FGF signaling functions in the hypodermis to regulate
advertisement

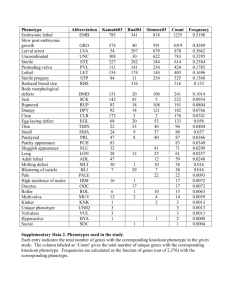
Research article 2595 FGF signaling functions in the hypodermis to regulate fluid balance in C. elegans Peng Huang and Michael J. Stern* Yale University School of Medicine, Department of Genetics, I-354 SHM, PO Box 208005, New Haven, CT 06520-8005, USA *Author for correspondence (e-mail: michael.stern@yale.edu) Accepted 17 February 2004 Development 131, 2595-2604 Published by The Company of Biologists 2004 doi:10.1242/dev.01135 Summary Signaling by the Caenorhabditis elegans fibroblast growth factor receptor EGL-15 is activated by LET-756, a fibroblast growth factor, and attenuated by CLR-1, a receptor tyrosine phosphatase. Hyperactive EGL-15 signaling results in a dramatic Clr phenotype characterized by the accumulation of clear fluid within the pseudocoelomic space, suggesting that regulated EGL-15 signaling is essential for fluid homeostasis in C. elegans. To determine the cellular focus of EGL-15 signaling, we identified an enhancer element (e15) within the egl-15 promoter, which is both necessary for the promoter activity and sufficient when duplicated to drive either egl-15 or clr1 rescue activity. This enhancer drives GFP expression in hypodermal cells. Consistent with this finding, immunofluorescence studies of EGL-15 indicate that EGL15 is expressed in hypodermal cells, and hypodermal promoters can drive full clr-1 and egl-15 rescue activity. Moreover, a mosaic analysis of mpk-1, which acts downstream of egl-15, suggests that its suppression of Clr (Soc) function is required in the hypodermis. These results suggest that EGL-15 and CLR-1 act in the hypodermis to regulate fluid homeostasis in worms. Introduction EGL-15 signaling leads to a spectrum of phenotypic consequences. Mutations that compromise the activity of CLR1 result in hyperactive EGL-15 signaling, leading to a dramatic Clear (Clr) phenotype characterized by the accumulation of clear fluid within the pseudocoelomic space (Kokel et al., 1998). A similar Clr phenotype can be observed in transgenic animals bearing arrays expressing a constitutively activated form of EGL-15, called EGL-15(neu*) (Kokel et al., 1998). Conversely, animals that completely lack egl-15 activity arrest early in larval development (Let), while severe hypomorphic egl-15 alleles result in a Scrawny (Scr) phenotype (DeVore et al., 1995). Genetic screens for suppressors of clr-1, or soc mutants, have identified multiple components in the EGL-15 signaling pathway, including EGL-15, SOC-1, SOC-2/SUR-8, SEM-5/GRB2 and LET-341/SOS (Borland et al., 2001; DeVore et al., 1995). In addition, a candidate gene approach has demonstrated the involvement of LET-60/RAS and members of the MAPK cascade in mediating the essential function of EGL-15 signaling (Borland et al., 2001; Schutzman et al., 2001). The Clr and Soc phenotypes suggest that EGL15 signaling is crucial to regulating fluid homeostasis in worms. Increased EGL-15 signaling leads to fluid accumulation and confers a Clr phenotype, while decreased EGL-15 signaling results in a Soc, Scr or Let phenotype. Although the Clr and Soc phenotypes have provided a powerful genetic tool for assembling the components of an EGL-15 signaling pathway, the biological basis of these phenotypes remains elusive. The accumulation of fluid within the pseudocoelom in Clr animals could be due to an imbalance between fluid intake and fluid excretion. Identifying the Fibroblast growth factor (FGF) receptors are a class of receptor tyrosine kinases that play pivotal roles in transducing extracellular cues to elicit diverse biological responses such as mitogenesis, angiogenesis, cell proliferation, differentiation and cell migration (Ullrich and Schlessinger, 1990). Signaling by FGF receptors is initiated by FGF ligand binding, which induces receptor dimerization, stimulation of its tyrosine kinase activity and the activation of the RAS/MAPK cascade. FGF signaling plays many important roles during normal development, and alterations in the level of FGF signaling have been implicated in human skeletal disorders (Webster and Donoghue, 1997) and tumorigenesis (Basilico and Moscatelli, 1992). In the nematode Caenorhabditis elegans, egl-15 encodes the FGF receptor and plays a crucial role in multiple aspects of development (Borland et al., 2001). Two major events mediated by EGL-15 are the migrations of the hermaphrodite sex myoblasts (SMs) and an early essential function (DeVore et al., 1995). There are two known FGFs in C. elegans, encoded by the genes egl-17 and let-756. EGL-17 is the chemoattractant that guides the migrations of SMs to their precise final positions (Burdine et al., 1998). LET-756 appears to be required for the essential function of EGL-15, as animals that lack LET-756 activity arrest at an early larval stage (Roubin et al., 1999), similar to the phenotype caused by lack of the EGL15 receptor (DeVore et al., 1995). The essential function of EGL-15 is attenuated by the action of a receptor tyrosine phosphatase (RTP) known as CLR-1 (Kokel et al., 1998). Genetic perturbation of the strength of Supplemental data available online Key words: FGF receptor, EGL-15, Hypodermis, Fluid balance 2596 Development 131 (11) cellular focus of EGL-15 activity would lead to a better understanding of the mechanism by which EGL-15 signaling regulates fluid balance in worms. Here we present evidence indicating that CLR-1 and EGL-15 function in the same cells, and that the EGL-15 signaling pathway acts in the hypodermis to regulate fluid homeostasis. Materials and methods Strains and genetics The wild-type C. elegans strain used was Bristol, N2. Standard methods were used for maintenance and manipulation of strains (Brenner, 1974). The following genes, mutations and chromosomal rearrangements were used: LGI: unc-74(e883), szT1(I;X)[lon2(e678)]; LGII: clr-1(e1745ts), clr-1(e2530); LGIII: dpy-17(e164), mpk-1(oz140), let-756(s2887), ncl-1(e1865), unc-36(e251), unc32(e189), sDp3(III;f); LGIV: soc-2(n1774), dpy-20(e1282ts); LGX: egl-15(n1456), egl-15(n1783). Three integrated arrays were obtained during this work: ayIs25[egl-15(+) (20 ng/µl); Pmyo-3::GFP (5 ng/µl)], ayIs26[Phsp16-2::let-756 (50 ng/µl); Pmyo-2::GFP (5 ng/µl)], and ayIs29[egl-15(+) (5 ng/µl); Pmyo-2::GFP (5 ng/µl); pGEM5Z (70 ng/µl)]. ayIs25 and ayIs26 were generated by Psoralen-UV mutagenesis (S. Clark, personal communication); ayIs29 was a spontaneous integrant. ayIs26 and ayIs29 were mapped to LGIII and LGIV, respectively. Promoter analysis of egl-15 A series of truncated egl-15 promoters was generated by PCR amplification. The size and the 5′ position of each promoter/5′UTR fragment (the number of each referring to its position relative to the initiating ATG) is as follows: 2.0 kb, –1993; 1.7 kb, –1694; 1.5 kb, –1530; 1.3 kb, –1332; 1.1 kb, –1168; 0.8 kb, –815. These promoters were inserted upstream of the clr-1 genomic coding sequence (NH#268) to replace the endogenous clr-1 promoter/5′UTR fragment. The resulting constructs were assayed for clr-1-rescuing activity as described (see below). To generate Pe15*2::GFP and Pe15*2::lacZ, e15 (from –1530 to –1296 in the egl-15 promoter) was inserted duplicated upstream of the minimal pes-10 promoter of