A “Green” Approach to Synthesis of trans-4
advertisement

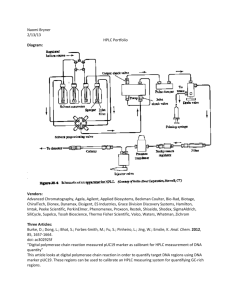
Chem. Educator 2014, 19, 347–350 347 A “Green” Approach to Synthesis of trans-4-Methoxycinnamic acid in the Undergraduate Teaching Laboratory Kristopher J. Keuseman* and Nicole C. Morrow Department of Natural & Applied Sciences, Mount Mercy University, Cedar Rapids, Iowa 52402, kkeuseman@mtmercy.edu Received August 5, 2014. Accepted September 25, 2014. Abstract: A green Knoevenagel reaction is described utilizing microwave irradiation. Students prepare trans-4methoxycinnamic acid from malonic acid and 4-methoxybenzaldehyde (p-anisaldehyde) with ammonium acetate as a catalyst under solvent-free conditions. The product is purified by recrystallization and analyzed by proton NMR. The proton NMR spectrum allows students to determine the substitution pattern of the benzene ring and relative stereochemistry of the double bond through examination of the splitting pattern and coupling constants. The principles of green chemistry are demonstrated through use of low hazard reactants and reagents. The solvents used in purification of the product are derived from renewable resources and are biodegradable. Furthermore, the use of solvent-free conditions and microwave heating demonstrate resource conservation and waste reduction techniques. O Introduction O + Carbonyl condensation reactions are often an important topic in second-semester sophomore organic chemistry. In the laboratory portion of the course, Aldol, Claisen, Dieckmann and Knoevenagel reactions [1] are often used to illustrate carbonyl chemistry. As part of our ongoing lab development, we sought a new experiment to teach carbonyl chemistry. Of the numerous possible target molecules, cinnamic acid and its derivatives stood out to us because of its biochemical relevance to lignin, which is a biopolymer that helps strengthens cell walls [2]. As part of the “shikimate pathway” [2], cinnamic acid is an important intermediate in plant metabolism. Cinnamic acids are also useful starting compounds for organic synthesis. The broad-spectrum antibiotic chloramphenicol [3] can be synthesized from a cinnamic acid derivative. The synthesis of cinnamic acid has previously been explored in the sophomore organic chemistry laboratory. In 1990 Kolb et al. [4] published a microscale experiment using benzaldehydes and malonic acid. The authors report satisfactory yields (50–80%) using the Verely-Doebner modification of the classic Knoevenagel reaction using pyridine and β-alanine to catalyze the reaction under reflux. Similarly, Harwood and co-workers describe a similar reaction using pyridine and piperidine [5]. In our hands the reaction described by Harwood and co-workers worked well, but we were not satisfied with the use of hazardous reagents (pyridine and piperidine are toxic and corrosive; pyridine is also considered a carcinogen). Therefore, we sought a more “green” application of the Knoevenagel reaction for our students. Kumar et al. reported [6] that cinnamic acids could be formed under solvent-free conditions using ammonium acetate as a catalyst with microwave irradiation (Scheme 1). Ammonium acetate is advantageous as a catalyst for the Knoevenagel reaction in the teaching environment because it’s a nonhazardous, inexpensive solid. Furthermore, use of solventfree conditions and microwave irradiation are both advantageous from the standpoint of green chemistry [7]: the HO O O H OH MeO NH4 OAc W OH MeO Scheme 1. lack of solvent prevents production of unnecessary waste and microwave irradiation can often accomplish reactions in a fraction of the time needed with conventional heating using much less energy. The reaction outlined by Kumar et al. appeared to fulfill the requirements for our lab experiment, so we used it as the basis for further work on its application to the teaching environment. The present report outlines the use of solvent-free Knoevenagel condensation under microwave irradiation in the undergraduate organic laboratory. The