Ch 8 & 9 review
advertisement

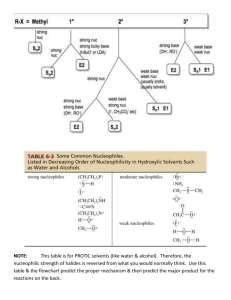
Loudon Chapter 8 & 9 Review: Substitutions and Eliminations CHEM 3311, Jacquie Richardson, Spring 2010 - Page 1 Chapter 9 covers reactions you can do with alkyl halides. For the most part, these break down into two categories: substitution and elimination. Substitution results in replacing the halogen with some other group. Elimination results in removing the halogen, along with a hydrogen on a neighboring atom (the β H), to create a new double bond. substitute eliminate Cl Cl Nu for Nu with base H the B H The molecule that’s having these reactions done to it is called the substrate, and the halogen on it is called the leaving group. The other molecule, the one responsible for changing the substrate, is called the base/nucleophile. That’s because it can play either of these roles. Remember, a base goes after an H only, so if an elimination is happening then it’s acting as a base. A nucleophile prefers to form an attachment to any other atom, so this is the job the base/nucleophile is doing when is causes a substitution. There are two different ways to perform each of these reactions. Substitutions are called either SN1 or SN2, depending on the mechanism. Eliminations are called E1 or E2. The number tells you how many molecules are involved in the rate-determining step; the “2”-type reactions are bimolecular (a.k.a. concerted) while the “1”-type reactions are unimolecular (a.k.a. stepwise). • SN2: the attacking nucleophile is forming a bond to the substrate at the same time the leaving group is detaching. The best way to picture this is as an umbrella turning inside-out. The nucleophile must do a backside attack, by attacking the carbon from the opposite face of where the leaving group is. This results in an inversion of geometry- for instance, if the leaving group had a bold bond before, then the new group will have a dashed bond. This also means that as long as the leaving group and the incoming group have the same priority ranking in CIP rules, the molecule will convert from R to S or vice-versa at the attacked carbon. Finally, since the carbon is trying to hold on to five atoms at once, space is at a premium. The fewer R groups and the more Hs there are on the central carbon, the faster this reaction will go. SN2 Br Nu Nu Br Nu Transition state- don't draw as part of the mech • E2: the attacking base is pulling off the H at the same time the leaving group is leaving. Since everything is happening at once, geometry is important here just like it is for SN2. You need antiperiplanar geometry: the four atoms (H, C, C, and the leaving group) are all in the same plane, with the H and the leaving group pointing opposite directions. Base E2 • H Br SN1: the leaving group falls off on its own, leaving a carbocation behind. Later, the nucleophile comes along and sticks on. Everything we know about carbocations applies here: more substituted is faster, stereochemical information Loudon Chapter 8 & 9 Review: Substitutions and Eliminations CHEM 3311, Jacquie Richardson, Spring 2010 - Page 2 is scrambled, and the molecule can undergo a rearrangement if it’s beneficial to do so. Also, since the carbocation is the highest-energy intermediate, forming it is the rate-determining step and the rate depends only on the concentration of substrate. Nu Nu Br • SN1 E1: the leaving group falls off, then the base comes along and pulls off an H. All the same characteristics apply as for SN1. Since these two reactions start the same way, E1 and SN1 usually occur as a mixture. Base H E1 H Br How can we predict which one of these four will actually happen? 1. Classify substrate as Me, 1o, 2o, 3o, or “honorary 3o” 2. Classify solvent as polar protic or polar aprotic 3. Classify base/nucleophile as strong or weak base 4. Classify base/nucleophile as good or poor nucleophile 5. Use this information to select which mechanism(s) will occur 6. Apply this mechanism to the substrate. Classifying the substrate is relatively straightforward – just group it based on how many R groups are attached to the carbon bearing the leaving group. One exception is that even if a molecule is primary or secondary, enough steric bulk attached to a carbon further away can cause it to act like a tertiary carbon. This is called the neopentyl effect. Br Br Br Br Br Me 1o 2o 3o "honorary 3o" Solvents fall into one of two categories for these reactions: either polar protic or polar aprotic. The difference is whether they can lose