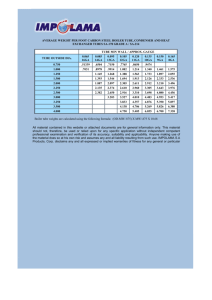
lab-book-sem-2 copy
advertisement

Modern Physics II Lab Manual University of Puget Sound Spring Semester 2011 2 Contents 1 Photelectric Effect 1.1 Procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.2 Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.3 Questions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 7 8 10 2 Millikan Oil Drop Experiment 11 2.1 Theory . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 2.2 Experiment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14 2.3 Writeup . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 3 Spreadsheet Energy Levels 17 3.1 Harmonic Oscillator . . . . . . . . . . . . . . . . . . . . . . . . . 19 3.2 The Finite Square Well . . . . . . . . . . . . . . . . . . . . . . . 21 3.3 A Triangular Potential . . . . . . . . . . . . . . . . . . . . . . . . 22 4 Measurement of e/m for the Electron 4.1 Doing the Experiment . . . . . . . . . 4.2 Analysis . . . . . . . . . . . . . . . . . 4.3 Writeup . . . . . . . . . . . . . . . . . 4.4 Hooking up the e/m Power Unit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 27 28 30 31 5 Spectroscopy 33 5.1 Calibration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 5.2 Hydrogen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35 5.3 Other spectral series . . . . . . . . . . . . . . . . . . . . . . . . . 36 6 X-Ray Diffraction 6.1 Procedure . . . . . . . . . . . . . . . . . 6.2 Measuring the Kα and Kβ wavelengths 6.3 Measurement of bond length . . . . . . 6.4 Analysis and Report . . . . . . . . . . . 6.4.1 Peak Angle Determination . . . . 3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37 39 40 41 42 42 4 CONTENTS 7 Radioactivity 7.1 Finding the Optimum GM Tube Voltage . . 7.2 Measuring the Background Count Rate . . 7.3 Determining the GM Tube Resolving Time 7.4 Measuring the Half-Life of 137 Ba . . . . . . 7.5 Qualitative Properties of α Radiation . . . 7.6 Beta Radiation . . . . . . . . . . . . . . . . 7.7 Gamma radiation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 46 47 48 49 51 51 52 8 Nuclear Spectroscopy 8.1 Operation and Calibration . . 8.2 Energy Scale and Resolution 8.3 Compton Scattering . . . . . 8.4 Using Gamma Spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 55 56 57 59 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . A Review of Error Analysis 61 A.1 The error from the statistical error on the mean . . . . . . . . . . 61 A.2 Error propagation . . . . . . . . . . . . . . . . . . . . . . . . . . 62 A.3 Combining several measurements of the same quantity . . . . . . 64 Laboratory 1 Photelectric Effect Equipment PASCO Photoelectric Effect Apparatus, including: − set of filters and apertures − Hg light source − mounting base rail − photodiode enclosure − power supply − current monitor and amplifier − PASCO experiment guide for photoelectric effect. Albert Einstein’s elucidation of the photoelectric effect (1905), together with Max Planck’s theory of blackbody radiation (1901) form the two seminal experiments that heralded the quantum theory. Heinrich Hertz had already observed in 1887 that, under certain conditions, light shined on metallic surfaces would result in the ejection of electrons, in amounts sufficient to constitute a measurable “photocurrent.” Philip Lenard carried out studies by 1902 that established the basic properties of the effect. Some of these properties conflicted with the predominant understanding of light up to that time – the wave theory. It was Einstein’s breakthrough view of light coming in quantized packets called photons, with energy that is directly proportional to the frequency, that was able to fully account for all properties of the effect. What you will be doing in this laboratory session is not very complicated – however it is very important that you understand the ramifications of the measurements youll be taking. There are three key properties of the photocurrent: 1. When a photocurrent is present, due to shining the “right kind” of light, it is proportional to the intensity of the light source. The brighter this light, the stronger the current will be. 2. The photocurrent appears instantly after the light is turned on – there is no time delay. 3. If the wavelength of the light falls below a certain minimum frequency then no photocurrent is obtained, regardless of the intensity. 5 6 LABORATORY 1. PHOTELECTRIC EFFECT The second property, and especially the third conflict with the wave theory of light. How can photoelectrons resist being emitted if sufficient power is delivered to the metal? In fact if the frequency of the light is below the threshold for emission of a photocurrent, the metal can absorb sufficient energy to melt into a pool of liquid, and yet no photoelectrons will be observed! Einstein’s new view of light suggested that, at the atomic level, light was interacting with the metal atoms on a quantum-by-quantum basis. Each light quantum, or photon has energy equal to: E = hν = hc , λ (1.1) where ν is the frequency of the light (in oscillations per second, or Hz), λ is its wavelength, and h is a constant of quantization called Plancks constant, that you will be measuring in this lab. Einstein considered the electrons of the metal atoms to have a certain minimal “binding energy,” called the “work function,” φ, which had to be delivered by an incident photon in order to liberate an electron. Once this minimum energy is “paid,” anything left over goes into kinetic energy of the free electron. Consider a metal sample that has been curved into a semicircular “chute” which is concentric with a “collector” wire that gathers electrons incident upon it, as illustrated in Figure 1.1. Light that is incident on this chute will eject electrons in all directions with respect to the inside surface. If an electron happens to emerge at nearly normal incidence there is a good chance it will make it to the center rod, where it contributes to a measurable photocurrent. If a potential difference is applied between the rod and the metal chute in such a polarity as to deter the electrons from the rod, only those electrons that are sufficiently energetic and that emerge from the chute surface heading almost exactly for the rod will make it there. And if the potential difference becomes too large, no electrons will have enough energy to register a current. Recall that a charge q accelerated through potential difference ∆V acquires kinetic energy K = q∆V , so the kinetic energy imparted to the photoelectrons by light of a single frequency can be measured by the maximum amount of opposing potential difference the photoelectrons are capable of overcoming as they move from the atom of the chute that they have been liberated from, to the “collector” rod, or anode.1 In this experiment you’ll be measuring this maximum opposing potential, or “stopping voltage,” V0 , as the amount of reverse-bias voltage that just causes the current in the photocell to turn off. This is a challenging measurement because the current can drop to extremely small levels (pico-Amperes) before it truly goes to zero. After all, as the stopping potential is approached it takes a very “lucky” electron - one that heads EXACTLY from the cathode to the anode - to overcome the reverse-bias potential. 1 The term “anode” refers to an electrode or a surface from which conventional current flows, and “cathode” refers to the electrode/surface that conventional current flows to. Because the electrons are negatively charged this makes the rod the anode. It also explains why, historically, beams of electrons were once referred to as “cathode rays” since they always emerge from the cathode. 1.1. PROCEDURE 7 incident light V – cathode + A anode (collector) photoelectron Figure 1.1: The phototube consists of a metal chute (cathode) from which electrons are ejected by the photoelectric effect, and a collector rod (anode) that is concentric with the chute. A reverse-bias voltage ∆V is applied that repels electrons from the rod. The number of electrons per unit time that nonetheless manage to reach the collector comprise the photocurrent measured by the ammeter, A. The photoelectric effect can be described by the Einstein equation, which expresses the kinetic energy of the liberated electron at the stopping potential as the difference between incoming photon energy and the work function: eV0 = hν − φ , (1.2) where e is the charge of the electron. Since the frequency and wavelength of light are related by c = νλ, this can be rearranged as: hc 1 φ − . (1.3) V0 = e λ e This equation suggests the overall strategy of this lab: you can measure the work function, φ, and also Planck’s constant, h (taking the charge of the electron and the speed as light as given) by measuring the stopping voltage as a function of inverse wavelength, for several different wavelengths. The sources for these wavelengths will be a low-pressure mercury vapor lamp, which emits prominently at five different wavelengths (including one ultraviolet line). Color filters are used to select individual wavelengths. 1.1 Procedure The self-contained apparatus comes with a mercury light source mounted on a rail and a receptor unit which has a built-in phototube containing a rounded chute (cathode) and a rod that runs down the center of the chute that collects the photocurrent (anode). Power supply and current monitoring units are connected 8 LABORATORY 1. PHOTELECTRIC EFFECT as described in the PASCO guide (note well the color coded cables). The current monitor greatly amplifies the photocurrent, while introducing relatively little noise – at its most sensitive setting the output reading is in units of 0.1 pA (10−13 A)! The apparatus allows you to apply voltages that are either reversebiased or forward biased. The three investigations you will carry out are outlined in detail in the PASCO guide, on pages 9–19: 1. Measurement of h from the dependence of V0 vs. 1/λ. You should do this for each one of the three different apertures. 2. Measurement of current vs. voltage for same frequency, different intensities (by using different-sized apertures). You can do this for any one of the wavelengths of mercury light given in Table 1.1 that you choose (not just the 436 nm line suggested in the PASCO writeup), using three different aperture sizes. 3. Measurement of current vs. voltage for different frequencies at constant intensity. Use the intermediate (4 mm) aperture and take data for the three visible lines of mercury: blue, green and orange. You will be writing up your results in report form IN LATEX, with one report for each group. You and your partner will share the grade for your report – make sure you cooperate on all aspects of data taking, analysis, and writing. Include a description of the theory of the photoelectric effect, a description of the apparatus and be clear in presenting your data and how you completed the analysis and error analysis. Guidelines for what to make sure to include for each investigations are given in the next section. Mercury produces a variety of spectral lines, including several lines that lie very close to each other (see, e.g., C. J. Sansonetti, M. L. Salit and J. Reader in Applied Optics 35 (1996) 74-77; or the CRC Handbook of Chemistry and Physics). Two of these are in the ultraviolet range - bleached white copier paper can reveal these lines, but the filters that come with the apparatus are very well attuned to these wavelengths. For our purposes well take the wavelengths of mercury as being in five separate groups: 1.2 Analysis Determine Planck’s constant from the slope of a linear fit through your data points from Experiment 1, and show the fitted line on a plot of V0 versus (1/λ). You can incorporate individual errors on V0 at each wavelength (which may differ from each other) by performing a weighted linear least–squares fit (formulas for this can be found in the appendix). This fit will provide you with the best-fit slope and intercept values and errors on those values. The slope and intercept, and the errors on them, translate directly into values and errors for h and φ. The value of φ you obtain will most likely not correspond to the work function of any known metal. Its value will be too small because when extremely small 1.2. ANALYSIS 9 line label UV 1 UV 2 blue green orange wavelength (nm) 365.22 404.93 435.74 546.1 578.15 2 Combination of four lines at 365.016 nm (relative intensity = 2800), 365.484 nm (300), 366.289 nm (80) and 366.328 nm (240). 3 Comb. of two lines at 404.657 nm (rel. int. = 1800) and 407.784 nm (150). 4 Comb. of two lines at 434.751 nm (rel. int. = 400) and 435.854 nm (4000). 5 Comb. of two lines at 576.961 nm (rel. int. = 240) and 579.067 nm (280). Table 1.1: Dominant spectral lines of mercury in the visible light range. currents are measured in an electrical circuit, there is some “interference” that occurs at every junction point between two metals that have different work functions (including wires, circuit traces - everything!) These differences in work functions are called “contact potentials”, or “Galvani potential differences” and they directly affect the measured value of stopping voltage. In order to ascertain the net contribution of the contact potentials in our circuit we would have to do more investigative work that is beyond the scope of this laboratory. Determine a value of h, and its error, from the data taken with each of the three apertures. Discuss the consistency of your values of h with the established value, 6.63 × 10−34 J · s. Is there any bias or inconsistency in your result? If so, do you understand what could have caused it? It is nice to put these on a plot of h versus aperture size, including a horizontal dashed line for the standard value of h. Take care in quoting values and errors. A common offense in dealing with errors is to cite too much precision. E.g., the statement h = 6.799 × 10−34 ± 5.99421 × 10−35 J · s NOT GOOD is 1) hard to read, 2) has differing precision between central value and error, and 3) contains excess significant figures. Use the error to guide you in the number of significant figures quoted (keep AT MOST two significant figures in the error), and write the final result in a way that makes the answer, and the error as clear and easy to interpret as possible: h = (6.80 ± 0.60) × 10−35 J · s MUCH BETTER For Experiment 2 include a single plot showing the current vs. voltage for each one of the three apertures, 2 mm, 4 mm and 8 mm. Discuss the behavior in this plot, comparing the similarities and differences in the three series. How is this behavior seen in the plot consistent/inconsistent with the wave theory of light? How is it consistent with the photon theory? You may want to “blow up” the area around the convergence point to address this. 10 LABORATORY 1. PHOTELECTRIC EFFECT For Experiments 3 include a single plot of showing the current vs. voltage for each one of the visible wavelengths from Table 1.1. Discuss the behavior you see in this plot, including which aspect(s) cannot be explained by the wave theory of light, and how the photon theory succeeds in explaining them. 1.3 Questions In your lab writeup discuss the following questions about the photoelectric effect and its significance in the history of physics: • What aspects of this phenomenon conflict with the wave theory of light, and how is this conflict resolved by the quantum theory proposed by Einstein? • What aspects of this phenomenon are consistent with the wave theory of light, and how are they consistent? Keep in mind the significance of what this experiment establishes. It is a fundamental paradigm shift in our picture of the universe, that light is composed of tiny packets called photons. It is NOT composed of a continuous waves. We almost never see any effects of the “granularity” of light because h is so small. Yet it is a fundamental fact that it is photons, and not continuous light waves, that interact with atoms. This is the essence of quantum electrodynamics, one of the most successful physical theories of the 20th century. Laboratory 2 Millikan Oil Drop Experiment Equipment PASCO Millikan experiment high voltage supply (500 V) two digital multimeters banana plug cables thermometer (to read room temperature) ringstands, to elevate apparatus “I have never begun a laboratory with more misgiving...” might be an apt paraphrase of the opening line of W. S. Maugham’s The Razor’s Edge appropriate for this lab. While it is central to the history of physics, and very simple in concept, this lab is challenging. If you care about detail, about quality and about how the measurements in your lab turn out you will do well at this. The experiment is a close rendition of the one performed by Robert A. Millikan over the course of several years in the beginning of the twentieth century, to determine the charge of the electron. Youll be using a commercially available unit made by Pasco Scientific (Model AP-8210), which is a great advance over previous editions of the experiment that sometimes left students (and instructors) very frustrated. In fact the advertisement for this Pasco apparatus claims “Typically, a careful student can achieve results within 3% or less of the accepted value.” The key word here, and with so many endeavors in life is “careful.” A copy of the essential pages of the Pasco guide for this experiment will be provided to you (pages 1-9, and 19 and 20), which includes a derivation of the equations for converting rising and falling velocities into charges on the droplets. I’ll go through the essential elements of this theory in the next section, but you should expand on these in your writeup to give a complete derivation. Extra suggestions for executing the lab are also given below, as well as systematic error considerations and a discussion of other essential elements to include in your writeup. 