procedure is a modification of the published procedure of Kumar and coworkers [6]. Experimental Procedure General: Microwave irradiation (µW) was performed using an Amana Radarange household microwave oven model RS415T produced in 1990 (frequency = 2450 MHz, maximum power = 1500 W). Melting points (uncorrected) were determined using a Mel-Temp melting point apparatus fitted with a Fisher Scientific digital temperature probe. All reagents were used as received from the supplier. Absolute ethanol was 200 proof ethyl alcohol (Fisher Scientific) and 95% ethanol was 190 proof ethyl alcohol (EMD). Proton NMR data was collected on an Anasazi Ef 90 FT-NMR operating at 90.019 MHz. Samples for NMR experiments were prepared using approximately 40 mg of compound in ca. 0.5 mL of DMSO-d6, to which was added one drop of tetramethylsilane (TMS). Infrared spectroscopy was conducted using a Thermo Scientific Nicolet iS5 instrument; data was collected by attenuated total reflection (ATR). E-3-(4-Methoxyphenyl)prop-2-enoic acid (trans-4methoxycinnamic acid) [830-09-1]. Malonic acid [141-82-2], 3.6 g (35 mmol), and 2.7 g (35 mmol) of ammonium acetate [211-162-9] were ground together in a mortar and pestle until a sticky solid was formed without large clumps of either reactant. This solid was transferred to a 125- or 250-mL Erlenmeyer flask. 4- © 2014 The Chemical Educator, S1430-4171(14)12588-6, Published 11/05/2014, 10.1333/s00897142588a, 19140347.pdf 348 Chem. Educator, Vol. 19, 2014 Methoxybenzaldehyde (p-anisaldehyde, [123-11-5]), 4.5 mL (37 mmol), was added to the solid reactants and swirled by hand until the mixture began liquefying, was homogeneous, and evenly coated the bottom of the flask. A small beaker (50-, 80- or 100-mL) was inverted over the top of the flask to act as a loose cover. The reaction mixture was placed in a 1000-mL beaker containing ca. 200 mL of chromatographic-grade alumina. The Erlenmeyer flask was pushed down into the alumina powder so that the level of the alumina outside the flask was at or above the level of reactants inside the flask. The beaker containing the alumina and reaction flask was then placed inside the microwave oven, which was located inside a fume hood. The reaction was heated with microwave irradiation for 3 minutes at power setting 1 (10% irradiation time). The reaction became a honey-colored viscous liquid with the formation of bubbles during the reaction. The viscous liquid quickly solidified on cooling to give a waxy, yellow, opaque solid. In some instances the reaction mixture formed a solid during heating. To isolate the product, the yellow-orange crude product was completely dissolved in a minimum amount of hot glacial acetic acid (ca. 8 mL) on a hot plate. Three-times the volume of 95% ethanol (ca. 24 mL) was subsequently added after the solution was removed from the hot plate. The crude product began to precipitate on cooling to room temperature; precipitation was completed in an ice-water bath (solution was cooled to 5 °C). The resulting yellow solid was isolated through vacuum filtration, washed with cold 95% ethanol and immediately purified by recrystallization or air dried until the next lab period. A spectroscopic quality product was obtained by purifying the compound through recrystallization from 95% or absolute ethanol. Typical student chemical yield was 0.70–1.5 g (11–24%), off-white to orange crystalline solid (needles); melting point, 181–184 °C (abs. ethanol). 