20 questions on heme on midterm majority on sickle cell anemia
advertisement

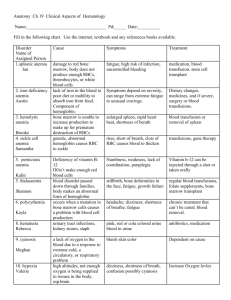
Chapter 26: Hematological System, Page 1 of 19 20 Heme questions on midterm majority on sickle cell anemia and leukemia. NORMAL VALUES (RANGE) RBC: 3.7-6.3 Hgb: 11-22 WBC: 5-20 Plts: 200,000-260,000 Hct: 54-40 RBCs Average life cycle is 120 days. Bone marrow is continually producing new cells. ANEMIA Decrease in the total number of RBCs and or Hgb for age/sex. Occurs due to the following factors: ↓ RBC production; or ↑ RBC destruction. As a result of this decrease, the oxygen carrying capacity of the blood is altered and decreased oxygen is available for tissue perfusion. When Hgb falls enough to cause symptoms, the symptoms are due to tissue hypoxia but cyanosis does not occur. Cyanosis is a result of respiratory distress. Cardiac failure (CHF) will occur when Hgb reaches 2.5-5. Children have a high tolerance for very low Hgb, but a gradual fall is better tolerated; the more rapid the drop, the more severe the symptoms. CLINICAL MANIFESTATIONS OF ANEMIA Depends on the severity of the anemia and can include: Pallor Pale waxy skin (a waxy pallor is seen with severe anemia) Evidence of CHF (with severe anemia) Pancotopenia (petichae, purpura) Bleeding (guaiac + stool and gastocult) Muscle weakness Fatigue o Frequent resting o Shortness of breath o Poor sucking in infants Tachycardia on exertion Headache, dizziness Irritability Slow thought process, decreased attention span Apathy Depression Chapter 26: Hematological System, Page 2 of 19 SICKLE CELL ANEMIA Autosomal recessive disorder Defect in hemoglobin molecule Cells become sickle shaped and rigid RBCs lose the ability to adapt shape to surroundings. Sickling may be triggered by fever and emotional or physical stress SICKLE CELL TRAIT A child with sickle cell trait has inherited only one abnormal HgS gene, o only 45% of cells are affected, the rest are normal Hgb. These kids are usually asymptomatic with a low Hgb. These kids need to have genetic counseling when they are old enough to think about marriage and having children. DIAGNOSIS OF SICKLE CELL Almost all diagnoses are made from the newborn screening test or at birth. In a high-risk child blood tests are done immediately after birth. PATHOLOGY OF SICKLE CELL ANEMIA Hemoglobin in the RBC develops a sickle or crescent shape; these cells are rigid and obstruct capillary blood flow, leading to congestion and tissue hypoxia. Clinically this hypoxia causes additional sickling and extensive infarctions. HgS changes to a sickled form in conditions of: o low oxygenation (e.g. high altitudes, small un-pressurized airplanes) o dehydration o hypoxia o acidosis; and o fever When deoxygenated (above conditions) resolve, the cell resumes its normal round shape. This repeated sickling shortens the RBCs’ survival and results in a chronic hemolytic anemia. Sickle sell anemia manifests as a chronic, hemolytic anemia in which sudden, severe and life threating complications are caused by acute intravascular sickling of RBCs with severe pain and organ dysfunction. CLINICAL MANIFESTATIONS - THE CHILD WITH SICKLE CELL ANEMIA Children with sickle cell anemia start exhibiting symptoms as young as 4 months of age when adult Hgb is replaced by HgS (Hemoglobin Sickle). o At this time they start having splenic dysfunction (RBCs getting stuck in capillaries of spleen). This sickling within the spleen results in infarction of splenic tissue; by age 2-4 the spleen becomes unable to filter microorganisms from the blood; the high microorganism count in the blood promotes the release of a large amount of phagocyte cells. Their spleen is essentially non-functional o This functional asplenia leaves children effectively immunocompromised, very susceptible to overwhelming infections (s. pneumonia, hep b, influenza) and ↓ tolerance to fevers Any child with fever must be evaluated IMMEDIATELY This chronic well compensated anemia with increased hemolytic causes jaundice, pallor, cardiomyopathy, hemosideosis (increased storage of iron in liver, spleen, bone marrow, kidneys, lymph nodes). Bone marrow: increased production of RBCs Chapter 26: Hematological System, Page 3 of 19 SICKLED RBCs Sickled cells cause microvascular obstruction (vaso-occlusive crisis), which leads to tissue ischemia and infarction. Hypoxia, acidosis, hypo- or hyperthermia, and dehydration add to the sickling. This repeated process causes the complications of sickle cell anemia. BODY SYSTEMS AFFECTED BY SICKLE CELL ANEMIA Brain: CVA – paralysis, death Eyes: Retinopathy – blindness Lungs: Pneumonia Abdomen: Pain, hepatomegaly, splenomegaly Skeletal: Joint pain, bone pain, osteomyelitis Skin: Chronic ulcers, poor wound healing SICKLE CELL CRISIS ** We must know all of these ** Vaso-occlusive crisis: Most common type