11 12 LABORATORY 2. MILLIKAN OIL DROP EXPERIMENT IMPORTANT: It is essential that you read the guide material from Pasco and this handout before coming to do this lab. This will increase your efficiency in the lab and satisfaction in doing the experiment. 2.1 Theory The experiment depends on careful measurement of the time it takes for a small charged droplet of oil to fall under gravity only – no electric field applied, and the time it takes for the same droplet to rise under the combined effects of upward electric force and downward gravitational forces. The first case is characterized by the “falling velocity,” vf , and the second by the “rising velocity,” vr . Falling velocity gives information about how big the droplet is. According to Stokes’ Law, for very small droplet velocities (much less than 1 cm/sec) the drag force due to the viscosity of air is linearly proportional to the velocity, v and to the droplet radius, a, according to: Fdrag = 6πη0 av . (2.1) In this equation, η0 is called the viscosity coefficient and it has units N·s/m2 . When the droplet is falling at constant terminal velocity vf , the upward drag force balances the droplet’s weight so the net force on it equals zero, and we can say: mg = 6πη0 avf . (2.2) The viscosity coefficient η0 depends on the air temperature according to the plot given in Appendix A on page 19 of the PASCO writeup. In order to monitor the temperature (which increases slightly, the longer the light bulb is turned on), a thermistor is installed in the chamber’s lower plate. Record its resistance at regular intervals as you do the measurement, so that you can make the proper adjustment to the viscosity coefficient. Charged droplets can be made to rise by applying a voltage of the correct polarity across the two chamber plates (droplets from the atomizer may be neutral, or positively or negatively charged). An approximately-uniform electric field of strength E is created, that can move the droplet of charge q upward. A new terminal velocity when rising, vr , is reached when the sum of the electrical, gravitational and drag forces is again zero: 0 = qE − mg − 6πη0 avr or: qE = mg + 6πη0 avr . (2.3) Elimination of the constants 6πη0 a between equations 2.2 and 2.3 gives the charge on the droplet: q= mgd (vf + vr ) mg (vf + vr ) = , Evf vf V (2.4) 2.1. THEORY 13 where the electric field strength between the upper and lower chamber plates is related to the distance of separation, d and voltage difference, V between them by: Ed = V . This simple equation is the root of the measurement. All quantities on the right-hand side are known, or measured except for the droplet mass, m. The rest of the derivation involves finding the droplet’s mass from its falling velocity in equation 2.2 by using the mass density of the oil and Stokes’s law. The oil used in this experiment has a fairly uniform density which is listed 3 in the Pasco writeup as ρ = 886 kg/m . However from measuring a couple of bottles I’ve found instead the value: 3 ρ = (880 ± 5) kg/m . (2.5) This also agrees with a value of 881 kg/m3 obtained in followup discussions with Pasco representative Steve Meschia. You should use the value for ρ given in equation 2.5 for your analysis, and its error for assessing systematic uncertainty (discussed further below). The density, mass, and radius of the droplet are related by m = (4/3)ρπa3 . Combining this with Stokes’s law in equation 2.2 above, we get that the droplet’s radius is related to its density and falling velocity by: r 9η0 vf . (2.6) a= 2gρ Millikan showed that Stokes’ law is not exactly correct, because the droplets in the experiment are small enough that their size is comparable to the mean free path between colliding air molecules. This means the viscous drag is slightly weaker because the droplet sees more empty space between colliding air molecules. An adjustment is made for this effect by replacing the coefficient of viscosity, η0 with an “effective” value that depends on the droplet radius a, and the air pressure p as: ! 1 ηeff = η0 , (2.7) b 1 + pa where b is a correction factor that is experimentally determined and given on page 2 of the Pasco writeup as: b = 6.17 × 10−4 (cm of Hg)·cm (you will need to convert this into MKS units). Observe that in this equation, η0 is reduced by inclusion of the factor b/pa in the denominator. For large droplet sizes, a, this effect is small. It is important to have a feel for how significant this correction factor is or, equivalently, how large the droplets are. IN YOUR WRITEUP, include a plot or give a table of the correction factor for η0 in equation 2.7 as a function of a over a range of radii representative of the values encountered in your experiment. Be sure to discuss the approximate range of droplet sizes you used in your measurement, and to discuss how significant the correction is. The corrected viscosity value should replace the uncorrected η0 in equation 2.6 for the droplet radius. Once you do this, solve the resulting expression for the adjusted estimation of the droplet radius. IN YOUR WRITEUP, show 14 LABORATORY 2. MILLIKAN OIL DROP EXPERIMENT that this results in a quadratic equation for a, and explain why the only feasible root is: s 2 b 9η0 vf b a= + − . (2.8) 2p 2gρ 2p Now we have the essential elements for the analysis, that you can incorporate directly into a spreadsheet: 1. Estimate the droplet radius, a from the falling velocity vf when no electric field is applied (equation 2.8). 2. Determine the droplet mass from m = (4/3)ρπa3 . 3. Combine the droplet mass with the falling velocity, vf , the rising velocity, vr , the plate potential difference, V and the separation distance, d, to get the charge on the droplet according to equation 2.4. These are the same steps enumerated on page 9 of the PASCO writeup. The derivation performed on page 2 of their writeup is tedious if arranged into a single equation as in their equation (10), and it is unnecessarily complicated. I suggest you set up separate columns in your spreadsheet for each of these three steps, to convert each pair of values for a droplet’s falling and rising velocities into charge on the droplet in a sequence of easy-to-follow steps. 2.2 Experiment When this experiment is done successfully, it both demonstrates charge quantization and also gives a value for the fundamental unit of charge. You should approach it in the same way: DO NOT ASSUME charge is quantized, rather focus your efforts on collecting very good charge information for a small number of droplets (at least ten), and watch the charge quantization evidence happen of its own accord. Here are some practical suggestions for the setup of the experiment: 1. Understand the apparatus before attempting to take any measurements. Make sure it is level. Make sure to record the number of the apparatus you used, and use the same one for all your measurements. 2. The high voltage should remain set to 500 volts (as read by the digital multimeter) so that you need only turn on the switch. WHILE YOU WON’T BE ADJUSTING THE HIGH VOLTAGE HERE (IT STAYS AT 500 V) BE CAREFUL AROUND IT! 3. The entire apparatus is mounted on ring stands so that you can set it to a comfortable height for taking readings. BE CAREFUL NOT TO KNOCK IT OVER, AND BE CAREFUL IF YOU RAISE/LOWER IT NOT TO 2.2. EXPERIMENT 15 DROP IT, AND TO ENSURE THE SET SCREWS ARE FULLY SECURED AT THE END OF ANY ADJUSTMENT. Always check the leveling bubble that is built into the unit, to see that the apparatus is level, before you start taking measurements. 4. It really helps to follow the suggestions of the guide from PASCO, in particular regarding initial focus using the wire (you won’t need to do this much – presumably with several groups using the apparatus it will stay around optimum focus for the center of the chamber). Once you have the focus set, remember to put the small “droplet hole cover” into place (see Figure 5 in the Pasco writeup). The problem here is NOT in getting too few droplets into the chamber, but in getting too many of them. One sharp squeeze of the atomizer is usually enough to create many droplets, followed by a slow squeeze to push enough droplets in through the hole into the viewing chamber (with the chamber lever moved to “Spray Droplet Position”). If you see a diffuse haze of light in the chamber that makes seeing any individual drops difficult this is usually because the chamber is filled with too many droplets, most of which are out of focus but are scattering diffuse light into the eyepiece. In this case clear the chamber (after you have turned the high voltage off) and start over. Once you have the right number of droplets in the chamber turn the lever to the “OFF” position. 5. Please make sure to turn everything off when you are finished: • the high voltage power supply, • the halogen light bulb (unplug it), and • the digital multimeters. Search for droplets that have small net charge on them, by toggling the polarity control, and seeking slowly-moving droplets. The approximate rise and fall times for sufficiently small, and singly-charged droplets are about 20–30 seconds over 10 small grid divisions. After the chamber has been filled initially, you can clear out droplets that are multiply charged by leaving the plate voltage on for a few seconds (the highly-charged droplets crash into the plates, while the singly-charged ones haven’t enough time to get there). You can greatly improve your measurement precision by holding on to one drop for a long time, and taking several measurements of the fall and rise velocities. In the end you can take the average of each and use it to calculate the charge. Do this for at least 10 droplets. Then organize the results into groups in order of ascending charge value. A histogram is the ideal way to present your charge data, to illustrate charge quantization, and to show how you extract your final value for the fundamental charge. THE GOAL OF THIS LAB IS TO BE ABLE TO SEE EVIDENCE OF CHARGE QUANTIZATION WITHOUT ASSUMING IT. THIS MEANS YOU HAVE TO BE CAREFUL ABOUT USING ONLY THE VERY BEST DATA POINTS IN YOUR PLOT. METICULOUSNESS IS VERY IMPORTANT! 16 LABORATORY 2. MILLIKAN OIL DROP EXPERIMENT Sometimes your droplet changes charge in the middle of a sequence of measurements, due to interactions with cosmic ray radiation. See if you can hold on to the same droplet and get more measurements with its new charge. Multiple measurements of the rise and fall velocities are crucial for improving the precision of charge determination. Do you see evidence of charge quantization in your data? Comment. If so, see if you can extract the size of the elementary charge unit, e – the charge of the electron. You can determine a systematic error on this value by using the error you will have estimated for how well you measured the plate separation, d, the uncertainty in the oil density in equation 2.5, and any other common parameters in equation 2.4 above. Some parameters such as oil density, ρ, are “buried” deep inside the complicated expression for the droplet charge so that classical error analysis expressions (which require finding a derivative) can be cumbersome. In such cases an easy way to determine how much a change in the density changes the determined charge is by manually inserting slightly larger, and slightly lower values of ρ into some of the calculations in your spreadsheet and observing how much it changes the charge. If you do see evidence of charge quantization, you can take the elementary charge determined from each droplet (dividing by 2, 3, etc. as needed for droplets with charges of 2e, 3e, etc.) and combine into a single value, and give the statistical error on this value. An important consistency check is obtained by quoting the values of e obtained by each of these groups, as well as the overall combined value. 2.3 Writeup This is a demanding experiment, so you have two weeks to work on it. However, it is very rewarding if you put in the care and effort to take good measurements. Think of the ramifications of what you are doing here: a singly-charged droplet moving inside the viewing chamber shows the effect of the smallest building block of an atom! Be sure to include responses to the individual points (“IN YOUR WRITEUP”) stated above. When you quote a final value for the electron charge, you can separate the systematic error effects (uncertainty in the charge plate separation, d, uncertainty in oil density, ρ, temperature uncertainty, etc.) from statistical error (the natural spread of your values for e). For example, e = [1.75 ± 0.25 (stat.) ± 0.35 (syst.)] × 10−19 C . This is useful to show at a glance whether your experiment is “statistics limited” or “systematics limited” – whether it would pay to take more data, or whether a refinement or redesign of the experiment is first called for. Comment on any ways in which you might improve the experiment if you were to do it again, or were given the task of building a better oil drop experiment. A good source for historical background on this experiment and some of the controversy surrounding Millikan’s measurement is given in Wikipedia. Laboratory 3 Spreadsheet Energy Levels The goal of this numerical exercise is to get a better feel for wave functions and the Schrödinger equation, for how quantization arises whenever a particle is confined in a potential, and for how the energy level spacing depends on the shape of the potential. You will be finding the eigenfunction solutions, and their eigenvalues (the bound state energies) for three different potentials. This lab is an EXCEL exercise, and you should work together with your lab partner to complete all the investigations, to discuss and understand your results and to put all the information together in your writeup. The Schrödinger equation for stationary state solutions in one dimension is a second order differential equation in the position, x: − h̄2 d2 ψ + U (x)ψ(x) = Eψ(x) . 