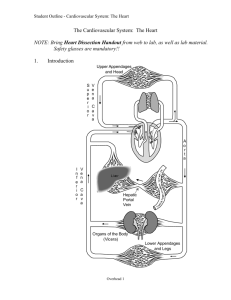
Chambers, valves, conduction system and coronary
advertisement

LSS Cardiovascular System Alexandra Burke-Smith Chambers, valves, conduction system and coronary circulation CVS 1 - Karen McCarthy (k.mccarthy@imperial.ac.uk) 1. Describe the circulatory pathway through the heart and be able to identify and name the vessels that enter the heart and the vessels that leave the heart 2. Explain the spatial relationships of left heart chambers relative to right heart chambers, the differences between atria and ventricles and the structural differences between right and left ventricles 3. Identify and label the components of septum (atrial septum, ventricular septum, membranous septum) 4. Identify and label the heart valves and their locations, and state the structural similarities and differences 5. Describe the components of the conduction system 6. Outline the coronary circulation and be able to identify the main coronary arteries and cardiac veins Cardiac Position and Borders The long axis of the heart is at an angle to the long axis (midline) of the body, with the apex (formed by the INFEROLATERAL part of the left ventricle- the bottom part furthest away from the midline of the body) in the left side of the body (Approx 2/3rds of the heart lies in the left side of the body) The heart lies between the STERNUM and the SPINE o The sternum is ANTERIOR to the right ventricle o The spine is POSTERIOR to the left atrium Functionally consists of two pumps separated by a partition. Each pump consists of an atrium, ventricle separated by a valve. o The right pump receives deoxygenated blood from the body and sends it to the lungs, and the left pump receives oxygenated blood from the lungs and sends it to the body The heart can be thought of as having 5 surfaces: o Posterior surface (which is the base of the heart) o Anterior surface (which also then forms the apex) o Right pulmonary surface (facing the right lung) o Left pulmonary surface (facing the left lung) o Diaphragmatic surface (facing the diaphragm) The posterior surface The posterior surface consists of: left atrium Small portion of the right atrium PROXIMAL (beginning) parts of the great veins: o Superior vena cava (enters top right atrium- delivering blood from body). Also known as superior caval vein o Inferior vena cava (enters bottom right atrium- delivering blood from body). Also known as inferior caval vein. o Coronary sinus (enters right atrium medial to the inferior vena cava opening delivering deoxygenated blood draining from the coronary veins, i.e. from the heart itself) o Pulmonary veins (enter either side of left atrium- delivering blood from lungs) There are four pulmonary veins: o Right upper 1 LSS Cardiovascular System o o o Alexandra Burke-Smith Right lower Left upper Left lower The anterior surface The anterior surface consists of: o Mainly the right ventricle o Some of right atrium o Some of left ventricle The pulmonary trunk emerges from the right ventricle, and divided into the left and right pulmonary artery o The pulmonary trunk has a central position, and a spiral relationship with the aorta, which emerges from the left atrium Cardiac Chambers The four chambers of the heart are separated by interatrial, interventricular and atrioventricular septa: Atrial Chambers Right Atrium - Venous Sinus: superior vena cava, inferior vena cava and coronary sinus o The inferior vena cava is guarded by the EUSTACHIAN VALVE, and the coronary sinus is also guarded by a valve - The right atrium can be divided into two continuous spaces, divided by the TERMINAL CREST (also known as CRISTA TERMINALS) - Characteristic PECTINATE muscle bundles cover the walls of the atrium on the triangular appendage in the space anterior to the crest, known as the RIGHT AURICLE - In the space posterior to the crest has smooth, thin walls and both venae cavae and the coronary sinus empty into this space Interatrial septum - The right and left atriums are separated by the interatrial septum - A depression/infolding rim in the septum (just above the ORIFICE of the inferior vena cava in the RIGHT ATRIUM) is clear – this is the OVAL FOSSA o The oval fossa effectively is a flat valve, which prevents blood flow directly between the atrial chambers o The oval fossa marks a location important for foetal circulation as it allows oxygenated blood to bypass the non-functioning lungs and enter the right atrium passing directly to the left atrium o however when the lungs begin to function, this hole in the septum is supposed to close, however a defect in this closure often occurs which leads to shunting of the blood between atrial chambers o the ARTERY TO THE SINUS NODE can course within the depression/infolding between the two chambers Left Atrium - Posterior half is smooth and receives blood from the four pulmonary veins - Anterior half is contiuous with the LEFT AURICLE, and contains pectinate muscles. However in the left atrium, there is no distinct separation (like the terminal crest) between these two halves 2 LSS Cardiovascular System - Alexandra Burke-Smith The crescent shape of the oval fossa is apparent in the anterior wall of left atrium, and is known as the VALVE OF FORAMEN OVALE (which prevents blood flow from left to right atrium) o However this valve may not completely fuse with the oval fossa in adults, which may leave a passage between the two atrial chambers, leaving a PATENT FORAMEN OVALE (effectively a hole in the septum, leading to shunting of blood between the atria) Ventricular Chambers Both ventricular chambers have an INLET, APICAL and OUTLET components, and constant apical TRABECULATIONS. Right Ventricle - Forms most of the anterior surface of the heart, and a portion of the diaphragmatic surface o Inlet component – the tricuspid valve and the atrioventricular septum o Apical component – trabecular portion o Outlet component - infidulum - It is to the right of the right atrium, and in front of and left of the RIGHT ATRIVENTRICULAR ORIFICE; blood therefore enters the ventricle moving in a horizontal and forward direction - Outflow portion: PULMONARY INFIDIBULUM– leads to the pulmonary trunk – has smooth walls - Inflow portion wall has substantial complex muscle structures called COURSE TRABECULATIONS; these are either attached continuously to the walls forming ridges, or attached at both ends forming bridges - Trabeculations which are only attached at one end to the ventricular surface and the other end are attached to CHORDAE TENDONAEA (fibrous tendon-like chords which connect to the free edges of the tricuspid valve), are also known as PAPILLARY muscles. There are 3 types of papillary muscles in the right ventricle depending on their point of origin o ANTERIOR – largest, arises from the anterior wall o POSTERIOR – arise from the posterior wall o SEPTAL (medial)– most inconsistent as either small or absent, but allow chorea tendineae to emerge directly from the interventricular septum - The SEPTOMARGINAL TRABECULA/MODERATOR BAND: single trabecula which forms a bridge between the lower portion of the IV septum and the base of the anterior papillary muscle, carrying the right atrioventricular bundle to the anterior wall of the right ventricle during cardiac conduction o SEPTOPARIETAL Trabeculations extend from the anterior surface of the moderator band to the wall of the ventricle - any of the individual muscle structures within the right ventricle have the potential to become atrophied, or necrose following a myocardial infarction Interventricular Septum - The left ventricle is some-what posterior to the right ventricle, so the interventricular septum forms some of the posterior wall of the right ventricle (and is to the left). - The septum is described as having two parts: o Muscular o Membranous - The muscular part is thick and forms the major part - The membranous part is thin and forms the upper part of the septum - A third part of the septum may be considered to be atrioventricular as its superior location places it between the left ventricle and the right atrium Left Ventricle - Contributes to the anterior, diaphragmatic and left pulmonary surfaces of the heart - Forms the apex of the heart 3 LSS Cardiovascular System - Alexandra Burke-Smith Blood enters from the left atrium through the LEFT ATRIVENTRICULAR ORIFICE; flows in a left forward direction to the apex Chamber is conical, longer than the right ventricle and has a thicker layer of MYOCARIUM Inlet – holds the atrioventricular valve Outflow tract (AORTIC VESTIBULE) is posterior to the infidibulum of the right ventricle and leads to the AORTA Apical component: Trabeculations are fine/delicate in comparison with right ventricle Papillary muscles are larger than those of the right ventricle, and consist of only ANTERIOR (anterolateral) and POSTERIOR (postero-medial) muscles Cardiac Valves All cardiac valve leaflets are composed of highly organised connective tissue fibres. The connective tissues (collagen and elastic) are arranged in layers along fibroblasts. This organisation provides strength to the leaflet. The entire valve is surrounded by a layer of endothelial cells. The free edges are thicker allowing cushioning of the leaflets. Atrioventricular valves o MITRAL valve o TRICUSPID valve o At the atrioventricular junctions o Separated from each other by the interventricular septum Arterial valves o AORTIC valve o SEMI-LUNAR PULMONARY valve o Formed from the cushions of tissue within the developing outflow tracts from each ventricle o Have TRI-LEAFLET structures; each leaflet is separated by an inter-leaflet triangle of fibrous tissue o There is then a cross-over/spiralling of the great arteries: aorta and pulmonary trunk On the right side - Tricuspid Valve Atrioventricular valve- at the right atrioventricular orifice Closed during ventricular contraction Consists of three CUSPS; the base of each is secured to a fibrous ring that surrounds the orifice o The cusps are continuous with each other near their bases; these sites are termed COMMISSURES - The free margins of each cusp are attached to the chordate tendineae which arise from the tips of the papillary muscles o Tendinaea from TWO papillary muscles attach to each cusp, which ensures proper closing of the valve during contraction – therefore blood exits the right ventricle and enters the pulmonary trunk and is prevented from moving back into the right atrium - The cusps are named based on their relative position in the right ventricle o ANTEROSUPERIOR cusp o MURAL (posterior) cusp o SEPTAL cusp - During the filling of the right ventricle (ATRIAL SYSTOLE), the tricuspid valve is open, and the 3 cusps project into the right ventricle Pulmonary valve - Arterial valve – at the opening of the pulmonary trunk 4 LSS Cardiovascular System - - Alexandra Burke-Smith At the apex of the SUBPUMLONARY INFUNDIBULUM (muscle sleeve within the right ventricle which supports the valve) Consists of 3 SEMILUNAR CUSPS with free edges projecting upwards into the lumen of the pulmonary trunk o LEFT cusp o RIGHT cusp o ANTERIOR cusp Between the cusps, there are interleaflet triangles which make sure there are no gaps between the cusps Acts as SINUTUBULAR JUNCTION: junction between myocardium and arterial tissue Prevents flow of blood back into the right ventricle during VENTRICULAR DIASTOLE On the left side - Mitral Valve Atrio-ventricular valve – at left atrioventricular orifice Closed during ventricular systole Also known as BICUSPID VALVE – consists of two cusps: o ANTERIOR (aortic) cusp – this has FIBROUS CONTINUITY with the aorta o POSTERIOR (mural) cusp - Bases of the cusps secured at commissures, and are coordinated by the action of two papillary muscles attached by tendinuous chords: o ANTERO-LATERAL muscle o POSTERO-MEDIAL muscle Aortic Valve - Arterial valve – central location compared to other cardiac valves - Similar in structure to pulmonary valve; 3 semilunar cusps with free edge projecting upwards into aortic lumen - Between the semilunar cusps and the wall of the ASCENDING aorta are: o RIGHT aortic sinus – origin of right CORONARY ARTERY o LEFT aortic sinus – origin of left CORONARY ARTERY o POSTERIOR aortic sinus – also known as the NON-CORONARY sinus - Membranous septum is situated between the right aortic and non-coronary sinus - Function: similar to pulmonary valve; in addition as blood recoils during VENTRICULAR DIASTOLE, the blood fills the aortic sinuses, forcing blood into the coronary arteries The Coronary Arteries: - Right Coronary Artery Originates from the right aortic sinus Located along right atrioventricular groove Gives rise to the POSTERIOR DESCENDING ARTERY (PDA) – also known as the inferior interventricular artery Left Coronary Artery - Originates from left aortic sinus - Divides into the: o CIRCUMFLEX ARTERY – located in the left atrioventricular groove o LEFT ANTERIOIR DESCENDING (LAD) – also known as the superior interventricular artery Septum perforator – supplies the septum, feeding the conduction system Conduction System & Membranous Septum 5 LSS Cardiovascular System Alexandra Burke-Smith SAN is located at the junction of the Superior Vena Cava and the right atrium – cells histologically distinct from surrounding myocardium, but not insulated. Origin of electrical impulse Conduction of impulse from SAN does not run in CONDUCTION TRACTS, but the orientation of the normal myocardial fibres within the right atrium directs the impulse towards the atrioventricular node The atrioventricular node within the right atrium is situated between in the tricuspid valve, Eustachian valve and the Right coronary sinus o Is at the apex of the TRIANGLE OF KOCH – surrounded by central fibrous body adjacent to the membranous septum o Specialised myocardium known as the BUNDLE OF HIS – which separates into right and left bundle branches. o Conduction passes down the bundles through the membranous septum to the PURKINJE FIBRES 6 LSS Cardiovascular System Alexandra Burke-Smith 7 LSS Cardiovascular System Alexandra Burke-Smith Mechanical Properties of the Heart I CVS 2 - Dr Ken MacLeod (k.macleod@imperial.ac.uk) 1. Describe the relationship between ventricular wall tension, chamber radius, and chamber pressure (Law of Laplace) 2. List the sequence of events from excitation that bring about contraction then relaxation of a ventricular cell 3. State Starling’s Law of the Heart 4. Explain the mechanisms underlying Starling’s Law of the Heart 5. Use a graph to compare the length-tension relationships for cardiac and skeletal muscle 6. Explain the concepts of preload and afterload Importance of Calcium with regards to contraction of the whole heart Single cell structure Described by Sidney Ringer Ventricular cells 100 μm long and 15 μm wide T-tubules (transverse tubules) are finger-like invaginations from the cell surface T-tubule openings up to 200 nm in diameter Carry surface depolarisation deep into the cell and are spaced (approx. 2 μm apart) so that a T-tubule lies alongside each Z line of every myofibril. Lace-like structure; sarcoplasmic reticulum surround the T-tubules and myofibrils, also store Ca Major components of Myocytes are myofibrils and mitochondria Excitation-Contraction coupling On excitation; influx of Ca into the myocate vie L-TYPE CA CHANNELS occurs Ca then binds to intracellular SR-Ca release channels, causing them to change conformation o This change in conformation leads to INDUCED CALCIUM RELEASE from the sarcoplasmic reticulum Ca release then causes contraction of the myocyte. This is completely dependent on the presence of extracellular Ca Relaxation: o Intracellular Ca is taken up into the SR by Ca-ATPase (also known as SERCA) ready to be released again o Ca is also removed from the myocyte by Na/Ca exchanger, which uses the energy gradient from sodium to expel Ca into the extracellular matrix Contraction Force Force production and Intracellular Calcium There is a SIGNOIDAL relationship between Log of the cytoplasmic Ca concentration and the % of maximum force produced Length-tension Relation Consider Isometric (no shortening) contraction At a stretched length, a larger contraction is produced which leads to an increased force production (this is true up to a certain point, after which further stretching reduces the force produced) – ACTIVE FORCE production 8 LSS Cardiovascular System Alexandra Burke-Smith When stretched, muscle exerts a PASSIVE force which is indicated by the indicated by the increase in the BASELINE force before contraction Comparison with Skeletal muscle o Cardiac muscle more resistant to stretch, therefore the PASSIVE force is reduced o Less compliant than skeletal muscle due to properties of the extracellular matrix and cytoskeleton o Cardiac muscle is very unlikely to be over-stretched, as PERICARDIUM limits stretch, therefore only the ascending limb of the length-tension relation is important for cardiac muscle Concepts of Pre-load and After-load Pre-load: weight that stretches muscle before it is stimulated to contract After-load: weight not apparent to muscle in resting state, and only encounteres when the muscle has started to contract Isotonic (shortening) contraction Inverse linear relationship between afterload and shortening Almost linear inverse relationship between afterload and velocity of shortening If pre-load increases, there is an initial enhanced stretch, which increases the ability of the muscle to produce more force by shifting the graph to the right, i.e. a greater afterload will result in more shortening than before In-vivo correlates of pre-load As blood fills the ventricles during the relaxation phase (or diastole) of the cardiac cycle it stretches the resting ventricular walls The stretch or filling determines the preload on the ventricles before ejection Preload is dependent upon venous return to the heart Exercise increases pre-load Measures of preload: end-diastolic volume, end diastolic pressure, right atrial pressure In-vivo correlates of after-load Definition: The load against which the left ventricle ejects blood after opening of the aortic valve A simple measure of afterload is the diastolic arterial blood pressure Any increase in afterload decreases the amount of isotonic shortening that occurs and decreases the velocity of shortening I.e. small ventricular filling leads to a smaller contraction as the ventricular cardiac muscle responds less effectively to the afterload of the arteriol blood pressure 9 LSS Cardiovascular System Alexandra Burke-Smith Ways to alter contraction of the heart Intrinsic Mechanisms o Frank-Starling Relationship o Rate-induced regulation Extrinsic Mechanisms o Autonomic Nervous System o Endocrine System o Blood gases & pH Intrinsic Mechanisms Frank-Starling relationship - Definition: increased diastolic fibre length increases ventricular contraction - Consequence: ventricles pump greater stroke volume so that, at equilibrium, cardiac output exactly balances the augmented (increased) venous return - Two influencing factors: o Changes in the number of myofilament cross bridges that attach o Changes in the Ca sensitivity of the myofilaments - Changes in the number of cross bridges o At optimum sarcomere length: maximum INTERDIGITATION between thick and thin filaments o At shorter lengths than optimal, the actin filaments overlap on themselves therefore reducing the number of myosin cross bridges that can be made o At increased lengths than optimal, there is reduced overlap between myosin and actin, therefore reducing the number of cross-bridges that can form - Ca Sensitivity o Changes with change in sarcomere length o At longer sarcomere lengths, the affinity of Troponin C for Ca is increased o Therefore less Ca is required for the same amount of force produced WORK DONE Stroke work: work done by the heart to eject blood under pressure into the aorta and pulmonary artery Stroke volume (SV): volume of blood ejected during each stroke by each ventricle o This is greatly influenced by afterload Pressure (P): pressure at which blood is ejected o Greatly influenced by strcture Stroke Work = SV x P 10 LSS Cardiovascular System Alexandra Burke-Smith Law of Laplace States: when the pressure within a cylinder is held constant, the tension on its walls increases with increasing radius. o Therefore if pressure and tension (wall stress) are to remain constant, wall thickness must be increased or radius of the cylinder must be decreased T = PR/h o o o o T = wall tension P = internal pressure R = cylindrical radius h = height/length of cylinder Physiological relevance: Radius of curvature of walls of Left ventricle less than that of Right ventricle allowing LV to generate higher pressures with similar wall stress to combat the higher aortic blood pressure than pulmonary bp Facilitates late ejection Wall stress kept low in giraffe by long, narrow, thick-walled ventricle In frog, where pressures are low the ventricle is almost spherical Failing hearts often become dilated which decreases pressure generation and ejection of blood and increases wall stress by increasing the radius 11 LSS Cardiovascular System Alexandra Burke-Smith Cardiac Electrophysiology I: Electrical Activity of the Heart CVS 3 - Dr Frank Harrison (f.harrison@imperial.ac.uk) 1. Describe the main structures of the human heart. 2. Describe the structure of a typical cardiac ventricular myocyte. 3. Briefly describe the pathways of the heart that subserve the normal orderly passage of electrical activity through it. 4. Sketch an intracellular action potential for a) a sino-atrial node cell b) an atrial cell c) a ventricular cell. 5. State that the sino-atrial (SA) node is the normal pacemaker and explain why and how this is so. 6. Describe how activity in the SA node spreads to both atria. 7. Explain why transmission of electrical activity from the atria to the ventricles normally only occurs at the atrioventricular (A-V) node. 8. Describe how electrical activity is transmitted to all parts of the ventricles through the Bundle of His and the Purkinje fibres. 9. Explain why the ventricular action potential has a long duration and relate this to the function of the ventricles. 10. Describe the ECG waveforms using the conventional PQRST nomenclature, and state the electrical events that each represents. Structure of the human heart Cardiac myocytes Small cells attached to adjacent ones by end-to-end junctions known as INTERCALATED DISCS o Also small gap junctions with LOW electrical resistance Action potentials spread between cells and then the cells act together – SYNCYTIUM Contain actin and myosin as contractile proteins The conduction system (NB: numbers on diagram indicate time at which depolarisation occurs) 5 principle components: Sino-Atrial Node (SAN): - strip of modified muscle cells on the posterior wall of the right atrium which is the site of excitation signal generation Atrio-Ventricular Node (AVN): - Collection of specialised cells which forms a bridge of conducting tissue over the non-conductive ring within the atrio-ventricular septum Bundle of His: - Also known as the atrio-ventricular bundle. It is a direct continuation of the AVN – a bundle of rapidly conduction tissue which follows along the lower border of the membranous part of the interventricular septum – conveying electrical activity from the AVN down the septum. 