Carbon 42 (2004) 83–94
www.elsevier.com/locate/carbon
Adsorption of organic molecules from aqueous solutions
on carbon materials q
Carlos Moreno-Castilla
*
Departamento de Quımica Inorg
anica, Facultad de Ciencias, Universidad de Granada, Campus Fuentenueva, 18071 Granada, Spain
Received 22 July 2003; accepted 30 September 2003
Abstract
Adsorption of organic molecules from dilute aqueous solutions on carbon materials is a complex interplay between non-electrostatic and electrostatic interactions. Non-electrostatic interactions are essentially due to dispersion and hydrophobic interactions,
whereas the electrostatic or coulombic interactions appear with electrolytes when they are ionized at the experimental conditions
used. Both interactions depend on the characteristics of the adsorbent and the adsorptive and the solution chemistry. Among them
the carbon surface chemistry has a great influence on both electrostatic and non-electrostatic interactions, and can be considered one
of the main factors in the adsorption mechanism from dilute aqueous solutions. In this paper the current knowledge about the
fundamental factors that control the adsorption process from aqueous phase will be presented.
Ó 2003 Elsevier Ltd. All rights reserved.
Keywords: C. Adsorption; D. Adsorption properties; Surface oxygen complexes
1. Introduction
2. Adsorption from dilute solutions: generalities
Adsorption of organic solutes from the aqueous
phase is a very important application of powdered and
granular activated carbons. This covers a wide spectrum
of systems such as drinking water and waste water
treatments, and applications in the food, beverage,
pharmaceutical and chemical industries. Activated carbon adsorption has been cited by the US Environmental
Protection Agency as one of the best available environmental control technologies [1].
In spite of the large market for activated carbon, the
specific mechanisms by which the adsorption of many
compounds, especially organic compounds, takes place
on this adsorbent are still ambiguous. This is because
liquid-phase adsorption is a more complicated process
than gas or vapour-phase adsorption [2].
Thus, current knowledge about the fundamental
factors that control the adsorption process from the
aqueous phase will be presented, giving examples of the
different situations.
When studying adsorption from solutions on solids it
is convenient to differentiate between ‘‘adsorption from
dilute solution’’ and ‘‘adsorption from binary and
multicomponent mixtures covering the entire mole
fraction scale’’. To judge by the number of papers
published annually on adsorption from dilute solution,
this subject is more important than the adsorption from
binary mixtures. Therefore, reference will be made
hereafter to the adsorption from dilute aqueous solutions.
The main differences between adsorption from the gas
phase and liquid phase are as follows [3]:
q
Plenary lecture delivered at the International Conference on
Carbon, Carbon’03, held in Oviedo (Spain) July 6–10, 2003.
*
Tel.: +34-958-243-323; fax: +34-958-248-526.
E-mail address: cmoreno@ugr.es (C. Moreno-Castilla).
0008-6223/$ - see front matter Ó 2003 Elsevier Ltd. All rights reserved.
doi:10.1016/j.carbon.2003.09.022
(i) A solution is typically a system of more than one
component. In real cases, there are at least two substances that can adsorb. For a dilute solution, adsorption of one type of molecule (say A) involves
replacement of the other (B). Thus, adsorption
from solution is essentially an exchange process,
and hence molecules adsorb not only because they
are attracted by solids but also because the solution
may reject them. A typical illustration is that the attachment of hydrophobic molecules on hydrophobic adsorbents from aqueous solutions is mainly
C. Moreno-Castilla / Carbon 42 (2004) 83–94
driven by their dislike of water and not by their attraction to the surface.
(ii) Isotherms from solution may exhibit non-ideality,
not only because of lateral interactions between adsorbed molecules but also because of non-ideality in
the solution.
(iii) Multilayer adsorption from solution is less common than from the gas phase, because of the stronger screening interaction forces in condensed fluids.
Adsorption isotherms are normally developed to
evaluate the capacity of activated carbons for the adsorption of a particular molecule. They constitute the
first experimental information, which is generally used
as a tool to discriminate among different activated carbons and thereby choose the most appropriate one for a
particular application. The shape of the isotherms is the
first experimental tool to diagnose the nature of a specific adsorption phenomenon, and it is expedient to
classify the most common types phenomenologically [3].
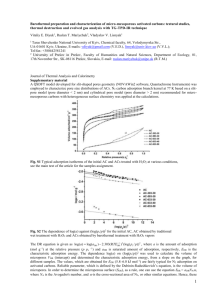
There are several types of isotherms but those mainly
found in carbon materials are the five depicted in Fig. 1.
Long-linear isotherms are not common in adsorption
on carbons but are found in the initial part of all isotherms on homogeneous surfaces. The Langmuir type
(L) frequently occurs, even when the premises of the
Langmuir theory are not satisfied. Type F, typical for
heterogeneous surfaces, is perhaps the most common.
High-affinity isotherms are characterised by a very steep
initial rise, followed by a pseudo-plateau. Sigmoidal
isotherms have been obtained with homogeneous surfaces such as the graphitised carbon blacks Graphon
and V3G.
Statistically, adsorption from dilute solutions is simple because the solvent can be interpreted as primitive,
that is to say as a structureless continuum [3]. Therefore,
all the equations derived from monolayer gas adsorption
remain valid after replacing pressure by concentration
and modifying the dimensions of some parameters.
Effluent concentration
84
C0
C0
Ci
Ci
1
*
Breakthrough volume
*2
Effluent volume treated
Fig. 2. Progression of the adsorption front through the adsorber bed.
The mass transfer zone is depicted in grey color.
Some of these equations, such as the Langmuir and
Dubinin–Astakhov, are widely used to determine the
adsorption capacity of activated carbons.