2m dx2 (3.1) Here, ψ(x) is the wave function representing the particle of mass m immersed in a potential energy function U (x) and E, which is a number and not a function of x, is the particle’s energy. A bound-state particle is confined mostly to the region of space where E > U (x) (unbound particles have E > U (x) everywhere). In this case the differential equation assumes the form: d2 ψ ∝ −ψ(x) . (3.2) dx2 We already have experience with a differential equation like this, from our work with harmonic oscillators, and so we know that the solution has to be oscillatory. In regions where E < U (x), the “—” sign in equation 3.2 disappears, giving an exponential solution instead. We can understand this behavior mathematically by recalling that the curvature of a function f (x) is related to the second derivative by f 00 (x) κ= (3.3) 3/2 (1 + [f 0 (x)]2 ) (the curvature is equal to the inverse of the radius of curvature). The sign of the curvature is determined purely by the sign of the second derivative. If the 17 18 LABORATORY 3. SPREADSHEET ENERGY LEVELS curvature is positive, the curve has increasing slope values (like a parabola that opens upward), and if it is negative the curvature is like that of a parabola opening downward. What the Schrödinger equation in the form of equation 3.2 is telling us, then, is that if ψ(x) is positive (above the axis) then the curve heads downward, back towards the axis and if ψ(x) is negative (below the axis) then the curve heads upward, again back toward the axis. This tendency to head back to the axis, regardless of which side of it the function is on, is the root of oscillatory behavior – it explains why the bound state solutions are wave-like regardless of the exact form of the potential function. The three binding potential energy functions you will investigate in this exercise are: 1. A harmonic oscillator potential, U (x) = 21 κx2 , with spring constant κ = 13, 456.665 N/m; 2. An finite square well of width L = 6.1321295 × 10−10 m, that is exactly 30.000000 eV deep; and 3. A triangular potential, U (x) = a|x|, with a = 2.6784462 × 10−6 m. The reason for the high precision is discussed more below. You will be considering an electron bound within each of these potentials. You already know the energy levels and wave functions that the Schrödinger equation gives for the first one, so it will serve as a “test drive” for your numerical method of solution. The second potential was discussed in class, but we never calculated any actual wave functions or exact energies for it. You’ll do that in this exercise, for a specific example of the square well. The last potential is one for which we don’t have solutions, but you’ll find them out numerically, using EXCEL. Once you have them you will compare and discuss the energy level structure of these three potentials for the five lowest-energy solutions in each case. Since we have EXCEL at our command we may as well use high-precision CODATA1 (2008) values for physical constants: m = 9.109 382 15(45) × 10−31 kg h = 6.626 068 96(33) × 10−34 J · s e = 1.602 176 487(40) × 10−19 C h̄ = 1.054 571 628(53) × 10−34 J · s The last two digits, in parentheses, give the uncertainties on the values in the last two places of the quoted values. E.g., m = (9.109 382 15 ± 0.000 000 45) × 10−31 kg . Start by setting up the numerical solution for the harmonic oscillator potential, which is one for which we already know the solutions. 1 The Committee on Data for Science and Technology, of the National Institute of Standards (NIST). See: http://physics.nist.gov/cuu/Constants/. 3.1. HARMONIC OSCILLATOR 3.1 19 Harmonic Oscillator The equation for the wave function is − h̄2 d2 ψ 1 2 + κx ψ(x) = Eψ(x) . 2m dx2 2 (3.4) The greek letter kappa, κ, is used for the spring constant instead of k so as not to be confused with the wave number. For this example use κ = 13, 456.665 N/m. You have seen the mathematical solution of this problem in your text. First, a variable transformation was made: r r 2πmν mω κ x= x , where ω 2 = . (3.5) y≡ h̄ h̄ m This transformation scales from x values that have very small numerical values to the dimensionless variable y which has values that are not so tiny. The differential equation 3.4 then becomes ψyy + α − y 2 ψ = 0 , (3.6) 2 d ψ 2E where α ≡ 2E hν = h̄ω . The symbol ψyy = dy 2 is a common shorthand notation for derivatives. Since the potential energy function is symmetric about y = 0 the wave function solutions for this equation must be either symmetric (even functions in y, ψ(−y) = ψ(y)) or antisymmetric (odd functions: ψ(−y) = −ψ(y)). We’ll make use of this fact by working only with the region y > 0 (you can just reflect the solution into the region y < 0 to get the rest of the wave function). The variable y automatically changes x (which is on the order of 10−10 m) to a more convenient (and dimensionless) distance scale, as the spring constant and mass involved give: y = 3.240176 × 1010 m−1 x . (3.7) Furthermore if we agree to measure the energy in eV, then with the given spring constant, which gives h̄ω = 1.2817412 × 10−17 J = 80.000000eV, we can put equation 3.6 into the form: 1 2 ψyy + ε−y ψ =0 , (3.8) 40 where ε replaces α (which contains the energy E) in order to remind us that it is the energy measured directly in eV, so that the dimensionless quantity 2E 1 α = 2E hν ⇒ 80 eV ⇒ 40 ε. As we have discussed, when a particle is bounded by a potential this differential equation has finite solutions for only a discrete set of energy eigenvalues, ε. However, there is nothing like seeing this for yourself by setting up a spreadsheet solution. Now set up a spreadsheet to generate solutions for ψ(y) from equation3.8. Start your solution at y = 0, and propagate the wave function and its derivatives 20 LABORATORY 3. SPREADSHEET ENERGY LEVELS (this means at least four columns, for y, ψ(y), ψy (y), and ψyy (y)) in short steps of about ∆y = 0.01 or so in y, up to y value of about 5, according to the propagation equations: 1 2 ψyy (y) = y − ε ψ(y) , (3.9) 40 ψy (y + ∆y) = ψy (y) + ψyy (y)∆y , and (3.10) 1 2 ψ (y + ∆y) = ψ(y) + ψy (y)∆y + ψyy (y) (∆y) . (3.11) 2 Make sure that you understand the propagation strategy here: for any given value of y, the second derivative is determined FOR THE CURRENT y VALUE by the first equation; this second derivative plus the first derivative value FOR THE CURRENT y VALUE determine the first derivative FOR THE NEXT y VALUE according to the second equation; and lastly the first and second derivatives FOR THE CURRENT y VALUE plus the current ψ values determine ψ FOR THE NEXT y VALUE according to the last equation. If you think through the logic of this for a minute you will see that two pieces of information are missing, in this “zipper” strategy for propagating y, ψ, and its derivatives: the values of ψ and ψy FOR THE CURRENT y VALUE (look closely at the left hand side arguments in equations 3.9, 3.10 and 3.11). These must be input – they are the initial conditions, or constants of integration. It will be convenient to leave separate cells in the spreadsheet for the input energy, ε and for the starting values, the fundamental constants, and for ψ(0) and ψy (0) of the wave function at y = 0 so that you can change these easily. The last two will be adjusted to generate the symmetric and antisymmetric solutions: SYMMETRIC: ψ(0) = positive value ψy (0) = 0 (odd number of “bumps”) ANITSYMMETRIC: ψ(0) = 0 ψy (0) = positive value (even number of “bumps”) Set up a spread sheet that goes from y = 0 to y = 5 in steps of 0.01, and try some different energy values and initial starting conditions. Don’t worry about what exact starting values ψ(0) and ψy (0) you use (I usually just set them to 1) – we’ll do the normalization later. “Shop around” for energy eigenvalues that give finite solutions and their corresponding wave functions. Remember the guiding principle: The wave function must go to zero at large y. To help you find these it’s convenient to set up two small graphs on your spreadsheet, one for the wave function out to about y = 2, and another that goes all the way out to the end (y = 5). Find the energy eigenfunctions and wave functions for the ground state (one bump, symmetric) and the first excited state (two bumps, antisymmetric). At first you will see a graph where the wave function takes off wildly to infinity at the upper end. Keep searching if the 3.2. THE FINITE SQUARE WELL 21 divergence at y = 5 changes from positive infinite to negative infinite - you’ve crossed an eigenvalue! Go back and “home in” on it. 1. Find the energy values of the first four bound states, and compare them with the predictions of the known solution, En = n + 21 hν. Notice the acute sensitivity to getting the energy just exactly right – anything even slightly above or below a “magic values” leads to strongly divergent solutions. In fact to get a solution convergent out to arbitrarily large y you would have to continue the searching process to infinite precision in the energy eigenvalue! Also notice that, without knowing anything about Hermite polynomials or the detailed solution given in your text: ψn (y) = 2mν h̄ 1/4 n 1/2 (2 n!) Hn (y)e −y 2 /2 1 , En = n + hν , 2 (3.12) and even without a priori knowledge that finite solutions exist only for certain discrete energy values, your numerical method reproduces all of these results. You can confirm this by overplotting ψ0 (y) and ψ1 (y) from equation 3.12 on your numerical solutions to see that the shapes agree. You’ll need to scale your wave function to do this – try adjusting your initial value ψ(0) for the numerical ψ0 solution, or the initial value ψy (0) for the ψ1 solution, until you can get them to fit on top of each other. 2. Plot the first two wave functions you found numerically, together with the corresponding exact solutions from equation 3.12 above. Scale the former so they fit against the numerical solutions. Do their shapes agree? Since we know that we have to normalize the numerical wave functions anyway, this confirms we have found the same exact solutions. 3.2 The Finite Square Well The potential for this part will be a square well of width L centered at x = 0: x < −L/2 30 eV , 0, −L/2 < x < +L/2 V (x) = (3.13) 30 eV , x > L/2 where the width is L = 6.1321295 × 10−10 m. As a point of comparison, and to motivate the choice of L, first calculate the energy levels (in eV) that you would expect for an infinitely-deep square well (whose solutions you know) of this same width. How would you expect the energy levels of the finite well to differ from these? About how many bound state energy levels would you expect to be contained in this finite well? Now set up your spread sheet just as you did in the previous section, and find them. Make the height of the square well an adjustable parameter in your spreadsheet. Use an increment size of and start at x = 0, the middle of the 22 LABORATORY 3. SPREADSHEET ENERGY LEVELS well. We can do this again due to the symmetry with respect to the center of the well, so that we know there are only symmetric and antisymmetric solutions as with the harmonic oscillator. With a spread sheet that goes from x = 0 to x = 10−9 m in a thousand steps of 10−12 m, try different energy values and initial starting conditions, and “shop around” for the energy eigenvalues that give finite solutions. Again it’s useful to have two small graphs on your spreadsheet, one for the wave function out to about x = 4 × 10−10 m (400 steps – to just beyond the right wall), and another that goes all the way out to the end. Find the energy eigenfunctions and wave functions for the ground state (one bump, symmetric) and the first excited state (two bumps, antisymmetric). Do their energy values differ, with respect to the values for the infinitely-deep square well, in the way you expect? Do the wave functions look reasonable? 3. Find the energy values of all the bound states, and discuss how they compare with the infinitely-deep square well. 4. Find the normalized wave functions for the second and the fifth energy eigenvalues, and plot them together with the normalized wave functions for the infinitely-deep square well. Give a brief discussion of the comparison of the two. For the finite well, determine the probability of the electron being found in the classically forbidden region, for each of the bound state solutions. Since you have left the height of the finite well as an adjustable parameter, and since you know what the answers for the energy levels are for the infinitelydeep square well, you can check the functioning of your spreadsheet program by increasing the (adjustable) height of your well to a big value (say 100,000 eV). 5. Discuss what happens when you do this what large value of the well height did you use? What is the new ground state energy? Does it agree with the trend you expected? 3.3 A Triangular Potential Now that you have confidence with your spreadsheet method of solution, use it to find the energy eigenvalues for the triangular potential: V (x) = a|x| , with a = 2.6784462 × 10−6 m. (3.14) Technically this means there are two separate Schroödinger equations for the regions of negative and positive x: 2 2 2 2 h̄ d ψ − 2m dx2 − axψ = Eψ , for x < 0 , and (3.15) h̄ d ψ − 2m dx2 + axψ = Eψ , for x > 0 . We don’t have a solution for this potential in our textbook. Even if we did, it wouldn’t be particularly enlightening because it involves special functions 3.3. A TRIANGULAR POTENTIAL 23 called “Airy functions”, that we would want to plot anyway in order to understand them. We can find them easily and automatically with your spreadsheet solution. 6. Discuss what you expect the wave functions to look like, in both wavelength and amplitude variation over the breadth of the potential. Once again, due to the symmetry of the potential we expect either symmetric or antisymmetric wave functions to result. So we can work with only the region x > 0 as before, and either reflect or antireflect to get the solution for x < 0. The latter of the equations 3.15 can be rewritten as: E 2ma x− ψ . (3.16) ψxx = 2 a h̄ Following the method used to solve the harmonic oscillator, and also in order to simplify the spreadsheet calculation by scaling down the distance and energy, introduce the variable change to get a dimensionless variable y: y = βx ≡ 2ma h̄2 1/3 x = 7.5988854 × 1010 m−1 x . (3.17) With this definition equation 3.16, transformed to an equation in y, becomes: ε ψyy = y − ψ , (3.18) b where b≡ a = β h̄2 a2 2m 1/3 = 3.52478827 × 10−17 J = 220.000000 eV (3.19) is the new energy scaling factor. This again allows you to use the energy parameter ε measured directly in eV. The reworked differential equation you’ll use in your spreadsheet is then: ε ψyy = y − ψ . (3.20) 220 eV As with all bound state solutions (and this potential as specified has ONLY bound state solutions), convergent solutions exist for only discrete energy eigenvalues, ε. Modify your spreadsheet to find solutions for this potential. You can start at y = 0 and use short steps of about ∆y = 0.1. You can figure out what upper bound in y is appropriate. Determine the energy values of the first five bound state energy levels and their corresponding wave functions. 7. Discuss the energies of the lowest five eigenfunctions in eV in your writeup and comment on the spacing between them – is it even, increasing, or decreasing? Is this what you might expect? Do the wave functions look like what you predicted? Discuss. 24 LABORATORY 3. SPREADSHEET ENERGY LEVELS Once you have the energy eigenvalues, you can adjust the non-zero starting value of ψ(0) or ψy (0) that you used to generate the solution, in order to get the integral of the probability density to be 0.5 for the range y > 0. 8. Include plots of the wave functions in your writeup for the first five energy eigenfunctions (for y < 0 you can just reflect the wave function across zero). 