12 LSS Cardiovascular System Alexandra Burke-Smith Bundle Branches: - RIGHT bundle branch – continues on the right side of the interventricular septum towards the apex of the right ventricle - LEFT bundle branch – passes to the left side of the muscular interventricular septum and descends to the apex of the left ventricle Purkinje Fibres: - Subendocardial plexus of conduction cells – fibres located on the endocardium which penetrate the muscle walls of the ventricles including the papillary muscles NB: the conduction system ensures ventricular mass conducts as simultaneously as possible to maximise force production – i.e. maximise the systolic ventricular blood pressure. Contraction is spontaneous – this is different to skeletal muscle which requires electrical activity in the motor nerve supplying it. Depolarisation of the heart - Atrial depolarisation Excitation signal is generated at the SAN, and spread across the atria (right to left) The atria contract Wave of depolarisation meets the AVN, depolarising it There is a delay between atrial depolarisation and ventricular depolarisation – this is due to the AVN Ventricular depolarisation Wave of depolarisation flows from the AVN down the bundle of His and the two bundle branches to the purkinje fibres This leads to complete ventricular depolarisation, which causes the ventricles to contract Action Potentials Generated Pacemaker Cells (SAN) Pacemaker cells have a natural rhythm of approx 70 action potentials (and hence heartbeats) per minute They have a resting potential of approx -65mV, but this is not stable therefore becomes MORE negative. This is known as the PRE-POTENTIAL, and is seen on the graph as the slope as the potential increases towards -50mV o At -50mV, a full action potential is generated o Cause of the pre-potential: there is a special inward Na+ current into pacemaker cells, along with a decrease in the membrane permeability to K+, i.e. increase Na+ influx, decreased K+ efflux Influence of the Sympathetic Nervous System, e.g. ADRENALINE o Increases Na+ influx o Seen by an increase in the pre-potential slope, therefore the threshold potential is reached more rapidly – therefore heart rate is increased Influence of the Parasympathetic Nervous System, e.g. ACTEYLCHOLINE o Reduces Na+ influx o Seen by a decrease in the pre-potential slope, therefore the threshold is reached more slowly and heart rate is decreased 13 LSS Cardiovascular System Alexandra Burke-Smith Atrial Cells Resting potential is stable at -100mV Duration of action potential approx 100ms PLATEAU phase is seen as curve of repolarisation Ventricular Cells Resting potential stable – approx -90mV Duration – approx 250 ms (> Atrial cells) The plateau phase of the action potential is very long (200 ms) o this is due to a Ca2+ influx through ventricular voltage-gated channels o at approx -35mV, the channels open leading to a Ca2+ influx, which delays repolarisation AV Nodal Cells Similar to ventricular cells, but pre-potential slopes seen Under “heart-block”, the AV nodal cells become pacemaker cells, as the normal action potential spread from atria to ventricles does not occur Timing of Ventricular cell action potential and isometric force Due to the Ca2+ influx leading to a long plateau phase (and hence long membrane refractory period where another action potential cannot be generated), the contractile force starts to relax during the refractory period This prevents the production of a FUSED tetanus This is designed for the pumping action of the heart – pumping requires regular relaxation –activation, not continuous activation Ca2+ Excitatory Action Potential Coupling Caused by the influx of Ca2+ into ventricular cells, thus delaying repolarisation This increases the strength of contraction Therefore a possible treatment for heart failure – drug that increases the ventricular intracellular Ca2+ concentration, leading to more powerful contractions – DIGOXIN Angina treatment: o Angina – vascular supply to cardiac muscle decreases, oxygen supply reduced o Treatment – Ca2+ ion blockers, e.g. berapamil 14 LSS Cardiovascular System Alexandra Burke-Smith ECG Introduction Depolarisation Waves Detected as a change in the potential difference between two electrodes When a wave of depolarisation is moving TOWARDS the POSITIVE electrode, it is seen as an UPWARD deflection When a wave of depolarisation is moving AWAY from the POSITIVE electrode, it is seen as a DOWNWARD deflection If the wave of depolarisation is at right angles to the axis of the two electrodes, there should be no deflection – however the sensitivity of ECG measurements means often a BIPHASIC EQUIPOTENTIAL is seen instead of a stable potential with no deflection EGC Waveform Atrial Depolarisation – P WAVE - Depolarisation wave spreads across the atrial myocytes from the SAN - Mean direction of spread is right to left, this causes an electric field - Seen as small upward deflection Ventricular Depolarisation – QRS Complex - Wave of depolarisation reaches the AVN - The AVN is surrounded by junctional fibres with a lower conduction velocity – leads to delay between atrial depolarisation and ventricular depolarisation (approx 100ms) o During this delay, the atria contract and expel blood into the ventricles - Septum depolarisation of the bundle of his spreads towards the apex of the heart and then along the purkinje fibres o The depolarisation has a mean direction – known as the MEAN FRONTAL PLANE AXIS OF THE VENTRICLES (towards the left apex) o The endocardium depolarises before the epicardium - The apex of the heart then contracts slightly before the base, but the whole ventricular depolarisation lasts about 40ms - Atrial repolarisation also occurs during this time, but it is negligible in comparison to ventricular depolarisation Ventricular Repolarisation – T wave Note: the mean direction of the ventricular depolarisation along the MFPA is because the stroke volume of the ventricles is the same, but the systolic blood pressures are different. The right ventricle has a thinner wall and has a systolic blood pressure of about 25/12mmHg The left ventricle has a thicker wall and works against a larger pressure gradient, therefore needs to do more work and hence requires more electrical activity. Its systolic blood pressure is about 120/80mmHg 15 LSS Cardiovascular System Alexandra Burke-Smith Cardiac Electrophysiology II: Understanding the ECG CVS 4 - Dr Frank Harrison (f.harrison@imperial.ac.uk) 1. Describe how the recordings of the six standard limb leads are obtained from the four electrodes attached to the limbs 2. Explain briefly the principles underlying the concept of Einthoven's Triangle 3. Appreciate why the magnitude and direction of components of the ECG vary from lead to lead 4. Know the normal physiological range of the mean frontal plane axis 5. Understand what is meant by the terms left and right axis deviation, and how these conditions may occur 6. Describe how the recordings of the six pre-cordial (chest) leads are obtained 7. State how the information obtained from the chest leads is different from that derived from the limb leads 8. Explain why the magnitude and direction of the components of the ECG vary as the recording electrode is moved across the chest from V1 to V6 Attachment of Electrodes Right foot – zero reference point o Point of comparison so that potentials can be generated o Also removes effect of background electrical noise Left foot, right and left arm – positions available for recording signals from other electrodes Standard Limb Leads An equilateral triangle is considered between the right arm, left arm and the left foot, where the heart lies in the centre – this is known as EINTHOVEN’S TRIANGLE There are three leads; each denoted using ROMAN NUMERALS: o Lead I – comparison between RIGHT ARM and LEFT ARM (where the left arm is considered to be the positive electrode) o Lead II – comparison between RIGHT ARM and LEFT FOOT (where the left foot is considered to be the positive electrode) o Lead III – comparison between the LEFT ARM and LEFT FOOT (where the left foot is considered to be the positive electrode) Lead I is then considered to be at 0° The positive pole of Lead II is therefore considered to be +60° to the positive pole of Lead I The positive pole of Lead III is therefore considered to be +120° to the positive pole of Lead I +ve NB: angles BELOW the 0° line are considered POSITIVE 0° +ve +ve Augmented Limb Leads Couple the standard limb leads to form augmented vectors – aV 120° 60° 3 augmented leads, denoted by letters: o aV-R: the right arm is considered the positive electrode, and the negative electrode is considered to be half way between the left arm and left foot 16 LSS Cardiovascular System Alexandra Burke-Smith o aV-L: the left arm is considered the positive electrode, and the negative electrode is considered to be half way between the right arm and left foot o aV-F: the left foot is considered the positive electrode, and the negative electrode is considered to be half way between the right and left arm the readings from these leads will be > standard limb lead readings as they are coupled considering einthoven’s triangle, where Lead I is 0° o aV-R is at -150° (150° above the 0° line) o aV-L is at -30° (30° above the 0° line) o aV-F is at +90° (90° below the 0° line, i.e. perpendicular) Combining Limb Leads Hexagonal Reference System Diagrammatic representation of the 6 limb leads Arranged in 3 pairs of 2 leads which are at right angles to each other: o Lead I and AVF o Lead II and AVL o Lead III and AVR The Mean Frontal Plane Axis The mean vector/direction of wave depolarisation in the ventricles is towards the apex of the LEFT ventricle This may be: o along the axis of lead I, i.e. at 0° o in the direction of AVF, i.e. at +90° Remember, when a wave of depolarisation is moving TOWARDS the positive electrode it causes an UPWARD deflection. When it is moving AWAY from the positive electrode it causes a DOWNWARD deflection. Waveforms recorded in the leads 0 indicates no deflection, i.e. the wave of depolarisation is at 90° to the MFPA + indicates upward deflection, i.e. the wave of depolarisation is towards the positive electrode of the MFPA (++ is larger deflection) - indicates downward deflection, i.e. the wave of depolarisation is away from the positive electrode of the MFPA 17 LSS Cardiovascular System MFPA 0° 90° Lead I ++ 0 Alexandra Burke-Smith Lead II + + Lead III + Lead AVL + - Lead AVR - Lead AVF 0 ++ Why do waveforms vary in size? Consider SOH CAH TOA If lead is exactly on MFPA, the signal will be max size (i.e. the angle between the lead and MFPA is 0, and cos0 = 1 = max) The fraction of the max signal obtained in each lead can be calculated using SOH CAH TOA (if right angled triangles are drawn) Equipotential and Negative Waves The value of cos 90o is zero. o Hence the value of (MPFA cos 90o) is also zero. o This explains why a lead with its axis at right angles to the MFPA show no signal (or a small equipotential). Cosines of angles between 90o and 270o are negative. o Thus when a lead is more than 90o to MFPA the ECG will show downward (negative) rather than upward deflections. Range of MFPA The normal range of the MFPA is between -30° and +90°, and may vary between patients o This depends on the orientation of the heart in the chest If a patient has an MFPA that is more negative than -30°, they are exhibiting LEFT AXIS DEVIATION (enlarged left ventricle e.g. aortic stenosis) If a patient has an MFPA that is more positive than +90°, they are exhibiting RIGHT AXIS DEVIATION (enlarged right ventricle which could be pulmonary disease) Location of Chest Electrodes Designation as V1 – V6 (Arabic numberals) V1 on one right side of sternum, V2 – V6 All electrodes are positive Septum depolarisation occurs first, and is from left to right MFPA is then in the right to left direction QRS complex o V6 records small wave of depolarisation AWAY from the electrode, then large wave TOWARDS (qR wave seen in diagram) o V1 records a small wave of depolarisation TOWARDS the electrode, then a large wave AWAY (rS wave seen in diagram) o These combine to form the QRS complex seen on an ECG V3 records a BIPHASIC (both direction) wave known as the TRANSITION ZONE The ECG then combines the recordings at the 6 chest electrodes, with the hexagonal reference system for the 6 leads (standard + augmented) 18 LSS Cardiovascular System Alexandra Burke-Smith The Microcirculation CVS 5 - Dr Chris John (c.john@imperial.ac.uk) 1. Describe the branching structure of the microvasculature 2. List the three types of capillary and order them in terms of their permeability to water and small lipophobic solutes 3. Describe the factors controlling capillary blood flow 4. Explain the functional importance of capillary density 5. Identify the different mechanisms by which solute is transported between blood and tissue (depending on size and lipid solubility). 6. Explain how the “Starling forces” influence fluid transfer across the capillary wall 7. Describe the origin of lymph fluid. 8. Understand how clinical oedema arises Introduction Microcirculation: the circulation for every individual tissue/organ in the body Consists of: o 1st order arterioles o Terminal arterioles o Capillary o Pericytic (post-capillary) venule o Venule Surrounded by large amounts of smooth muscle within the blood vessel walls Blood Flow The overall aim of the cardiovascular system is to achieve adequate blood flow through the capillaries Blood flow rate: volume of blood passing through a vessel per unit time o F = ΔP / R o ΔP = pressure gradient o R = vascular resistance ΔP = pressure gradient o The pressure at the beginning of arterioles vs. Pressure at the beginning of capillaries determines blood flow rate through tissues o Increase ΔP, increase blood flow rate R = vascular resistance o Hindrance to blood flow due to friction between moving fluid and stationary vascular walls o Influencing factors: Blood viscosity – is relatively constant though Vessel length – increased length, increased resistance (although length is relatively constant) Vessel radius – variable (R α 1/r4, therefore if the radius is halved, the resistance is increased 16x) Summary o Increased blood pressure in major arteries increased blood flow rate, increased pressure gradient, decreased vascular resistance o Arteriolar vasoconstriction decreased blood flow rate, decreased pressure gradient, increased vascular resistance 19 LSS Cardiovascular System Alexandra Burke-Smith Microvessels - Arterioles Major resistance vessels Pressure gradient = 93mmHg (mean arteriole pressure=MAP) 37mmHg F = ΔP / R o If 93mmHg is the mean arteriole pressure, as blood flows through an organ, by the time it reaches the end of the venule, the pressure is considered 0 o without this pressure difference, blood would not reach the capillary beds and would accumulate in the arterioles o therefore FORGAN = MAP/ RORGAN Vasoconstriction/Vasodilation the STATE OF TONE of the arterioles within that tissue also determine flow: o VASOCONSTRICTION – decreases the radius, increases vascular resistance therefore decreases flow rate o VASODILATION – increases the radius, decreases the resistance therefore increases flow rate o State of tone is usually a state of PARTIAL CONSTRICTION at rest Vasoconstriction and vasodilation can happen simultaneously in different tissues The radii of arterioles are adjusted independently to accomplish two functions: o Match blood flow to the metabolic needs of specific tissues – regulated by local INTRINSIC controls o Help regulate arterial blood pressure – regulated by EXTRINSIC controls 1. Matching blood flow to the metabolic needs of specific tissues (depending on the body’s momentary needs) Chemical response: - ACTIVE HYPEREMIA - E.g. skeletal muscle during initial exercise - Increase metabolism therefore increase glucose requirement and oxygen consumption - This is sensed by tissue, i.e. if oxygen concentration falls, REFLEX VASODILATION occurs Physical response: - E.g. Reduced blood temperature on superficial structures, e.g. the skin o Sensed locally, then in order to decrease blood flow to tissue REBOUND VASOCONSTRICTION occurs to divert blood from the tissue - E.g. 2. Physical stretch o AUTOREGULATORY response to physical stretch of arterioles o MYOGENIC VASOCONSTRICTION occurs (e.g. the gut during exercise) – this increases the vascular resistance hence reducing blood flow 2. Help regulate arterial blood pressure Apply F = ΔP / R to the entire circulation o Cardiac Output (CO) can be seen as F o Mean Arterial Pressure (MAP) can be seen as ΔP o Total Peripheral Resistance (TRP) can be seen as R CO = MAP/TRP , therefore MAP = CO x TRP o Hence if you control resistance in all tissues, you can control and maintain the MAP Neural Control - Cardiovascular Control Center (CCC) in the Medulla (part of brain stem) sends a profound VASOCONSTRICTION SIGNAL which decreases blood flow to all organs 20 LSS Cardiovascular System - Alexandra Burke-Smith This can be used after significant blood loss, as it preserves the MAP but is not a good long term system, as it leads to dysfunction and infarction α receptors within the periphery and β receptors within the heart respond to this neural signal o β receptors especially important as they can result in an increase in heart rate Hormonal Control - Vasoconstrictors: o Vasopressin – Posterior Pituitary Gland o Angiotensin II – Lungs - Hormones which act on α and β receptors to increase sympathetic activity o Adrenaline o Noradrenaline – both from adrenal glands Microvessels – Capillaries Capillary exchange – delivery of metabolic substrate to the cells of the organism Design is specific – accentuates function: o Single cell wall (1 micrometer diameter) o Diameter of lumen (7 micrometers) o Extensive branching increases surface area Therefore capillaries are ideally suited to enhance diffusion (via FICK’S LAW): o Minimise diffusion distance o Maximum diffusion time o Maximise surface area Capillary Network depends on: o Highly metabolically active tissues – denser capillary networks o E.g. skeletal muscle, myocardium, brain, lung o However not all capillaries dilated at once, e.g. at rest only 10% of capillaries are dilated in skeletal muscle Structure Continuous - Most common – continuous flattened endothelial cells with water-filled gap junctions - As blood flows through the capillary: o Nutrients diffuse across junctions o LIPO molecules diffuse across cells o Transport proteins present to transport larger molecules into tissues - BLOOD BRAIN BARRIER: modified continuous capillary – very tight gap junctions reduce capacity for a large number of small molecules diffusing into the brain tissue – more selective control of transport of substances into tissues Fenestrated - Circular FENESTRAE (circular holes approx 80 nm large) allow slightly larger molecules to leave the blood and enter the tissues - E.g. in the glomerulus in kidney nephron Discontinuous - Very large gap junctions, therefore large molecules i.e. White blood cells can leave blood and enter tissues (and vice versa) - E.g. in bone marrow 21 LSS Cardiovascular System Alexandra Burke-Smith Fluid movement BULK FLOW – a volume of protein free plasma filters out of the capillary, mixes with the surrounding interstitial fluid (IF) and is reabsorbed Two important forces affect the bulk flow: the STARLING FORCES: o HYDROSTATIC PRESSURE – derived from the heart, and drives fluid into surrounding tissues o ONCOTIC PRESSURE – derived from the fact there is an increased concentration of plasma proteins in the blood, but not in the surrounding IF. This generates an osmotic force pulling water back into the capillaries Starling’s Hypothesis: o "... there must be a balance between the hydrostatic pressure of the blood in the capillaries and the osmotic attraction of the blood for the surrounding fluids. " o " ... and whereas capillary pressure determines transudation, the osmotic pressure of the proteins of the serum determines absorption." The oncotic pressure along the capillary remains relatively constant, but there are changes in the hydrostatic pressure: o The hydrostatic pressure at the venous end of the capillary is < the arterial end o ULTRAFILTRATION - When the pressure inside the capillary > in the IF, there is a net loss of fluid into the surrounding tissues (Hydrostatic pressure > Oncotic pressure) – this occurs at the arterial end o REABSORPTION - When the oncotic pressure > hydrostatic pressure, there is a net reabsorption of fluid back into the capillary. This occurs at the venous end. There is a net loss of fluid from the capillaries, as the oncotic pressure is never great enough to reabsorb all the fluid lost by ultrafiltration o Therefore a mechanism is required for the return of this loss of fluid to the capillaries – this is the role of the LYMPHATIC SYSTEM The Lymphatic System Initial Lymphatics LYMPHATIC CAPILLARIES are interwoven with capillaries These are blind-ended, therefore do not form complete loop therefore fluid which enters cannot leave The excess fluid in the capillaries is then drained back into the blood All excess fluid is eventually drained into the blood by the lymphatic system Lymph Nodes Important for immune surveillance Filled with immune cells; excess fluid passes through the lymph nodes before draining into the blood SPLEEN – organ acts as giant lymph node Lymph Flow No heart – relies on skeletal muscle contraction etc Areas where lymphatic system returns fluid, i.e. drainage ducts: o Right lymphatic duct o Thoracic duct o Right & left subclavian veins 3L/day returned from the lymphatic system into the blood If the rate of production of fluid > rate of return, this leads to the accumulation of fluid within the tissues = OEDEMA Parasitic blockage of lymph nodes may also lead to Oedema, e.g. ELEPHANTIASIS 22 LSS Cardiovascular System Alexandra Burke-Smith ECG: Identifying some basic disturbances of cardiac rhythm CVS 6 - Dr Sanjay Prasad (s.prasad@imperial.ac.uk) 1. 2. 3. 4. 