Batch equilibrium tests are often complemented by
dynamic column studies to determine system size requirements, contact time and carbon usage rates. These
parameters can be obtained from the breakthrough
curves. To study these curves it is usual to apply a
macro-approach method based on the mass transfer
zone concept (MTZ). The basic principles of this concept are depicted in Fig. 2. The MTZ is the region of the
bed between the already-saturated activated carbon and
the point where the concentration of the adsorptive in
the aqueous phase is at the maximum acceptable limit
in the effluent stream. Breakthrough is reached when the
leading edge of the zone advances to the end of the bed
(point 2 in Fig. 2). The height of this zone is a measure
of the adsorption efficiency of the bed. The lower the
height, the more efficient is the carbon bed. Under the
appropriate experimental conditions, this parameter is
independent of the bed depth and generally shows little
variation with in the initial concentration.
Adsorption measurements should preferably be amplified by microcalorimetry, such as immersion and flow
microcalorimetry. These techniques can give additional
information about the surface nature of the adsorbent
and the mode or mechanism of adsorption.
3. Factors that control the adsorption process
Adsorption is a spontaneous process that takes place
if the free energy of adsorption, DGads , is negative. There
is a wide range of energies contributing to the free energy of adsorption, which can be grouped into nonelectrostatic and electrostatic.
Fig. 1. Most common adsorption isotherms found from dilute aqueous solutions on carbon materials.
DGads ¼ DGnon-elect: þ DGelect:
C. Moreno-Castilla / Carbon 42 (2004) 83–94
Although at atomic level all ionic and molecular interactions can be interpreted as ‘‘electric’’, this term is
restricted to coulombic interactions and all other interactions are termed non-electrostatic, whatever their origin.
Electrostatic interactions appear when the adsorptive
is an electrolyte that is dissociated or protonated in
aqueous solution under the experimental conditions
used. These interactions, that can be either attractive or
repulsive, strongly depend on the charge densities for
both the carbon surface and the adsorptive molecule
and on the ionic strength of the solution, as we shall see
later. The non-electrostatic interactions are always attractive, and can include van der Waals forces, hydrophobic interactions and hydrogen bonding. Factors that
influence the adsorption process are the characteristics
of the adsorbent and adsorptive, the solution chemistry
and the adsorption temperature. These factors will now
be discussed.
3.1. Characteristics of the adsorbent
The characteristics of carbon that affect the adsorption process are the pore texture, surface chemistry and
mineral matter content. The adsorption capacity of
carbon materials is not related in a simple form with
their surface area and porosity. The adsorption capacity
will depend on the accessibility of the organic molecules
to the inner surface of the adsorbent, which depends on
their size. Thus, under appropriate experimental conditions, small molecules such as phenol can access
micropores, natural organic matter (NOM) can access
mesopores, and bacteria can have access to macropores.
Activated carbon fibres have received increasing attention in recent years as a better adsorbent than
granular activated carbons, because they normally
present much higher adsorption kinetics and adsorption
capacity (Fig. 3). Activated carbon fibres only have
micropores, which are directly accessible from the external surface of the fibre. Thus, adsorptive molecules
reach adsorption sites through micropores without the
85
additional diffusion resistance of macropores, which is
usually the rate-controlling step in the case of granular
adsorbents.
The surface chemistry of activated carbons essentially
depends on their heteroatom content, mainly on their
surface oxygen complex content. They determine the
charge of the surface, its hydrophobicity, and the electronic density of the graphene layers. Thus, when a solid
such as a carbon material is immersed in an aqueous
solution, it develops a surface charge that comes from
the dissociation of surface groups or the adsorption of
ions from solution [2]. This surface charge (Fig. 4) will
depend on the solution pH and the surface characteristics of the carbon. A negative charge results from the
dissociation of surface oxygen complexes of acid character such as carboxyl and phenolic groups. Therefore,
these surface acid sites are of Br€
onsted type. The origin
of the positive surface charge is more uncertain because,
in carbons without nitrogen functionalities, it can be due
to surface oxygen complexes of basic character like pyrones or chromenes, or to the existence of electron-rich
regions within the graphene layers acting as Lewis basic
centres, which accept protons from the aqueous solution.
An indication that surface basicity can be mainly due
to these electron-rich regions is that, the net enthalpy of
neutralization of the total basic sites decreases with the
oxygen content of the surface (Fig. 5), whereas the net
enthalpy of neutralization of total acid sites increases [4].
Results obtained with activated carbons of different
origins can be explained by the fact that oxygen complexes reduce the electronic density of the graphene
layers and consequently reduce the basicity of the carbon surface.
The surface charge can be determined by electrokinetics or titration methods. These are complementary
methods, especially in the case of granular porous
Xm (mg ads/g carbon)
25
Norit R 0.8
ACF
20
15
F400
10
5
0
0
2
4
6
8
t (days)
Fig. 3. Adsorption kinetics of aminetriazol on an activated carbon
fibre (ACF) and activated carbons (F400 and Norit R 0.8).
Fig. 4. Macroscopic representation of the features of carbon surface
chemistry. From [2].
86
C. Moreno-Castilla / Carbon 42 (2004) 83–94
Amount adsorbed
(mg/g)
Micropore volume (ml/g)
0.4
200
0.5
0.6
0.7
100
0
0
5
10
15
% Oxygen content
carbons [2]. The first method primarily measures the
surface charge at the more external surface of the particles whereas the second one provides a measure of the
total surface charge. The pH at which the external surface charge is zero is referred to as the isoelectric point,
pHIEP , while the total surface charge is zero at the point
of zero charge, pHPZC .
In addition, surface oxygen complexes also affect the
surface hydrophobicity, which determines the hydrophobic interaction. Hydrophobic interaction or, more
specifically, hydrophobic bonding describes the unusually strong attraction between hydrophobic molecules
and hydrophobic carbon surfaces. The surface hydrophobicity of carbon materials can be determined, for
instance, by measuring their contact angle with test
liquids of known surface tension, by inverse gas chromatography or by water adsorption.
Hydrophobic bonding occurs exclusively in aqueous
solution: in fact it mainly comes from the strong tendency of water molecules to associate with each other by
hydrogen bonding and from the characteristic structure
of water molecules. Hydrophobic bonding plays an
important role in interface and colloid science [5]. For
instance, the increasing adsorbability of aliphatic acid
molecules with increasing hydrocarbon length, also
known as Traube’s rule, is essentially due to the increasing hydrophobic effect [6].