9. In your conclusion show the energies of the first five levels of all three potentials, and discuss the variation of the spacings between them. Also feel free to give any discussion about things you have learned in this exercise about eigenfunction, quantization, etc. Laboratory 4 Measurement of e/m for the Electron Although we take the electron for granted nowadays, it was not “discovered” until the end of the 19th century. During the 1890’s, several people studied cathode rays in vacuum tubes and gas discharge tubes. Electric fields and magnetic fields could deflect cathode rays, so it seemed that cathode rays contained rapidly moving charged particles. The question was what kind of particles they were. Were they charged atoms (ions) or something else? Two important experiments helped settle the matter. In 1899, J. J. Thomson measured e/m, the ratio of the charge to the mass of the particles. And in 1909, Robert A. Millikan accurately determined e, the charge of the particles (as you did in a previous lab). Taken together, these two results allow one to calculate m, the mass of the particles, which turned out to be about 1,800 times smaller than the mass of the smallest atom, hydrogen. This result confirmed the idea that the particles, which we now call electrons are parts of, and not whole atoms. For his role, we usually credit J. J. Thomson with “discovering the electron.” In this experiment you will be measuring e/m in an experiment similar to that of J. J. Thomson. Your apparatus includes a vacuum tube and a set of coils that produce a magnetic field. Figure 4.1 below shows what goes on inside the vacuum tube. Practice your right hand rule and see if you agree with the path shown, given the directions indicated for the velocity and magnetic field. The apparatus produces electrons from a hot filament, by thermionic emission. Starting with essentially no velocity, the electrons accelerate toward a metal plate held at a large positive voltage Va (called the “plate voltage”). Some of the electrons pass through a hole in the plate. The magnetic field causes the electrons to move in a circular path of radius r. After being accelerated the electrons are subjected to an adjustable focusing, controlled by the “grid voltage.” They then emerge from the hole in the metal plate you see in the center of the concentric ring pattern and continue to move in half of a circular path. The radius of this path can be adjusted so that the 25 26 LABORATORY 4. MEASUREMENT OF E/M FOR THE ELECTRON B (into page) r Va hot filament 6.1 V Figure 4.1: Schematic representation of the apparatus for measuring e/m for the electron. The magnetic field is uniform, and pointing into the page, everywhere along the path of the electron which initially moves straight up after being accelerated by the potential Va . electrons hit one of the fluorescent ring patterns on the metal plate. The radii of these rings are known to be 0.50, 1.0, 1.5, and 2.0 cm. The kinetic energy each electron gains as it accelerates from the filament through the voltage rise Va to the plate is eVa : 1 mv 2 . (4.1) 2 There is technically a small voltage drop across the filament (up to 6 Volts) depending on where the electrons originate from. But we can ignore this effect, as it averages to zero since the 6 Volts powering the filament is alternating current. Thus it is correct to take the filament at zero voltage with respect to Va. The magnetic field, B (discussed in more detail below) is perpendicular to the velocity of the electrons, and causes them to move in a circle of radius r: eVa = mv 2 . (4.2) r You should convince yourself that you can eliminate v between equations 4.1 and 4.2 to find: e 2Va = 2 2 . (4.3) m B r The two coils of wire surrounding the vacuum tube are called “Helmholtz coils.” In this arrangement the coils of wire are separated by an amount equal to the radius of either coil. This configuration produces a surprisingly uniform magnetic field in the central region between the coils, given by NI 8µ0 N I ≈ 8.9918 × 10−7 T · m/A , (4.4) B=√ R 125R evB = 4.1. DOING THE EXPERIMENT 27 2rmeas 2.54 mm 2ractual Figure 4.2: Illustration of the offset of the origin of the accelerated beam of electrons. The electrons accelerated by the potential Va begin bending in a circle for approximately 2.54 mm before entering into the visible part of the tube. where B is measured in tesla (T), N is the number of turns of wire in either one of the twin coils, I is the current in amperes flowing through them, and R is the coil radius in meters. For the CENCO 71267 apparatus N equals 119. You can measure R directly (but consider where you should measure it!). An ammeter must be included in the coil supply in order to measure the current. At this point you will have the magnetic field, B, the radius, r of the path the electrons travel, and the supply voltage as measured directly by another multimeter. One final adjustment must be made, as the electrons emerge from the accelerating plates into the magnetic field region at a position about 2.54 mm below the center of the circular hole in the circular target plate so the center of curvature lies slightly below the target plate, as seen in Figure 4.2. Show that the value of the radius you measure by sight, rmeas , can be adjusted to very nearly the correct value for this relatively small shift by using1 2 2 2 ractual ≈ (rmeas ) + (0.00254/2) . (4.5) The final working formula for the experiment is then e 2Va h i . = 2 m B 2 r2 + (1.27 × 10−3 m) 4.1 (4.6) Doing the Experiment Your apparatus has power supplies that provide electricity to power: (1) the filament, (2) the accelerating voltage, (3) the focusing potential, and (4) the Helmholtz coils. They are already connected, but you should check it out to make sure you understand which wire goes where. A connection diagram is given at the end of this writeup. The filament voltage is already set to about 6.3 VAC (the outermost two taps under “FILAMENT” you can check it with the DMM after turning the unit on). This voltage should not be adjusted (by moving the plugs to different taps) – if the filament burns out the bulb is lost, and they are very expensive to replace! Turn on the power to the unit, which powers 1 This disagrees with CENCO’s writeup by a factor 1/4, but it is the correct form 28 LABORATORY 4. MEASUREMENT OF E/M FOR THE ELECTRON up simultaneously the plate, grid and coil supplies. The leftmost knob adjusts the plate (accelerating) voltage, Va , and should be measured by a multimeter. The second knob adjusts the grid, or focusing voltage, and the rightmost one adjusts the coil current (there should be an ammeter there, set to 10 A scale, to measure the current for your magnetic field calculation). Turn the rightmost knob off, to deactivate the magnetic field at the start. Turn off the lights, and see the slight glow of the filament under the circular plate. By adjusting the plate voltage (40-80 Volts) and grid voltage you should see a very thin ray of electrons going straight up inside the tube. Adjust the grid to focus this beam. Then, you are ready to take measurements. Since you can vary the accelerating voltage and current to the coil independently you can obtain e/m points for a variety of values of Va and I. In order to keep the equipment functioning properly, please 1. DO NOT EXCEED 170 VOLTS ON THE ACCELERATING (“PLATE”) POWER SUPPLY. 2. DO NOT ALTER THE FILAMENT POWER HOOKUPS. Do not leave the filament on for more than 20 minutes at a time. This should be enought time if you plan your measurements. 3. DO NOT EXCEED 5 AMPERES OF COIL CURRENT. Generally, you will work with less than this. TURN THE MAGNET POWER OFF WHEN NOT MAKING MEASUREMENTS. The coils heat up quickly at high currents, and can melt. NEVER LEAVE THE MAGNET ON MORE THAN SEVERAL MINUTES AT A TIME. 4. WHEN FINISHED TURN ALL CURRENTS AND VOLTAGES TO ZERO BEFORE TURNING OFF THE INTEGRATED POWER SUPPLY UNIT. Leave the filament hookups as they are. 4.2 Analysis Find the overall value for e/m using all your data points. You can find the statistical error using the standard deviation of all your data points. Is your value consistent with the standard known result? Discuss the effect of Earth’s magnetic field on your measurement. How does its magnitude compare to the B field generated by the Helmholtz coils? The intensity of BEarth is about 5.4 × 10−5 T around these parts, with 20 µT horizontal component (pointing North, of course) and 50 µT vertical component. What would you do to reduce the effect of BEarth , in terms of orienting the apparatus? Now check the values you obtained for e/m as a function of Va , I, and R. Ideally they should be constant all the way across. However this experiment has several potential sources of error that illustrate the difference between two types of experimental error statistical and systematic. STATISTICAL errors refer to those which simply come from an inability to make measurements with perfect 4.2. ANALYSIS 29 precision. For example, measuring the coil radius involves eyeing a ruler, and measuring r involves making sure you hit a ring “dead center.” Presumably, the more times you measure these, the more the average of all your measurements approaches the “right” answer. SYSTEMATIC errors do not get smaller the more times you measure something. These can be very dangerous if not accounted for. For example if the multimeters you have used to measure current and voltage were simply not calibrated correctly, no amount of re-measuring or averaging would help to give the right answer, and so there is an “inaccuracy” which cannot be reduced in size or eliminated without an alteration of the experimental procedure and/or apparatus. [By the way: we assume the meters are OK here!] How many standard deviations away from the accepted value of e/m is your mean value? A rule of thumb is that if your value is more than about two standard deviations from the accepted value, the disagreement is likely NOT due to STATISTICAL errors in your measurement, but due to some SYSTEMATIC error(s). Here are some ways to look at your data to give clues about possible error sources. • Plot e/m versus the accelerating voltage Va . Do you notice a trend? Does this graph tell you anything about your experiment, in particular, what might be going on when the accelerating voltage is small? Can you formulate a hypothesis about what might be going on here in terms of kinetic energy lost to collisions in the tube? • Plot e/m versus the current I in the Helmholtz coils. Do you notice a trend? If yes, does this graph tell you anything about your experiment, in particular, what might be going on when the magnetic field from the Helmholtz coils is small? This allows you a direct check on the effect of Earth’s magnetic field in your experiment. If either of these plots show a trend, you have identified a source of systematic error. In this case, do you still think that the most accurate experimental value you can extract would be found by simply taking the average of ALL of your data points ? If not, what do you think a better approach might be? What part of your experiment contributes the greatest STATISTICAL error? Estimate how large this error is, and the effect it has on the final error for e/m. A way to approach this is to estimate typical percentage errors in the relevant experimentally measured quantities, such as B, I and Va , and then propagate them through the calculation. What part of your experiment contributes the greatest SYSTEMATIC error? Try to estimate how large this error is, and explain how this source of error might result in an incorrect value for e/m. In light of the above (i.e. lack or presence of trends in the data plots), would you revise your final value for e/m? In the event that systematic errors are well-identified, the final measurement value is often quoted in the format: e = x.xx ± y.yy (stat.) ± (syst.) m 30 LABORATORY 4. MEASUREMENT OF E/M FOR THE ELECTRON where the sizes of the statistical and systematic errors are given separately. This allows one to see at a glance where the predominant limitations to the measurement lie – in limited statistics, or in some inherent limitation of the experimental design. 4.3 Writeup You should write a standard laboratory report for this lab. In addition to carefully and concisely describing everything you did, include the following elements in your writeup: 1. Derive Equation 4.4 for the magnetic field from a pair of Helmholtz coils. This arrangement is famous, as an inexpensive way to produce a uniform magnetic field over a reasonably large range. Find the general expression for the magnetic field of these coils on the axis of symmetry between them, as a function of distance z away from the center point. Make a plot of Bz (z) for the region between the coils and include it in your writeup. Find the first and second derivates, dBz /dz and d2 Bz /dz 2 , and evaluate them at the middle point, z = 0, and discuss the plot and the derivatives in your writeup. 2. The mass of the proton is 1.67 × 10−27 kg. What is q/m for a proton? What would you have to do to this apparatus to in order to do this experiment with protons? Give conjectures that are reasonable, that you might be able to implement for modest cost. 4.4. HOOKING UP THE E/M POWER UNIT 4.4 31 Hooking up the e/m Power Unit The CENCO power unit supplies three voltages and the current for the Helmholtz coil. These are shown in Figure 4.3. Common ground is used for the three voltages. • FILAMENT SUPPLY - This is a non-adjustable AC voltage, which you can tap as any one of several settings from 2.1 V to 6.3 V. From left to right, the steps between the four taps are 2.2 V, 3.2 V, and 1.2 V. We are tapping all the way across (setting “6”) to get our filament operating voltage as 6.6 V. NEVER USE MORE THAN THIS IF THE FILAMENT BURNS OUT THE TUBE IS USELESS. [Other tap settings indicated on the unit are 4.4 VAC for setting “4” and 5.5 VAC for setting “5.”] • GRID VOLTAGE - This is a beam focusing voltage that goes from zero up to about 80 V. Adjust this by sight to give the tightest beam, depending on the Plate Voltage. Typical values are around 20-30 V. • PLATE VOLTAGE This is the accelerating voltage for the beam, adjustable from zero up to about 400 V. • COIL CURRENT This is a high-current supply to power the Helmholtz coils. It “kicks in” at about 1.5 A, and is adjustable to about 6 A. The current is best monitored with a multimeter (set to 10 A scale!) for improved accuracy, as the exact value of the magnetic field depends upon it. USE THE HIGH-END CURRENTS ONLY FOR SHORT DURATIONS! POWER SUPPLY UNIT COIL CURRENT PLATE 1.5 – 6 A FILAMENT up to GRID (connects to 6.3 VAC 500 VDC ~ 20 VDC back side) e/m UNIT filament common ground Figure 4.3: Hookup diagram for connecting the CENCON e/m Power Unit to the coil/tube unit. The various supplies are described in the text. plate grid 32 LABORATORY 4. MEASUREMENT OF E/M FOR THE ELECTRON Laboratory 5 Spectroscopy Equipment Gaertner-Peck Spectrometer Sodium light source High-quality diffraction grating, with mount Spectral tubes, including H, Hg, He and Ne In this experiment you will be measuring the wavelengths of the spectral series for various gases using a high-precision spectroscopic measuring apparatus. Spectroscopy has played a central role in the evolution of modern physics, and is today an indispensable tool for astronomers and physicists. Each individual chemical element produces its own unique “fingerprint,” a series of spectral lines which are different from any other element. Furthermore these spectral lines are subject to modifications under different conditions that influence the electronic energy level structure, or also if the source is moving with respect to us. In the former case we have phenomena such as the Zeeman effect (spectral line separation caused by a magnetic field), or shifts due to molecular bonding, strained crystal lattices, etc. In the latter case Doppler spectroscopy is used widely in measuring motions of distant stars, and has been used to establish the expansion of the universe, and the discovery of extrasolar planets. And of course there is the remarkable observation of the element helium in the Sun’s light, before it was ever found here on Earth. You’ll be using a Gaertner-Peck spectrometer that allows a high degree of resolution between nearby lines in a series. A high-quality diffraction grating will split input light into sprectral components. In this experiment a sodium light source will be taken as a reference source whose wavelengths we know, and this light source will be used to precisely determine the ruling spacing of the grating. This step is called the “calibration.” After this value has been determined you’ll be measuring the spectral series of hydrogen, mercury and a noble gas (helium, neon, argon or krypton - your choice). There is a theoretical formula for predicting the measured wavelengths of the spectral lines of hydrogen, and you’ll use this formula to determine a value for the Rydberg constant, RH . No such simple formula exists for other gases, however, and you will be comparing 33 34 LABORATORY 5. SPECTROSCOPY adjustable collimating slit diffraction grating light source COLLIMATOR ARM PRISM TABLE TELESCOPE ARM m =1 LEFT observer Figure 5.1: An overhead view of the Gaertner-Peck spectrometer. The telescope arm swings around in a table whose axis coincides with the center of the prism table, to accurately measure the angular deflections of spectral lines. your results with the known spectral lines that have been established by more sophisticated instruments. A diagram of spectrometer is given in Figure 5.1. Incoming light is collimated with an adjustable slit control that is vertically oriented, and which allows you to narrow the slit to give extremely fine lines actually of light. These are focused by a lens in the collimator arm and passed on to a diffraction grating, which splits the colors into different series on the right or left sides. A telescope arm is positioned with fine adjustment controls and a vernier scale to give precise locations in angle of the spectral components. The diffraction grating has very fine lines etched onto a transparent surface. In addition to passing a large portion of the light straight through, it also causes secondary maxima to appear at angles θ which correspond to the wavelength of the light and the order of the maximum, n, according to the familiar diffraction formula: d sin θ = mλ . (5.1) The order of diffraction can be any integer value m that is small enough to give a value of sin θ in this equation that is less than one. In order to use this relation we must have an accurate determination of the spacing between the lines, d, on the grating. 