of sickle cell anemia crisis; can occur in any organ. Mild-severe pain, low grade fever. Usually occurs in the long bones of arms/legs but can occur anywhere - VERY Painful Hand-foot syndrome: Causes infarction of bones in hands/feet with soft tissue swelling. Takes a few weeks to resolve, most common in ages 6 months – 2 years. Acute chest syndrome: Clinically presents as pneumonia. Develops over several hours with chest pain, congestion, wet cough, tachypnea, tachycardia, temp >102º, decreased breath sounds, rales. Believed to be caused by the sickling of cells in the small blood vessels in the lungs; can cause severe hypoxia. Will cause chronic lung damage as the child reaches adulthood. Splenic sequestration crisis: Occurs between 2 months – 5 years. Severe, RAPID decrease in Hgb secondary to pooling of child’s RBCs and sickling within the spleen and liver. Will result in shock (hypovolemic) and cardiovascular collapse. The rupture of the spleen will cause massive hemorrhage and rapid death SPLENIC SEQUESTRATION Life-threatening / death within hours Pooling of blood in the spleen Signs include profound anemia, hypovolemia, and shock Abdominal distention, pallor, dyspnea, tachycardia, and hypotension To prevent splenic sequestration most children with sickle cell anemia have an elective splenectomy at an early age. This practice varies from center to center. A child who has had a splenectomy will need to be on prophylactic antibiotic therapy for life Chapter 26: Hematological System, Page 4 of 19 TREATMENT FOR SICKLE CELL ANEMIA CRISIS Oxygen o Do not give 100% oxygen; will cause cells to sickle faster. Give 50-60%. o O2 during a crisis will prevent further sickling of cells but will not reverse cells already sickled in crisis as it cannot reach those cells. o Prolonged O2 therapy will depress bone marrow production and make anemia worse. Hydration Pain management: PCA pump- Morphine sulfate IV. Transfusions of P(acked)RBCs Bed rest during acute phase, HEAT to affected areas, and passive ROM exercises. Routine RBC transfusions to maintain Hgb >10mg and suppress overproduction by bone marrow. Splenectomy: ↑ lifespan of RBCs and ↓ need for transfusions. CONSIDERATIONS IN SICKLE CELL ANEMIA Delayed puberty and physical growth (but adult height and sexual maturation will happen) Congestion in glomerular capillaries leads to kidney necrosis, scarring hematuria, enuresis, inability to concentrate urine. From chronic hypoxia—weakening of bones and osteomyelitis are common; aseptic necrosis of the femoral head is very common. CNS infarction resulting in CVA. Sickle cell anemia is the leading cause of stroke in children Chelating therapy with deferoxamine/Desferal (an iron chelating agent) to prevent accumulation of iron in tissues/organs, administer with RBC transfusions. Frequent assessment: parents must be taught how to expertly manage disease and when child needs hospitalization. In the child with a spleen, continually measure abdomen for splenic enlargement. IRON DEFICIENCY ANEMIA Most common type of anemia in children. The main causes are: Increased demands for RBC production (rapid growth) Poor nutritional intake Common in the 9-24 month old child as they transition between iron fortified formula and “regular” food Common cause of childhood anemia is excessive milk intake at expense of other iron-rich foods. Children only need two glasses (16 oz) of milk per day. Less if they are eating cheese or yogurt. TREATMENT OF IRON DEFICIENCY ANEMIA In mild to moderate anemia a trial of iron (Fe) 3-6mg/kg/qd for 6 months Empty stomach (best time at bedtime with Vit C to aid in absorption) Do not give with milk Stool color change to tarry black Teach parents sources of iron: spinach, raisins, organ meats, molasses, dark green leafy vegetables, Total™ cereal Chapter 26: Hematological System, Page 5 of 19 APLASTIC [BONE MARROW] ANEMIA Aplastic anemia is a deficiency of the blood cells that results from the failure of the bone marrow to produce adequate numbers of circulating blood cells. It may be congenital or acquired. RBCs, WBCs and platelets are all simultaneously depressed (pancytopenia), resulting in profound anemia, leukopenia, and thrombocytopenia. Thought to be an autoimmune disorder. Congenital Aplastic anemia is known as Fanconi’s anemia. It is a rare autosomal recessive disorder consisting of multiple congenital anomalies: o Patchy brown skin discoloration due to melanin deposition o Microcephaly o Horse shoe shaped kidney o Absent thumbs Acquired Aplastic anemia in children is either idiopathic or from a drug reaction. Common causes include: o Chemotherapeutic drugs: ionizing radiation o Antibiotics: chlorinphenicol, sulfa drugs, cytotoxic drugs, tetracycline, NSAIDs, cimitedine o Industrial and household products: benzene solvents, lead, insecticides o Viral