly. Because the pathways for the excretion of excess iron are limited, there is a tendency in hemolytic anemias for abnormal amounts of iron to accumulate, giving rise to systemic hemosiderosis or, in very severe cases, secondary hemochromatosis. Hemolytic anemia may be acquired or inherited (see classification) Hereditary spherocytosis Hereditary spherocytosis results from abnormalities of cytoskeletal proteins such as spectrin, ankyrin and others. In general, patients with dominant hereditary spherocytosis have only mild deficiency and mild to moderate hemolysis. Cytoskeletal defect renders erythrocyte spheroidal, less deformable, and vulnerable to splenic destruction. The major clinical features of this disorder are anemia, splenomegaly and jaundice. Jaundice is due to an increased concentration of unconjugated bilirubin in plasma. Hemolytic disease due to erythrocyte enzyme defects Hemolytic disease due to erythrocyte enzyme defects: glucose-6-phosphate dehydrogenase deficiency. Abnormalities in the hexose monophosphate shunt or in the glutathione metabolism resulting from deficient or impaired enzyme function reduce the ability of red cells to protect themselves against oxidative injuries and lead to hemolytic disorders. The most important is a hereditary deficiency of glucose-6-phosphate dehydrogenase (G-6-PD). G-6-PD reduces NADP to NADPH which is used to convert oxidized glutathion to the reduced form. Exposure to oxidants causes oxidation of the sulfhydryl groups of the globin chains. This leads to denaturation of hemoglobin and formation of precipitates with following membrane damage and intravascular hemolysis. Sickle Cell Disease The abnormal HbS gene. Sickle cell trait confers some protection on the erythrocyte against infection with Plasmodium falciparum; this selective advantage is believed to have favored the persistence of the abnormal HbS gene in malaria-endemic areas. Sickle cell disease represents the homozygous (S/S) state. Pathology A single point mutation dictates replacement of the normal glutamic acid at position 6 in the beta chain with valine. The result is HbS. This amino acid substitution is on the surface of the molecule and results in a tendency to polymerization, yielding semisolid crystalline structures called tactoids, under conditions of decreased oxygen tension. The degree of anoxia required to induce tactoids is small in homozygous (S/S) patients (sickle cell disease), in whom the red cells contain up to 80% HbS, and greater in heterozygous (A/S) patients (sickle cell trait), in whom the red cells contain about 30% HbS and 70% HbA. Tactoid formation causes (1) decreased solubility of hemoglobin, (2) change in shape of the erythrocyte to a sickle cell, and (3) decreased deformability of the erythrocyte. Change in shape is due to interaction between the tactoids and the spectrin-actin cytoskeleton. Initially, sickling reverses when oxygenation improves, but with repeated anoxia, the erythrocyte assumes a permanently sickled shape. Decreased deformability leads to phagocytosis in the splenic and liver sinusoids. Clinical Features The onset of sickle cell disease is in early infancy, as HbS replaces HbF, and death often occurred during early adult life. With improvements in management, many patients now survive longer. Patients present with evidence of chronic extravascular hemolysis and severe anemia. Growth retardation is common, as is heart failure (high-output type). Mild hemolytic jaundice with absent urinary bilirubin and increased fecal and urinary urobilinogen are usual. The bone marrow shows marked compensatory normoblastic hyperplasia, often leading to expansion of the marrow cavity in bones and causing bony deformities (tower skull and hair-on-end appearance on skull x-rays). Chronic leg ulcers that fail to heal are a typical feature of sickle cell disease but are of unknown pathogenesis. Diagnosis Sickle cells in peripheral blood smears are diagnostic but not always present. Addition of metabisulfite to the blood smears induces sickling (metabisulfite sickle preparation). Patients with sickle cell disease have over 80% HbS in the blood with absent HbA; Hemolytic Disease of the Newborn Hemolytic disease of the fetus and newborn is a normocytic, normochromic anemia seen in an Rhpositive fetus or infant born to an Rh-negative mother who has previously been exposed to Rh-positive blood, and has therefore developed antibodies to the Rh antigen. The development of antibodies usually occurs only after multiple maternal exposures to the antigen; these may occur during previous pregnancies, abortions, or miscarriages, or during amniocentesis. The maternal antibodies, usually IgG, are transferred to the fetus through the placenta and attack fetal red blood cells, leading to excessive red cell lysis and anemia. If the condition is mild, the maternal circulation effectively eliminates the waste products of hemoglobin metabolism, including bilirubin, for the fetus, and it suffers few ill effects in utero. Occasionally, maternal destruction of the fetal cells may be excessive, leading to a severe anemia and hydrops fetalis, a fatal condition characterized by massive edema and heart failure. After delivery in the less affected infant, clinical signs of anemia may occur. More significant in the neonatal period is the development of severe jaundice, as the breakdown products of hemoglobin are ineffectively cleared by the infant's immature liver. A dramatic elevation in bilirubin can lead to a significant neurologic disorder, called kernicterus, as the unconjugated bilirubin precipitates out in the infant's brainstem, causing brain damage. Hemolytic disease of the newborn in response to Rh incompatibility is uncommon and has become rarer with fewer pregnancies experienced by each woman and important prophylactic interventions. More common than Rh incompatibility is ABO incompatibility. In this condition, maternal antibodies are produced as a result of ABO incompatibility, even during a first pregnancy. The presence of antibodies against the A or B antigens seldom leads to full-blown newborn hemolytic disease. Clinical Manifestations Mild hemolytic disease may be relatively asymptomatic, with slight hepatomegaly and minimally elevated bilirubin. Moderate and severe diseases manifest with pronounced signs of anemia. Hyperbilirubinemia, resulting