1H-NMR (DMSO-d6, 90 MHz, ): 3.81 (s, 3H, methoxy group), 6.40 (d, J = 16 Hz, 1H, vinylic H), 6.98 (d, J = 8.7 Hz, 2H, CH arom.), 7.58 (d, J = 16 Hz, 1H, vinylic H), 7.65 (d, J = 8.7 Hz, 2H, CH arom.). IR (ATR): ca. 2550 cm–1( OH), 1672 cm–1 (ν C=O), 821 cm–1 ( oop-arom.). Results and Discussion Microwave energy has become a very useful tool for synthetic chemistry [8], but it presents some challenges for the undergraduate laboratory course. It is best practice to use a microwave reactor specifically designed for chemical synthesis, however, these units are often cost-prohibitive. To minimize the risks associated with use of a domestic microwave oven, we have chosen to use low-hazard, highboiling compounds and an open reaction vessel to prevent pressure build-up. We also placed the reaction in a bed of alumina powder [9] to moderate the temperature. Instead of using benzaldehyde, we chose to use 4-methoxybenzaldehyde (p-anisaldehyde) because of its lower hazard level. According to section 2 of the Safety Data Sheet (SDS) available from Sigma-Aldrich, benzaldehyde is a flammable liquid and toxic [10], whereas 4-methoxybenzaldehyde is listed as not hazardous [11]. Kumar and coworkers report near quantitative yield (98%) [6] for synthesis of 4-methoxycinnamic acid under microwave irradiation; however, our results do not confirm this finding. We have found that typical student yields (before recrystallization from ethanol) are 2.25–3.65 g (36–58%). Despite this, the average student can still perform this reaction on the 35 millimole-scale with reasonable success using normal laboratory glassware. Because of the simplicity of the reaction set-up, short reaction time and simple work-up, this experiment generally does not take a full 3-hour lab period. In our program this experiment is conducted when a previous Keuseman et al. experiment needs to be completed or another experiment is also begun, to fill the lab period. During the development stage for this lab experiment we found that increasing the microwave irradiation time beyond 3 minutes had little effect on product yield, but the increased time produced a product that is much darker in color. Because household microwave ovens can vary considerably in performance, we recommend that the experiment be repeated by the instructor to adjust the irradiation time and power settings to the specific oven used. Our initial investigations repeated the procedure of Kumar et al. by pouring the still-liquid reaction mixture into water after the irradiation period was complete. In our experience, this would be somewhat difficult in a teaching lab because the product often solidifies very soon after removal from the microwave (or is a solid when removed). If the product has solidified, water can be poured onto the product and triturated with a glass rod prior to suction filtration. In either case, an insoluble substance is commonly present in the crude product necessitating gravity filtration during recrystallization [6] from ethanol. With the aqueous treatment the recrystallized product is varying shades of yellow or orange with melting point 181183 °C (95% ethanol). Ultimately, we determined that the aqueous work-up was of little benefit. To simplify the procedure we eliminated the aqueous workup and crystallized the crude product directly after microwave irradiation from hot glacial acetic acid and ethanol. The crude product is completely soluble in hot acetic acid, but addition of three-times its volume of ethanol begins precipitation of the product. Cooling to room temperature and then further to 5 °C in an ice-water bath provides the product. After precipitation from acetic acid-ethanol, the compound melted at 173–182 °C. Recrystallization from absolute (or 95%) ethanol raises the melting point to 181–184 °C. A search of the current literature found a wide range of melting points reported for 4methoxycinnamic acid: Aslam et al. [12] reported 169–173 °C, while Gupta and coworkers [13] reported 198–200 °C. For comparison purposes during development of this experiment, 4-methoxycinnamic acid was also synthesized as described by Harwood and co-workers [5] (Scheme 2). O O piperidine + HO O O H OH MeO OH pyridine, MeO Scheme 2. After two recrystallizations from absolute ethanol, the product was a white needle-shaped crystalline solid having a melting point of 180–183 °C. A mixed melting point of this product with 4-methoxycinnamic acid synthesized using the current procedure was 180–182 °C. Despite the differences in color between the products of reactions in Scheme 1 and Scheme 2, the mixed melting