infectious process: HPV, Hep A/B/C, EBV, or overwhelming infection CLINICAL MANIFESTATIONS OF APLASTIC ANEMIA Depends on the degree of bone marrow failure Weakness Purpura (bleeding into tissue) Petichae (pinpoint lesions) Bleeding from MM Epistaxis Fatigue Pallor LABORATORY RESULTS IN APLASTIC ANEMIA Leukopenia Marked neutropenia Thrombocytopenia Very low Hgb and HcT Onset is usually insidious; bone marrow aspiration reveals yellow fatty deposits in place of bone marrow. Predominance of lymphocytes, plasma cells and mast cells TREATMENT OF APLASTIC ANEMIA Therapy is directed at: 1. Immunosuppressive therapy (ALG, ATG) to remove the immunologic functions at work; and 2. Replacement of the bone marrow. Bone marrow transplant should be done ASAP after diagnosis if suitable donor is available Immunosuppressant agents: ALG (Anti-lymphocyte globulin) or ATG (anti-thymocyte globulin) suppresses T-cell autoimmune response. Colony-stimulating factors to enhance bone marrow production Cyclosporine (anti-rejection drug) may be used in non-responders as well as high dose steroids and chemotherapy. Disease has an extremely poor prognosis and a very high mortality Chapter 26: Hematological System, Page 6 of 19 DISSEMINATED INTRAVASCULAR COAGULATION (DIC) Normal homeostasis is a delicate balance between bleeding and clotting, in DIC this homeostasis is altered and the patient is simultaneously bleeding and clotting. Intravascular activation of both the clotting and bleeding factor lead to abnormal bleeding and excessive clot formation in the small vessels throughout the body. This process, if not reversed, leads to multisystem organ failure and death. DIC is not a primary disorder but occurs as a result of a variety of alterations in health. Some causes of DIC are: o burns o cancer o tissue hypoxia o asphyxia o heat stroke o snake bites o transfusion reaction o ARDS o liver disease o shock o trauma o viral illnesses o sepsis o congenital infections can occur after child birth (from hypovolemic shock caused by excessive bleeding) ASSESSMENT OF THE PATIENT IN DIC The most obvious clinical feature of DIC is bleeding. Renal involvement = hematuria, oliguria, and anuria. Pulmonary involvement = hemoptysis, tachypnea, dyspnea and chest pain. Cutaneous involvement = petechiae, ecchymosis, jaundice, acrocyanosis and gangrene. MEDICAL & NURSING TREATMENT OF DIC PROMPT RECOGNITION and reversal of the condition that is causing the DIC, hypoxia, acidosis, perfusion abnormalities. If DIC still persists, transfusion of FFP (fresh frozen plasma) and platelets. There is significant mortality associated with DIC β- THALASSEMIA The Thalassemias are a group of inherited blood disorders of defective Hgb synthesis characterized by anemia that can be mild or severe. β –Thalassemia (also known as Cooley’s anemia) is the most common form. Most commonly occur in people of Mediterranean descent but are also found in Middle Eastern Asian, Asian (mainly Chinese), and African populations. Caused by deficiency in the synthesis of the β-polypeptide chain of the Hbg molecule. Severe hemolytic anemia occurs. To compensate the bone marrow increase production of RBCs unless suppressed with RBC transfusions The breakdown products of hemoglobin will accumulate in the blood causing jaundice and be excreted in the urine causing the urine to become dark brown in color. Chapter 26: Hematological System, Page 7 of 19 TYPES OF THALASSEMIA Thalassemia minor Produces mild anemia Thalassemia Intermedia Produces severe anemia Produces severe anemia and is incompatible with life Thalassemia Major without continued RBC transfusion's PATHOLOGY OF THALASSEMIA Anemia is SEVERE and bone marrow cannot compensate Child goes into CHF and has significant growth failure Bone marrow produces large numbers of immature cells in an attempt to compensate. These cells have a very short life span and are very thin and abnormally shaped. Aplastic crisis can occur after any infection (even 1 day of infection can suppress bone marrow) and without WBCs, sepsis may result. Splenomegaly: is usually associated with increased workload (such as in hemolytic anemias), which suggests that it is a response to hyperfunction. It is therefore not surprising that splenomegaly is associated with any disease process that involves abnormal red blood cells being destroyed in the spleen. The spleen becomes so large it interferes with other abdominal organs and respiratory Chronic hypoxia produces headache, bone pain from expansion of bone marrow cavities with pathologic fractures Listlessness Anorexia Hemosiderosis from continued transfusions (increased iron store in liver, heart, lymph tissue, spleen, bone marrow, kidneys) Retarded physical growth and sexual maturation; endocrine system is very sensitive to iron toxicity and secondary sexual characteristics