pPD97.78 and pPD95.21 (a gift from A. Fire), respectively between the HindIII and StuI sites. The resulting constructs (NH#1100 and NH#1090, respectively) were injected at 20 ng/µl into dpy-20 animals with pMH86 [dpy-20(+)] as the cotransformation marker at 50 ng/µl. Tissue-specific expression of egl-15 and clr-1 The promoters used in tissue-specific expression of egl-15 and clr1 included: Pdpy-7, Prol-6 (hypodermal expression); Punc-54 (body wall muscle expression); Paex-3, Punc-14, Psnb-1 (neuronal expression) (Gilleard et al., 1997; Iwasaki et al., 1997; Mello et al., 1991; Nonet et al., 1998; Ogura et al., 1997; Okkema et al., 1993). These promoters were obtained by PCR amplification to yield fragments that span the following regulatory regions (the numbers indicate the base pair relative to the initiating ATG): Pdpy-7, from –609 to –1; Prol-6, from –976 to –1; Punc-54, from –1428 to –1; Paex-3, from –1464 to –1; Punc-14, from –1738 to –1; Psnb-1, from –3330 to –1. For the egl-15 and clr-1 promoter swapping experiments, Pegl-15 is from –1993 to –1, and Pclr-1 is from –2422 to –1. P∆pes-10, Pe15*1 and Pe15*2, which contain zero, one and two copies of e15 fused to the minimal pes-10 promoter, respectively, were generated by PCR amplification using NH#1100 as a template (see above). To generate heterologous egl-15, clr-1, and egl-15(neu*) constructs, the promoter fragments were digested with appropriate restriction enzymes, and inserted upstream of the egl-15 genomic coding region (NH#150), the clr-1 genomic coding region (NH#268) and the egl15(neu*) construct (NH#526), respectively, to replace the endogenous promoters. Research article Transgenic rescue assays clr-1(Clr) rescue assay clr-1-rescuing activity was assayed in a clr-1(e1745ts) background. Animals were injected with tester DNA at 50 ng/µl and the cotransformation marker rol-6(su1006) in plasmid pRF4 at 100 ng/µl. Transgenic lines were obtained at the permissive temperature (15°C) based on the Roller phenotype. A minimum of 12 Rol animals from each line were tested for rescue of the Clr phenotype by shifting animals at the L3-L4 stage to the non-permissive temperature (25°C). clr-1-rescued animals are non-Clr at 25°C; non-rescued animals are Clr at 25°C. clr-1-rescuing activity is classified into four categories based on the percentage of rescued animals: strong (++), >75% rescued; intermediate (+), 25-75% rescued; weak (+/–), <25% rescued; or no rescue (–), 0% rescued. egl-15(Soc) rescue assay egl-15(Soc)-rescuing activity was assayed in a clr-1(e1745ts); egl15(n1783) background. Animals were injected with tester DNA at 20 ng/µl and the co-transformation marker rol-6(su1006) in plasmid pRF4 at 100 ng/µl. Transgenic lines were obtained at the permissive temperature (15°C) based on the Roller phenotype, and scored as described above for the clr-1(Clr) assay. egl-15-rescued animals are Clr (non-Soc) at 25°C; non-rescued animals are non-Clr (Soc) at 25°C. egl-15-rescuing activity is classified into four categories similar to the clr-1-rescuing assay (see above): strong (++), intermediate (+), weak (+/–) and no rescue (–). egl-15(Let) rescue assay egl-15(Let)-rescuing activity was assayed in a unc-74/szT1; egl-15(n1456)/szT1 background. Animals were injected with tester DNA at 20 ng/µl and the co-transformation marker pJKL449.1[Pmyo-2::GFP] at 5 ng/µl. Transgenic lines were obtained based on GFP expression in the pharynx. The balanced strain normally does not segregate any non-arrested Unc progeny; rescue is scored by the presence of viable Unc progeny derived from the nonUnc parental strain. egl-15(neu*) assay egl-15(neu*) constructs were injected into wild-type animals at 20 ng/µl with the co-transformation marker pJKL449.1[Pmyo-2::GFP] at 5 ng/µl. The phenotype of GFP-positive F1 transformants was examined. Immunofluorescent staining Mixed-stage populations of each strain were fixed and stained according to the protocol of Finney and Ruvkun (Finney and Ruvkin, 1990). Affinity-purified rabbit anti-EGL-15 antibodies (Pop) were used at a concentration of 1:10; the secondary antibody was Alexa Fluor 546-conjugated mouse anti-rabbit antibody (Molecular Probes) diluted at 1:250. Mosaic analysis Mosaic analysis was carried out as previously described in strains bearing sDp3, a free chromosomal duplication (Hedgecock and Herman, 1995). ncl-1 was used as a cell-autonomous marker (Hedgecock and Herman, 1995). Cells displaying an Ncl phenotype were assumed to have lost sDp3. The point of sDp3 loss within each mosaic animal was inferred by assuming the minimum loss of sDp3 consistent with the Ncl phenotype pattern of each animal. The following representative cells from each lineage were scored for the Ncl phenotype: CANL/R, RID, ALA, ASKL, ADLL (from AB.al); MI, I5, ALML/R, BDUL/R (from AB.ar); ASIL, excretory pore, HSNL, PHBL, QL, V5L (from AB.pla); excretory duct, excretory cell, K, repD, PHAL, hyp8/9, repVL, U, F, imL, hyp10 (from AB.plp); ASKR, ADLR, ASIR, QR, V5R, HSNR, PHBR (from AB.pra); PHAR, hyp8/9, repVR, B, body muscle, hyp10 (from AB.prp); vccL/R, dccL/R, dtcA/P, anchor cell, M4, imR, body muscles (from EGL-15 signaling in the hypodermis 2597 MS); hyp11, body muscles (from C); body muscles (from D). Intestinal cells do not display a Ncl phenotype. Animals that have lost sDp3 in the MS-lineage could possibly have lost sDp3 also in the E lineage. mpk-1 mosaics We screened approximately 1500 Unc or weakly Unc progeny of clr1(e1745ts); mpk-1 ncl-1 unc-36; sDp3 at the L3-L4 stage for Ncl mosaics using Nomarski optics at the permissive temperature (15°C). Eighteen mosaic animals were identified; 17 of these animals had sDp3 loss in AB descendants, and one animal was P1(–). These animals were recovered and shifted to the non-permissive temperature (25°C). The phenotype was subsequently determined after at least 10 hours at 25°C. To identify P1 mosaics, we shifted approximately 50,000 synchronized progeny of clr-1(e1745ts); mpk-1 ncl-1 unc-36; sDp3 at the L3-L4 stage to the non-permissive temperature (25°C), and screened for animals that were Soc non-Unc or semi-Clr non-Unc using a dissecting microscope after 10 hours at 25°C and then for Ncl mosaics using Nomarski optics. We identified two semi-Clr non-Unc and one Soc non-Unc animals with losses in P1. P1 losses are expected in approximately one out of every 400 random animals (Hedgecock and Herman, 1995). Thus, we expected to identify approximately 125 P1(–) mosaics in our screen. These mosaic animals should all be phenotypically Soc non-Unc if loss of mpk-1 activity in the P1 lineage were sufficient to suppress the Clr phenotype. Therefore, it is likely that the vast majority of P1(–) animals had already turned Clr by the time we screened them after 10 hours at 25°C. To test the possibility that loss in P1 might merely delay a strong Clr phenotype to mosaic animals, we screened for P1 mosaic animals by looking for an intermediate phenotype at an earlier time after transfer to the restrictive temperature. We screened an additional 82,000 synchronized animals after 4-5 hours at 25°C using a dissecting microscope, looking for animals that were semi-Clr nonUnc, and then for Ncl mosaics using Nomarski optics. Eight P1(–) animals were identified this way; they were