5.1 Calibration We will take sodium light as a source whose wavelengths are very well known (these may have been precisely determined, for example, using an interferometer). Sodium has two very strong spectral lines that are usually indiscernible because they are very close together. With this instrument you should be able to resolve them easily. Turn on the sodium light. It takes about 10 minutes or so for it to warm up to the point where all sodium in the low-pressure tube 5.2. HYDROGEN 35 to vaporize, at which point you will see the familiar yellow color that lights parking lots around the world . The spectral lines are separated by less than one nanometer, at wavelengths 588.9950 and 589.5924 nm, which we will take as given. Make sure the diffraction grating is well centered and that it faces the light coming from the slit/collimator arm at normal incidence. Make sure that is properly fastened down if it moves while you are making your measurements you will have to start all over. Then measure the angles of the two separated lines, both to the right and the left sides, and for both first and second order diffraction maxima. The sighting tube for the apparatus has fine crosshairs that you can use in concert with the slit width adjustment to determine the angles of the lines very precisely. You should make sure your apparatus has cross hairs oriented in an “X” pattern, and not a “+” pattern – that latter makes it more difficult to precisely find the centers of fine lines since the vertical line partly occludes them (ask Marcus or me to adjust them if they are not already set in this way). The vernier angular measurement takes a little getting used to. It allows you to measure angles down to 1 arc minute of precision. Make sure you understand how this works by asking for guidance if it’s not clear to you - your measurements depend on it! These angles will allow you eight separate measurements of the grating spacing, d, given the wavelengths for the sodium lines assumed above (both left and right sides, and two orders of diffraction with two spectral lines in each one). Are your values consistent? Determine both your best estimate for d and the uncertainty on it, which you can find from the standard deviation of your measurements. 5.2 Hydrogen Because of the brightness of the sodium lamps the first part could be done in a lighted room. This is not the case with the following measurements, as they involve tube sources for the light that are far fainter. You may want to draw the window blinds down, or to use one of the small side rooms on the east side that allow you to shut the door and turn off the lights. For hydrogen, mercury and the noble gas that follow you will have to develop your own working procedure for how you’ll get the vertical crosshair centered on the line you are measuring. It may involve periodically turning on the overhead light just long enough to see where it is with respect to the line, or shining a modest amount of light towards your viewing tube in order to show up the crosshairs, etc. Find a method that works best for you. Having your lab partner assist with lighting, writing down information, etc. will be a great help for this. The object of this section of the experiment is to measure the Rydberg constant for hydrogen. Recall that Rydberg was able to show that hydrogen’s 36 LABORATORY 5. SPECTROSCOPY spectral lines could be predicted by the empirical formula 1 1 1 − 2 , = RH λkn n2 k (5.2) where each of the observed spectral lines, of wavelength λkn , corresponds to a pair of integer values, n and k > n, with the Rydberg constant, RH , as a common conversion factor. As you know from class, spectral lines correspond to transitions from an energy level k to a lower energy level n (n < k). The visible lines of hydrogen have the integer n = 2, corresponding to transitions ending on the second energy level. This can be rearranged to solve for the Rydberg constant, 2 2 2 2 m k n 1 k n = . (5.3) RH = λkn k 2 − n2 d sin θ k 2 − n2 From equation 5.3 RH is seen to depend entirely on the measured quantities d and θ, assuming the values m, n, and k are unambiguously determined integers. Thus you can extract several measurements of RH , which you can use to quantify your experimental error. Make sure you quote very clearly your final value for RH with its error, and describe how you found your error, and discuss the consistency of your measurements. 5.3 Other spectral series Use the spectrometer to measure the wavelengths of the spectral lines of one of the noble gases. Use as many values as you can get for each line, by using not just first but also second order measurements (if possible), and both right and left side measurements. Make sure to give an error estimation for each of these. Compare your series to established values for the element you chose from tables such as the CRC Handbook for Chemistry and Physics, or online values, being sure to cite which reference you used. Note also that the CRC Handbook gives line intensity information that you can use to compare to what you observe in your readings. Laboratory 6 X-Ray Diffraction Equipment Tel-X-ometer 580M X-ray diffraction apparatus Tel-X 2590 Driver Motor, Digicounter and Ratemeter units Computer interface unit, tube current monitor cables Computer with software installed The planes of atoms in highly-symmetric crystals can diffract electromagnetic radiation of wavelengths similar to the spacings between planes of atoms in the crystal lattice. Ionic-bonded crystals such as NaCl, KCl, RbCl and LiF have very simple lattice structures, with a fraction of nanometer between atoms which makes them very suitable for diffracting X-rays with wavelengths of 100300 pm. The Bragg diffraction law says that if an X-ray of wavelength λ is incident at angle θ on a crystal with layers of spacing d as shown in Figure 6.1, then diffraction maxima result if the path length difference equals an integer number of wavelengths: nλ = 2d sin θ . (6.1) θ θ d Figure 6.1: Bragg scattering in a regular crystal, with d equal to the distance between successive planes of ions. 37 38 LABORATORY 6. X-RAY DIFFRACTION Make sure you understand the derivation of this result, by examining the path length difference between the two rays shown and seeing that the equation simply requires this difference to be an integral number of wavelengths. The index, n, gives the order of the diffraction maximum: n = 1 is a path difference of one wavelength, n = 2 for two wavelengths path difference, and so on. The goal of this laboratory is to achieve familiarity with the production and use of X-rays by measuring the lattice spacings in ionically-bonded crystals from the Bragg scattering peaks. It will be very reminiscent of your experience with optical spectroscopy, but with peaks in the X-ray region instead of the visible light range. Familiarize yourself with the Tel-X-ometer 580M apparatus. Observe that it has an interlock device that allows X-rays to be created only with the protective cover engaged and locked into the center position. The X-rays cannot penetrate the protective cover. Inside the cover is an X-ray production tube. This tube has a heated filament that liberates electrons by thermionic emission, and also lights up when in use. The liberated electrons are accelerated by a 20 kV or 30 kV potential (selectable by the red switch on the top of the unit). X-rays are created by bombarding a piece of copper with the accelerated electrons. The Xray tube’s envelope is made of lead glass, that allows visible light to pass through but not X-rays, except for a “port” on the front of the tube. X-rays pass through this port to a target mounted in the center of the rotating measuring apparatus. Photons of specific energies/wavelengths result when electrons on the innermost shells of the copper atoms are dislodged by the bombarding electrons, and the vacancies in those shells are filled by outer-shell electrons falling into their place. Usually these will be n = 2 electrons falling into n = 1 holes (Kα X-rays) or n = 3 electrons falling into the n = 1 holes (Kβ X-rays). Which one will be of higher energy/shorter wavelength? Which one do you expect to occur more often? There are more series, such as the L, M , etc. series of transitions resulting when n = 2, n = 3 etc. electrons are dislodged and outer shell electrons fall into their place. But their energies are much lower, and the resulting wavelengths are too long for crystallography studies (for example, the Lα line has energy 0.93 keV, or wavelength 1.3 nm, about ten times the Kα wavelength). Observe the gearing of the center-mounted “chamfered” crystal mounting post, with respect to that of the measuring arm that holds the Geiger Muller tube: it turns exactly half the angular distance of the measurement arm. Thus the angular displacements you read around the perimeter of the table are 2θ, where θ is the angle of either incoming or outgoing X-rays with respect to the crystal surface as shown in Figure 6.1. Make sure you understand how the geometry of this figure relates to the X-ray source, the crystal, and the measuring unit of the Tel-X-ometer. The exercise of determining the spacing between planes of atoms in a crystal is then one of finding “peaks” in the rate of diffraction, as a function of angular position with respect to the crystal face. The Geiger-Muller tube is a standard device for measuring radiation in the form of either high-energy photons or energetic charged particles. It consists of a fine wire centered inside a metal cylinder with a suitable gas between them (various mixtures are used, usually an inert gas such as a argon with a 6.1. PROCEDURE 39 small amount of a hydrocarbon such as methane). A high voltage difference is applied between the fine wire and cylinder, that creates a high electric field strength between them. The passage of an ionizing photon or charged particle initiates an “avalanche” process – an electron is dislodged from an atom of gas by a penetrating photon or charged particle, and is accelerated away from its parent ion strongly enough that it gains sufficient energy to dislodge secondary electrons from other gas molecules, which in turn liberate tertiary electrons, etc. The “gain” on these tubes is typically in the 104 range (the number of electrons liberated and collected at the anode wire as the result of a single initial ionization). The collected charge registers as a pulse/count, and in a short time (< 1 ms) the tube is “cleared” and ready to register another “hit.” The GM tube can be powered by one of three TEL-Atomic units, the Ratemeter, the Digicounter, or the Tel-X-Driver unit. In all three cases the single coaxial cable running to the tube both electrifies it (the voltage used for these GM tubes is around 425 V) and also reads out the pulses registered by avalanche activity in the tube. The Ratemeter and Digicounter units have been used for performing this experiment in previous years (recording the count rates by hand!). While you’ll be using the new Tel-X-Driver unit for most of your work, the other units are useful aids in setting up the experiment and checking alignment. When the Ratemeter powers the GM tube, the radiation level can be monitored as audible “clicks” or as a DC signal that you can send to an XY plotter. This can be used to establish the approximate angular positions of peaks. It also monitors the current of the bombarding electrons inside the X-ray tube, which is a measure of the flux of X-rays emitted by the tube. We usually assume this flux is constant if this current constant. The Digicounter can be used to obtain high accuracy angular positions by precisely counting the rates. It can be used in either single-measurement mode (use the red RESET button to start it) or in continuous mode, where it counts for intervals of selected duration (red LED light on), pauses a few seconds (so you can read out the rate number manually), and resets to count in the next interval. However, the Digicounter does not provide monitoring of the X-ray tube current. In this laboratory we will use the Tel-X-Driver unit to record data. The driver unit controls a stepping motor, and is under computer control so that it can complete scans of rate activity, versus angle, in step as small as 1/6th of a degree. Additionally, the software controlling the stepper unit controls the GM tube high voltage and readout, and also monitors (and records) the X-ray tube current. 6.1 Procedure This section describes the procedure for checking alignment and proper function of the X-ray apparatus. It may not be necessary to perform all these steps, if the equipment was recently used or previously aligned. The Teltron manual entitled “The Production, Properties and Uses of X-rays” has much more detail 40 LABORATORY 6. X-RAY DIFFRACTION on these steps. First check for alignment of the measuring arm with respect to the center post. Lift the hood up and, after ensuring the stepper motor apparatus is not engaged with the central gear, move the measuring arm to zero degrees. With all collimators and GM tube removed from the measuring arm, sight through the measuring arm to check that the center post is aligned straight with respect to the X-ray tube. The apparatus has rulings on the center table base that should line up approximately with the 90 degree marks on both sides next to them. If you see misalignment, make sure to ask Marcus or myself for assistance in correcting it. Crystals of several different types can be mounted into the center post for study. All crystals used in this study are strongly-bonded ionic compounds such as NaCl (“halite”), LiF, RbCl, etc., and all of them have color-painted ends to keep them all straight. Please take care with these crystals - they are carefully grown and cleaved in order to provide pure, well-formed crystal samples with clean faces for very clear X-ray diffraction peaks. Mount a LiF crystal (colorcoded blue) into the center post, and make sure it is secured but DO NOT OVER TIGHTEN. Install two collimators in the measuring arm: a 3 mm slot collimator (TEL 562.016) at Experimental Station 18 (first slot) and a 1 mm collimator (TEL 562.015) at ES 18. Use the 30 kV setting for accelerating voltage (red selector switch to left of crystal post). Rotate the measuring arm to zero degrees (the GM tube is not in place). With the hood still up, turn the key on and turn the timer knob so the filament heats and lights up (no X-rays can be produced when the hood is up). Sight through the 1 mm collimator and make sure the crystal is well-centered with respect to the copper surface shining light through the 1 mm collimator at the basic port. Once it is, you can insert the GM tube in ES 26. It may fit snugly against the collimator in ES 18, so insert gently. Close the hood and engage the interlock. Connect the GM tube cable to the Digicounter and turn it on. Plug the cable into the “Tube Current Monitor” of the unit and connect into the “Ext” of the Ratemeter unit. It should be reading 80 − 90 µA. The GM tube voltage is controlled by the knob in the upper left, and should be left at 425-430 V. Push the red button to start the X-rays (the red “X-rays ON” indicator under the hood lights up). Check that a vigorous count rate is observed within half a degree of 2θ = 45.0◦ , and note what this maximum is, in counts per second (cps). This should fall off dramatically as you move a small amount away from this position. It is the Cu Kα peak. 6.2 Measuring the Kα and Kβ wavelengths NaCl has face-centered cubic crystal structure without one-to-one assignment of any ion Na+ to a particular Cl− ion. Instead each ion is equally paired to the six oppositely-charged ions on orthogonal axes around it. So if the density of salt is known then the bond length follows directly (there is no possibility for extra space between as there is in a covalent molecular solid). Derive this bond 6.3. MEASUREMENT OF BOND LENGTH 41 length, using a precise value for the density of crystalline salt, or halite (see if you can find it to at least four significant figures). You should get something close to 0.282 nm. This known bond spacing, and the regularity of the salt crystal allow it to be used as a diffraction grating to find the wavelength. With the same set of collimators in place make a complete scan of the rates as a function of 2θ, going from 20 degrees up to about 120 degrees. Try to locate three orders of diffraction maxima. The angles are very narrowly defined, and you should be able to see Kα and Kβ lines separately, even for n = 1. Your plot will look something like Figure D14.8 on page 17 of the manual (this also gives you an idea of where to concentrate your attention). The quality of data you take depends on how much time you spend at each position, and how many positions you record. A strategy you might consider is taking 10 readings of 10 seconds duration spaced at 100 intervals around peak positions; and do the same for every 1 degree interval outside this region for 2◦ angles under 30◦ ; while in the high-angle “flat” plateau areas that are not near any peak you can get by with one rate measurement every 2◦ . By taking ten measurements at the same angle you can form the mean central value and use the standard deviation, divided by the square root of the number of measurements, as the error on that mean value. Extract a peak position and uncertainty for each of diffraction maximum. A suggestion for how to do this is given below. Once you have these use the Bragg formula to find the corresponding wavelengths, making sure have the correct order index, m, included. You will have two, and possibly three independent measurements of each of the copper Kα and Kβ wavelengths with their uncertainties. You can combine each of these sets to quote your final single result for these two wavelengths, with your uncertainty (by error propagation). Make sure you include all this information in a table in your writeup. 