are delayed. CLINICAL MANIFESTATIONS OF THALASSEMIA Folic acid deficiency Frequent epistaxis (nosebleeds) Osteoporosis Retarded and delayed growth/ Delayed sexual maturation Susceptible to pathologic fracture Chronic CHF from myocardial fibrosis (from excessive iron) Murmurs Fibrotic pancreas (often resulting in DM) Darkening of skin Facial deformities: o enlarged head o Prominent forehead due to frontal and parietal bone bossing o Prominent cheek bones o Broadened and depressed bridge on nose o Enlarged maxilla with protruding front teeth o Eyes: Mongolian slant, epicantheal folds TREATMENT OF THALASSEMIA Child is transfusion dependant from end of first year of life (sometimes earlier) Transfused q3 weeks with 10-20mL/kg of leukocyte-depleted PRBCs to suppress bone marrow Frequent transfusions decrease cardiac involvement and splenomegaly, increases metabolic expenditures allowing normal activity. Also allows bone to heal and grow normally Chapter 26: Hematological System, Page 8 of 19 Splenectomy is considered when transfusion requirement exceed 250mL/kg/year Bone marrow transplantation has been used in infants with some success It is a progressive, fatal disease with death occurring in the early-mid 20’s. NURSING INTERVENTIONS Assist child and family in dealing with a fatal, chronic illness that requires extensive treatment and frequent hospitalizations. ITP – IDIOPATHIC THROMBOCYTOPENIC PURPURA Idiopathic = cause is unknown Thrombocytopenic = blood does not have enough platelets Purpura = excessive bleeding / bruising ITP - PATHOLOGY Immune reaction to platelets; antibodies see platelets as bacteria and work to eliminate them. Antibodies bind to the platelets resulting in the autoimmune destruction of platelets in the spleen. Other than thrombocytopenia, other blood counts are normal. Preceded by a viral illness o URI o Live vaccine: MMR, Varicella o Mononucleosis NURSING INTERVENTIONS IN THE CHILD WITH ITP No rectal temperatures Drug therapy o Glucocorticoids to prevent further platelet destruction. o Immunoglobulin to prevent further platelet destruction. o Don’t administer any form of aspirin or ibuprofen (NSAID) for pain. o Acetaminophen (Tylenol) for pain relief. Dip all urine and guaiac all stool (for blood) Closely monitor neurological status Closely monitor for any s/s of bleeding (mm, sclera, epistaxis, hematuria, blood in stool) CLINICAL MANIFESTATIONS OF ITP Children typically present 1-4 weeks after a history of a viral illness (chicken pox, measles, rubella) or immunization with abrupt onset petichae and ecchymosis and bleeding from mucous membranes. Bleeding is severe after trauma. Severe internal bleeding is rare but can occur. CVA can occur when platelet count is less than 10,000. TREATMENT OF ITP Approximately 80% of cases of acute ITP resolve spontaneously within 6 months, some relapse/become chronic. Clinically significant bleeding or severe thrombocytopenia (<20,000) is treated with high dose steroids, IVIG, and platelet transfusions. Steroids and IVIG will decrease the duration of severe thrombocytopenia and decrease the rate of antibody coated platelets but does not diminish the production of antiplatelet antibodies. Does not affect the long term course of the diseases process. Chapter 26: Hematological System, Page 9 of 19 Chronic ITP (6-12 months of chronic thrombocytopenia) is treated with IVIG and or splenectomy. Repeated treatments with IVIG have proven to delay splenectomy. A splenectomy will produce remission in 70-80% of cases of chronic ITP. HEMOPHILIA Hereditary X linked bleeding disorders in affected males and carrier females. A daughter who inherits the gene from her father has a 50% chance of passing the defective gene on to her sons; 1/3 of hemophiliacs have no family history of the disorder. It results from a deficiency in one of the specific factors required to coagulate blood. There are 2 types: 1. Hemophilia A: caused by a deficiency in factor 8 and accounts for 80% of all hemophilia (about 1/5000 male births) 2. Hemophilia B: Known as Christmas disease is caused by a deficiency in factor 9. Factor 8 is produced by the liver and is necessary for the formation of thromboplastin, which is crucial in the formation of a normal fibrin clot. Severity of each individual’s disorder is determined by the degree of clotting factor deficiency. CLINICAL MANIFESTATIONS OF HEMOPHILIA Bleeding tendency = mild to severe. Children with 1% factor 8 deficiencies may have little to no bleeding with trauma and no spontaneous bleeding. Children with hemophilia usually do not manifest symptoms until they are at lease 6 months old (as they become mobile and incur minor injuries). The first manifestation may be with circumcision or from a slight fall or bruise. o Easy bruising o Epistaxis o Hematuria Hemorrhage into a bone or joint (hemarthrosis) is