recovered at 25°C, and their terminal phenotype was analyzed after at least 10 hours at 25°C. In addition to mosaic animals, we identified a number of animals in which the duplication had broken down. In the first screen we identified six Soc non-Unc animals, and in the modified screen we identified 23 Soc non-Unc animals that probably represent breakdown products of sDp3. Their classification as duplication breakdown events was based on one of the three following criteria: (1) the Ncl phenotype pattern of the animal was not consistent with its Soc nonUnc phenotype; (2) the Ncl phenotype pattern and the Soc non-Unc phenotype of the animal was contradictory to our previous observations; or (3) the animal had a complicated pattern of sDp3 loss. Since all of these animals were sterile, progeny testing was not possible to verify their classification. let-756 mosaics We screened approximately 10,000 progeny of dpy-17 let-756 ncl-1 unc-36; sDp3 hermaphrodites for animals that were Unc non-Dpy or Dpy non-Unc using a dissecting microscope. These animals were then screened for Ncl mosaics using Nomarski optics. We identified 80 Unc non-Dpy animals, all of which had losses in the AB lineage, as expected (see Fig. 5 and Table S1 at http://dev.biologists.org/ supplemental/). Some of them were recovered, and the phenotypes of their progeny were checked to verify that the parents were mosaic animals rather than the result of duplication breakdown events. We identified only one Dpy Unc, two Dpy non-Unc, and two semi-Dpy non-Unc animals from the screen. The Ncl phenotype of these animals was determined using Nomarski optics. These animals were recovered, the phenotypes of their progeny determined, and the genotype of the let-756 locus of the mosaic animals and their progeny analyzed by single-worm PCR. Because the genotypes of these animals and their progeny did not correlate with their phenotypes, these Dpy animals were classified as the result of duplication breakdown events. We further screened approximately 750 randomly selected L3-L4 animals for mosaics directly based on their Ncl phenotype. We identified 31 mosaic animals using this approach, and the Ncl phenotype patterns are represented in Fig. S1 (http://dev.biologists.org/supplemental/). Results Genetic interactions between let-756, egl-15 and clr-1 Since EGL-15 is the sole FGF receptor in C. elegans, it is reasonable to hypothesize that the LET-756 FGF acts as one of its ligands. Nonetheless, support for this hypothesis is based solely on the similarity of their lethal null phenotypes and the scrawny phenotypes of severe hypomorphs (Roubin et al., 1999). While these data are consistent with the hypothesis that these genes act in the same pathway, the general and nonspecific nature of these phenotypes does not offer strong evidence that LET-756 functions as a ligand for EGL-15. To test this hypothesis more rigorously, we sought additional genetic evidence that would support the hypothesis that these components act as a ligand–receptor pair. If LET-756 acts as a ligand for EGL-15, then constitutive activation of EGL-15 might bypass the requirement for LET756, and overexpression of LET-756 might stimulate the more specific Clr phenotype characteristic of EGL-15 hyperactivation. Consistent with the first prediction, the Let phenotype of let-756(s2887) could be suppressed by the overexpression of wild-type EGL-15 (Fig. 1D) or by the lowlevel expression of hyperactive EGL-15(neu*) (data not shown). Furthermore, overexpression of LET-756 can cause a Clr phenotype that is dependent on EGL-15 signaling. let-756 was overexpressed using the heat shock promoter Phsp16-2 (Stringham et al., 1992) to drive expression of LET-756. Heat shock of animals bearing a transgenic array containing Phsp16-2::let-756 in a background overexpressing egl-15 resulted in a dramatic Clr phenotype (Fig. 1E). This phenotype was completely suppressed when the same experiment was performed in a soc-2 mutant background (Fig. 1F). Moreover, heat shock of the same array in the absence of overexpressed egl-15 results in animals that do not display any obvious phenotypes (data not shown). These experiments show that overexpression of LET-756 can confer a Clr phenotype that requires EGL-15 and some of the same components that mediate EGL-15 signaling. These results show that LET-756 can regulate the same processes as EGL-15, supporting the hypothesis that LET-756 is the FGF ligand for this function of EGL-15. To establish the relationship between LET-756, an activating ligand for EGL-15, and CLR-1, a negative regulator of EGL15 signaling, we tested genetic interactions between these two components. Our results show that progressive reduction of clr-1 function promotes increasing suppression of the let756 phenotype by increasing net EGL-15 signaling. The temperature-sensitive allele clr-1(e1745ts) suppressed the lethality of let-756(s2887) homozygotes at the permissive temperature (15°C), but the animals appeared scrawny (Fig. 1G), a phenotype indicative of compromised EGL-15 signaling (Borland et al., 2001; DeVore et al., 1995). Further reduction of clr-1 activity, achieved by placing clr-1(e1745ts) in trans to the null allele clr-1(e2530), resulted in essentially wild-typelooking animals at the permissive temperature (15°C) (Fig. 2598 Development 131 (11) Research article Fig. 1. Genetic interactions between let-756, egl-15 and clr-1. (A) Wild type. (B) let-756 unc-32 arrested L1 larva. (C) clr-1(e2530) Clr hermaphrodite. (D) let-756 unc-32; ayIs25[egl-15(+)] Scr hermaphrodite. (E) ayIs26[Phsp16-2::let-756]; ayIs25[egl-15(+)] Clr hermaphrodite. (F) ayIs26[Phsp16-2::let-756]; soc-2; ayIs25[egl-15(+)] Soc hermaphrodite. (G) clr-1(e1745ts); let-756 unc-32 Scr hermaphrodite, at 15°C. (H) clr-1(e1745ts)/clr-1(e2530); let-756 unc-32 non-Clr non-Scr hermaphrodite, at 15°C. (I) clr-1(e2530); let-756 unc-32 viable semi-Clr hermaphrodite. All animals were photographed as adults. (E-F) Animals were heat shocked for 30 minutes at 37°C, and allowed to recover at 20°C for 5.5 hours. The Clr phenotype in (C) and (E) can be discerned from the appearance of the intestine floating within the fluid-filled pseudocoelomic cavity. Abbreviated genotypes: 0, null allele; ts, temperature-sensitive allele; ++, transgenic overexpression. Scale bar: 200 µm. 1H). Finally, the double-null strain clr-1(e2530); let756(s2887) is viable and partially Clr (Fig. 1I), demonstrating that the loss of the negative regulator not only can bypass the requirement for LET-756 but also allow hyperactivity of basal levels of EGL-15. These results are consistent with a model in which there is a balance established by the actions of LET-756 and CLR-1 on EGL-15 activity, with the basal, unregulated activity of EGL-15 resulting in a slight gain-of-function phenotype that is independent of the LET-756 ligand. Rescue analysis indicates EGL-15 functions in the hypodermis Interchangeability of the egl-15 promoter and the clr-1 promoter Both EGL-15 and CLR-1 are membrane-associated receptors (DeVore et al., 1995; Kokel et al., 1998). Since the intracellular phosphatase activity of CLR-1 is absolutely required for its negative regulation of EGL-15 signaling (Kokel et al., 1998), it is likely that both CLR-1 and EGL-15 