Medical Device Product Verification and Validation
advertisement

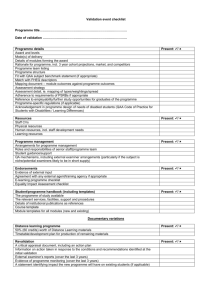
[ Device Validation Forum. John E. Lincoln Medical Device Product Verification and Validation John E. Lincoln “Device Validation Forum” discusses regulatory requirements, scientific principles, strategies, and approaches associated with medical device validation that are useful to practitioners. We intend this column to be a valuable resource for daily work applications. The key objective for this column: Useful information. Reader comments, questions, and suggestions are needed to help us fulfill our objective for this column. Please send your comments and suggestions to column coordinator John E. Lincoln at jel@ jelincoln.com or to journal coordinating editor Susan Haigney at shaigney@advanstar.com. dures [SOPs], and manufacturing process) from the development stage to the production floor (i.e., “technology transfer”) are major sources of product quality problems. The FDA’s design control and medical device current good manufacturing practice (CGMP) requirements (1) were designed, among other things, to reduce or minimize this risk. And they will, if they are properly and conscientiously applied. INTRODUCTION Figure 1: Product validation. This issue of the “Device Validation Forum” discusses the issues and considerations for successful verification and validation (V&V) of a new or substantially changed medical device. Product validation is different from process, equipment, or software validation, although any or all may be involved in a product’s development and included in a product’s design history file (DHF). Such product validation must address the US Food and Drug Administration’s design control elements of 21 CFR 820.30 (1, 2). These design elements are required by FDA to document the development and marketing of a new medical device. The purpose of product validation activities (see Figure 1) is to prove by a series of verification and test activities that the product’s design (input) requirements have been met by the design output (DO) and the resultant device. Next to the product design itself, change control and the transfer of product defining elements (i.e., the device master record elements— drawings, specifications, standard operating proce- For more Author gxpandjvt.com/bios 58 Journal The design verification requirements per 21 CFR820.30(f) are as follows: “Each manufacturer shall establish and maintain procedures for verifying User Needs, Standards, Guidance Verifications Design Review Design Input Design Review Design Process Design Review Design Output Design Review Finished Medical Device [ ABOUT THE AUTHOR John E. Lincoln is principal of J.E. Lincoln and Associates (www.jelincoln.com), a global consulting company with more than 29 years experience serving US FDA-regulated industries. He may be reached by e-mail at jel@jelincoln.com or by phone (toll free) at 888.882.4655. information, go to V&V DEFINITIONS of Validation T echnology [Spring 2010] iv thome.com John E. Lincoln. the device design. Design verification shall confirm that the design output meets the design input requirements. The results of design verification, including identification of the design, method(s), the date and the individual(s) performing the verification, shall be documented in the DHF [Design History File].” The design validation requirements per 21 CFR 820.30(g) are as follows: “Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate. The results of the design validation, including identification of the design, methods(s), the date, and the individuals(s) performing the validation, shall be documented in the DHF.” The design transfer requirements per 21 CFR 820.30(h) are as follows: “Each manufacturer shall establish and maintain procedures to ensure that the device design is correctly translated into production specifications.” THE NEW PRODUCT DEVELOPMENT SYSTEM New or substantially changed medical products need to be developed under design control procedures. Minor changes can be dealt with under a company’s change control procedures. The documentation supporting such changes may be pulled from laboratory notebooks, red-lined documents attached to change orders (e.g., drawings, schematics, specifications, etc.), or may be part of formal component product verification or test, research and development (R&D), engineering, validation protocols or test reports, and process or equipment protocols and test reports. Any change, minor or major, must be considered in light of its impact on completed or future V&V activities. SOPs must be developed as guidelines to assure certain minimum elements are addressed and included in test data to meet reproducibility and traceability concerns, and maintain adherence to the scientific method, as well as standardize history, documentation and product or process verification or validation requirements. Documentation should be prepared with sufficient detail so that a person having the requisite training or gxpandjv t.com experience could replicate the data or test using only the written documentation as a guide. INITIAL DESIGN INPUTS NECESSARY FOR PRODUCT DESIGN VALIDATION In order to prove that design inputs (DI) have been met by design outputs, a determination of the specific design inputs is the first obvious step after a general decision has been made to develop a new product. FDA requires that design control be instituted somewhere between the research and development stage. A helpful definition for such a start date is when pure research is shifted to