the most frequent form of internal bleeding, especially in the knees, elbows, and ankles, which results in bone changes and crippling deformities with loss of ROM after years of bleeding episodes. Bleeding in mouth, neck or thorax can compromise airway. Intracranial bleeding is a major cause of death in hemophiliacs. Bleeding around spinal cord may result in paralysis. Early s/s of a bleed may include tingling, stiffness, aching and decreased ROM followed by warmth, redness, swelling, and pain; also headache, slurred speech, loss of consciousness (from cerebral bleeding), and black tarry stools (from GI bleeding). PTT very prolonged, however bleeding time is normal because platelet function is normal. Hemophiliacs do not bleed faster; it just takes them longer to clot. MEDICAL TREATMENT OF HEMOPHILIA Primary prophylaxis w/ factor 8 concentrate on a regular basis before joint damage occurs. Secondary prophylaxis w/ factor 8 concentrate after child has first bleeding episode, given 3x week. IV infusion of factor 8 clotting factor concentrate immediately after a bleeding episode/injury lessens the complications. Parents are taught how to start IV and how to administer drug; child will self-administer when they are old enough. DDAVP (synthetic vasopressin) for mild hemophilia raises factor 8 levels; stimulates a glycoprotein needed for platelet adherence to a vascular clot in milder hemophilia. No aspirin. Acetaminophen or ibuprofen are effective in relieving pain r/t synovitis Chapter 26: Hematological System, Page 10 of 19 For active bleeds: Elevate extremity and immobilize. Apply pressure as with other types of active bleeding for 10-15 minutes Apply ice to area Follow with active ROM exercise to help absorb blood from joint to minimize and prevent bone and muscle changes that limit movement. RICE: Rest, Ice, Compression, Elevation. Control of weight helps lessen strain on joints, decreasing potential for bleeding episodes. NURSING INTERVENTIONS TO MINIMIZE INJURY TO THE CHILD WITH HEMOPHILIA The earlier a bleeding episode is recognized, the more effectively it can be treated. Prevent bleeding episodes by decreasing risk of injury. Make environment as safe as possible especially during infancy and during the toddler years. Use protective equipment / Advise school Child will not be able to play ANY sports with the potential for contact (football, lacrosse, baseball, wrestling, basketball, hockey, soccer); golf, tennis and swimming are permitted. Use soft toothbrush or sponge tipped brush to prevent oral bleeding. Child should wear medic-alert bracelet. Family should always have supply of Factor 8 concentrate and IV supplies with them wherever they go, and whenever they travel they should know the location of the nearest pediatric emergency room. Child and family need to be well educated to recognize situations where bleeding is a potential (dental work, immunizations etc) JOINT DYSFUNCTION IN HEMOPHILIA PEDIATRIC MALIGNANCIES 1% of all cancers Involves tissues of: o CNS, bone, muscle, endothelial tissue Rapid growth of cancers Chapter 26: Hematological System, Page 11 of 19 CLASSIFICATION OF TUMORS Embryonal tumor = arises from embryonic tissue Lymphomas = lymphatic tissue Leukemias = blood Sarcoma = seen in bone, cartilage, nerve and fat SURGERY The oldest form of cancer treatment Surgery plays important role in initial diagnosis: biopsy of primary tumor. Excision of tumor when possible Facilitating treatment: insertion of catheters for long-term treatment CARDINAL SIGNS OF CANCER Unusual mass or swelling Unexplained paleness and loss of energy Spontaneous bruising Prolonged, unexplained fever Headaches in morning; Vomiting in morning Sudden eye or vision changes Excessive – rapid weight loss. DIAGNOSTIC TESTS X-ray Skeletal survey CT scan Ultrasound MRI Bone marrow aspiration Biopsy: identify cell to determine type of treatment TREATMENT MODALITIES Determined by: Type of cancer Location Extent of disease CHEMOTHERAPY Primary treatment modality used to cure many pediatric cancers. Chemotherapy is the use of drugs to destroy cancer cells. The destruction is accomplished by inhibiting cells within the body to divide, which eventually leads to cell death. Can be given in addition to another form of therapy such as radiation or surgery (combination chemo). Drugs may be administered before surgery to reduce size of tumor. Chemotherapy is used after surgery or radiation therapy to prevent relapse. Chapter 26: Hematological System, Page 12 of 19 GOALS OF CHEMOTHERAPY Reducing the primary tumor size Destroying cancer cells Preventing metastases and microscopic spread of the disease ADMINISTRATION Chemotherapy can be given PO, sub-q, IM, IV, intrathecally, or rectally. o Oral route used if drug is well absorbed and non-irritating to the GI tract o Sub-q or IM: slow