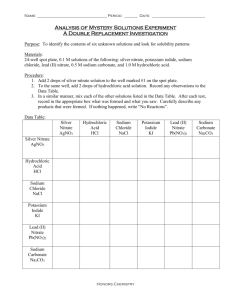
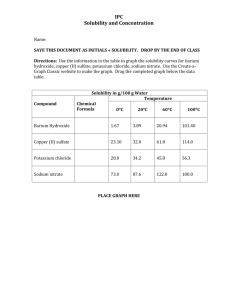
No-waste manual for Educational Institutions
advertisement

No- Waste Lab Manual for Educational Institutions Prepared by College of the Redwoods Under a Grant From California Department of Toxic Substances Control WE'VE CHANGED! On July 17, 1991, the California Environmental Protection Agency officially came into existence and the Toxic Substances Control Program became the Department of Toxic Substances Control under that Agency. The Toxics Program is no longer affiliated with the Department of Health Services or the Health and Welfare Agency. The wording within this particular document has not been changed to indicate this new affiliation. The new mailing address follows: Department of Toxic Substances Control Office of Pollution Prevention and Technology Development 400 P Street, 4th Floor P.O. Box 806 Sacramento, CA 95812-0806 (916) 322-3670 93 83948 NOWASTE LAB MANUAL - A procedure that eliminates Toxic Waste Production from Introductory Chemistry Laboratory Courses. ABSTRACT The feasibility of producing a laboratory manual for introductory chemistry courses which incorporates procedures which produce little or no toxic waste has been demonstrated. This is accomplished by the use of consecutive chemical reactions, so that the product of one reaction is used as the starting material for the next. In this fashion, all hazardous chemical products are retained in the cycle, and benign waste, which serves as a bleed on the system, is converted into educationally useful products. Another major benefit derived from this procedure is a substantial reduction in the expense of administering laboratory courses, an expense of This provides a strong incentive for the adoption of concern to educators. the described approach. Other advantages are accrued. The school becomes a role model of waste recycling and thus influences student attitudes toward our environment. Student interest in chemistry is strengthened by creation of a purpose, meaningfulness and connection to their concept of the "real world". work as they learn. The newly acquired chemical knowledge is put to useful Conservation of dwindling resources is aided. The goal is to significantly reduce or eliminate the generation of hazardous wastes by our school laboratories. Acknowledgements The author would like to express gratitude to Karen Prentice, Chris Schomers, and Louise Jensen for their help and encouragement. This report was submitted in fulfillment of Award Number 87-TO137 by the Department of Health Services, Toxic Substances Control Division under the sponsorship of the Department. Work was completed as of April 30th 1989. Disclaimer The statements and conclusions of this report are those of the Grantee and not necessarily those of the State of California. The mention of commercial products, their source, or their use in connection with material reported herein is not meant to be construed as either an actual or implied endorsement of such products. _- TABLE OF CONTENTS ............................................... Recommendations ....................................................... Introduction .......................................................... Summary and Conclusions PAGE 1 3 4 Discussion ............................................................. Experimental Outline (Schematic) .................................... Experimental Details ................................................ 1 . Preparation of Oxygen ......................................... 2 . Preparation of Hydrogen ....................................... 3 . Chemistry of Alkaline Earths .................................. 4 . Determination of an Empirical Formula ......................... . 5 . Chemistry of the Halogens ..................................... 6 . Reactions of Ions in Solution ................................. 7 . Types of Chemical Reactions I ................................. 8. Types of Chemical Reactions I1 ................................ 9. Activity Series ............................................... 10. Titration ..................................................... General . 12 . 13 . 14. 15 . 16 . 17 . 18. 19 . 20 . 21 . 22 . 23 . 11 .. ......................................... Fractional Crystallization .................................... Chemistry of Aluminum ......................................... Chemistry of Lead ............................................. Solubility of Salts and Saturation ............................ Preparation of Lead Nitrate and Iodine from Lead Iodide ....... Chemistry of Chromium ............. Chemical Separation of Zinc ................................... Redox Titration ............................................... Electrolysis of Zinc Sulfate .................................. Ion Exchange. Recovery of Oxalic Acid ......................... Kinetics. Clock Reaction ...................................... Synthesis of Methyl Benzoate .................................. 24 . Saponification of Methyl Benzoate ............................. 25 . Preparation of Iodine ......................................... . 27 . 26 Preparation of Silver Nitrate from Silver Halides .................... ................................... 10 14 51 51 54 58 60 61 66 67 67 71 72 73 73 75 76 77 78 79 84 84 85 85 86 87 87 88 Preparation of Ammonium Iodide from Iodine 89 Synthesis of Sodium Nitrate 91 TABLE OF CONTENTS PAGE . 29 . 30 . 31 . 32 . ...................................... Distillation ................................................ Separation of Salts ......................................... Preparation of Alumina ...................................... Preparation of Bleach ....................................... 33 . Applications of Industrial Chemicals ........................ 34 . Hydrolysis of Salts ......................................... 35 . Determination of Purity by Melting Point .................... Waste Products .................................................... Cost Reduction .................................................... Impurity Buildup .................................................. Storage Space ..................................................... 28 Kjeldahl Determination ............................... Outline of Introductory and Physical Chemistry Experiments ................ Chemicals and Costs of a Typical Chemistry Manual ...... Costs of Chemicals in NOWASTE Manual ................... fnnovative Nature of NOWASTE MANUAL Appendix I Appendix I1 Appendix 111 Appendix IV Suggested Names for Solids and Solutions Used in NOWASTE Manual ............. 92 93 93 94 94 95 95 95 96 99 102 104 105 107 111 113 114 SUMMARY AND CONCLUSIONS A feasibility study has confirmed the practicality of developing a laboratory manual for introductory chemistry courses of a novel nature. This manual is intended to minimize or eliminate the production of toxic wastes by using the products of one experiment as the starting materials for the same or other experiments. In this manner, the toxic chemicals are retained in the cycle of experiments. As a valuable dividend, the costs of conducting laboratory classes are reduced, since the toxic chemicals are not discarded and do not have to be repurchased in amounts formerly required. This cost reduction can serve as a strong incentive for adoption of this methodology by our institutions, particularly at a time when the rising costs of laboratory classes are of concern to all administrators. Other advantages are accrued. disposal. The school becomes a role model of waste The pattern of responsibility for waste is implanted in the student mind. Student interest in chemistry in the sense of meaningfulness, purpose and continuity is expected to be enhanced. The student's chemical knowledge is immediately put to use in a real and worthwhile cause. An experiment in which iodine is carried through a series of reactions producing remarkably different products, and then is regenerated, has an obvious educational advantage. eliminated. Costs of hazardous waste disposal are essentially Conservation of irreplaceable resources is achieved. This study was carried out by a combination of desk research and laboratory work. A concerted review of pertinent literature and published laboratory manuals was conducted. This process helped to define the types of experiments necessary in introductory chemistry courses and recent tendencies in emphasis of topics. Having defined the scope of necessary experiments, procedures were created that could accomplish the stated goals. These have been tested in the laboratory to the point of establishing their suitability for intended purpose. There have been several approaches to design of the experiments. Sometimes solutions can be used repeatedly as in the electrochemical cell voltage and crystallization experiments. In other cases, an element may be used in a succession of chemical reactions to produce a useful final product, for example, plaster of paris. - 1 - The chemistry of aluminum is demonstrated by carrying aluminum through a series of reactions and then converting it in the final experiment to activated alumina, which is then used as a desiccant in future experiments. In general, however, the approach has been to devise a series of reactions which retain the chemicals within a cycle. This method represents a general approach to the design of laboratory experiments for educational purposes. Since the procedures virtually eliminate hazardous waste production and at the same time-reduce expenses to a significant degree, it is felt that this approach may well represent the method of experimental design for academic purposes in the future. In conclusion, the feasibility of utilizing a novel approach consisting of consecutive chemical reactions has been demonstrated. Further work will concern perfection of the methods to the point that they may be easily and safely performed by our beginning students of chemistry. Beyond that goal, appIications to general chemistry, organic chemistry and analytical chemistry courses are envisioned. - 2 - RECOMHENDATIONS A feasibility study has demonstrated that the concept of a basic chemistry laboratory manual which uses toxic chemical products from experiments as starting materials for others is a practical concept. following tasks remain to complete the work. The procedures must be cor- rected and modified to fit laboratory time requirements. term reuse of chemicals must be ascertained. The Effect of long Final design of the experiments must be completed. Exact quantities of chemicals necessary for the procedures must be determined. the manual, must be written, proofread and finally pub- lished. Efforts to achieve adoption of the manual must be carried out. Later, similar manuals should be devised for general, organic and analytical chemistry courses. - 3 - INTRODUCTION The purpose of this project is to determine the feasibility of a novel approach which eliminates the production of toxic chemicals or waste products from chemistry laboratory classes. The experiments are designed in such a manner that toxic chemicals are recycled to other or the same experiments. Build-up of impurities is eliminated by bleeding the salts formed and converting them to either chemicals used in the cycles (e.g., bleach, nitric acid) or other products which are educationally useful (e.g., of paris, desiccants). plaster A laboratory manual will be written which is intended for use in our highschools and community colleges. The ultimate goal is to extend the use of this method to general (college) chemistry, non-science major chemistry, organic chemistry, and perhaps analytical chemistry courses. It is helpful to understand the general background of this project from the point of view of a chemistry teacher. All laboratory procedures used for educational purposes produce chemical wastes. At present, the means of disposal of these chemicals is in sanitary sewage systems and landfills, and ocassionally, particularly in the case of organic wastes, incineration. The moral and, increasingly legal, responsibilities2 dictate that sanitary sewage or sanitary landfill disposal are unsatisfactory methods in the long term. They are, nevertheless, the only options available at this writing. For example, a 1988 publication by the American Chemical Society recommends sanitary sewage disposal for copper salts, alcohols, organic acids, acetone, barium sulfate, and all filtrates produced after precipitation of the sulfides of lead, cobalt and silver with sodium sulfide. It also reccrnnnends ahat the precipitates be disposed of in sanitary landfills. 3 Versions of this procedure are found in other references.4 1 McKusik, B.C., J Chem. Educ., 61, A152 (1984). 2 for example, U.S. Environmental Protection Agency (EPA) regulations under the Resource Conservation and Recovery Act (RCRA), 45 Federal Register, 10 May 1980, and subsequent amendments; Title 40 Code of Federal Regulations, Chapter 1. 3 Summerlin, L.R., Ealy, Jr., J.L. and Borgford, C.L., "Chemical Demonstrations, A Sourcebook for Teachers", Vol 1, American Chemical Society, Washington, D.C., 1988. Browne, L.M., and Weir, G.L., "Prudent Practices for Disposal of Chemicals from Laboratories", National Academy, Washington, D.C., 1983., Armour, M.A., Browne, L.M., and Weir,. G.L., "Hazardous Chemicals Information and Disposal Guides", 2nd edition, University of Alberta, 1984, p.165. 4 for example, see Armour, M.A., -_ - 4 - There have been a few suggestions at chemical recycle, the theme of this text. Walton suggests designing an experiment which "illustrates a disposal method of a product from a previous experiment". A multistep organic synthesis has been devised by Stradling and Gage in which acetophenone is carried through several reactions, the products being used as starting materials until eventually an end product is produced which must be discarded.2 Neckers, et al, describe recyle of organic solvents, and a sequential experiment to reduce the costs of maintaining an organic chemistry laboratory.3 Schumm has suggested a series of experiments using alum synthesized from scrap aluminum.4 Other than these limited suggestions, use of microscale experiments, or disposal by landfill or sanitary sewer, there have been no alternatives offered to the chemistry instructor. of solution at the present time. This presents a problem of great urgency Failure to provide a solution will permit undesirable practices to be perpetuated. This project departs completely from the disposal approach. All toxic chemicals are continually reused; losses occur only from accidents or tracequantities not removed from solution. Even non-toxic wastes are retained and processed to produce chemicals useable in the experimental cycle, or useful educationally. This project has consisted of three phases: the desk research, and the laboratory research. The literature search, The product of this work is an outline of experiments to be incorporated into a laboratory manual, after further research has established the practicality and efficacy of the proposed experiments. The literature search provided information on previous related work and familiarization with a number of existing laboratory manuals. The search was also invaluable in providing the germ of innovative experiments in the proposed mode. J Chem Educ., A69 (1987) 1 Walton, W.A., 2 Stradling, S. and Gage, C., J Chem. Educ., 1116 (1985) 3 Neckers, D.C., Duncan, M.B., Gamor, J. and Grasse, P.B., J. Chem. Educ., 54, 690 (1977). 4 Schumm, M.K., "Recycling Materials and Organic Identification Without Instruments", Montgomery College, Roclnrille, M.D., 20850, Paper MC4-02, AMW, paper presented at the Seventh Biennial Conference in Chemical Education, August, 1982. - 5 - The following laboratory manuals were investigated by listing each experiment, its subject and a brief expository paragraph on the subject. Thirteen manuals were analyzed in this fashion. They were: 1. "Laboratory Kanual" by Joseph E. Davis, W. Keith McNab, A.L. McClellan, and Paul R. OConner, 1st Edition, D.C. Heath and Co., Lexington, Mass., 1983. 2. "Laboratory Manual for Chemistry" by Maxine Wagner, 1st Edition, Allyn and Bacon, Inc., Newton, Mass., 1983. 3. "Foundations of Chemistry in the Laboratory" by Morris Wein, Leo R. Best and Robert L. Miner, 5th Edition, Brooks/Cole Pub. Co., Monterey, Calif., 1982. 4. "Laboratory Experiments" by Charles M. Wynn and Gary A. Joppich, 2nd Edition, Wadsworth Pub. Co., Belmont, Calif., 1981. 5. "Chemistry in the Lab" by William L. Masterson, Emil J. Slovinski, and Edward T. Walford, 1st Edition, Holt, Rinehart and Winston, New York, N.Y., 6. 1980. "Laboratory Chemistry" by L. Neal Carmichael and David F. Haines, 1st Edition, Charles E. Merrill Pub. Co., Columbus, Ohio, 1979. 7. "Laboratory Manual" by Phyllis M. Dietz, Robert L. Telefsen, Robert W. Parry and Luke E. Steiner, 2nd Edition, PrenticeHall, Inc., Englewood Cliffs, N.J., 1975. 8. "Laboratory Experiments", by L. Neil1 Carmichael and David F. Haines, Charles E. Merrill Pub. Co., Columbus, Ohio, 1979. The above manuals are intended for high school and community college introductory chemistry courses. In addition, the following general chemistry manuals were examined. 9. "Encounters in Experimental Chemistry" by William L. Jolly, 1st Edition, Harcourt Brace Jovanovich, Inc., New York, N.Y. 1972. 10. "Chemistry in the Laboratory" by Charles W.J. Scaife and O.T. Beachley, Jr., 1st Edition, Saunders College Pub. Co., Philadelphia, Pa., 1987. -_ 11. "Basic Laboratory Studies in College Chemistry", by Grace R. Hered, 7th Edition, D.C. Heath and Co., Lexington, Mass., 1980 - 6 - 12. "Laboratory Manual for General Chemistry" by James E. Brady and Jo. A. Beran, 2nd Edition, John Wiley and Sons, New York, 1982. 13. "Fundamentals of Chemistry" by Frank Brescia, John Arents, Herbert Neislick, Amos Turk and Eugene Weiner, Academic Press, Inc. 1980. From this survey, a familiarity was gained with the subject matter covered and with the educational approach. Some specific information was New York, N.Y., For example it was determined that 52% of the experiments in the introductory manuals involved chemical reactions and that the most frequent obtained. topics covered, in order of decreasing number of experiments were stoichiometric relationships, organic chemistry, electrochemistry, equilibrium, qualitative analysis, redox, kinetics, various reactions of ions, chemistry of halogens, types of chemical reactions, empirical formulas, titration, and hydrates. Egghty per cent of the topics covered were in these categories. These analyses were used as guidelines in the design of this manual. In the general survey of literature pertinent to the subject of introductory chemical courses, easily the most valuable source proved to be the Journal of Chemical Education, published by the American Chemical Society. This journal contained many articles specific to this topic, as is demonstrated by the number of refernces in the text. helpful. Two texts proved to be particularly These were "Qualitative Analysis" by "herald Moeller, 1st Edition, McGraw-Hill, N.Y., 1958, and "General Chemistry" by Henry F. Holtzclaw, Jr., William R. Robinson and William 11. Nebergall, 7th Edition, D.C. Heath and Co., Lexington, Mass., 1984. inorganic chemical reactions. These are excellent references for general And lastly, the "Handbook of Chemistry and Physics", published each year by the Chemical Rubber Co., Cleveland, Ohio, was invaluable in providing, in particular, data on solubilities of inorganic compounds. The general approach to the methods developed within this report is best explained by contrasting conventional experiments with those developed here. In almost every experiment investigated in conventional procedures. experiments are performed non-sequentially. For example, if the experiment is designed to illustrate the activity series of the metals, the instructions __ would be, for example: - 7 - 1) add copper metal to an aqueous solution of silver nitrate and observe the results (silver is precipitated and the copper goes into solution). Discard the mixture. add lead metal to an aqueous solution of copper nitrate and observe the 2) results (copper precipitates and lead goes into solution). Discard the mixture. 3) add iron metal to a solution of lead nitrate and observe the results (lead precipitates and iron goes into solution). Discard the mixture. ........etc. In this process, waste products of copper, silver nitrate, copper nitrate, lead, lead nitrate, iron, and iron nitrate have been created. Contrast this with the approach used in the method of this report. 1) add copper metal to an aqueous solution of silver nitrate and observe . the results. After the reaction is complete, remove the unreacted copper and precipitated silver (for details see Experiment 9. 2) add lead metal to the copper nitrate solution from 1) and observe the results. After the reaction is complete, remove the unreacted lead and precipitated copper. 3) add iron metal to the lead nitrate solution from 2) etc. In other words, by carrying out the reactions sequentially, the products are minimized. reactions. The second phase is to use all products in other chemical In the case above silver produced eventually returns to the cycle as silver nitrate, copper as copper nitrate, etc. So that, in this case, they create a closed cycle. There are some cases where the cycle is not closed, and the starting material ends up as a product useful in some way to the students. . In this manner aluminum is carried through a series of reactions to finally exit the cycle as alumina, which is then used as a desiccant in future experiments. Calcium metal is carried through sequential reactions eventually exiting as plaster of paris, from which the students fashion an object such as a mold. These products serve as a bleed to the system to prevent buildup of impurities. The general scheme, then, is to create a sequence of reactions which either retains the species within a cycle, or utilizes it as a useful -_ product. This is contrasted with the conventional approach, which carries - 8 - out reactions separately and in a non-connected fashion, and thus produces a multitude of waste products. The educational advantage to the sequential approach should be noted here; the connection between chemicals and reactions becomes much more pervasive. The work carried out here is limited to introductory chemistry courses. The concept can be expanded to include general (college level), organic and analytical chemistry courses. It may be somewhat applicable to certain phases of the biological sciences, perhaps in cooperation with the chemistry department. More generally, it is a way of thinking that should have applicability to a number of laboratory procedures. - 9 - DISCUSSION Chemistry laboratory instruction is an essential part of a chemistry education. But a chemistry laboratory generates toxic chemical waste which must be eliminated in some manner. Historically this waste has been discharged into the sewers but this practice is now regarded as unsatisfactory and, in some cases, illegal. Concern for the environment is reflected in the passage of the Tanner Bill, AB 2948, requiring each county to assess its hazardous waste streams and adopt alternatives to land disposal, the Safe Drinking and Toxic Enforcement Act of 1986 which prohibits discharge of carcinogens or teratogens into water or land, and Farr Assembly 3i11 685, which provides grants to programs demonstrating hazardous waste source reduction, recycling, or treatment technologies. In any case, toxic waste discharge of any kind is undesirable environmentally, economically and conservationally. This project was conceived to address this issue, eliminating toxic waste disposal entirely by integrating the toxic chemicals into a cyclical laboratory instructional process which uses products of one experiment as a starting point for the same or other experiment. Considerable attention has been given recently to the problem of toxic waste from academic laboratories. Solutions suggested have been either reduction of amounts of chemicals employed for instruction to the microscale range or disposal by commercial haulers, form of concentration. usually after some Neither of these methods is entirely satisfactory. \ Experiments on a micro scale do not have the visual impact that macro scale experiments have, in the same way that a 6 inch TV screen is:.less'irhpressive than a 22 inch screen. Further, special techniques must be taught to inex- perienced students in order to achieve results equivalent to the macro scale. This is why micro scale experiments have been used only to a limited extent in the past. Disposal of concentrated chemical wastes adds an additional cost to the laboratory instruction, and concentration of wastes requires additional handling. Further, it is precisely this land contamination which we are attempting to avoid. 