purposeful development, often by the commitment of major dollars by senior management to commercialize a research project. Such commercialization can involve the development of a product to the point of the following: •In-house production and sale •Outsourced production •Sale or spin off of the prototype •Any combination of the above. Marketing is often the driving force for such product development, which can take the form of any of the following: •A new product •A substantially changed old product •A line extension to a family of products. With this formal “start” date, the development of product documentation, under design or change control, starts. This would include a formal design specification, which by definition would include design inputs and design requirements. The marketing or product manager may provide much of the initial design inputs, based on past product experience, and include additional information from focus groups and other consumer research. This is especially true of marketing needs based on competitive products, field requirements, and desires. Concurrent with initial product development is the development of at least a draft Product Hazard Analysis and Risk Management File and Report, per International Organization for Standardization (ISO) 14971 (3) and/or International Conference on Harmonisation (ICH) Q9 (4). This usually involves at a minimum a failure mode, effects, and criticality analysis (FMECA) for design, process and use, and any software (see reference 5 for a discussion of product risk management). Journal of Validation T echnology [Spring 2010] 59 Device Validation Forum. FLESHING OUT DESIGN INPUTS THE PRODUCT VALIDATION PLAN The engineering department will start adding industry standards requirements and adjust inputs to limitations to marketing requirements and the developing risk management file. These are based on what is “doable,” given the current state of the material and process “art,” company and/or outsourcing capabilities, and budget constraints. They will start on prototype development, models or “kludges,” drawings, schematics, specifications, materials, much of the labeling (including instructions for use), and do some feasibility testing, all with marketing, production and manufacturing engineering, and quality assurance (QA) and regulatory assurance (RA) input. Engineering will then determine the type of more formal or structured bench and functional testing, packaging and shipping testing, environmental testing, and manufacturing process and equipment testing and validation required, based on the product, its intended use, and use environment. This is often based on a product’s hazard analysis and risk management file and report, per ISO 14971 and ICH Q9. Engineering refers to applicable ISO, Association for the Advancement of Medical Instrumentation (AAMI), American National Standards Institute (ANSI), European Standards (EN) (6), and other standards. QA/RA will then add quality and regulatory issues and additional test requirements (i.e., biocompatibility and sterility) to the list, as required by the product, its intended use, and use environment and any applicable standards and/or guidance documents. They will review current good manufacturing practice (CGMP) issues for manufacture, controlled manufacturing, or clean room requirements, test procedures and equipment and sourcing, and human factors or use environment issues. They will then evaluate, assist, and/or actually prepare much of the product and process validation documentation, especially to ensure compliance to regulatory and product quality requirements. A project leader with a product development team will then drive the development of a formal product specification that addresses these realistic and achievable design inputs. These specifications will help drive the project per the milestones and tasks described in “Medical Device Development Under Design Control” in the Winter 2010 issue of the Journal of Validation Technology (2), including the periodic project design reviews and the developing V&V requirements. This list of product design inputs, specifications, and requirements now also becomes the basis for developing a product validation plan. The plan will address not only product validation but also the associated manufacturing process, production equipment, and test equipment V&V requirements. These include calibration, PM, spare parts, documentation, site preparation (part of installation qualifications [IQs]), design of experiments (DOEs) (often part of operational qualifications [OQs]), Cpk (process capabilities, centered), gauge repeatability and reproducibility (GRR), and similar. It will include timelines and departmental responsibilities. Time spent in careful up-front project planning and management will prevent key activities from not being considered early on, “dropping through the cracks,” and surfacing late in the project, if at all, delaying the project unnecessarily, and/or yielding a less than robust finished product. Some product V&V activities can start early on, such as material selection, material-to-material (including any bonding agent(s)) compatibility, material biocompatibility, hemolysis, cytotoxicity, sterilization V&V, etc. Other V&V activities have to wait until a viable prototype or a close to finished product has been developed. FDA correctly demands that functional, stability, and other such product testing and verification activities be performed on product as close to the final production version as possible, including being built by the