From this it is derived that it is important to plot the
adsorption isotherms obtained for adsorptives with
different solubilities against the concentration relative to
that in saturated solution. This normalisation eliminates
the differences in hydrophobicity between the adsorptive
molecules, so that the normalised isotherms more truly
reflect the affinity for the surface [5,6].
In general, an increase in the oxygen content of carbon brings about a decrease in its hydrophobicity. Thus,
Pendleton et al. [7] showed (Fig. 6) that the adsorption
of dodecanoic acid on different activated carbons linearly decreased when the oxygen content of the carbo-
naceous adsorbent increased and that there was no
relation with the micropore volume of the adsorbent.
Water molecules are bound to the surface oxygen
complexes by H-bonds, reducing the accessibility of the
hydrophobic aliphatic chain of dodecanoic acid to the
hydrophobic parts of the carbon surface.
This model is consistent with that found for the decrease in methyl isoborneol (MIB) adsorption when the
oxygen content of carbons increased [8]. In this case, the
average enthalpy of displacement of water by MIB (Fig.
7) decreased when the concentration of hydrophilic sites
on the surface increased. This finding suggests that it is
more difficult for MIB molecules to displace water
molecules from carbon surfaces when their oxygen
content increases. Results obtained show that the enthalpy of displacement reaches a limiting value when the
number of hydrophilic sites is higher than about 0.7
mmol/g.
Finally, surface oxygen complexes also affect the
electronic density of the graphene layers [9]. This in turn
would affect the dispersion interactions between the
carbon surface and the adsorptive molecules. For instance, carboxyl groups fixed at the edges of the
graphene layers have the ability to withdraw electrons,
whereas phenolic groups release electrons.
-50
Enthalpy of displacement
(kJ/mol)
Fig. 5. Variation of the net enthalpy of neutralization DH (NaOH)net ,
D; and DH (HCl)net , j, with the total amount of oxygen on the surface.
From [4].
Fig. 6. Relationship between the maximum amount of dodecanoic
acid adsorbed on different carbons with their oxygen content and
micropore volume. Adapted from [7].
-40
-30
-20
-10
0
0
0.4
0.8
Hydrophilic sites (mmol/g)
1.2
Fig. 7. Enthalpy of displacement of water by 2-methylisoborneol
(MIB) versus hydrophilic sites on activated carbons. Adapted from [8].
C. Moreno-Castilla / Carbon 42 (2004) 83–94
AC
AC–OH
AC–COOH
HOMO (eV)
LUMO (eV)
)6.19
)5.85
)6.61
)1.35
)1.09
)1.92
Thus, Tamon and Okazaki [10] determined by a semiempirical quantum chemical method the HOMO and
LUMO levels of a model aromatic molecule such as a
non-substituted phenanthroperylene cluster (AC), and
the same cluster substituted with two phenolic groups
(AC–OH) and with two carboxyl groups (AC–COOH).
Results are shown in Table 1 and indicate that the
withdrawing groups, such as carboxyl groups, decrease
HOMO and LUMO levels with respect to the original
carbon cluster, whereas the donating groups increase
these levels.
Finally, the other characteristics of the adsorbent that
control the adsorption process is its mineral matter
content. This has in general a deleterious effect on the
adsorption because it can block the porosity of the
carbon matrix and can preferentially adsorb water due
to its hydrophilic character, in this case reducing the
adsorption of the adsorptive.
3.2. Characteristics of the adsorptive
Among the characteristics of the adsorptive that
mainly influence the adsorption process are its molecular size, solubility, pKa and the nature of the substituent
if they are aromatic. The molecular size controls the
accessibility to the pores of the carbon and the solubility
determines the hydrophobic interactions. The pKa controls the dissociation of the adsorptive if it is an electrolyte. This parameter is closely related to the solution
pH and its importance will be discussed later on.
The substituent of the aromatic ring of the adsorptive
molecules, as in the case of the substituent of the
graphene layers, can withdraw or release electrons from
it, which would affect the dispersion interactions between the aromatic ring of the adsorbate and the
graphene layers of the adsorbent.
To date, the adsorption of phenolic compounds on
carbon materials and the effect of surface oxygen complexes on phenol uptake have been the most studied
adsorption processes [2]. Thus, it has long been known
that the increase in surface acidity of activated carbons,
produced after their oxidation, brings about a decrease
in the amount of phenol adsorbed from diluted aqueous
solutions. To show this phenomenon (Table 2) sample A
was oxidized to increase its surface acidity. Heat treatment of the oxidized sample progressively decreased the
Table 2
Surface area and surface acidity of activated carbons with different
degrees of oxidation. From [11]
Sample A
N2 surface
area (m2 /g)
Surface acidity
(meq/g)
As received
As received oxidized
Oxidized and heat treated
at (°C):
300
400
500
700
950
975
503
0.12
5.93
603
606
681
640
675
4.79
2.74
1.60
0.34
Nil
surface acidity due to the removal of mainly carboxyl
and phenolic groups [11]. A sample heat treated at 950
°C had no acidity and its surface area was lower than
that of as-received sample. Fig. 8 shows the adsorption
isotherms of phenol on the above carbons. There was a
large decrease in phenol uptake after oxidation. This
phenol uptake progressively increased as the surface
acidity decreased, and the oxidised sample heat treated
at 950 °C had the same adsorption capacity as the asreceived one, despite the lower surface area of the former sample compared with the latter.
Three mechanisms have been mainly proposed to
explain this behaviour, namely: the p–p dispersion interaction mechanism, the hydrogen bonding formation
mechanism and the electron donor–acceptor complex
mechanism.
The first two mechanisms were proposed by Coughlin
and Ezra [9] in 1968, and the third mechanism was
proposed by Mattson et al. [12] in 1969. At that moment
it was known that phenol was adsorbed in flat position
on the graphene layers, and in this situation the adsorption driving forces would be due to p–p dispersion
interactions between the aromatic ring of phenol and the
aromatic structure of the graphene layers.