1 "Prudent Practices for Disposal of Chemicals from Laboratories", National _, Research Council, National Academy Press, Washington, D.C., 1983 2 See, for example, A m o u r , M., J. Chem. Educ., 65, A64 (1988); Armour, K., Browne, L.M. and Weir, G.L. , J. Chem. Educ. , 6T; A93 (1985). - 10 - ? This project was conceived to address the toxic waste issue, and eliminate the waste disposal problem by integrating these chemicals into a cyclical process whereby products of one experiment are used as starting points for the same or another experiment. In designing these experiments, a number of considerations were kept in mind. These considerations were: 1) Types of experiments to be emphasized. 2) Safety of the experiments. 3) Creation of interesting, educational and entertaining experiments. 4 ) Use of economical and easily available materials. 1. Types of experiments A survey of a number of modern introductory level laboratory manuals was conducted to determine those topics most frequently covered. and authors of these manuals are listed in the introduction. Titles In addition, the chemical education literature was researched to ascertain modern trends There appears to be a major movement back .in chemistry laboratory education. to descriptive chemistry, taught almost exclusively prior to the 1940's. By descriptive chemistry is meant syntheses, reactions, commercial processes of chemicals. In other words, the students need to understand that facts come before theories. A tendency away from descriptive chemistry began with Linus Pauling's classic "Nature of the Chemical Bond" which introduced a great many theoretical concepts (e.g., etc.) electronegativity, orbital hybridization, into the field of chemistry and dramatically altered the mode of present- ation.2 This has continued up to the present time. There is now a feeling among educators that the theoretical emphasis must yield somewhat to the descriptive aspect of the ~ c i e n c e .This ~ tendency has been followed in the design of the present manual, which is ideally suited to this mode of presentation. 2. Safety of the experiments. The safety of the experiments will be given careful consideration. Cautionary warnings will be given whenever hazardous chemicals are to be used. __ A full section on safety consideration will appear at the beginning 1 Zuckermann, J.J., J. Chem. Educ., 63, 829 (1986) 2 Pauling, L., "The Nature of the Chemical bond", 3rd Ed., Cornel1 University Press, Ithaca, N.Y., 1960 3. for example, see Whisnant, D.M., J. Chem. Educ., 2,792 (19821, "What Happened to Descriptive Chemistry?", Editorial, J.Chem. Educ., 915 (1985) and Zuckermann, J.J., J. Chem. Educ., 63, 829 (1986). 62, - 11 - of the manual. Apparatus set-ups are and will be designed to trap all noxious gases or vapors evolved. Substitutions of less harmful chemicals for those known to be dangerous have been made when possible. Thus, cyclohexane has been substituted for carbon tetrachloride in several experiments. The use of potassium chlorate, a potentially explosive compound when dry, has been avoided in the preparation of oxygen. In some cases where important chemistry is involved, such as that of chromium, a known carcinogen, appropriate measures such as wearing of gloves, cautionary statements, and strict supervision will be emphasized. A large number of hazardous compounds result from the combin- ations of the chemical elements, and it is a purpose of chemistry to examine the nature of these substances. Since this is unavoidable, the best approach is the teaching of safe and appropriate procedures, much as we instruct students in driver's training or woodshop in safe procedures with potentially hazardous equipment. 3. * Creation of interesting, educational and entertaining experiments. If this manual is to be used extensively, it must not only eliminate toxic waste and save money, but it must provoke interest of the teachers and students. The author has attempted to do this by including experiments with colorful reactions, such as the chromium series of experiments or the reactions of copper, striking phenomena such as the crystallization of alum, the formation of iodine from lead iodide, or the clock reaction, and practical descriptive experiments such as the manufacture of plaster of paris and chemical recovery techniques such as the recovery of zinc sulfate (beautiful fractal crystalline growth of zinc is observed from a colorless solution) or fractional distillation. In addition, a number of interesting laboratory techniques and student built pieces of equipment have been introduced or suggested. 4. Use of economic and easily available materials. An effort has been made to incorporate the use of readily available materials in the course in order to reduce expense and to provide student connections to "real-world" experience. This is reflected in the use of bleach, aluminum foil, vinegar, salt, etc. Using the guidelines outlined above, research was conducted to conceive and carry out a laboratory program containing sequentially designed experiments. The feasibility of such an approach has now been demonstrated. The possible consequences of teaching an introductory course in chemistry with a laboratory course designed in this manner are worth analysis. - 12 - First, let's consider student interest in chemistry. An often heard comment from beginning students is that chemistry has no connection with the real world. If they are reminded that the stuff of nature is all chemical, they remain unconvinced. The chemicals they ordinarily use in the laboratory experiments come out of bottles not found in any store, and the products from their experiments are quickly disposed of to be seen no more. Contrast that experience with possible student reaction using the sequential approach. The chemicals used now are chemicals they have themselves produced. products of their labors are used for further work. familiar substances are used: These In addition, ordinary copper wire, baking soda, bleach, aluminum foil. The connections begin to be made. Chemistry becomes "real-world". From an educators point of view, these connections are invaluable. Further, students experience their newly acquired skills being used to improve their world. They are accomplishing a goal, as well as becoming .educated in the process. And they are learning an attitude: responsible for one's actions. One must be In the experience of the author, most students have an environmental awareness and should respond very positively to this program. And the school then sets the pattern. model. - 13 - Our schools should be the role SCHEMATIC OUTLINE OF CYCLICAL EXPERIMENTS The following symbols are used in the outline of experiments: denotes an experiment number. For example, [SI indicates Experiment 14. EtOH denotes ethyl alcohol MeOH denotes methyl alcohol HOAC denotes acetic acid 0 denotes the phenyl group T denotes a gas 4 A denotes a solid 0 denotes that the compounds are by-products or unreacted species. denotes heat This outline will be helpful in following the discussion, and providing a quick summary of the experiments. This will be followed by a narrative. This summary and discussion represent only the descriptive chemistry experiments that are proposed to be included in the manual. Other types of experi- ments, including those of an introductory nature on physical chemistry experiments, will not be discussed in this report. of these are included in Appendix I. - 14 - A list and brief summary DISCUSSION: Note: Experimental Outline All chemicals should be assumed to be in aqueous solution unless specified as solid by ,& Experiment 1: H202 . Preparation of Oxygen Fe 3 O2t ___) Fe f Purpose : This experiment illustrates the preparation and properties of oxygen, and the action of a catalyst. Description: Oxygen is generated from 3% hydrogen peroxide and an iron nail as catalyst. The reaction mixture is heated. Oxygen is collected over water and tested in the usual manner. Comments : This experiment eliminates the hazards of conventional preparation of oxygen from potassium chlorate (explosion potential) or 9% hydrogen peroxide (chemical burn potential), as well as the necessity of disposing of manganese dioxide, the conventional catalyst. Costs of the experiment are reduced. - 15 - Experiment 2: Preparation of Hydrogen Purpose: This experiment illustrates the preparation and properties of hydrogen. Description: Hydrogen is prepared by the action of clean aluminum, prepared by pickling in hydrochloric acid, on sulfuric acid. The hydrogen is collected over water and conventionally tested. Comments : Use of zinc metal in this preparation is avoided completely by the substitution of aluminum. Aluminum is rendered reactive to dilute sulfuric acid by pretreatment with hydrochloric acid which removes the protective oxide coating. Scrap aluminum from cans, foil, or other source work well. The necessity of dealing with zinc (and its inevitable companion cadmidin the comparatively large quantities required for this experiment is eliminated. - 16 - Experiment 3: Chemistry of the Alkaline Earths Purpose: This experiment acquaints the student with some properties of the Group I1 metals and their compounds. Description: Reactions of calcium and magnesium with water are carried out and the resulting hydroxides are tested with litmus and then dissolved in hydrochloric acid. The chlorides are reacted with sulfuric acid. Calcium sulfate precipitates. Magnesium sulfate does not. The products from both experiments go to 1301. In set 2, the metals are shown to combine directly with iodine. Unreacted iodine is converted to iodide with sodium thiosulfate. The carbonates are precipitated. The iodide-containing filtrates - and the precipitates in are processed in [25] [a]. - 17 - Experiment 4 : Determination of an Empirical Formula Purpose : As stated. Description: Ammonium iodide is reacted with lead nitrate to produce lead iodide. The formula of lead iodide can be calculated from the known percentage of lead in the nitrate and the mass of lead iodide obtained. The lead iodide is converted 16]. back to lead nitrate and iodine in [- - 18 - Experiment 5: Chemistry of the Halogens - 19 - Purpose : This experiment illustrates some chemical properties of the halogens as well as techniques that may be employed to separate halides, and to identify halides. Description: In Set 1 the silver halides of iodine and chlorine are precipitated to illustrate the classic test for halide ion. The mixtures are heated to coagulate the halides and filtered. The filtrates go to [=I. The precipitates are treated with ammonium hydroxide on'-ihe filter. The silver chloride dissolves, the silver iodide does not, illustrating a halide separation technique. The ammoniacal solution of silver chloride and the silver iodide precipitate are treated with hydrochloric acid. The silver chloride reforms. The silver halides go to [-I- . In Set 2, ammonium halides of iodine and chlorine are treated with chlorine generated in situ from acidified bleach. The free halogens are extracted into cyclohexane, yielding a yellow and a violet organic layer, respectively. Phase separation is acheived by passage of the mixture through a water-wetted filter paper. The organic phase is retained by the paper. The aqueous phase goes to waste acid. The organic phases are then treated with aqueous thiosulfate which converts the halogens to the halide ions. The mixture is phase separated, the or anic layer going to and the aqueous layer going to [z] - Comments : By confining the experiments to chlorine and iodine, the extremes of the behavior of typical halogens are illustrated. Fluoride ion is avoided as atypical (see Discussion) and because of its toxicity. Bromide ion is avoided because of the complications introduced into the procedures by its inclusion. The experiment may prove to be too long for a one hour period, and may be brobea:ppi.~nto;-.twa sacperhnts. - 20 - Experiment 6: Reactions of Ions in Solution ORANGE-RED BLACK PALE BLUE YELLOW Purpose : This experiment gives a sense of characteristic precipitates of the various ionic combinations. Description: Salt solutions are mixed and the reactions observed. These are carried out in quantities of several drops on spot plates containing eight depressions. In this manner, the student is able to keep track of the various reactions. The waste products are minimized. Comments : The reactants are in such small amounts that the waste produced is minimal (see Discussion). The waste may be accumulated over a period of years, and processed as a student project. - 21 - Experiment 7: Types of Chemical Reactions I: Combination, Double Replacement Purpose: As stated. Description: is ground Copper obtained the previous year from in a mortar, placed in an evaporating dish covered with aluminum foil and heated under an atmosphere of oxygen to produce copEr oxide. The oxygen is generated as described in [ 1 ] fron 3% hydrogen peroxide. The oxygen thus produced is conducted through a hole in the foil and provides a blanket of oxygen for the copper as it is heated. The hot copper reacts with the oxygen to produce copper oxide. This is allowed to cool and dissolved in hydrochloric acid to produce copper chloride for the next experiment. C O ~ t :S [E] The union of the copper with oxygen exemplifies a combination reaction. The copper oxide is then dissolved in hydrochloric acid in a double replacement reaction. - 22 - Experiment 8 : Types of Chemical Reactions 11: Double Replacement, Decomposition, Single Replacement Description: Copper (11) chloride from [7] is carefully neutralized with sodium hydroxide and then rendered basic to precipitate copper hydroxide. This is heated and the green hydroxide is converted to black copper (11) oxide in a decomposition reaction. The oxide is dissolved in sulfuric acid to produce the sulfate. This is treated with aluminum metal preconditioned with hydrochloric acid to yield copper metal This is removed by filtration and goes to [TI. The filtrate goes Bo]. The reclamation of the copper m z a l illustrates a single replacement reaction. Comments: This experiment produces a number of striking chemical change and a final product of copper, the starting material for [ 7 1 . - 23 - Experiment 9: Activity Series Purpose : This experiment demonstrates the Activity Series of metals in a sequential manner. Description: In sequence copper replaces silver, lead replaces copper, aluminum replaces lead. By heating the solutions, the replacement reactions can be performed in sequence in about one hour. Copper wire is added to silver nitrate solution and heated to precipitate silver. The wire is removed from the mixture, washed and set aside. The mixture is filtered to yield copper nitrate solution and silver metal. This sequence is repeated with the aqueous copper nitrate and lead (lead in the form of shot is useful here). The lead shot is washed free of copper and removed. 'Phe aqueous lead nitrate is heated with scrap aluminum (soda or beer cans) and the process repeated. The aluminum is pretreated with hydrochloric acid. The wire, lead sinkers and scrap aluminumAre reused the following year. The silver powder goes to 1171. The copper powder goes to [TI. - The lead powder g o e s 7 0 [-I. - Comments : The Activity Series is well visualized in this experiment as each more active metal replaces the less active. - 24 - Experiment 10: Titration Purpose: This experiment illustrates the principles of acid-base titration. Description: Sodium hydroxide is standardized with oxalic acid. The standardized solution is then used to standardize hydrochloric acid and determine the percentage of acetic acid in vinegar. Comments: This experiment is mentioned here because the sodium oxalate produced is used as raw material for which produces oxalic acid for reuse in this experiment. [z] - 25 - Experiment 11: Chemistry of Chromium K2C1-207 HOAc H202 K2cr041 (HOAc) HoAc Purpose: , I- (HOAc Cr05 (H202) ( KOAc ) K2Cr207 (HOAc) Cr(OAc13 ( HOAc ) - 1 } KOH, KCr02 (KOAC 1 [El The chemistry of a transition metal is explored. Hydrogen peroxide is shown to act as both an oxidizing and reducing agent . Description: ~ Potassium dichromate is treated with acidic 3 % hydrogen peroxide to form the deep blue peroxo complex. Heat converts this complex to red chromium acetate. Potassium hydroxide precipitates green chromium hydroxide which with excess base is converted to potassium chromite. Basic hydrogen peroxide oxidizes this to yellow potassium chromate which is converted by acetic acid to orange potassium dichromate. Comments : This is a visually interesting experiment because of the color changes. The varied chemical reactions are shown to eventually produce the starting material. Several oxidation states of chromium are achieved. - 26 - Experiment 12: K2Cr207 (KOAc) (HOAc) Fractional Crystallization 1 evaporate chill filter Purpose : This experiment will recover the potassium dichromate and illustrate a chemical separation technique. Description: Aqueous potassium dichromate solution obtained from is concentrated by evaporation and chilled to produce crystals of sparingly soluble potassium dichromate. The crystals are washed with ethanol and dried. The mother liquor is retained and added to the next years dichromate solution (see diagram). Every five years or so the mother liquor should be processed as follows: the solution is treated with acidic hydrogen peroxide to convert all chromium to the trivalent form and this is then replaced with aluminum, the chromium precipitating the solid. The filtrate goes to and the chromium in the third is dissolved in acetic acid and used in [A] step. [z[ [z] - 27 - Experiment 13: The Chemistry of Aluminum, an Amphoteric Metal Purpose : The chemistry and amphoteric properties of aluminum are demonstrated. Description: Aluminum is reacted with sulfuric acid to illustrate its metallic properties. It is then treated with potassium hydroxide to illustrate its amphoteric properties. Sulfuric acid is added to the basic reaction mixture to re-precipitate aluminum hydroxide. The product is used to prepare alum. Comments: The prepared alum may be used in experiments demonstrating stoichiometry of a hydrate and melling point determination or in the preparation of alumina [32]. - - 28 - Experiment 1 4 : Chemistry of Lead [COLOFUESS] [WHITE] [CHOCOLATE] [WHITE] filter Purpose : As stated. Description: Lead powder from -3 reacted with nitric acid to produce lead nitrate. these series of reactions are carried out in a reaction flask equipped with thistle tube and gas exit. The gases conducted through two more flasks containing sodium hydroxide which act as nitrogen dioxide absorbers in this first reaction. The absorbed nitrogen dioxide goes to [=I. [z] - The lead nitrate is carefully neutralized with sodium hydroxide to precipitate lead hydroxide, which is oxidized with bleach to yield lead dioxide. This is converted to lead chloride. Chlorine produced by lead (IV) oxide oxidation of chloride ion is reduced to chloride by sodium thiosulfate. The lead chloride is converted to the less soluble lead iodide with ammonium iodide. The lead iodide goes to [E]. - - 29 - Experiment 15: Solubility of Salts and Saturation: Demonstration seed NaOAc & -- NaOAc T Purpose : The principles of unsaturated, saturated and supersaturated solutions are discussed and the striking phenomenon of rapid crystallization from supersaturated solutions is demonstrated. Description: This experiment represents a closed cycle. Supersaturated solutions of sodium acetate and sodium thiosulfate are retained. When used, the solutions are heated to dissolve all salts and then seeded to produce large crystals of sodium thiosulfate and sodium acetate. Heats of crystallization are observed. Solutions may be used indefinitely. - 30 - Experiment 16: Preparation of Lead Nitrate and Iodine from Lead Iodide Purpose : . Description: This experiment demonstrates some chemistry of lead and iodine, and illustrates chemical synthesis. In the process, lead nitrate and iodine are recovered. Lead iodide is treated with nitric acid in a flask equipped with thistle tube and gas exit tube. The evolving gas is conducted into a flask containing water. The water traps any iodine which escapes the reaction flask as vapor. Heat is applied if necessary to initiate reaction. At first nothing happens, and then the lead iodide begins to disappear, the solution turns brown and violet vapors of iodine are observed as well as brown nitrogen dioxide. The lead nitrate-iodine solution is chilled and filtered to produce a brown solution of lead nitrate and solid iodine. The iodine goes to [ g ] . The brown lead nitrate solution is extracted with cyFiohexane to yield a colorless nitrate and iodine in cyclohexane. The lead nitrate goes and the cyclohexane to (291. to - [XI - 31 - Experiment 17: Preparation of Silver Nitrate from Silver Halides Purpose: The experiment demonstrates the economically important reclamation of silver from insoluble silver halides. Description: Any mixtures containing silver are treated with zinc and sulfuric acid to produce silver metal. The silver is separated by filtration and dissolved in nitric acid to produce silver nitrate. The filtrate is processed in [UI. Comments : The silver nitrate produced here is used in This experiment serves to further purify the silver by chloride formation and subsequent dissolution in ammonia. [z]. - 32 - Experiment 18: Chemical Separation of Zinc Z ~ C JO ~ Na I 7 - 1191 Purpose: This experiment illustrates a chemical separation technique by precipitation of the carbonate of zinc in order to isolate the zinc from other chemicals. Description: Aqueous zinc from all sources is treated with sodium carbonate to precipitate zinc carbonate which is separated by filtration. The Zinc carbonate goes to [TO]. - The 25]. iodide-containing filtrate goes to [ - - 33 - Experiment 19: Redox Titration m4= Purpose : This experiment will demonstrate a redox titration. It also serves todetermine the concentration of iodide ion produced by [ 2 5 ] the previous year to assure that exsss -I . ammonium iodide is used in silver precipitation in [i5 Description: A redox titration is carried out using bleach as the oxidizing agent and starch indicator. The solution is back titrated with sodium thiosulfate to reduce the iodine and from the data the amount of iodide ion originally presentis determined. The chemical products are processed in [ZJ. - 34 - Experiment 20: Electrolysis of Zinc Sulfate ZnC03 H2S04 , ZnSOq CO21 elec. v znh >- , H2S04 wash _j [IT] [Tg Purpose : The experiment illustrates the processof electrolysis and reclaims the zinc necessary for Description: Zinc carbonate and sulfuric acid are charged to a beaker and the zinc sulfate electrolyzed using platinum or carbon electrodes. Beautiful fractal crystals of zinc are produced. A battery charger is used as a source of D.C. current. The zinc is in a form such that a large amount of surface area/mass is pEsent, which renders it particularly suitable €or [LA. [LA. - 35 - Experiment 21: Na2C204 Ion Exchange: cation H2C204 exchange?. Recovery of Oxalic Acid . chilr evap H2C204 $. wash dry ’ [El Purpose : This experiment introduces ion exchange to the student and utilizes it to recover oxalic acid. Description: The sodium oxalate solution from the titration experiment is passed through a cation exchanger to convert sodium acetate to oxalic acid. This is then purified by The fractional crystallization and reused in mother liquor is evaporated to dryness and ignited to carbon dioxide and water. Alternatively, the ion exchange may be skipped and the sodium oxalate evaporated to dryness and ignited. The ash would then be dissolved in water and go to .