point data suggests that the identity and purity of the samples is the same. Product identity was further established by proton NMR spectroscopy. Recrystallized samples of product synthesized by students (via Scheme 1) were analyzed and compared to the recrystallized product synthesized using the procedure of Harwood and co-workers (Scheme 2) [5], as well as literature [14] data. The proton NMR spectra of samples synthesized in our lab and by our students (see Supporting Data) are consistent with the spectral data reported by Duarte and coworkers [14], except in the absence of the carboxyl proton © 2014 The Chemical Educator, S1430-4171(14)12588-6, Published 11/05/2014, 10.1333/s00897142588a, 19140347.pdf A “Green” Approach to Synthesis of trans-4-Methoxycinnamic acid... Chem. Educator, Vol. 19, 2014 349 product is a para disubstituted benzene derivative, because the protons are observed as a pair of doublets. Conclusion Synthesis of trans-4-methoxycinnamic acid as outlined here can be used to strengthen the organic chemistry laboratory curriculum by providing an example of a carbonyl condensation reaction and illustrate principles of green chemistry [7]. Other possible educational opportunities of this experiment include: recrystallization, melting point and mixed melting points, Infrared spectroscopy and proton NMR acquisition and interpretation. The product compound is of relevance to biochemistry, natural products chemistry and medicinal chemistry, thus providing an opportunity for “buyin” with students having interest in these subjects. Figure 1. 1H-NMR (90 MHz) spectrum showing overlap (in box) between vinyl and aromatic protons in 4-methoxycinnamic acid; the upfield peaks correspond to the signals for the other pair of aromatic protons (6.98 ppm, J = 8.7 Hz) and the other vinyl proton (6.40 ppm, J = 16 Hz). signal from spectra taken in our lab. The phenomena of hydrogen bonding and proton exchange make observation of carboxyl protons (and other acid hydrogens) difficult and chemical shift values unpredictable [15]. As a result, under the conditions used to record our spectra, the signal of the acidic carboxyl proton was not sufficiently intense or too broad (large halfwidth) [15] to be observed. The presence of the carboxylic acid functional group can be established by the IR spectrum. The spectrum shows a broad signal centered around 2550 cm–1 corresponding to the O–H stretch of a carboxylic acid. Due to conjugation with the -system, the C=O stretch of the carboxyl group is shifted to lower frequency and is found at 1672 cm–1. The IR spectrum also supports the para substitution pattern of the aromatic ring, characterized by the strong absorption band at 821 cm–1 attributed to the out-of-plane bending mode [16]. The proton NMR spectrum of 4-methoxycinnamic acid is not complicated and can be interpreted by students. The most confusing aspect of the spectrum is the overlap of signals in the aromatic region in the 90 MHz spectrum [17]. The overlap of the benzylic vinyl proton and the signal for a pair of aromatic protons presents an opportunity to apply knowledge of H-H coupling constants (Figure 1). Normally the coupling constant for vinyl hydrogens is large (about 16 Hz), whereas the coupling constant between aromatic hydrogens in this case is about half as much (ca. 8.7 Hz). Even without dividers or numerical values (in Hz) for these peaks, some students can visually compare the coupling constants in the overlap region to the other well-resolved peaks and determine which peaks are part of each doublet in the overlap region. The magnitude of the coupling constant between the vinyl protons is also useful in establishing the geometry of the C=C bond. Typically the coupling constants for vinyl protons are quite dependent on relative stereochemistry: for cis protons 3J = 6–15 Hz and for trans protons 3J = 11–18 [16]. The observed coupling constant of 16 Hz for 4-methoxycinnamic acid, therefore, indicates a predominantly trans geometry in the purified product. Finally, one