Thus, Coughlin and Ezra proposed that acidic surface oxygen groups, which are located at the edges of the
A
1.5
Phenol uptake (µ mol/g)
Table 1
Energies of the highest occupied molecular orbital (HOMO) and
lowest unoccupied molecular orbital (LUMO) for model carbon
clusters. Adapted from [10]
87
A-ox 950º
A-ox 700º
1.0
A-ox 500º
0.5
A-ox 400º
A-ox 300º
A-ox
0.0
0
100
200
300
400
Equilibrium concentration (ppm)
500
Fig. 8. Adsorption isotherms of phenol on activated carbons with
different degrees of oxidation. From [11].
C. Moreno-Castilla / Carbon 42 (2004) 83–94
3.5
3.0
GRAPHITE
2.5
2.0
1.5
1.0
0.5
GRAPHITE-0.79% B
0.0
0
5
10
15
20
Equilibrium concentration (ppm)
25
Fig. 9. Adsorption of phenol on graphite substitutionally doped with
B and undoped. From [11].
1.5
1.5
1.0
1.0
0.5
0.5
0.0
0.0
100
200
Equilibrium conc. (µmole/l)
Phenol uptake (µmole/m2)
doped graphite samples (Fig. 9). Results show that the
presence of substitutional boron in the lattice of polycrystalline graphite, which removes p-electrons from the
solid, results in a lowering of the phenol uptake from
water.
These authors also indicated that both phenol and
water can compete to form H-bonds with surface oxygen groups, such as carboxyl groups. In this competition
water molecules are preferentially bound by H-bonds
[11].
This idea of competition between phenol and water
molecules for H-bonding to surface oxygen groups
suggested that phenol uptake from its cyclohexane solution would be favoured by the surface oxygen complexes. This is shown in Fig. 10. When adsorption of
phenol per unit cyclohexane area is plotted for two heattreated carbons, the carbon with higher surface acidity
exhibited a higher phenol uptake.
More recently, Pinto and co-workers [14] revisited
this issue by studying the adsorption of phenol aniline,
nitrobenzene and benzoic acid from both aqueous and
cyclohexanic solutions. Although the interpretation of
Phenol uptake (mmole/g)
basal planes, remove electrons from the p-electron system, creating positive holes in the conducting p-band of
the graphitic planes. This would lead to weaker interactions between the p-electrons of the phenol aromatic
ring and the p-electrons of the basal planes, therefore
reducing the phenol uptake.
Coughlin also proposed that the bonding of water
molecules to the oxide functional groups by H-bonding
can play an important role in the uptake of phenolic
compounds. In this case, Coughlin adopted Dubinin’s
proposal that water molecules adsorbed to oxygen
groups become secondary adsorption centres, which
retain other water molecules by means of H-bonds. As a
result, complexes of associated water form within the
pores of a carbon adsorbent. These complexes could
prevent the migration of organic molecules to a large
portion of the active surface area. This mechanism was
ruled out by Coughlin, whose results indicated that
surface oxygen complexes had no influence on phenol
uptake from concentrated solutions (in the second plateau of the isotherms).
Mattson and coworkers suggested that aromatic
compounds adsorb on carbons by a donor–acceptor
complex mechanism, with the carbonyl oxygen of the
carbon surface acting as the electron donor and the aromatic ring of the adsorbate acting as the acceptor.
Once the carbonyl groups are exhausted, the aromatic
compounds form donor–acceptor complexes with the
rings of the basal plane. Thus, Mattson and co-workers
explained the decrease in phenol uptake after carbon
oxidation as due to the oxidation of carbonyl groups to
carboxyl groups. As a result, the electron donor–acceptor complexes cannot be formed.
At this point it is interesting to indicate that Coughlin
and Ezra pointed out in their key paper [9] that ‘‘evidence for any single explanation (of the decrease of
phenol uptake with increase in surface acidity) is not
overwhelming’’ and added that ‘‘it is hoped that
continuing research will shed more light on this question’’. However, this was not, unfortunately, the case in
many instances, as can be seen in the recent review
published by Radovic et al. [2].
Since the initial proposals by Coughlin and Mattson,
many published papers have attempted to elucidate the
most appropriate mechanism to explain the adsorption
of phenolic compounds, and in general aromatic compounds on carbon materials. Thus, perhaps one of the
latest papers published on this topic to date was the one
published by Haydar et al. that appeared in 2003 entitled ‘‘Adsorption of p-nitrophenol on an activated carbon with different oxidations’’ [13]. This paper gives
some experimental evidence for the p–p dispersion interaction mechanism.
Perhaps the first experimental evidence of this
mechanism was given by Mahajan et al. [11] in their
study of phenol adsorption on graphite and boron-
Phenol uptake (mmol/g)
88
0.0
Fig. 10. Effect of surface oxygen complexes on adsorption of phenol
from cyclohexanic solution. Oxidized carbon heat treated at 300 °C, ,
and 400 °C, d, SBET with cyclohexane 420 and 500 m2 /g, respectively.
Surface acidity: 4.79 and 2.74 meq/g, respectively. From [11].
C. Moreno-Castilla / Carbon 42 (2004) 83–94
60
Enthalpy of exchange
(-kJ/mol)
the authors of the p–p dispersion interactions is confused, their experimental results seem to indicate that
the adsorption mechanism is both by water H-bonding
with carboxyl groups and p–p dispersion interactions
between the aromatic ring of the adsorptive and the
graphene layers. These authors also concluded that
Mattson’s mechanism is not the driving force for the
adsorption of aromatics on activated carbons.
One of the weak points of the Mattson mechanism is
that there is much experimental evidence that although
oxidation of carbons increases their concentration of
CO2 -evolving groups, the CO-evolving groups also increase or remain essentially unchanged.
Additional experimental evidence of the influence of
water adsorption comes from the enthalpy of immersion
into water of activated carbons, which reflects the specific and non-specific interactions between the liquid and
the solid [15] as shown in Fig. 11. The study of a large
number of carbons oxidised to different degrees reveals a
simple correlation between the enthalpy of immersion,
the oxygen content of the surface, the basic groups titrated by HCl, the micropore filling and the wetting of
the external surface. Thus, the specific interaction between the surface oxygen and water is 12.1 kJ/mol oxygen, involving an average of two water molecules per
oxygen.
