Journal of Alloys and Compounds 334 (2002) 27–33
L
www.elsevier.com / locate / jallcom
Theoretical approach of phase selection in refractory metals and alloys
a
b
c,
C. Berne , M. Sluiter , A. Pasturel *
a
´
Commissariat a` l’ Energie Atomique, Direction de la Recherche Technologique, Departement
des Techniques des Energies Nouvelles,
17 Rue des Martyrs, 38054 Grenoble-Cedex 09, France
b
Institute for Materials Research, Tohoku University, Sendai 980 -8577, Japan
c
´
´ , Maison des Magisteres
`
Laboratoire de Physique et Modelisation
des Milieux Condenses
BP 166 CNRS, 38042 Grenoble-Cedex 09, France
Received 17 June 2001; accepted 13 July 2001
Abstract
First-principles total energy calculations are used to examine the phase stability of the Frank–Kasper (FK) structures. The sequence of
FK phases observed as a function of composition in many transition metal binary systems is shown to have a band-filling origin. We show
that the alloying effects can be treated using a first-principles statistical thermodynamics approach and first results are given for Re–Ta
and Re–W sigma alloys. 2002 Elsevier Science B.V. All rights reserved.
Keywords: Transition metal compounds; Crystal structure; Thermodynamic modelling
1. Introduction
To study the problem of phase formation in alloys, one
has to understand what kind of crystalline structure and
atomic configuration an alloy may probably exhibit at a
given composition and temperature. This is the purpose of
theoretical studies which deal with the prediction of alloy
phase diagrams.
Until recently, most theories were based on phenomenological models. The stability of many structures has been
discussed using geometrical arguments based on space
filling and size effects. Chemical or electronic effects have
been also used. For instance, the Hume–Rothery rules
relate the stability of several structures to the number of
valence electrons per atom. Finally, some electronegativity
scales have been introduced. To study the effects of
composition and temperature, most approaches have been
based on phenomenological thermodynamic models using
regular solution models or various improvements on them.
Recent research shows that it is now possible to deal
with these problems from a microscopic theory of the
cohesive properties of alloys. This microscopic theory is
based on first-principles total energy calculations and on
the development of efficient thermodynamic tools [1,2].
One can easily understand that the use of methods starting
¨
from the Schrodinger
equation and from general thermo*Corresponding author.
E-mail address: pasturel@belledonne.polycnrs-gre.fr (A. Pasturel).
dynamics depends on the complexity of alloys systems;
this explains why alloys based upon single underlying
crystalline structures such as bcc and fcc, for which a large
variety of ordered states can exist, have received much
attention in recent years.
On the contrary few studies have been undertaken on
fundamental grounds to treat alloys based on complex
crystallographic arrangements as in the case of the socalled Frank–Kasper phases (FK) [3]. For transition metal
based alloys, the starting point is the phenomenological
approach formulated by Watson and Bennett [4]; in this
approach, it is shown that the average filling of the d band
is important in establishing which alloys form FK phases
except the Laves phases for which the high degree of
atomic order is usually the result of a dominant size factor.
Previous theoretical works based on tight-binding arguments [5] suggest that electronic effects and more particularly the d-band filling are crucial to explain the
stability of the A15-based structure. To study ordering
effects in A15-based alloys, Turchi and Finel [6] propose
to couple tight-binding calculations in the context of the
generalized perturbation method (GPM) [7] with a thermodynamic variational method such as the cluster variation
method (CVM) [8,9].
To address the problem of FK phases stability using a
first-principles statistical thermodynamics approach, we
propose a twofold contribution.
In a first step, total energy calculations of several
transition metals are performed to analyze how different
0925-8388 / 02 / $ – see front matter 2002 Elsevier Science B.V. All rights reserved.
PII: S0925-8388( 01 )01773-X
28
C. Berne et al. / Journal of Alloys and Compounds 334 (2002) 27 – 33
FK structures, namely A15, sigma and chi, compete with
simple structures such as bcc, fcc and hcp as a function of
the band filling; more particularly, these calculations allow
us to provide an energetic scale or a degree of metastability for these structures which are not observed in elemental
metals but occur frequently in refractory transition metal
based alloys [10].
In a second step, the alloying effects are discussed in
terms of the sequence of occurrence of these FK phases as
a function of composition. We will also show how we can
use total energy calculations to study ordering–disordering
phenomena; we will focus our attention on the sigma phase
which displays different composition limits according to
the studied systems.
In the following pages, we briefly present the firstprinciples based method (Sec. I) used to determine the
structural stability in pure transition metals. Then in Sec.