process, equipment, and personnel involved in actual production. The less that is the case, the more the documentation must explain the differences and test compensations made with rationale, and the higher the risk of incomplete V&V data. While such stipulations are often viewed as burdensome by a product development team and even by management, a review of product recalls and a “reading between the lines,” indicates that failure to do so can lead to future recalls or other field problems and resulting liability issues. Companies have also taken a major financial hit due to developing products that can only be assembled by engineers or technicians, rather than the planned for production line operators, effectively pricing the product off the market. THE PRODUCT V&V PROTOCOL AND TEST REPORT The product validation protocol or test report should be a controlled document, having a test report num- 60 Journal of Validation T echnology [Spring 2010] iv thome.com John E. Lincoln. ber or protocol identification number assigned by QA or document control. Ideally, the calibration of process, production, and test instrumentation should be performed before and after the validation and be included in the V&V file. This is to ensure accuracy of readings that not only validate the product but usually are used to establish equipment parameters for production, as called out in SOPs, and also any on-unit labeling and the instructions for use (IFUs). The validation package should have pre-established and referenced acceptance criteria and both pre-approval and post-approval signature blocks. Lab notebooks and formal protocol or test reports should follow a defined format, such as the following suggested listing. SUGGESTED KEY PROTOCOL ELEMENTS The following is presented as one possible format and recommended sequence, as applicable, for the product validation protocol or test report file: •Protocol or test report number •Title •Approvals. A single signature may suffice for most routine tests or verifications; complex validations may require more reviews and approvals, including pre- and post-approvals. •Purpose. Brief statement or rationale for the protocol and reasoning as to its chosen development •Equipment. Listing of all non-consumable equipment, tools, instruments, fixtures, etc., used to test and run the protocol, including description, model, S/N, calibration numbers, and expiration dates •Material. Listing of all consumables used in the protocol. Include description, part number (P/N) or item/catalog number, lot number, sterile load number and quantity, etc. •Procedure. A step-by-step explanation or written narrative of the test and the actual test protocol (i.e., how to run the test), written in such a way that it could be duplicated by someone else with the necessary technical expertise but unfamiliar with the procedure; include any set-up drawings, instructions, sample check sheets or data sheets, etc. This step-by-step narrative should be as follows: •Product V&V. See Figure 1 and design control SOP for product V&V steps, to include bench tests, lab tests, and any animal or human clinical studies (IDE required). •Equipment or process V&V. Includes the following: gxpandjv t.com ✦✦Design qualification (DQ) (Optional). Formal documentation of criteria considered in the purchase and/or manufacture of a piece of equipment, often supplied with a purchase order ✦✦Installation qualification (IQ). Documented functional requirements of the equipment and how requirements were met during installation, often in checklist format (see design control SOP) ✦✦Operational qualification (OQ). V&V steps, test cases, or design of experiments (DOE) to establish the key operating parameters, settings, and their tolerance ranges, and verify their proper operation; may also include hazard awareness of critical control points (HACCP), Ishikawa (Fishbone) diagrams, flow charts, process maps, and similar tools ✦✦Performance qualification (PQ). Challenge process or equipment under validation, usually with a minimum of three runs, under “worst case” conditions (i.e., different shifts, different material lots, environmental, other varying inputs, etc.); verify reproducibility and repeatability of the equipment or process—its capability to consistently produce desired results; should be risk based (i.e., tied to a product risk document where possible) and include at least one “unplanned” power outage to see how process or equipment recovers or re-initializes. When several PQs are run (the old, de facto “standard” was a minimum of three runs; now the number should be based on the number of inputs and risks involved), the basic blank PQ may be copied and sequentially numbered prior to running and filling out (e.g., PQ 1 of 3, PQ 2 of 3, and PQ 3 of 3, and so on) ✦✦Software (product, process, equipment, and hardware) V&V. See also the company’s design control, or software V&V SOPs ✦✦Quality management system (QMS) software (and hardware) V&V. V&V to be performed under requirements of 21 CFR Part 11, “Electronic Records/Electronic Signatures” ✦✦Test case format. OQs and PQs should be performed using the suggested test case format shown in Figure 2. There may be numerous test cases under each key element of the process or equipment being validated. •Results. List the actual/proven test results, data, and reference the data sheets, check sheets, etc.