act in the same cells to mediate their downstream effects. We tested this hypothesis using promoter swapping experiments to see whether the egl15 promoter and the clr-1 promoter could interchangeably provide the functions of the endogenous promoters. When the clr-1 promoter was used to drive egl-15 expression (Pclr-1::egl15), the resulting temporal and spatial expression of egl-15 was able to rescue both strong and weak egl-15 loss-of-function phenotypes. This construct could rescue the Soc phenotype caused by egl-15(n1783) as well as the Let phenotype caused by the null allele egl-15(n1456) (Table 1A). Conversely, when the egl-15 promoter was used to drive clr-1 expression (Pegl-15::clr-1), the resulting temporal and spatial expression of clr-1 was able to rescue the Clr phenotype of clr-1(e1745ts) (Table 2A). In addition, a construct in which the clr-1 promoter drives egl-15(neu*) expression (Pclr-1::egl-15(neu*)) could confer a dominant Clr phenotype similar to that seen when egl15(neu*) was expressed from the egl-15 promoter (Table 1A). The interchangeability of these two promoters suggests that CLR-1 and EGL-15 probably function in the same cells to carry out their normal functions. It is interesting to note that the clr-1 promoter appears to be weaker than the egl-15 promoter. This is reflected both in the lower penetrance of the Clr phenotype when Pclr-1 is used to drive expression of egl15(neu*) as well as the weaker rescue of egl-15(Soc) by Pclr-1::egl-15 compared with Pegl-15::egl-15 (Table 1A). Identification of the egl-15 enhancer element e15 We have taken several approaches to address the cellular focus of the essential function of EGL-15. The first of these approaches was to identify an enhancer element in the egl-15 promoter that is necessary and sufficient for rescue of both egl15 and clr-1. To look for important regulatory elements within the egl-15 promoter, we fused truncated egl-15 promoters to the clr-1 coding sequence, and tested their ability to rescue the clr-1 mutant phenotype (Fig. 2). The degree of clr-1 rescue activity is a sensitive readout for expression in the appropriate subset of cells. Full-scale clr-1 rescue activity was found using promoter fragments as small as 1.5 kb (Fig. 2). Smaller promoters, such as the 1.3 kb or the 1.1 kb egl-15 promoters, EGL-15 signaling in the hypodermis 2599 Table 1. Tissue-specific rescue of egl-15 Extent of rescue of egl-15(Soc) lines† n Rescued egl-15(Let) lines‡ % Clr phenotype in egl-15(neu*) F1s§ (n) 3 5 5/5 2/2 84.2% (329) 25.3% (645) 6 3 0 6 5 7 ND¶ ND 4/4 ND ND 84.3% (637) 0 3 15 0 12 4 12 15 1 12 2/2 2/2 2/2†† 1/1 0/2 99.6% (479) 94.4% (621) 0.4% (275) 66.8% (277) 0% (369) Transgene promoter‡‡ ++ + +/– -- (A) Interchangeability of the egl-15 and clr-1 promoters Pegl-15 Pclr-1 3 0 0 3 0 2 0 0 (B) Expression of egl-15 driven by the e15 element P∆pes-10 Pe15*1 Pe15*2 0 0 7 0 1 0 0 1 0 (C) Tissue-specific expression of egl-15 Pdpy-7 (hypodermis) Prol-6 (hypodermis) Paex-3 (neurons) Punc-14 (neurons) Punc-54 (body wall muscles) 4** 4 0 1 0 0 4 0 0 0 0 1 0 0 0 ‡‡Major site of expression of the tissue-specific promoter is indicated in parentheses. †egl-15(Soc) rescue assays were performed in a clr-1(e1745ts); egl-15(n1783) background. At the non-permissive temperature (25ºC), egl-15-rescued animals are Clr, whereas non-rescued animals retain the Soc phenotype. Rescuing activity is classified as strong (++), intermediate (+), weak (+/–), or no rescue (–). n, the number of transgenic lines scored. ‡Transgenic lines were isolated in an unc-74/szT1; egl-15(n1456)/szT1 background; lines were scored as rescued if the parental line could segregate a viable unc-74; egl-15(n1456) strain bearing the extrachromosomal array. §Promoter::egl-15(neu*) constructs were injected into wild-type animals. F1 transformants were identified by expression of the P myo-2::GFP co-transformation marker, and the percentage of transformants with the Clr phenotype was determined. n, the number of F1 transformants. ¶ND, not determined. **clr-1(e1745ts); egl-15(n1783); ayEx[Pdpy-7::egl-15] animals were Clr even at the permissive temperature (15°C). ††Rescued animals from both lines were Scrawny. Table 2. Tissue-specific rescue of clr-1 (A) Interchangeability of the egl-15 and clr-1 promoters (B) Expression of clr-1 driven by the e15 element (C) Tissue-specific expression of clr-1 Extent of rescue of clr-1 lines† Transgene promoter§ ++ + +/– -- n Pclr-1 Pegl-15 7 6 0 0 0 0 0 0 7 6 P∆pes-10 Pe15*1 Pe15*2 0 0 14 0 4 0 2 3 0 8 1 0 10 8 14 Pdpy-7 ‡ (hypodermis) Prol-6 (hypodermis) Paex-3 (neurons) Punc-14 (neurons) Psnb-1 (neurons) Punc-54 (body wall muscles) 4 14 1 2 3 0 4 0 3 0 0 0 1 0 7 0 0 4 3 0 4 0 0 2 12 14 15 2 3 6 §Major site of expression of the tissue-specific promoter is indicated in parentheses. †clr-1 rescue assays were performed in a clr-1(e1745ts) background. clr-1 rescuing activity is classified as strong (++), intermediate (+), weak (+/–), and no rescue (- -). n, the number of transgenic lines scored. ‡P dpy-7::clr-1 was injected at 5 ng/µl; all other constructs were injected at 50 ng/µl. show decreased rescue activity (Fig. 2). These results suggest that the 234 bp DNA segment that is the difference between the 1.5 kb and 1.3 kb promoters (e15, from –1530 to –1296) is necessary for maximal egl-15 promoter activity (Fig. 2). To test whether e15 is also sufficient for egl-15 and clr-1 rescue, we fused one or two copies of e15 to the pes-10 minimal promoter (P∆pes-10). The resulting artificial promoters (named Pe15*1 and Pe15*2, respectively) were used to drive either egl-15 or clr-1 expression. Strikingly, the duplicated e15 enhancer, Pe15*2, was able to drive full-scale rescue activity of egl-15, similar to the endogenous egl-15 promoter (Table 1B). One copy of the e15 enhancer, Pe15*1, showed a reduced extent of rescue activity, while the minimal promoter, P∆pes-10, showed only background levels of rescue activity (Table 1B). Since a single e15 element is not sufficient to drive EGL-15 expression to confer full rescue activity, additional elements might be present between the 1.1 kb and 0.8 kb promoters to help drive normal expression of EGL-15. The significant rescue activity found for the 1.1 kb promoter fragment is consistent with this hypothesis (Fig. 2). Similar results were obtained using the clr-1 rescue assay (Table 2B), consistent with the hypothesis that CLR-1 and EGl-15 act in the same cells. Additionally, the egl-15(neu*) transgene under the control of Pe15*2 conferred a Clr phenotype to transgenic animals with a penetrance comparable to that of egl-15(neu*) driven by the egl-15 promoter (Table 1B). This analysis thus identified an enhancer element that is necessary for the egl-15 promoter. Furthermore, when present in two copies, this 2600 Development 131 (11) Research article Pegl-15 ::clr-1 constructs Fig. 2. Identification of the e15 enhancer element within the egl-15 promoter. egl-15 promoter fragments, from 2.0 kb to 0.8 kb, were used to drive expression of the clr-1 genomic coding region. Rescue assays were performed in a clr-1(e1745ts) background. Rescue activity is classified as strong (++), intermediate (+), weak (+/–), or no rescue (– –). n, the total number of transgenic lines scored. The location