Phase Transfer Catalysis and Solvent
advertisement

Eric Dammer
Dr. Jose Baretto
Organic Chemistry I
June 21, 2001
Lab 3: Regioselective electrostatic environments of inert immiscible or miscible solvents enable reactivity
across solution interfaces and provide a means of identification and separation through a combination of
solvation effects and shifts in primary resonance contributors of certain molecules such as methyl red.
Abstract
Of interest is the location and relocation of solute due to mechanisms enabling mobility across the
solution interface of immiscible solvents. Successful phase transfer of otherwise incompatible or more
poorly soluble potential reagent solutes into a common solvent layer makes possible the wide array of
syntheses that have been a cornucopia for the industrial production of various organic chemicals of
practical and economic importance.
Procedures testing the viability of extraction by phase separation of pure molecular components of
bimolecular mixtures are examined. These procedures are extrapolated and adapted for tests of the
aforementioned assisted phase transfer of the same test solutes (consistently, methylene blue and methyl
red) as enabled by the detergents SDS and CTAB. Each detergent is chosen to specifically target phase
transfer of the counterion or most ionic substituent of the target ion or molecule for phase transfer,
respective to the azobenzene dyes referenced above. The result is a successfully catalyzed removal of the
target species from the phase in which the lowest energy state of the molecule would otherwise be
achieved. Thus, there is a step up in the free energy of the target solute molecules successfully transferred
for potential exposure to yet higher energy states once in a common phase with other reagents intended for
reaction with the transferred species. While additional energies for the reactions just mentioned are not
considered in this procedure, it is currently conjectured that the transfer of molecules to specific locations
near similar solution interfaces with some additional mechanisms of molecular selectivity may allow highly
specialized synthesis of organics that could be mimetic with respect to the mechanisms of biochemical
pathways. This analogy is strengthened by the similarity of the employed detergents’ hydrophobic
hydrocarbon chains and hydrophilic polar heads to the structure of phospholipid bilayers that are the
centerpiece of the fluid-mosaic model explaining the functional structure of eukaryotic cell membranes.
Finally, procedures employing the same polar or nonpolar electrostatic solvent interactions are
used to separate samples, usually for identification purposes in thin layer chromatography (TLC). The
procedure here is concerned with discovering the characteristics of ideal solvents, which turn out to be
compatible mixtures of solvents that in conjunction are moderately polar, when the plate is similarly polar,
as in the specific silica gel plate coating used in this case.
Dammer, 2
Data
i. Extraction
In the extraction procedure, either methylene blue or methyl red was added to a test tube filled
with two immiscible solvents in equal volumetric proportion. The two sets of solvents were water and
hexane or water and methylene chloride. In each case, the denser solvent comprised the solvent layer
below the solution interface in the tube. In all, four different solute-solvent combinations were produced
when either methyl red or methylene blue azobenzene dye was added. The four tubes were stopped, shaken
and observed before being centrifuged for a short period after which additional observations were noted. In
table one, the lighter solvent in either set is in the left column.
Solute
Methylene Blue
Methyl Red
Table 1: Visible Solvation Effects in Immiscible Solvent Pairs
Hexane
/ /
Water
Water
/ /
Uncolored, clear
Blue
Blue
CH2Cl2
Uncolored,
clear
Yellow
Pink, slightly cloudy
Clouded
Orange
Uncolored, clear
Deeper Yellow
Blue
Pink-orange
Blue
Less clouded
Slightly blue
Orange
After Centrifuge
Methylene Blue
Methyl Red
Characteristics of the nonprotic solvents relative to water as well as the polar silicate surface of
each test tube could be gauged by the meniscus at the solvent-solvent interface; methylene chloride is less
adhesive to the polar silicate tube surface in that a convex bulge upward pushed into the aqueous phase. In
the case of hexane, a concave depression at the solvent surface was similar to an aqueous meniscus,
indicating some polar intermolecular interaction between the hexane and glass.
It is clear that methyl red preferred the denser aprotic organic chloride solvent to water in that the
water in this set did not maintain any red color, even with gentle mixing. The shift from red to orange is of
interest as an indication of shift in intramolecular electronic interaction. Methylene blue leaves the polar
water phase with difficulty, if at all. Only after centrifugal force pulled the heavier solute molecules down
into the methylene chloride did a slight blue coloration appear in methylene chloride while none became
visible in hexane.