More recently, MacDonald and Evans [16] measured
the enthalpy of exchange of phenol–water from diluted
aqueous solutions on BPL carbons with different oxygen
contents. They found (Fig. 12) that phenol exchange
enthalpy is inversely proportional to the net molar enthalpy of immersion into water. This indicates that the
strong preference of water to H-bond to surface oxygen
complexes lowers the phenol exchange enthalpy from
aqueous solution.
The two mechanisms proposed by Coughlin can
better explain many of the experimental results obtained
to date. However, an electron donor–acceptor mecha-
50
45
2.0
3.0
4.0
Enthalpy of immersion (-kJ/mol)
5.0
Fig. 12. The enthalpy of exchange for phenol–water versus the molar
enthalpy of immersion in water for a series of BPL carbons. Adapted
from [16].
nism cannot be ruled out. Thus, it is well known that
adsorption of phenolic compounds is partly physical
and partly chemical. The physisorbed part can be desorbed by treating with different solvents or by heat
treatments in inert atmosphere. However, the chemisorbed part cannot be desorbed, even at high temperatures, and it is converted to light gases and heavy
products that evolve from the surface of the carbon, and
also to a polymeric carbon residue that remains on the
surface. This residue reduces the adsorption capacity of
thermally regenerated activated carbons, as depicted in
Fig. 13, which shows that the adsorption capacity for
different phenolic compounds decreased when the regeneration cycle increased [17].
Better regeneration results could be achieved by
treating the exhausted carbons with liquid water at 150
atm and 350 °C in the absence of oxygen. Some of the
results obtained in a recent study [18] of the regeneration
of exhausted carbons with ortho-chlorophenol (OCP)
are shown in Table 3. In no case was there any loss of
carbon due to the treatment. The efficacy of the treatment was slightly above 100% for the first regeneration
cycles. However, for successive regeneration cycles there
210
90
70
50
30
30
50
70
90
110
12.1[O] + 10.3[meq.HCl] +
+ 0.8(N am-2.0[O] - 1.0[meq.HCl]) + Se×0.031
Fig. 11. Correlation between the enthalpy of immersion into water,
the total surface oxygen, the basic groups titrated with HCl, the micropore filling and the wetting of the non-porous surface area. From
[15].
Adsoption capacity (mg/g)
_ ∆ H(H O) / J g-1
i
2
55
40
1.0
110
10
10
89
140
70
0
0
1
2
3
4
5
Number of cycles
6
7
Fig. 13. Variation of the adsorption capacity of the activated carbon
AP-10 as a function of the adsorption–regeneration cycle. Phenol, D;
m-aminophenol, ; p-cresol, ; p-nitrophenol, }. From [19].
90
C. Moreno-Castilla / Carbon 42 (2004) 83–94
Table 3
Amount of ortho-chlorophenol adsorbed and regeneration yield in
successive adsorption–regeneration cycles. Regeneration conditions:
liquid water at 350 °C and 150 atm. Adapted from [18]
Run number
Amount adsorbed
(mmol/g)
Regeneration
yield (%)
H-30
0
1
2
3
1.63
1.69
1.61
1.58
104
99
97
0
1
2
3
0.92
0.95
0.96
0.87
104
105
95
0
1
2
3
1.05
1.07
1.09
1.04
103
104
99
H-13
RO-0.8
was a slight decrease in the OCP adsorbed, because,
according to the authors, part of it was strongly adsorbed and was not completely removed from the carbon surface.
All these data indicate that chemisorbed phenolic
compounds are strongly bound by other forces than
those of dispersion. These would likely involve chargetransfer complexes in which the direction of the electron
transfer could be either similar to that proposed by
Mattson or in a reverse direction.
3.3. Solution chemistry and adsorption temperature
Factors from solution chemistry that influence the
adsorption process are the solution pH and ionic
strength. Solution pH is one of the key factors that
control the adsorption process of organic weak electrolytes and polyelectrolytes on carbon materials because it controls the electrostatic interactions between
the adsorbent and the adsorbate. Thus, solution pH
determines the carbon surface charge and the dissociation or protonation of the electrolyte. At a solution pH
lower than the pHPZC or the pHIEP , the total or external
surface charge, respectively, will be on average positive,
whereas at a higher solution pH they will be negative.
In addition, the solution pH also controls the dissociation or ionization of the electrolyte through its pKa .
Thus, for instance, acidic electrolytes will be dissociated
at pH > pKa . Therefore, the solution pH controls the
adsorptive–adsorbent and adsorptive–adsorptive electrostatic interactions, which can have a profound effect
on the adsorption process.
Thus, the adsorption of substituted phenols on activated carbon depends on the solution pH (Fig. 14). At
acidic pH, the uptake was maximal because phenols
were undissociated and the dispersion interactions predominated. At basic pH, however, the uptake was lower
140
mg/g
Activated
carbon
165
115
90
65
2
7
12
pH
Fig. 14. Adsorption capacity of activated carbon CP-10 as a function
of solution pH. , Phenol (pKa ¼ 9.96); , m-chlorophenol
(pKa ¼ 8.80); D, p-nitrophenol (pKa ¼ 7.13). From [19].
because of electrostatic repulsions between the negative
surface charge and the phenolate anions and between
phenolate–phenolate anions in solution. The pH at
which the uptake decreased was found to be dependent
on the adsorptive pKa and the difference between the
pHPZC and the pHIEP [19].
Solution pH also affects the characteristics of carbon
beds, as shown in Table 4. Thus, adsorption of OCP is
favoured at acidic pH, as illustrated by the higher values
of the breakthrough volumes and the lower values of the
height of the mass transfer zone found at this pH. The
lower adsorption at basic pH is due, as explained before,
to the repulsion between the net negatively charged
carbon surface and the ortho-chlorophenolate anions
[20].