a proton readily under the conditions of these reactions. Protic solvents are generally things like ROH, RCO2H, or H2O. Aprotic solvents are generally the ones that go by acronyms: DMSO, DMF, THF, and acetone. In general, polar protic solvents favor E1 and SN1 reactions, while polar aprotic solvents favor E2 and SN2 reactions. Usually this preference is not strong enough to control the choice of reaction, but be careful – using a polar protic solvent with a very strong base can often cause the base to react with the solvent first, which creates a different base/nucleophile that will then go on to react with the molecule. Strong bases are technically anything with a pKa greater than or close to 15.7, the pKa of water. In general anything with a minus charge on C, N, or O is strong, unless there’s additional stabilization coming from somewhere (resonance, etc.). If it’s a strong base then it’s assumed to be a good nucleophile, unless it’s very bulky. Loudon Chapter 8 & 9 Review: Substitutions and Eliminations CHEM 3311, Jacquie Richardson, Spring 2010 - Page 3 Weak bases are anything with a pKa below 15.7. Weak base/good nucleophiles fall into two categories: those at the stronger end of the weak base categories like N3- (pKa of 9.4) and CN- (pKa of 4.7), and those with a minus charge on large atoms (I, Br or S). Weak base/poor nucleophiles are anything outside of this category, including molecules with no minus charge at all. Good Nucleophiles Poor Nucleophiles Strong Bases Weak Bases HO-, RO- (if R isn’t bulky), RC≡C- I-, Br-, HS-, RS-, CN-, N3- O (tBuO-) Cl-, F-, RCO2-, H2O, ROH, RCO2H N (LDA) If you have a strong base: You must do SN2 or E2 only! Carbocations (from E1 or SN1) can’t exist in the presence of strong bases. E2 is the default, unless both the substrate and the base/nucleophile are unhindered. • If you have a strong base/good nucleophile and… o substrate is Me or 1o, SN2 is favored o substrate is 2o, SN2/E2 mix is favored o substrate is 3o or very bulky, E2 is favored • If you have a strong base/poor nucleophile and… o substrate is Me, SN2 is favored o substrate is 1o, 2o, or 3o, E2 is favored If you have a weak base: E1/SN1 mix is the default, but SN2 can happen under the right circumstances. • If you have a weak base/good nucleophile and… o substrate is Me, 1o, or 2o, SN2 is favored o substrate is 3o or very bulky, SN1/E1 mix is favored • If you have a weak base/poor nucleophile and… o substrate is Me or 1o, SN2 is favored o substrate is 2o, 3o, or very bulky, SN1/E1 mix is favored A warning: none of this is set in stone. If you have a weak base that’s getting close to being a strong base, it might produce a little product from acting like a strong base, and the rest of its product from acting like a weak base. Now we can go through a couple of examples: Br NaOH H2O OH + Here, you’ve got a 2o substrate, a protic solvent, and a strong base/good nucleophile. This combination gives you a mixture of E2 and SN2 as the favored outcome. Using SN2 gives you the first product shown, and E2 gives you the second product. Loudon Chapter 8 & 9 Review: Substitutions and Eliminations CHEM 3311, Jacquie Richardson, Spring 2010 - Page 4 H2 O Br OH Here, the solvent is also working as the base/nucleophile. This is called “solvolysis”. You have a 1o substrate, protic solvent, and weak base/poor nucleophile, giving you an SN2 outcome. A few other things to consider: • Regiochemistry of eliminations. Sometimes there are multiple β Hs to choose from. Usually the most substituted alkene will be the major because it’s the most stable. This is called Zaitsev’s rule. However, sometimes you can force the reaction to produce only the anti-Zaitsev product by using a big, bulky base like tert-butoxide. Anti-Zaitsev product tBuOK H H Br EtOK • • • Zaitsev product - usually major Nucleophilicity can change depending on conditions. This is because stronger bases can hydrogen bond to protic solvents, which greatly decreases their availability for attacking the substrate. In general, larger atoms are better nucleophiles in protic solvents and worse nucleophiles in aprotic solvents, and the opposite is true for small atoms. This means that the rate will follow different trends under different conditions. However, the trends listed above for good Nu/poor Nu should be good enough to predict the general outcome of the reaction. Leaving group ability and equilibrium. The best leaving groups are the weakest bases. So if all else is equal, kicking out a bigger