II, we discuss the phase stability properties of pure
transition metals for A15, sigma, chi, fcc, bcc and hcp
structures. Sec. III is devoted to alloying effects and more
particularly we will emphasize how first-principles calculations can be used to tackle ordering–disordering phenomena in complex structures.
2. General method
2.1. Frank–Kasper structures
It is well known that the FK phases are built entirely out
of tetrahedral packing units [3]. In contrast, the fcc
structure consists of both tetrahedral and octahedral units
while the bcc structure is an intermediate case with the
octahedra containing second-neighbor bonds. The tetrahedral packing in FK phases leads to characteristic polyhedra which are labeled CN12, CN14, CN15, and CN16,
where the numbers refer to the coordination number of the
atom centering the polyhedron.
The three A15, sigma and chi structures contain all the
characteristic polyhedra of the FK phases. The simplest
example of FK phases is the cubic A15 structure since it
has eight atoms per unit cell. Two of these are surrounded
by CN12 polyhedra, which are icosahedra, and the remaining 6 by CN14 polyhedra. The sigma phase is more
complex since it contains 30 atoms and in this tetragonal
structure, there are 5 crystallographically different sites,
commonly designated by A, B, C, D, and E. The coordination shells around the various sites are either icosahedral
(around A and D sites) or are of CN14 (around C and E
sites) or CN15 (around B site) type. Finally the chi
structure contains 58 atoms distributed on four crystallographically different sites designated by A, B, C and D.
The coordination shell around the D site is icosahedral
whereas the A and B sites are surrounded by CN16
polyhedra; the remaining C sites have a coordination shell
made up of 13 neighbors which are not tetrahedrally
coordinated. Strictly speaking, therefore, this structure
should not be considered as a member of the FK structures.
However, studies on alloy chemistry and ordering indicate
that the same general principles hold for chi- and sigmarelated structures.
2.2. Total energy calculations
To compute total energies of several 4d and 5d transition metals, we used the Vienna ab initio simulation
program (VASP) [11] which is based on the following
principles. A finite-temperature density-functional approximation is used to solve the generalized Kohn–Sham
equations with an efficient iterative matrix diagonalization
scheme based on a conjugate gradient technique and
optimized density mixing routines. Within this framework,
the free energy is the variational functional and a fractional
occupancy of the eigenstates is allowed which eliminates
all instabilities resulting from level crossing and quasidegeneracies in the vicinity of the Fermi level in metallic
systems.
The exchange–correlation functional given by Ceperley
and Alder is used in the parametrization of Perdew and
Zunger [12]. Non-local exchange-correlation effects are
considered in the form of generalized-gradient corrections
(GGCs). We use the Perdew–Wang functional [13]. The
GGCs are applied self-consistently in the construction of
the pseudopotentials as well as in the calculations of the
Kohn–Sham ground state. The calculations are performed
using fully non-local optimized ultrasoft pseudopotentials
[14] allowing small energy cut-offs. In this work, particular care was taken for the total-energy convergence
with regard to the k-space integration. The Methfessel–
Paxton k points technique [15] with a smearing parameter
of 0.10 eV was used. The number of irreducible k points
was 182, 190, 150, 56, 18 and 20 for fcc, bcc, hcp, A15,
sigma and chi respectively.
All the parameters of the different studied unit cells as
well as the possible cell internal coordinates were optimized using a conjugate gradient method [11].
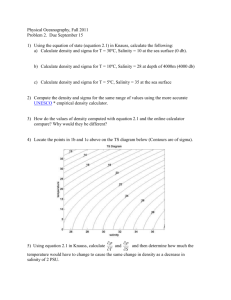
3. Stability in pure transition metals
The total energies were calculated for bcc, fcc, hcp,
A15, sigma and chi structures and for Nb, Mo, Tc, Ru, Ta,
W, Re, Os metals. As already discussed, the three A15,
sigma and chi structures contain all the characteristic
polyhedra of the tetrahedral packing with a percentage of
icosahedral sites which is 25, 33, and 41% for A15, sigma
and chi respectively.
Results are illustrated in Fig. 1. The first spectacular
result is the relative stability of the FK phases for valence
electron per atom ratios (e /a) ranging from 4.5 to 6.5.
More particularly, the A15 structure is found to strongly
compete with the bcc structure for Nb and Mo metals as
C. Berne et al. / Journal of Alloys and Compounds 334 (2002) 27 – 33
29
deviations from such symmetry are small enough to expect
that to a first approximation the bonding of CN12 atoms is
the same as the one which would exist under exact
symmetry of the icosahedron point group. Thus it appears
that the icosahedral sites show a predominance for atoms
with filled or nearly filled, or else empty d shells. Atoms
with d shells close to half filled are mostly found in
coordinations of lower symmetry where the d orbitals may
hybridize to form directed bonds.