5. State the normal duration and amplitude of the components of the ECG waveform Recognize normal sinus rhythm and bradycardia and tachycardia on the ECG Recognize common abnormalities of cardiac conduction Recognize common patterns suggesting acute myocardial infarction on the ECG Adopt a systematic approach to ECG interpretation The ECG Waveform 1 big square = 5mm, and represents a time interval of 0.2seconds, and a potential of 0.5mV I little square = 1mm, and represents a time interval of 0.04seconds, and a potential of 0.1mV Normal heart rate = 60-100bpm To calculate heart rate: o Count number of squares between each QRS complex and divide 300 by this number o Count number of QRS complexes in 10 seconds, and multiply this number by 6 P wave – represents atrial depolarisation - Duration: <0.11 s - Amplitude: <2.5 mm in Lead II (Right Arm Left foot) PR interval – represents time taken for the wave of depolarisation to migrate from one side of the AVN to the other (the AVN acts like a safety valve to separate atrial and ventricular systole) - Duration: from 0.12-0.20s QRS complex/interval – represents ventricular depolarisation o Duration: < 0.12s o Amplitude: R wave is recorded in chest electrode V6 - <25mm (MFPA range -30 - +90 degrees) 23 LSS Cardiovascular System Alexandra Burke-Smith Q Wave o Duration: <0.04s o Amplitude: 25% of total QRS complex amplitude, but in OPPOSITE DIRECTION; downward deflection indicates depolarisation wave moving away from the recording positive electrode QT interval o Duration: from 0.38-0.42s ST segment o Roughly the same as the PR interval T wave – represents ventricular repolarisation o May appear inverted in Lead III, Lead AVR, or chest electrodes V1 &V2 – but this does not mean abnormal Checking an ECG 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. Is it the correct recording – i.e. the right patient Identify leads Check calibration and speed of paper Identify the rhythm Look at QRS axis Look at P wave Look at PR interval Look at QRS complex Determine position of ST segment Calculate the QT interval Look at T wave Check Common Arrhythmias Bradycardia (<60bpm) Tachycardia (>100bpm) Cardiac Conduction abnormalities o Supraventricular arrhythmias (AVN or above) Atrial fibrillation Atrial flutter AVNRT o Ventricular arrhythmias (Below AVN) Ventricular tachycardia Ventricular fibrillation Sinus Tachycardia P waves have normal morphology; towards positive electrode of Lead II (RA LF; similar to axis of heart), and REVERSE direction of Lead AVR Atrial rate: 100-200bpm Ventricular rate: 100-200bpm (regular rhythm) One P wave precedes every QRS complex (normal) 24 LSS Cardiovascular System Alexandra Burke-Smith Atrial Fibrillation P waves absent, replaced by oscillating baseline fibrillation waves Atrial rate: 350-600bpm – all atrial myocytes are firing rapidly and irregularly, therefore atrial systole not completely efficient therefore blood pools in the atria – compromises cardiac output and increases risk of stroke Ventricular rate: 100-180bpm – rapid atrial rate means ventricles rapidly depolarise leading to a narrow QRS complex (NARROW COMPLEX RHYTHM DISTURBANCE), but since atrial systole is irregular, the ventricular rate is also irregular. Atrial Flutter P wave morphology abnormal: undulating saw-toothed baseline flutter waves “re-entry” circuit develops in right atrium there atrial systole is irregular and rapid Atrial rate: 250-350bpm Characterised by occasional depolarisation of the AVN, therefore the heartbeat is variable but rhythm is regular Ventricular rate: 150bpm (often with a 2:1 AV block, but also sometimes 4:1) Pre-excitation Syndrome Abnormal physical pathway develops where conducting tissue connects the atria and ventricles via an accessory pathway other than the AVN This leads to depolarisation of the ventricles early This leads to a slurring of the QRS complex, and a lack of regulation by the AVN, therefore electrical activity is conducted faster leading to an increased heart rate. 25 LSS Cardiovascular System Alexandra Burke-Smith Normal ECG Reading from V5 Heart Block - AV Nodal Block 1st degree – PR interval is lengthened >0.20s 2nd degree – one or more of the atrial impulses fail to conduct to the ventricles, i.e. a non-conducted P wave No QRS complex o Type 1 (Mobitz I/ Wenckebach) – disease of AV node – progressive lengthening of PR interval on consecutive heartbeats followed by a blocked P wave (i.e. no QRS complex), then the PR interval resets and the cycle repeats o Type 2 (Mobitz II/ Hay) – disease of the His-Pukinje conduction system – intermittently nonconducted P waves not preceded by PR interval lengthening and not followed by PR Interval shortening 3rd Degree – complete heart block in which the impulses generated in the SAN does not propagate to the ventricles; an accessory pacemaker in the ventricles will then cause systole (escape rhythm), resulting in two independently regular rhythms Second o The P waves with a regular P to P interval represents the first rhythm. o The QRS complexes with a regular R to R interval represent the second rhythm. Rhythm: R-R intervals o The PR interval will be variable, as the hallmark of complete heart block is no apparent relationship First Rhythm: between P waves and QRS complexes. P-P intervals Normal QRS complex seen in V1 & V2 26 LSS Cardiovascular System Alexandra Burke-Smith Heart Block – Bundle Branch Blocks Normal Impulse Conduction: SAN – AVN – Bundle of His – BUNDLE BRANCHES – Purkinje fibres Bundle branch and purkinje fibre depolarisation seen as QRS complex, therefore conduction black represented by change in the QRS complex Septum depolarisation usually in left to right direction The left bundle branch then leads to the anterior and posterior fascicles Two ECG changes seen: o QRS complex widens (>0.12s) – when the conduction pathway is blacked, it takes longer for the electrical signal to pass throughout the ventricles o QRS morphology changes – depending on which lead, and right vs. Left branch block Right BBB - The left bundle branch depolarizes normally, but the right bundle has a conduction block - wide QRS complex assumes a unique, virtually diagnostic shape in those leads overlying the right ventricle (V1 and V2) o this is seen as RSR complex instead of a QRS complex – like “rabbit ears” Left BBB - The right ventricle depolarises first, therefore the wide QRS complex assumes a characteristic change in shape in those leads opposite the left ventricle (right ventricular leads - V1 and V2). - This is seen as broad deep negative S waves Ventricular Tachycardia Medical emergency Irregular rapid contraction of the ventricles, not as a result of depolarisation through the normal AVN and rapid conduction system This leads to a broad QRS complex Unstable rhythm disturbance; often occurs in the middle/just after MI May lead to cardiac ischemia 27 LSS Cardiovascular System Alexandra Burke-Smith Ventricular Fibrillation Most commonly identified arrhythmia in cardiac arrest patients. This arrhythmia is a severe derangement of the heartbeat that usually ends in death within minutes The ventricular muscle twitches randomly, rather than contracting in a coordinated fashion (from the apex of the heart to the outflow of the ventricles), and so the ventricles fail to pump blood into the arteries and into systemic circulation. Use The ECG made easy, 150 ECG problems, ECG in practice. Author: John Hampton. Published by Churchill Livingstone for revision 28 LSS Cardiovascular System Alexandra Burke-Smith Blood Pressure and Flow CVS 7 - Professor Alun Hughes (a.hughes@imperial.ac.uk) 1. Understand the role and design of the normal circulation 2. Be able to describe physical factors influencing flow., know ‘Ohms law for the circulation’ and the principles of the Poiseuille’s equation 3. Be able to describe physical factors acting on blood vessels and know the Laplace equation, 4. Know the basic mechanisms by which flow of blood and transmural pressure influence blood vessel structure and function 5. Understand how standing (gravity) affects the circulation 6. Understand how the compliance of the aorta and elastic arteries affect the pulse pressure. The CVS & Circulation Role of the Circulation • • • • To transport blood around the body (gases, nutrients, metabolites, ions, hormones, heat) Flow is achieved by the action of a muscular pump (heart) propelling blood through a network of tubes (blood vessels). The pump generates a pressure gradient that drives bulk flow of blood through the network of blood vessels The circulation consists of two such pumps (left and right ventricle) which are physically coupled and pump through the systemic and pulmonary circulations respectively. At the capillary level gas and nutrient exchanged is accomplished by diffusion. Diffusion is crucial for movement of materials through tissues, but is only effective over short distances so a capillary needs to be ~10m from every cell. This necessitates a highly branched structure Structure of the circulation • Highly specialised - Consists of different vessel types which have distinct structures highly appropriate for their function. o Large ELASTIC arteries – act as conduits and dampening vessels o Small MUSCULAR arteries o Arterioles – have extensive smooth muscle in their walls so they can regulate their diameter and resistance to blood flow o Capillaries – very numerous and have thin walls to facilitate transport and diffusion o Venules o Medium sized & large veins – highly compliant vessels which act as a reservoir for blood volume Design of the CVS • • • In essence consists of two pumps and circuits: o Pulmonary (RV LA) o Systemic (LV RA) The cardiac output from both ventricles must be the same, despite to different in pressures within the two circulatory systems o Otherwise blood will tend to pool The relative areas and volumes within each circulatory system are also relatively equal o Relative cross sectional area – primarily capillaries; related to exchange function o Blood volume (total 5L) – primarily veins and venules; related to reservoir function 29 LSS Cardiovascular System • • Alexandra Burke-Smith The diameter of the blood vessels changes dramatically from the aorta (25mm in man) to the capillaries (5m = 0.005mm). o As a result of the change in diameter and the expansion of components of the vascular system due to branching there are large changes in the cross-sectional area of the vasculature at different levels. o There are billions of capillaries and this segments resents by far the largest cross-sectional are of the circulation. This presents a huge surface area for exchange to take place. o Although the volume in a single capillary is tiny, the equivalent of the whole cardiac output passes through the capillary bed every minute. The majority of blood volume is contained within the venous part of the circulation. o Regulation of the capacitance of the veins and venules regulates how much blood is stored and influences venous return to the heart and ventricular work via the Frank-Starling effect in the heart. Why does blood flow? • • • Blood pressure – the force that drives the circulation First measured in a horse by STEVEN HALES, where blood rises 8ft above the crural artery when a brass pipe is inserted and then the ligature on the artery is removed A simplified model of the mare’s circulation in Hales experiment is used to explain blood pressure o This is a very simple model of the circulation but it is useful in understanding how the system works. o It assumes that the action of the heart (pump) has established a pressure in the tank (the aorta) equivalent to 8 ft of water (as measured by Hales) – this is P1 o This drives a steady flow (Q) through the circulation. o The branching vessels of the circulation are simplified into a single long rigid pipe for the purposes of this model. o Pressure drops along this pipe due to viscous losses of energy (friction), so that the pressure measured at the end (P2) is lower than at P1 – this pressure difference drives the flow (Q). o At the end of the circulation the system empties into the right atrium of the heart which is almost at atmospheric pressure. Resistance • • • Links blood flow and pressure In its simplest form the circulation can be equated to an electrical circuit: o The pressure difference (P) is equivalent to the potential difference (V) o The fluid/volumetric flow (Q) is equivalent to current flow (I) o The fluid resistance (R) equates to the electrical resistance (R). Ohm’s law can be used to describe the relationship between V, I and R or P, Q and R: o V = IR o Therefore, ΔP = QR (this is DARCY’S LAW) The hemodynamic determinants of mean blood pressure (MBP) • In the circulation (systemic & pulmonary) Ohm’s relationship between pressure, flow and resistance can be restated in physiological terms as: o ΔP MBP 30 LSS Cardiovascular System o o • • • Alexandra Burke-Smith Q CO R PVR Therefore MBP = CO x PVR, where: o MBP = mean blood pressure o CO = cardiac output (=stroke volume x heart rate) o PVR = peripheral vascular resistance (like TPR – total peripheral resistance) This relationship is an approximation since flow is the circulation is not steady (due to the intermittent pumping of the heart) and blood vessels are not rigid. o Nevertheless it is a simple and useful relationship which is applicable in many situations. o The relationship between pressure and flow can be used to estimate the resistance of the circulation using estimates of cardiac output as bulk flow/unit time and the difference between mean arterial and venous pressure as the pressure drop across the circulation. Remember it is pressure drop not absolute pressure itself that drive flow. o If this is done for the systemic and pulmonary circulation then it is clear that the resistance of the pulmonary circulation is substantially less than the systemic. o Physiologically, regulation of flow is achieved by variation in resistance while blood pressure remains relatively constant. Pressure • • • • • Pressure is not constant in the circulation - It falls due to the resistance to blood flow provided by the blood vessels. The distribution of pressure throughout the circulation is illustrated in the figure. o Both the maximum (systolic) and minimum (diastolic) pressures are illustrated. o It is important to remember that it is the pressure difference between points in the circulation that drives flow not the absolute pressure. The magnitude of oscillation in pressure (pulse pressure) is damped in the smaller arteries and arterioles. The diagram also illustrates that the major site of resistance (i.e. major region of pressure drop) is in small muscular arteries (<0.5mm internal diameter) and arterioles. Note that the pulmonary circulation operates at lower pressures but shows a broadly similar distribution of pressure across the difference components of the circulation. Why is there resistance to Blood flow? LAMINAR FLOW - In the normal circulation flow is laminar, i.e. the fluid behaves as if it flows in layers or streamlines. - Laminar flow can be demonstrated by injecting a dye into fluid, showing the existence of a clearly defined streamline. VISCOSITY - Dynamic Viscosity (µ) is a measure of the resistance of a fluid to deform under shear stress. - Resistance arises as a result of the resistance due to friction between fluid laminae moving at different velocities. SHEAR 31 LSS Cardiovascular System - Alexandra Burke-Smith A force per unit area (the pressure difference) is needed to move the fluid in opposition to viscosity. The flow velocity on the surface of the vessel wall is zero (so called NO SLIP CONDITION) but in a flowing fluid, the velocity of each lamina increases progressively as you move further way from the wall. o The spatial velocity gradient is called the shear rate (s) s = du/dr S = shear rate U = velocity of blood flow R = radial dimension o The shear rate multiplied by the dynamic viscosity is the shear stress (τ). The shear stress near the wall is believed to be an important influence on endothelial function in health and disease. τ = (du/dr) µ µ = is dynamic viscosity Poiseuille’s law and Vessel calibre • • • • Experiments performed by Jean Poiseuille (1797-1869) in long glass tubes elucidated the relationship between pressure and laminar flow (i.e. resistance) in long straight tubes. Subsequently the theoretical basis of this relationship was derived by Wiedman, and Neumann and Hagenbach. The resistance to flow in a long straight rigid tube depends on the viscosity of the fluid (µ), the length of the tube (L) and the radius of the tube (r) and is described by Poiseuille’s equation: o µ= fluid viscosity o L = vessel length o r = vessel radius o d= diameter o ΔP = pressure difference (P1-P2) o Q = volumetric flow o ΔP/Q = resistance This equation emphasizes the importance of arterial diameter as a determinant of resistance. Consequently relatively small changes in vascular tone (vasoconstriction/vasodilatation) can achieve marked changes in flow. Due to the 4th order relationship between diameter and flow, relatively small changes in diameter have marked effects on flow. o This explains why active changes in arterial and arteriolar diameter are so important in the physiological regulation of blood flow and blood pressure. Distribution of blood flow to organs • • During exercise, there is a change in the distribution of blood flow to different organs Dilation of the arteries and arterioles feeding skeletal muscle results in a ~30fold increase in muscle blood flow. 32 LSS Cardiovascular System Alexandra Burke-Smith Blood Pressure • • • • Conventionally blood pressure measurements are made in the arm. Blood pressure varies over the cardiac cycle with a peak in systole and a minimum in diastole. o SYSTOLIC blood pressure (SBP) o DIASTOLIC blood pressure (DBP) o PULSE PRESSURE (PP) = SBP – DBP o MEAN blood pressure = ~ DBP + 1/3 PP The systolic (SBP) and diastolic pressure (DBP) are usually recorded in clinic as SBP/DBP (e.g. 110/70). Values of blood pressure vary widely in a community. o High levels of blood pressure are termed hypertension. Ventricular vs. Aortic pressure • • • Shown by Wigger’s diagram During systole, the aortic valve opens due to the difference in pressure between the ventricles and the aorta (marked by AO) Note the difference in ventricular and aortic pressure in diastole. o Once the pressure gradient between the aorta and ventricles is reversed, the aortic valve closes o Once the aortic valve closes ventricular pressure falls rapidly but aortic pressure only falls slowly in diastole o This can be explained by the elasticity of the aorta and large arteries which act to ‘buffer’ the change in pulse pressure. Arterial compliance and pulse pressure • • • The ability of the aorta and the elastic arteries to buffer or damp the oscillation in blood pressure is often termed a WINDKESSEL (German for air chamber) In systole more blood is ejected into the aorta and large elastic arteries than leaves them. This distends these vessels. o ~40% of the stroke volume is stored by the elastic arteries. o In effect some of the pressure energy generated in systole is converted to elastic energy in the artery wall which is stored during systole. Once the heart ceases ejection and the aortic valve closes, pressure starts to fall. Consequently the walls of the aorta and elastic arteries recoil and the elastic energy is reconverted into pressure and the stored volume is discharged. o This process damps the magnitude of pressure change and accounts (to a large extent) for the diastolic component of arterial pressure. 33 LSS Cardiovascular System o Alexandra Burke-Smith It also accounts for the maintenance of flow in the microcirculation during diastole. If arterial compliance is reduced (i.e. arteries get stiffer e.g. with age) then this mechanism is less able to damp the fluctuation in pressure and pulse pressure increases. What is the effect of the blood pressure on the vessel wall? • • • • Pressure difference between two locations in the circulation is important for flow, but the pressure inside the vessel (TRANSMURAL pressure) determines the distension of the vessel wall o This pressure causes a tension in the wall (T) The relationship between transmural pressure and wall tension is determined by LAPLACE’S LAW: o T = Pr where P= transmural pressure, r=radius of vessel, T= tension The CIRCUMFERENTIAL STRESS (σ) is determined by the tension caused by transmural pressure and the wall thickness: o σ = Pr / h where h = wall thickness o Therefore larger arteries which have higher pressures require increased wall thickness The relationship between the transmural pressure and the vessel volume depends on the ELASTICITY of the vessel, and is known as COMPLIANCE o In extreme cases over a prolonged period in a weakened vessel high circumferential stress can cause a balloon like distension (ANEURYSM) or even rupture. Aneurysms • • • When compliance of a vessel fails, and a weakened vessel is exposed to high circumferential stress a balloonlike dilation can occur This is a tensile-strength failure Aneurysms are prone to rupture, which could lead to exsanguinations Compliance properties of arteries and veins • • The elastic properties of blood vessels depend mainly on structural proteins, elastin and collagen. o Elastin is much more distensible than collagen. o The combination of elastin and collagen in vessels results in a non-linear relationship between vessel pressure and volume (i.e. non-linear compliance). The elastic properties of arteries and veins differ and this is important for their function. o Veins are highly compliant at low pressures while arteries are compliant over a wider pressure range. o This means that relatively small changes in venous pressure distend veins and increase the volume of blood stored in them. This is important when the pressure in veins changes for example on standing. Gravity and venous pressure • • In man (and other bipeds) the venous reservoir is not always at the same level as the heart. On standing gravity increases pressure in the lower limbs (~80mmHg). Since veins are compliant this increases the volume of blood in these vessels and (transiently) reduces the venous volume returning to the heart. o This would reduce cardiac output and blood pressure if there were no compensatory response. o When at heart level venous pressure = central venous pressure The effect of gravity and posture affects the transmural pressure in all vessels, but at any particular location the gradient of pressure from large artery to capillary to vein is maintained so flow still occurs in the same way. 34 LSS Cardiovascular System • • • • • Alexandra Burke-Smith The major effect of gravity is on the distensible veins in the leg and the volume of blood contained in them. Veins act as an important reservoir for blood. This is because despite the low pressures, vein walls are relatively thin and compliant therefore they accommodate large volumes of blood (~2/3 total blood volume) at low pressures. o This reservoir/compliance function is physiologically regulated. Vein walls, although thin, do contain smooth muscle. o The role of this muscle is to stiffen the wall i.e. reduce compliance. A number of mechanisms act to limit the effect of blood pooling in the lower limb veins on the circulation: o Activation of the sympathetic nervous system to: constrict venous smooth muscle and ‘stiffen’ the veins. constrict arteries to increase resistance and maintain blood pressure increase heart rate + force of contraction and maintain cardiac output o Myogenic venoconstriction (in response to elevated venous pressure) to ‘stiffen’ veins o Use of muscle and respiratory ‘pumps’ to improve venous return Nevertheless cerebral blood flow falls on standing Failure of these mechanisms causes fainting (SYNCOPE) Muscle and Respiratory “Pumps” • • • Return of blood to the heart during upright posture is assisted by the contraction of skeletal muscle in the lower limb which compresses veins within the muscle and forces blood back to the heart This is called the muscle pump. Another mechanism called the respiratory pump also assists venous return. o During respiration expansion of the chest and diaphragm causes a negative pressure within the thorax which effectively sucks blood into the central veins by reducing the extra-vascular pressure in the thorax and increasing it in the abdominal cavity. Both the skeletal and respiratory pump depend on the presence of valves in the veins outside the chest to prevent retrograde flow. o Incompetent valves cause dilated superficial veins in the leg (VARICOSE VEINS) o Prolonged elevation of venous pressure, even with intact compensatory mechanisms, causes OEDEMA in feet 35 LSS Cardiovascular System Alexandra Burke-Smith Blood vessel order, function, and specialisation of cells in the CVS CVS 8 - Dr Adrian H Chester (a.chester@imperial.ac.uk) 1. Appreciate the function of the endothelium as a generator of hormones that regulate vascular and cardiac muscle form and function 2. Describe ways in which the endothelium can be stimulated and how this results in release of the named hormones: NO, prostacyclin, endothelin-1. 