of the egl-15 enhancer (e15, from –1530 to –1296) lies between the dashed lines. Pegl-15 Promoter Size Extent of Rescue of clr-1 lines –– ++ + +/– n clr-1 2.0 kb 6 0 0 0 6 clr-1 1.7 kb 6 0 0 0 6 clr-1 1.5 kb 4 0 1 0 5 clr-1 1.3 kb 1 1 1 2 5 clr-1 1.1 kb 2 0 3 2 7 clr-1 0.8 kb 0 0 4 7 11 e15 element can drive functionally robust expression in cells relevant to the essential function of egl-15. To determine the expression pattern of the e15 enhancer, we constructed GFP and lacZ reporters under the control of Pe15*2. The Pe15*2::GFP reporter is expressed in the major hypodermis (Fig. 3A), persisting throughout larval development and into the adult stage. Pe15*2::GFP was not seen in the lateral hypodermal blast cells (the seam cells) (Fig. 3A). An identical expression pattern was observed using the Pe15*2::lacZ reporter (data not shown). The strong hypodermal expression driven by the e15 enhancer suggests that an important site of EGL-15 and CLR-1 function is in hypodermal cells. Tissue-specific expression of EGL-15 and CLR-1 To test the hypothesis that EGL-15 and CLR-1 function in the hypodermis, we used tissue-specific promoters to drive either EGL-15 or CLR-1 expression in mutant rescue assays. When we expressed EGL-15 from either of two hypodermal promoters, Pdpy-7 (Gilleard et al., 1997) and Prol-6 (Mello et al., 1991), we observed robust rescue of egl-15 mutant phenotypes (Table 1C). By contrast, expression of EGL-15 in all body wall muscles from the unc-54 promoter (Okkema et al., 1993) failed to show any rescue activity (Table 1C). These data further support the hypothesis that EGL-15 functions in the hypodermis. In addition to the rescue of the Soc and Let phenotypes, hypodermal expression of EGL-15(neu*) is also able to confer a highly penetrant Clr phenotype (Table 1C). These results further suggest that the Clr, Soc and Let phenotypes are caused by perturbation of the same process in the same tissue. Interestingly, pan-neuronal expression of EGL-15 could also show strong rescue activity. Expression of EGL-15 by the unc-14 promoter (Ogura et al., 1997) can rescue egl-15 phenotypes, and transgenes containing Punc-14::egl15(neu*) can confer the Clr phenotype (Table 1C). By contrast, expression of EGL-15 from another pan-neuronal promoter derived from the aex-3 gene (Iwasaki et al., 1997) failed to rescue the egl-15(Soc) phenotype, and Paex-3::egl-15(neu*) transgenes also failed to confer the dominant Clr phenotype (Table 1C). Although Paex-3::egl-15 was found to rescue the Let phenotype of egl-15(n1456), the rescued animals were scrawny, a phenotype indicative of compromised egl-15 signaling (Table 1C). Immunofluorescence studies using antiEGL-15 antibodies have shown that neuronal EGL–15 expression in animals with Punc-14::egl-15 and Paex-3::egl-15 Fig. 3. EGL-15 is expressed in the hypodermis. (A) GFP expression pattern of Pe15*2::GFP animals. GFP is seen in the hyp7 syncytium, and excluded from the lateral seam cells (*). (B) Immunofluorescence staining of EGL-15 in the strain ayIs29[egl15(+)] using anti-EGL-15 antibodies. Staining is observed in the hyp7 syncytium, but absent in the seam cells. The punctate staining is highly variable, and has not been assigned to any defined anatomical structure. Animals shown were photographed at the L1 (A) and L2 (B) stage. Anterior is to the left. Scale bar: 50 µm. transgenes is comparable (data not shown). These data suggest that EGL-15 expression in the neurons may not be the relevant site of expression that confers rescue activity, but rather that low level expression elsewhere might be responsible for the rescue results. We performed similar tissue-specific transgene rescue experiments using these same promoters to drive expression of clr-1. The same subset of promoters that restored egl-15 function could also restore clr-1 function (Tables 1C and 2C), further suggesting that EGL-15 and CLR-1 act in the same tissues. Since hypodermal expression of CLR-1 also shows robust rescue activity (Table 2C), the hypodermis is also likely to be the site where CLR-1 normally functions to regulate fluid balance. However, based on these heterologous rescue experiments, it remains possible that the nervous system might EGL-15 signaling in the hypodermis 2601 zygote (P0) Clr semi-Soc Soc AB P1 A P L A R P A EMS L P A R P A MS P A P2 E P C A P3 P D P4 Fig. 4. Summary of mpk-1 mosaics. The mosaic analysis was carried out in a strain of genotype clr-1(e1745ts); mpk-1 ncl-1 unc-36; sDp3. Patterns of duplication loss are represented by different colors: red, loss in a single lineage; green, losses in a single lineage and some independent cell(s); blue, loss in multiple independent lineages. Terminal phenotypes were determined at the non-permissive temperature (25°C) and represented by different shapes: 䊏, Clr; 䉱, semi-Soc; 䊉, Soc. be a relevant site for the functions of EGL-15 and CLR-1, since neuronal promoters such as the unc-14 (Ogura et al., 1997) and snb-1 promoters (Nonet et al., 1998) can rescue the clr-1 mutant phenotype (Table 2C). Expression analysis of egl-15 To determine where EGL-15 is normally expressed, we carried out immunofluorescence studies using anti-EGL-15 antibodies. We were unable to detect endogenous EGL-15 in wild-type animals, probably due to the low-level expression of the endogenous receptor. Therefore, we generated a strain overexpressing a chromosomally integrated egl-15(+) array (ayIs29), and performed immunofluorescence staining on this strain using affinity-purified anti-EGL-15 antibodies. EGL-15 expression was observed in hypodermal cells as well as the sex myoblasts, the type I vulval muscles and some unidentified neurons in the head (Fig. 3B and data not shown; a more detailed analysis will be reported elsewhere). The staining is specific to EGL-15, since no signal is observed with secondary antibody alone (data not shown). Hypodermal expression is obvious throughout all four larval stages, with stronger expression in the early larval stages. A similar pattern of EGL15 expression has been observed for many other arrays containing transgenic egl-15 constructs (data not shown). The expression in the hypodermis is very similar to that observed in Pe15*2::GFP animals, in which expression was observed in the major hypodermis but was excluded from the seam cells (Fig. 3A). A similar expression pattern can be observed in rescued egl-15(null) animals expressing an egl-15 transgene driven by a hypodermal promoter (Prol-6 and Pdpy-7) (data not shown). Hypodermal expression of EGL-15 is also reported in animals expressing Pegl-15::lacZ (Hope Lab Expression Pattern Database: http://129.11.204.86:591/default.htm), consistent with the antibody staining that we have observed. Mosaic analysis of the Soc function of mpk-1 The data described above support the hypothesis that the Let, Clr and Soc phenotypes of egl-15 and clr-1 mutants are due to various perturbations of a single process that occurs in the same place for both EGL-15 and CLR-1. The cellular focus of the Soc function can thus provide additional insight into the site of action of this pathway. Multiple downstream components of the EGL-15 signaling pathway have been identified based on the Soc phenotype (DeVore et al., 1995; Schutzman et al., 2001). To