more point of interpretation can be made from this spectrum, namely, the substitution pattern of the benzene ring. The splitting pattern of the aromatic protons indicates that the Acknowledgements. The authors would like to thank the organic chemistry students who helped develop this laboratory experiment and Dr. Joe Nguyen for assistance in manuscript preparation. We would also like to thank Mount Mercy University, the R. J. McElroy Trust and the Roy J. Carver Charitable Trust for generous direct and indirect financial support. Supporting Materials. One supporting file is available. (http://dx.doi.org/10.1333/s00897142588a). References and Notes 1. Some recent examples: (a) Balija, A. M.; Reynolds, A. M. J. Chem. Ed. 2013, 90, 1100–1102. DOI: 10.1021/ed300454d (b) Horta, J. E. J. Chem. Ed. 2011, 88, 1014–1015. DOI: 10.1021/ed1000944 (c) Mohring, J. R.; Hammond, C. N.; Schatz, P. F.; Davidson, T. A. J. Chem. Ed. 2009, 86(2), 234–239. DOI: 10.1021/ed086p234 (d) Murphee, S. S.; Kappe, C. O. J. Chem. Educ. 2009, 86(2), 227–229. DOI: 10.1021/ed086p227 (e) Esteb, J. J.; Fravel, B.; Magers, J.; McNulty, L.; O’Reilly, S.; Wilson, A. M. Chem. Educator 2007, 12(5), 324–326. DOI: 10.1333/s00897072069a. 2. Dewick, P. M. Medicinal Natural Products, A Biosynthetic Approach, third edition; John Wiley & Sons Ltd.: Chichester, UK, 2009. 3. Wu, G.-Z.; Tormos, W. I. Asymmetric process for preparing florfenicol, thiamphenicol chloramphenicol and oxazoline intermediates. US 5352832 A, Oct. 4, 1994. 4. Kolb, K. E.; Field, K. W.; Shatz, P. F. J. Chem. Ed. 1990, 67(12), A304. DOI: 10.1021/ed067pA304. 5. Harwood, L. M.; Moody, C. J.; Percy, J. M. Experimental Organic Chemistry, Standard and Microscale, second edition; Blackwell Science Ltd.: Oxford, UK, 1999. 6. Kumar, H. M. S.; Subbareddy, B. V.; Anjaneyulu, S.; Yadav, J. S. Synth. Commun. 1998, 28(20), 3811–3815. DOI: 10.1080/00397919808004934. 7. Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998. 8. Kappe, C. O. Agnew. Chem. 2004, 43, 6250–6284. DOI: 10.1002/anie.200400655. 9. Karchgaudhuri, N.; De, A.; Kumar Mitra, A. J. Chem. Res. 2002, 180–183. DOI: 10.3184/030823402103171591 10. Sigma-Aldrich Co. LLC. Material Safety Data Sheet W212709, 2014. Sigma-Aldrish USA Home. http://www.sigmaaldrich.com/MSDS/MSDS/DisplayMSDSPage.do? country=US&language=en&productNumber=W212709&brand=AL DRICH&PageToGoToURL=http%3A%2F%2Fwww.sigmaaldrich.c © 2014 The Chemical Educator, S1430-4171(14)12588-6, Published 11/05/2014, 10.1333/s00897142588a, 19140347.pdf 350 Chem. Educator, Vol. 19, 2014 om%2Fcatalog%2Fproduct%2Faldrich%2Fw212709%3Flang%3De n (accessed July 25, 2014). 11. Sigma-Aldrich Co. LLC. Material Safety Data Sheet W267020, 2014. Sigma-Aldrich USA Home. http://www.sigmaaldrich.com/MSDS/MSDS/DisplayMSDSPage.do? country=US&language=en&productNumber=W267020&brand=AL DRICH&PageToGoToURL=http%3A%2F%2Fwww.sigmaaldrich.c om%2Fcatalog%2Fproduct%2Faldrich%2Fw267020%3Flang%3De n (accessed June 25, 2014). 12. Aslam, M.; Stansbury, W. F.; Reiter, R. J.; Larkin, D. R. J. Org. Chem. 1997, 62, 1550–1552. DOI: 10.1021/jo962029k. 13. Gupta, M.; Gupta, R.; Anand, M. Beilstein J. Org. Chem. [Online] 2009, 5(68). DOI: 10.3762/bjoc.5.68. Keuseman et al. 14. Duarte, C. M.; Verli, H.; Araújo-Júnior, J. X.; Medeiros, I. A.; Barreiro, E. J.; Fraga, C. Eur. J. Pharm. Sci. 2004, 23, 363–369. DOI: 10.1016j.ejps.2004.08.011. 15. Macomber, R. S. A Complete Introduction to Modern NMR Spectroscopy. In A Complete Introduction to Modern NMR Spectroscopy; John Wiley & Sons: New York, 1998. 16. Pavia, D. L.; Lampman, G. M.; Kriz, G. S. Introduction to Spectroscopy, A Guide for Students of Organic Chemistry, third ediction; Brooks/Cole: Belmont, CA, 2001. 17. Note: Higher field strength NMR resolves these peaks. For comparison, the 300 MHz spectrum can be viewed on the SigmaAldrich website: http://www.sigmaaldrich.com/spectra/fnmr/FNMR000074.PDF (accessed Oct. 7, 2014). © 2014 The Chemical Educator, S1430-4171(14)12588-6, Published 11/05/2014, 10.1333/s00897142588a, 19140347.pdf