from excessive red cell lysis, may occur, leading to jaundice. Complications Kernicterus. Severe anemia may cause heart failure. Hydrops fetalis. Affected fetuses often abort spontaneously at approximately 17 weeks' gestation. In one study, 10% of school-aged children who had received in utero transfusions for severe Rh incompatibility showed neurologic abnormalities, most likely related to asphyxia and anemia at birth. Transfusion Reaction A transfusion reaction is an immune-mediated destruction of incompatible red blood cells received in a blood transfusion. Transfusion reactions against donated white blood cells occur more frequently, but are typically mild. Although host and donor blood antigens are always identified (typed) for ABO and Rh compatibility before a transfusion is given, an error in red blood cell typing or a mix-up in the blood supplies may occur. Transfusion reactions may also develop as a result of an immune reaction to bacteria transferred in contaminated blood products. Clinical Manifestations Immediate, life-threatening reactions occur with ABO incompatibility. Manifestations include immediate flushing of the face, a feeling of warmth in the vein receiving the blood, fever and chills, Chest, flank, or low back pain, abdominal pain with nausea and vomiting, decreased blood pressure with increased heart rate, dyspnea (a sensation of breathing difficulty), transfusion reactions against white blood cells are milder and usually include fever and occasionally chills. Complications Renal failure may result from red blood cell casts and hemoglobin obstruction of the nephrons. Malaria It has been estimated that 200 million persons suffer from this infectious disease; it is one of the most widespread afflictions of humans. Malaria is endemic in Asia and Africa, but with widespread jet travel, cases are now reported all over the world. In malaria, red cell lysis occurs episodically upon release of proliferating merozoites from infected red cells. Intermittent fever coincides with hemolysis every 48 hours (tertian fever—Plasmodium vivax, Plasmodium falciparum) or every 72 hours (quartan fever—Plasmodium malariae). Red cell debris is cleared by the reticuloendothelial system, and splenomegaly is common. Complications include blackwater fever, glomerulonephritis, and cerebral malaria. In most cases of malaria, the malarial parasite can be identified within red cells in peripheral blood smears, and the different species of plasmodia can be identified by their morphologic features. III Anemia due to blood loss Acute Blood Loss The acute bleeding phase. Immediately following an acute bleeding, the hematocrit is normal because there has not yet been time for hemodilution. In the acute bleeding phase, blood values— including red cell count, hemoglobin, and hematocrit—are normal because equivalent amounts of red cells, hemoglobin, and plasma are lost. The hemodilution phase. With acute blood loss, water shifts from the interstitial to the intravascular compartment causing hemodilution. Within hours, the effect of this dilution is seen as a progressive decrease in red cell count, hemoglobin, and hematocrit in the peripheral blood. The amount of decrease of these values depends on the amount and rate of blood loss and the effectiveness of renal compensation. Soon after acute blood loss, the red blood cells appear normal in size and color. The regenerative phase. Reduction in the oxygenation of kidneys triggers the production of erithropoietin with the following stimulation of erythropoiesis. During this regenerative phase, the bone marrow shows erythroid hyperplasia and depletion of iron stores, and the peripheral blood shows a reticulocytosis proportionate to the increased rate of erythrocyte production. Reticulocytes begin to increase in number in the first two days. However, it takes 3 to 6 days for erythroid hyperplasia to appear and 7 to 10 days before the erythropoietic response is maximal, producing reticulocyte counts up to 20 to 30%. The anemia is therefore temporary. The body's iron stores are replenished over the next few months. In patients who have depleted iron stores and have a marginal dietary intake of iron, an episode of acute hemorrhage may precipitate iron deficiency anemia (hypochromia, microcytosis). Chronic Blood Loss Chronic bleeding is compensated for initially by erythroid hyperplasia of the bone marrow and increased production of erythrocytes. This persists until iron stores have been depleted, at which time iron deficiency prevents adequate compensation. Anemia due to chronic blood loss is therefore a form of iron deficiency anemia. POLYCYTHEMIA Polycythemia, or erythrocytosis, as it is sometimes referred to, denotes an increased concentration of RBCs, usually with a corresponding increase in Hb level. Such an increase may be relative, when there is hemoconcentration caused by decreased plasma volume, or absolute, when there is an increase in total RBC mass. Relative polycythemia results from any cause of dehydration, such as deprivation of water, prolonged vomiting, diarrhea, or excessive use of diuretics. Absolute polycythemia is said to be primary when the increase in RBC mass results from an autonomous proliferation of the myeloid stem cells and secondary when the RBC progenitors are normal but proliferate in response to increased levels of erythropoietin. Primary polycythemia (polycythemia vera) is one of several expressions of clonal, neoplastic proliferation of myeloid stem cells and is therefore considered later in chapter with other myeloproliferative disorders. Secondary polycythemias may be caused by an increase in erythropoietin secretion that is physiologically appropriate or by an inappropriate (pathologic) secretion of erythropoietin. PATHOPHYSIOLOGIC CLASSIFICATION OF POLYCYTHEMIA. 1. Relative Reduced plasma volume (hemoconcentration) 2. Absolute 1) Primary: Abnormal proliferation of myeloid stem cells, normal or low erythropoietin levels (polycythemia vera); inherited activating mutations in the erythropoietin receptor (rare) 2) Secondary: Increased erythropoietin levels - Appropriate: lung disease, high-altitude living, cyanotic heart disease - Inappropriate: erythropoietin-secreting tumors (e.g., renal cell carcinoma, hepatoma, cerebellar hemangioblastoma); surreptitious erythropoietin use (e.g., in endurance athletes)