The effect of pH and the nature of functional groups
of both the aromatic adsorptive and the adsorbent on
the adsorption process have recently been investigated
by Radovic et al. [21]. For this purpose, they used an asreceived activated carbon oxidized with nitric acid and
nitrided with ammonia to study the adsorption of aniline and nitrobenzene, which are, respectively, electrondonating and -withdrawing groups. Some of the results
obtained from the adsorption of aniline at different pHs
are shown in Table 5. At pH 2, anilinium cations were
predominant in solution and the surface charge was
positive. Carbon oxidation enhanced adsorption with
respect to the as-received carbon, whereas nitriding deTable 4
Characteristics of activated carbon beds when removing ortho-chlorophenol at pHs 2 and 10. Particle size between 0.15 and 0.25 mm.
From [20]
Sample
pH ¼ 2
3
C-2
C-13
C-24
pH ¼ 10
VB (cm )
HMTZ (cm)
VB (cm3 )
HMTZ (cm)
4450
6500
10,650
1.02
0.95
0.48
550
1820
2800
2.02
1.54
1.32
C. Moreno-Castilla / Carbon 42 (2004) 83–94
Table 5
Aniline uptake (mmol/g) at a relative concentration of 103 on Norit
GCW carbon at different solution pHs
Carbon
As received
Oxidized
Nitrided
8.0a
2.6
8.9
pH ¼ 2
pH ¼ 11
pH ¼ pHPZC
0.48
1.14
0.33
1.10
0.86
0.86
1.10
1.67
0.80
Aniline pKa ¼ 4.6. Adapted from [21].
a
pHPZC values.
creased it. The authors indicated that in this case the
electrostatic adsorbent/adsorbate interactions were
more important than the dispersion interactions. Thus,
oxidation shifted the pHPZC to a value close to the solution pH, whereas nitriding increased the pHPZC , enhancing the adsorbate–adsorbent interactions.
At pH 11, the surface charge was predominantly
negative and aniline was not protonated in solution. In
this case, oxidation and nitriding were detrimental.
These results can be explained because dispersion interactions predominate and are not favoured, because
the presence of some oxygen and nitrogen-containing
functional groups withdraws electrons from the graphene layers. However, in this case the preferential Hbonding of water to some oxygen and nitrogen surface
functional groups can also explain these results.
At a solution pH equal to the pHPZC , aniline uptake
was relatively high on all adsorbents. According to the
authors this is consistent with the minimized electrostatic repulsions.
Some of the results obtained with nitrobenzene are
shown in Table 6. In this case solution pH has little
effect on equilibrium uptakes because nitrobenzene
molecules are the dominant species across the entire
range of solution pH. Therefore the effect of surface
chemistry is much more important. Across the pH range
the as-received carbon exhibits the highest uptake. The
authors explained these results as due to the dispersion interactions, which are dominant in this case.
Thus, surface oxygen and nitrogen complexes withdraw electrons from the graphene layers decreasing the
p–p interactions. However, as commented above, the
H-bonding of water to some surface functionalities can
also play an important role in this case. Maximal uptake
Table 6
Nitrobenzene uptake (mmol/g) at a relative concentration of 2.102 on
Norit GCW carbon at different solution pHs
Carbon
As received
Oxidized
Nitrided
8.0a
2.6
8.9
Adapted from [21].
a
pHPZC values.
pH ¼ 2
pH ¼ 11
pH ¼ pHPZC
2.43
0.78
1.30
2.61
0.70
1.65
3.04
0.95
1.91
91
is obtained at pH ¼ pHPZC because dispersion interactions are maximized.
These results, together with others found in the literature, indicate that functionalization of either the adsorptive or the adsorbent, which increases the p-electron
density, leads to enhanced or stronger adsorption when
the adsorption process is governed by dispersion forces.
The converse is also true.
Ionic strength is the other key factor that controls the
electrostatic interactions. Thus, these interactions, either
attractive or repulsive, can be reduced by increasing the
ionic strength of the solution. This is due to a screening
effect of the surface charge produced by the added salt.
Therefore, when the electrostatic interaction between
the surface and the adsorptive is repulsive, or the surface
concentration is sufficiently high, an increase in ionic
strength will increase the adsorption. Conversely, when
the electrostatic interactions are attractive, or the surface concentration is sufficiently low, an increase of the
ionic strength will diminish the adsorption. These effects
have been shown to take place with many weak organic
electrolytes and polyelectrolytes, for instance in the case
of bisphenol A and NOM [22,23].
In the case of bisphenol A, Fig. 15 shows that its
uptake increased with the ionic strength [22]. At solution
pH 7, the surface charge would be on average positively
charged and screened by the increase in the electrolyte
concentration, resulting in an enhancement of the bisphenol uptake.
NOM is found in varying concentrations in all natural water sources. It is a complex mixture of compounds varying from small hydrophilic acids, proteins
and amino acids to larger humic and fulvic acids.
Newcombe and Drikas [23] have studied the adsorption
of NOM (with nominal molecular weight between 500
and 3000 Da) on different activated carbons. Fig. 16
shows the adsorption of NOM at pH 7 on carbon W. At
this pH, both the carbon surface and NOM are negatively charged. At very low surface coverage direct
surface–adsorbate interactions should predominate.
An increase in salt concentration effectively screens repulsive interactions, resulting in increased adsorption.
In the case of carbon C, Fig. 17, the adsorption of
NOM was carried out at pH 4. In these conditions the
carbon surface will be positively charged whereas NOM
will be negatively charged. The adsorption isotherms
show an intersection point indicating a transition from a
screening-reduced to a screening-enhanced regime. At
low surface concentrations below the intersection point
of the isotherms, attractive interactions between the
adsorbent and the adsorbate are screened by an increase
in salt concentration, decreasing the adsorption. Above
the intersection point, where the surface concentration is
higher, salt screens the repulsion between charged segments of the polyelectrolyte and between adsorbed
NOM and NOM in solution, increasing the adsorption.