— facts, not opinions Journal of Validation T echnology [Spring 2010] 61 Device Validation Forum. Figure 2: Suggested test case format. SUGGESTED TEST CASE FORMAT Generally used with OQs and PQs (IQs are usually in a check list format) other follow-on protocols, lab notebook test write-ups, deviations, ECOs/non-conforming material reports (NCMRs), rework instructions, or where required, to ensure proper device history, and reproducibility or repeatability of test data or the process. CHANGE MANAGEMENT DURING V&V #_. [Operation]: Risk: [describe operation and risk / rationale – tie to appropriate risk document, e.g., in Risk Management File / Report, per ISO 14971:2007]. Test Case: [brief operation description and test case verification step]. Verification Element Expected Outcome Observed Outcome [list specific item(s) within the operation that is to be tested] [list the expected results to be expected from the operational element(s) that is being tested] [describe the actual, observed result(s) from the operational element when it was run] Tested by: ___________________ Date: ______________ Verified by: __________________ Date: ______________ •Conclusions. State obvious and provable conclusions that can be drawn and proven from the data obtained, documented, and collated under the results section. Can include statistical tests and reasonable inferences (identified as such) from the data and results •Attachments (Optional). Copies of any supplemental info (e.g., SOPs used, references, manuals, additional calibration, test data, work sheets, sample documents, etc.) Copies of controlled documents should be stamped “Copy.” Merely referencing the document or copying it and including it in the validation package or file is a matter of preference, balancing ease of use of the validation file versus added bulk. Consideration should be given to the ability to retrieve archived documents years later when questions come up or an audit of the file is conducted. Simple test and verification reports may not require all the protocol elements. Some validation sections, such as IQs, may be handled by a checklist or analysis. Ensure that all information is documented in such a way that it can, years later, be understood, explained, defended (even in a court of law), and/or replicated, including the documents, methodology, and its data, results, and conclusions. The assigned protocol number is to be referenced in 62 Journal of Validation T echnology [Spring 2010] The management of product and/or process changes is always important, but especially during the V&V process. As mentioned, while change is initiated to improve upon some aspect of a product or the V&V process, it often introduces other problems. These are often undetected initially, or even throughout the remainder of the development and production process. The following are types of changes that may occur: •Minor changes. May be documented by retaining the existing protocol number and only adding the revision as an addendum, referencing the original protocol and number. At a minimum, a lab book study will be performed to ensure no process change has occurred; and a written rationale will be added to the documentation file or included in the lab book as to why no revalidation or recertification was deemed necessary. Define in a company SOP •Major changes. May require that a new protocol number be issued. If so, then any referencing documentation must be annotated or changed to reflect the new protocol number. This may also be the case when a too-early prototype product (or process) was used in a V&V activity, often to speed up the V&V process, but having the opposite effect. Again, define in a company SOP. Major product or process changes must be revalidated by formal protocol (not a lab book study) •Determination of what constitutes a major or minor process change is made by senior management, or designee, usually QA, and should be defined in a company SOP. CALIBRATION OF TEST INSTRUMENTATION All applicable gauges and instrumentation will be calibrated or re-calibrated prior to initiation of any V&V activity. Proof of such will be added to the V&V file. Instrumentation or gauges not requiring calibration (usually due to some downstream calibrated instruments or gauges) or verification steps should be labeled as such (e.g., non-calibrated instrument [NCI]), with an explanation as to rationale and referenced accordingly in the V&V file. iv thome.com John E. Lincoln. VALIDATION PROTOCOL TEST CASES APPROVALS A product V&V will include test data from independent labs, especially dealing with product biocompatibility, particulate, bioburden, and/or sterility issues. It may include test data from other labs as to electromagnetic compatibility (EMC) or ground leakage and other electrical data, as applicable to the specific product. However, most of the in-house portion of a V&V protocol, especially the OQs and the PQs involved in production and test equipment as well as product functional testing and much software testing if applicable, will be made up of a series of test cases. Generally test cases are derived from the specific elements needing testing, for example the following: •Human factors—intuitive; ease of use issues •Product start-up and initialization •Product functional tests, including purposeful “mistakes” •Product shutdown—orderly and un-planned •Software, if applicable •Product recovery from unintentional power outages, if applicable •Environmental (e.g., heat, cold, humidity cycling) •Sterilization process, residuals, if applicable •Accelerated aging •Shake and drop •Cross-country shipping •Product sterility (different from sterilization above). Pre-approvals, if required, can be obtained at any time during the test cycle. However, if the