*..-:::. [a [E]. [,so]:.. - - 36 - Experiment 22: Kinetics: ~ a I 0 3 NaHS03, starch Clock Reaction 12.starch (Na2S04) boil ' I2 4 (K2SO4) chill f ilte? 124 K2SO4 I2 [ZI cyclthexane phase separate Purpose : This experiment will demonstrate the effects of temperature and concentration of reactants on the speed of a chemical reaction. Description: Potassium iodate and sodium bisulfite are reacted in the presence of starch. A deep blue suddenly appears after some seconds. This delayed reaction permits the observation of the effects of time and concentration on the time of color production. The starch-iodine adduct is destroyed by boiling which precipitates iodine. This product is chilled and filtered to remove solid iodine, and the filtrate is extracted with cyclohexane to r z o v e dissolved iodine. Solid and dissolved iodine go to &I. Comments : Students are always intrigued by the sudden appearance of color in this reaction. Boiling of the solution then causes the color to vanish. - 37 - Experiment 23: 0-c r 0 - Synthesis of Methyl Benzoate MeOH + H2S04 OH Phase -te 0-f - ()Me Na2C03 (MeOH) ( 2s04 ) MeOH 0-C . 0-C ** - OMe MeOH Na2S04 04, - ONa 0 ] - 0-CP- OMe (Org.) - ONa 1 I b a wash 0-c" 0-C"' filter MeOH Na2S043 -Me -OH = ____) 1241 [301 Purpose : This experiment permits the student to carry out a simple organic reaction which generates a pleasant smelling product. Description: Benzoic acid and methyl alcohol are placed in a large test tube equipped with a glass tube and stopper to act as an air condenser. Sulfuric acid is carefully added to the mixture. The tube is placed in a boiling water bath and the mixture is reflexed for about one-half hour. The mixture is cooled, washed with sodium carbonate and phase separated. The organic layer is crude methyl benzoate. This is washed with water. The aqueous phases are combined and acidified to produce unreacted benzoic acid. - 38 - Experiment 24: Saponification of Methyl Benzoate 0 0-C* - OMe NaoH , 0 $-C 4 0 - ONa (MeOH) H2S04 ~ 0-c4 - OH cool filter MeOH [ 2 3 1 Na2C03+ MeO€I]+ to neutral ~iiiSO4 Purpose: Another simple organic chemistry experiment that can be performed by introductory students. The chemistry of ester formation is punctuated by its reverse process in this experiment. Description: The ester is placed in a flask equipped with an air condenser with sodium hydroxide (2M) and refluxed for 30 minutes or until the mixture is homogeneous. The mixture is then cooled with running water and carefully acidified and heated to boiling to drive off methanol. It is then again cooled in running w s e r and filtered. The recovered benzoic acid goes to [231. - - 39 - ral, Experiment 25: (NH4Cl) (NaC1) Preparation of Iodine I filter Purpose : Description: (NaCl) cyclohexan phase sepa?ate .This experiment-demonstrates a-commercial process for the preparabion of iodine from seaweed and brine, and serves to separate iodine from-salts. Iodine and iodide ion from all sources are combined and treated with nitrite ion to preferentially oxidize iodide to iodine. The iodine is removed by filtration and goes The filtrate is extracted with cyclohexane to and phase separated to remove dissolvg iodine. The cyclohexane-iodinesolution goes to [La].The extracted filtrate goes to [E]. [z]. - 40 - Experiment 26: Preparation of Ammonium Iodide from Iodine Purpose: This experiment illustrates a chemical synthesis and serves to convert the iodine generated in previous experiments to ammonium iodide, and iodide salt useful in our experimental scheme. Description: Iodine is converted directly to ammonium iodide by reaction with hydrogen peroxide and ammonium hydroxide by mixing at ambient temperature. The cyclohexane layer is removed by phase separation and goes to [ 2 9 ] . The ammonium iodide is freed of ammonia by boiling and then is rendered slightly acidic to neutralize any ammonia remaining. This anrmmnium iodide solution is used the 4 I. following year in [- - 41 - Experiment 27: Synthesis of Sodium Nitrate from Nitrogen (11) Oxide: Demonstration Purpose : This experiment illustrates some chemistry of nitrogen and isolates nitrates produced in previous experiments, Description: A l l nitrate solutions from previous experiments are combined and after iodide ion is removed are concentrated. Reaction with copper is initiated by heat, as evidenced by evolution of brown nitrogen dioxide. The reaction is carried out in a flask equipped with thistle tube and gas exit, which is conducted through two other traps containing cold aqueous sodium hydroxide. The nitrogen dioxide is absorbed, producing sodium nitrate and nitrite. Unreacted copper is removed with forceps and the reacted copper is precipitated by the addition of iron o r aluminum. The solution is filtered to remove the copper, and the filtrate goes to [z]. Comments : This experiment tends to concentrate the nitrates and nitrites so that they may be more easily handled in This experiment may best be a demonstration experimenL 28]. lasting several periods and done in connection with [ - [E]. - 42 - Experiment 28: Kje.fdahl Determination: Demonstration Purpose : This experiment illustrates a classical analytical procedure, and converts nitrates to harmless ammonium salts. Description: The nitrates and nitrites are heated in strong base with an aluminum catalyst to convert them to ammonia. The ammonia is t r a m e d in waste base using the apparatus described in [ 7 1 . Students then titrate the unreacted sulfuric acid and determine the amount of nitrogen originally present in the solution. The digestion portion of the experiment is performed by the instructor as it involves concentrated base. - 43 - Experiment 29: Distillation Cyclohexane ( impurities) distill methanol ethanol (impurities) distill cyclohexane (impurities) methano1 ethanol (impurities) ___) A >- l-5- 1 bo-] k51 - 112.1 bo-1 Purpose : This experiment illustrates a physical method of purification of liquids. Description: Using student-built condensers and distillation columns, cyclohexane is separated from any impurities. Methanol and ethanol are separated by fractional distillation. Connnents : The bottoms from these distillations may be added to the distillation charge for the following year. They are expected to be minimal. The separation of methanol from ethanol is not critical. The methanol is used to synthesize methyl benzoate and any ethyl benzoate formed will not interfere with the experiment in any way. The ethanol is used in conjunction with the fractional crystallization of potassium dichromate. The presence of methanol in the ethanol used will not affect the results of that experiment. - 44 - Experiment 30: Separation of Salts WASTE ACID WASTE BASE to neutral’ filter K+ Mg2+ K+ Description: reduce ,Na2S044 All waste acids and waste bases are combined carefully and neutralized with sulfuric acid or sodium hydroxide as needed. Calcium sulfate preckitates, is removed by filtration and goes to Experiment b 3 ] . The solution is basicified with sodium hydroxide andTeated. Ammonia is expelled and is absorbed in dilute sulfuric acid with phenolpthalein added to indicate exhaustion. Ferric hydroxide and magnesium hydroxide coprecipitate, are removed by filtration, and dissolved in sulfuric acid. The sulfates are treated with activated (that is, pretreated with hydrochloric acid) aluminum. The undissolved aluminum is removed and washed, and the solution heated t o allow any remaining aluminum to dissolve. The precipitated and the iron is removed by filtration and goes to sulfates of aluminum and magnesium go to [AI. m, - 45 - Experiment 31: Preparation of Alumina, an Abrasive, Desiccant and Catalyst Purpose: This experiment illustrates the industrial preparation of alumina and serves to process aluminum from various experiments. Description: Aluminum compounds from various experiments are neutralized with sulfuric acid to the point at which aluminum hydroxide precipitates. After filtration of a small amount of the mixture, the filtrate is tested for completeness of precipitation. When precipitation is complete the aluminum hydroxide is removed by filtration and ignited to alumina. A small amount of magnesium oxide will also be present. - 46 - Experiment 32: NaCl Preparation of Bleach: 1 Demonstration NaClO Purpose: As stated Description: The preparation of bleach is demonstrated by electrolyzing the two salts to yield chlorine and alkali metal hydroxides which react together to yield bleach. This is used to bleach cloth and flowers. The destruction of microorganisms by bleach is demonstrated with a culture of protozoans and a microscope. The bleach is then combined with stock bleach. - 47 - Experiment 33: Applications of Industrial Chemicals CAS04 . 2H20 ( W 4 )2SO4 ALUMINA Fe 02 I__) A , , 2CaS04 . H20 Plant Plaster of Paris [CELLS ] Fiber [PAPER H20 Fez03 [CAST] 1 [ALUMINA-H70 ] Linseed Oil 0 ~ ALUMINA [PAINT ] Purpose: This experiment will illustrate some practical applications of various chemicals produced from Experiment Description: Gypsum is heated to produce plaster of paris from which a mold is constructed. Ammonium sulfate is utilized as a fertilizer for a laboratory plant. Sodium and potassium sulfates are used to size paper. Alumina is used as a desiccant, and iron powder is oxidized to iron oxide from which rouge or paint is manufactured. Comments: The amounts of these products produced in a year is small. These may be collected over a period of several years, and one of the above projects carried out per term. - 48 - Experiment 3 4 : Hydrolysis of Salts Purpose : Determination of the pH of salts Description: Dilute (0.1M) solution of various salts are tested for pH. General rules are developed to predict basicity, neutrality or acidity. Suggested salts are ammonium sulfate, calcium nitrate, sodium chloride, potassium acetate, zinc sulfate, sodium iodide, ammonium chloride, potassium aluminum sulfate. Comments : These salts are not used up in the experiment, and may be used repeatedly each term. - 49 - Experiment 35: Determination of Purity by Melting Point Oxalic Acid(C0OH)z from [El (COOH)2 dissolve) melting point in H20 determination evaporate cool, filter (COOHI2 co2 ignire’H20 Purpose: The purpose of this experiment is to illustrate how the purity of an organic solid may be determined by melting point. This serves to identify the purity of the oxalic acid produced from [XI - and to repurify it as necessary. Description: The melting point of the oxalic acid produced in is determined by placing it in a capillary tube along with another tube filled with oxalic acid of the desired purity. The tubes are attached to a thermometer, placed in an oil bath and heated until they melt. If the oxalic acid is not of sufficient purity for it is recrystallized and the purity established by repeating the described procedure. [u] [E], - 50 - DISCUSSION: EXPERIMENTAL DETAILS In this discussion, the various experiments proposed for the laboratory manual will be discussed and examined. along with the reasons and experimental results that led to their tentative adoption. Letters and numbers in parentheses refer to the laboratory notebook reference, for example (G-1-77-11) refers to the second experiment in laboratory notebook 1, page 77. Experiment 1: Preparation of Oxygen The reaction initially considered for the preparation of oxygen was the classic manganese dioxide catalyzed thermal decomposition of potassium chlorate. The compound is mixed with a small amount of manganese dioxide, placed in a test tube and heated to yield oxygen, which is collected over water and tested in various ways to determine its chemical properties. procedure initially proposed was as follows: The after carrying out the above described procedure, heating would be continued to quantitatively convert all of the potassium chlorate to its thermal decomposition product, potassium chloride. Evidence of completion of reaction would be the cessation of oxygen evolution. The residue would then be extracted with water to remove the potassium chloride, and the remaining manganese dioxide would be removed by filtration and reused the next year for the same reaction. This thermal decomposition of potassium chlorate has been in common useage for decades as the preferred method of oxygen preparation. however, a serious drawback. It has, Potassium chlorate can react explosively if heated in contact with reducing agents, such as dust, cork or rubber, all possible contaminants in an introductory class. It has been suggested that the largest contributor to chemical accidents in academic laboratories is the decomposition of a-chloraEe! - SinaeisafetyLis: of :.primary-cnncern. other methods of oxygen preparation were investigated. A laboratory preparation of oxygen from hydrogen peroxide is described by Hein, Best, and Miner.2 Thirty percent hydrogen peroxide is diluted to 9Xw and its decomposition is catalyzed by manganese dioxide. The method, howevern still possesses a safety risk, and that is the possibility of --chemical burns resulting from skin contact of peroxide in this concentration. 1 2 Burns, C.J., J.Chem. Educ.,Z, 508 (19561, Winderlich, R.J., J.Chem. Educ., 27, 670 (1950). Hein, Best, and Miner, "Foundation of Chemistry in the Laboratory", 6th Edition, Brooks-Cole Publishing Co., p.21 51 - - Consequently, an investigation was undertaken to determine if nonhazardous commercially available 3 % hydrogen ~ peroxide could be used to generate oxygen. A preparation was attempted (6-1-77-1), following the procedure outlined by Hein et a1 but substituting 3% peroxide for the 9%. Into a 250cc flask equipped with thistle tube and gas exit tube was placed 25 mls H20 and approximately 0.5g of manganese dioxide. The mixture was heated (here the procedure departed from Hein) and 3% hydrogen peroxide was added in 15-30 ml portions as needed to keep approximately one hour. Evolution was somewhat sluggish, even with the heat. An additional pro- blem was that manganese dioxide was occasionally forced up the thistle tube a few centimeters due to surges in pressure, and there was sone danger of plugging the tube, which acts as a pressure relief device. The procedure was repeated (G-1-77-11] but without preliminary addition of the 25mls of water. This improved the oxygen evolution rate. An addi- tional flask was added with an inlet tube reaching near the bottom of the flask. to act as a trap for any water that distilled from the reaction mixture. This second flask was charged with 50cc of water. It also served as a monitor of the rate of oxygen evolution, as the bubbles of oxygen were easily observed. With the elimination of the initial 25mls of water charged to the first experiment, oxygen evolution was more rapid and four 150 mls bottles were filled in approximately 45 minutes. Four 15Oml bottles is This procedure still had the . disadvantage of the necessity of reclaiming the catalyst and the attendant sufficient quantity for the usual experiments. problem of potential plugging of the thistle tube. Wikoff and Brown3 report a preparation of oxygen using yeast as a catalyst. Their preparation called for yeast cake. This method was explored. substituting dry yeast cake macerated with water to make a slurry (G-1-78-1). The reaction flask was charged with 15Occ of hydrogen peroxide, and the macerated yeast slurry added to the peroxide. to avoid denaturing the yeast. The mixture was not heated The reaction produced several bottles of oxygen in approximately 30 minutes time but the reaction was sluggish and other methods were explored. Heavy metals will catalyze the decomposition of hydrogen peroxide. With _this in mind, a homogeneous catalytic reaction was investigated using copper chloride as a catalyst. 3 Using the apparatus previously described, Wikoff, H.L. and Brown, J.B., J. Chem Educ., - 52 - 2, 1.434 (1926) approximately 0.5g of the chemical was dissolved in 150cc of 3% hydrogen peroxide, and the mixture gently heated. Evolution of oxygen was not vigorous, and the preparation (G-1-78-11] was discontinued. Also investigated was the acid catalyzed decomposition of hydrogen peroxide using iodide ion as ~ a t a l y s t .Approximately ~ 0.5g of potassium iodide was charged to 150cc of 3% hydrogen peroxide and 20cc of 3m H2SO4 added by means of the thistle tube (G-1-80-11). No visible reaction occurred. The mixture was warmed to about 30C but there was still little evidence of reaction. The feasibility of decomposing 3% hydrogen peroxide using Fe/Fe304 as catalyst was investigated by charging 15Omls of hydrogen peroxide to the reaction flask previously described and adding a rusty iron nail. action mixture was gently heated. The re- Oxygen expelled was again passed through a second flask containing 5Oml of water to act as a trap for any steam generated and to serve as an indication of oxygen flow. As soon as heat was applied, bubbles of oxygen were observed to form on the surface of the nail, and evolution of oxygen was vigorous. Four 150cc bottles of oxygen were collected in about 15 minutes. The reaction was repeated by a laboratory assistant with the same results (61-79-11). This preparation appears satisfactory and is an inno- vative and improved procedure over the classical method for the following reasons : Safety hazards are considerably reduced over the thermal decomposition of potassium chlorate or generation from concentrated peroxide solutions. The expense of the catalyst is essentially eliminated. The 3% hydrogen peroxide is easily available. The catalyst is easily removed from the solution without filtration. The reaction product is water. The oxygen formation on the catalyst is easily observed and is convincing evidence to the student of catalytic activity of the iron. 4 KOtz, J.C. and Purcell, K.F., "Chemistry and Chemical Reactivity" 1st Edition, Saunders College Publishing CO., Philadelphia, PA. 1987, p.475 - 53 - Experiment 2: Preparation of Hydrogen Hydrogen may be conveniently prepared in the laboratory by displacement from a dilute acid by a metal, electrolysis of water, or reaction of an active metal with water. The amounts of hydrogen produced by the latter methods are generally insufficient in the usual allotted time to be conveniently tested by the introductory student and the method of choice, for this reason,is usually the reaction of zinc with acid, and that is the method that is considered here. A typical preparation5 is as follows: Zinc metal is placed in a flask equipped with a thistle tube and gas outlet tube, covered with water, and concentrated sulfuric acid added slowly until generation of hydrogen ensues. The hydrogen is collected by water displacement in a pneumatic trough. The major work carried out on this experimental method concerned not the hydrogen preparation itself, but the reclamation of the zinc from the zinc salts produced in the reaction. Two methods were considered, the electrolysis of zinc salt solutions and the precipitation of the hydroxide followed by the reduction of the hydroxide to zinc metal with carbon at elevated temperature. Of the two methods the former is the simpler and was the method of choice. For an introductory chemistry laboratory there are two simple sources of DC current for electrolysis, an automobile battery or an inexpensive battery charger ($30 - $50) available from auto supply houses. All experi- ments conducted here used a Viking Model VA 7612 Solid State Battery Charger on the 12V setting. A typical hydrogen preparation was carried out by charging 10.7g mossy zinc to a flask, covering the zinc with water (approximately 50cc) and adding lOcc of concentrated sulfuric acid. vigorous. Evolution of hydrogen was The reaction of the zinc was completed by the addition of 5cc more sulfuric acid. The reaction mixture was filtered to remove suspended particulate matter to yield a colorless filtrate. The following series of experiments dealt with finding a practical simple method of recovering zinc from the zinc sulfate solution by electrol- yeis. In the first experiment (G1-34-11), the zinc sulfate was electro- €yzed using the battery charger and two electrodes consisting of pencil , "Laboratory Manual for Chemistry". 