3. Describe in general terms the renin-angiotension system and know how its major components regulate vascular function 4. Describe how the following work: i. low dose aspirin ii. calcium channel blockers iii. nitrovasodilators 5. Appreciate why these drugs carry side effect risks along with their therapeutic benefits CVS Overview • • Consists of the heart, blood and blood vessels Function - rapid convective transport of: o Oxygen o Glucose o Amino acids o Fatty acids o Vitamins o Water o As well as removal of: Carbon dioxide Urea Creatine o Also homeostasis: Hormone delivery Temperature regulation reproduction Arterial and venous structure - Artery Regularly shaped lumen Thick muscular wall consisting of MEDIA and ADEVENTITIA Lined by endothelium and connective tissue Vein Possible irregular shaped lumen Lined by endothelium and connective tissue Thin muscular wall; very thin media and thin adventitia Note: the vascular system forms a continuous circuit between the left and right side of the heart, consisting of arteries, arterioles, capillaries, venules and veins 36 LSS Cardiovascular System Alexandra Burke-Smith The Endothelium Endothelial mediators of vascular function • Mediators target smooth muscle, myocytes, and platelets: o Nitric oxide o Prostacyclin o thromboxane o Endothelin-1 o Angiotensin II Effects of Nitric Oxide (NO) Effects of Prostacycline (PGI2) Effects of Thromboxane (TXA2) Effects of Endothelin-1 (ET-1) Effects of Angiotensin II (Ang II) Smooth muscle – Relaxation & Inhibition of growth Smooth muscle – Relaxation & Inhibition of growth Smooth muscle – contraction Smooth muscle – contraction & weak stimulation of growth Smooth muscle – contraction & stimulation of growth Myocytes Increased blood flow & Enhance contractility Myocytes Increased blood flow Platelets – inhibit aggregation Myocytes – reduce blood flow Platelets – stimulates aggregation Platelets – inhibit aggregation Myocytes reduced blood flow & Enhance contractility Myocytes - reduced blood flow, remodelling & fibrosis Platelets – no effect Platelets – no effect Control of Vascular Tone • • • Balance between vasodilation and vasoconstriction Vasodilators: NO, PGI2 Vasoconstrictors: ET-1. TXA2, Ang II Endothelial Hormones Nitric Oxide • • • • • Release is induced by physical force, as well as by acetylcholine or hormones Precursor is L-ARGININE, which is cleaved by e-NOS (NITRIC OXIDE SYNTHASE enzyme present in all endothelial cells, whose function is Ca2+ dependent) o ACh binding on endothelial receptors activates the phospholipase C 2nd messenger pathway, which results in an increase in Ca2+ which activates the e-NOS enzyme The NO then migrates to target (smooth muscle), where is converted to cyclic-GMP by SOLUBLE GUANYLYL CYCLASE (s-GC) o The cyclic-GMP activates PROTEIN KINASE G (PKG), which causes vasodilation and a decrease in Ca2+ The c-GMP has a short half life, as it is rapidly broken down by PHOSPHOESTERASES This is known as FLOW-INDUCED VASODILATION, and is a response to increased sheer stress in blood vessels. It is important in: o Thermoregulation o Penile erection 37 LSS Cardiovascular System Alexandra Burke-Smith Prostacyclin & Thromboxane • • • • • Precursor is ARACHIDONIC ACID, and is converted under the influence of CYCLO-OXYGENASE (COX) There are two ISOFORMS of COX: o COX 1 – active in a healthy CVS o COX 2 – active in an unhealthy CVS, e.g. in CVD related to pain and inflammation COX 1 and COX 2 act on the arachnidonic acid to convert it first into PGG2 and then PGH2 PROSTACYCLIN SYNTHASE then acts on the PGH2 to synthesise the prostaglandins, the most important being PGI2 o Prostacyclin acts on IP receptors and eventually causes vasodilation and inhibition of platelets – this can reduce ATHEROSCLEROSIS THROMBOXANE SYNTHASE acts on the PGH2 to synthesis thromboxane o TXA2 also is converted to TXB2 o thromboxane acts on TP receptors and causes vasoconstriction and stimulation of platelets – this increases atherosclerosis Endothelin-1 • • • • • • • • Endothelin-1 is a very potent vasoconstrictor It has a polypeptide chain consisting of 21 amino acids specific conformation due to the bods between CYSTEINE molecules At times of pathophysiological insult, transcription of the PREPRO-ET1 mRNA occurs, which is then translated to form the precursor PREPRO-ET1 PREPRO-ET1 is then converted to PRO-ET1, which is then finally converted to ET-1 by ECE-1 (endothelin converting enzyme 1) Transcriptional control: o Inhibition – Prostacyclin Nitric oxide ANP, Heparin, HGF & EGF o Stimulation – Adrenaline Ang II Vasopressin Steroids IL-1, TGF-β, Endotoxin, endothelin, VECF, tacrolimus, CsA Endothelin Receptors o On smooth muscle cells ETA – leads to contraction of the smooth muscle ETB – leads to contraction of the smooth muscle o On endothelial cells ETB – binding causes NO to be produced, which then in turn acts on the smooth muscle cells with a vasodilatory effect Inhibition of ET-1 pathway produces vasodilation, therefore can have therapeutic potential The Renin-Angiotensin System • • Renin (an enzyme from the kidneys produced in response to a decrease in blood pressure) converts ANGIOTENSINOGEN (from the liver) ANGIOTENSIN I ACE (ace converting enzyme) converts angiotensin I to ANGIOTENSIN II 38 LSS Cardiovascular System Alexandra Burke-Smith o • Also simultaneously degrades BRADYKININ on epithelial cells (acts on beta-1 receptors to release valodilators) Angiotensin II causes vasoconstriction by acting on AT I receptors o Also causes renal salt absorption Effects of Angiotensin II ACE Inhibitors • • E.g. CAPTORIL Reduce blood pressure by blocking the action of ACE so that no angiotensin II is produced (which acts on AT1 receptors to cause vasoconstriction) o Also prevents the breakdown on bradykinin into inactive fragments (bradykinin then stimulates the production of Nitric Oxide involving NOS; this leads to vasodilation via a cyclic GMP pathway) Aspirin • • Works by balancing the effects of thromboxane and prostacyclin Effects of 75mg of aspirin over 7 days: o Prostacyclin production reduction of 10% each day o Reduction of thromboxane production 10% more each day (i.e. -10%, -20%, -30% etc) Pharmacology of NO • • • NO donors – e.g. nitroglycerine, nitroprusside E-NOS activators – e.g. endothelium-dependent vasodilators Phosphodiesterase inhibitors – e.g. Viagra, zaprinast How do Nitro-vasodilators work? • • Increase [NO] within smooth muscle cells, where it is converted to cyclic-GMP by SOLUBLE GUANYLYL CYCLASE (s-GC) The cyclic-GMP activates PROTEIN KINASE G (PKG), which causes vasodilation and a decrease in Ca2+ How does Viagra work? • • Phosphodiesterase inhibitor Phosphodiesterase breaks down cyclic GMP, therefore its inhibition leads to excess cyclic GMP activity vasodilation 39 LSS Cardiovascular System Alexandra Burke-Smith Calcium Channel Blockers • • • • • • • • • • Drugs that do not act by modifuing endogenous receptors or enzymes, but increase or decrease intracellular Ca2+ by affecting the entry of calcium into the cell Increase intracellular Ca2+ o Endothelin o Angiotensin II o Thromboxane Decrease intracellular Ca2+ o Nitric oxide o Prostacyclin They decrease intracellular calcium levels by blocking the voltage gated calcium channels in cardiac and blood vessel muscle. The voltage-gated channels mediate calcium influx in response to membrane depolarisation o Regulate intracellular processes such as contraction, secretion, neurotransmission and gene expression o Activity is essential to couple electrical signals in the cell surface to physiological events in cells This means that upon stimulation, less calcium flows into cells (negative INOTROPIC effect) and so the muscles contract less. o In cardiac muscle, this results in a decrease in cardiac output by decreasing the heart rate and stroke volume. o In smooth muscle around vessels, this decreases total peripheral resistance and so results in vasodilation. o Overall result is a decrease in blood pressure. They prevent coronary artery vasospasm, which makes them very useful in the treatment of variant angina the affinity of the blockers for the channel is directly related to the membrane potential of the target cells o in smooth muscle, vasodilation occurs at -50mV o in myocytes, negative inotropic effects are seen at more negative potentials of -80mV Dihydropyridines – e.g. NIFEDIPINE o Used to decrease systemic vascular resistance and arterial blood pressure but not in angina as reduced cardiac output may lead to reflex tachycardia Phenylalkylamine – e.g. VERAPAMIL o Selective for myocytes, reducing myocardial oxygen demang o Used to treat angina but is not a potent vasodilator Side Effects • • • • • • • Our body often uses the same chemical to regulate more than one process Interaction between different systems in the body Unfortunately, drugs are not always as selective Tissue specific distribution of receptors It is also a fact that two people taking the same medicine can have very different experiences E.g. 1 Viagra o There are 5 types of phosphodiesterase enzymes o Expression varies between tissues therefore different side effects may be observed E.g. 2 Prostacyclin/thromboxane synthesis o Arachidonic acid may also be converted to LEUKOTRIENES instead of PGG2 by LIPO-OXYGENASE o This may cause asthma is 3-5% of patients 40 LSS Cardiovascular System Alexandra Burke-Smith Mechanical Properties of the Heart II CVS 9 - Ken MacLeod (k.macleod@imperial.ac.uk) 1. Describe the mechanical events of the cardiac cycle 2. Use a graph to correlate electrocardiographic events and pressure events of the atria, ventricles, aorta and pulmonary artery 3. Indicate on the graph the phases of the cardiac cycle and the corresponding pressure changes, valve openings and closures 4. Define and state normal values for right and left ventricular end-diastolic volume, end-systolic volume, stroke volume, end-diastolic pressure and peak systolic pressure 5. State the origin of the heart sounds 6. Provide the mathematical equation for ejection fraction 7. Define cardiac output and indicate its determinants 8. Construct simple pressure-volume diagrams from the events during the cardiac cycle and annotate these graphs appropriately Introduction Preload: the stretch or filling of the ventricles before they contract Afterload: the load/pressure against which the ventricles eject blood after opening of the aortic/pulmonary valve The ventricular heart beat is divided into two main phases: o Diastole: ventricular relaxation during which the ventricles fill with blood Split into 4 sub-phases o Systole: ventricular contraction when blood is pumped into the arteries Split into 2 sub-phases Cardiac cycle: a description of mechanical and electrical events, volume changes and sounds associated with the heart beat. It consists of: o Atrial systole (resulting in the end diastolic volume) o Isovolumetric ventricular contraction o Ventricular ejection Rapid ejection Reduced ejection o Isovolumetric ventricular relaxation o Late diastole Rapid ventricular filling Reduced ventricular filling 41 LSS Cardiovascular System Alexandra Burke-Smith ATRIAL SYSTOLE Mechanical events Changes in pressure & volume Just prior to atrial systole, blood flows passively through the open atrioventricular valves (tricuspid and mitral) Atrial depolarisation contraction of atria, which “tops off” the volume of blood in the ventricles green – aortic pressure yellow – atrial pressure Electrocardiogram SAN activation depolarisation of atria (seen as P wave) red – ventricular pressure white – ventricular volume As atria contract, the “a wave” can be seen on the yellow graph due to the increase in atrial pressure Blood is also pushed back into jugular vein, causing a wave in jugular venous pulse Heart sounds No heart sound should be heard, but 4th heart sound may be heard as an abnormality – occurs in congestive heart failure, pulmonary embolism or tricuspid incompetence 42 LSS Cardiovascular System Alexandra Burke-Smith ISOVOLUMETRIC CONTRACTION Mechanical events Changes in pressure & volume Occurs just as the ventricles depolarise – is the interval between AV valve closing and semi-lunar valve (aortic and pulmonary) opening green – aortic pressure yellow – atrial pressure Electrocardiogram Ventricular depolarisation marked by QRS complex red – ventricular pressure white – ventricular volume the AV valves close as the ventricular pressure exceeds the atrial pressure Since the AV and semi-lunar valves are closed, there is no movement of blood out of the ventricles, just an increase in pressure approaching the aortic pressure Heart sounds Consider the heart sound to be LUB- DUB Ventricular depolarisation is the 1st heart sound (lub) – this is due to the closure of the AV valve with associated vibrations 43 LSS Cardiovascular System Alexandra Burke-Smith RAPID EJECTION Mechanical events Changes in pressure & volume Ventricular muscle walls undergo ISOTONIC contraction, pushing blood out of the ventricles Semi-lunar valves open green – aortic pressure yellow – atrial pressure red – ventricular pressure white – ventricular volume Electrocardiogram as the ventricles contract, the pressure within them exceeds the pressure in the aorta and pulmonary arteries when the semi-lunar valves open, the volume of the ventricles decreases the right ventricular contraction pushes the tricuspid valve slightly into the atrium, creating a small wave into the jugular vein – “c wave” observed in yellow graph Heart sounds No changes No heart sounds 44 LSS Cardiovascular System Alexandra Burke-Smith REDUCED EJECTION Mechanical events Changes in pressure & volume Marks the end of ventricular systole Aortic and pulmonary valves begin to close green – aortic pressure yellow – atrial pressure red – ventricular pressure white – ventricular volume Electrocardiogram as the blood flow from the ventricles decreases, the ventricular volume decreases more slowly as the pressure in the ventricles fall blow that in the arteries, blood begins to flow back causing the semi-lunar valves to close Heart sounds Ventricular repolarisation marked by T wave No heart sounds 45 LSS Cardiovascular System Alexandra Burke-Smith ISOVOLUMETRIC RELAXATION Mechanical events Beginning of diastole Aortic and pulmonary valves shut completely AV valves remain closed Atria fill with blood Electrocardiogram No changes Changes in pressure & volume green – aortic pressure yellow – atrial pressure red – ventricular pressure white – ventricular volume atrial pressure rises as volume of blood in atria increases blood pushing on the tricuspid valve gives a second jugular pulse (“v wave” on yellow graph) as the aortic valve shuts, there is a rebound pressure wave against the valve as the distended aortic wall relaxes. This recoil reduces the aortic pressure and is seen as the DICHROTIC NOTCH on the green graph Heart sounds 2nd heart sound (dub) occurs when aortic and pulmonary valves close 46 LSS Cardiovascular System Alexandra Burke-Smith RAPID FILLING (late diastole) Mechanical events Changes in pressure & volume AV valves open, and the blood flows rapidly (although passively) into the ventricles green – aortic pressure yellow – atrial pressure Electrocardiogram No changes red – ventricular pressure white – ventricular volume the ventricular volume increases, as the atrial pressure falls Heart sounds 3rd heart sound abnormal – can signify turbulent ventricular filling due to severe hypertension or mitral incompetence 47 LSS Cardiovascular System Alexandra Burke-Smith REDUCED FILLING (late diastole) Mechanical events Changes in pressure & volume Called DIASTASIS Ventricles fill more slowly as pressure difference between atria and ventricles decreases Electrocardiogram green – aortic pressure yellow – atrial pressure No changes red – ventricular pressure white – ventricular volume ventricular volume increases more slowly Heart sounds No heart sounds 48 LSS Cardiovascular System Alexandra Burke-Smith Summary of the Cardiac Cycle – the Wiggers diagram Pressure Volume Loops pressures Typical pressure of the systemic circulation – 120/80mmHg Typical pressure of the pulmonary circulation – 25/5mmHg PAWP – pulmonary artery wedge pressure o Taken from a branch of pulmonary artery when the back pressure has been occluded o Elevation can indicate left ventricle failure, mitral insufficiency, mitral stenosis 49 LSS Cardiovascular System Alexandra Burke-Smith Pressure-volume loop in heart 1 2 3 4 X1 – end diastolic volume, i.e. the preload after max ventricular filling o Blood filling the ventricle during diastole determines the preload that stretches the resting ventricle X2- The blood pressures encountered in great vessels (aorta and pulmonary artery) represent the afterload Between X2 and X3, isotonic contraction of the ventricles occurs Frank-Starling Relationship The pressure-volume loop can be fitted into the frankstarling graph The straight line of the active force is equal to the end-systolic pressure line Increasing preload increases stroke volume (increasing X1 increases the width of the loop) Increases afterload decreases stroke volume o There is a greater pressure to overcome in order to open the aortic valve, therefore X2 increases and less shortening occurs Cardiac Contractility Definition: Contractile capability (or strength of contraction) of the heart Simple measure of cardiac contractility is ejection fraction Contractility is increased by sympathetic stimulation o Beta-adrenoreceptor activation increase cyclic AMP phosphorylation of key Ca2+ handling proteins Ca2+ channels open for longer increased Ca2+ in cytoplasm increased force of contraction 50 LSS Cardiovascular System Alexandra Burke-Smith Family of different Frank-Starling relations as cardiac contractility changes During exercise contractility is increased due to increased sympathetic activity During exercise end diastolic volume is increased due to changes in the peripheral circulation (venoconstriction and muscle pump) 51 LSS Cardiovascular System Alexandra Burke-Smith The sympathetic nervous system & reninangiotensin system CVS 10 - Dr Mike Schachter (m.schachter@imperial.ac.uk) 1. 2. 3. 4. 5. 6. 7. 8. Describe the principles of the organisation of the sympathetic nervous system Describe the synthesis, release and removal of the neurotransmitter, noradrenaline Outline the types of adrenoreceptor in the sympathetic nervous system Evaluate the cardiovascular effects of infusion of some common adrenergic agonists Describe the principles of the organisation of the renin-angiotensin-aldosterone system Describe the biosynthetic pathway for angiotensin II synthesis Evaluate the individual roles the SNS and RAS play in modulating the behaviour of the CVS Recognize some of the pharmacological concepts involved in how important sympathetic neurotransmitters interact with receptors to evoke downstream effects The autonomic nervous system Consists of the parasympathetic and sympathetic nervous systems Sympathetic nervous system is organised around the thoracic and lumbar spinal cord o There is no sympathetic innervation in the bronchi, but practically everywhere else Cardiovascular control Baroreceptors in carotid sinus and aortic arch sensitive to stretch (increased stretch increased frequency of impulses to hypothalamic autonomic centre) Increased frequency of impulses reduced inhibition of sympathetic activity from solitary tract nucleus increased blood pressure through increased vasoconstriction o α1 receptors at end of pre-ganglionic neurone o α2 receptors in arterioles o β2 receptors in heart via vagus nerve Effector nerves Sympathetic outflow - Paravertebral sympthatic chain ganglion – neurotransmitter is acetylcholine therefore is a cholinergic receptor - Post-ganglionic fibre contains lots of noradrenaline vesicles which are released on depolarisation, binding to the adrenergic receptor on the effect organ. o The NA is then either taken up by the neurone and repackages, or taken up by the effector organ and broken down by COMT Parasympathetic outflow - Parasympathetic ganglia are in or near effector organ, and involve acertylcholine - Effector organ also has cholinergic receptor, which binds to the acetylcholine released by the postganglionic neurone. o This acetylcholine is then recycled 52 LSS Cardiovascular System Alexandra Burke-Smith Catecholamines Noradrenaline and adrenaline are synthesised in the terminal VARISCOSITY (see neurotransmitter lecture) They are then removed from the neuroeffector junctional synapse via uptake systems: o Neuronal reuptake and recycling, or degradation into deaminated metabolites by MAO o Extraneuronal uptake into effector organ and degradation by COMT or MAO Adrenoceptors Two groups of effects: Excitatory effects on smooth muscle (constriction) o alpha-adrenoceptor-mediated o lead to an increase in intracellular calcium Relaxant effects on smooth muscle, stimulatory effects on heart o beta-adrenoceptor-mediated o lead to an increase in cyclic AMP, which causes an increase in Ca2+ in the heart but a decrease in smooth muscle β- receptors β1 adrenoceptors located on: o cardiac muscle o smooth muscle of the GI tract β2 adrenoceptors located on: o bronchial, vascular and uterine smooth muscle β3 adrenoceptors located on: o adipocytes o smooth muscle of GI tract α- receptors α1-adrenoceptors: located post-synaptically i.e. predominantly on effector cells o important in mediating constriction of resistance vessels 53 LSS Cardiovascular System Alexandra Burke-Smith α2 -adrenoceptors: located on presynaptic nerve terminal membrane o their activation by released transmitter causes negative feedback inhibition of further transmitter release o some are post-synaptic on vascular smooth muscle Receptor coupling α1-adrenoceptors - coupled with G-protein linked receptor which activates the phosphlipase C pathway, which leads to an increase in free Ca2+ and activated protein kinases (involving IP3 and DAG) α2-adrenoceptors - coupled with beta-receptor - activates adenyl cyclase, which converts ATP cyclic AMP leading to a decrease in intracellular Ca2+ Effects of catecholamines on activation of adrenoceptors - Natural Noradrenaline – α1, α2, β1 Adrenaline – α1, α2, β1, β2 Dopamine – weak effects at α1, β1, but has own receptors Synthetic - Isoprenaline - β1, β2 (unselective beta-agonist) - Phenylephrin - α1 (selective alpha-agonist) CVS effects (10 g/min infused IV) Effect on: Noradrenaline Systolic BP ↑↑↑ Diastolic BP ↑↑ Mean BP ↑↑ Heart Rate ↓ Adrenaline ↑↑ ↓ ↑ ↑ isoprenaline ↑ ↓↓ ↓ or → ↑↑ NB: noradrenaline acts on the beta receptors in the heart to increase cardiac output, and the alpha receptors in the periphery to increase total peripheral resistance Response of major vascular beds Skin (alpha receptors) o Noradrenaline – constriction o Adrenaline – constriction o Isoprenaline – no effect Visceral (alpha receptors) o Noradrenaline – constriction o Adrenaline – constriction o Isoprenaline – no effect (slight dilation) Renal (alpha & beta receptors) – constriction o Noradrenaline – constriction o Adrenaline – constriction o Isoprenaline – no effect (slight dilation) Coronary (alpha & beta1 receptors) – dilation 54 LSS Cardiovascular System Alexandra Burke-Smith o Noradrenaline – dilation o Adrenaline – dilation o Isoprenaline – dilation Skeletal muscle (alpha & beta2 receptors) o Noradrenaline – constriction o Adrenaline – dilation o Isoprenaline – dilation The Renin-Angiotensin System NB: angiotensin II can also be converted to angiotensin III by aminopeptidase Stimuli for renin release A decrease in the renal perfusion pressure, a decrease of blood pressure in the pre-glomerular vessels. A decrease in arterial blood pressure. Haemorrhage, salt and water loss, hypotension (low blood pressure). A change in Cl- (or Na+) ion concentration. β1-receptor activation in the kidney (sympathetic nervous system). NaCl reabsorption at the macula densa (which are a group of cells in the glomerulus). 