6.3 Measurement of bond length Reversing the philosophy of your first measurement, now assume the wavelengths of the copper peaks are λα 0.154 nm and λβ = 0.138 nm.1 Repeat the scan procedure for peak structure for one of the other ionic crystals – KCl, RbCl or LiF. These are in labeled tubes and are also color coded. Please make sure to put them back in their tubes and into the box when you are finished. In this part, assuming the values for the copper X-ray wavelengths you will deduce, instead, the bond length and its uncertainty. 1 The exact picture, important for very high resolution measurements, is more intricate: the Kα line consists of Kα1 and Kα2 lines separated by about 0.4 pm, and the value of 154 pm is an average of these two. Similarly there are several lines for the Kβ series. The different energies result from a small splitting of the energy degeneracy between s, p, d, etc. levels in higher-n shells that occurs in many-electron atoms. 42 LABORATORY 6. X-RAY DIFFRACTION 6.4 Analysis and Report Once you plot the count rate vs. angle you’ll see the issues of peak position determination, and possible background subtraction that are involved. Exactly how you carry these out is up to you. You can use a weighted average (discussed in the subsection below), or even a fit (parabolic, Gaussian, etc.) to the individual peaks. Whichever method you use, be sure to assign errors to the angular positions you determine for each peak. Use these, in turn, to give a combined average AND ERROR on either the wavelengths (Part 1) or the the lattice spacing (Part 2). In your report give a basic discussion of the apparatus and theory of operation for the experiment, and include plots of the count rate data as a function of angle. Discuss how you analyzed the data, and how you determined the errors on the final quoted values in each part. In addition answer the following questions, at some appropriate place in the report (most would fit well into the introduction). 1. When you go to have an X-ray done at the doctor’s or dentist’s office, how are they produced? What energy/wavelength do they correspond to? 2. Does the GM tube record any hits when there are no X-rays on? There is an inescapable cosmic ray radiation background, which you should be able to detect. How big is this rate (cps)? Will it affect your results? Discuss. 3. The avalanche pulses recorded as hits by the GM tube have a small, but non-zero time duration. This is included in the quoted “dead time” of 100 µs per pulse (which also includes any electronics reset, or “dead time”). What maximum count rate could the GM tube produce? What would happen if the activity rate exceeded this? Does this “dead time” have a significant effect on altering the count rates you record, perhaps by underestimating them? Discuss. 6.4.1 Peak Angle Determination Suppose that you have identified a peak somewhere by several successive angular readings of enhanced rate vs. angle, and you want to extract a precise angular value for the peak together with an error for that value, as shown in Figure 6.2. In this case the counting rate is shown as a function of location x. One way to get an estimate for the peak position is to fit a quadratic (you will need at least three points to do this) in EXCEL (TRENDLINE option). From the values of the parameters in this fit you can quickly get a peak location. However due to noise, etc. the peak isn’t particularly well fit by a quadratic, or even a Gaussian distribution. A simple ad hoc method is to do a sort of weighted approximation of rate vs. angle for the peak location. Suppose that the locations are given by xi and that the rate counts at these positions are ri . As usual with random events occuring at a consistent average rate, we’ll assume the count rates follow a Poisson distribution, with uncertainty equal to the standard deviation which 6.4. ANALYSIS AND REPORT 43 45 40 35 Count rate, r 30 25 20 15 10 5 0 3 3.5 4 4.5 5 5.5 6 Position, x Figure 6.2: Hypothetical data and counting uncertainties for a peak in counting rate. √ is the square root of the count rate, ri . We can fit for a peak position by 2 minimizing the χ function defined by: χ2 = X [ri (xi − x̄)]2 X 2 = ri (xi − x̄) , √ 2 ri (6.2) where x̄ will be the best estimate of the central location position. Proceeding by setting dχ2 /dx̄ = 0 gives the result: P ri xi x̄ = P . (6.3) ri From this the χ2 value at the best-fit peak location x̄ can be found, and the uncertainty on this angle can be taken through either standard error propagation applied to equation 6.2 or by finding the variation in angle from x̄ that increases χ2 by 1 unit from its minimum. 44 LABORATORY 6. X-RAY DIFFRACTION Laboratory 7 Radioactivity Equipment SpecTech ST360 Interface Unit Geiger-Müller Tube with BNC cable 137 Cs/137 Ba Eluting Kit Computer with ST360 software installed Slotted holder station with tray Various radioactive sources Set of absorber plates We are constantly and inescapably immersed in radioactivity from various sources: electromagnetic radiation from the Sun, TV and radio stations, and cell phones; cosmic rays (charged particles) from outer space; and even radioactive sources that are built into our own bodies (about 1 in 10,000 of all potassium atoms are radioactive 40 K, and roughly 1 in a trillion carbon atoms are radioactive 14 C). In this lab you will investigate the various types of radioactivity, becoming familiar with their detection and also their penetrating properties. There are three fundamental types of emissions from radioactive nuclei: • Alpha radiation: the emission of a 4 He nucleus, a particulary tightly bound combination of two protons and two neutrons, common in the highest-A elements. • Beta radiation: the emission of an electron (or positron) which changes the Z value of the decaying nucleus by +1 (or 1) and is common in nuclei with very small (or very large) values of Z/N , the ratio of number of protons to number of neutrons. • Gamma radiation: the emission of a high-energy photon directly from the nucleus, usually as a by-product when a decay daughter nucleus is born as an excited nuclear state. As a point of distinction, γ rays are emitted from nuclei (and tend to be in the MeV range) and X-rays are produced from inner-shell electrons (and tend to be in the keV range). As you will see these radiations are quite different in nature. 45 46 LABORATORY 7. RADIOACTIVITY IMPORTANT: Although all the radioactive sources in this lab are NRCexempted due to their relatively low activity level, please limit your exposure to them. Keep them in their plastic cases in the box of sources until you are ready to use them. Be sure to put them away when you are done. NO FOOD OR DRINK ALLOWED WHEN DOING THIS LAB!! Familiarize yourself with the ST360 apparatus. Turn the unit on (the power switch is on the back), and open the ST360 application by double clicking on the icon which controls it (it may open automatically - if there isn’t an icon on the desktop, look under: Programs → Spectech → Options → ST360). This program controls the GM tube high voltage and records its count rates. BE CAREFUL WITH THE GM TUBE. There is a thin mica window on the end (to allow all radiation types including alpha to be detected) WHICH IS VERY FRAGILE. If the GM tube isn’t mounted into the holder station already, carefully remove its red protective plastic end cover before inserting into the top of the measuring station, and connect the BNC cable for HV/readout to the ST360 back panel plug. When you are done taking measurements, make sure to turn the HV to zero (from the program menu), turn the ST360 unit off, and make sure all sources are returned to the box. You can leave the GM tube in the holder. There are 7 parts to this lab, to be done over the course of TWO WEEKS: 1. Map the Geiger-Muller “plateau” to determine the tube’s optimum operating voltage. 2. Measure the background count rate. 3. Determine the GM tube’s “resolving time,” also known as “dead time.” 4. Measure the half-life of 137 Ba. 5. Qualitative familiarization with α radiation using the 210 Po source. 6. Study of the penetrating properties of β radiation using the sources and absorber plates. 90 7. Study of the penetrating properties of γ radiation using the and 137 Cs sources. 54 Sr or Mn, 204 109 Tl Cd I suggest you do parts 1–4 the first week and parts 5–7 in the second week. 7.1 Finding the Optimum GM Tube Voltage All GM tubes have their own optimum operating voltages, depending on the size and geometry of the tube. The Tel-X-ometer unit has a smaller GM tube which operates at about 425 V. The GM tube used in the ST360 is larger and has a higher operating voltage. In this first part you’ll observe its operating characteristics and assess what the best operating voltage is. 7.2. MEASURING THE BACKGROUND COUNT RATE 47 Place a 90 Sr source into the clear plastic holding tray (with the hole facing up/foil label facing down) and install it into slot 2 of the measuring station. Power up the ST360 and active its control panel (ST360 desktop icon). In the Setup menu use the “HV Setting” option to control the high voltage. The “Preset” menu allows you to take multiple runs (a series of count measurements) for varying amounts of time. These are recorded into a data buffer you see in the window, which you can write out as a file for use in EXCEL. Also notice the button with a little “erase,” which clears the program’s data window. Set the high voltage to 650 V and take a short run of 10 seconds to see if there are any counts. Then gradually move up to 700 V, looking for where counts begin to register. Once you have found the “threshold” take a series of points from just below threshold voltage, up to 1100 V, measuring the count rate at each one. The program will do this automatically for you: 1. Set the step option “On” in the “Step Voltage Enable” frame (“Setup” menu). 2. In “HV Setting” set the step size to around 20 V, depending on your scan. E.g., if you are starting from 660 V (first point with zero counts), you can do 25 runs with a 20 V increment, which would take you from 660 V through 1140 V. 3. In the “Preset” menu, set the time duration for each step to 30 seconds (under “Preset Time”). 4. Set the number of runs to 25 (under “Runs”). You should see a window for number of counts and data for all the runs and the settings for high voltage and run parameters [If this window is not up, go to the “View” option and select “Counts”]. Hit the “Erase” button to remove any previous data, and then hit the green diamond button to start taking data. Save a copy of the data, and then open EXCEL and read it in (read in the “.TSV” file using the “IMPORT DATA” tab). You can now plot the count rate as a function of voltage, and look for the “GM plateau.” You want a voltage that is in the middle of a fairly stable count rate region. What is the best operating voltage? Be sure to include a plot of this in your report, AND INDICATE WHICH GM TUBE NUMBER YOU USED. One way to assess the stability of the operating voltage in the central region of the plateau is to measure the plateau’s slope. A “good” plateau can be considered to have less than 10% variation in count rate (per second) per 100 V. Does your GM tube have a “good” plateau? Will this value be the same for all the other tubes in the lab? Will it be the same for this tube 10 years from now? Discuss. 7.2 Measuring the Background Count Rate Cosmic rays and natural background radiation are an inescapable source of background that you must quantify and then subtract from any radioactivity 48 LABORATORY 7. RADIOACTIVITY measurements. Remove any sources from the area of the test station and set the high voltage to the optimal operating voltage you determined in the previous section (also turn off the “Step Voltage Enable” feature). Take twenty runs of 30 seconds duration, and combine these into an averaged background count rate (per second) with error. The distribution for a randomly-occurring count rate from a stable source, such as that from this background or from a radioactive source, should a Poisson distribution, which we discussed briefly last Fall: PPoisson (n : µ) = µn −µ e , n! (7.1) where P (n : µ) gives the probability of observing n counts when expecting an overall average rate equal to µ. Make a histogram of the count rates you observed for the twenty 30-second intervals, and overplot the expectation from Poisson statistics to see if they agree. I suggest you use bin intervals of 2 counts, to get more statistics. Make sure to use 20 × P (n : µ) to match your sample size of 20 runs. You can overplot your measurements (points with error bars) with the Poisson prediction (solid curve). Do they agree? Quantify the level of agreement by giving a fit χ2 with NDOF. 7.3 Determining the GM Tube Resolving Time When ionizing radiation traverses the interior of the GM tube, the strong electric field causes electrons and ionized gas particles to separate, rather than quickly recombining as they would in a zero electric field. The liberated electrons accelerate, gaining enough energy to knock off more electrons, resulting in an “avalanche effect” of a significant number of electrons reaching the central cathode wire. This collection of charge registers as a count. The amount of time it takes for this entire process to occur, which is also equal to the minimum amount of time necessary between tracks in order to be able to record a second distinct count, is called the “resolving time,” or the “dead time.” If a second track follows too soon after the first, the tube registers only one count, of a longer pulse duration. You must estimate this resolving time, so you can make corrections for it when the count rates are very high. Let the true rate of ionizing tracks traversing the tube per second be R. Because of the resolving time, τ , the number of counts registered by the electronics is a reduced value, R0 < R. Out of one second, the total dead time is R0 τ so the counting efficiency will be (1 − R0 τ ). [It is assumed that the count rate is not enormous, so there are very few “overlapping” dead hits.] Then the reduction of count rate from its true value R to R0 arises from this efficiency factor: R0 = R (1 − R0 τ ) . (7.2) The strategy for measuring τ is to use a pair of fairly intense calibrating sources, and to observe the difference in the sum of the count rates obtained by 7.4. MEASURING THE HALF-LIFE OF 137 BA 49 measuring each source individually, compared to the rate measured when both are present at the same time. Let the TRUE count rates be called R1 , R2 and R12 , and suppose these are some fixed values. Then from equation 7.2 we have: R10 = R1 (1 − R10 τ ) , R20 = R2 (1 − R20 τ ) , and 0 R12 0 = R12 (1 − R12 τ) . By definition, a fourth equation also holds for the true rates: R12 = R1 + R2 . (7.3) 0 The above constitute a set of four equations in the four unknowns R10 , R20 , R12 and τ . They can be solved for τ , under the assumption that the difference 0 is much smaller than any individual in measured count rates, R10 + R20 − R12 measured count rate. Show that when this assumption is made, you obtain the following approximation for the resolving time: τ= 0 R10 + R20 − R12 . 