1st Edition, Allyn and Bacon, Inc., Newton, Mass. , 1983, p. 89 5 Wagner, M. - 54 - graphite. No effort was made to hold current density or concentration constant. Hydrogen was evolved at the anode, and a dark zinc deposit formed at the cathode, but rather slowly. The procedure was repeated (G-1-34-1111 but continued for a period of approximately 5 hours. At the end of this period the pencil graphite electrodes had disintegrated to the point that one of the electrodes was no longer immersed in the solution. A search was made for more durable electrodes. Two strips of 304 stainless steel were obtained and the procedure described above was repeated (G-1-68-11). The strip used as the anode slowly dissolved and the solution became colored, presumably due to cobalt or nickel present in the steel. Since these metals are undesirable con- taminants, this procedure was discontinued. However, use of stainless for the cathode was promising. Several carbon rods used in arc welding were obtained. These consist of a carbon rod plated with copper with approximately a centimeter of bare carbon at the tip. Using these rods as electrodes, the anode again dis- integrated as the electrolysis was continued (Gl-68-11). A platinum elec- trode was then used as an anode, and the carbon rod as the cathode (G-1-69-1). This arrangement proved satisfactory. When two platinum electrodes were used (G1-69-111), constant useage and removal of zinc from the cathode caused mechanical damage to the electrode, with the platinum eventually becoming separated from the copper wire attached to it. As the zinc is removed from solution by electrolysis, it deposits in beautiful fractal crystals resembling ferns. As this deposit grows and branches, it tends to grow toward the anode, which could cause eventual ehort circuiting of the charger. was sought to prevent this. zinc cathode from the anode. Consequently, a simple inexpensive method A porous cup (6-1-70-1) was used to isolate the Since a quantity of electrolytic zinc produced by this method was needed for work on zinc reduction of silver halides (see Experiment 17), electrolysis of zinc solutions was carried out intermittently over a period of several weeks. The porous cup was frequently removed from the solution and set in a beaker. Zinc sulfate crystals frequently formed on the surface of the cup and presumably within the pores. In any case, after about two weeks useage the porcelain cup began to - 55 - disintegrate. This may have been caused by mechanical strain due to internal crystal formation in the pores. If this were the case, immersion in water when not in use would prevent this. In any case, isolation of the electrodes were sought. A simple salt bridge between cells has been suggested by Howell, et al. 6 A length of 1/8 in. ID (1/32 wall thickness) amber latex tubing was filled with a hot solution of 3% agar in 0.1M zinc sulfate and the solution allowed to cool as gel formation occurred (G-1-71-11]. This cell bridge was used to span two beakers containing zinc sulfate, an electrode in each. Inadequate electrical current was obtained between the cells as judged by the rate of hydrogen evolution from the anode. In another experiment (6-1-71-1) a 6 inch length of tubing was filled with the zinc sulfate solution and plugged with cotton at both ends and placed in two beakers with electrodes. Once again, current density was severely diminished. An experiment (G-1-68-1 and G-1-70-11) was conducted in which a beaker was roughly divided into two cell compartments with a piece of polycarbonate plastic cut loosely to shape. The fit permitted solution circulation but isolated the anode from the cathode directly. An aquarium aerator provided agitation of the solution to minimize electrode polarization. Fractal growth of the zinc in this arrangement was virtually eliminated and the zinc growth took place directly on the cathode. This technique appeared to work well. Having achieved a simple method of zinc recovery by electrolysis, an experiment was conducted to determine the rate of zinc recovery. At this point, consideration was given to avoiding the entire problem of zinc reclamation by employing aluminum instead of zinc. Zinc has pre- sumably been the metal of choice in the past because of its high rate of reactivity, which is necessary to produce the quantity of hydrogen required in the short amount of laboratory time generally allotted to beginning classes. Aluminum has a lower reduction potential (-1.66V) than zinc (-0.76) and consequently should have a high reactivity with dilute acid, as well as producing a relatively non-toxic product, alumina. Aluminum is ubiquitous and scrap may be obtained at essentially no cost (scrap cans, foil, etc.). It does have an oxide coating which renders it unreactive to most dilute 6 Howell, B . S . , Cobb, v.S. and Hacksima, R.A., - 56 - J Chem Educ., 60, 273 (1983) acids. This coating is easily removed by a quick pretreatment in hydro- It is then quite reactive toward dilute sulfuric acid. These experiments demonstrate that fact. A sample of aluminum foil was placed in about lOOcc of lOXw sulfuric acid. No reaction was apparent. In another beaker was placed another lOOcc chloric acid. of sulfuric acid, but about 3ml of hydrochloric acid were added. Hydrogen evolution began almost immediately. In a 250cc flask was placed approximately lg of aluminum foil and sufficient water was added to cover the foil (G-1-67-1). Two ml of conc. sulfuric acid was added to the flask. No reaction was observed. hydrochloric acid was added. No reaction was visible. Five drops of 6M Eventually after addition of a total of 12cc of conc. H2SO4 and 10 drops of 6M HC1 was added, evolution of hydrogen began and built in intensity. The evolution became quite satisfactory for a laboratory preparation. In another experiment (G-1-67-111), aluminum foil was immersed in 6M HC1 until hydrogen evolution commenced (approximately 10 seconds). It was then placed in 10% w sulfuric acid at which point hydrogen evolution became quite vigorous. These experiments have led to the tentative design of Experiment 2 in which aluminum metal is pretreated with hydrochloric acid for a few seconds to remove the oxide coating and then reacted with dilute sulfuric The reason that hydrochloric acid is not acid in the conventional manner. used for the reaction is that fumes from this acid are noxious, and since hydrogen is prepared early in the beginning chemistry course, good laboratory skills have not yet been developed, and danger of hydrogen chloride fumes escaping into the room is greater. In any event, it is proposed that aluminum be substituted for zinc in this preparation because of two distinct advantages. It is much less expensive (at todays prices mossy zinc sells for about $30/lb., aluminum for $.30/lb.) and produces a relatively non-toxic product, aluminum sulfate, which can be converted t o alumina, a major component of earth. The procedures developed for zinc recovery by electrolysis will not -- be for nought, however, but will be utilized in Experiment 20 for recovery of zinc produced from silver halide reduction. - 57 - Experiment 3: Chemistry of the Alkaline Earths This experiment has been developed to utilize a number of reactions of the alkaline earth metals and their compounds in a sequential fashion so that the reactions illustrate the various properties. been divided into two sets. The experiment has In Set 1, calcium and magnesium are reacted with water to produce the hydroxides, which are shown to be bases by litmus paper, and insoluble in water. These are dissolved in hydrochloric acid to demonstrate the solubilities of the chlorides, and calcium sulfate is precipitated with sulfuric acid, whereas magnesium sulfate remains in solution, illustrating a means of separation of the two metals. The resulting mixtures go to Experiment 30. In Set 2 , calcium and magnesium metals are reacted directly with iodine to illustrate the direct combination of the alkaline earths with a halogen. The reaction mixtures are filtered to remove unreacted iodine and the iodides are treated with sodium carbonate to precipitate the carbonates and the sodium iodide is removed by filtration. The carbonates are then dissolved in acid to illustrate their solubility in that medium, and the product goes to waste acid. A l l of the reactions described are straight-forward with the exception of the reactions of the alkali metals with iodine. To investigate this reaction 0.24g magnesium and 3.00g iodine were placed in an evaporating dish with the intention of adding 15cc of water. As the water was being added to the mixture the reaction was initiated, resulting in spectacular emission of purple iodine vapor resulting from the heat of reaction. This would make a good demonstration reaction (61-107-1). The reaction was repeated with the following modifications (6-1-107-11). Iodine (3.03g10.015 mole) was added with agitation. The mixture immediately turned dark brown, and a vigorous reaction ensued with evolution of heat. The dark reaction mixture was filtered to yield a brown filtrate containing the magnesium iodide and a trace of dissolved iodine. The solution was converted to colorless with several drops of 1M sodium thiosulfate and the colorless magnesium iodide solution was ready for treatment with sodium garbonate. - 58 - The reaction of calcium with iodine was carried out as follows (61-108-1). Iodine (3.05g10.012 moles) was placed in lOml of water in a 5Oml flask and calcium turnings (0.60g=0.01 moles) added. An exothermic reaction ensued. The resulting mixture was yellow with a small amount of precipitate calcium hydroxide (white) evident. The mixture was filtered to yield a yellow filtrate which was decolorized with several drops of 0.1M sodium thiosulfate solution. Presence of iodide ion in the filtrate was confirmed by precip- itation of lead iodide by reaction with 1M lead nitrate. The two methods described appear satisfactory for the demonstration of the reactions of calcium and magnesium with iodine, Set 2. - 59 - Experiment 4: Determination of An Empirical Formula In this experiment, lead nitrate is reacted with ammonium iodide obtained from Experiment 26 to yield lead iodide, which is removed by filtration, dried, and weighed. From the data obtained and the amount of lead known to be in the lead nitrate, the empirical formula of lead iodide may be calculated. reaction produces as a by-product potassium nitrate. The In the early stages of this project, an effort was made to eliminate nitrates from the experimental schemes. Consequently, consideration was given to reacting lead chloride instead of lead nitrate with potassium iodide. chloride and lead nitrate. The products would then be potassium Although relatively insoluble in cold water (O.O95g/cc at 20°C), lead chloride increases in solubility considerably in hot water (3.34g/cc at 100°C). rhus, if the concentration is held to this level or below at 100°C, the lead will be in solution. * T o investigate this reaction, 0.75% of lead chloride was added to 5Omls of water and 5Omls of 3% sodium iodide, and the mixture brought to a boil (61-41-1). Addition of 25mls of 1M sodium iodide immediately produced the deep yellow lead iodide. The mixture was decanted and the precipitate washed with three 5Oml portions of hot water and the suspension filtered. was dried and weighed, A recovery of 90.3% was obtained, losses apparently due to solubility of lead iodide in hot water. using 25ml boiling water. losses (61-41-11). the lead iodide The experiment was repeated A recovery of 100% was achieved, including solubility This approach to eliminate nitrate ion, was eventually rejected as cumbersome, and the original experiment using lead nitrate was adopted, subsequent to methods being developed for the handling of nitrates (Experiment 27) and iodides (Experiment 25). 7 "Handbook of Chemistry and Physics", Weast, R.C. and Selby, S.M. Editors, 47th Edition, Chemical Rubber Co., Cleveland, 1964, P.B-187. -_ - 60 - Experiment 5: Chemistry of the Halogens The reader is referred to the Experimental Outline to aid in following this discussion. This type of experiment, which surveys a number of chemical reactions and produces many products, proved to be the most difficult sort of experiment to adopt to the methods used in this report. The author will attempt to guide the reader through the processes which eventually led to the proposed scheme. The original intention was to divide the experiment into four sets of reactions: 1) reaction of halides with calcium ion; this set would contrast the insolubility in water of calcium fluoride to the solubility of the other calcium halides, 2) reaction of halide ion with silver nitrate; this set would contrast the solubility of silver fluoride with the insolubility of the other silver halides, 3) reaction of insoluble silver halides with ammonia to aillustrate the varying solubilities, 4) oxidation of halides to the free halogen with chlorine. Methods used here were those used for each experiment discussed in this report. These were A), create a sequence of reactions instead of the conventional approach of a number of one-step reactions to minimize the number of products necessary to deal with and B), incorporate the reaction products in the rest of the cycle of experiments. Set 11, above was first considered. In the original scheme, sodium fluoride, chloride, bromide and iodide would be reacted with calcium nitrate in aqueous solution. These reactions would yield only one insoluble compound, calcium fluoride, and thus illustrate the marked difference in behavior of the fluoride ion. The problem with this is that calcium fluoride is very difficult to incorporate into further reactions because of its very low solubility and lack of reactivity. The following plans were considered. First, react calcium fluoride with sulfuric acid to produce hydrogen fluoride as a demonstration reaction. This was dismissed as too hazardous. Second, convert calcium fluoride to silicon tetrafluoride with silicon dioxide and sulfuric acid, dismissed for the same reason as the first. Third, bottle the calcium fluoride for some future experiment, rejected as antithetical to the purposes __ of this report. -61 - Since none of these alternatives were desirable, the decision was made to avoid use of fluoride ion entirely. This is justifiable on the basis that the chemistry of fluoride ion is so different from the other halides that it is placed in a different group for purposes of qualitative analysis. reason Set 1) was rejected. Next considered was Set 2. For this In this series of reactions the original intention was to react silver nitrate with sodium fluoride, chloride, bromide, and iodide. The silver salts of the latter three halides precipitate, the fluoride does not. experiment. Elimination of fluoride from condsideration-simplifiesthe Further, if the precipitated silver halides can be easily removed by filtration, then Set 2 could be combined with Set 3 , the reactions of the silver halides with ammonium hydroxide. To investigate the filterability of the silver halides, 5mls of 0.1M silver nitrate was combined with 5mls 1.OM sodium chloride, and the reaction repe'ated with the bromide and iodide. The three resulting mixtures were placed in a boiling water bath for about 5 minutes and the mixtures filtered. The ageing of the precipitates in the water bath was effective and the resulting curdy precipitates were satisfactorily separated from the mixtures by filtration (61-23-1). The satisfactory isolation of the precipitates by filtration meant that they could be treated with ammonia on the filter, and the solubility of the silver halides observed. Silver chloride is completely soluble, silver bromide much less so and the iodide is insoluble. The partial solubility of silver bromide in the amounts of ammonium hydroxide which are practical to use here presents a problem in that some of the bromide will appear in the filtrate and some will remain on the filter. Bromine poses another problem; it com- plicates procedures later used (see Ehperiments 25 and261 for halogen recovery. The decision was made to restrict the halogens investigated to chlorine and iodine. This seems reasonable, the two representing extremes of halogen behavior of the representative halogens, fluorine being atypical. Separation and recovery schemes are considerably simplified. In this fashion the sequence of reactions for Set 1 were conceived. These are the following. The silver halides of chlorine and iodine are precipitated, removed by filtration, and treated with ammonium hydroxide. - 62 - Silver chloride dissolves, silver iodide does not. The dissolved silver chloride is regenerated from ammoniacal solution by acidification with hydrochloric acid. The undissolved silver iodide is washed from the filter paper into hydrochloric acid to demonstrate its insolubility in that medium. The resulting silver halides are processed in Experiment 17. Consideration was then given to the oxidation of halides by chlorine (originally Set 4). This is an important reaction in halogen chemistry, since it is used to prepare iodine and bromine industrially. 8Bleach (aqueous solution of sodium hypochlorite) was chosen as an inexpensive source of chlorine. Reaction of bleach with an acid will produce chlorine in situ, and thus eliminate the hazards of handling chlorine water. The intent of the experiment is to demonstrate the colors of chlorine and iodine in an organic solvent. This can be used as a test for halogens. For example, in carbon tetrachloride, chlorine is yellow, bromine is red, and iodine is .violet. The experiment also demonstrates the oxidizing power of chlorine with respect to halogens of greater atomic number. To carry out this experiment, bleach and an organic solvent (cyclohexane) are added to an aqueous solution of the halide. acid causes the liberation of chlorine. Addition of dilute sulfuric In the case of the chloride, the liberated chlorine preferentially extracts into the organic phase, yielding a yellow solution. In the case of the iodide, the chlorine oxidizes the halogen to free iodine, which extracts into the cyclohexane layer as a violet solution. The organic phases may be separated from the aqueous phase using a simple technique developed. on the funnel. A filter paper is prewetted with water while When a mixture of a water phase and a water-insoluble phase is poured onto the paper, the aqueous phase easily passes through and the organic phase is retained by the paper. The organic phase may then be re- covered by punching a hole in the filter paper and allowing the phase to drain into a container or by simply pouring it from the filter. This method is used in this report whenever there is a need for phase separation. The cyclohexane solutions of halides thus prepared are shaken with a dilute sodium thiosulfate solution to reduce the halogens to halides. The halides and cyclohexane solutions of halogens are processed in Experiment 25. This procedure becomes Set 2. 8 Holzclaw, Jr., H.F., Robinson, W.R. and Nebergall, W.H., Chemistry, 7th Edition, 1984, p.569 - 63 - General A number of experiments were conducted concerning Set 2. In (61-25-1) the suitability of phase separation using filter paper was investigated. Two mls of 0.1M solutions of fluoride, bromide and iodide were treated with 20 drops of chlorine water and 2mls of carbon tetrachloride. phases became, respectively, clear, red, and violet. The organic When the water-carbon tetrachloride mixture was poured on the filter some phase separation took place, but the carbon tetrachloride somewhat hindered the passage of the aqueous phase since it is denser than water and settles to the bottom of the cone. Nevertheless, it worked well enough to be promising. Carbon tetrachloride is now considered undesirable as a reagent for the introductory laboratory, due to its carcinogenicity and particularly because of its immediate toxic effect on the kidneys, and the possibility of absorbtion through the skin in toxic q~antities.~ Consequently substitutes for carbon tetrachloride were investigated. . Experiment (61-24-11) repeated the procedure described above (61-25-1) but using kerosene as the organic solvent. This worked well as the extractant but bromine and chlorine solutions were observed to fade in color with time indicating possible halogenation of the hydrocarbons, an undesirable side reaction producing toxic compounds. As a result, kerosene was not used in later experiments. In (G1-28-I), mineral oil, a petroleium fraction, was used in a repetition of (Gl-25-11. This proved to have the disadvantage of kerosene in that halogen solutions decolorized on standing, and in addition it was difficult to remove from glassware using conventional cleaning procedures. On (61-24-IV), cyclohexane was used as extractant, and this appears to be the most acceptable of the carbon tetrachloride altematives tested. Use of bleach as a chlorine generator was investigated as follows (Gl-24-111). Two m l s of a 0.1M sodium iodide solution was mixed with 2mls of bleach and 2mls of kerosene, acidified with several drops of dilute sulfuric acid and shaken. colorless. The kerosene became violet in color and the lower layer The procedure was repeated, using 0.1M sodium bromide. kerosene layer became red. The The mixture was phase separated with pre- moistened filter paper. The experiments described were carried out to develop the procedures -_ used in Set 2 and described earlier. The final proposed procedure was 9 MeinLloow,J., Keeffe, J.R., and Bemstein, R.L., J.Chem. Educ., 5 8 , All (1981). - 64 - carried out for Sets 1 and 2 (61-291). The total elapsed time was about 4 5 minutes, so it was decided to add another reaction set. selective oxidation of iodide ion by acidic nitrite ion. (Gl-261) demonstrates this reaction. This involved An experiment To 2Omls of 0.1M sodium iodide was added 2mls of 6M nitric acid, 8mls of 0.1M sodium nitrite, and 40 mls of cyclohexane. The mixture was agitated and then allowed to phase separate. Addition of further nitrite produced no more iodine, as judged by the color of the mixture. reaction. Repetition of the procedure with chloride ion produced no This selective oxidation was chosen as Set 3 of the halogen exper- iment. In (61-31-1) the complete experiment was carried out in about 50 minutes, a practical duration. A later modification to the experiment was the sub- stitution of the ammonium salts of the halides for the sodium salts, since ammonium iodide is generated from Experiment 26. - 65 - Experiment 6 : Reactions of Ions in Solution This experiment is conducted to illustrate a variety of insoluble substances produced in solution. The proposed reactions are: 1. reaction of silver nitrate with sodium chloride to produce silver chloride. 2. reaction of mercury (11) chloride with sodium carbonate to produce mercury (11) carbonate. 3 . reaction of barium chloride with sodium sulfate to produce barium sulfate. 4 . reaction of lead nitrate with potassium chromate to produce lead chromate. 5. reaction of mercury (11) chloride with potassium iodide to produce mercury - (11) iodide. 6 . reaction of copper (11) chloride with sodium sulfite to produce copper (11) sulfide. 7 . reaction of calcium chloride with sodium carbonate to produce copper carbonate. 8:reaction of barium chloride with potassium chromate to produce barium chromate. A large number of diverse products are generated in an experiment of this type, making it difficult to fit into the proposed method of this report. The final solution to this dilemma was to reduce the experiment to the semimicro scale so that the amounts of chemicals produced as waste are miniscule. The reactions are carried out on a spot plate, which helps the students to keep track of the reactions. The total amount of chemicals produced per year were estimated as follows. There are eight reactions. Assuming 0.1M solutions and 2 drops of each chemical, an average formula weight of 200g/mole, a class of 30 consisting of 15 students pairslexperiment, the amount of chemicals produced would be: - 8 reactions x 15 experiments x 2 chemicals x 2 drops x ml experiment term reaction chemical 20 drops x Liter lOOOml x 0.1 mole liter x 200g mole - 0.48 chemicals term The amount of waste generated in 200 years of a chemistry class is thus 192g. - 66 - Experiments 7 and 8: Types of Chemical Reactions I and I1 An experiment often referred to as the copper cycle has been widely employedlOas an experiment in introductory chemistry. This procedure consists of a sequence of steps beginning and ending with metallic copper, and generally consisting of the following steps: copper is dissolved in nitric acid, the hydroxide precipitated by addition of sodium hydroxide, the hydroxide converted to copper oxide by heat, the copper oxide converted to copper sulfate with dilute sulfuric acid, the copper precipitated by addition of zinc, the excess zinc dissolved by heat, the copper removed by filtration, dried and weighed. The experiment is generally used to demonstrate the principle of conservation of mass, as near quantitative yields of copper are obtained. "his is the only procedure found by the author in the literature, a portion of which was adaptable to the methods of this report directly, due to its sequential nature. Here it is used to illustrate the basic types of ionic reactions; combination, decomposition, single replacement and double replacement. The first reaction, an oxidation-reduction reaction of copper with nitric acid, does not fit easily into the four basic catagories described. Consequently another reaction of copper metal was sought which would be relatively simple and which would illustrate a combination reaction. Copper is known to combine directly with aqueous ammonia to produce the deep blue tetramine copper (11) ion. This reaction is catalyzed by the presence of air. l1To investigate this reaction approximately 5g of copper foil was immersed in lOOml of concentrated ammonium hydroxide (Gl-8-11). Air was sparged into the solution, and evolved ammonia was trapped in an adjacent flask containing dilute sulfuric acid. overnight. The reaction was allowed to proceed The next morning a deep blue solution resulted containing some precipitated copper hydroxide. The reaction makes an excellent demonstration reaction because of the beautiful color generated. But the conversion of copper (27.4%) was not complete enough to warrant using this reaction as a first step. 10 Condike, G.F., J.Cjem Educ.,X, 615 (1975); Weiner, E., "Fundamentals of Chemistry; Laboratory Studies", 4th Edition, Academic Press, New York, 1980 pp141-151; Davis, Jr.,J.B., MacNab,K.W., McClellan, L.L., and __ O'Conner, P.R., "Laboratory Manual for Chemistry", 1st Edition, D.C. Heath and Co., Lexington, Mass., 1982, pps.58-60, Todd, D. and Hobey, W.D., J Chem Educ.,%, 177, (1985). 