92
C. Moreno-Castilla / Carbon 42 (2004) 83–94
Table 7
Surface area (m2 /g) of different carbons from adsorption isotherms
applying DRK equation and immersion calorimetry
300
X (mg/g)
200
Carbon
STotal a
Sphenol
Sm-chlorophenol
PLW
AP-5
CP-5
1097
404
477
1016 (1037)b
453
596
1050 (1047)
453
522
From [24].
a
From CO2 at 273 K.
b
From immersion calorimetry.
100
0
0
40
Ce (mg/l)
80
Fig. 15. Adsorption isotherms of bisphenol A on activated carbon A
(pHPZC ¼ 10) in the absence of NaCl, d, and in the presence of NaCl:
0.01 M, N; 0.1 M, j. Solution pH ¼ 7.
0.30 M NaCl
60
40
20
0.01 M NaCl
0
0
4
8
12
16
Solution Concentration (mg/l)
Fig. 16. Adsorption of NOM on carbon W (pHPZC ¼ 4.5) at two ionic
strengths and at solution pH ¼ 7. Adapted from [23].
Amount adsorbed (mg/g)
80
0.30 M NaCl
60
40
0.01 M NaCl
400
20
0
0
4
8
12
Solution Concentration (mg/l)
16
Fig. 17. Adsorption of NOM on carbon C (pHPZC ¼ 7.5) at two ionic
strengths and at solution pH ¼ 4. Adapted from [23].
Therefore, both solution pH and ionic strength control the electrostatic interactions between the adsorbent
and the adsorptive.
When the adsorption of organic molecules is governed by non-electrostatic interactions, such as p–p
dispersion or hydrophobic interactions, the area of the
adsorbent occupied by the adsorbate depends on the
Amount adsorbed (mg/g)
Amount adsorbed (mg/g)
80
porosity of the former and the molecular size of the
latter. Thus, it has been shown [24] in adsorption from
diluted aqueous solution and immersion calorimetry
measurements (Table 7) that phenol and metachlorophenol are adsorbed as monolayers by both porous and
non-porous carbons with basic surface properties,
provided that the adsorptive is undissociated at the solution pH. This did not apply where molecular sieve
effects reduced the accessibility of the micropore system.
In the case of the adsorption of NOM (Fig. 18) in the
molecular weight range between 500 and 3000 on different activated carbons at solution pH 3, where electrostatic interactions are minimal and non-electrostatic
interactions predominate, the amount adsorbed depends
on the volume of pores between 0.8 and 50 nm [23].
Another example is the adsorption of trichloroethene
(Fig. 19) from highly diluted aqueous solutions on activated carbon fibres and granular activated carbons
with similar surface chemistry [25]. In this case, the trichloroethene adsorption on the more hydrophobic carbons was primarily controlled by pore volume in the
0.7–1 nm width range. According to the authors, this
micropore range is between 1.3 to 1.8 times the trichloroethene kinetic diameter (0.56 nm).
With regard to the effect of temperature on the adsorption, an increasing uptake of organic molecules is
expected when the adsorption temperature decreases
because adsorption is a spontaneous process. However,
200
0
0
0.2
0.4
0.6
Pore volume (cm3/g)
between 0.8 - 50 nm
0.8
Fig. 18. Relationship between NOM adsorption capacity and available adsorption volume for different activated carbons at solution
pH ¼ 3. Adapted from [23].
C. Moreno-Castilla / Carbon 42 (2004) 83–94
Amount adsorbed (mg/g)
40
30
AAW
AW
HAW
G219
F600
20
10
0.1
0.15
0.2
0.25
Pore volume in the 0.7 - 1nm
width range (cm3/g)
Fig. 19. Effect of pore volume on trichloroethene adsorption. Adapted
from [25].
some examples have been reported where the amount
adsorbed increased with temperature. Thus, for instance, it has recently been reported [26] that the adsorption of paracetamol from diluted aqueous solution
increased with adsorption temperature. This effect was
independent of the type of carbon, its surface chemistry
or solution pH. The authors explained this behaviour as
due to phase changes in the crystal form of the adsorptive.
93
face by stronger interactions than the dispersion ones.
This likely involves an electron donor–acceptor or
charge-transfer mechanism. Thus, further research
would be needed in this area.
On the other hand, it has been shown that the surface
of the carbon material is more effectively used if nonelectrostatic interactions are the driving force for the
adsorption. This condition can be reached by controlling the surface chemistry of carbon, and the pH and
ionic strength of the solution.
Finally, some research trends on this topic are the
study of competitive adsorption, for instance between
NOM and micropollutants such as pesticides; adsorption of bacteria which in turn can catalyse the decomposition of the adsorptive; adsorption–desorption of
drugs for medical applications and modifications of the
adsorptive depending on the solution and carbon surface chemistry.
Acknowledgements
The author wants to acknowledge to Drs. Jose Rivera-Utrilla and Victoria L
opez-Ram
on for fruitfull
discussions about the manuscript. Financial support
from the Ministerio de Ciencia y Tecnologıa and Feder
is also acknowledged.
4. Conclusions
References
Adsorption of organic molecules from dilute aqueous
solutions on carbon materials is a complex interplay
between electrostatic and non-electrostatic interactions.
Electrostatic interactions appear with electrolytes, essentially when they are ionized at the experimental
conditions used. Both interactions depend on the characteristics of the adsorbent and the adsorptive, and the
solution chemistry. Non-electrostatic interactions are
essentially due to dispersion and hydrophobic interactions. The surface chemistry of the carbon has a great
influence on both electrostatic and non-electrostatic interactions, and can be considered the main factor in the
adsorption mechanism from dilute aqueous solutions.
Aromatic compounds are physisorbed on carbon
materials essentially by dispersion interactions between
the p-electrons of the aromatic ring and those of the
graphene layers. Functionalization of either the adsorbent or the adsorptive profoundly affects these dispersion interactions. In addition, the presence on carbon
surfaces of chemical functionalities that can give rise to
H-bonds with water can effectively reduce the adsorption of the adsorptive molecules.