55 LSS Cardiovascular System Alexandra Burke-Smith What happens when blood pressure decreases? Pharmacologic manipulation of renin release Loop diuretics – block NaCl reabsorption at macula densa NSAIDs – block renin release via inhibition of COX ACE inhibitors – block the synthesis of Ang II AT1 blockers (Ang II receptor antagonists) – block vasoconstriction and aldosterone synthesis and secretion o E.g. losartan, valsartan Alpha2 and beta1 blockers - block receptor activation in the kidney, inhibiting renin release AT1 ./ Ang II type I receptors Are G-protein coupled; Gi and Gq. It also couples to phospholipase A2. The AT1 receptors are located in the blood vessels, brain, adrenal glands, the kidneys and the heart. Activation of the AT1 receptors works to increase the blood pressure, and stimulate aldosterone secretion Effects of angiotensin II Peripheral resistance: o Direct vasoconstriction. o There is enhanced action of peripheral noradrenaline. Increased norandrenaline release. Decreased noradrenaline uptake. o increased sympathetic discharge (CNS). o release of catecholamines from the adrenal glands. - These all act to produce a rapid pressor response. Renal Function: o Direct effects to increase Na+ reabsorption in the proximal tubule. o Synthesis and release of aldosterone from the adrenal cortex. o Altered renal haemodynamics. Renal vasoconstriction. Enhanced noradrenaline effects on the kidney. 56 LSS Cardiovascular System - Alexandra Burke-Smith These all act to produce a slow pressor response. Cardiovascular structure: - Haemodynamic effects: o Increased preload and afterload. o Increased vascular wall tension. - - Non-haemodynamic effects: o Increased expression of proto-oncogenes. o Increased production of growth factors. o Increased synthesis of extracellular matrix proteins. These all act to induce vascular and cardiac hypertrophy and remodelling. Pharmacology of ACE Inhibition ACE is needed to convert Angiotensin I to II. Angiotensin II, remember, increases blood pressure (by vasoconstriction and stimulation of the SNS). o Therefore inhibition of ACE will prevent angiotensin II production, and so ACE inhibition reduces blood pressure. At the same time, a local hormone called bradykinin is also broken down by ACE. o Bradykinin is important local vasodilating hormone. o ACE inhibition therefore stops the bradykinin from being broken down, and so the bradykinin therefore will have vasodilating effects, and so here ACE inhibition further acts to reduce the blood pressure AT2 ./ Ang II type 2 receptor antagonist actions: No effects on the bradykinin system. Selectively blocks the effects of Angiotensin II. o Pressor effects. o Stimulation of the noradrenaline system. o Secretion of aldosterone. o Effects on renal vasculature. o Growth-promoting effects on the cardiac and vascular tissue. Uricosuric (increased amount of uric acid in the urine) effect. Aldosterone Physiological effects – maintains body content of Na+, K+ (and water) o Increases Na+ (and hence water) retention o Increases K+ (and H+) excretion Location of receptors o Previously known Kidneys 57 LSS Cardiovascular System o Alexandra Burke-Smith Recently discovered Brain Heart Vessels Pathophysiologic effects in CVD Myocardial fibrosis and necrosis Inflammation, vascular fibrosis and injury Prothrombotic effects – impaired fibrinolysis Central hypertensive effects Endothelial dysfunction Autonomic dysfunction: o Catecholamine potentiation o Decreased heart rate variability Ventricular arrhythmias Sodium retension Potassium and magnesium loss Effects of stress On both the sympathoadrenal system, and the renin-angriotensin system: Increased blood pressue Increased heart rate Increased Na+/water retention Increased coagulation Decreased fibrinolysis Increased platelet activation 58 LSS Cardiovascular System Alexandra Burke-Smith Regulation of the CVS CVS 11 - Dr Ken MacLeod (k.macleod@imperial.ac.uk) 1. 2. 3. 4. 5. 6. 7. Describe the local mechanisms that regulate blood flow Describe how blood vessel diameter and heart rate are controlled by the autonomic nervous system Describe how the autonomic nervous system changes the force of contraction of the heart State the location of the baroreceptors Define cardiac output, stroke volume and mean systemic arterial pressure and state their determinants Indicate, using simple flow diagrams, how baroreceptors control blood pressure Describe the changes in impulse activity in the carotid sinus nerve, parasympathetic and sympathetic nerves to the heart and sympathetic vasoconstrictor nerves that take place following an increase or decrease in mean blood pressure 8. Construct an integrated picture of the various systems that control blood pressure and be able to apply this to specific clinical examples involving blood loss or fluid overload Key Equations Stroke Volume = End diastolic volume – End systolic volume Cardiac Output = Heart Rate x Stroke Volume (SV = EDV – ESV) (CO = HR x SV) Mean Systemic Arterial Pressure = Cardiac Output x Total Peripheral Resistance (MSBP = CO x TPR) Design of the CVS Systemic & pulmonary circulations Right heart lungs left heart Veins have capacitance (act as store of 61% of blood) Venous volume distribution affected by o peripheral venous “tone” o gravity o skeletal muscle pump o breathing (decreased pressure in thoracic cavity) Central venous pressure (mean pressure in the right atrium) determines the amount of blood flowing back to the heart. The amount of blood flowing back to the heart determines stroke volume (using Starling’s Law of the Heart) Flow control Veins – constriction determines compliance and venous return Arterioles – constriction determines: o Blood flow to organs o Mean arterial blood pressure o The pattern of distribution of blood to organs (especially important during exercise) Flow is changed primarily by changing vessel radius (F=P/R, and R is inversely proportional to r4) 59 LSS Cardiovascular System Alexandra Burke-Smith Ways of regulating blood flow Local mechanisms Autoregulation – the intrinsic capacity to compensate for changes in perfusion pressure by changing vascular resistance - Without autoregulation, a decrease in pressure will result in a decrease in blood flow and a slight increase in resistance - With autoregulation, passive constriction as intravascular pressure falls results in an increase in the blood flow close to the initial level, as well as a decrease in resistance - Result of 2 theories/mechanisms: o MYOGENIC THEORY - smooth muscle fibres respond to tension in the vessel wall; e.g. as pressure rises muscle fibres contract; stretch-sensitive Ca2+ channels probably involved o METABOLIC THEORY - as blood flow decreases “metabolites” accumulate and vessels dilate; when flow increases “metabolites” are washed away. Involves e.g. CO2, H+, adenosine, K+ o SERATONIN RELEASE – injury results in a serotonin release from platelets which causes local constriction Endothelial release – substances released from the lining of vessels - NITRIC OXIDE – endothelium-derived relaxing factor synthesised from arginine o Plays a key role in vasodilation - PROSTACYCLIN & THROMBOXANE A2 – relative amounts for clotting o Vasodilator and vasoconstrictor respectively - ENDOTHELINS – potent vasoconstrictors Systemic regulation Circulating hormones affecting the vascular system: Kinins o e.g. bradykinin, have complex interactions with renin-angiotensin system; relax vascular smooth muscle ANP o Atrial natriuretic peptide - secreted from the cardiac atria, vasodilator o Circulating vasoconstrictors ADH – antidiuretic hormone (also known as vasopressin) secreted from posterior pituitary, o noradrenaline released from adrenal medulla, o angiotensin II formed by increased renin secretion from kidney The Autonomic Nervous System (ANS) The sympathetic nervous system is important in controlling the circulation o Fibres originate in thoracic and lumbar nerves o Short pre-ganglionic fibres with long post-ganglionic fibres that release noradrenaline The parasympathetic nervous system is important in regulating heart rate, but has no effect on vessel radius o Fibres originate in cranial and sacral nerves o Long pre-ganglionic fibres with short post-ganglionic fibres that release acetylcholine At all pre-ganglionic fibres, acetylcholine is released SNS INNNERVATION TO BLOOD VESSELS: Sympathetic nerve fibers innervate all vessels except capillaries and precapillary sphincters and some metarterioles. Large veins and the heart are also sympathetically innervated 60 LSS Cardiovascular System Alexandra Burke-Smith Distribution of sympathetic fibres is variable. There are more innervating the vessels supplying kidneys, gut, spleen, and skin and fewer innervating skeletal muscle and the brain. Noradrenaline preferentially binds α1-adrenoceptors to cause smooth muscle contraction and vasoconstriction. Circulating adrenaline binds with high affinity to smooth muscle β2-adrenoceptors to cause vasodilation in some organs; however, the effect of adrenaline is very concentration-dependent. o While it has a higher affinity for β2 than α1 or α2-adrenoceptors, at high concentrations it does bind to αlpha adrenoceptors, which can override the vasodilatory effects of β2-adrenoceptor stimulation and produce vasoconstriction. The Vasomotor Centre (VMC) Located in medulla and pons (brainstem) Composed of: o Vasoconstrictor area (PRESSOR) o Vasodilator area (DEPRESSOR) o Cardioregulatory inhibitory area Transmits impulses distally through the spinal cord to almost all blood vessels Affected by many higher centres of the brain, e.g. the hypothalamus (may exert excitatory or inhibitory effect) Lateral portions – control heart activity by influencing heart rate and contractility Medial portion – transmits signals via vagus nerve to heart; tends to decrease heart rate Nervous control of blood vessel diameter Blood vessels receive sympathetic post-ganglionic innervation o Neurotransmitter involved = noradrenaline There is always some level of tonic activity, known as the BASELINE (basal level of constriction) Control of nerve activity can accomplish dilation or constriction by changing levels of sympathetic nerve discharge: o Depressor – inhibits sympathetic activity o Pressor – stimulates increased sympathetic activity There is generally no parasympathetic innervation to the vascular system Summary – control of vessel radius 61 LSS Cardiovascular System Alexandra Burke-Smith Cardiac Innervation Heart rate is controlled by changing the pulse rate of SAN activity, both via sympathetic and parasympathetic nerves Heart rate is increased by: o increasing activity of sympathetic nerves to heart o decreasing activity of parasympathetic nerves to heart o increasing plasma adrenaline Controlling force of contraction SNS also influences contractility Force of contraction is increased in two ways: o Intrinsic mechanisms o Extrinsic mechanisms Increased Ca2+ influx Increase Ca2+ uptake into intracellular stores EXTRINSIC MECHANISMS Noradrenaline binds to the beta1adrenoreceptor present on the membrane of myocytes Binding causes the increase in cyclic AMP which activates PKA (protein kinase A) This activation leads to the phosphorylation of Ca2+ handling proteins and channels, e.g. the L type Ca channel The channel is then open for longer, which leads to a greater delivery of Ca2+ to myofilaments, increasing force of contraction Controlling stroke volume Extrinsic control (to increase stroke volume) o Increase activity of sympathetic nerves to heart o Increase plasma adrenaline Intrinsic control (to increase stroke volume) o Increased end diastolic ventricular volume (due to Starling’s law – increased venous return increased stretch & preload increased force). This in turn is increased by: Increased respiratory movements a decrease in intrathoracic pressure Increased venous return 62 LSS Cardiovascular System Alexandra Burke-Smith Increased atrial pressure (also increased by increased venous return) Integration of regulation Fight or flight response Leads to: Increased circulating catecholamines Increased respiratory movements Increased sympathetic activity Providing Feedback Feedback mechanisms can be summarised has consisting of 5 components: o Set point o Comparator o Output o Controlled variable o Sensor A disturbance to the controlled variable is the stimulator of the feedback mechanism SET POINT (determined within CNS) COMPARATOR (within CNS) OUTPUT (SNS, PNS, Ang II, ADH/Vasopressin) CONTROLLED VARIABLE (Arterial Blood Pressure) SENSOR (baroreceptors) DISTURBANCE Baroreceptors Afferent neuron cell bodies from the internal carotid arteries to the brain via the GLOSSOPHARYNGEAL NERVE Afferent neuron cell bodies from aortic arch to brain via VAGUS NERVE Both the glossopharyngeal and vagus nerve input lead to increased activity in the VMC Increased blood pressure increased afferent activity to brain (although increase is signoidal) 63 LSS Cardiovascular System o o o Alexandra Burke-Smith Carotid sinus baroreceptors respond to pressures between 60 and 180 mmHg. Baroreceptors respond to changes in arterial pressure. Baroreceptors reflex is most sensitive at pressures around 90 – 100 mmHg. Reciprocal innervation Afferent input from baroreceptors to VMC o Stimulus of parasympathetic nerves to heart o Inhibition of sympathetic innervation to heart arterioles and veins (causing a decrease in tonic activity) Effects of increased blood pressure Increased afferent input via vagus nerve to VMC in medulla oblongata o Increased parasympathetic stimulation of the heart via vagus nerve Decreased heart rate decreased blood pressure o Decreased sympathetic stimulation of the heart decreased heart rate & stroke volume decreased cardiac output decreased blood pressure Also via sympathetic chain, there is decreased sympathetic stimulation to the blood vessels, which produces vasodilation (this may cause a redistribution of blood supply to the different organs) Carotid sinus nerve activity Decreased blood pressure reduced stretch of baroreceptors o Decreased afferent activity to VMC via carotid sinus nerve o Decreased efferent activity via vagus nerve to SAN (parasympathetic) increased heart rate o Increased sympathetic activity via cardiac nerve to ventricle increased heart rate & increased contractility o Increased sympathetic activity via vasoconstrictor nerves to resistance vessels (arterioles) & capacitance vessels (veins) increased constriction Increased blood pressure (reverse occurs) Sympathetic vasoconstrictor nerves allow the control of venous return Haemorrhage reduced blood volume reduced venous pressure and return to heart reduced atrial pressure reduced end diastolic volume reduced stroke volume and cardiac output DECREASED blood pressure (therefore sequence of events same as above) 64 LSS Cardiovascular System Alexandra Burke-Smith Summary - Maintaining arterial blood pressure SUMMARY Local mechanisms regulating blood flow o Autoregulation, the endothelium and paracrine effects Systemic regulation by hormones o circulating vasoconstrictors, kinins, ANP Neural regulatory mechanisms o Nervous control of blood vessel diameter o Cardiac innervation, changing cardiac contraction o Baroreceptors o Reciprocal innervation Integration of control systems 65 LSS Cardiovascular System Alexandra Burke-Smith Responses to Cardiovascular Stress CVS 12 - Dr Chris John (c.john@imperial.ac.uk) 1. Describe the cardiovascular problems associated with: i. movement from a supine to standing position ii. exercise iii. haemorrhage 2. Explain how the components of the cardiovascular system respond to these various challenges Change in Posture Problem: movement from supine to standing position is a sever challenge to the human circulation The Vertical Position In a foot capillary, the usual blood pressure resulting from cardiac contraction ~25mmHg On standing, there is the additional effect of gravity on a column of blood, which causes the pressure to increase to 105mmHg (+80mmHg) On standing, there is a decrease in blood pressure in areas above the heart, and an increase in the areas below the heart Lying Flat Standing Arterial Pressures Heart – 100 Heart – 100 Standing also increases the hydrostatic pressure in the blood (mmHg) Head – 95 Head – 55 vessels in the legs; blood pools in the veins and they are easily Feet - 95 Feet - 195 distended due to their thin muscular wall Venous Pressures Heart – 1 Heart – 1 (mmHg) Head – 5 Head – -35 If hydrostatic pressure becomes greater than oncotic pressure, Feet 5 Feet - 105 fluid is forced into the surrounding tissue beds – this reduces EEFECTIVE circulating blood volume decreased blood pressure END RESULT o Reduced venous return decreased end-diastolic volume o Decreased stroke volume o TRANSIENT HYPOTENSION Compensatory Mechanisms Decrease in blood pressure is detected by arterial baroreceptors in the carotid sinus and aortic arch decreased firing to VMC Max baroreceptor sensitivity occurs near normal mean arterial blood pressure Effects of decrease blood pressure reduced afferent input via vagus nerve to VMC in medulla oblongata o reduced parasympathetic stimulation of the heart via vagus nerve increased heart rate increased blood pressure o reduced inhibition of sympathetic stimulation of the heart increased heart rate & stroke volume increased cardiac output increased blood pressure Also via sympathetic chain, there is reduced inhibition of sympathetic stimulation to the blood vessels, which produces vasoconstriction (this may cause a redistribution of blood supply to the different organs) 66 LSS Cardiovascular System Alexandra Burke-Smith Haemorrhage Problem: reduction in ACTUAL circulating blood volume Compensatory mechanisms similar to that as with change of posture: Reduced baroreceptor firing o Increased heart rate o Increased heart contractility (helps to maintain CO) o Increased TPR (via organ specific vasoconstriction) Extra Compensatory mechanisms Autotransfusion - Reduces the hydrostatic pressure, while the oncotic pressure remains the same: o Reduces ultrafiltration from blood o Increases reabsorption of fluid from interstitial fluid - This bulks up blood volume using extracellular fluid and no erythrocytes - Decreased urinary output ADH/Vasopressin release from pituitary water retention in collecting duct Angiotensin II synthesis decreased renal blood flow Aldosterone production increased Na+ and therefore water retention Haemorrhage Volumes <10% blood loss (~500ml) compensation via BP variation 20-30% blood loss (1-1.5l) hypotension, but with maintained tissue perfusion 30-40% blood loss (1.5-2l) shock; tissue perfusion not maintained which leads to tissue dysfunction and possible infarction Tissue resuscitation can be used initially as treatment, but then a blood transfusion should be performed Exercise Problem: significantly increased blood flow is required to certain tissues (heart, lungs and skeletal muscle), but total peripheral resistance decreases, which may reduce mean arterial blood pressure (MABP = CO x TPR) Exercise increases blood flow, metabolism and oxygen usage within tissues, leading to vasodilation ACTIVE HYPERAEMIA Control Mechanisms - Afferent input to medullary cardiovascular center o “preprogrammed” pattern o Muscle chemoreceptors Efferent output to heart, veins and arterioles o Via autonomic nervous system Control of TPR Increases sympathetic activity in GI tract and kidney profound vasoconstriction Decreased sympathetic activity in heart, lungs, skeletal muscle and skin vasodilation Net result: o Reduced TPR o Increased CO 67 LSS Cardiovascular System o o Alexandra Burke-Smith Increased blood flow to muscles, heart, lungs Reduced blood flow to GI tract and kidneys Control of CO - Reduced parasympathetic activity and increase sympathetic activity increased stroke volume and increased heart rate - If stroke volume increases, venous return must increase – this is due increased force of contraction by the skeletal muscle pump, and increased breathing (which reduces the pressure in thoracic cavity) - There are also NEGATIVE EFFECTS: o Reduced plasma volume opposes increased venous return o There is increased capillary pressure across muscle walls o Loss of salt and water due to sweat - Net result: o increased heart rate o increased contractility o increased venous return increased stroke volume o increased cardiac output Overall result - CO increases because of: o Increased heart rate o Increased contractility o Increased venous return - TPR decreases because of: o Increased vasodilation o (also increased vasodilation in GI tract and kidneys) - However increase in CO > decrease in TPR, therefore there is an overall INCREASE IN BP 68 LSS Cardiovascular System Alexandra Burke-Smith Basic Haemostasis CVS 13 - Dr Jim Crawley (j.crawley@imperial.ac.uk) 1. Describe in outline the normal haemostatic mechanisms including the interaction of vessel wall, platelets, clotting factors and fibrinolytic system 2. Describe the causes of bleeding disorders 3. Describe the types of bleeding seen in different haemostatic disorders 4. Describe in outline how coagulation is regulated by the natural anticoagulant pathways Haemostasis “the biochemical process that enables both the specific and regulated cessation of bleeding in response to vascular insult” Response to a challenge to the vascular system Role – o to prevent blood loss from intact and injured vessels o enable tissue repair o modulate inflammation Important clinical applications – o Diagnosis of bleeding disorders o Treatment of bleeding disorders o Identification of risks for thrombotic disease o Treatment of thrombotic disease o Monitoring of anticoagulant