Therefore transition metals like Nb, Mo, Ta or W prefer
occupying CN15 and CN16 positions which are of lower
symmetry whereas metals like Re or Os tend to occupy the
CN12 position. A mixture of both elements may occur on
CN14 (CN13) positions and this will be discussed in Sec.
III.
4. Alloying effects in Frank–Kasper phases
4.1. Sequence of the Frank–Kasper phases
Fig. 1. Structural energy differences for (a) 4d metals and (b) for 5d
metals.
well as for Ta and W metals. Let us mention that this
peculiar behavior of the A15 phase has been used to
interpret the occurrence of a metastable phase in Ta metal
brought on by drop-tube undercooling [16]. The electron
concentration found in our calculations is very similar to
the one obtained by Phillips and Carlsson [17] or by
Watson and Bennett [4]. However, our study emphasizes
that, in this range, the FK structures strongly compete with
the bcc and not with the fcc as assumed by Phillips and
Carlsson [17].
The second important result of the present calculations
is that all of the lines in Fig. 1 intersect at one particular
band filling, namely e /a56.7. As a consequence, the
sequence A15-sigma-chi also depends on the band filling.
As these three FK phases display a different percentage of
icosahedral sites, i.e. CN12, it is tempting to relate their
relative structural stability with this percentage. Indeed
perturbations conforming to the point group of the
icosahedron do not affect the degeneracy of the atomic d
levels whereas for all of the other coordination symmetries
the d-level degeneracy is partly broken. Actually, the
surroundings of CN12 atoms do not exactly conform to the
point group of the icosahedron. However, the effective
Results presented in Sec. II can be used to understand
the sequence of FK phases in transition metal based alloys.
For instance, if a chi phase exists at a given composition in
an alloy, a composition change which reduces the e /a ratio
could possibly give rise to a new phase such as a sigma
phase, an A15 phase, or a bcc solid solution, but not an
hcp phase. Reciprocally, if the e /a ratio increases an hcp
phase may occur. If the same arguments are applied to a
sigma phase a decreasing e /a may lead to the occurrence
of A15 or bcc phases but not to chi or hcp phases. Thus,
with increasing the electron concentration, the following
succession of phases may exist: bcc-A15-sigma-chi-hcpfcc. They are many binary alloys of 4d and 5d transition
elements in which these sequences can be checked [10].
For instance, the sequence bcc-sigma-chi-hcp can be found
in the Ta–Os, Ta–Re, W–Re, Nb–Re, and Mo–Re systems. The sequence bcc-A15-sigma-hcp occurs in the Mo–
Os, Mo–Tc, and Nb–Rh systems. In the Nb–Os system
the complete sequence bcc-A15-sigma-chi-hcp occurs.
However, if this approach provides a nice interpretation
of the sequence of the FK phases as a function of electron
concentration, it does not predict their actual occurrence
for a given alloy. To try to answer this question, it is
necessary to treat alloying effects in these structures.
Another difficulty arises from the fact that some FK phases
like sigma or chi do not have a definite stoichiometry.
Hence their occurrence varies with composition and thus
with electron concentration. We present now a first-principles based treatment of the alloying effects in the sigma
phase.
4.2. Alloying mechanism in the sigma phase
When dealing with the general problem of phase stability in an alloy from a microscopic description, one has to
C. Berne et al. / Journal of Alloys and Compounds 334 (2002) 27 – 33
30
combine electronic structure calculations and statistical
studies. This key may be played by an Ising Hamiltonian
for which the energetics associated with changing local
atomic configurations is parametrized by a set of effective
cluster interactions (ECI’s). Total energies of a large
number of configurations are used to extract these effective
cluster interactions following the Connolly–Williams
method (CWM) [18]. The ECI Vi for a cluster i is
calculated with:
Ov SE
a
a
OV j D 5 min
n
a
form
2
2
i
a
i
(1)
i 51
where j is the cluster correlation function as defined in Eq.
(10) of Ref. [19]; v represents the weights assigned to
each structure. Formation energies Eform are extracted from
the total energies Etot by substracting the concentration
weighted total energies of pure constituents with the lattice
structure.