systemic release o IV push (administered by Oncologist, not a nurse), piggyback or intravenous infusion RADIATION THERAPY The use of ionizing radiation to break apart bonds within a cell causing cell damage and death. External beam therapy accounts for the majority of radiation treatments in children. Problems: radiation beams cannot distinguish between malignant cells and healthy cells. May lead to developmental delays in a young child. BONE MARROW TRANSPLANT Involves giving patients high doses of chemotherapy and / or radiation therapy to eradicate disease and then rescuing the patient with a source of stem cells. This wipes out their WBC/immune system and leaves them vulnerable to overwhelming sepsis. THREE TYPES OF BMT Allogeneic Donor – matched donor Autologous Donor – patient’s stored cells or identical twin Umbilical Cord Blood – just starting to be used Stem Cell transplant (?) GENE THERAPY Use of gene therapy in the treatment of childhood cancer is promising yet complex and still in early phases of clinical application. IMMUNOSUPPRESSION AND INFECTION Children with cancer become immune impaired from a number of causes: o Lymphocyte production is altered o Splenic dysfunction can prevent maturation of blood cells and alteration in inflammatory response. o Cancer therapy can decrease immunoglobulin concentrations. o Often children don’t die of cancer, they die of infection. NEUTROPENIA Significant neutropenia can develop during chemotherapy, creating an increased risk of infection in the child with cancer. Neutropenia occurs when the absolute neutrophil count decreases below 500. Neutrophils usually make up 50-70% of circulating white blood cells and serve as the primary defence against infections by destroying bacteria in the blood. Chapter 26: Hematological System, Page 13 of 19 CHILDHOOD LEUKEMIA Leukemia is the most common form of pediatric malignancy; it is cancer of the blood-forming bone marrow, characterized by the proliferation of immature, abnormal WBCs; peak incidence in the pediatric population is between 2-6 years of age. There are multiple types of leukemia but the most common in children are: ALL – Acute Lymphocytic Leukemia AML – Acute Myeloid Leukemia PROGNOSIS Initial WBC most significant The higher the count the poorer the outcomes - >100,000 WBC count = poor outcome - Age of child , < 2 or > 10 = poor prognosis Girls do better than boys ALL Has a much better prognosis then AML 25% of all pediatric cancers and 75% of all pediatric leukemias; more common in males and caucasians. ETIOLOGY OF LEUKEMIA The specific causes of leukemia are not well understood, some genetic factors are believed to play a role in some types (children with certain chromosomal disorders such as children with Down syndrome have an increased risk of ALL). Leukemia occurs when the stem cells in the bone marrow produce immature WBCs. These cells proliferate rapidly causing the bone marrow to fill with abnormal WBCs, these abnormal cells then leave the bone marrow and go into systemic circulation and replace the “normal” WBC’s. Protective functions of cellular and humeral immunity are reduced, leaving the body defenseless. The malignant WBCs rapidly fill the bone marrow, replacing stem cells that produce RBCs and platelets. The result is leukemia “clones”, which results in severe anemia. Leukemic cells demonstrate the same neoplastic properties as solid cancers or tumors. They invade highly vascular organs (spleen/liver). The main consequence of the Leukemic cells pushing out the other cells are: Anemia from decreased RBC Infection from neutropenia Decreased platelets = increased bleeding Invasion of bone marrow causes weakness of bone marrow/bones with an increase in fractures. SEVERE bone pain results. Spleen, liver, lymph glands show high degree of infiltration, enlargement and eventually fibrosis. Infiltration of CNS causes ↑ ICP, meningeal irritation Leukemic cells may invade testes, kidneys, prostrate, ovaries, GI tract and spinal column. Chapter 26: Hematological System, Page 14 of 19 PRESENTING S/S OF LEUKEMIA #1 large joint or bone pain Fever Pallor Bleeding Lethargy, malaise Anorexia Petichae, frank bleeding and bone pain are cardinal signs of bone marrow failure Hepatosplenomegaly (r/t infiltration) Lymphadenopathy (r/t infiltration) If cells invade the CNS: headache, vomiting, papilledema, 6th cranial nerve paralysis (unable to move eyes laterally) DIAGNOSIS OF LEUKEMIA Peripheral blood smear Bone marrow aspiration, analysis Lumbar puncture TREATMENT OF LEUKEMIA (ALL TYPES) Treatment is divided into 3 phases: 1. Remission Induction: begins on diagnosis. Massive dose of chemotherapeutic agents are used to kill all possible leukemic cells. Lasts 4-6 weeks. 