drugs Haemostatic plug formation Is the response to injury to endothelial cell lining. Consists of 4 stages: I. II. III. IV. I. Vessel constriction Primary haemostasis – formation of an unstable platelet plug Secondary haemostasis – stabilisation of the plug with fibrin Vessel repair and dissolution of clot Vessel Constriction Vascular smooth muscle cells contract locally, limiting blood flow to the injured vessels This is a LOCAL CONTRACTILE RESPONSE to injury, and is mainly important in small blood vessels A normal vessel wall consists of: o A layers of endothelial cells – ANTICOAGULANT barrier, which again consists of anticoagulant proteins: GAGs – glycosaminoglycan TFPI - tissue factor pathway inhibitor TM - thrombomodulin EPCR - endothelial protein C receptor o Subendothelium which is PROCOAGULANT and consists of: Elastin Collagen VSMC (tissue factor) – vascular smooth muscle cells Fibroblasts (tissue factor) 69 LSS Cardiovascular System II. Alexandra Burke-Smith Other vital components of haemostasis circulate in the blood, in a quiescent state: o Platelets o Clotting factors o Plasma proteins Within a few seconds of injury, these components endeavour to minimise blood loss via local constriction Primary haemostasis – formation of an unstable platelet plug PLATELETS: o Circulate in blood o Derived from MEGAKARYOCYTES in the bone marrow o Each megakaryocyte produces a large number of platelets o Have a granulated cytoplasm, and are highly specialised ANUCLEAR PLASMA CELLS o Many different ultrastructural features (ass seen in diagram) Formation of the plug consists of: o Platelet ADHESION – recruitment of platelets from flowing blood to site of injury o Platelet AGGREGATION – formation of the plug o Between adhesion and aggregation, it can also be said that the platelets undergo ADHESION: o Within the blood, there are circulating platelets and VWF (von Willebrand factor – a glycoprotein) o These do not interact, as the VWF are in a globular conformation therefore their binding sites are hidden from the platelets – binding sites are called Gp1b (membrane glycoprotein Ib) o Vascular injury damages the endothelium and exposes the sub-endothelial matrix which consists of collagen o The sub-endothelial collagen then binds to VWF, recruiting them to the endothelial surface The rheological (flowing force) shear forces of flowing blood through the vessel then unravels the VWF on the endothelial surface o Unravelled VWF has exposes binding site (Gp1b) therefore the platelets bind o The platelets can also bind directly to the exposed collaged via Gp1a, but this is only under LOW shear forces This binding recruits the platelets to the site of vessel damage ACTIVATION – conversion from a passive to an interactive functional cell o Change shape (spreads and flattens) o Change membrane composition o Present new proteins on their surface (GpIIb/GpIIIa) o The platelets bound to the collagen or VWF release ADP and thromboxane – these activate the platelets Collagen and thrombin also activate platelets AGGREGATION o Activated platelets bind more tightly to the collagen and VWF via GpIIb/IIIa GpIIb/IIIa also binds fibrinogen, which develops the platelet plug o The platelet plug helps slow bleeding and provides a surface for coagulation 70 LSS Cardiovascular System III. Alexandra Burke-Smith Secondary haemostasis – stabilisation of the plug with fibrin Also known as blood coagulation Stops blood loss Complex biochemical process Components involved from: o Liver – most plasma haemostatic proteins o Endothelial cells – VWF. TM, TFPI o Megakaryocytes – VWF, FV These clotting factors circulate as inactive precursors (ZYMOGENS) – then activated by specific proteolysis (to form either as SERINE PROTEASE ZYMOGENS or COFACTORS) Zymogens (inactive) Serine proteases Cofactors Inhibitors Prothrombin (contains GIa domain) FVII (contains GIa domain) thrombin TF TFPI (Kunitz-type) FVIIa (contains GIa domain) FVa FIX (contains GIa domain) FIXa (contains GIa domain) FVIIIa FX (contains GIa domain) FXa (contains GIa domain) FXIa FXIIa Protein C (serine protease) (contains GIa domain) Protein S (cofactor for APC) (contains GIa domain) Antithrombin (serpin) FXI FXII FXIII TISSUE FACTOR PATHWAY Consists of two pathways; intrinsic and extrinsic: o o Intrinsic pathway – initiated when FXII is activated (not biologically as important) FVIIIa is the only cofactor, all other activated clotting factors are serine proteases Extrinsic pathway – primary driver of clotting cascade. Steps include: 1) Initiated when TF on surface of cells (which normally do not come into contact with blood) are exposed to plasma clotting factors o TF + FVII TF-FVIIa complex 2) TF-FVIIa then activates FIX and FX 3) FXa activates prothrombin (ProT) inefficiently leading to the generation of trace amounts of thrombin. 71 LSS Cardiovascular System Alexandra Burke-Smith 4) Thrombin can then activate FVIII and FV, which function as non-enzymatic cofactors for FIXa and FXa, respectively. 5) FIXa-FVIIIa catalyses the conversion of increased quantities of FXa 6) FXa-FVa catalyse enhanced generation of thrombin (more efficient by bypassing initial step) 7) Thrombin at the site of vessel damage converts fibrinogen (Fbg) to fibrin (Fbn), which is the molecular scaffold of a clot. NOTE: cascade or amplification system zymogens which are converted to proteinases & cofactors which need to be activated at surfaces The surface might be platelets (Pl) which localise and accelerate the reactions The trigger to initiate coagulation in vivo is tissue factor Although FXII can be activated to FXIIa, this is mainly an in vitro reaction, useful for some diagnostic tests IV. Vessel repair and dissolution of clot Inhibitory coagulation mechanisms help keep clotting to the site of vessel injury, therefore deficiencies these mechanisms are PRO-THROMBOTIC: - TFPI (Tissue Factor Pathway Inhibitor) Targets initial tissue factor of extrinsic pathway, as well as FXa This leads to the formation of a TF/FVIIa/Xa/TFPI complex This shuts down the coagulation pathway The Protein C anticoagulant pathway (Activated Protein C/APC & Protein S) - APC Protein S, which targets FVIIIa and FVa - Generated thrombin then binds to THROMBOMODULIN of endothelial cells, which shuts down any further thrombin generation Antithrombin - SERPIN (serine protease inhibitor) - Inhibits FIXa, FXa and thrombin, completely shutting down the cascade NB: HEPARIN accelerates the action of antithrombin, therefore is used for immediate anticoagulation in venous thrombosis and pulmonary embolism FIBRINOLYSIS Restores vessel integrity PLASMINOGEN binds to the fibrin clot, and via TPA (tissue plasminogen activator) is converted to PLASMIN Plasmin then cleaves and degrades the fibrin clot into soluble fragments known as FDP (fibrin degradation products), which are then cleared by the liver. o FDP is elevated in DIC (disseminated intravascular coagulation) tPA and a bacterial activator; STREPTOKINASE, are used in therapeutic thrombolysis for Myocardial Infarction, ischaemic stroke etc. these are known as CLOT BUSTERS 72 LSS Cardiovascular System Alexandra Burke-Smith Clinical Haemostasis and Thrombosis CVS 14 - Professor Mike Laffan (m.laffan@imperial.ac.uk) 1. 2. 3. 4. 5. 6. Describe what is meant by abnormal bleeding Describe patterns of abnormal bleeding with examples Describe the manifestations of venous thrombosis List the main risk factors for venous thrombosis Give a rough estimate of its prevalence Be acquainted with the principles of treatment of venous thrombosis NB: normal haemostasis is a state of equilibrium between: Fibrinolytic factors & anticoagulant proteins Coagulation factors & platelets Abnormal Bleeding – the result of an INCREASE in fibrinolytic factors and anticoagulant proteins, and a DECREASE in coagulation factors and platelets Characteristics • • • • Spontaneous Out of proportion to the trauma/injury Unduly prolonged Restarts after appearing to stop Examples • • • • • Epistaxis not stopped by 10 mins compression or requiring medical attention/transfusion. Cutaneous haemorrhage or bruising without apparent trauma (esp. multiple/large). Prolonged (>15 mins) bleeding from trivial wounds, or in oral cavity or recurring spontaneously in 7 days after wound. Spontaneous GI bleeding leading to anaemia. Menorrhagia requiring treatment or leading to anaemia, not due to structural lesions of the uterus. Heavy, prolonged or recurrent bleeding after surgery or dental extractions. Defects of Primary Haemostasis (the formation of an unstable platelet plug) See previous lecture for details on primary haemostasis Deficiency/defect Collagen within subendothelial matrix of vessel wall Von willebrand factor platelets Examples Steroid therapy, age Von willebrand disease (genetic) Aspirin &other drugs, thrombocytopenia Patterns of bleeding • • • • • • Easy bruising Nosebleeds (prolonged:>20 mins) Gum bleeding (prolonged) Menorrhagia Bleeding after trauma/surgery Petechiae (specific for thrombocytopenia) o Typical of thrombocytopenia (decreased platelets) – small blood spots 73 LSS Cardiovascular System Alexandra Burke-Smith Defects of secondary haemostasis (fibrin mesh formation/coagulation) See previous lecture for details on secondary haemostasis Definition: Deficiency or defect of coagulation factors I-XIII Common examples include: • Haemophilia: FVIII or FIX (hereditary due to genetic defect) • Liver disease (acquired – most coagulation factors are made in the liver) • Drugs (warfarin – inhibits synthesis) • Dilution • Consumption (DIC – disseminated intravascular coagulation) (acquired) Acquired coagulation disorders – DIC • • • • • • Generalised activation of coagulation – Tissue factor Haemostasis then takes place WITHIN blood vessels, and throughout the general circulation Associated with sepsis, major tissue damage, inflammation Consumes and depletes coagulation factors & platelets Activation of fibrinolysis depletes fibrinogen Consequences: o Widespread bleeding - from iv lines, bruising, internal o Organ failure – due to deposition of fibrin in vessels Patterns of bleeding • • • • • • • • Often delayed (after primary haemostasis) Deeper: joints and muscles Not from small cuts etc. Nosebleeds rare Bleeding after trauma/surgery After i/m injections ECCHYMOSIS – easy bruising o Virtually all bleeding disorders HAEMARTHROSIS – spontaneous bleeding into joints o Hallmark of haemophilia o Increases pressure in joints o Very painful and damaging Defects of clot stability – excess fibrinolysis • • Can be used in therapy to break down clots after MI (but this must be done carefully as can lead to haemorrhage) Due to either: o Excess FIBRINOLYTIC components – plasma, tPA Can occur with some tumours o Deficient ANTIFIBRINOLYTIC components – antiplasmin Can have a genetic antiplasmin deficiency NB: ANTICOAGULANT excess is usually only due to therapeutic administration, e.g. heparin or hirudin 74 LSS Cardiovascular System Alexandra Burke-Smith Thrombosis – result of a DECREASE in fibrinolytic factors and anticoagulant proteins, with an INCREASE in coagulation factors and platelets What is thrombosis? • • • • • ‘Intravascular coagulation’ ‘Inappropriate coagulation’ Coagulation inside a blood vessel ‘Coagulation not preceded by bleeding’ Thrombi may be Venous or Arterial Effects of thrombosis • • Obstructed flow of blood o Artery – myocardial infarction, stroke, limb ischaemia o Vein – pain and swelling Embolism o Venous embolise to lungs (pulmonary embolus) o Arterial emboli, usually from heart, may cause stroke or limb ischaemia Venous Thrombo-embolism • • • • Deep Vein Thrombosis (DVT) – venous return of blood is obstructed o Causes painful, swollen legs Pulmonary embolism – causes shortness of breath, chest pain, may lead to sudden death Prevalence o 1 in 1000-1,000 per annum (with incidence doubling each decade) o However is cause of 10% of hospital deaths, and is a preventable cause of death 25000 preventable deaths per annum Consequences: o Death- VT mortality 5% o Recurrence - 20% in first 2 years and 4%pa thereafter o THROMBOPHLEBITIC syndrome Severe TPS in 23% at 2 years (11% with stockings) o Pulmonary hypertension - 4% at 2 years Why do some people get thrombosis? • • • Genetic constitution Effect of age and previous events, illnesses, medication Acute stimulus VIRCHOW’S TRIAD: the 3 contibutory factors to thrombosis • Blood - dominant in venous thrombosis o Deficiency of anticoagulant proteins – antithrombin, protein C, protein S o Increased coagulant proteins & activity – Factor VIII Factor II &others 75 LSS Cardiovascular System • • Alexandra Burke-Smith Factor V Leiden (increased activity due to activated protein C resistance) Thrombocytosis (increased platelets) vessel wall - dominant in arterial thrombosis o Many proteins active in coagulation are expressed on the surface of endothelial cells and their expression altered in inflammation Thrombomodulin Tissue factor Tissue factor pathway inhibitor flow - complex, contributes to both o reduced flow (STASIS) increases the risk of venous thrombosis. This can occur due to: surgery fracture long haul flight bed rest Thrombophilia – increased risk of thrombosis • • Clinical: o Thrombosis at young age o ‘idiopathic thrombosis’ o Multiple thromboses o Thrombosis whilst anticoagulated Laboratory o Identifiable cause of increased risk AT deficiency Factor V Leiden global measures of coagulation activity. Absolute and Relative Risks 76 LSS Cardiovascular System Alexandra Burke-Smith Acquired risks for thrombosis • Numerous conditions will alter blood coagulation, vessel wall and/or flow to precipitate thrombosis or make it more likely, e.g. o Oral contraceptive pill o Pregnancy o Malignancy o Surgery o Inflammatory response Therapy and venous thrombosis • • Treatment o LYSE CLOTS – e.g. using TPA (this presents with a high risk of bleeding) o LIMIT RECCURENCE/EXTENSTION Increase anticoagulant activity - e.g: heparin (immediate acting, parenteral) Lower Procoagulant factors – e.g. warfarin (oral, slow acting for long term therapy) Prevention (NICE Guidelines 2010) o Assess individual risk and circumstantial risk o All patients admitted should have VTE risk assessment o Give prophylactic antithrombotic therapy o E.g. heparin for in-patients o TED stockings 77 LSS Cardiovascular System Alexandra Burke-Smith Introduction to Atherosclerosis CVS 15 - Dorian O. Haskard (d.haskard@imperial.ac.uk) Recognize: 1. The changes in the walls of arteries that lead to atherosclerosis. 2. How atherosclerosis leads to clinical manifestations 3. The time-course of atherosclerosis Areas of Medicine involved/affected by atherosclerosis: Neurology – Cerebrovascular disease Acute medicine – heart attack, stroke Metabolic medicine – lipids Endocrinology – diabetes Vascular surgery – revascularization Cardiac surgery – revascularization Cardiology – coronary disease Atherosclerosis Noted by Rudolph Virchow in 1952 Term coined by Felix Marchand in 1904 There are 6 stages involved: I. Lesion prone location within a coronary artery - Smooth muscle within the vessel wall undergoes ADAPTIVE THICKENING II. Type II lesion - Macrophage foam cells appear III. Type III lesion (pre-atheroma) - Small pools of extracellular lipids form within the vessel wall IV. Type IV lesion (atheroma) - Macrophage foam cells form NECROTIC CORE (consisting of dead cells and extracellular lipids) V. Type V lesion (fibroatheroma) - The necrotic core becomes encased by a FIBROUS CAP), which separates coagulation factors from tissue factors VI. Type VI l lesion (complicated lesion) - The fibrous thickening may reduce blood flow - Atheroma may break down, causing a thrombus, fissure and/or haematoma Risk factors - Potentially modifiable: Smoking Lipids Blood pressure Diabetes Obesity 78 LSS Cardiovascular System - Alexandra Burke-Smith Lack of exercise Not modifiable Age Sex Genetic background NB: smoking, hypertension and high cholesterol can be considered the BIG 3, and combined lead to a 16x increased risk Paradigms (ideas) of pathogenesis Inevitable consequence of ageing The cholesterol hypothesis Inflammation and immunity The cholesterol hypothesis 1904 N.N. Anitschkow – studied advanced plaque in a rabbit fed cholesterol for 124 o This lead to the formation of a fibrous plaque surrounding foam cells, and a necrotic core consisting of cholesterol crystals and calcium The cholesterol controversy: “the view that raised plasma cholesterol is per se a cause of coronary heart disease is untenable” BMJ 1976 Evidence: in favour of cholesterol as a major aetiological factor o EXPERIMENTAL – e.g. rabbits, mice o CLINICAL GENETIC – e.g. familial hypercholesterolaemia o EPIDEMIOLOGICAL – e.g. Framlingham; a long standing epidemiological survey o INTERVENTIONAL – e.g. randomised controlled trials of statins Foam Cells LDLs deposit in the SUBINTIMAL SPACE (intima is the component of the subendothelial layer of vessel wall, between the endothelium and the internal elastic lamina) o The vessel wall consists of many layers (from lumen outwards) Endothelium Subendothelium – Intima Internal elastic lamina Media Adventitia The LDLs then bind to MATRIX PROTEOGLYCANS o NATIVE LDLs are encased with a phospholipid case by apoB o The native LDL is then oxidised and modified to form a MODIFIED apoB The modified apoB is recognised as NON-SELF by macrophages, and is taken up by SCAVENGER RECEPTORS to form a MACROPHAGIC FOAM CELL The foam cells then release inflammatory mediators (e.g. cytokines, chemokines & oxidised phospholipids) and/or lead to cell death o This damage causes increased recruit and adhesion of MONOCYTES by the endothelium With normal levels of oxidised LDLS, HOMEOSTATIC DEBRIS DISPOSAL occurs With INCREASED levels of oxidised LDLs, this leads to an INFLAMMATORY RESPONSE 79 LSS Cardiovascular System Alexandra Burke-Smith Inflammatory basis of atherosclerosis Increased levels of activated macrophagic foam cells lead to release of: Free radicals Proteases VSMC growth factors Angiogenic factors Apoptosis Why is atherosclerosis focal? The branches and curvatures of blood vessels are more likely to be “HOT SPOTS” for atheroma formation Blood flow is FAST, LAMINAR, SHEAR At these hot-spots, blood flow is more disorganised The endothelium is very sensitive to these changes in blood flow Terminology of Atherosclerosis Stenosis – the gradual loss of lumenal diameter leading to critical reduction in blood flow o This can be observed by ANGIOGRAPHY Ischaemia – insufficient blood supply to meet metabolic demands of tissues leads to hypoxic cellular dysfunction o Typically experienced as pain on exertion, e.g. angina pectoris (heart pain), intermittent claudication (in legs) Atheroslcerotic plaque – the localised area of fat deposition and tissue breakdown (necrosis) within the arterial wall Plaque erosion – the breakdown of endothelial lining of the lesion WITHOUT full rupture of the fibrous cap Plaque rupture – the breakdown of the fibrous cap of tissue separating plaque from blood Effects of plaque erosion/rupture - Plaque growth Platelet recruitment (involving adhesion and degranulation) and blood coagulation at site This may lead to silent non-occlusive thrombus and plaque growth Occlusive thrombosis Blood coagulation at the site of rupture may lead to an occlusive thrombus and cessation of blood flow Embolism Definition – the dislodgment of solid material (e.g. platelet plug, thrombus, cholesterol-rich plaque contents) into the arterial circulation leading to occlusion at distant sites Consequences depend on the size of the embolus and the target organ of the arterial circulation (e.g. brain, eye, bowel, limbs) Effects of arterial occlusion Transient occlusion – short ischaemia from an occlusion that spontaneously resolves o E.g. in brain: TIA, in eye: amaurosis fugax Infarction – the death of tissue due to unresolved ischaemia o E.g. in heart: MI (heart attack)., in brain: CVA-cerebrovascular attack (stroke) 80 LSS Cardiovascular System Alexandra Burke-Smith Natural history of atherosclerosis Development from: Normal vessel intermediate lesions advances lesions complications (stenosis, rupture etc) This may occur over a period of >60 years: o Normal vessel – neonatal o Intermediate lesions – 40 yrs o Advanced lesions – 50 yrs o Complications - >60 yrs Glasgow Phenomenon – describes the early expansion of arteries before constriction of the lumen occurs (also known as POSITIVE ARTERIAL REMODELLING) o This is not seen by an angiogram Window of opportunity for CLINICAL INTERVENTION o Used at the complications stage (>60 yrs) o Secondary prevention o Catheter based interventions o Revascularisation surgery o Treatment of heart failure Window of opportunity for PRIMARY PREVENTION o Used during the development of advanced lesions from intermediate lesions o Life-style changes o Risk factor management Aim is to shift away from clinical intervention to primary prevention Pathogenesis Atherosclerosis – a chronic inflammatory response in the walls of arteries, in large part in reaction to the deposition of lipoproteins (plasma proteins that carry cholesterol and triglycerides) The main cell types involved: Vascular endothelial cells White blood cells (leukocytes) o Particularly monocytes/macrophages Platelets Vascular smooth muscle cells 81 LSS Cardiovascular System Alexandra Burke-Smith Vascular endothelium in atherosclerosis CVS 16 - Anna M. Randi (a.randi@imperial.ac.uk) Recognize: 1. The importance of the vascular endothelium for the health of blood vessels 2. The role of endothelium in regulating permeability and leukocyte recruitment 3. The role of the vascular endothelium in atherosclerosis a. Regulation of permeability and leukocyte recruitment b. The role of flow in regulating endothelial function c. Endothelial aging: cell senescence in atherosclerosis d. Pro and cons of angiogenesis in atherosclerosis Structure of Arteries & Veins 3 layers (except capillaries and venules): TUNICA INTIMA - Endothelium - Basement membrane - Lamina propria (smooth muscle and connective tissue) - Internal elastic membrane TUNICA MEDIA - Smooth muscle cells - External elastic membrane TUNICA ADVENTITIA - Vasa vasorum - Nerves Vascular endothelium Surface separating blood from other tissues Very extensive - Surface area >1000m2, Weight >100g Acts as a vital barrier separating blood from tissues Formed by a monolayer of cells, one cell deep o CONTACT INHIBITION – mechanism at cell junctions which stops further cell growth Very flat cells, about 1-2 micrometres thick and 10-20 micrometers in diameter Not all cells are the same In vivo, cells live a long life and have a low proliferation rate (unless new vessels are required) Endothelial cells Regulates: Thrombosis & haemostasis - Antithrombotic factors - Procoagulant factors Angiogenesis - Growth factors - Matrix proteins Vascular tone & permeability - Vasodilator factors - Vasoconstricting factors 82 LSS Cardiovascular System Alexandra Burke-Smith Inflammation - Adhesion molecules - Inflammatory mediators Regulation of endothelial homeostasis At rest, there is a homeostatic balance between different factors: o Anti-inflammatory & pro-inflammatory o Anti-thrombotic & pro-thrombotic o Anti-proliferative & pro-angiogenic