investigate the cellular focus of the Soc function, we carried out a mosaic analysis of one of the soc genes, mpk-1, encoding MAP kinase. mpk-1 had already been shown to be amenable to mosaic analysis using the well-characterized free duplication sDp3 and the cell-autonomous lineage marker ncl1 (Church et al., 1995). To analyze its Soc phenotype, we constructed the strain clr-1(e1745ts); mpk-1 ncl-1 unc-36; sDp3 for mosaic analysis. The sDp3 free duplication carries a wild-type copy of each of the mpk-1, ncl-1 and unc-36 genes. At the restrictive temperature (25°C), clr-1(e1745ts) mutants are Clr, but animals homozygous for the additional mutation mpk-1(oz140) are Soc with complete penetrance. Therefore, non-mosaic animals that carry the duplication [P0(+)] are Clr at the restrictive temperature (25°C), since the wild-type copy of the mpk-1 gene on the duplication allows hyperactive EGL15 signaling to cause fluid accumulation. By contrast, lack of MPK-1 in animals that lose the duplication [P0(–)] prevents the transduction of hyperactive EGL-15 signaling, resulting in a Soc phenotype. unc-36 is known to function in the AB.p lineage (Kenyon, 1986; Yuan and Horvitz, 1990). It was included to identify animals that had not inherited the duplication [P0(–)] by their Unc, Soc phenotype, and to facilitate the identification of animals with mosaic losses in descendants of the AB blastomere. We identified 17 animals with sDp3 losses in AB descendants at the permissive temperature (15°C) (see Materials and methods), including two AB(–), two AB.a(–) and three AB.p(–) animals, as well as animals with losses within the AB.p sublineage (Fig. 4). Interestingly, all these animals displayed the Clr phenotype when shifted to the restrictive temperature (25°C). These results suggest that loss of mpk-1 activity in the AB lineage is not sufficient to suppress the Clr phenotype. Therefore, mpk-1(+) activity within P1 descendants can function to prevent the Soc phenotype. 2602 Development 131 (11) Research article zygote (P0) AB P1 A P L A R P A EMS L P A R P A MS P A P2 E P C A P3 P D P4 Fig. 5. Summary of let-756 mosaics in the AB lineage. The mosaic analysis was carried out in the background of dpy-17 let-756 ncl-1 unc-36; sDp3. Mosaic animals were identified based on their Unc non-Dpy phenotypes. Patterns of duplication loss are represented by different colors: red, loss in a single lineage; green, losses in a single lineage and some independent cell(s); blue, losses in multiple independent lineages. If P1 losses would confer a Soc phenotype, then P1(–) mosaic animals would be easily identified phenotypically as Soc non-Unc. However, Soc non-Unc mosaic animals were extremely rare (see Materials and methods). Strikingly, nine additional P1 mosaic animals that were identified by alternative approaches (see Materials and methods) displayed a Clr phenotype at the restrictive temperature (25°C) (Fig. 4). We conclude that loss of mpk-1 activity in the P1 lineage is also not sufficient to suppress the Clr phenotype. Therefore, the presence of mpk-1(+) activity within either the AB lineage or the P1 lineage can prevent the Soc phenotype, and, conversely, loss of mpk-1 activity in both the AB and P1 lineage is required for the suppression of the Clr phenotype. Since the hypodermis has significant contributions from both the AB and the P1 lineage, the results of our mpk-1 mosaic analysis are consistent with MPK-1 acting in the hypodermis to carry out its Soc function. Mosaic analysis of let-756 We also performed a mosaic analysis of let-756 to identify the site where its expression is required for its essential function. Loss of LET-756 at this site would result in lethality, thereby prohibiting the identification of this class of mosaics. Since LET-756 is predicted to be a ligand, such an analysis could determine its site of expression rather than its site of action. Similar to the mpk-1 mosaics, we took advantage of the free chromosomal duplication sDp3, which also carries a wild-type copy of let-756. Mutations in unc-36 and dpy-17 were included to help facilitate the identification of specific classes of potential mosaic animals. dpy-17 is required in the P1 lineage for a portion of its activity (Kenyon, 1986; Yuan and Horvitz, 1990). Therefore, AB(–) or AB.p(–) animals are Unc non-Dpy, while P1(–) animals are semi-Dpy non-Unc. We first screened dpy-17 let-756 ncl-1 unc-36; sDp3 animals for either Unc non-Dpy or Dpy non-Unc mosaic animals. Unc non-Dpy animals were identified as expected, and the vast majority of these animals had losses in either AB, AB.p, or descendants in the AB.p lineage (Fig. 5 and Table S1 at http://dev.biologists.org/supplemental/). Besides being Unc, these mosaic animals were completely wild type, suggesting that lack of let-756(+) expression in the AB lineage is not essential for viability. By contrast, Dpy non-Unc animals were extremely rare, and the few Dpy non-Unc animals identified turned out to result from the breakdown of the free duplication (see Materials and methods). This striking finding suggests that the cellular focus of let-756 is not easily separable from that of dpy-17. DPY-17 is thought to be required in the P1 lineage (Kenyon, 1986; Yuan and Horvitz, 1990). Based on the lack of P1 losses, we conclude that LET-756 expression, like DPY-17, is likely required in the P1 lineage. Consistent with our mosaic analysis, a Plet-756::GFP reporter shows robust expression in the body wall muscles (Bülow et al., 2004), all but one of which is P1-derived. We also screened randomly for mosaic animals using the Ncl lineage marker. As expected, losses within the AB lineage were identified, while no P1(–) mosaic animals were identified (see Fig. S1 at http://dev.biologists.org/supplemental/). These data are consistent with a requirement for LET-756 expression in cells derived from the P1 lineage. Interestingly, mosaic animals with losses in the entire MS lineage and part of the C lineage can also be identified (see Fig. S1 at http://dev.biologists.org/ supplemental/). Since both the MS and the C lineages contribute a significant portion of the body wall muscles in the animal (35% and 40%, respectively), this result suggests that LET-756 expression is not required in all the body wall muscles. Discussion The Clr phenotype of hyperactive EGL-15 signaling in C. elegans has been powerfully exploited to identify components of an FGF signaling pathway. Here we present evidence supporting the hypothesis that EGL-15 and CLR-1 act together in the same cells to mediate their downstream effects. These effects appear to play a physiological role in fluid homeostasis, as opposed to a developmental role. Such a physiological role for EGL-15 is supported by the rapid phenotypic response of clr-1(e1745ts) mutants when shifted to the non-permissive temperature. Further support derives from the fact that this response can occur at any time during larval development. EGL-15 signaling in the hypodermis 2603 To understand the biological function of the CLR-1/EGL-15 pathway, we analyzed the site of action of EGL-15 signaling components involved in this function of EGL-15. Three lines of evidence indicate that EGL-15 signaling acts in the hypodermis to regulate fluid balance. First, EGL-15 is