2R10 R20 (7.4) The values inserted into this equation are the observed counting rates, not the true ones. For example, if we recorded R10 = 540 counts/sec, R20 0 R12 = 640 counts/sec, and = 1020 counts/sec, then the inferred resolving time is 0.230 ms. This would imply complete saturation if the source were “hot enough” so as to approach 4000 or more tracks per second impinging on the GM tube. Among the source collection is a set of calibrating sources, in the shape of half circles. Insert first one half-source into the tray, together with the blank half circle (this helps keep the sources in exactly the same geometrical location with respect to the end of the GM tube). Take 10 runs of data at 30 seconds per run. Repeat for the second half-source in the location of the blank half circle, and the blank half circle in place of the the first half-source. Then repeat for the blank half-source replaced by the first half-source, so that both half sources occupy the tray. MAKE SURE YOU REMOVE ANY SOURCES NOT BEING MEASURED AWAY FROM THE AREA BEFORE TAKING DATA. A reasonable resolving time is in the range of a couple hundred microseconds. Be sure to state clearly your estimated resolving time and its uncertainty. 7.4 Measuring the Half-Life of 137 Ba In this part you’ll observe the exponential nature of the activity for a shortlived source of radiation and compare it to an exponential decay curve, and 50 LABORATORY 7. RADIOACTIVITY then extract its half-life from the data. The decay population of a sample of N radioactive atoms is characterized by the lifetime, τ (not the same as the resolving time!), which is the time constant in the exponential; and the closely related half-life t1/2 which is the time taken for one half of the atoms to decay. These are related by: N (t) = N0 e −t/τ = N0 e −λt t/t1/2 1 = N0 2 (7.5) where the decay constant, λ is related to the half-life by λ = ln 2/t1/2 . The rate at which the population decreases is the count rate you observe in the detector (when adjusted for angular acceptance). It is called the activity: R=− dN = λN0 e−λt = λN . dt (7.6) This is the law of radioactive decay – the activity, or decay rate is directly proportional to the number of radioactive atoms preset. The rate decreases in time with exactly the same exponential decay form as the number of radioactive atoms left in the overall population. The source we will use is the metastable isotope, 137 Ba∗ , which decays to (stable) 137 Ba by gamma emission. This is an example of a nuclear energy level transition which has a relatively long lifetime (“metastable”) because a transition rule for nuclear energy level transitions is being violated. As you will see the half-life is of the order of a few minutes. We obtain samples of it by a clever process using elution which means “the removal of adsorbed material by means of a solvent.” To generate a sample of 137 Ba∗ we use a canister of 137 Cs, which is a longer-lived isotope (t1/2 = 30.1 years) that decays to barium-137 by beta emission. When radioactive cesium decays into barium its chemical properties change from those of a Group 1 element to a Group 2 element. The eluting solution (salt water with a bit of HCl) takes advantage of this to strip out the freshly-formed 137 Ba∗ atoms, leaving the undecayed Cs-137 atoms of the substrate behind. Most of the time (94.4%) the beta decay of cesium-137 is to the metastable excited state of barium-137. Look up the details of this decay online, and find out what the energy of the photon is that is emitted from the decay of the metastable state.1 This is the energy of the photons your GM tube will be detecting. You should use 30 runs of 30 seconds each. Make sure the GM tube voltage is at the optimum level you determined in Part 1. First measure the background rate as you did in Part 2, taking 10 runs of 30 seconds. You can use the resolving time from Part 3 for adjusting the number of counts. When you are ready to start, obtain a small sample of 10 to 12 drops of Ba-137m isotope in solution from Marcus or myself. [We will dispense this into a small aluminum planchet for you – DO NOT do this on your own.] Immediately place the planchet with 1 One of the handiest reference sources is from the Korean Atomic Energy Research Institute, at: http://atom.kaeri.re.kr/. 7.5. QUALITATIVE PROPERTIES OF α RADIATION 51 solution into the plastic holding tray, mount it into the second shelf of the testing station and start the run. After the run has finished you can use EXCEL to analyze the data, and from a plot of vs. time, you can get the decay constant. Apply corrections to your data the “dead time” in the GM tube (using the correction factor you found in Part 3, and equation 7.2), and for the backround rate. Once you have completed this you can use linear regression with a plot of ln(count rate) to find the half-life and its error. Make sure to lay out clearly how you do your data analysis — the order of steps, the logic, the quality of the fit result, etc. Is your measured value consistent with the standard value? Include a plot of your data and the fit result, and your measured half-life, with error, in your lab writeup. 7.5 Qualitative Properties of α Radiation In this part you will carry out qualitative studies of the penetration of alpha particles using a 210 Po source. Alpha particles are enormous in their effective size, compared to electrons or photons. Additionally, the alpha will grab one or two electrons as soon as it can (to become a helium atom) which helps stop its progress even faster. Check the background count rate by taking five 30-second runs of data with the GM tube at its optimal voltage setting WITH NO SOURCE IN PLACE. Then insert the 210 Po source into the top shelf, making sure that the foil/label side is DOWN (the visible hole faces the GM tube). This is a short-lived isotope (t1/2 = 138.4 days) so make sure you use one of the new sources (2008). This shelf is situated only 1.5 cm from the GM tube front face. Record the count rate for 30 seconds. Next, insert a single sheet of paper between the source and the GM tube, and count the rate for another 30 seconds. Finally, remove the paper and move the holder down to slot 2 and record for 30 seconds more. Discuss the penetrating properties of alpha radiation. Does this mean sources of alpha radiation pose no significant health risk? If not, then why is radon decay in basement areas considered a hazard? In 2006, Russian political refugee Alexander Litvinenko died from poisoning by 210 Po – how was this possible, considering the feeble penetration of α particles? 7.6 Beta Radiation Beta particles are electrons emitted from the nucleus during decay, with energies that are typically fractions of MeV. Due to their high energies, when such electrons enter matter they lose energy principally by Coulomb interactions with the heavy nuclei of the atoms in the material. The process is called bremsstrahlung (“braking”) radiation, and is discussed briefly on page 68 of your text. The reduction in intensity of a beam of electrons entering matter is characterized by 52 LABORATORY 7. RADIOACTIVITY the equation I(x) = I0 e−x/X0 , (7.7) where X0 is called the radiation length. For this part you will do a quantitative study to estimate the radiation length of electrons in aluminum using a 90 Sr source. First, use explore the penetrating characteristics of β radiation in different materials by placing the source in slot 2 and experimenting with absorbers of different composition from the set. Is paper a good attenuator? Plastic? Foil? Lead? How would you characterize what goes into making a good β radiation absorber? Discuss this in your report. Now leave the 90 Sr source in slot 2 and measure the count rates per second for source alone, and then trying the various thicknesses of aluminum in the absorber set. You can take 5 runs of 30 seconds for each of the steps. Plot the logarithm of count rate as a function of thickness of aluminum in order to measure X0 (include a plot with your report). You should find a value of about 0.10 cm. The attenuation length depends on the energy of the electrons. What is the energy of the beta rays from your source? How does the attenuation length change, as the energy is increased? Which process(es) of electron energy loss dominates at lower energy, and which at high energy? Be sure to discuss this in your report. 7.7 Gamma radiation Photons interact, and lose energy when interacting with matter by the three mechanisms we discussed in Chapter 2 of your text: the photoelectric effect, Compton scattering, and pair production. The dominating effect(s) depend on which energy range the photon is in. Taking as a scaling factor the electron rest energy, me c2 = 0.511 MeV we can express the photon energy by x = hν/me c2 . At lowest energies (x less than about 0.05) the photoelectric effect is dominant. At highest energies, x > 2, pair production takes over. Most gamma radiation falls between these thresholds, however and so Compton scattering is the principle mode of interaction. There are three gamma sources: 109 Cd, 137 Cs and 54 Mn. Look up the energies of the gamma rays emitted by these sources. Then characterize the general effectiveness of various absorbers on attenuating the radiation emitted by these sources by using your absorber set. Does the energy of the gamma rays have a significant effect on their penetration ability? Finally, use the 54 Mn source to find the radiation length in lead, for the energy of gamma rays emitted by this nucleus. You can proceed in the same way as the radiation length determination in the previous section. A value around 1 cm is expected here. How does your value compare? Again, include a plot with the fit in your report. Laboratory 8 Nuclear Spectroscopy Equipment Spectech NaI scintillator and photomultiplier tube Set of 8 radioactive sources for gamma ray spectroscopy Computer with UC30 software installed The Geiger-Muller tube is a very useful device for detecting radiation. However it gives us little direct information about the characteristics of that radiation. In this experiment you’ll use a different type of detector that is well suited for not only sensing the presence of gamma radiation, but also for giving information about the energy of the gamma rays. This detector, illustrated in Figure 8.1, comes as an integrated, sealed package comprising a NaI crystal encased in an aluminum tube, with a light connection to a photomultiplier tube. The basic mechanism of detection is scintillation – the emission of light when bombarded by radiation – which you already encountered with the phosphorescent rings in the e/m experiment. Here, the NaI (with a small admixture of thallium) is the medium in which the scintillation occurs, under the stimulus of gamma radiation. The remarkable property of this process in NaI is that the number of scintillation photons produced is proportional to the gamma radiation energy. The NaI crystal must remain sealed because it degrades under the influence of humidity when exposed to air. The inside of the aluminum shield that covers it has a highly reflective material that causes nearly all photons that result from scintillation to enter the photomultiplier tube, where the signals are significantly amplified. You should include a thorough explanation of the NaI scintillator and photomultiplier tube in your report. You will need to research the mechanisms of operation yourself (remember to include citations). A manual for the UC30 unit will be made available to you, which includes at least a good start in this direction. It also has useful reference material for carrying out the investigations outlined below. 53 54 LABORATORY 8. NUCLEAR SPECTROSCOPY high-voltage cable readout cable photomultiplier tube NaI crystal, encased in Al Pb shield and mount Figure 8.1: The integrated NaI crystal unit from SpecTech, which is operated by the UCS30 computer interface. Gamma rays that are emitted during nuclear decay do not change A, Z or N but instead carry off energy from the transition of a daughter decay nucleus from an excited state to a lower energy state or the ground state. As an example of this consider the decay of 137 Cs, for which a decay diagram is shown in Figure 8.2. This is a beta decay process and the daughter is 137 Ba. A small fraction, 5.6% of the decays, result in the emission of an electron (and electron antineutrino) and a daughter 137 Ba nucleus in the ground state. However, it is more likely the case that the daughter barium nucleus is created in an excited state – this happens the other 94.4% of the time. The excited-state barium nucleus is “metastable” (has an unusually long lifetime). Its decay to the ground state results in the emission of a 0.611 MeV gamma ray, which you detected with a GM tube in your previous lab in order to measure the half-life of this transition. This process is called an isomeric transition (IT). The NaI detector senses the presence of 137 Ba∗ , and hence 137 Cs, by detecting this gamma ray. There are numerous other decay processes resulting in the emission of gamma rays of unique energy, and they constitute “fingerprints” for the presence of these isotopes, similar to the way in which spectral lines can be used to identify which elements are present in atomic spectroscopy. By providing energy information the NaI crystal allows a similar identification process for certain nuclei, and this technique is called nuclear spectroscopy. Not all radioactive isotopes decays yield gamma rays – this requires an IT or EC (electron capture) in their decay chain. Hence, only these isotopes can be detected. In this laboratory you will examine some of them with this technique. 