Infection, injury etc, may temporarily tip the balance leading to an activated endothelium, but then will return to its resting state The endothelium may be activated by: o Smoking o Viruses o Mechanical stress o Inflammation o High blood pressure o Oxidised LDLs o High glucose This activation leads to: o Thrombosis o Senescence o Increased permeability o Leukocyte recruitment Chronic activation of the endothelium may also lead to ATHEROSCLEROSIS Role of the endothelium in atherosclerosis Vascular permeability The endothelium regulates the flux of fluids and molecules from blood to tissues and vice versa Increased permeability results in leakage of plasma proteins through the junctions into the subendothelial space: o This causes lipoprotein trapping and oxidative modification o The modified LDLs may then be taken up by macrophages forming foam cells, which causes chronic inflammation Leukocyte recruitment Contact inhibition at endothelial junctions regulates the movement of leukocytes from blood into tissues The activation of the endothelium LEUKOCYTE ADHESION CASCADE o Capture o Rolling o Arrest o Adhesion o Intravascular crawling o Paracellular and transcellular transmigration This involves: o selectins (E, P & L-selectins) o chemoattractants (MCP-1, IL-8) 83 LSS Cardiovascular System Alexandra Burke-Smith o ICAM-1, VCAM-1 o Integrins (LFA-1, VLA-4) VESSEL DIFFERENCES: o Capillary – endothelial cells surrounded by basement membrane and pre-capillary cells (perycites) o Post-capillary venule – structure similar to capillaries but more pericytes o Artery – 3 thick layers rich in cells and extracellular matrix Recruitment of blood leukocytes into tissues takes place normally during INFLAMMATION o Leukocytes adhere in POST-CAPILLARY VENULES In ATHEROSCLEROSIS, leukocytes adhere to the activated endothelium of LARGE ARTERIES and get stuck in the subendothelial space (in the smooth muscle layer) o Newly formed post-capillary venules at the base of developing lesions provide a further portal for leukocyte entry Blood flow Laminar Flow Occurs normally Streamlined Outermost layer moving slowest and centre moving fastest Leads to high shear stress Antithrombotic Antimigration Antigrowth Promotes: o NO production o Factors that inhibit coagulation, leukocyte adhesion, smooth muscle cell proliferation o Endothelial survival Disturbed flow Occurs at branch-points Blood flow interrupted Fluid passes a constriction, sharp turn, rough surface etc Leads to low shear stress Prothrombotic Promigration Progrowth Promotes: o Coagulation, leukocyte adhesion, smooth muscle cell proliferation o Endothelial apoptosis Senescence REPLICATIVE SENESCENCE: the limited proliferative capacity of human cells in culture In response to stress and damage, CELLULAR SENESCENCE locks cells in a permanent form of growth arrest This is linked to progressive shortening and dysfunction of TELOMERES (ends of chromosomes) Senescent cells have a distinct morphology and acquire specific markers, e.g. beta-gal OXIDATIVE SENESCENCE is telomere independent In atherosclerotic lesions, vascular cells have morphological features of senescence o associated with beta-gal activity within arteries CONSEQUENCES of endothelial senescence: 84 LSS Cardiovascular System Alexandra Burke-Smith o o Can be induced by CV risk factors, such as oxidative stress, that induced increased cell replicatio n to replace dead or damaged cells These changes result in a pro-inflammatory, Angiogenesis Formation of new blood vessels by sprouting from pre-existing vessels Initiated by pro-angiogenic stimulation of a pre-existing mature blood vessel Involves a large number of cells Important in many diseases e.g. cancer Cascade of events lead by specialised endothelial tip cells Cascade involves: o Angiogenic factor production o Release of factor o Extracellular receptor binding Intracellular signalling occurs o Extracellular activation Endothelial matrix degradation o Extracellular proliferation o Directional migration o Extracellular matrix remodelling o Tube formation o Loop formation o Vascular stabilization THE JANUS PARADOX: o Angiogenesis promotes plaque growth, but can be used therapeutically to induce new formation in ischaemic tissues Delivers growth factors and stem cells to the ischaemic region to induce new vessel growth Pathogenesis of Atherosclerosis: Inflammation Model Endothelial dysfunction leads to: o Increased endothelial permeability o Leukocyte migration and adhesion This leads to fatty-streak formation and foam-cell formation There is then a formation of an advances, complicated lesion of atherosclerosis o Macrophage accumulation & formation of a necrotic core 85 LSS Cardiovascular System o Alexandra Burke-Smith Angiogenesis also occurs 86 LSS Cardiovascular System Alexandra Burke-Smith Atherosclerosis Pathology: the role of lipids, macrophages and vascular smooth muscle CVS 17 - Dr Joseph J Boyle (j.boyle@imperial.ac.uk) 1. 2. 3. 4. Recognize: How lipoproteins that are deposited in the arterial wall stimulate macrophage functions The protective and deleterious functions of macrophages that result in a chronic inflammatory response in the vessel wall The way in which vascular smooth muscle cells (VSMC) remodel the structure of the artery and protect plaque integrity The different roles of macrophages and VSMC in plaque instability Macrophages • • • • • • • • • White blood cells can injure host tissue if they are activated excessively or inappropriately In atherosclerosis, the main inflammatory cells are macrophages Macrophages are derived from blood monocytes There are many different types of macrophages Macrophage subtypes are regulated by combinations of transcription factors binding to regulatory sequences on DNA We do not yet understand the regulation Two main classes - resident or inflammatory macrophages Inflammatory macrophages adapted to kill microorganisms (germs) Resident macrophages – normally homeostatic o suppressed inflammatory activity o Alveolar resident macrophages (surfactant lipid homeostasis) o Osteoclasts (calcium and phosphate homeostasis) o Spleen (iron homeostasis) Lipoproteins • Low density lipoprotein (LDL) o ‘bad’cholesterol o synthesised in liver o carries cholesterol from liver to rest of body including arteries • High density lipoprotein (HDL) o ‘good cholesterol’ o carries cholesterol from ‘peripheral tissues’ including arteries back to liver (=“reverse cholesterol transport”) Oxidised LDL(s), modified LDL(s) o Due to action of free radicals on LDL (see later) o Not one single substance o families of highly inflammatory and toxic forms of LDL found in vessel walls. • Sub-endothelial trapping of LDL • Low density lipoproteins (LDL) leak through the endothelial barrier by uncertain mechanisms 87 I. LSS Cardiovascular System • • Alexandra Burke-Smith LDL is trapped by binding to sticky matrix carbohydrates (proteoglycans) in the sub-endothelial layer Trapped LDL is susceptible to modification Modification of trapped LDL • • • • Best studied modification is oxidation Chemically represents partial burning LDL becomes oxidatively modified by free radicals Oxidised LDL is phagocytosed by macrophages to form a foam cell - this stimulates chronic inflammation Familial Hypelipidemia (FH) • • • • Autosomal recessive genetic disease Massively elevated cholesterol (20mmol/L) failure to clear LDL from blood xanthomas (fatty cholesterol lumps visible on skin) and early atherosclerosis o if untreated fatal myocardial infarction before age 20 Cholesterol Homeostasis • • • • • HMGCoA reductase is the rate limiting enzyme of cholesterol synthesis SREBP (sterol response element binding protein) o Activated by low cholesterol o Activates gene for HMGCoA reductase o If LDL receptor (LDLR) is working properly, increased LDL levels inactivate liver SREBP and reduce cholesterol synthesis This is Negative feedback In LDLR-negative patients, macrophages accumulate cholesterol In atherosclerotic lesions, there is a second LDL receptor - not under feedback control o Called the ‘scavenger receptor’ since it hoovers up chemically modified LDL o Now known that scavenger receptors are a family of pathogen receptors that ‘accidentally’ bind oxidised LDL Macrophage Scavenger receptors - Receptor A Expresses CD204 Binds to oxidised LDL Binds to Gram-positive bacteria like Staphylococci and Streptococci Binds to dead cells - Receptor B Expresses CD36 Binds to oxidised LDL Binds to malaria parasites Binds to dead cells Macrophages within atherosclerotic plaques Generate of free radicals that further oxidise lipoproteins • Macrophage Oxidative enzymes • Can modify native LDL modified LDL • NADPH Oxidase 88 LSS Cardiovascular System • Alexandra Burke-Smith o superoxide O2MYELOPEROXIDASE o HOCl hypochlorous acid (bleach) from ROS + Clo HONOO Peroxynitrite II. Phagocytose/scavenge modified lipoproteins, and become foam cells III. Become activated by modified lipoproteins/free intracellular cholesterol to express/secrete a. Inflammatory mediators (eg TNFa, IL-1, MCP-1) that recruit more monocytes • Cytokines – protein immune hormones that activate endothelial cell adhesion molecules o Interleukin-1 upregulates vascular cell adhesion molecule 1 VCAM-1 o VCAM-1 mediates tight monocyte binding o Atherosclerosis is reduced in mice without IL-1 or VCAM-1 • Chemokines - small proteins chemoattractant to monocytes o Monocyte chemotactic protein-1 (MCP-1) o MCP-1 binds to a monocyte G-protein coupled receptor CCR2 o Atherosclerosis is reduced in MCP-1 or CCR2 deficient mice • Positive feedback loop / vicious cycle leading to self-perpetuating inflammation b. Chemoattractants and growth factors for Vascular Smooth Muscle Cells (VSMC) • Macrophages release complementary protein growth factors that recruit VSMC and stimulate them to proliferate and deposit extracellular matrix, reducing their contractile filaments • Platelet derived growth factor o Vascular smooth muscle cell chemotaxis o Vascular smooth muscle cell survival o Vascular smooth muscle cell division (mitosis) • Transforming growth factor beta o Increased collagen synthesis o Matrix deposition c. Proteinases that degrade tissue (e.g. the fibrous cap) • Metalloproteinases (=MMPs) o Family of ~28 homologous enzymes o Activate each other by proteolysis o Degrades collagen o Catalytic mechanism based on Zinc • Vulnerable yet stable plaques have certain characteristics: o Large soft eccentric lipid-rich necrotic core o Thin fibrous cap o Reduced VSMC and collagen content o Increased VSMC apoptosis o Infiltrate of activated macrophages expressing MMPs d. Tissue factor that stimulates coagulation upon contact with blood • Crosstalk between inflammation and coagulation: healing wound need to clot blood and fight infection • Tissue factor (TF) is a 263 amino acid membrane protein expressed on activated macrophages • TF initiates the coagulation cascade • Macrophage TF is increased in atherosclerosis 89 IV. LSS Cardiovascular System • Alexandra Burke-Smith Erosion/rupture of the fibrous cap leads to access of the plasma coagulation cascade to macrophage tissue factor with consequent thrombosis Die by apoptosis – contributing to the lipid-rich core of the plaque • Oxidised LDL derived metabolites are toxic e.g. 7-keto-cholesterol • Macrophage foam cells have protective systems that maintain survival in face of toxic lipid loading • Once overwhelmed, macrophages die via apoptosis • Releases macrophage tissue factor and toxic lipids into the ‘central death zone’ called lipid necrotic core o Thrombogenic and toxic material accumulates, walled off by the fibrous plaque, until plaque ruptures which causes it to meet blood SUMMARY • • • Macrophages are the major inflammatory cell type in atherosclerosis protective functions in the plaque o Clearing debris (modified lipoproteins, dead cells) o Stimulating “wound healing” response involving VSMCs deleterious functions o Release of free radicals that modify LDL o Recruitment of further monocytes via cytokines and chemokines o Expression of MMPs that may destabilise the fibrous cap o Expression of tissue factor that can stimulate thrombosis 90 LSS Cardiovascular System Alexandra Burke-Smith CHD, Angina and MI – investigation and treatment CVS 18 - Professor Peter Collins (p.collins@imperial.ac.uk) 1. 2. 3. 4. State the main cardiac factors which give rise to chest pain State the main clinical investigations that help diagnose angina State some of the drug treatments for angina Define myocardial ischemia and its pathophysiology Causes of Coronary Heart Disease • • • • • • • • Risk factors may be classified as non-modifiable or modifiable Non-modifiable: o Gender o Age o Menopause o Family history o Ethnicity Modifiable: o Hypertension o Diabetes o Smoking o Lipids o Lifestyle o Obesity o Oestrogen status The major modifiable CHD risk factors are: o Smoking o Hypertension o Dyslipidaemia o A cluster of these 3 risk factors is associated with a 16x increased risk for CHD The cause of CHD has nothing to do with the heart muscle itself, but is a disease of the coronary arteries which supply blood to cardiac muscle known as ATHEROSCLEROSIS Atherosclerosis consist of 3 phases: o Initiation o Progression o Complication There is now a substantial body of research that suggests the origins of atherosclerosis begin at an early age. o the extent of atherosclerosis increases progressively with advancing age o CHD is highly prevalent and that even relatively young individuals may have a substantial plaque burden that will require aggressive intervention. aim of treatment of CHD is to shift away from aggressive intervention, + replace it wil primary prevention. The AHA statement is an example of diet/ lifestyle modification recommendations for prevention: o Aim for a healthy body weight** o Consume an overall healthy diet** o Be physically active** o Aim for the recommended lipid profile o Aim for a normal blood pressure o Aim for a normal glucose level o Avoid use of and exposure to tobacco products 91 LSS Cardiovascular System Alexandra Burke-Smith Body Weight • • • In order to maintain a healthy body weight, we need a energy balance between energy consumed and used on a day to day basis Our modern lifestyle can be considered a “toxic environment” which promotes a POSITIVE energy balance of too much consumed and too little used o The increased consumption of energy is due to large portion sizes, high fat/energy/glycaemic index foods, soft drinks with high sugar levels, the high accessibility/variety of foods, relatively low cost of food and advertising of food o The decreased use of energy can be said to be caused by TV, online shopping, car travel, elevators, “energy saving” devices, i.e. less physical activity is required to carry out daily activities Moderate weight-loss is associated will reduced blood pressure and lipid counts o Moderate weight loss may also allow a reduction or discontinuation of anti-hypertensive and antidiabetic medications Myocardial ischaemia • • • • It is defined as a lack of oxygen to the heart muscle. o Myocardial pertaining to the heart muscle, and ischaemia pertaining to lack of oxygen. causes a fixed narrowing in the coronary arteries, due to fatty build up (i.e. atherosclerosis). It also causes spasm of the blood vessels. Myocardial ischaemia is caused by an imbalance between myocardial blood flow and metabolic demand, where blood supply does not equal demand. o The coronary artery lumen must be reduced by more than 75% to significantly affect myocardial blood supply. Factors affecting coronary blood flow: o Aortic blood pressure – a decreased blood pressure reduces coronary flow. o Myocardial work – exercise increases coronary flow. o Coronary artery narrowing – fixed narrowing (such as a “fatty plaque”), or an acute plaque change (due to rupture or haemorrhage), or a blood clot in the vessel (thrombus), or vasoconstriction. o Aortic valve dysfunction. o Increased right atrial pressure. Angina Pectoris • • • Chest discomfort due to myocardial ischaemia typically associated with coronary artery disease Types of angina include: o STABLE - angina occurring over several weeks without major deterioration, although symptoms may vary considerable over time (e.g.. with exertion, stress) o UNSTABLE - Abruptly worsening angina or new angina at low work load. This leads to a decreased exercise capacity. o VARIANT (prinzmetal) - spontaneous (i.e. no precipitating cause) angina with ST elevation on ECG o SYNDROME X –Rare. Angina with objective evidence of myocardial ischaemia in the absence of evident coronary atherosclerosis or epicardial (large vessel) disease Other terminology used when describing angina: o DECUBITUS – chest pain when lying flat (in bed) o NOCTURNAL – at night o ACUTE CORONARY INSUFFICIENCY – unstable o CRESCENDO – increasing in frequency and severity 92 LSS Cardiovascular System Alexandra Burke-Smith Cardiac Chest pain • • • Diagnosis often made from history, but examination confirms Information obtained O/E: o Site o Character o Mode of onset - sudden or gradual o Progression o Radiation – e.g. left arm, neck etc o Positional bodily function o Precipitating or relieving factors - does anything make it better or worse? o Any current treatment o Depth of pain There are different causes of cardiac chest pain, with distinct features: o MYOCARDIAL INFARCTION Sudden Severe Sweating o AORTIC DISSECTION Tearing pain In abdomen or back o PERICARDITIS Pain sharp & sudden Pain while breathing Postural NB: only 20% of MI are preceded by angina, but 50% are followed by angina Investigations of Angina • • • • • • Good history: o Past medical history o Previous illness o Rheumatic fever o Diabetes o Hypertension o Previous treatment, hospitalisation o Medication o Allergies Blood pressure Lipids ECG o Exercise ECG – valuable Disease severity correlates with degree of ST depression There is also a crude correlation between exercise capacility and prognosis after MI Stress test angiography Role of imaging in CAD (computer aided diagnosis) • Diagnosis 93 LSS Cardiovascular System • • • • • • • • • Alexandra Burke-Smith Coronary anatomy & function Myocardial anatomy & function Valve anatomy & function Objective assessment of symptoms Disease severity & burden Acute & chronic risk assessment Myocardial viability, stunning & hibernation Guiding revascularisation Monitoring therapy Subspecialty Cardiac Imaging • • • • Echocardiography o Rest o Stress o Specialist (e.g. trans-oesophageal) Radionuclide imaging o Myocardial perfusion scintigraphy o Radionuclide ventriculography Magnetic resonance imaging X-ray computed tomography o Coronary calcium imaging o Percutaneous transluminal coronary angioplasty (PTCA) Tube inserted into an artery at the groin and directed towards the coronary arteries A dye is injected and X-rays of the heart and coronary arteries are taken No benefit on longevity Treatment • • Aims of therapy: o Reduce morbidity and mortality o Eliminate angina with minimal adverse effect allowing the patient to return to normal activities Overview of management: o Education and risk factor management – The patient needs to have the diagnosis explained to them, given a basic understanding of the underlying cause(s), what steps they can take to improve the condition, and how to recognise the symptoms of ACS/MI Risk factor assessment should be carried out and steps taken to modify those identified. These include poor diet, physical inactivity, cig smoking, hypercholesterolaemia, DM or insulin resistance syndrome, and hypertension o o o o Anti-platelet therapy – should be initiated from the outset Where ASA is contraindicated, clopidogrel, which is a platelet ADP-ase inhibitor, should be used. Anti-anginal drug treatment – should be started on follow-up arranged to monitor symptom relief and adverse effects follow-up revascularisation therapy (PCI or CABG) Where there is failure of drug treatment or prognostic indication (from angiography), revascularisation should be considered 94 LSS Cardiovascular System Alexandra Burke-Smith Anti-anginal drugs Beta-blockers Atenolol, metoprolol, bisoprolol Calcium antagonists Dihydropyridines (nifedipine, amlodipine) Others (diltiazem, verapamil) Nitrates GTN, long-acting nitrates (ISMN, ISDN) Nicorandil Your choice of a drug or combination will be based on: • evidence from clinical trials • the drug’s efficacy on an individual patient’s angina • presence of adverse side effects and the patient’s ability to tolerate them • the existence of co-morbidities that would make one drug more preferable than the other, for example: o using a combination of beta-blocker and Ca antagonists to treat angina and hypertension o avoidance of nitrates when there is outflow tract obstruction o choosing diltaizem or verapamil in preference to beta-blockers when you suspect vasospastic angina Coronary Angioplasty • • • • The coronary blood vessels that supply the heart are vital as even under resting conditions the oxygen supply from the resting blood flow is almost completely exhausted. Thus, the only way to increase oxygen supply in times of higher demand is to increase flow. The supply of blood to the cardiac muscle is vital for survival thus any disruption in the coronary circulation is likely to cause problems. If coronary artery disease is present the coronary vessels can be narrowed restricting the supply of blood during periods of higher demand. If coronary flow is reduced to the point that the myocardium it supplies becomes hypoxic, angina pectoris develops. In severe angina, sufferers experience pain at rest and not just on exertion. In most cases flow is reduced because of an atherosclerotic plaque. If the myocardial ischaemia is severe and/or prolonged irreversible changes occur resulting in myocardial infarction. If coronary disease is symptomatic treatment is by revascularisation either by angiography or coronary artery bypass grafting. Despite recent advances angina and myocardial infarction remain common in the developed world and are a significant cause of morbidity and mortality. Summary of treatment • • • • Anti-anginal treatment is only a part of the overall management of angina Beta-blockers 1st-line, then choose a combination that suits the patient and his co-morbidities (if exist) Use at least three agents where possible before accepting failure of medical therapy Consider revascularisation at any time if prognostically indicated or if drug treatment fails Myocardial Infarction • • • Caused by coronary thrombosis with full occlusion of a coronary artery o Coronary thrombosis in turn is caused by atherosclerosis When an atheroma plaque ruptures (known as a plaque fissure), two things may occur: o The fissure may heal, forming an INTRA-INTIMAL thrombus o The fissure may form an INTRA-INTIMAL and INTRA-LUMINAL thrombus An intra-luminal thrombus may in turn form a complete OCCLUSIVE thrombus, which leads to myocardial infarction 95 LSS Cardiovascular System Alexandra Burke-Smith Hypertension CVS 19 - Professor Alun Hughes (a.hughes@imperial.ac.uk) Recognize that: 1. Blood pressure levels are continuously distributed in a population 2. Blood pressure in individuals is associated with increased risk of cardiovascular disease - particularly strokes, heart attacks and heart failure 3. The definition of hypertension is arbitrary and is based on the balance of the risks of elevated blood pressure versus the risks of investigation and treatment 4. Established (essential/primary) hypertension is due to elevated peripheral vascular resistance 5. The increase in peripheral resistance in hypertension is due to active and structural narrowing of small arteries and loss of capillaries (rarefaction) 6. 