Strongly adsorbed aromatic compounds on the carbon surface, which cannot be desorbed even at high
temperatures, would probably be adsorbed on the sur-
[1] Derbyshire F, Jagtoyen M, Andrews R, Rao A, Martın-Gull
on I,
Grulke E. Carbon materials in environmental applications. In:
Radovic LR, editor. Chemistry and physics of carbon, Vol. 27.
Marcel Dekker: New York; 2001. p. 1–66.
[2] Radovic LR, Moreno-Castilla C, Rivera-Utrilla J. Carbon materials as adsorbents in aqueous solutions. In: Radovic LR, editor.
Chemistry and physics of carbon, Vol. 27. Marcel Dekker: New
York; 2000. p. 227–405.
[3] Lyklema J. Fundamentals of interface and colloid science, Vol. II.
solid–liquid interfaces. New York: Academic Press; 1995.
[4] L
opez-Ram
on MV, Stoeckli F, Moreno-Castilla C, CarrascoMarın F. On the characterization of acidic and basic surface sites
on carbon by various techniques. Carbon 1999;37:1215–21.
[5] Lyklema J. Fundamentals of interface and colloid science, Vol. I.
fundamentals. New York: Academic Press; 1993.
[6] Mattson JS, Mark Jr HB. Activated carbon: surface chemistry
and adsorption from solution. New York: Marcel Dekker; 1971.
[7] Pendleton P, Wu SH, Badalyan A. Activated carbon oxygen
content influence on water and surfactant adsorption. J Colloid
Interface Sci 2002;246:235–40.
[8] Pendleton P, Wong SH, Schumann R, Levay G, Denoyel R,
Rouquerol J. Properties of activated carbon controlling 2-methylisoborneol adsorption. Carbon 1997;35:1141–9.
[9] Coughlin R, Ezra FS. Role of surface acidity in the adsorption of
organic pollutants on the surface of carbon. Environ Sci Technol
1968;2:291–7.
[10] Tamon H, Okazaki M. Desorption characteristics of aromatic
compounds in aqueous solution on solid adsorbents. J Colloid
Interface Sci 1996;179:181–7.
94
C. Moreno-Castilla / Carbon 42 (2004) 83–94
[11] Mahajan OP, Moreno-Castilla C, Walker Jr PL. Surface-treated
activated carbon for removal of phenol from water. Sep Sci
Technol 1980;15:1733–52.
[12] Mattson JS, Mark Jr HB, Malbin MD, Weber Jr WJ, Crittenden
JC. Surface chemistry of active carbon: specific adsorption of
phenols. J Colloid Interface Sci 1969;31:116–30.
[13] Haydar S, Ferro-Garcıa MA, Rivera-Utrilla J, Joly JP. Adsorption of p-nitrophenol on an activated carbon with different
oxidations. Carbon 2003;41:387–95.
[14] Franz M, Arafat HA, Pinto NG. Effect of chemical surface
heterogeneity on the adsorption mechanism of dissolved aromatics on activated carbon. Carbon 2000;38:1807–19.
[15] L
opez-Ram
on MV, Stoeckli F, Moreno-Castilla C, CarrascoMarın F. Specific and non-specific interactions of water molecules
with carbon surfaces from immersion calorimetry. Carbon
2000;38:825–9.
[16] MacDonald JAF, Evans MJB. Adsorption and enthalpy of
phenol on BPL carbon. Carbon 2002;40:703–7.
[17] Moreno-Castilla C, Rivera-Utrilla J, Joly JP, L
opez-Ram
on MV,
Ferro-Garcıa MA, Carrasco-Marın F. Thermal regeneration of an
activated carbon exhausted with different substituted phenols.
Carbon 1995;33:1417–23.
[18] Rivera-Utrilla J, Ferro-Garcıa MA, Bautista-Toledo I, SanchezJimenez C, Salvador F, Merchan MD. Regeneration of orthochlorophenol-exhausted activated carbons with liquid water at
high pressure and temperature. Water Res 2003;37:1905–11.
[19] Moreno-Castilla C, Rivera-Utrilla J, L
opez-Ram
on MV, Carrasco-Marın F. Adsorption of some substituted phenols on
activated carbons from a bituminous coal. Carbon 1995;33:845–51.
[20] Rivera-Utrilla J, Utrera-Hidalgo E, Ferro-Garcıa MA, MorenoCastilla C. Comparison of activated carbons prepared from
agricultural raw materials and Spanish lignites when removing
chlorophenols from aqueous solutions. Carbon 1991;29:613–9.
[21] Radovic LR, Silva IF, Ume JI, Menendez JA, Le
on y Le
on C,
Scaroni AW. An experimental and theoretical study of the
adsorption of aromatics possessing electron-withdrawing and
electron-donating functional groups by chemically modified
activated carbons. Carbon 1997;35:1339–48.
[22] Rivera-Utrilla J, Bautista-Toledo I, Ferro-Garcıa MA, MorenoCastilla C. Bisphenol A removal from water by activated carbon.
Effects of the carbon characteristics and solution chemistry.
Extended abstracts (in CD format) Carbon’03 (publishers Linares-Solano A, Cazorla-Amor
os D). Oviedo, Spain. July 2003.
[23] Newcombe G, Drikas M. Adsorption of NOM onto activated
carbon: electrostatic and non-electrostatic effects. Carbon
1997;35:1239–50.
[24] Stoeckli F, L
opez-Ram
on MV, Moreno-Castilla C. The adsorption of phenolic compounds from aqueous solutions by activated
carbons described by the Dubinin–Astakhov equation. Langmuir
2001;17:3301–6.
[25] Li L, Quinlivan PA, Knappe DRU. Effects of activated carbon
surface chemistry and pore structure on the adsorption of organic
contaminants from aqueous solution. Carbon 2002;40:2085–100.
[26] Terzyk AP, Rychlicki G, Biniak S, Lukaszewicz JP. New
correlations between the composition of the surface layer of
carbon and its physicochemical properties exposed while paracetamol is adsorbed at different temperatures and pH. J Colloid
Interface Sci 2003;257:13–30.