protocol is changed during the pre-approval cycle, any additional requirements must be addressed and added to the test requirements, with explanation, prior to routing the protocol for post-approval. The greater the risk or cost of changes in the protocol, the greater the need for consensus and support, and hence the stronger the argument for pre-approval prior to start. If the test method is non-controversial, the protocol is a repeat of a previously approved or agreed-to protocol, a recognized industry standard test, etc., or, if the purpose of the protocol is merely to gather data, is a preliminary test prior to the process-defining protocol(s), or test methodology is common for purpose intended to trained personnel, pre-approval may not even be necessary. In such a case, if additional testing is deemed necessary during the post-approval sign-off phase, it can be performed and added as an addendum. Post-approvals are completed once all aspects of the V&V test report are completed, including any changes. The completed protocol is routed to document control for filing and retention. Changed product or product produced by equipment or processes subject to verification, validation, or re-verification/revalidation must be quarantined and cannot be released until post-approval sign-offs are completed. The composition of the list of such tests is determined by the product itself, its use environment, FDA and Europe’s Medical Device Directive (MDD) requirements, applicable standards, and similar. For additional considerations, see the list in the “Device Validation Forum” column in the Journal of Validation Technology, Winter 2010 issue (2). Such test cases should include the following (see Figure 2): •A descriptive title •A brief description of the test being preformed •Risk justification. In risk-based V&V activities (and most should be risk based), the test case may reference the specific line item in a product risk management file, or FMEA, FMECA, and/or fault tree analysis (FTA) •The V&V element •The expected outcome •The actual observed outcome •Test and verification signatures and dates. gxpandjv t.com REVALIDATION Revalidation is required whenever a major change occurs to a product or process, or after moving any sensitive process equipment, unless a written extension or exception is granted, with rationale. Such rationale can be included as part of the original validation protocol. Consideration should be made as to whether or not it is to be company policy to re-verify or validate a product or process not on an arbitrary time-based interval (e.g., every year), but as defined by degree of changes, risk analysis, documented. This should be defined in the master validation plan and/ or SOP. CONCLUSION Product verification and validation activities should not be short changed. While there is usually tremendous pressure to get the new product out, the time and effort spent upfront in planning and executing Journal of Validation T echnology [Spring 2010] 63 Device Validation Forum. a product V&V project will greatly assist in setting up a robust manufacturing process and delivering a product that meets customer requirements, stated, unstated, and required by industry standards, while minimizing fall-off, scrap, rework, risks of recall, and other liability or warranty issues. Lessons learned from reviewing a company’s own product development and corrective action and preventive action (CAPA) history, the product’s risk management file, FDA adverse events data bases such as MAUDE for similar products’ problems, and even other industries’ (even non-medical) recall experiences, will assist in developing a product validation plan that provides such a robust manufacturing process and product. REFERENCES 1. FDA, 21 CFR 820, Quality System Regulation, sections 21 CFR 820.70, 820.75, and 820.170. 2. Lincoln, John E., “Medical Device Development Under Design Control,” Journal of Validation Technology, Vol. 16, No. 1, Winter 2010. 3. ISO, ISO 14971:2007, Medical Devices—Application of Risk Management to Medical Devices. 4. ICH, Guidance for Industry, Q9 Quality Risk Management, June 2006. 5. Lincoln, John E., “Product Risk Management Under ISO 14971:2007,” Journal of Validation Technology, Vol. 15, No. 4, Autumn 2009. 6. EC, Council Directive 93/42/EEC of 14 June 1993 Concerning Medical Devices, amended by M1-M5, latest dated 21.9.2007. JVT 64 Journal of Validation T echnology [Spring 2010] ARTICLE ACRONYM LISTING AAMI Advancement of Medical Instrumentation ANSI American National Standards Institute CFR Code of Federal Regulations (US) CGMP Current Good Manufacturing Practice DHFDesign History File DIDesign Input DODesign Output DOEDesign of Experiments, or Designed Experiments DQDesign Qualification ECOEngineering Change Order EMCElectromagnetic Compatibility ENEuropean Standards FDAUS Food and Drug Administration FMEAFailure Modes, Effects Analysis FMECAFailure Modes, Effects, and Criticality Analysis FTAFault Tree Analysis GMPGood Manufacturing Practice GRRGauge Repeatability and Reproducibility ICHInternational Conference on Harmonisation IFUsInstructions for Use IQInstallation Qualification ISOInternational Organization for Standardization MDDMedical Device Directive (EU) NCINon [or Not] Calibrated Instrument NCMRNon-Conforming Material Report OQOperational Qualification PMPreventive Maintenance P/NPart Number PQPerformance Qualification QAQuality Assurance QMSQuality Management System QSQuality System(s) R&DResearch and Development SOPStandard Operating Procedure V&VVerification and Validation iv thome.com