90-95% of cases of hypertension have no identifiable cause – this is termed primary or essential hypertension 7. Secondary hypertension is rare, but important causes include renal disease, oral contraceptive use, tumours secreting aldosterone (Conn's syndrome), and tumours secreting catecholamines (pheochromocytoma) 8. The cause of essential/primary hypertension is unknown but the strongest evidence implicates renal abnormalities and/or excessive activity of the sympathetic nervous system Epidemiology of Hypertension Affects ~1 billion worldwide BP distribution is unimodal and any distinction between normal and abnormal is arbitrary Hypertension definition: the level of blood pressure above which investigation and treatment do more good than harm Prevalence of hypertension increases with advancing age o At young ages, the prevalence was higher in males than in females; from age 60, however, the trend was reversed, with prevalence higher in women than in men. o Pulse pressure also rises with age, therefore the majority of people >60 yrs are expected to be hypertensive; by current definitions o The reasons for gender differences in BP are not known, although it has been suggested (but not proven) that estrogen may be responsible for lower BP in younger women There is an association between hypertension and increased risk of stroke o However there is no threshold for BP risk BP causes disease in the whole population, not jst hypertensive individuals o 50% of the attributanle burden of hypertension falls to the left of the solid line on the graph opposite (yellow half of graph) Pathophysiology of Primary Hypertension 96 LSS Cardiovascular System Alexandra Burke-Smith Classification Primary hypertension – 90-95% of cases o Also known as essential hypertension Secondary hypertension – 5% of cases o Identifiable causes Renal disease, including renal artery stenosis Tumours secreting aldosterone (Conn’s syndrome) Tumours secreting catecholamines (pheochromocytoma) Oral contraceptive pill Pre-eclampsia (pregnancy associated hypertension) Rare genetic causes, e.g. Liddle’s syndrome Aetiology Genetics – twin and other studies suggest 30-50% of variation in blood pressure is attributable to genetic variation - Monogenic (Rare) – causes <1% of hypertension o Liddle’s syndrome – mutation in amiloride-sensitive tubular epithelial Na channel o Apparent mineralocorticoid excess – mutation in 11beta-hydroxysteroid dehydrogenase - Complex polygenic (common) o Multiple genes with small effects o May have appositive or negative effect o Interaction with sex, other genes, environment Environment - Dietary salt (sodium) o A low salt environment is associated with a decrease in blood pressure in both men and women - Obesity - Alcohol - Pre-natal environment (birth weight) - Pregnancy (pre-eclampsia) Haemodynamics BP = CO x PVR Typically, established hypertension is associated with: o Uniformly increased peripheral resistance o Reduced arterial compliance o Normal cardiac output o Normal blood and extracellular volume Elevated PVR is accounted for by: o Active narrowing of arteries – vasoconstriction o Structural narrowing of arteries – growth and remodelling o Loss of capillaries - rarefaction 97 LSS Cardiovascular System Alexandra Burke-Smith Candidate causes - Kidney Key role in BP regulation Relation to salt intake and reabsorption Sympathetic nervous system Evidence linking high sympathetic sympathetic activity to the development of hypertension Endocrine/paracrine factors There is inconsistent evidence, but some suggests a link to hypertension The Kidney The kidney exerts a major influence on BP – Guton’s concept of ‘infinite gain’ of renal sodium/water/BP regulation Impaired renal function or blood flow is the commonest secondary cause of hypertension (e.g. renal parenchymal disease, renal artery stenosis), Most monogenic causes of hypertension affect renal Na+ excretion Salt intake is strongly linked with blood pressures of human populations. Populations with low salt have low population blood pressures and no rise in BP with age. Animals with reduced renal Na+ handling (genetic or experimental) develop hypertension. Excess salt intake in many animals results in elevated blood pressure In rats hypertension can be ‘transplanted’ with the kidney, there is similar, though incomplete data, in man The major risks attributable to elevated blood pressure coronary heart disease stroke peripheral vascular disease/atheromatous disease heart failure atrial fibrillation dementia /cognitive impairment retinopathy increase in left ventricular wall mass (LVMI) and changes in chamber size thickened walls (hypertrophy) of large arteries and acceleration of atherosclerosis arterial rupture or dilations (aneurysms). This can lead to thrombosis or haemorrhage (e.g. strokes) retina illustrates microvascular damage in hypertension. There is thickening of the wall of small arteries, arteriolar narrowing, vasospasm, impaired perfusion and increased leakage into the surrounding tissue microvasculature damage – o reduction in capillary density impaired perfusion and increased PVR o elevated capillary pressure damage and leakage Renal dysfunction is common in hypertension (e.g. increased (micro)albumin excretion in urine). o Extreme (accelerated/malignant hypertension) is now rare but leads to progressive renal failure 98 LSS Cardiovascular System Alexandra Burke-Smith Pathophysiology of Heart Failure CVS 20 - Professor Peter Collins (p.collins@imperial.ac.uk) 1. 2. 3. 4. 5. Provide an up-to-date definition of heart failure Appreciate the epidemiology and prognosis for heart failure Explain in general terms the underlying pathophysiology producing heart failure List the symptoms and signs of heart failure and appreciate suitable investigations to assist in its diagnosis Appreciate the principles of current treatments Overview Origins of Heart failure the heart is unable to maintain an appropriate blood pressure without support Evolutionary origin – the body response similar to that of exercise and haemorrhage Definition: a clinical syndrome caused by an abnormalilty of the heart and recognised by a characteristic pattern of haemodynamic, renal, neural and hormonal responses Epidemiology Prevalence 1 - 3 %; 10% in those over 75 years Incidence 0.5 - 1.5 % per annum Prognosis worse than cancer. 50% dead in 3 y In community mean age 76 y. Men: women is 50:50 5% of acute hospital admissions and 10% bed occupancy 40% readmission rate in one year 40% of hospital admissions mortality in one year Approximately 2% of total health budget Signs and symptoms - - Symptoms – subjective, expressed by patient Ankle swelling Exertional breathlessness Fatigue Orthopnoea PND Nocturia Anorexia + Weight loss Signs – objectives, observed by doctor CLINICAL (O/E) o Tachycardia o Decreased pulse volume o Pulsus alternans o Increased Jugular venous pressure o Pitting Oedema o Rales o Hepatomegaly o Ascites INVESTIGATIONS o X-ray 99 LSS Cardiovascular System o o o o o Alexandra Burke-Smith Echocardiogram Radionuclide ventriculography Ambulatory ECG monitoring Exercise test (VO2) Cardiac catheter Nature of heart failure Patient is breathless, tired and retains fluid Heart is damaged Heart less effective as a pump Marked neurohormonal activation Quality of life is poor Life expectancy reduced Cardiac X-ray comparison Normal X-ray ↓ Abnormal X-ray ↓ The NYHA Classification of Functional Capacity 100 LSS Cardiovascular System Alexandra Burke-Smith Progression Following onset, there is a relatively stable progression, where acute coronary events or sudden death may occur. Following this progression, deterioration from mild to severe heart failure occurs – eventually leading to death Syndromes of heart failure Entity – synonym/variant Acute heart failure – pulmonary oedema Circulatory collapse – cardiogenic shock (poor peripheral perfusion, oliguria, hypotension) Chronic heart failure – untreated, congestive, undulating, treated, compensated Causes of heart failure General causes Arrythmias Valve disease Pericardial disease Congenital heart disease Myocardial disease Myocardial diseases Coronary artery disease Cardiomyopathy o Dilated (DCM) o specific or idiopathic (IDCM) o hypertrophic (HCM, HOCM, ASH) o restrictive o arrhythmic right ventricular caropmyopathy (ARVC) Hypertension Drugs o Beta-blockers o Calcium antagonists o Anti-arrhythmics Other/unknown Heart Disease Coronary heart disease is the leading cause of death in Europe Modern treatment increases survival Survivors are left with a damaged heart 50% of all survivors develop heart failure Deaths due to heart attacks are declining but due to heart failure are increasing The population is ageing and heart failure is commoner in old age Only a minority of patients with heart failure are receiving the latest drugs 101 LSS Cardiovascular System Alexandra Burke-Smith Myocardial infarction Of the left ventricle Initially – baseline area at risk before infarction Within hours – infarct expansion Within days – left ventricular expansion due to thinning of wall Thinning due to cell slippage, hypertrophy, loss of cells and fibrosis Cardiomyopathy Definition: heart disease in the absence of a known cause and particularly coronary artery disease, valve disease, and hypertension Cause of approx 5% of heart failure in a population Types: o Hypertrophic cardiomyopathy (HCM) o Dilated cardiomyopathy (DCM) o Restrictive cardiomyopathy o Arrhythmic right ventricular cardiomyopathy (ARVC) Causes of dilated CM Idiopathic dilated cardiomyopathy Genetic and/or Familial cardiomyopathies Infectious causes o Viruses & HIV o Mycobacteria o Rickettsia o Fungus o Bacteria o Parasites Toxins and poisons o Ethanol o Metals o Cocaine o Carbon dioxide or hypoxia Drugs o Chemotherapeutic agents o antiviral agents Metabolic disorders o Nutritional deficiencies and endocrine diseases Collagen disorders, autoimmune cardiomyopathies Peri-partum cardiomyopathy, neuromuscular disorders Causes of restrictive CM Associated with fibrosis o Diastolic dysfunction – Elderly Hypertrophy Ischaemia Scleroderma Infiltrative disorders 102 LSS Cardiovascular System Alexandra Burke-Smith o Amyloidosis o sarcoid disease o inborn errors of metabolism o neoplasia Storage disorders o Haemochromatosis and haemosiderosis o Fabry disease o glycogen storage disease Endomyocardial disorders o Endomyocardial fibrosis o hypereosinophilic syndrome o carcinoid, metastases, radiation damage Causes of death Progression of heart failure o Increased myocardial wall stress o Increased retention of sodium and water Sudden death o Opportunistic arrhythmia o Acute coronary event (often undiagnosed) Cardiac event e.g. myocardial infarction Other cardiovascular event e.g. stroke, PVD Non cardiovascular cause Hormonal mediators in heart failure - Constrictors Noradrenaline Renin/angiotensin II Endothelin Vasopressin NPY Dilators ANP Prostaglandin E2 & metabolites EDRF Dopamine CGRP Growth factors Insulin TNF alpha Growth hormone Angiotensin II Catecholamine Nitric oxide Cytokines Oxygen radicals 103 LSS Cardiovascular System Alexandra Burke-Smith Inflammatory markers and cytokines involved All cell types Interleukin-1b Interleukin-6 Tissue necrosis factor alpha Heart Troponin T Troponin I Vessel wall ICAM-1 VCAM-1 E-selectin P-selectin Macrophages Lipoprotein-associated phospholipase A2 Secretory phospholipase A2 Liver C-reactive protein Fibrinogen Serum Amyloid A Adipose Management and treatment of heart failure Management algorithm for heart failure Establish that patient has heart failure Determine aetiology of heart failure Identify concomitant disease relevant to heart failure Assess severity of symptoms Predict prognosis Anticipate complications Choose appropriate treatment Monitor progress and tailor treatment Objectives of treatment Prevention o Of myocardial damage – occurrence, progression and further episodes o Reoccurrence – symptoms, fluid accumulation, hospitalisation Relief of symptoms and signs o Eliminate oedema and fluid retention o Increase exercise capacity o Reduce fatigue and breathlessness Prognosis o Reduce mortality 104 LSS Cardiovascular System Alexandra Burke-Smith Management Eliminate causes and precipitating factors - Limit/avoid alcohol Prevent myocardial ischaemia -Treat hypertension Prevent paroxysmal arrhythmias - Reduce salt intake Check drugs prescribed and taken - Assess lipids Encourage exercise - Flu immunisation Advice on lifestyle - Discourage smoking Treatment options Drugs o Diuretics o Beta-blockers o ACE inhibitors o Aspirins, statins, anticoagulants o Ang II receptor inhibitors, nitrates, hydralazin o Antiarrhythmics, IV inotropes Surgery, CABG or valve surgery Implantable cardioverter-defibrillator Maemofiltration, peritoneal dialysis or haemodialysis Aortic balloon pump, ventricular assist devices, cardiomyoplasty, volume reduction, transplantation Treatment of severe heart failure - IV drugs Diuretics Nitrates Positive inotropes Fluid control Haemofiltration Peritoneal dialysis haemodialysis Devices ICD or pacing Intraaortic balloon pump Ventricular assist device Total artificial heart Surgery CABG for “hibernation” Valve surgery Cardiomyoplasty Volume reduction/restriction transplantation 105 LSS Cardiovascular System Alexandra Burke-Smith Practical 1 – Measurement of Systemic Arterial Blood Pressure Background information BP is a measurement routinely made for diagnostic purposes The pressure that is measured is pulsatile, having its maximum value during ventricular systole and its minimum immediately prior to the next cardiac systole Determined by physical factors (e.g. elasticity of arterial walls) and the physiological factors; stroke volume, heart rate + vascular peripheral resistance Learning objectives 1. Obtain an accurate measurement of systolic and diastolic blood pressures using a sphygmomanometer and stethoscope and state the values. 2. Explain how methodological factors can affect the accuracy of measurement of arterial blood pressure and take appropriate precautions to obtain “correct” values 3. Explain the principles of the methods used Method Can be measured directly by inserting needle/catheter attached to a pressure transducer into a peripheral – however this is invasive, and must be under aseptic conditions Sphygmomanometer – consists of an inextensible material cuff containing an inflatable bag o The cuff is wrapped around an extremity so that the inflatable bag lies between the cuff and the skin (directly over the artery to be compressed) The pressure in the cuff is raised until the artery is occluded, and then released at a rate of 2-3mmHg per second – as this occurs a series of Korotokoff sounds can be detected using a stethoscope placed over the artery Two assumptions are made: 1. The pressure in the limb tissue under the centres of the cuff is the same as that in the bag 2. The artery offers no resistance to collapse by external pressure This means that: o When pressure in bag > systolic BP – flow of blood prevented o When pressure in bad is between systolic and diastolic BP – blood flow periodically o When pressure < diastolic BP – flow of blood continuous Systolic pressure = transition from no flow to periodic flow (heard by stethoscope) Auscultation as the pressure drops: o Nothing o Series of taps (phase 1) o Murmur sound (phase 2) o Loud banging (phase 3) o Sudden softening/muffling (phase 4) o Cessation of all sound (phase 5) Systolic pressure corresponds to the start of phase 1, diastolic pressure to the start of phase 5 106 LSS Cardiovascular System Alexandra Burke-Smith Practical 2 – The Electrocardiogram Learning Objectives 1. Be familiar with the normal ECG waveform 2. Describe the precautions and conventions employed in recording the standard 12-lead human ECG 3. Know how the recordings of the six standard limb leads are obtained from the four electrodes attached to the limbs 4. Know how the recordings of the sex pre-cordial (chest) leads are obtained Why can we record ECGs? First, consider the heart as a triangular wedge of excitable cells in a large dish of saline with two electrodes placed at a distance from each end. An oscilloscope can then be used to trace the potential across this wedge. Initially, the muscles are in their resting state and although each cell has a resting membrane potential, there is no potential between the extracellular electrodes When the cells closest to electrode 1 depolarise, their membrane potential is reversed creating a wave of depolarisation, which acts as a dipole between the depolarised cells and those in their resting state. As a result electrode 2 becomes negative relative to electrode 1, resulting in an upward deflection Provided the electrodes are fixed, the magnitude of this deflection is determined by the voltage of the dipole itself. This is determined by the demand for depolarising current, which depends on the conduction velocity towards electrode 2 and the fact that the width of the wedge is increasing When all the cells are depolarised, the dipole ceases to exist no potential difference between the electrodes A wave of repolarisation then starts at those closest to electrode 1, which creates another dipole but with the poles reverse resulting in a downward deflection At the end of repolarisation, there is no potential difference. The deflection caused by repolarisation is longer but smaller than that caused by depolarisation. This is because the time course of repolarisation of myocardial cells is much longer than that of depolarisation. NB: the size + direction of the deflection recorded depends on the angle between the axis of the recording electrodes + the direction of the depolarising wave, therefore the 6 limb leads show different tracings. Remember that in this model, the wave of depolarisation is assumed to be going from electrode 1 towards electrode 2. Limb Leads Right foot – zero reference point Electrodes placed at right arm, left arm + left foot – assumed to form an equilateral triangle known as Einthoven’s triangle, with the heart lying in the centre Remember these leads are all in the same plane, therefore only record the electrical activity in this (frontal) plane 107 LSS Cardiovascular System Alexandra Burke-Smith Standard limb leads The three electrodes are connected to form bipolar electrodes. By convention the apparatus is connected to that when a wave of depolarisation moves towards the positive electrode, an upward deflection is recorded. Lead I RA LA (LA designated as positive) Lead II RA LF (LF designated as positive) Lead III LA LF (LF designated as positive) Remember any given wave of depolarisation and its derived vector will be seen from a different angle Augmented limb leads 3 additional leads designate one electrode as positive, and record the signal between this and the remaining two leads connected to form a negative electrode; situated halfway between the two connected points aVR RA (LA + LF) (RA designated as positive) aVL LA (RA + LF) (LA designated as positive) aVF LF (RA + LA) (LF designated as positive) This provides more angles from which the electrical signal can be recorded However again these are all on the same plane – to record the heart’s electrical activity from a plane at right to this using chest electrodes Chest leads (precordial) Firstly, the three standard limb leads are electronically connected together to form one indifferent negative electrode Then a unipolar positive exploring electrode is placed on the chest wall in 6 positions (labelled V1-V6 in Arabic numbers) Anatomical locations (NB: ICS = intercostal space) o V1 – right 4th ICS parasteneral o V2 – left 4th ICS parasteneral o V3 – left, midway between V2 + V4 o V4 – left 5th ICS, mid-clavicular line o V5 – left anterior axillary line on the same horizontal plane as V4 o V6 – left mid-axillary line The ECG waveform 1. P wave: the intitial depolarisation of the SAN and its spread across the atria 2. PR Interval: After the P wave, the recording returns to the baseline. There is then an interval caused by the delay in conjuction thorugh the junctional fibres and AVN 3. QRS complex: depolarisation of the septum and ventricles. R wave is the first upward wave of the complex. Any downward deflection after the Q wave which goes below the baseline is called the S wave 4. There is then another interval 108 LSS Cardiovascular System Alexandra Burke-Smith 5. T wave: repolarisation of the ventricles The limb leads look at the mean ventricular depolarisation on the mean frontal plane axis of the ventricles (towards the apex of the left ventricle; depends on the oritentation of the heart in the thorax and the relative muscle masses of the ventricles) from different angles This means that they will all show a different form of the ECG wave It is by examining these different waveforms that it is possible to estimate the MFPA of the ventricles The same considerations apply to the chest leads, which are all positive electrodes: o The first part of the ventricles is to depolarise the septum, which occurs from left to right o The second part is ventricular depolarisation, where again the left ventricle makes the greater contribution Lead V1 will therefor show a “right ventricular complex” – small septal depolarisation moving towards the electrode gives a small upward r wave, followed by a deep S wave caused by ventricular depolarisation Lead V6 will show a “left ventricular complex” – small q wave (septal depolarisation) followed by large upright R wave (ventricular depolarisation) Lead V3 will show a “transition zone” characterised by biphasic equipotential (waves are equal in magnitude but opposite in direction) The electrocardiograph Consists of two essential parts: o Amplifier – needed to magnify the very small potential differences produced at the surface of the body by the electrical activity of the heart o Recording system – gives a visual, permanent display of these recordings. Consists of a high-density thermal print head that records the ECG on special heat sensitive paper The international standards for the gain (amplification) and paper speed of an ECG: o Gain – a signal of 1mV = deflection of 10mm o Speed – 25mm per second, therefore large square=0.2s, little square=0.04s Normal ECG recording shown on next page Analysis ECG is normally analysed for: Rate – mean heart rate Conduction of excitation – considered in two phases: o PR Interval – measures time required for activity to propagate through atria, AVN and bundle of His o QRS duration – gives the time required for excitation to spread throughout the ventricles Duration of electrical systole – estimated by measuring the QT interval Comparison with Normal values - for PR, QS + QT interval Estimated mean frontal plan axis 109 LSS Cardiovascular System Alexandra Burke-Smith 110