11 Holzclaw, Jr., H.F., Robinson, W.R, and Nebergail, W.H., "General Chemistry" 7th Edition, 1984, p860. - 67 - Copper reacts with hydrogen peroxide and acetic acid to produce copper A procedure is described by Umans and deVos.12Two to 3mls of 20% acetate. acetic acid is mixed with 2 to 3mls of 5I hydrogen peroxide and warmed gently without boiling. The reaction is initiated by the addition of copper. The procedure was carried out as described, using copper powder obtained by a single replacement reaction of copper sulfate with iron. This results in a fine copper powder which has a large amount of surface area, and is typical of the copper that will be produced in the experiments in this report. copper was further reduced with a mortar and pestle. The Carrying out the reaction as described, a bluish-green solution was obtained overnight. The unreacted copper was removed from solution by filtration, washed with acetone, dried, A conversion of 3.4% was obtained, too low to be practicable here. and weighed. Copper will react directly with sulfur to produce copper (I) and copper (11) sulfides. 13A reaction was carried out using copper powder prepared as described above (61-83-1). Copper (2.93g) and sulfur (3.01g) were intimately mixed by grinding the two together in a mortar and pestle. The mixture was placed in an evaporating dish, and an attempt was made to initiate reaction by striking a match and plunging it into the mixture, but without success. The mixture was placed on a hot plate and heated for about 3 minutes on high heat. A struck match plunged into the mixture immediately initiated the reaction. The initial incendiary point of black copper sulfide spread to the entire dish in about 30 The reaction was rapid and the yield essentially quantitative (5.90g seconds. copper sulfide). Unfortunately, the reaction does have disadvantages. Sulfur dioxide is evolved, and sulfides are created which can generate poisonous hydrogen sulfide in the presence of acid. Sulfides can be oxidized in acidic media to sulfur. 14This can be affected by permanganate, iodide, dichromate, nitrate ion, etc. in the presence of hydrogen ion. (61-84-1). warmed. A reaction was investigated using 0.5g of copper sulfide Approximately 5cc of 6M nitric acid was added and the mixture Evolution of nitrogen dioxide was observed. sulfide was detected. No odor of hydrogen The reaction mixture was filtered, and the residue, which had the appearance of sulfur was ignited and burned with evolution of sulfur dioxide, 12 Umans, T. and deVos, W., J.Chem. Educ., 2, 52 (1982). 13 Mahan, B . H . , "University Chemistry", 3rd Edition, Addison, Wesley Pub. Co., Menlo Park, Ca. (1975) p710 14 Moeller, T., "Qualitative Analysis", 1st Edition, McGraw-Hill, New York 1958, p221 - 68 - The above procedure is cumbersome. sought. A more satisfactory reaction was Copper will react with the oxygen of the air or pure oxygen to yield copper oxide. It was felt that since the processes in this manual produce a finely divided copper, it might present enough surface to yield good conversions to copper oxide in a reasonable amount of time. If, instead of air, pure oxygen were used as a reactant, the reaction time should be shortened. To acheive this, copper powder (2.07g) was placed in the center of a 2cm. diameter glass tube 20cm. long and stoppered at either end (61-84-11). An oxygen generator described in Experiment supplied oxygen. The copper in the tube was heated with a Meker burner, and the oxygen generator, charged with 3% hydrogen peroxide and a rusty nail, was heated to generate oxygen. The copper in the tube immediately blackened on the surface. The reaction was carried out in 45 minutes. The tube was allowed to cool and the product removed, placed in a beaker, and 5Omls 6M hydrochloric acid added. The mixture was examined 2 days later. At that time, the solid was completely dissolved and a green copper (11) chloride solution was obtained. The above reaction was repeated with the following modifications (61-81 I). The equipment was simplified by replacement of the glass tube with an evaporating dish to contain the copper. Aluminum foil was placed over the dish, form-fitted around the top, and a small hole made in the center to permit entry of the tube containing oxygen. A flask acting as steam trap and bubbler was installed between the oxygen generation flask and the dish containing the copper. Oxygen from the generator was conducted through a tube in this flask which terminated close to the bottom of the flask, which was charged with 5Omls of water. A gas exit tube permitted the oxygen to exit the trap and enter the vapor space above the copper in the dish. excess oxygen escaped to the atmosphere. Since the foil was a loose fit, The dish was charged with 2.21g copper powder, heated, and oxygen passed over the copper for 30 minutes. The dish was allowed to cool, and the contents transferred to a 250cc flask, and 5Oml of 6M hydrochloric acid was added. t o yield a The residue dissolved completely gum solution of copper (11) chloride. This conversion of copper to copper oxide (combination) and then to copper chloride (double replacement) is quite satisfactory for the purposes Intended. The two reactions require about 50 minutes for completion. I 69 - It now remained to employ the standard sequence of reactions of the copper cycle to complete the experiment. 1. These are Conversion of the chloride to copper hydroxide with sodium hydroxide (double displacement). 2. Conversion of the hydroxide to the oxide by heat (decomposition). 3. Conversion of the oxide to the sulfate with sulfuric acid (double replacement). 4. Conversion of the sulfate to copper metal with aluminum (single replacement). This series of reactions was carried out in toto. and physical changes is quite striking (61-87-1). made to the procedure. The sequence of chemical A further modification was An experiment (61-87 11) was carried out to determine if aluminum (oxide coating removed by treatment with hydrochloric acid) could be ‘substituted for zinc (a more toxic and expensive element). Pretreated aluminum (log) was added to 1 5 h l of a solution of copper sulfate and heated. Conversion of the copper sulfate (5Xw aqueous) was complete in about 5 minutes, as evidenced by the change of color from blue to colorless. The excess aluminum was removed by forceps and 25mls of 1M H2SO4 was added to dissolve remaining unreacted aluminum. The mixture was heated for about 5 minutes to complete reaction, and the reaction mixture filtered to yield 3.0g copper. A summary of the reaction sequence follows: 1) copper metal is treated with oxygen to yield the oxide (combination). 2) the oxide is converted to the chloride with hydrochloric acid (double replacement). 3) the chloride is converted to the hydroxide with sodium hydroxide (double replacement). 4) the hydroxide is heated to yield the oxide (decomposition). 5) the oxide treated with sulfuric acid yields copper sulfate (double replacement). 6) the copper sulfate reacts with aluminum to give copper (single replacement). The reaction sequence occupies about 2 50 minute lab periods. It illustrates the four types of simple ionic reactions, conservation of mass, and some chemical properties of copper and its compounds. - 70 - Experiment 9: Activity Series A typical activity series experiment is found in Hein, et all5 In six separate experiments, copper is reacted with silver nitrate, lead with copper nitrate, zinc with lead nitrate, zinc with magnesium sulfate, copper with sulfuric acid, and zinc with sulfuric acid. The approach used in the work reported here is the creation of a sequence of reactions so that products are minimized. on paper. A number of different reaction schemes were initially proposed Most were quite complicated. It was then realized that reduction of this experiment to a sequence of reactions in which a lesser metal is replaced by the more active would not only greatly simplify the scheme, but make the point of the experiment much more accessible to the student. The most promising sequence was: . 1) silver nitrate plus copper yields copper nitrate plus silver. 2) copper nitrate plus lead yields lead nitrate plus copper. 3) lead nitrate plus zinc yields zinc nitrate plus lead. The zinc would ultimately be recovered from the nitrate salt by electrolysis. The silver is eventually converted to silver nitrate, the copper to copper nitrate, and the lead to lead nitrate. The cycle is complete, the increasing activity of the metals easily comprehended. There are several problems with the scheme. Can the reactions be completed in a 55 minute period? Can the unreacted more active metals used in the single replacement be separated from the less active metal produced from the reacting salt? For example, suppose an excess of copper powder is added to silver nitrate to produce a finely divided silver, the copper being in excess to ensure completeness of reaction. and copper. Then the mixture contains both finely divided silver These are not easily separable. An excess of the more active metal i s necessary for completeness of reaction, otherwise the reaction mixture is made even more complicated by containing unreacted salt. problems to be addressed. These were the Replacement reactions are usually carried out at ambient temperature in An experiment was carried out to explore the effect of temperature increase on the reaction rate (61-48-1). Five ml of 0.1M silver introductory courses. nitrate were placed in a test tube and copper in the form of rolled up copper screen was added (2g). 15 The mixture was placed in a water bath at 100°C for Hein, M., Best, L.R., and Miner, R.L., "Foundations of Chemistry in the Laboratory", 5th Edition, Brooks/Cole Publishing Co., Monterey, Ca. 1982, p.75. - 71 - approx,mately complete. 10 minutes. At the end of this period, the reaction appeared The unreacted copper was removed with forceps and washed free of silver with water. The copper was set aside to dry on a paper towel. The reaction mixture containing the silver and copper nitrate was filtered, and the recovered silver treated on the filter with 6M nitric acid, yielding silver nitrate. The filtrate, containing copper nitrate, was treated with lead shot and placed in a boiling water bath and the tube occasionally removed and shaken. The mixture became colorless in about 10 minutes, indicating completeness of reaction. The lead was removed with forceps and washed free of copper with water into the mixture. retrieve the copper powder. The mixture was then filtered to To the lead nitrate solution was added a strip of zinc metal and the sequence repeated. The lead was obtained as a powder. The products resulting from the reaction sequence are then zinc nitrate, silver powder, copper powder, and lead powder, as well as unreacted pieces of copper, lead, and zinc, which are reused. The zinc from the zinc nitrate may be recovered by electrolysis (Experiment20). verted to silver nitrate in Experimentl7. The silver powder will be con- The copper powder will be used as starting material for the copper cycle, Experiment 7. The lead powder will be used as starting material for Experiment 1 4 , The Chemistry of Lead. The problem of separation of the excess of active metal from the precipitated less active metal is resolved by using the more active metal in bulk(i.e., not powdered) form. The former may then be easily retrieved with forceps, and washed free of the less active metal. laboratory time (maxi” The problem of limited time is assumed to be 55 minutes) is overcome by conducting the reactions near the boiling point of water. Another modification made to the reaction scheme is the use of aluminum or iron to substitute for zinc, which reduces the amount of zinc necessary to recover by electrolysis (Experiment 20 1. Experiment 10: Titration In this experiment, oxalic acid is used as a primary standard to determine the normality of sodium hydroxide, used as a titrant in the experiment. This experiment is mentioned here since oxalic acid is part of a cycle represented by--hperiments10, 21, and 36. - 72 - Experiments 11 and 12: Chemistry of Chromium and Fractional Crystallization A sequence of reactions involving chromium was sought, which could be incorporated into a cycle. These reactions should be visually interesting to the beginning student, and demonstrate solubilities, chemical properties, and, ideally, several oxidation states of the element. Solutions of chromate ion are bright yellow. acidified, the orange dichromate ion is formed. When these solutions are This is a powerful oxidizing agent and is reduced readily to chromium (III)!6 Reaction in acidic solution of chromate or dichromate with hydrogen peroxide yields a peroxo compound (contains 0-0 bonds). atures but This compound is comparatively stable at low temper- decomposes in water slowly to yield chromium (111) ion and oxygen. Excess acid favors this. '17Chromium is amphoteric and with excess base chromium hydroxide is converted to sodium chromite. 18Alkaline peroxide will oxidize chromite ion to chromate ion. These reactions suggest a sequence of experiments that might be performed. The first sequence attempted was as follows: 1) sodium chromate is converted to sodium dichromate with sulfuric acid. 2) sodium dichromate is converted to the peroxo complex with acidic hydrogen peroxide. 3) chromium peroxo complex is converted to chromium sulfate with heat. 4) chromium sulfate is reacted with sodium hydroxide to produce first chromium hydroxide, and then with further base sodium chromite. 5) sodium chromite is converted to sodium chromate with alkaline hydrogen peroxide. This sequence of reactions was attempted in (61-36 I). However, it was soon realized that potassium dichromate is a much less soluble salt than either sodium or potassium dichromate. Solubilities are sodium dichromate: 238g/lOOcc at OOC, potassium dichromate: 4.9g/lOOcc at O o , sodium chromate: 50g/lOOcc at 10°C.19Step 1) was then modified to 1) potassium dichromate is converted to potassium chromate with potassium hydroxide. The resulting chromate ion will then be in basic solution, and this must be made acidic for the peroxide reduction to chromium (111) ion. If the acid chosen here is and Humiston, G.E., "General Chemistry", 3rd Edition, John Wiley and Sons, New York, 1975, p671 17 Jolly, W.L., "Encounters in Experimental Chemistry", 1st Edition, Harcourt, Brace Jovanovich, Inc., New York, 1972, p134. 18 Moeller, T. , "Qualitative Analysis", 1st Edition, McGraw Hill, New York, 1958, p.221 19 "Handbook of Chemistry and Physics", Weast, R.C. and Selby, S.M., Editors, 47th Edition, Chemical rubber Co., Cleveland, Ohio, 1964 , p.B-203 and B-223. 73 16 Brady, J.E., - - acetic instead of sulfuric as used in the previous scheme, then the potassium acetate (solubility 253g/lOOcc at 20°C) will be much less likely to cocrystallize in the final step with the potassium dichromate than would potassium sulfate (solubility 12g/lOOcc at 25OC). Therefore acetic acid was substituted for sulfuric acid, formerly introduced in Step 1). Finally, a Step 6) is added in which potassium chromate is converted to dichromate with acetic acid, and the potassium dichromate is isolated by crystallization. The modified reaction scheme then becomes: 1) potassium dichromate is converted to potassium chromate with potassium hydroxide. 2) potassium chromate is converted to the chromium peroxo complex with acetic acid and hydrogen peroxide. 3) chromium peroxo complex is converted to chromium acetate with heat. 4) chromium acetate is reacted with excess sodium hydroxide to eventually produce potassium chromite. 5) potassium chromite is converted to potassium chromate with alkaline hydrogen peroxide. After several modifications the experiment described below (61-111 I) was carried out, and represents a successful application of the sequential reaction descussed. Potassium dichromate (2.82g) was placed in 5Oml of water in a 50Oml beaker. Five cc of glacial acetic acid was added and then 50cc of 3% hydrogen peroxide. then reddish black. The solution turned dark blue immediately, then black, Oxygen evolved. of oxygen was evolved. The mixture was heated and a small amount Fifty mls of 6M KOH were added and the solution turned an emerald greem (chromium (111) ). Fifty m l s of hydrogen peroxide were added. The solution, over about a three minute period, turned from emerald green to light green to gold in color. The mixture was heated to reduce the solution to about fifty mls in volume. It was cooled and 27mls of glacial acetic acid added at which point the solution turned from gold to orange indicating conversion of the chromate to dichromate. added. Three m l s more of acetic acid were The volume of the solution was now 70 mls. This was chilled and filtered. The dichromate was washed with two 25ml portions of ethanol and dried on low heat to constant weight. seating at 118% yield. The yield of potassium dichromate was 3.338, repre- The obvious contaminant was potassium acetate, which will not interfere with the cyclical use of the potassium dichromate. This contaminant can undoubtedly be reduced by reduction in the amount of potassium hydroxide used. - 74 - The mother liquor from the experiment is added to the next year's crude potassium dichromate. Ever so often, the mother liquor should be treated with hydrogen peroxide to reduce the chromium to chromium (111), the hydroxide precipitated and removed by filtration and added to the chromium acetate of Experiment11 hydroxide. , making sure sufficient acetic acid is present to dissolve the This procedure will rid the cycle of excess potassium acetate. Alternately the chromium can be reduced to chromium (111) and precipitated with aluminum and the potassium acetate removed by filtration. The complete experiment occupies about 2 hours and may be separated into 2 experiments, the first being the series of chemical reactions and the second the fractional crystallization of the potassium dichromate. The reaction sequence exhibits chromium in two oxidation states and colored compounds of yellow, orange, deep blue, green and red. The sequence of the two experiments is a complete cycle. Experiment 13: The Chemistry of Aluminum, an Amphoteric Wetal This procedure was developed by a combination of a sequence of reactions of aluminum and its compounds, partly based on a procedure described by Summerlin, et a1.20A sequential scheme was developed for aluminum in which the metal is reacted with potassium hydroxide to produce hydrogen and potassium aluminate, this converted to hydroxide with sulfuric acid and with excess to alum, potassium aluminum sulfate, which precipitates in large crystals. -_ This procedure illustrates an industrial process. 20 Summerlin, L.R., Borgford, C.L. and Ealy, J.B., "Chemical Demonstrations", 1987, p54. Vol 2, American Chemical Society, Washington, D.C., - 75 - Experiment 1 4 : The Chemistry of Lead Lead is easily brought into solution with nitric acid in a reaction impressive to students, in which nitrogen dioxide is evolved. is one of the few soluble lead compounds, lead (11) nitrate. The product Lead (IV) oxide can be synthesized by oxidation with bleach of lead (11) hydroxide. compound is a chocolate brown color. This This compound in acidic solution is a strong oxidizing agent, capable of oxidizing chloride ion to chlorine.fO Thus lead (IV) oxide is converted to lead (11) chloride with hydrochloric acid. Lead chloride in the presence of iodide ion will form the less soluble lead iodide.21The lead iodide is the link that can complete the cycle. The lead iodide is converted to lead nitrate in Experiment 16, and then to lead metal in Experiment 9 , completing the cycle. The complete cycle is then lead (silver metal) to lead nitrate (colorless solution) to lead hydroxide (white solid.) to lead (IV) oxide (chocolate-brown solid) to lead chloride (white solid) to lead iodide (yellow solid) to lead nitrate (Experiment 1 4 ) to lead (Experiment 9 ). The set of reactions would illustrate a number of compounds of lead, interesting physical and chemical changes, the two oxidation states of lead, and the oxidizing power of lead dioxide. To investigate this series of reactions, 0.5g of lead foil was covered in a beaker with concentrated nitric acid and heated to initiate reaction. A total of 5 d of nitric acid was added to dissolve all of the lead. Fifty m l s of water was added and the solution heated until all of the nitrogen dioxide was expelled, as indicated by the color of the solution. Twenty ml of 6M potassium hydroxide was added to precipitate lead hydroxide, which appeared as a creamy white precipitate. When heated this formed curds and settled. Ten m l s of bleach were added and the precipitate immediately darkened and upon further heating turned chocolate brown, indicating formation of lead dioxide. acid. To this was carefully added, in the hood, 3Omls of 6M hydrochloric Considerable chlorine was evolved and upon heating the mixture slowly turned from brown to off-white (lead chloride). was added. One gram of sodium iodide At first a yellow precipitate formed but this turned t o white and brown iodine was apparent, formed by oxidation of iodide by the chlorine present. 20 Brady, J.E. and Humiston, G.E., "General Chemistry", 3rd Edition, John Wiley and Sons, New York, 1975, p. 21 Moeller, T., "Qualitative Analysis", 1st Edition, McGraw-Hill, New York, 1958, p.277 - 76 - A small amount (0.5g) of sodium thiosulfate was added which immediately caused a beautiful yellow precipitate to form, and the supernatent liquid to become clear. The lead iodide was removed by filtration (61-109 I). Because of the evolution of nitrogen (IV) oxide and chlorine from the reaction mixture, the following modifications were made to the above procedure. The reaction was conducted in a 2.5cm ID test tube equipped with thistle tube and gas exit tube. The gas exit was conducted to a trap consisting of a 250cc flask equipped with an inlet tube terminating near the bottom, and a gas exit The trap was initially charged with 10% sodium hydroxide to trap tube. oxides. nitrogen One half gram of lead was charged to the tube and 5ml of concentrated hydrochloric acid added to the flask. oxides and complete the reaction. Heat was applied to expel the nitrogen The sodium hydroxide was removed from the flask and replaced with 1M sodium thiosulfate to absorb chlorine. Six molar hydrochloric acid (3Omls) was added and the tube heated until no more chlorine whs evolved. Approximately 2mls of 1M sodium thiosulfate was added via the thistle tube to reduce all dissolved chlorine. the tube and 1.0 g sodium iodide added. The stopper was removed from The mixture was heated. The yellow precipitate of lead iodide formed. This reaction was further modified to essentially eliminate chlorine evolution as follows: the above procedure was repeated, reducing the amount of lead charged to 0.25g. The sequence of reactions was carried out, halving the amounts of reagents added. After the lead (IV) oxide was formed, 0.5g of sodium thiosulfate was added to the mixture to reduce chlorine as formed. Hydrochloric acid was then added to yield lead chloride without evolution of chlorine. The rest of the reaction was carried as described above. Experiment 15: Solubility of Salts and Saturation: Demonstration This is an instance in which the solutions may be used indefinitely. In this experiment, solutions of sodium thiosulfate and of sodium acetate are heated unci1 all the solid is dissolved. They are then cooled to room temp- erature to supersaturate them, and they are ready for demonstration of saturation, supersaturation, supercooling, and heat of crystallization. A thermometer is placed in the solution and equilibrated. The solutions are then seeded with well-formed crystals whereupon a shower of crystal forms and a riee in temperature is observed. The solutions are then stored for the following year. - 77 - Experiment 16: Preparation of Lead Nitrate This conversion of lead iodide to lead nitrate and iodine was considered one of the key experiments to the success of this method. Lead iodide is produced in Experiments 4 and 14. Lead nitrate is used in Experiments 4 and 9 . The iodine produced from the process here is converted to ammonium iodide, which is used in Experiments 4, 5 and 19. The first method to be explored was the attempted oxidation of lead iodide with nitric acid. One gram of lead nitrate was dissolved in 5Omls of water. To this was added 2.20g of potassium iodide in 5Omls of water. The mixture was stirred and lead iodide was allowed to settle, removed by filtration, and washed with two 25ml portions of water, two 25ml portions of ethanol and dried with a heat gun (61-42 I). Lead iodide thus prepared was placed in a 250 ml flask with 25mls of water. Twenty five mls of concentrated nitric acid was added to the mixture. there was no visible sign of reaction. At first The flask was placed in a boiling water bath and in about 3 minutes the violet vapor of iodine was observed in the vapor space. The lead iodide dissolved with evolution of iodine vapor. minutes the reaction mixture subsided. mixture. In several Crystals of iodine were suspended in the In the first attempts, the iodine was removed from the reaction mixture by extraction with kerosene, mineral oil, cyclohexane, followed by phase separation. This had the disadvantage of requiring considerable volume of the organic extractant and this procedure was modified. The reaction mixture was chilled to ice temperature, the iodine removed by filtration, and the filtrate extracted with the organic solvent. Extraction in this fashion produced a clear colorless solution of lead nitrate, a precipitate of solid iodine, and an violet organic phase containing dissolved iodine. A quantitative experiment was conducted to determine the yield of lead nitrate with several concentrations of nitric acid, 15M, 6M, and 3M. was prepared by dissolving 2.03g lead nitrate in 25ml water. was removed by filtration, washed, and dried. Lead iodide The precipitate Three portions, 0.7Og each, were treated with two ml each of the three concentrations of nitric acid. tative yields ( 0 . 