expressed in the hypodermis, as shown by immunofluorescence as well as an analysis of the e15 enhancer. Second, hypodermal expression of either EGL-15 or CLR-1 by tissue-specific promoters can restore their normal functions. Finally, our results of a mosaic analysis of mpk-1, which acts downstream of egl-15, are consistent with MPK-1 acting in the hypodermis to carry out its Soc function. Fluid homeostasis results from maintaining the balance between fluid inflow and outflow rates. To date, laser ablation experiments and mutant analysis have implicated the excretory system and the canal-associated (CAN) neurons as important regulators of fluid balance in C. elegans. The excretory system consists of four cells: the excretory cell, the excretory duct cell, the excretory pore cell and the binucleate gland cell (Nelson et al., 1983). Laser ablation of the excretory cell, the duct cell or the pore cell results in a Clr phenotype (Nelson and Riddle, 1984). A mosaic analysis of let-60 ras lent further support for the role of the excretory system in fluid regulation. The rod-like dead larvae resulting from severely compromised LET23(EGFR)/LET-60(RAS)/MAP kinase signaling display a dramatic Clr phenotype that appears to be due to the failure to specify the fate of the excretory duct cell (Yochem et al., 1997). Although more severe in its lethal phenotype, the fluid accumulation in the pseudocoelom is very similar to that seen in clr-1 mutants. Ablation of the CAN neurons also results in a Clr phenotype (Forrester et al., 1998). This is further supported by the analysis of ceh-10 null mutants that display both a Clr phenotype and CAN neurons that fail to migrate and differentiate properly (Forrester et al., 1998). The tight association of the CAN neurons with the canals of the excretory cell suggests that these cells work together to regulate fluid efflux. Here we present evidence indicating that EGL-15 signaling controls fluid homeostasis by acting in the hypodermis. Interestingly, the excretory canals form extensive gap junctions to the hypodermis (Nelson et al., 1983), suggesting a mechanism that might couple their influences on maintaining fluid balance. Mosaic analysis of mpk-1 not only suggests action of EGL15 signaling in the hypodermis, but also strongly discounts the importance of EGL-15 activity in the excretory system. Our analysis suggests that MPK-1 acts in cells derived from both the AB and P1 lineages to carry out its Soc function. Therefore, the cellular focus of mpk-1 could either be in multiple tissues or in a tissue that derives from both the AB and P1 lineages. The hypodermis has major contributions from both the AB and P1 lineages (Sulston et al., 1983) and similar mosaic results have been interpreted as supporting function in the hypodermis (Herman and Hedgecock, 1990). Consistent with this hypothesis, tissue-specific expression of EGL-15 and CLR-1 shows that hypodermal expression is sufficient to restore their normal functions. Importantly, all four cells in the excretory system are derived from the AB.p sublineage. Since lack of mpk-1 activity in either AB or AB.p is not sufficient to suppress the Clr phenotype caused by a clr-1 mutation, EGL-15 signaling does not appear to act in the excretory system exclusively. Tissue-specific transgene expression also strongly suggests hypodermal action, despite some indications that neuronal expression might be important. Supporting a hypodermal site of action, two different hypodermal promoters (Pdpy-7 and Prol-6) can drive strong rescue activity for all related EGL-15 functions as well as CLR-1 function. We have also shown by immunofluorescence that a major site of EGL-15 expression is in the hypodermis. Furthermore, an essential enhancer element from the egl-15 promoter, e15, which is sufficient for robust transgenic rescue of EGL-15 functions when duplicated, drives expression in the hypodermis. Combined with the mosaic results, we believe that the most likely site of action for the essential function of EGL-15 is in the hypodermis. Interestingly, however, we also observed strong transgenic rescue for a number of pan-neural promoters that have been reported to be neuronal-specific. Two out of three pan-neural promoters (Punc-14 and Psnb-1) showed strong transgenic rescue, while the third, Paex-3, showed no rescue activity. Both the aex3 and the unc-14 promoters drove strong expression of EGL15 in neurons, as assayed using immunohistochemistry. Thus, it appears that it is not EGL-15 expression in the neurons that accounts for the observed transgenic rescue, but rather that low level expression elsewhere might confer rescue activity. The mpk-1 mosaic analysis further refutes a pan-neural site of action of EGL-15. Although 97% of all neurons, including the CAN neurons, are derived from the AB lineage (Sulston et al., 1983), loss of mpk-1 activity in the AB lineage alone is not sufficient to confer a Soc phenotype. Specific action in the CAN neurons is even more strongly refuted by the mosaic data, since these cells derive exclusively from the AB lineage. This evidence is further bolstered by our results that expression of EGL-15 in the CAN neurons, using either the ceh-10 promoter or the ceh-23 promoter (Forrester et al., 1998; Svendsen and McGhee, 1995), failed to rescue the Soc phenotype of egl15(n1783) (P.H. and M.J.S., unpublished). The combined data from all these approaches strongly support EGL-15 signaling acting in the hypodermis to regulate fluid balance. It is interesting to note that the same RAS/MAPK cascade is exploited in two distinct processes in maintaining fluid homeostasis in worms: a developmental role in the specification of the excretory duct cell fate regulated by EGF signaling (Yochem et al., 1997) and a physiological role to control fluid influx regulated by FGF signaling. The use of a receptor-mediated response pathway allows responding to fluid imbalance by regulating the availability of the appropriate EGL-15 ligand. Our data support the function of the LET-756 FGF as the ligand for this function of EGL-15. To understand how LET-756 regulates EGL-15 signaling, we performed a mosaic analysis of let-756 to determine its cellular source. Our data indicate that LET-756 expression is not required in the AB lineage. Since all the cells that have been previously implicated in fluid homeostasis, such as the excretory system and the CAN neurons, are derived from the AB lineage, this result suggests that these cells are unlikely to be the cellular source of LET-756. Instead, the cellular focus of let-756 is tightly associated with that of dpy-17, which is required in P1-derived cells for a portion of its activity (Kenyon, 1986; Yuan and Horvitz, 1990). Interestingly, a let756 reporter is expressed extensively in body wall muscles (Bülow et al., 2004), which derive predominantly from P1. Understanding the regulation of LET-756 in response to different physiological conditions may lead to new insights into how C. elegans maintains its fluid balance. 