Eq. (1) is solved using a singular value decomposition
(SVD) procedure. As in the spirit of Ref. [20], the sets of
equations corresponding to thermodynamically stable
structure are given more weight than the equations pertaining to unstable structures. The weighting in the SVD
procedure provides an alternative to the linear optimization
technique for ensuring that the cluster expansion reproduces the proper sequence of structures energy. An
additional benefit of weighting the lowest energy states
more heavily in the CWM method is that it makes the CE
convergence more rapid.
Let us consider the case of the sigma phase. For a
AB-sigma alloy, the five inequivalent sites of the sigma
phase make 2 5 532 possible distributions of A and B
atoms. These 32 configurations are not superstructures of
the sigma phase because they all have the same space
group symmetry. In addition, 10 sigma superstructures
were considered that allowed us to determine the pair
interactions between sites of the same type (i.e. B–B,
C–C, D–D, and E–E type pairs). The formation energies
of these 42 configurations are computed with a full
geometry relaxation including both the optimization of the
a and c parameters of the tetragonal unit cell and the
optimization of the internal atomic coordinates. The SVD
algorithm is used to extract effective interactions [21]. A
set of 29 and 27 interactions was shown to reproduce the
formation energies with a root mean square error of 0.06
kJ / atom for systems like the Re–W and the Re–Ta
respectively. This error is well within the precision of the
total energy calculations and is much smaller than the
smallest difference between the formation energies of any
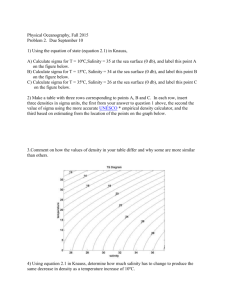
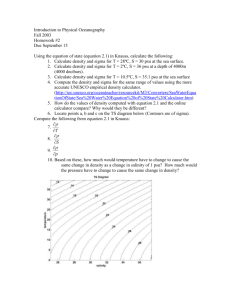
of the 32 configurations used in the CWM (see Figs. 2–3).
The interactions are used to compute the configurational
entropy and the associated Helmholtz free energy as a
function of composition and temperature within the CVM.
The site occupancy of the 5 inequivalent sites are also
determined in these calculations. In the present case, the
Fig. 2. Formation energy (in kJ / atom) of Re–W relaxed atomic configurations with the sigma structure: as computed with VASP (1); as
computed from the effective interactions (1). Circles emphasize the 10
configurations that are sigma superstructures. The most stable configurations have been connected with a solid line and the occupancy of the A,
B, C, D, and E sites have been indicated.
tetrahedron approximation is a natural choice since the
sigma phase is tetrahedrally close packed. For this structure, the tetrahedron approximation leads to 17 tetrahedron
maximal clusters and a total of 71 correlation functions
[22]
Figs. 4–5 show the site preference for two systems,
namely Re–Ta and Re–W systems, at T5500 K. The first
comment is that the site occupancy as a function of W or
Ta composition has the same sequence for both systems.
First the B site happens to be filled with W (or Ta) and
takes most W (or Ta) content; then come sites C and E, and
finally the A and D sites are filled. Results of Sec. II allow
us to understand the clear preference for W or Ta to
Fig. 3. Formation energy (in kJ / atom) of Re–Ta relaxed atomic configurations with the sigma structure: as computed with VASP (1); as
computed from the effective interactions (1). Circles emphasize the 10
configurations that are sigma superstructures. The most stable configurations have been connected with a solid line and the occupancy of the A,
B, C, D, and E sites have been indicated.
C. Berne et al. / Journal of Alloys and Compounds 334 (2002) 27 – 33
31
Fig. 4. Computed occupancy of the inequivalent sites in the Re–W sigma phase at T5500 K.
occupy the lower symmetry B, C and E sites and for Re to
occupy the icosahedral A and D sites. As discussed in Ref.
[21], more subtle alloying effects are necessary to understand the sequence between the sites of lower symmetry,
namely B, C and E sites. For instance, another E–C–B
sequence was found in the Fe–Cr sigma alloy [22].
In Figs. 6–7, the site occupancy of both Re 0.6 W0.4 and
Re 0.6 Ta 0.4 alloys as a function of temperature is shown. At
high temperatures, the site occupancy very slowly reaches
the value corresponding to the random composition. This
limit occupancy being determined purely by the alloy
composition, in this case the limit of each site is 60% Re.
However, this limit is not obtained because no order–
disorder transformation due to the removal of symmetry
elements in going to the chemically randomized configuration takes place in the present calculations. The occupancy
of B, A and D sites reaches the high temperature limit
more rapidly in Re–W than in Re–Ta, due to weaker
alloying effects in Re–W system. In this case, the temperature effects on the ordering mechanism are more pronounced. When the temperature is lowered, the fraction of
W (Ta) on B site increases to reach an occupancy of 100%
around 500 K for Re–W and 2000 K for Re–Ta. For the
same temperatures, the A and D sites display an opposite
behavior which gives a Re occupancy of 100%. For both
alloys, the C and E sites have an important mixed
occupancy which is more or less independent of the
temperature.