5 4 g ) of iodine were obtained in each case. Quanti- It was necessary to heat the 3M nitric acid a little longer to complete reaction, but all reactions were complete in several minutes. - 78 - These reactions were carried out in a 250 ml flask equipped with thistle tube and gas outlet. The gas was conducted through two flasks equipped with inlet tube leading to near the bottom of each flask, and a gas exit tube. These flasks were each filled with lOOmls of cold water. This reaction produces lead nitrate which is evaporated to dryness and used in Experiments 4 a n d 9, and iodine, which is converted to ammonium iodide used in Experiments4 Experiment 17: , 5 and19 . Preparation of Silver Nitrate from Silver Halides Because of the high cost of silver, a number of articles have appeared in the literature concerning reclamation of silver from its various compounds. any of these methods use high temperature reactions (500-10OO0C), or hazardous material (cyanides, silver-ammonia solutions, concentrated base or aqua regia). 25-27 Experiment 5 produces both silver chloride and silver iodide, and a method was.sought to easily convert these compounds to silver, from which silver nitrate map be easily obtained. iodide to iodine. MoellerZ8reports that copper (11) selectively oxidizes Copper (I) iodide, a rather unreactive solid is unfortunately also produced in the process and would present problems of incorporation into the experimental cycle. An industrial method of iodine preparation consists of the following scheme: A silver nitrate solution is added in sufficient quantity to brine to precipitate only silver iodide, which is removed by filtration and treated with clean steel scrap to form metallic silver and a solution of iron (11) iodide. ,The iron (XI) iodide is then treated with chlorine to yield ferric chloride and iodine.29 This approach seemed promising, that is, treatment of silver iodide(and silver chloride) with iron to yield silver and iron (11) iodide. The iron (11) iodide could be used instead of other iodide salts (sodium or potassium iodide were originally considered) for the reaction in kperiments 4, 5, and 19. The first step of the reaction investigated was the conversion of the silver chloride to the less soluble silver iodide, which was known to react with iron to yield silver. 22 Wilbanks, B.L., J Chem. Educ. 2,347 (1953) 23 Steed, S . P . and Hayes, J.M., J. Chem. Educ. 49, 156 (1972) 24 Bush, J.B., and Diehl, H. , J. Chem. Educ., 5 c 54 (1979) 25 A m o u r , M/A., J. Chem. Educ., 65, A66 (1988) J. Chem. Educ., 63, 537 (1986) 26 Rawait, J.P. and Kamoonpuri, S.I.M., 27 Hill, J.W. and Bellows, L., J. Chem. Educ., 63, 357 (1986) 28 Moeller, t., "Qualitative Analysis", McGraw-Hill, N.Y., 1958, p231 29 Kotz, J.C. and Purcell, K . F . , "Chemistry and Chemical Reactivity", 1st Ed. Saunders College Publixhing Co., Philadelphia, Pa., 1987 p851 79 - - Fifty mls of 0 . 1 M silver nitrate were treated with 70 mls of 0.1M hydrochloric acid to yield a white precipitate of silver chloride. of 0.1M silver iodide was added to the light grey residue. heated on the hot plate for 10 minutes and then filtered. obtained. One hundred mls The mixture was A dark filtrate was To determine if this color was due to iodine, a portion of the filtrate was extracted with mineral oil to yield a clear aqueous layer and a violet upper layer, confirming iodine, probably formed by nitrate ion oxidation (61-9 I). To the precipitate of silver chloride was The experiment was repeated. added 10Omls of 0.1M sodium iodide. of conversion to silver iodide. A yellow-gray mass was obtained indicative The mixture was brought to a boil to complete conversion of the silver chloride to silver iodide. removed from the filter and placed in 25mls of water. The precipitate was washed, Two ungalvanized iron nails, cleansed of rust by pickling in hydrochloric acid, were added to the precipitate and the mixture heated on a hot plate for 10 minutes and then filtered. The dark filtrate was extracted with mineral oil to yield a clear aqueous layer and again a violet upper layer, indicating the presence of a small amount of free iodine. It was felt that the iodine produced in the above reaction was due to nitrate ion oxidation. The procedure was altered to include this step (61-13-A2) and the darkening due to iodide oxidation was eliminated. Silver iodide conversion was determined in several experiments and the Silver nitrate (0.77g) was dissolved in 5Omls following is typi,cal (61-10-1). of distilled water and a solution of 0.99g of sodium iodide in 5Omls of water was added with stirring. Light yellow silver iodide precipitated immediately. The silver iodide precipitate was placed The mixture was heated and filtered. in 50mls of water. Three steel screws were cleaned by brief immersion in hydrochloric acid, rinsed, and added to the silver iodide. the mixture was A black deposit of silver formed immediately agitated with a magnetic stirrer. around the metal. The mixture stood for several days. At the end of this period a grey-black precipitate remained. The steel screws were removed with forceps and washed with water. cold. The precipitate was treated with 15mls 6M HNO3 There was no visible reaction. nitrogen (IV) oxide was evident. The mixture was warmed and evolution of Within about 2 minutes the black precipitate haii disappeared and in its place a small amount of yellow-white precipitate remained (unreacted silver iodide). The mixture was cooled and filtered. - 80 - The weight of unreacted silver iodide remaining on the filter was 0.09g representing a 91.5% conversion of silver iodide. orating the silver nitrate solution to dryness. An This was checked by evapamount of 0.77g of silver nitrate representing an 80.5% conversion was obtained. The lower yield was attributable to some loss due to spattering of the silver nitrate. The procedure described above was repeated with steel wool, cleansed by pickling with hydrochloric acid, rinsed, and immersed in 5Omls of water containing silver iodide prepared from 0.75g of silver nitrate. Silver chloride recovered amounted to 0.49g, representing a 75.4% conversion of silver iodide (Gl-74 I). Faust30reports a method of recovery of silver chloride residues by reaction with zinc in acid solution, based on procedures previously reported in the literature. 3l,32Meyer reports that zinc and iron both reduce silver iodide to silver, but that its reduction is less easy and less complete than in the case with silver chloride. The procedure described by Faust f o r silver chloride is as follows: A suspension of the compound in water is vigorously stirred with granular zinc and this is added until only a flocculent gray solid remains in the reaction vessel. Once gas evolution ceases, the mixture is filtered and the grey silver residue is washed with an aqueous solution of 25% sulfuric acid followed by water. The reaction is reported to be complete in 5 minutes. The procedure was carried out, using electrolytic zinc produced from Experiment 20. This zinc is in the form of metallic threads of zinc, or fractals and presents considerable surface area as does granular zinc, recom- mended in the Faust procedure. In a typical experiment, 0.47g of silver nitrate was dissolved in 5Omls of water and added to 1.02g of sodium iodide in 5 h l s The precipitate silver iodide was heated to coagulate the precipitate, cooled, and lomls concentrated sulfuric acid. The solution turned an ochre water. color, probably caused by nitrate oxidation of a small amount of iodide ion. 30 Faust, D.F., J. Chem. Educ., 61, 924 (1984) Chem Abst., 19, 1827 (1925) 31 Meyer, A . , Chem. Ztg., 49, 22?-(1925), 32 Mellor, J.W., "A Comprehensive Treatise on Organic and Theoretical Chemistry", V01.111, Longmans, Green and Co., N.Y., 1923, p.314 See Reference - 81 - The mixture was agitated by air sparging. (2.09g) was added. Electrolytic zinc from Experiment Immediately a black scum formed on the surface of the In approximately 30 seconds, chunks of black substance were evident. mixture. No yellow-white precipitate of silver iodide was evident (61-15 I). The reaction mixture was decanted and the supernatent filtered. A golden mother liquor and grey precipitate were obtained. To the precipitate was added lOml of 6M nitric acid. The mixture was warmed on the hot plate. No reaction was noted. Evolution of nitrogen (IV) oxide was observed and the dark grey pre- cipitate disappeared leaving a small amount of light grey precipitate. The mixture was filtered, washed with 25ml of water, then 25 ml of acetone and dried with a heat gun. The filtrate was tested with 6M hydrochloric acid and confirmed the presence of silver nitrate. The unreacted silver iodide- silver (light grey precipitate) was dried to constant weight to obtain 0.07g unregcted residue. Assuming the residue is silver iodide, this represents an 861 conversion of silver iodide. This reaction was immediately put to practical application (61-17 11). Filter paper containing silver residues contaminated with other substances (zinc and chromium for instance) were ashed to obtain 38.81g of black ash. This was placed in a 40Oml beaker, 15Oml of water and 3Oml of concentrated sulfuric acid were added. The mixture was placed on a steam bath and heated for one hour to dissolve any soluble metals. The mixture then stood overnight and the next day was washed with two lOOml portions of water. was added to a chromium/zinc waste container. The filtrate Four hundred m l s of water were used to transfer the solids from the filter to a 60Oml beaker. Nitric acid (25ml) was added and a vigorous evolution of nitrogen (IV) oxide ensued. The mixture was heated for about 30 minutes and filtered to yield a green (chromium) filtrate. Ten grams of sodium chloride in lOOcc of water was added to form large curds of silver chloride. The mixture was allowed to settle. A small amount of dilute hydrochloric acid confirmed completeness of precipitation of the silver. The precipitate was filtered and washed with several 5Oml portions of water. Enough 10% sulfuric acid was added to precipitate all the silver as the sulfate. The sulfate was removed by filtration, placed in a 250 ml beaker, and 100 mls of water and then lOmls of concentrated sulfuric acid added, followed by log of zinc metal. - 82 - The mixture was agitated and a black residue forned. The agitation was stopped because of excessive foaming. After approximately one hour the evolution of hydrogen subsided. An additional ml of concentrated sulfuric acid was added to insure completeness of reaction. The grey silver residue was removed by filtration. Nitric acid (6M) was added dropwise on the filter to convert the silver metal to the nitrate salt. The silver nitrate solution was evaporated to dryness to yield 2.64g of silver nitrate. The described method of zinc reduction seems quite satisfactory for the reclamation of silver from silver residues. It uses zinc produced from the experimental sequences, the conversions of silver iodide are good and the proAn experiment was carried out using a mixture of halides cedure is simple. to determine the conversion of the mixture. Three halides, sodium chloride, sodium bromide, and sodium iodide were weighed out in the amounts of 0.50g each. Silver nitrate in the amounts of l.OOg, 0.64g, and 0.44g were dissolved in 5Omls of water and the three halides added to each beaker respectively. The halides were combined and 3.10g sodium iodide was added to convert the yellowish white to the less soluble silver iodide. The mixture was digested over low heat for about 2 hours. period the precipitate was isolated and washed by decantation. After this One hundred fifty mls of water were added followed by 2.03g zinc produced by electrolysis (Experiment 20). Ten mls of concentrated sulfuric acid were added and the mixture stirred vigorously. After the bulk of the hydrogen was evolved, 3mls of sulfuric acid were added and the mixture heated until all evolution of hydrogen ceased. The mixture was then decanted and washed with 5Omls of water. The filtrate was saved for G-2-11-1. nitric acid. The precipitate wae treated with 33% When evolution of nitrogen (IV) oxide ceased the mixture was heated for about 10 minutes and then filtered. The precipitate (unreacted silver halides) was washed with acetone to remove water, and then air-dried to constant weight. The amount of unreacted halide was 0.45g giving a 79.2% conversion of silver halide (6-2-9-1). Recovery of silver as silver nitrate was carried out by adding sufficient nitric acid (12.5M) to dissolve the silver, and simply evaporating the nitric acid solution to dryness. When no more fumes were evolved, the solution was cooled, the crystals removed from the beaker and bottled. - 83 .. In summary, silver may be recovered from a mixture of silver halides by reduction of silver with zinc in the presence of sulfuric acid. The silver is dissolved in nitric acid and the silver nitrate obtained by heating the mixture until fumes of nitric acid are no longer expelled. be expected. Conversions of 75Xw may Unreacted silver halides are added. Experiment 18: Chemical Separation of Zinc Solutions of zinc sulfate, zinc iodide and zinc chloride combined with sulfuric and hydrochloric acids, and ammonium chloride are produced in Experiment 17. A convenient method for separation of the zinc appeared to be precipi- tation of an insoluble zinc compound, perhaps the carbonate or hydroxide. To explore this possibility, 15Omls of the filtrate from a silver halide-zinc reduction (Experiment 17) was treated with approximately 7Omls of 1 0 % ~sodium carbonate until foraming cleared. Twenty mls additional sodium carbonate solution was added to precipitate the zinc as zinc carbonate. ous mixture was heated for 10 minutes considerabie settling of the solid. t3 The white gelatin- coagulate the precipitate. This caused The mixture was filtered through Whatmann 54 filter paper, yielding a white precipitate resembling mashed potatoes. The zinc carbonate was dissolved in 1.5M sulfuric acid (17mls) to give a colorless solution of zinc sulfate ready for electrolysis (G-2-11-1). This procedure represents a satisfactory method for isolation of zinc from its contaminants and the experiment consumes about 45 minutes. Experiment 19: Redox Titration This experiment accomplishes the very practical purpose of determining the percentage of ammonium iodide in the solution produced from Experiment 26, so that an excess of iodide ion may be added to lead nitrate in Experiment This is critical to the quantiAt the same time, the student is introduced t o precipitate all of the lead as the iodide. tative nature of Experiment 4. to an oxidation-reduction titration. In this titration, an excess of sodium hypochlorite is added to the iodide solution and acidified. Starch indicator immediately reacts with free iodine to yield the deep blue starch-iodine guest-host compound. The iodine is back titrated with thiosulfate and the percentage iodide is determined. Alternately, alkaline hydrogen peroxide may be used as the oxidant with the remainder of the procedure being identical. The iodide present in the titration mixture is reclaimed in Experiment 25. - 84 - Experiment 20: Electrolysis of Zinc Sulfate The methods developed in Experiment 2 are utilized here to reclaim zinc from zinc sulfate. The procedure consists of electrolyzing the zinc sulfate solution with a 12V source (a battery charger is convenient) using a carbon welder's rod as the cathode and a platinum electrode as the anode. Zinc in the form of delicate fern-like shapes is produced, which presents a large amount of surface area and works well in Experiment 17, in which silver halides ara reduced by zinc to silver metal. Experiment 21: Ion Exchange, Recovery of Oxalic Acid The experiment involves reclamation of oxalic acid converted to the sodium salt in Experiment 10 by titration with sodium hydroxide. In this scheme, the sodium oxalate is passed through a cation exchange resin in the hydrogen form to produce the oxalic acid. The column is then regenerated with acid and the sodium expelled as sodium chloride. To exmaine the effectiveness of this procedure DOWEX 50W-X8, H+ form was charged to a column by slow addition of 2ml/min. of a slurry of the resin. Several column volumes (25ml) of 3M HC1 were passed through the column, followed by sufficient distilled water to remove completely all the free chloride as tested by silver nitrate. A 1.06g sample of oxalic acid was placed in a 250 cc flask, 5Omls water added, and the mixture taken to the phenolpthalein end point with 0.5M sodium hydroxide. A flask containing a drop of 0.5M sodium hydroxide and phenolpthalein indicator was placed under the column. Change in color of the indicator signals the presence of acid. oxalate was charged to the columns. The sodium Thirty-five m l s of effluent remained pink but after addition of 4Omls the mixture in the receiver was colorless. A different receiver was placed under the column and approximately lOOmls of effluent was collected. Titration with 0.5M sodium hydroxide showed 1.Og of oxalic acid to be present in the effluent for a yield of 94.5%. - 85 - Experiment 22: Kinetics: Clock Reaction In this classical reaction3? iodate ion is reduced to iodine which is reduced to iodide ion until the reductant is exhausted. reacts with starch to form a colored complex. The iodine then The concentration of reactants may be adjusted so that the color change takes place only after a number of minutes have expired. The sudden color change is impressive (colorless to deep blue) and permits the kinetics of the reaction to be explored. Effect of time and temperature may be determined, and a pseudo rate constant and activation energy may be calculated from the data. Destruction of the starch-iodine inclusion adduct is necessary to isolate the iodine from the reaction mixture at the conclusion of the experiment. For example, iodine does not extract into an organic phase in which it is soluble, when bound to the starch. An experiment was carried out to determine if hydrolysis of the starch by sodium hydroxide would destroy the complex (61-99-1). Five g of sodium hydroxide was added to 15Omls of the reaction product from the clock reaction,prepared by addition of 5Oml of a solution containing 4.3g potassium iodate per liter to 50 ml of a solution containing 4g of soluble starch, 0.2g of sodium hydrogen sulfate and 5nl of 1M sulfuric acid diluted to 1 liter. faded to colorless. The solution was allowed to stand, and the blue Further literature research revealed that the hydroxide ion reacts with starch-iodine complex to yield iodide and iodate ion, both colorless. 34 In another experiment, the starch-iodine complex was heated to determine if this would destroy the complex(G1-89-1). in (61-99-1). became The complex was prepared as The mixture after boiling for about 30 seconds suddenly colorless. Either of these two methods is satisfactory for complex destruction. The iodine may then be recovered from the solutions by chilling to ice temperatures and filtering. This removes the bulk of the iodine. The brown filtrate is then extracted with cyclohexane to remove the last traces of iodine (61-99-111). Summerlin, L.R. and Ealy, Jr., J.L., "Chemical Demonstrations", Vol 1, 2nd Edition, American Chemical Society __ Washington D.C., 1988, p.107 34 Ophandt, C.E., J. Chem. Educ., 64, 9, 808, 1987. 33 See for example: - 86 - Experiment 23: Synthesis of Methyl Benzoate Preparation of an ester is a standard experiment for an introductory class in chemistry, since it is easily accomplished and produces a goodsmelling product, sometimes identifiable. of a 2 - This preparation becomes part experiment cycle in which the benzoic acid produced in Experiment becomes the raw material for this experiment. Methyl benzoate is prepared according to standard procedures .35 Modifying the procedure used in Cason and Rapoport, 2.5g of benzoic acid and 5.Oml methanol were placed in a-large test tube and 8 drops of concentrated sulfuric acid were added with constant swirling. was fitted with stopper and 20cm x 5 The test tube I D glass tube as reflux condenser. By suspending the tube above a boiling water bath, a gentle reflux was sustained for 30 minutes. The mixture was washed with 25mls of 10% sodium carbonate and phase separated. The crude methyl benzoate was washed with a 25ml portion of water and again phase separated. The sodium carbonate layer was carefully neutralized with 6M hydrochloric acid to produce 0.70 of unreacted benzoic acid. obtained ( G l - 1 0 2 - 1 ) . A 72% yield ( 2 . 0 0 g ) of methyl benzoate was This crude ester was then used in the hydralysfs experiment. Experiment 24: Hydrolysis of an Ester The ester prepared in Experiment 23 is saponified here to yield the original alcohol and acid. by the following experiment. The procedure is standard and is exemplified The amyl acetate ( 2 . 