8.1. OPERATION AND CALIBRATION 55 Beta ray: Max.E(keV) 1176( 1) 892.1( -) 514.03(23) Gamma ray: Energy(keV) 283.5( 1) 661.657( 3) Avg.E(keV) 416.264(72) 300.570(68) 174.320(61) Intensity(rel) 5.6( 2) 5.8E-4( 8) 94.4( 2) Spin 7/2+ 3/2+ 1/2+ 11/2- Intensity(rel) 5.8E-4( 8) 85.1( 2) Figure 8.2: Decay diagram for 137 Cs decay. IMPORTANT: Although all the radioactive sources in this lab are NRCexempted due to their relatively low activity level, please limit your exposure to them. Keep them in their plastic cases in the box of sources until you are ready to use them. Be sure to put them away when you are done. Make sure to turn off the high voltage of the UC30 when you are finished. NO FOOD OR DRINK ALLOWED WHEN DOING THIS LAB!! 8.1 Operation and Calibration Turn on the power for the UC30 interface unit and then open the UC30 software using the desktop icon. A display panel will open which allows you to control the unit, calibrate it, take data, determine peak positions and write output files. During measurements the photomultiplier amplifies light signals from the NaI crystal, producing electrical pulses that are proportional to the energy of the incident gamma ray. These electrical signals are converted into a digital signal by means of a 1024-channel ADC (analog-to-digital converter). The amount of amplification that happens between an incident gamma ray and the resulting pulse height (which is an ADC channel number between 0 and 1023) depends on two parameters: the high voltage of the PMT and the gain of the amplifying electronics that gather the PMT signal before conversion to a digital value. These two parameters “set the energy scale” and it is important to verify/calibrate this conversion process before data can be taken. There are two ways to calibrate: 1. Autocalibration: The UC30 has an autocalibration which uses signals from a 137 Cs source. The autocalibration will systematically adjust both the 56 LABORATORY 8. NUCLEAR SPECTROSCOPY high voltage and gain setting in small steps (you can watch the progress on your screen), moving these up until it sees a “healthy” peak for the 661.6 keV photon associated with this decay. It then calculates the scaling factor for the ADC and changes the digital scale along the bottom of the display into an energy display. This method of calibration is useful for studying lower-energy gamma rays, since the calibration puts the 661.6 keV peak at the upper end of the ADC range. Gamma rays of large energy (larger than 1 MeV or so) will be “off scale” and will not be registered at all. 2. Calibration using known peak positions: You can use a reference source(s) to tell the software how to set the ADC scaling. This method allows you to set the PMT and gain factors to smaller values so that higher-energy gamma rays can be measured. A suggested approach is to use as references the 22 Na source (photons at 511 keV and 1274.5 keV) and the 54 Mn source (photon at 834.8 keV), to perform a “3-peak” calibration. Use a high voltage setting of about 775 V with coarse gain of 4 and fine gain of about 1.6, and take data so that you can see these three peaks clearly, and in the range that you want them to be (you can put both sources at the same time). After you have gathered some data, stop the run and set “ROI’s” (regions of interest) at these locations. Select 3-peak calibration and enter these three peaks and the known energies in the popup menus. Whichever approach you take it is suggested that you periodically check the calibration, especially if you start a new session on a different day, by using the 137 Cs source and verifying the peak location at 661.6 keV. If the position your scaling gives differs by more than 10 keV you should recalibrate. The software will determine peak position (“cetnroid”) and width (FWHM) information, by setting ROI’s, accessed with a right click of the mouse. You can print out a summary of peak information using the DISPLAY menu. If you want to save a spectrum for later use with EXCEL or Mathematica, make sure to save it in tab-separated-value format (extension names “.tsv”), which is NOT the default given in the menu that appears when you use the SAVE command. IMPORTANT: When you are finished please be sure all sources are put away, and that the UC30 high voltage has been turned off. You can do this with the “SETTINGS” tab, where you see the HV ON/OFF radio buttons. Then turn off the power on the interface box. 8.2 Energy Scale and Resolution Complete a 3-point calibration as outlined above. For each one of the peaks you identify in this part of the experiment note the measured peak energy, the ADC channel numbers, and also the full width at half maximum (FWHM), which you can obtain by setting a “region of interest” (ROI). Find the energies and 8.3. COMPTON SCATTERING 57 FWHM values for the 137 Cs and 60 Co sources. You should now have gamma rays of six different energies. Plot the energy vs. ADC channel number. Is it a purely linear relationship? You might try a quadratic fit, which might do better in the case where there is a small nonlinearity across the range of the ADC. As you have noticed already, the width of the peaks increases with the energy. The number of photoelectrons from the PMT varies about a central value in a purely “random walk” manner, which means the shape of a single gamma ray peak is very nearly Gaussian. The width is expected to increase in a manner proportional to the square root of the energy. This relationship is usually expressed in the form: √ E ∆E ∝ , (8.1) E E where for ∆E we can use the FWHM. In order to linearize this, consider instead the square of the quantity on the left, which should be fitted by a line: 2 ∆E 1 =m +b . (8.2) E E Make a plot of the quantity on the left, vs. 1/E, and see how well it fits a line and also whether the intercept is consistent with zero, and discuss these in your report. Improving the energy resolution (making m as small as possible) is an important effort in scintillation detectors. 8.3 Compton Scattering We can understand more about the structure of the typical gamma ray spectrum by recall the mechanisms of Compton scattering, and X-ray production which you studied earlier this semester. In Compton scattering an incident photon of wavelength λ0 scatters from an electron of mass me (considered to be at rest) and emerges with a different wavelength λ0 and with an angle of deflection θ with respect to the original direction of travel. The change in wavelength is given by the relationship for Compton scattering, λ0 − λ0 = h (1 − cos θ) = λC (1 − cos θ) , me c (8.3) where λC = 2.426 pm is called the Compton wavelength. Looking at spectra you have taken so far, it appears that most of the time a gamma ray enters the NaI and undergoes a cascade whereby all its energy is converted into photoelectrons detected by the ADC. However, if the gamma ray undergoes Compton scattering then a different photon of energy E 0 given by: 1 1 1 − = (1 − cos θ) E0 E0 me c2 (8.4) is created, and a high-energy electron is created. When this occurs the photon may exit undetected and the electron may be detected instead. There is evidence 58 LABORATORY 8. NUCLEAR SPECTROSCOPY for this occurring in your data, and the process is noteworthy because it forms an inevitable background in many of the spectra you record. The maximum energy electrons can acquire corresponds to a backscatter event, θ = 180◦ . In this case the electron picks up energy equal to the difference between the incoming and outgoing photons: Emax (electron) = 2E02 , 2E0 + me c2 (8.5) and the scattered photon achieves its minimum (“backscatter”) energy: E0 me c2 Emin (photon) = . 2E0 + me c2 (8.6) An example spectrum for 137 Cs is shown in Figure 8.3. From the photon energy X-ray peaks 100000 Frequency 10000 Cs-137 spectrum backscatter “peak” @ 185 keV plateau main peak @ 662 keV Compton edge 1000 100 10 1 0 200 400 600 800 Energy (keV) Figure 8.3: A spectrum plot from the 137 Cs source, with various features as discussed in the text. Note that the frequency uses a log scale. of 661.6 keV, we predict a “Compton edge” at 477 keV. This is the highest energy that ejected electrons can have. In this spectrum only minor evidence of a backscattered photon peak at 185 keV is seen. There are also X-rays peaks at approximately 34 keV and 80 keV. The first is consistent with a Kα emission from cesium or barium. These could be produced, of course, by scattered electrons liberating an inner shell electron in the material of the sample itself. The second is consistent with a much heavier element – what do you suppose might be causing it? 8.4. USING GAMMA SPECTROSCOPY 59 Use another gamma ray source, such as 54 Mn, and repeat this exercise of identifying the Compton scattering features in your spectrum, and include a plot in your report. 8.4 Using Gamma Spectroscopy There is a source labeled “UNKNOWN” in the set of eight gamma ray sources. Record a spectrum for this material and see if you can identify what’s present there. The printed manual for the UC30 has useful reference material for this in the appendices, especially the lists in Appendices E and F. Give your best guess at what’s there, and see if you can say anything at all about the activities of the unknowns in that sample. Finally, if you wish, you may use the spectrometer to check for either environmental sources in the matter that’s in the lab area (tables, chairs, etc.) or other samples of material you may wish to bring in. Scintillator detectors are used principally in particle physics detectors, and also in forensics applications. In the latter case, while most atoms of a given material are not radioactive, they can be “activated” by flooding them with energetic neutrons at a reactor facility. The resulting sample will then contain neutron-heavy radioactive isotopes of the elements comprising the material which usually have signature gamma rays associated with their decays. This technique is called “neutron activation analysis,” and it can be applied successfully with very small samples of a material of unknown composition (it is the subject of homework problem 12-42). 60 LABORATORY 8. NUCLEAR SPECTROSCOPY Appendix A Review of Error Analysis In this course an ”error” does not mean a mistake, or an admission of deficiency in your measurement. Nor does it mean the difference between the measurement of a quantity that you made and some ”established value” of the same quantity. ”Error” means your own best estimate of the precision of your measurement. Although I refer to this quantity as ”error” it is completely synonymous with ”uncertainty.” This section reviews some of the techniques for estimating the error of your measurement, including the combination of error contributions from multiple effects. A.1 The error from the statistical error on the mean One of the most reliable ways to estimate statistical error is to repeat a measurement many times. Since you won’t get exactly the same answer every time you make a measurement, you will probably get a cluster of measurements about some central value. If these measurements are ”normally” distributed you’ll get something that looks like a Gaussian ”bump.” The average, or mean value is taken as your best estimate of the quantity you are measuring, and the error is taken as the error on the mean, which equals the standard deviation divided by the square root of the number of measurements: σ ε= √ N (A.1) As an example consider the distribution of ten measurements of the value of electron’s charge-to-mass ratio shown by the histogram in Figure. Note that this method requires a minimum number of measurements in order to justify treating the distribution using normal statistics (this minimum is quoted, variously, from 5 to 7). And note also this assumes your data are 61 62 APPENDIX A. REVIEW OF ERROR ANALYSIS distributed normally1 . Always look at the distribution of your data in the form of histogram! Are they distributed in a typical ”bump” fashion? Or are there values that are a long way outside where the majority lie (called ”outliers”)? If there are, find out what happened with these points! Remember, the procedure outlined here will always give you values of the mean and the error on the mean – it is up to you to decided whether it makes sense to do this with the distribution of values you have. Once you have determined the error, a convenient graphical comparison of your result to a reference value can be made by plotting your measurement as a point with error bars and the reference value as a straight line. Consistency at the 1σ level is indicated by the error bar overlapping the reference line. For the value of e/m for the electron from the above example, the comparison is shown in Figure. This figure also allows an illustration of the difference between two descriptions of a measurement that are often confused, or misunderstood. • Precision refers to the size of the error on a measurement. • Accuracy refers to the degree of consistency with a more established result, or standard value of the quantity being measured. In Figure you can see that a result can have either attribute without the other. A result may be precise but inaccurate, due to, e.g., undiscovered systematic error effects. It is also possible for a result to be accurate but imprecise due to making too few measurements, or exaggerated estimations of error contributions that cause the error bars to be larger than they should be. Ideally you should achieve both precision and accuracy – error bars should, to the best of your evaluations and estimations, truly represent 1σ confidence levels. A.2 Error propagation Occasionally you may need to find the error on a derived quantity, which depends on one or more measurements you made and whose errors you have estimated. For example, when using the Bragg scattering formula, d sin θ = mλ to estimating the lattice spacing, d, of a crystal one would measure the peak location θ to an error of εθ , that results from scattering of radiation of wavelength λ corresponding to order of diffraction m. The measurement of d is obtained from the measured peak angle θ, but what is the corresponding error on d? In most cases this error is found simply by treating the input measurement error(s) as differential inputs in a Taylor series expansion as a function of the measured quantities. For example if we have a function f (x), then in the vicinity of a point x0 the value of f is approximated by f (x) ≈ f (x0 ) + df (x − x0 ) dx 1 The term ”normal” has precise meaning in terms of a Gaussian distribution, as discussed in the second Appendix. A.2. ERROR PROPAGATION 63 so that small excursions εx about value x0 translate into excursions of the function f of size εf about the central value f (x0 ) that are obtained from the scaling factor df /dx evaluated at x0 : df εf = εx . dx The absolute value is used because we are not concerned with the sign of the derivative for plus-or-minus variations. This example shows how to treat the error with respect to variation of a single dependent quantity. If there are several dependent quantities, x, y, z, etc. the same idea holds in an extension of this rule: 2 2 2 ∂f ∂f ∂f 2 2 2 (εf ) = (εx ) + (εy ) + (εz )2 + ... (A.2) ∂x ∂y ∂z Notice that errors are combined in quadrature – this is correct only if the errors in the different measurements are uncorrelated, and allows for the fact that the maximal deviation in one dependent variable does not generally occur in concert with maximal deviation in another. Notice also that we have used only firstorder deviations (first derivatives) in these estimations. This approximation is generally valid for cases where the error is much smaller in magnitude than the quantity being measured (otherwise, it’s not much of a measurement, is it?). Here are three special cases of the general relationship above that occur frequently in laboratory analyses: our 1. Direct addition or subtraction of measured quantities results in a combined error that is the quadrature sum of the participating measurements. For example: f (x, y, z) = ax + by + cz has error found from: 2 2 2 (εf )2 = (aεx ) + (bεy ) + (cεz ) . (A.3) 2. Direct multiplication or division of measured quantities results in a combined fractional error that is the quadrature sum of the fractional errors of the participating measurements. For example: f (x, y, z) = xy z has error found from: 2 2 εf εx 2 εy εz 2 = + + . f x y z (A.4) 3. Power-law dependences multiply the error on the measured quantity by the powers of the exponents. For example, if: f (x, y) = xy 3 64 APPENDIX A. REVIEW OF ERROR ANALYSIS then: εf f 2 = ε 2 x x 2 εy . + 3 y (A.5) Therefore, large powers exacerbate the dependence of the overall error on that particular measurement. In this case, the relative precision of the measurement of y has three times the effect of the relative precision of the measurement of x in determining the overall precision of the derived quantity. In the example of the Bragg scattering formula above we have a functional relation that is not directly treated as one of these special cases. If the interplanar separation distance d is considered a function only of θ, d= then mλ sin θ dd mλ cos θ εd = εθ = = d cot θ . dθ sin2 θ The last form is especially revealing – since the same distance d results from measurement at any order of diffraction, it says that if angular uncertainties are the same at all angles of measurement then greater precision on d is obtained from the (higher order) diffraction angles closest to 90 degrees. A.3 Combining several measurements of the same quantity Several measurements of the same quantity, xi , i = 1, ...N , each with error εi can be combined into a single measurement: PN PN xi /ε2i i=1 wi xi = hxi = Pi=1 PN N 2 i=1 1/εi i=1 wi (A.6) provided that none of the errors are correlated. The inverse-error-squared is often referred to as the weight of the measurement, since a relatively smaller error results in more ”power” in determining the final overall value. The combined error is then: v v u uN N u X uX t 2 1/εi = t wi . (A.7) εx = 1/ i=1 I=1 The overall error is always smaller than the smallest of the individual measurement errors – you can only improve by adding more measurements.