While results of the mixing were relatively unchanged by centrifugal separation other than
clearing of clouded, or partially emulsified solvents, there was a notable exception: hexane color due to
dissolved methyl red deepened. The Beer-Lambert law relates absorption of light at any particular
wavelength by any agent in solution with molar concentration. An inference may be made by the higher
intensity of yellow that more methyl red had entered the hexane phase. These trends were confirmed stable
over a one-day period in which the yellow hexane increased in color intensity while water in the same tubes
became a lighter pink.
The reason for a color shift from red to yellow in hexane-dissolved methyl red and a middling
shift into orange transmittance of the same solute molecule in the considerably denser and more polar
methylene chloride can be clarified through analysis of the variable effects of solvation on the electronic
character of the methyl red molecule.
Dammer, 3
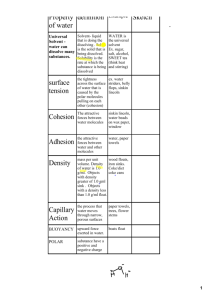
Fig. 1: Methyl red ionic tautomers, basic anion, and possible solvent-induced neutral
molecular structures.
resonance
Tautomers under
acidic conditions
pH shift
azo bond torsion
Anion in basic
conditions
pKa shift
Neutral molecule
It appears that solvation effects may raise the pKa of the methyl red molecule due to increased
nucleophilicity of oxygen relative to a nonpolar solvent like hexame. At the same time, the nucleophilicity
of the basic nitrogen atom in the molecule would be decreased in that nearby charges in a nonpolar solvent
phase would be electrostatically encouraged to exchange the proton. Thus, as time passed and though the
introduction of higher energy fluid kinetics in the centrifuge, the diagramed higher energy conformer
(center, right) would statistically occur. If occurring at or near the solution interface, the torsional strain
and increased electrostatic attraction may both be satisfied through rearrangement to the neutral molecule,
effectively possible within hexane. It is not clear from this experiment whether hydrogen exchange takes
place due to this effect, or whether there is an incipient neutral molecule with some torsional strain
preferred in the hexane. Because hexane is an unbranched alkane of some length, it should undergo
temporary induced dipoles due to the proximity of charged species especially like the methyl red
zwitterion. This makes cross-interface motility possible for methyl red with respect to the water-hexane
interface. The supplemental effect on the methyl red ion is a destabilization of its charged groups such that
pKa shifts higher as described, encouraging a neutral state of the molecule. Notice that both in the yellow
basic anion and in this neutral form of methyl red, there is one primary resonance contributor in which all
eight double bonds are conjugated. On the other hand, one resonance of the red zwitterion in acidic
conditions does not have this complete conjugation.
Comparing the concentration of red to yellow resonance structures in any phase is made possible
by Lambert’s Law, relating concentration to absorptivity with respect to the integral, or uniformly mixed
area, of the phase through which light passes. Apparently, the less conjugated resonance contributor
predominates in acid conditions, as inferred by the overall lower transmittance (and directly corresponding
lower absorption, on the yellow-to-red range of the visible spectrum). Interestingly, methylene chloride
could be stabilizing the more conjugated resonance contributor under the existing acidic conditions, so that
concentration of both is approximately equal, giving the orange hybrid. Its more compact spherical,
Dammer, 4
tetrahedral geometry also allows greater solvation effects of either zwitterion resonance structure, so this
seems a likely explanation.
An alternative explanation is that the methylene chloride is encouraging formation of the
effectively neutral form of methylene red in the same manner as hexane; however, methylene chloride has a
considerable net dipole that would not favor as great of a pKa shift as the highly nonpolar, but temporarily
polarizable, hexane chains, so this seems a likely explanation of the partial yellow character of the phase in
the presence of methyl red.
Certainly, the methylene chloride phase was not polar enough to effectively dissolve methylene
blue, the quaternary ammonium salt pictured to the right. This ion has one effective resonance structure
because nitrogen is more electronegative than
sulfur, the other possible charge carrier in the
molecule.
Based on the observations, it becomes
apparent that a fast, efficient partial separation of
methylene blue and methyl red can be
accomplished across the water/methylene
chloride interface. For a highly selective,
complete separation, it would be best to inject a
mixture of the two at the hexane-water interface.