0 g ) was placed in a 125ml Erlenmeyer flask with 2Oml of 2M sodium hydroxide and the flask fitted with a reflux tube. The sample was heated for 30 minutes at which time the sample was homogeneous. The solution was cooled and acidified with 6M hydrochloric acid and heated to boiling. It was then cooled and filtered and the benzoic acid (1.62g) washed with cold water (6-1-103-1). The benzoic acid is used in Experiment 23. -. See, for example, Cason, J. and Rapoport, H., "Basic Experimental Organic Chemistry", Prentice-Hall, Inc., Englewood Cliffs, N.J., 1964 p.90 - 87 - Experiment 25: Preparation of Iodine The filtrates from Experiments 3, 4, 18 and 19 contain iodides. The primary purpose of this experiment is to recover these water soluble iodides as water insoluble iodine, and thus afford a means of separation from the other components of the mixtures, which include ammonium chloride, sodium chloride, sodium sulfate, and sulfuric acid. The oxidation of iodide may be affected with either sodium nitrite and nitric acid, or with acidic hydrogen peroxide. To investigate the usefulness of sodium nitrite, 15Omls of the filtrates obtained from Experiments 3, 4, 18, and 19 were treated with 2 g of sodium nitrite and 25mls of 6M nitric acid. and iodine precipitated. filtered. The mixture immediately turned brown The mixture was cooled to ice temperature and A brown filtrate was obtained which was extracted with cyclohexane to yield a colorless filtrate and a violet cyclohexane solution (G2-21-1). Iodine (0.82 g) was obtained as a solid. The reaction was repeated using 5Omls of 3% sodium hypochlorite and 25mls of 3M H2SO4. solution Iodine (0.79 g) was obtained after chilling and filtering the . - a0 - Experiment 26: Preparation of Ammonium Iodide from Iodine This is a key experiment in the cyclical schenes explored in this report. Iodide ion and iodine are produced in a number of procedures and these species must be converted to a suitable soluble iodide that may be conveniently used in the cycle. The final procedure adopted is the reaction of ammonium hydroxide and hydrogen peroxide with iodine to yield ammonium iodide. The experiments that led to this final adoption will be described. The first reaction explored was the reaction of calcium hydroxide with iodine. The expected reaction products were calcium iodide and a precipitate of calcium iodate. The solubility of calcium iodate is 0.20g/lOOcc at 15OC whereas calcium iodide is soluble to the extent of 209g/lOOcc at 20OC. The precipitation of the iodate from the solution was expected to drive the reaction to the right. Filtration of the mixture should yield calcium iodate as a solid and a solution of calcium iodide. . At this point in the experimental program, it was intended to extract all iodine into an organic phase as the means of isolation of the element. For this reason, the calcium hydroxide reaction was carried out heterogeneously with a kerosene solution of iodine (G-1-53-1). Ten g of calcium oxide was suspended in water and agitated for approximately one hour to prepare a saturated calcium hydroxide solution. the clear limewater decanted. The solution was allowed to settle and Fifty mls of a 3% solution of iodine in kerosene was mixed with 5Omls of limewater. minutes. The mixture was agitated for 30 At the end of this period the solution was partially emulsified. It was placed in a boiling water bath which partially broke the emulsion. The experiment was modified. Twenty-five mls of 3% iodine in cyclohexane was briefly shaken with 25mls of limewater (6-1-53-11). of the iodine disappeared almost immediately. upper layer and a clear lower layer. The violet color In this case a white turbid The upper layer was heated in a water bath to demulsify it, but it remained turbid and this approach was abandoned. Iodine is reduced by sulfite ion in the presence of acid.36The reaction products of the direct reaction with sodium sulfite in the presence of sulfuric acid would be sodium iodide, sodium sulfate and any sulfuric acid and sodium sulfite in excess. The sodium iodide could be separated by the following scheme. Excess sodium sulfite would be destroyed with acid to 36 Hoeller, T., "Qualitative Analysis", 1st Edition, McGraw-Hill, New York, 1958, p.216 - a9 - yield sodium sulfate and sulfur dioxide. The excess acid could then be neutralized with sodium hydroxide to yield sodium sulfate. The two com- ponents of the mixture at this stage would be sodium sulfate and sodium iodide. Solubilities of these two compounds are greatly different in ethanol. Sodium iodide is reportedly soluble to the extent of 43g/lOOcc whereas sodium sulfate is reportedly insoluble .37Extraction of this mixture with ethanol after evaporation to dryness should then produce an alcoholic solution of sodium iodide. This entire procedure was explored. Iodine (0.5g) was placed in a test tube and l h l s of 1M sodium sulfite added. Addition of 5mls of 3M sulfuric acid with agitation brought about solution of the iodine, indicating reduction. The resulting solution was yellow, probably due to the presence of a small amount of triodide (6-1-57-1). To determine the efficacy of separation of sodium iodide from sodium sulfate, 2.01g of sodium iodide and 1.52g of sodium sulfate were placed in a 25Oml beaker and extracted with l 0 h l s ethanol by agitation for 45 minutes. The solution was allowed to settle and the ethanol solution re- moved by decantation. dried. The precipitate was washed with 25mls of ethanol and The yield of remaining sodium sulfate was 98.0% (1.49g), illustrating a nearly perfect separation of the iodide from the sulfate. Because the sulfite reduction and selective extraction with ethanol for preparation of sodium iodide is rather involved, other procedures for pre38 paration of iodide salts from iodine were investigated. Moeller reports sulfide ion will reduce iodine to iodide. Sulfur and sodium hydroxide are To examine this reaction, 5Omls of 8.1M sodium sulfide solution was added to 5Omls of 1% iodine in kerosene and the reaction mixture the by-products. sparged to agitate the solution. The kerosene lost color almost immediately. The lower layer was yellow and the upper kerosene layer colorless. After phase separation, the solution was boiled to coagulate the sulfur, but this was not successful. of it. Passage through charcoal helped to remove the bulk At this point the procedure was abandoned. 37 "Eandbook of Chemistry and Physics", Weast., R.C and Selby, S.M., Editors, 7th Edition, Chemical Rubber Co., Cleveland, 1964, P.B224 and B-226. 38 Moeller, T., Qualitative Analysis", 1st Edition, McGraw-Hill, New York 1958, P.221 - 90 - Iodine will react directly with zinc dust to produce zinc iodide.39 Iodine (1.Og) was dissolved in 5Oml This reaction was explored as follows: kerosene and lOOcc of water in a 25Oml flask. Electrolytic zinc produced from Experiment 20 was added and the mixture sparged with air overnight to agitate the mixture. The next morning the iodine had vanished and a brownish kerosene layer and clear lower aqueous layer remained. complete reaction. This indicated This reaction looked quite promising since the zinc iodide contained no by-products, and therefore no further separation scheme was necessary (6-1-58-1). Another promising reaction was the conversion of iodine to ammonium iodide with ammonium hydroxide and hydrogen peroxide?' carried out in the following manner. This procedure was One gram of iodine was reacted with 28mls of 10% ammonium hydroxide (double the stoichiometric quantity) and 6Omls of 3% hydrogen peroxide. oxygen was evolved. Most of the iodine dissolved readily and Another 2Oml of 3% hydrogen peroxide was added. iodine completely disappeared and the reaction mixture became yellow. All The solution was evaporated on the steam bath to yield colorless crystals in near quantitative yield (2.25g, 98,6% yield) (6-1-56-I). The ammonium hydroxide/hydrogen peroxide reduction of iodine to ammonium iodide procedure has been adapted because of the ease and rapidity of the reaction, and the lack of by-products other than water. This reaction is used to convert iodine from all sources to a conveniently used iodide salt, which is used in Experiments 4, 5, 14 and 19. Experiment 27: Synthesis of Sodium Nitrate from Nitrogen (IV) Oxide This experiment is designed to isolate the nitrates and nitrites from admixtures by converting them to nitrogen (IV) oxides by means of copper and heat. They are reabsorbed in sodium hydroxide to produce a solution of sodium nitrate in sodium hydroxide. This resulting basic solution of nitrate ion may then be used in the classical quantitative analysis of nitrate ion by reduction to ammonia using aluminum as the catalyst. This destroys the nitrate and nitrite ion and converts them to the much less toxic ammonium ion. The procedure illustrates some nitrogen oxide chemistry and an indus- trial method for the preparation of nitrates. 39 The evolved gases may also "Handbook of Preparative Inorganic Chemistry", ed. G. Brauer, 2nd Edition, Academic Press, New York, I, 1963. - 91 - be absorbed in water to illustrate the preparation of nitric acid. The nitric acid is then reacted with an excess of base to prepare the solution for Experiment 27, or the prepared nitric acid may be reused in the various experiments after appropriate concentration. An experiment was carried out to explore to nitrogen (IV) oxide reaction. Sodium nitrate (Z.OOg), nitric acid (1.OOg) and sodium hydroxide (1.1Og) were placed in a 25Oml flask, dissolved in lOOmls of water, and 5.00g of copper foil added to the mixture. A stopper equipped with gas outlet tube was placed in in the flask, and the evolved gas conducted through a tube near the bottom of the second flask containing lOOmls of 0.5M sodium hydroxide. second flask contained a vent to the atmosphere. This Fifteen nls of concentrated sulfuric acid was added to the first flask containing the nitrates, and evolution of nitrogen (IV) oxide began immediately. The reaction was allowed to proceed overnight, at which time the reaction had ceased and a blue solution of copper sulfate was obtained. No odor of escaping nitrogen (IV) oxide was ever detected during the procedure. To confirm the presence of nitrate in the sodium hydroxide absorber, the solution was made strongly acidic by addition of 3Omls of concentrated sulfuric acid and 5.0g of copper foil was added. The mixture was heated and the copper partially dissolved to yield a solution of copper sulfate with evolution of nitrogen dioxide. - This appears to be a satisfactory method for nitrate removal from other salts. Experiment 28: Kjeldahl Determination of Nitrogen This standard method of nitrogen determination may now be applied to the product of Experiment 27. necessary. The usual digestion procedures are un- In this procedure, nitrogen compounds are reacted with strong sodium hydroxide and a catalyst, in this case aluminum, to yield ammonia. The ammonia is usually absorbed in standardized acid, and back titrated with standard base to yield the nitrogen content. be expected here. No difficulties are to This procedure serves to convert the toxic nitrates to the lees toxic ammonia, which ends up as ammonium sulfate, a well known fertilizer. - 92 - Experiment 29: Fractional Distillation In this experiment students use a self-constructed distillation column and condenser to purify cyclohexane and ethanol contaminated with water. The principles of distillation are discussed. Clean separations are not a prerequisite and the major contaminant, water, will not interfere in moderate proportions with subsequent use of the chemicals. Experiment 30: Separation of Salts In this experiment, the waste acids, bases, and salts are combined, brought to neutrality with either sodium hydroxide or sulfuric acid as needed and processed by this and Experiments 32 and 33 to yield chemicals which may be utilized in several projects (see Experiment 34). This serves to bleed the system. . The various ions in the combined mixture are sodium, calcium, potassium, ammonium, iron (III), magnesium, chloride, and sulfate. precipitates and is removed by filtration. Calcium sulfate Addition of sodium hydroxide precipitates iron and magnesium hydroxides which, after heating the mixture to coagulate the precipitates, are removed by filtration. In addition, heating of the basic solution removes ammonium ion as ammonia. This is absorbed in dilute sulfuric acid to produce ammonium sulfate, used in Experiment 33. The precipitated ferric and magnesium hydroxides are dissolved in acid and heated with activated aluminum to precipitate iron. After digestion to remove unreacted aluminum, the mixture is filtered to isolate the iron, which is washed with water and dried. to Experiment 31. This goes to Experiment 33. The filtrate goes The filtrate from the precipitated iron and magnesium hydroxide is reduced in volume, chilled and the crystallized sodium and potassium sulfates are removed by filtration. Solubilities of potassium chloride and sodium chloride are listed as 34.7g/lOOcc water at 20°C and 35.7g/lOOcc water at O°C respectively, and for potassium sulfate and sodium sulfate decahydrate, 12g/lOOcc water at 25OC and llg/lOOcc water at ll°C respectively. If the separation proves difficult, the chlorides may be separated from the sulfates by use of a cation exchange resin. This experiment illustrates some principles of chemical separations of common cations. No experimental work has yet been done on this set of reactions. They are all well known and no major problems in the separations are expected. - 93 - Experiment 31: Preparation of Alumina, an Abrasive, Desiccant, and Catalyst A l l products containing aluminum are conbined, which includes aluminum sulfate, alum, and sodium aluminate. Magnesium sulfate is also present, and is immediately precipitated as magnesium hydroxide. The mixture is quite basic, and the pH is adjusted with sulfuric acid to the point at which aluminum hydroxide also precipitates. A small portion of the mixture is filtered and the filtrate tested for completeness of precipitation. When enough sulfuric acid has been added to precipitate all of the aluminum, the mixture is filtered, the precipitate washed and the solid evaporated to dryness and ignited to alumina and magnesia. This goes to Experiment 33. The filtrate contains potassium sulfate which also goes to Experiment 33. The point of the experiment is to prepare a mixture consisting largely of alumina which will be employed as a desiccant in Experiment 33. Experiment 32: Preparation of Bleach In this experiment sodium and potassium chloride are electrolyzed with 12V DC current to produce chlorine and sodium and potassium hydroxides. This procedure simulates the industrial preparation of bleach. The generated chlorine reacts with the hydroxides to yield potassium and sodium hypochlorite, the active ingredient in commercial bleach. Since a fair amount of chlorine is generated, this reaction should be demonstrated by the instructor. The bleach thus generated may be used to destroy microorganisms, and'its action observed by aid of a microscope. Objects such as cloth may be bleached. The apparatus used in this experiment is identical to that used in the electrolysis of zinc sulfate, Experiment 20. - 94 - Experiment 33: Applications of Industrial Chemicals This experiment utilizes the non-recycled chemicals produced in the entire series. Plaster of Paris is created from calcium sulfate dihydrate by ignition to dicalcium monohydrate. The students then use this to form a mold, and perhaps cast a reproduction in lead. Ammonium sulfate is used to fertilize a plant which has been grown from seed using ammonium sulfate as the sole nitrogen source. sizing to make paper. Sodium and potassium sulfates are used as The mixture of alumina and magnesium oxide is used as a dessiccant to demonstrate that moist ammonium sulfate becomes dry in its presence, and then used as a general purpose desiccant f o r future laboratory work. Iron powder is used to demonstrate magnetic lines of force, and then donated to the physics department or used as a source of iron for the plant fertilized with ammonium sulfate; Another choice is to oxidize it to iron (111) oxide and use it to make red paint or rouge. It should be pointed out that the quantities of these chemicals that are produced by each class are rather small. For example, the amount of calcium sulfate dihydrate produced in a year is the order of magnitude of 5g/yr. As a consequence, the chemicals can be allowed to accumulate for several years, and one or two utilized per year year thereafter. Experiment 3 4 : Hydrolysis of Salts Solutions of salts are reused each term for pH determination. See the Experimental Outline for more details. Experiment 35: Determination of Purity by Melting Point This experiment serves to determine the purity of the oxalic acid produced in Experiment 21, and thus, whether a fractional crystallization is necessary. - 95 - DISCUSSION: Waste Products A number of waste products were generated from this work. These were isolated into the following categories. Zinc wasteIIodide Waste Iodide Waste Chromium Waste Iodate/Iodide Waste Copper Waste Lead Waste/Iodide Waste Cyclohexane Waste, Acetone Waste, Ethanol Waste, Kerosene Waste Silver Waste Acid Waste Base Waste The waste was collected in one gallon plastic jugs. Processes were developed for the recycling of these wastes, in general using procedures developed for the various experiments. These will be discussed in the order listed, and an example of the process used will be included. * a) Zinc Waste/Iodide Waste: Zinc iodide was produced in a number of cases, hence the iodide was a necessary contaminant. One gallon of zinc waste containing zinc sulfate, zinc chloride and zinc iodide was concentrated to about 5OOml. Three hundred mls of 3% hydrogen peroxide was added to produce a pre- cipitated iodine. The mixture was chilled with ice water and filtered into another one gallon plastic jug through an ordinary coffee filter. filtrate was obtained. acid to yield 4.218. A brown The iodine was dried in a desiccator over sulfuric The filtrate was extracted with 2 15Oml portions of kerosene to yield an essentially colorless filtrate and a violet kerosene solution of iodine. The kerosene solution (ca 30Omls) was treated with 40mls of 10% ammonium hydroxide and l 2 M s of 3% hydrogen peroxide. The phases were agitated with air supplied by an aquarium aerator overnight. The next morning a colorless (slightly yellow) cyclohexane layer was removed by phase separation, leaving a colorless aqueous phase containing ammonium iodide, unreacted hydrogen peroxide and ammonium hydroxide. dryness to yield 1.30g of ammonium iodide. This was boiled and evaporated to The clear zinc-containing aqueous layer, after extraction with cyclohexane, was electrolyzed with a 12 volt battery charger, a carbon cathode and platinum anode to yield 12.lg electrolytic zinc - a feathery product with considerable surface area. The cyclohexane was placed in a separate container to be recovered by distillation.(6-2-11-11). b) Iodide Wastes: Approximately one gallon of iodide waste containing ammonium iodide, sodium iodide, potassium iodide, iron iodide and free iodine - 96 - was distilled and the distillate trapped in a flask surrounded by an ice bath to trap the vapours. The volume of the solution was reduced to 50Omls. To this mixture was added 25g sodium nitrite and 25mls concentrated nitric acid. Iodine was immediately precipitated, the mixture chilled to ice temperature and filtered to remove the iodine. To the mixture was then added lOOg copper scrap and 25 mls concentrated sulfuric acid and the mixture heated to decompose nitrates. Nitrogen dioxide was evolved and absorbed in cold water to yield A yield of 12.55g iodine was obtained a mixture of nitric and nitrous acids. including distilled iodine and iodine formed after the nitriteinitrate oxidation (6-1-34-1). c) Chromium Waste: Three liters of this waste was concentrated by heat to a volume of about 1 liter. This was made strongly basic by the addition of 30g of potassium hydroxide. A flacculent precipitate was obtained which did not dissolve in base, presumably iron hydroxide. settle by decantation over several days. This was allowed to The bulk of the supernatent liquid was removed by decantation, and the precipitate filtered after the addition of 10 grams of filter aid. The solution obtained was treated with 30Omls of hydrogen peroxide in increments of 5Omls. The mixture was carefully heated to destroy excess peroxide and then made acidic with acetic acid as indicated by the change in color of solution from yellow to orange. acetic acid was added. Fifteen m l s excess Some potassium dichromate immediately precipitated. The mixture was reduced to about lOOmls of volume, chilled to ice temperature, and filtered to remove crystals of potassium dichromate. The crystals were washed with two 25ml portions of ethanol and oven dried. A yield of 23.7g of potassium dichromate was obtained. The mother liquor was saved and added to the waste dichromate container (6.2-36-1). d) Iodate/Iodide Waste: All solutions containing iodide/iodate wastes were treated with an excess of sodium thiosulfate to reduce all iodate to iodide. This solution was then combined with b) Iodide Waste and treated as described earlier. e) Copper Waste: Copper was removed from these solutions by single replacement with aluminum activated with hydrochloric acid. For example, to approximately 1 liter of copper waste was added 2Og of aluminum foil which had -_ been activated by previous immersion in concentrated hydrochloric acid. - 97 - The mixture was heated until all colot was gone. The foil was washed with water to remove precipitated copper and removed from the solution. Twenty- five mls of concentrated sulfuric acid was added to dissolve remaining aluminum, and the solution heated f o r approximately 1 hour. The mixture was then filtered to remove the copper, the copper washed on the filter with acetone to remove the water, and air dried to yield 24.6g red copper powder. the filtrate is at present being reduced to dryness by solar evaporation. f) Lead WasteIIodide Waste: This mixture contained solid lead iodide. Four liters of this mixture was treated with lOOmls of concentrated nitric acid and heated on the steam bath. The mixture was chilled to ice temperatures and the iodine removed by filtration, washed with water and dried over sulfuric acid to yield 11.6g iodine. to precipitate lead. The filtrate was treated with 15g aluminum foil Unreacted aluminum foil was washed to remove lead and removed from the solution. One hundred m l s of concentrated sulfuric acid was6added to the mixture to dissolve unreacted aluminum. heated for approximately one hour and filtered. The mixture was The silvery lead powder was washed with acetone and air dried to yield 13.3g of lead. The filtrate is being reduced to dryness by evaporation. g) Organic Wastes: All organic wastes were distilled separately to yield, respectively, purified cyclohexane, acetone, ethanol and kerosene. The distil- lations were carried out in a 5OOml round bottom boiling flask equipped with a 50cm Vigreaux column and condenser. h) Silver Waste: The processing of this waste has been reported in the Experimental Discussion of Experiment 17. i), j) Acid Waste/Base Waste: These were used to neutralize each other to pH 7 and are in the process of being evaporated to dryness. - 98 - DISCUSSION: Cost Reduction Reduction in costs of operating an introductory chemistry laboratory using the methods outlined in the NOWASTE manual were calculated by the following procedure. The steps in the calculation were as follows: The manual selected for comparison was "Laboratory Manual for Chemistry'' by Maxine Wagner, Allyn and Bacon, INc., Newton, Mass, 1983. 1. This manual was selected for three reasons: The author considers it to be an excellent manual, covering all major topics in the course. 2. The manual is used by our local high school 3. The manual contains a list of all chemicals and their amounts used. This is included as Appendix 111. The costs of the chemicals used for experiments which produce waste chemicals was calculated. Cost for the other items listed represent one-time only costs, since these items are used repeatedly (for example, copper, lead or aluminum pellets for density determinations). The total costs for one class of thirty students (15 student-pair teams) to purchase chemicals which are used each term came to $333 (see Appendix 11). The amounts of chemicals used in the NOWASTE manual were estimated and the costs of all chemicals calculated assuming a 10% and 20% loss in those chemicals which are recycled. These costs totalled $3.00 and $3.45 per student team (see Appendix 111). These costs were multiplied by 15 student teams to give $45.00 and $52.00 per class for the NOWASTE manual. Since the Wagner manual contains 39 experiments which produce chemical waste and the NOWASTE manual contains 34 experiments which produce chemical waste, the costs of the NOWASTE manual were prorated by 39 = 1.147 to yield a cost comparable to the Wagner manual. These 3F costs came to $S2/class for a 10% loss and $60/class for a 20% loss in recycled Chemicals. Savings in costs were then calculated as follows: 10% loss NOWASTE manual ($333 $52) x 100 84% reduction - 1 ($333 - $60) x ( $333 1 ( 20% loss NOWASTE manual $333 - 99 - - in cost. 100 = 82% reduction in cost. G) If costs of disposal by sanitary landfill are included in the estimates, savings are even greater. The costs of packing, transporting, and disposing of "lab p a c k ~ ' * ~ ~ai n"secure" EPA-regulated landfill has been estimated to be $23/gal in a 1985 publication. waste from school labs end up as solutions. Normally, chemical To estimate the costs of chemicals disposed as wastes produced from the Wagner manual, the following procedure was used: 1) The total waste in grams was calculated = 7300 g 2) This was prorated from 39 to 34 experiments for reasons described 7300 x 34 = 6350 g 39 3) Aqueous solutions of these chemicals of 25Xw were assumed. 