halogen is easier than kicking out a smaller one. This also means that some reactions just won’t product much product at equilibrium – trying to replace an F with an I is going to be tough under most conditions. Kinetic isotope effects. Breaking a C-D bond is a little harder than breaking a C-H bond, so if a proton is removed in the rate-determining step (i.e. SN2 or E2), then the reaction will be slower if there’s a D instead of an H there. A few other reactions from chapter 8 and 9 Organometallics: If you take an alkyl halide and expose it to a metal, it’s possible to replace the halogen with a metal atom. This doesn’t go by SN1 or SN2, but a singleelectron-transfer mechanism. The organometallic that’s formed has a big delta minus charge on carbon, which makes it act like a very, very strong base. For this reason, you have to do the reaction in an ether-type solvent like diethyl ether or THF. Br Mg, THF MgBr Grignard reagent Br Li, THF Li Organolithium Loudon Chapter 8 & 9 Review: Substitutions and Eliminations CHEM 3311, Jacquie Richardson, Spring 2010 - Page 5 These reagents can do lots of useful things which we won’t get into until O Chem 2. For now, we’ll only look at one reaction: acid-base reactions with anything protic – water, alcohols, carboxylic acids, etc. MgBr H + H-OR Strong base + -OR Weaker base Radical halogenations of alkanes: this is a way to create alkyl halides from alkanes (unlike doing it from alkenes, which we saw in chapter 5). Here, instead of adding an H and a Br to a double bond, we’re pulling an H off the molecule and replacing it with a halogen. No radical initiator is needed – the dihalogen will split into radicals on its own, as long as you use UV light (written as hν), or heat (written as ∆). 1) Initiation Br Br 2 Br 2) Propagation H Br Br Br Br + Br 3) Termination: any step that involves 2 radicals getting together, for example: This works best for chlorine and bromine. For iodine it’s too endothermic and takes forever to go, and for fluorine it’s too exothermic and goes out-of-control until most Hs are replaced. Bromine is significantly endothermic, so it’s slow and energy-poor. For this reason, bromine will carefully select which H to pull off so that only the most stable radicals are formed. The net result is that radical halogenations with Br2 only puts on a Br at the most substituted carbons. Chlorine, on the other hand, is much less endothermic and can afford to create radicals anywhere. It’s a lot less predictable and basically useless unless every H on the molecule is equivalent. Br2, hv Br Only major product Cl Cl2, hv Cl Cl Cl Cyclopropanation: Both the eliminations we looked at were β eliminations- you lose an H and an LG from adjacent carbons. It’s also possible to do an α elimination, where you lose an H and an LG from the same carbon. These only happen under specific circumstances. To make this happen, you need a substrate that’s good at stabilizing a minus charge, and a strong, bulky base. Loudon Chapter 8 & 9 Review: Substitutions and Eliminations CHEM 3311, Jacquie Richardson, Spring 2010 - Page 6 H Cl C Cl Cl tBuO- Cl C Cl Cl Cl C or Cl Cl C Cl dichlorocarbene This creates a carbene. It looks like a carbon with a minus charge and a plus charge at the same time, although you shouldn’t draw it this way. It’s a neutral carbon with both an empty orbital (so it’s a great electrophile) and a lone pair (so it’s a great nucleophile). The one reaction that we’ll see it doing involves both behaviors at the same time. Cl Cl C Cl HCCl3 tBuONa Cl This is the same mechanism as that simultaneous attack/back-attack thing we used to make bromonium ions, only the three-membered ring it makes is permanent. Since we’re making an all-carbon cyclopropane ring, this is called a cyclopropanation. This particular reaction creates a ring with two halogens hanging off one corner, but what if we don’t want those halogens? In that case, we can use Simmonds-Smith cyclopropanation instead. H I C H I Zn-Cu THF H I C H ZnI carbenoid Combining diiodomethane with zinc-copper couple creates something that’s very similar to a Grignard, but with Zn instead of Mg. It’s not a true carbene. It doesn’t have a full plus and minus charge, but it does have sizeable delta plus (due to the bond to iodine) and delta minus (due to the bond to zinc) charges. For this reason, it behaves like a milder carbene, and it called a carbenoid. CH2I2 Zn-Cu THF H H I ZnI H H Again, it does the same attack and back-attack to make a three-membered ring.