2. Consolidation (or intensification) therapy: once remission is achieved, intense treatment to eradicate any residual cells and prevent emergence of any resistant clones. In this phase CNS prophylaxis is given (intrathecal [injection into the spinal canal] chemotherapy). 3. Maintenance therapy: to maintain remission achieved in induction and maintained phase, this can last up to 2 years Bone marrow transplant is an option in AML. BMT is also an option in ALL after the first relapse t is not used in ALL during the first treatment phase because prognosis is excellent with chemo alone. SURVIVAL RATES Age of child , < 2 or >10 = poor prognosis ALL better than AML ALL 75- 80% cure rate WBC count at diagnosis (the higher the count, the poorer the prognosis) Length of time to produce remission: the more rapid response = better outcome Chapter 26: Hematological System, Page 15 of 19 NURSING INTERVENTIONS FOR THE CHILD WITH LEUKEMIA Nursing care is directed towards: Prevention of infection o No flowers, fruits or vegetables permitted as gifts from visitors; they harbor bacteria and fungi; thick-skinned fruits such as bananas and oranges are safer. Managing the symptoms of Leukemia Managing the side effects of the therapy Controlling pain - Morphine Encouraging “normal” growth and development Assist parents, family, and child with diagnostic workup, repeated blood drawing, bone marrow aspirations, lumbar punctures. Continuous pain evaluations Administer pain medication PREVENT INFECTION: strict hand-washing, aseptic technique, private room, continual monitoring of temperature. Sepsis is a serious risk to children with leukemia with devastating consequence. Administration of platelets to prevent bleeding All blood products must be CMV-negative and leukocyte depleted. Nutrition – high protein, high calories, bland, soft diet – this is very difficult during treatment as chemotherapy causes nausea and vomiting Small, frequent meals – if you can not get nutritious foods into child, let them eat whatever they want. Maintain oral hydration Frequent mouth care to prevent infection of MM. Ulceration of GI tract is very common; medicated mouthwash, soft toothbrush. Stomatitis and rectal ulcers can be so severe that the child refuses to eat and must receive TPN. Meticulous skin care of perianal area, diarrhea can be severe. When home, child must be isolated from other children (siblings only when ill), home schooling Alopecia or hair loss from side effects of chemo (and in some cases radiation). Child may choose to wear wig, hats, or nothing at all. Side effects of high dose therapy are very disturbing to children (moon face, weight gain). Refer child and family to child life to help Refer child, family and siblings to appropriate spiritual and psychological help. Get Pediatric Oncology social worker involved with child and family Help family find appropriate financial resources Late effects of chemo and radiation therapy in survivors may not manifest until years after cure (intellectual defects, infertility, and growth retardation). CNS TUMORS Second most common malignancy 65% have 5 year survival rate Most common tumors: o Astrocytomas 50% o Medulloblastomas 25% o Brain stem gliomas 10% Chapter 26: Hematological System, Page 16 of 19 CLINICAL MANIFESTATIONS OF CNS TUMOR Classic signs and symptoms are indicative of ↑ ICP. Pressure is due to tumor mass compressing vital structure, blockage of cerebrospinal fluid flow or tumor associated edema. o Gait changes / ataxia o Headache with or without vomiting o Blurred vision, or diplopia o Forceful vomiting upon rising in the morning. MANAGEMENT Surgery if tumor accessible Chemotherapy (chemical substances) Radiation (ionizing rays) is reserved for patient older than 2 years of age Survival rate based on location NEUROBLASTOMA Approximately 600 new cases a year. Embryonic tumor Average age of diagnosis is 2 years. Poorest survival rate 50 to 60% have metastasis at time of diagnosis. Highly invasive HUGE tumors. CLINICAL MANIFESTATIONS Depends on site of tumor Diagnosis o CT scan o Bone scan TREATMENT Determined by the stage of disease and age of child. Children who have localized disease and complete response to treatment are more likely to achieve a disease free state and long-term survival. OSTEOGENIC SARCOMA Malignant tumor of bone Peak incidence is in the second decade of life, when adolescents are gaining height rapidly. Approximately 20% have metastasis at diagnosis High rate of metastasis to lungs OSTEOSARCOMA TUMOR Femur has a large mass involving the metaphysis of bone. Tumor has destroyed the cortex. TREATMENT Limb salvage Amputation Chemotherapy Chapter 26: Hematological System, Page 17 of 19 EWING SARCOMA Tumor of flat bones o Pelvic, chest, vertebrae, ribs, scapulae Rare in children under 5 years RHABDOMYOSARCOMA Most common soft bone tissue tumor o Head and neck 40% o GU 20% o Extremities 20% o Trunk 15% TREATMENT Surgical removal