These results may be used to predict composition limits
for the sigma alloys. At very low temperature, we can
assume that only Re occupy A and D sites and only W (or
Ta) B site and a mixture of Re and W (or Ta) C and E
Fig. 5. Computed occupancy of the inequivalent sites in the Re–Ta sigma phase at T5500 K.
32
C. Berne et al. / Journal of Alloys and Compounds 334 (2002) 27 – 33
Fig. 6. Computed occupancy of the inequivalent sites in the Re 0.6 W0.4 sigma phase as a function of temperature.
Fig. 7. Computed occupancy of the inequivalent sites in the Re 0.6 Ta 0.4 sigma phase as a function of temperature.
sites; thus the predicted limits are 13–66% W (or Ta)
taking into account the multiplicity of the crystallographic
sites. In fact this approach provides an over-limit because
the true composition limits of the sigma phase are given by
the competition of the other equilibrium phases and more
particularly the bcc solid solution.
when increasing electron concentration which is found in
the binary metallic systems based on refractory metals.
The alloying effects are discussed more particularly in the
sigma phase. It has been shown that the site occupation in
such a complex structure displays a peculiar behavior as a
function of composition and / or temperature which can be
explained from the site-coordination numbers.
5. Conclusion
We have shown that the structural stability among the
FK phases depends on the band filling of pure transition
metals. This behavior is related to the occurrence of high
local coordination sites which are more favorable for
metals with d-shells close to half-filled. These results are
also used to interpret the sequence bcc-A15-sigma-chi-hcp
References
[1] D. de Fontaine, Solid State Phys. 34 (1979) 73.
[2] F. Ducastelle, in: F.R. de Boer, D.G. Pettifor (Eds.), Order and
Phase Stability in Alloys, Cohesion and Structure, Vol. 3, North
Holland, Amsterdam, 1991.
[3] A.K. Sinha, Prog. Mater. Sci. 15 (1973) 79.
C. Berne et al. / Journal of Alloys and Compounds 334 (2002) 27 – 33
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
[12]
[13]
[14]
R.E. Watson, L.H. Bennett, Acta Metall. 32 (1984) 477.
P.E.A. Turchi, G. Treglia, F. Ducastelle, J. Phys. F 13 (1983) 2543.
P.E.A. Turchi, A. Finel, Phys. Rev. B 46 (1992) 702.
F. Ducastelle, F. Gautier, J. Phys. F 6 (1976) 2039.
R. Kikuchi, Phys. Rev. 81 (1951) 998.
J.M. Sanchez, F. Ducastelle, D. Gratias, Physica A128 (1984) 334.
T.B. Massalski, H. Okamoto, P.R. Subramanian, L. Kacprzak (Eds.),
Binary Alloys Phase Diagrams, American Society of Metals,
Materials Park, OH, 1990.
G. Kresse, G. Furthmueller, Phys. Rev. B 54 (1996) 11169.
J.P. Perdew, A. Zunger, Phys. Rev. B 23 (1981) 5048.
J.P. Perdew, Y. Wang, Phys. Rev. B 33 (1986) 8800.
G. Kresse, J. Hafner, J. Phys.: Condens. Matter 6 (1994) 8245.
33
[15] M. Methfessel, A.T. Paxton, Phys. Rev. B 40 (1989) 3616.
[16] L. Cortella, B. Vinet, P.J. Desre, A. Pasturel, A.T. Paxton, M. van
Schilgaarde, Phy. Rev. Lett. 70 (1993) 1469.
[17] R. Philipps, A.E. Carlsson, Phys. Rev. B 42 (1990) 3345.
[18] J.W.D. Connolly, A.R. Williams, Phys. Rev. B 27 (1983) 5169.
[19] M. Sluiter, D. de Fontaine, X.Q. Guo, R. Podloucky, A.J. Freeman,
Phys. Rev. B 42 (1990) 10460.
[20] M. Sluiter, Y. Watanabe, D. de Fontaine, Y. Kawazoe, Phys. Rev. B
53 (1996) 6137.
[21] C. Berne, M. Sluiter, A. Pasturel, Phys. Rev. B (2001) in press.
[22] M. Sluiter, K. Esfarjani, Y. Kawazoe, Phys. Rev. Lett. 75 (1995)
3142.