2604 Development 131 (11) Based on the phenotypic analysis of mutations affecting EGL-15 signaling, EGL-15 activity in the hypodermis either functions to promote fluid intake or inhibit fluid excretion. This could be achieved by regulating the permeability of the hypodermis. Interestingly, FGF2–/–/FGF5–/– double-mutant mice show enhanced permeability of the blood–brain barrier, which is accompanied by reduced levels of intermediate filaments as well as tight junction proteins (Reuss et al., 2003). Alternatively, EGL-15 might regulate the activity of ion channels, which could provide the driving force for water movement. In mammals, the kidney is the major organ for maintenance of global fluid balance as well as ion homeostasis. Interestingly, FGF-23 is implicated in phosphate homeostasis in the kidney, which may similarly aid in maintaining systemic fluid balance. Overexpression of FGF-23, or a mutant form of FGF-23 that is resistant to proteolysis, results in inhibition of phosphate reabsorption by the type II sodium-dependent phosphate (Na/Pi) co-transporter in renal epithelial cells, leading to hypophosphatemia (Kumar, 2002). Our findings indicate that the EGL-15 FGFR signaling pathway is probably the master switch in the hypodermis regulating fluid balance in C. elegans, and provide an important foundation from which to explore mechanisms by which FGF signaling functions to regulate fluid balance. We thank R. Roubin for discussions, for providing preliminary expression data for let-756 reporters, and for several let-756 constructs. We also thank: O. Hobert for discussions and sharing unpublished results; D. Baillie for dpy-17 let-756 unc-32; sDp3; E. Lambie for providing mpk-1 ncl-1 unc-36; sDp3; C. Z. Borland for making the initial observation that Pegl-15::clr-1 can rescue clr-1; I. E. Sasson for help in the integration of ayIs25 and observing EGL-15 expression in the hypodermis; T. Lo for generating ayIs29; and M. R. Koelle, L. Cooley and members of our laboratory for a critical reading of this manuscript. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH. This work was supported by NIH grant GM50504 to M.J.S. References Basilico, C. and Moscatelli, D. (1992). The FGF family of growth factors and oncogenes. Adv. Cancer Res. 59, 115-165. Borland, C. Z., Schutzman, J. L. and Stern, M. J. (2001). Fibroblast growth factor signaling in Caenorhabditis elegans. BioEssays 23, 1120-1130. Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 7194. Bülow, H., Boulin, T. and Hobert, O. (2004). Differential functions of the C. elegans FGF receptor in axon outgrowth and maintenance of axon position. Neuron (in press). Burdine, R. D., Branda, C. S. and Stern, M. J. (1998). EGL-17(FGF) expression coordinates the attraction of the migrating sex myoblasts with vulval induction in C. elegans. Development 125, 1083-1093. Church, D. L., Guan, K.-L. and Lambie, E. J. (1995). Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 121, 2525-2535. DeVore, D. L., Horvitz, H. R. and Stern, M. J. (1995). An FGF receptor signaling pathway is required for the normal cell migrations of the sex myoblasts in C. elegans hermaphrodites. Cell 83, 611-620. Finney, M. and Ruvkun, G. (1990). The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell 63, 895-905. Forrester, W. C., Perens, E., Zallen, J. A. and Garriga, G. (1998). Identification of Caenorhabditis elegans genes required for neuronal differentiation and migration. Genetics 148, 151-165. Research article Gilleard, J. S., Barry, J. D. and Johnstone, I. L. (1997). cis regulatory requirements for hypodermal cell-specific expression of the Caenorhabditis elegans cuticle collagen gene dpy-7. Mol. Cell Biol. 17, 2301-2311. Hedgecock, E. M. and Herman, R. K. (1995). The ncl-1 gene and genetic mosaics of Caenorhabditis elegans. Genetics 141, 989-1006. Herman, R. K. and Hedgecock, E. M. (1990). Limitation of the size of the vulval primordium of Caenorhabditis elegans by lin-15 expression in surrounding hypodermis. Nature 348, 169-171. Iwasaki, K., Staunton, J., Saifee, O., Nonet, M. and Thomas, J. H. (1997). aex-3 encodes a novel regulator of presynaptic activity in C. elegans. Neuron 18, 613-622. Kenyon, C. (1986). A gene involved in the development of the posterior body region of C. elegans. Cell 46, 477-487. Kokel, M., Borland, C. Z., DeLong, L., Horvitz, H. R. and Stern, M. J. (1998). clr-1 encodes a receptor tyrosine phosphatase that negatively regulates an FGF receptor signaling pathway in Caenorhabditis elegans. Genes Dev. 12, 1425-1437. Kumar, R. (2002). New insights into phosphate homeostasis: fibroblast growth factor 23 and frizzled-related protein-4 are phosphaturic factors derived from tumors associated with osteomalacia. Curr. Opin. Nephrol. Hypertens. 11, 547-553. Mello, C. C., Kramer, J. M., Stinchcomb, D. and Ambros, V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. Nelson, F. K. and Riddle, D. L. (1984). Functional study of the Caenorhabditis elegans secretory-excretory system using laser microsurgery. J. Exp. Zool. 231, 45-56. Nelson, F. K., Albert, P. S. and Riddle, D. L. (1983). Fine structures of the Caenorhabditis elegans secretory-excretory system. J. Ultrastruct. Res. 82, 156-171. Nonet, M. L., Saifee, O., Zhao, H., Rand, J. B. and Wei, L. (1998). Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J. Neurosci. 18, 70-80. Ogura, K., Shirakawa, M., Barnes, T. M., Hekimi, S. and Ohshima, Y. (1997). The UNC-14 protein required for axonal elongation and guidance in Caenorhabditis elegans interacts with the serine/threonine kinase UNC51. Genes Dev. 11, 1801-1811. Okkema, P. G., Harrison, S. W., Plunger, V., Aryana, A. and Fire, A. (1993). Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics 135, 385-404. Reuss, B., Dono, R. and Unsicker, K. (2003). Functions of fibroblast growth factor (FGF)-2 and FGF-5 in astroglial differentiation and blood-brain barrier permeability: evidence from mouse mutants. J. Neurosci. 23, 64046412. Roubin, R., Vatcher, G., Popovici, C., Coulier, F., Thierry-Mieg, J., Pontarotti, P., Birnbaum, D., Baillie, D. and Thierry-Mieg, D. (1999). let-756, a C. elegans fgf essential for worm development. Oncogene 18, 6741-6747. Schutzman, J. L., Borland, C. Z., Newman, J. C., Robinson, M. K., Kokel, M. and Stern, M. J. (2001). The Caenorhabditis elegans EGL-15 signaling pathway implicates a DOS-like multisubstrate adaptor protein in fibroblast growth factor signal transduction. Mol. Cell Biol. 21, 8104-8116. Stringham, E. G., Dixon, D. K., Jones, D. and Candido, E. P. M. (1992). Temporal and spatial expression patterns of the small heat shock (hsp16) genes in transgenic Caenorhabditis elegans. Mol. Biol. Cell 3, 221-233. Sulston, J. E., Schierenberg, E., White, J. G. and Thomson, J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64-119. Svendsen, P. C. and McGhee, J. D. (1995). The C. elegans neuronally expressed homeobox gene ceh-10 is closely related to genes expressed in the vertebrate eye. Development 121, 1253-1262. Ullrich, A. and Schlessinger, J. (1990). Signal transduction by receptors with tyrosine kinase activity. Cell 61, 203-212. Webster, M. K. and Donoghue, D. J. (1997). FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 13, 178-182. Yochem, J., Sundaram, M. and Han, M. (1997). Ras is required for a limited number of cell fates and not for general proliferation in Caenorhabditis elegans. Mol. Cell Biol. 17, 2716-2722. Yuan, J. Y. and Horvitz, H. R. (1990). The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev. Biol. 138, 33-41.