of tumor Chemo based on tissue biopsy Radiation RETINOBLASTOMA Intraocular / embyronic tumor 1 in 16,000 + family history High incidence of other malignancies Dx: pupil reflex “cat eyes” TREATMENT Surgical enucleation of eye Genetic counseling Follow-up care up to 18 Years HIV Mother-to-child transmission/Vertical transmission Mother-to-child transmission of HIV occurs through pregnancy, labor, delivery or breastfeeding Vertical transmission accounts for 95% of all pediatric HIV cases. An estimated 2 million children were at risk for HIV infection through mother-to-child transmission in 2003. Without preventative interventions approximately 35% of infants born to HIV-positive mothers contract the virus. Some 15-20% of infections occur in pregnancy 50% occur during labor and delivery. Breastfeeding accounts for 33% of infant infections. HIV-infected mothers should avoid all breastfeeding. There is a need for greater awareness of the facts that HIV can pass from an infected mother to her child, and that measures exist to reduce the risk of transmission. Chapter 26: Hematological System, Page 18 of 19 VERTICAL TRANSMISSION From mother to child in utero, intra partum, post partum through infected breast milk. The transmission rate from mother to child without retroviral treatment during pregnancy is 1530%. There are several risk factors which increase the risk of transmission from mother-child: o Increased viral load of mother o Low CD4 count (mother) o A vaginal birth does not increase risk INFANTS BORN TO A HIV+ MOTHER Maternal antibodies to HIV will persist in the infant for as long as 18 months. The HIV ELISA is a screening test for the diagnosis of HIV infection. If this test is positive, it must be confirmed with a second test called the Western Blot, which is more specific and will confirm if someone is truly HIV positive. However, early HIV infection often results in a negative test. PCR (polymerase chain reaction) test for viral DNA can diagnose 95% of infected children by 1 month of age. PHYSICAL MANIFESTATIONS OF HIV IN CHILDREN The most common presenting clinical manifestations in children are: Failure to thrive Multiple bacterial infections: URI, pneumonia, chronic OM/ sinusitis Chronic diarrhea Significant developmental delays (HIV in CNS) Other manifestations include: Lymphadenopathy Hepatosplenomegaly Oral canditis, esophagitis Parititis LIP (lymphoid interstitial pnemonitis) Opportunistic infections: o PCP, pnemocystis carini pneumonia o MAC, mycobacterium avium-intracellular complex HIV encephalopathy Severe herpes simplex infections CMV Wasting syndrome MEDICAL MANAGEMENT OF HIV The goal of therapy is to slow the growth of the virus (viral load), prevent opportunistic infections and promote normal growth and development. Treatment currently involves antiretroviral therapy and the use of protease inhibitors which interfere with the virus ability to replicate itself (see handout for specific medications). Children with HIV are on multiple antiretroviral and protease inhibitors to prevent mutation and replication of the virus, these various medications work at various stages in the lifecycle of the virus. Chapter 26: Hematological System, Page 19 of 19 BLOOD PRODUCT TRANSFUSIONS (PRBCs, PLATELETS, FFP, IVIG) Product must be typed and cross matched for each individual patient, Patient must have ID band on Product must be checked by 2 RNs prior to transfusion. Patient needs 2 sets of IV tubing: o One with blood product and institution appropriate filtering/tubing in place. o One line with NS piggy backed into line as close to entry site as possible. ALL CHILDREN who have any type of immunosuppression, e.g. HIV, cancer, or any other immunodeficiency need leukocyte depleted, CMV– blood products. NO EXCEPTIONS. Blood transfusion reactions are caused by a complex antigen/antibody reaction to a protein found on blood cells. The more transfusions a pt has had the more likely they are to have a reaction BUT it can happen with the first transfusion. TRANSFUSION/HEMOLYTIC REACTION Refers to an immune response against transfused blood products. Antigens on the surface of blood cells are recognized as “foreign proteins” and can stimulate lymphocytes to produce antibodies to the red blood cell antigens. o Patient reports “feeling funny” or flushed o Flank pain o Chills o Bloody urine o Rash o Low blood pressure o Dizziness, fainting o Wheezing any other s/s respiratory distress o Changes in VS (T/BP/RR/HR) NURSING INTERVENTIONS IN A TRANSFUSION REACTION 1. Stop the blood transfusion. - Assess pt / VS 2. Start normal saline infusion. - Assess pt / VS 3. Obtain blood sample and urine specimen. - Assess pt / VS 4. Call the MD - Assess pt / VS 5. Return blood to blood bank. - Assess pt / VS 6. Document /transfusion reaction document/incident report - Assess pt / VS