6350 = 25,400 g solution. 0.25 4) A density of 1 . 5 ~was assumed. Volume = 25,400 x 15 = 38,lOOml ml or 3.8 L. or 38 qts = 40 qts. or 10 gallons. 0.946 5) 10 gallons x 23 = 230 gal Thus, disposal costs are close to 2/3 the costs of chemicals. above. 6) If disposal costs are included in teh costs of a conventional chemical lab then savings by use of the NOWASTE manual are increased to: - 10% less NOWASTE manual (333+230)-(52) 333+2 30 x 100 20% less NOWASTE manual (333+230)-(60) 333230 x 100 = 89% 90% In s u ~ ~ p p ~savings ry, in costs by use of the NOWASTE manual are estimated to be between 80%-90% of the chemical costs for the term. excludes costs of new equipment due to breakage, etc., This estimate encountered in all labs. There may be some minor capital costs incurred upon adoption of the NOWASTE A battery charger makes a good source of 12V DC current for the electrolysis experiment (Experiment 20). For example, a Viking Model VA 7612 manual. Solid State Battery charger is available at the local auto supply house for about $40. Twelve and 6V automobile batteries may also be used. A pair of platinum electrodes is also needed for Experiment 20 and are available, for 40 In a "lab pack", 1 gallon bottles of solutions are placed in 55 gallon drums with vermiculite filler. 41 Walton, W.A., J.Chem. Educ., 64, A669, (1987); "Less is Better"; ACS Task Force on RCRA: Washington D.C., - 100 - 1985 example, from Central Scientific Co., catalog number 81275 for $17.50. A set of 48 500 ml capacity plastic bottles will provide enough storage for all the solids and solutions generated in the NOWASTE procedure (see DiscussionStorage Space). These bottles are available, f o r example, from Van Waters and Rogers, catalog number 16126-278, at 6 for $16.45 or $131.60 for 48. Total costs for these purchases is approximately $200. Payout for this expense is approximately one semester. No special training for instructors is anticipated. are standard. All techniques used Familiarization with the manual is necessary, as with any new laboratory manual. Also, no special approval by any state agency is required for adoption of a new laboratory manual in high schools or community colleges in the State of C a l i f ~ r n i a . ~ ~ 42 -. Telephone conversation with James Eurst, Board of Directors, Fort Bragg Unified School District, 12/16/88 and with L. Kavanaugh, Dean of College, College of the Redwoods; Mendocino Branch, Fort Bragg, CA 12/17/88. - 101 - DISCUSSION: Impurity Buildup The possibility that impurities will build up in some of the cyclical It is extremely difficult to answer this question without actual experimental evidence, and this is one of the major problems that will be addressed in future work on this project. However, in general, some educated guesses may be put forth. The first point to be made is that _ _ the experiments in this manual which are cyclical in nature involve a sequence of reactions. These reactions systems must be examined. are characteristic for that particular element, and thus tend to be purification schemes. For example, let us follow lead through the cycle of reactions starting with Experiment 14. Outline of Experiments will help the reader follow this discussion. Lead might be accompanied by impurities of copper, antimony, arsenic, bismuth, gold and silver, since these elements are present as it is obtained from the blast furnace. The physical separation that occurs in this experiment is a filtration after addition of ammonium iodide in acidic medium (HC1). Assuming all elements mentioned above are present, this reaction would probably produce soluble copper since copper (11) iodide does not exist46 and copper (I) iodide is soluble in a~id,~~soluble antimony and bismuth since the iodides of these elements decompose in hot water,48and soluble arsenic, probably present at this stage as arsenic acid. Species accompanying the lead iodide as precipitates might be gold metal and silver iodide. The filtered lead iodide is next reacted with nitric acid in Experiment 16 to yield soluble lead nitrate and iodine as a precipitate. Here the gold would join the iodine as an insoluble residue (if present in sufficient quantity) as would the silver iodide. The lead nitrate has thus been freed of impurities arising from the lead. (The iodine which contains the gold and silver iodide is reacted in Experiment 26 with hydrogen peroxide and 'ammonium hydroxide to yield water soluble ammonium iodide.) The gold and silver iodide are insoluble in water and would appear as a precipitate at this point, easily removable by filtration. 46 Holtzclaw, Jr., H . F . , Robinson, W.R. and Nebergall, W.H., "General Chemistry", 7th Edition, D.C. Heath andCo., Lexington, Mass. (1984) 47 Ibid., p. 8 6 3 , p. 896 48 "Handbook of Chemistry and Physics", Weast, R.C. and Selby, S. M., Editors, 47th Zdition, Chemical Rubber C o . , Cleveland Ohio ( 1 9 6 4 ) p. B-174 - 102 - Consider chromium, present in Experiments 1 1 and 12. A common impurity is iron, originally present in the chromite ore in which chromium is usually found in nature. In Experiment 11, the chromium ion is treated with strong base to convert it to the hydroxide and then to the soluble chromite salt. Under these conditions, iron would appear as the insoluble iron hydroxide, which would not dissolve in excess base. This could be easily removed by filtration. Because of many possibilities of impurities introduced in the various reagents, and the variety of reactions which might occur under varying conditions, these sort of arguments must be viewed as speculative, and best settled by experimentation. Nevertheless, the general conclusion may be drawn that the series of reactions specific to the elements will tend to purify them. A second point to be made is that most chemicals used are of reagent grade purity, and as a consequence, impurity buildup to the point of interference should be quite slow. For example, the analysis of "Bakers Analyzed" copper to have maximum analyzed impurities of 0.01%. In order for these impurities to build up to a level of detectable interference, assuming they all interfere and a level of say lX, would require 100 cycles, assuming replacement of all the copper each cycle and no loss of impurities. This represents 100 terms of chemistry classes. A third point is that if it is determined that there is a buildup of some by-product or impurity over a period of years, then there is a good chance that a simple procedure can be formulated which can remove this impurity on occasion. In conclusion, only a series of repetitive runs on each cyclical experiment will determine f o r a certainty if this will be a problem in a particular case, and it is very likely that some preventative measure can be taken, once the problem is defined. - 103 - DISCUSSION: Storage Space All of the chemicals and solutions produced in the suggested procedures may be stored in 48 wide-mouthed bottles of 500 ml capacity. These have a 2.5 inch diameter and are conveniently stored in a 15" x 20" space (2 ft.2). These may be conveniently labelled on the top of the large cap so that they may be identified easily. It might be pointed out here that a great deal of the preparation normally required for a typical course is eliminated by this type of procedure in which the product of one experiment is the raw material for the next. Suggested names for the solutions are given in Appendix IV. During each exper'bent, the appropriate product bottles should be made available to the students by the instructor. The instructor should check each solution before it is placed in the product container. - 104 - DISCUSSION: Innovative Nature of NOWASTE Manual The innovative nature of the NOWASTE manual is essentially an innovation in method. The essence of this method is the arrangement of chemical reactions into a sequence, so that the product of one reaction produces the starttng material for the next reaction. By connecting reactions and entire experiments in this fashion, the number of chemicals used is reduced greatly. When entire experiments can be connected in cycles, then final products can be eliminated. This is the general approach of this manual. The result of this approach to the design of academic chemical experiments is the elimination of chemical waste, a substantial reduction of costs and, an improvement in meaning and meaningfulness of the experiments. The intent of this work is the creation of a method of academic experiment design, which can To be applied to numerous chemistry courses involving laboratory experiments. the.author’s knowledge, no such laboratory manual has ever been published. A number of innovations have been made with particular experiments. It should be realized that the purpose of an introductory chemistry manual is not to introduce new and unusual chemical reactions, but to demonstrate well-known and typical properties of the elements. The innovations of the individual experiments therefore lie in the details of the experiments, for example, the choice of a particularly colorful reaction to demonstrate chemical property. In this respect, the author considers the following experiments innovative individually for the reasons to be cited. Experiment 1 uses a novel catalyst, an iron nail, to generate oxygen from hydrogen peroxide. This has the advantage of demonstrating to the students that the nail catalyzes the reaction, as oxygen can be observed forming on the surface of the nail. Further, this experiment utilizes 3% hydrogen peroxide avoiding the hazards of the usual 9% hydrogen peroxide or potassium chlorate. Experiment 2 is novel in that it utilizes aluminum, an inexpensive metal, Instead of the conventionally employed zinc to produce hydrogen. Normally, aluminum is unreactive to sulfuric acid but,by a pretreatment in hydrochloric acid to remove the oxide coating, it is rendered reactive. Experiment 3 demonstrates conventional reactions of the alkaline earths in addition to an innovative direct reaction of alkaline earths with halogens. The consecutive nature of the reactions is novel. - 105 - Experiment 4 is novel in that it uses ammonium iodide instead of the conventional potassium iodide. Experiment 5 is novel in its use of conventional reactions of halogens in a consecutive fashion. Experiment 7 employs a reaction novel to the beginning course, oxidation of copper to its oxide. This reaction is possible because of the finely divided nature of the copper produced in Experiment 8, providing the necessary large amount of surface area, and because a simple apparatus has been developed to carry out the reaction. Experiment 8 is novel in that reactions of the well-known copper cycle reaction have been arranged to illustrate three basic chemical reactions, decomposition, single and double replacement. Experiment 9 is novel in that the activity series of the metals is demonstrated in a clear fashion due to the consecutive nature of the reaction. Experiment 11 is novel in its consecutive nature of the reactions. Experiment 14 illustrates some chemistry of lead effectively by utilizing consecutive reactions to produce a series of colorful chemical changes. Experiment 16 demonstrates a reaction novel to beginning chemistry courses, yet of a fundamental nature, and indispensable to the recovery of the lead and iodine. Experiment 20 demonstrates a technique of recovery of a rather active metal from solution. This experiment is novel to beginning chemistry courses and indispensable to the recovery of zinc. The nature of the zinc crystals thus formed permit their use in Experiment 17, the recovery of silver. Experiment 22 is novel in the destruction of the iodine-starch adduct and the consequent recovery of iodine. Experiment 25 is novel in that the nitrite oxidation is employed for the actual isolation of iodine from other components of the iodide mixtures. Experiment 26 is novel to introductory courses and enables waste iodides to eventually be converted to useable ammonium iodide. Experiment 27 is novel in the sense that it is used to separate all the nitrogen in the reaction mixture form the other compounds present. Experiment 30 is novel and employs various techniques to bring about --separationof salts. - 106 - APPENDIX I OUTLINE OF INTRODUCTORY AND PHYSICAL CHEMISTRY EXPERIMENTS Experiment A: Laboratory Equipment Construction I Purpose : Glass working and cork boring. Description: In this experiment students familiarize themselves with the bunsen burner, and are taught to bend glass and construct some simple chemical apparatus. Schematic : None Comments : Some major construction projects are referred to here. These are intended to evoke student interest, and can be used as special projects for single students, groups, or the entire class, or can be built by the teacher. Some will have been built by previous classes. Experiment €3: Laboratory Equipment Construction I1 More of the sane. Experiment C: Alcohol Burner, Scientific Observations Purpose : Use of lab equipment and scientific observation. Description: Observations on combustion are made. Water production is proven with cobalt chloride paper. Carbon dioxide production is demonstrated by its reaction with lime water. Use of the balance is introduced. Ethyl alcohol consumed by the burner is related to the temperature rise in a beaker of water. Conservation of matter and energy are introduced. Experiment D: Density Determination Purpose : Determination of a Physical Property Description: The balance and graduate are employed to determine the densities of several substances including several liquids and solids. An unknown may be introduced. Values obtained are compared with reported values in the literature. Comments : Might use different metals painted the same color and several colorless liquids to be identified by density. - 107 - APPENDIX I (cont.) Experiment E: Acids, Bases and Salts. Organic and Inorganic Compounds To demonstrate that some common substances can be easily Purpose : classified as organic and inorganic. The compounds are also classified as acids, bases, and salts. Description: Tests using litmus paper; tests are performed on table salt, vinegar, aspirin, ammonia, bleach, wood ashes, soap, etc. A simple ignition test is performed to determine if substances are organic or inorganic. Comments : Gives students a feeling for acids, bases and salts, organic and inorganic substances early in the course. Experiment F: Percent Oxygen in Air Purpose : To determine X v oxygen in air Description: A test tube with metric rule attached by a rubber band contains steel wool cleaned by short immersion in hydrochloric acid. this tube is inverted over water, and from initial and final volumes of gas the X v oxygen is determined. 02 + + WASTE Schematic : Fe Comment : The experiment is simple, inexpensive and rapid. Experiment G: Fe304 SALT Flame Tests To illustrate a rapid method of identification of certain Purpose : elements and principles of fireworks. Description: Using a platinum wire or cotton swabs (*tips) the flame colors of salts of calcium, strontium, lithium, copper and potassium are observed. Fireworks are discussed. Comments : Solutions of the salts are retained and reused. Experiment H: Characterization of Alum: After Dehydration. Melting Point and Mass Remaining Purpose : To illustrate a melting point determination, and determination of water or hydration. Description: Alum is one of a few inorganic compounds which have a low melting point for characterization. A simple melting point apparatus is assembled and the melting point is determined. The alum is ignited to remove water of hydration and weighed in a closed container to prevent rehydration. - 108 - APPENDIX I (cont.) Experiment I: Acid-Base Indicators Purpose : Indicate color change with Ph Description: ids Using a spot plate, differnet con entrations of and bases are tested with litnus, phenopthalein, and natural indicators of grape juice, cabbage juice and wisteria flower extract (or other colored flower). The ph ranges at which the color changes occur are established with universal indicator. Experiment J: Molar Volume of a Gas Purpose : As stated Description: Carbon dioxide is generated from dry ice, dried by passage through drierite and conducted to a tared plastic bag. When the bag is inflated it is weighed, and the molar volume at STP calculated from p,T,V and mass. Experiment K: Molecular Weight Determination from Molar Volume Purpose : As stated Description: Unknowns of isopropanol or ethanol are presented to the students as an unknown and are placed in a flask covered with aluminum foil with a pinhole. The tared flask is placed in boiling water and equilibrated at 100" C. The flask is allowed to cool and is weighed. The molecular mass is determined from p,V,T and mass of condensed vapor. Experiment L: Atomic Weight Determination from Specific Heat Purpose : Determination of specific heat and atomic weight estimation. Description: The specific heat of a metal is determined using double Styrofoam cups, a plastic lid, thermometer, water and the metal. The atomic weight of the metal is then calculated by the Law of Dulong and Petit. Experiment M: Conservation of Mass in Chemical Reactions Purpose: Illustrates conservation of Mass in two consecutive chemical reactions, one involving gas formation and the other precipita tion. Description: Dry ice produces carbon dioxide which is trapped in a plastic bag. The mass of the apparatus is determined before and after addition of the sulfuric acid. Aqueous calcium chloride is then added to precipitate calcium sulfate. All of the chemicals and apparatus are on the balance so that it may be established that no change in mass is observable. - 109 - APPENDIX I (cont.) Experiment N: Electrochemical Cells I Purpose : Self-manufactured batteries Description: A battery is made from a lemon using zinc and copper strips. The voltage is tested with a volt-odometer (if available). The student batteries are combined and used to set off a flash cube (a capacitor may be necessary). Using dialysis tubing, magnesium, copper and sodium sulfate, cells are constructed and used to run a radio and electrolyze zinc iodide (Experiment ). A few drops of Na2S203 restores the solution to colorless. A n impressive and fun experiment. Comments : The cells are disassembled and reused next time. Chemical Demonstrations, Vol 2, ACS, Washington, D.C., 1987, p.107. Reference: Experiment 0: Enthalpy Purpose : Determination of a Heat of Combustion Description: A calorimeter is assembled from an Erlenmeyer flask, thermometer and a coffee can to shield the flask from drafts. The calorimeter is calibrated by burning a weighed amount of methanol. The heating value of different types of wood may be determined. The heat of combustion of ethanol may be determined. Experiment P: Entropy (Demonstration) Purpose : To demonstrate the 2nd Law of Thermodynamics. Description: Stretched rubbers possess negative thermal expansion contraction is observed on heating, coefficients expansion on cooling. In the apparatus illustrated, the length of the rubber band is measured at two different temperatures. Rubber bands used for model airplane engines work well. The effect of having a rather heavy load slowly being raised nearly to its breaking point is a convincing demonstration of a change in entropy. Reference: Bader, M., - J.Chem. Educ., - 110 - z, 285, (1981) APPmIX 11 CHEMICALS AND .COSTS OF iTYPICAL CHEHISTRY MANUAL, "LABORATORY MANUAL FOR CHEMISTSY~~, by MAXINE WAGNER, 1 s t EDITION, ALLYN AND BACON, INC., NEWTON, MASS., 19R3 Ibt includa approximate qUraUUU for O m C b S of 900 mL 1% m L MO c acid. glacial M CIcnonc r- rrcu . . h l b w C U Ammonia water (NHI.H.0) Ammonium acetate* Ammonium sulfate-.A m y l alcohol M u m chloride dihydrate Buium hydroxide octohydnte M u m nitrate Barium Sulfate e b Calcium turnings Cdt5um carbonate chips W u m chloride JCISdum hydroxi& CIildum nitrate t e u a h y d n t c - ~ a k i u m sulfated P t u b o n disulfide -s - IS grams 15 grams 15 grains 30 grams 20 grams 1s grams 210 I 8 APPENDIX 111 Costs of Chemicals in NOWASTE Manual Chemical * Mass/Vol VWR Catalog No.$/g or ml cost loss 1 0% T O % Hydrogen Peroxide 3% 345 mls - 0.0020/ml 0.69 Aluminum Foil 13.0s - o.o022/g 0.03 Calcium turnings 0.4g 321262-4 0.2192/g 0.09 Hydrochloric Acid 9.8g JT9535-4 0.0065 /ml 0.06 Sulfuric Acid 20.2g JT968 1-4 O.O046/g 0.09 Magnesium 0.4g EM MxOO10-1 0.3445/g 0.14 Sodium Carbonate, 5.5g JT3604- 1 O.O486/g 0.25 Iodine 2.6g EM XXO125-1 0.2290/g (0.60) 0.06 0.12 Lead Nitrate JT 2322-1 0.1301/g (0.43) 0.04 0.08 Potassium Iodide 3.3g 0.4g JT 3165-11 O.O207/g Silver ‘Nitrate 0.2g JT 3426-6 1.541 7/g Ammonium Chloride 0.03 JT 0660-11 O.O435/g Bleach 18mls Sodium Thiosulfate 1.5g Sodium Nitrite 1.2g Cyclohexane 1 3 . 0 ~ ~ EX CX2290-7 Nitric Acid 5.3cc Copper Wire . - (0.29) 0.03 0.06 O.O007/ml 0.01 JT 3954-1 O.O442/g 0.06 EM SXO665-1 0.0573/g 0.07 0.0479 /ml (0.62) 0.06 0.12 EM NXO409-14 o.o112/g 0.06 4.2g JT 1736-1 O.O930/g (0.39) 0.08 0.16 Sodium Hydroxide 13.9g VN 6720-5 O.O214/g 0.30 Lead Foil 0.6g 36996-807 o.o220/g (0.01) oxalic Acid 0.5g JT 0230-1 O.O867/g (0.04) Potassium Dichromate 2.8s JT 3090-1 O.O534/g (0.15) 0.02 0.04 Acetic Acid 13.4g 0.0145/g 0.19 Potassium Hydroxide 8-48 EM AXOO73-14 EM PX1480-13 0.0527/g 0.44 Starch 0.02g JT 4006-4 0.1304/g Cation Exchange Dowex 50W-X8 5.0g 1927-1 O.O795/g (0.40) 0.04 0.08 Potassium Iodate 0.16g PX1500-2 0.2506/g Sodium Bisulfite 0.88 SXO345-1 O.O329/g 0.04 0.03 Benzoic Acid 2.58 HT EM EM EM BX0360-1 O.O830/g (0.21) 0.02 0.04 Methanol 5 . m JT 9070-11 O.OlSS/g (0.08) 0.10 0.20 * ONE CLASS OF THIRTY STUDENTS (15 STUDENT - 113 - - PAIR TEAMS) $3.00$3.45 APPENDIX IV Suggested Names for Solids and Solutions Used in NOWASTE Manual (numbers at end refer to experiments) 1. RECOVERED ALUM1"M 2. RECOVERED ACIDS TO 30 3. RECOVERED BASES TO 30 4. RECOVERED HALIDES TO 25 5. RECOVERED LEAD IODIDE TO 26 6. RECOVERED SILVER TO 17 7. RECOVERED CYCLOHEXANE TO 29 8. RECOVERED NITRATES TO 27 9. RECOVERED IODINE IN CYCLOHEXANE TO 26 10. EXPERIMENT 6: SALTS TO 31 WASTE PRODUCTS 11.. RECOVERED COPPER (11) CHLORIDE TO 8 12. RECLAIMED COPPER TO 7 13. RECLAIMED COPPER WIRE TO 9 14. RECLAIMED LEAD SHOT TO 9 15. RECLAIMED LEAD TO 14 16. RECOVERED SODIUM OXALATE TO 21 17. RECOVERED POTASSIUM DICHROMATE TO 12 18. RECOVERED POTASSIUM DICHROMATE TO 11 19. RECOVERED POTASSIUM DICHROMATE MOTHER LIQUOR TO 11 20. - aECOpEBED -LEADIIODIDE-TO-16 21. 22. SODIUM ACETATE AQUEOUS TO 15 23. RECLAIMED LEAD NITRATE TO 4 24. RECLAIMED SILVER NITRATE TO 5 25. RECOVERED ZINC TO 18 26. RECOVERED ZINC CARBONATE TO 20 27. RECLAIMED ZINC TO 17 28. RECLAIMED OXALIC ACID TO 10 29. RECLAIMED BENZOIC ACID TO 23 SODIUM THIOSULFATE AQUEOUS TO 15 30 t. RECOVERED IODINE TO 26 - 114 - APPENDIX IV (cont.) 31. RECLAIMED AMMONIUM IODIDE TO 4 32. RECLAIMED CYCLOHEXANE TO 5 33. RECLAIMED METHANOL TO 23 34. RECLAIMED ETHANOL TO 12 35. RECLAIMED AMMONIUM SULFATE TO 33 36. RECLAIMED SULFATES TO 31 37. RECLAIMED CHLORIDES TO 32 38. RECLAIMED ALUMINA TO 33 39. RECLAIMED BLEACH TO 5 40. RECLAIMED OXALIC ACID TO 10 - 115 -