Separation would be completed by pipetting out
the solvent of choice and replacing it with fresh
solvent as needed until no additional coloration
Figure 2: Methylene Blue
of the extracting solvent occurred. At that point,
the separated solvents could be boiled away to
give the separate dyes in dry form.
ii. Phase Transfer Catalysis
In a related procedure, phase transfer catalyzed separation of the same dyes was performed using
either of the two detergents sodium dodecylmethyl sulfate (SDS) or CTAB. Each is effectively equivalent
but donates detergent anions and cations, respectively. A long hydrophobic tail on each of the charged
detergent species makes them motile in either phase, and particularly so across the solvent-solvent interface
of the same immiscible solvent pairs already mentioned.
Table 2: The Effects of Phase-Transferring Catalysts on Specific Solvent-Phobic Species (After
Centrifuge)
Solutes
Hexane
/ /
Water
Water
/ /
CH2Cl2
Methylene Blue /
Dark Gel matrix,
Slightly blue
Light blue gradient
Dark blue
SDS
w/ surface hexane
liquid
Methyl Red /
Yellow, cloudy
Light, clear
Light yellow
Bright yellow
CTAB ~5 mg
wisps
In this phase transfer catalysis procedure, it became apparent that the water/methylene chloride
interface caused more common detergent effects, especially slow-moving suds, and a gradient from one
phase to the other in which these suds, or in the case of CTAB in hexane, wisps could be seen blurring what
was before a sharp interface of immiscible liquids. The interface also no longer defined color as sharply as
observed in simple extraction—instead gradients in color could be seen.
The most interesting effect observed occurred in the hexane with SDS-catalyzed transfer of
methylene blue. Apparently, the detergent could transport the charged molecule only with difficulty.
Initially, only 4.3 milligrams were added to the tube, corresponding to and only a minor grey tint appeared
in the hexane. When the molar concentration of this detergent was exactly doubled, however, the
centrifuged tube produced the observed matrix in the hexane phase. Interestingly, the hexane still separated
from the gel when the tube was shocked, indicating a matrix structure trapping some nonpolar hexane. It
seems that the detergent successfully transferred the chlorine counterion of the quaternary ammonium
cation into the hexane phase, establishing electrostatic potential across the solution interface that drove the
large, bulky methyl red into the low-density hexane. However, because of the polarity difference, the
deposited ions could only stabilize by forming a semi-repeating structure within and between hexane
Dammer, 5
molecules, leading to the observed gel-like matrix. Earlier, only small clumps of material formed in
relatively planar macromolecular arrangements, but the increased detergent concentration enabled the
growth of these quasi-crystals in three dimensions, exclusively in the nonpolar solvent. Judging from the
shock-sensitivity of the product, methylene blue and its chlorine counterion do not have molecular
geometry to pack tightly, and lack sufficiently rigid bond character to resist torsional strain to be capable of
forming crystals under standard conditions.
iii. Thin-Layer Chromatography
A variety of solvents and mixtures of miscible solvents was tested for effectiveness in running
samples of methyl red, methylene blue, and a mixture of these two characteristic dyes. The three plates and
solvent used for each by this group were identical and run as the same time, for a total of twenty-five
minutes. Observations here are for a volumetric 50/50 mixture of methylene chloride and methanol as the
solvent.
In the first minute of the run, the rate of movement of solute, carried by the solvent’s polar
capillary interaction with the plate, was established as differing between the blue streak and the red streak;
each additional minute of the run reinforced the observation of a steady rate of movement. This rate was
35 mm per minute for methyl red and ~30 mm per minute for methylene blue Considering some spots of
uneven application across the plate, it became obvious that these tended to move at a slower rate while
remaining connected by a vertical sweep in the thin line to the leading edge of sample, highest on either
edge of the plate. Total movement on the plate was 7 cm for methylene blue and 8 centimeters for methyl
red. Apparently, the methyl red zwitterion consistent with its unadjusted acidic environment provided two
charged “handles” for electrostatic such that it could more consistently bind with the polar solvent initially
crossing any sample streak as it moved up the plate.
The collective data for other groups including the above results indicates that there were three
general categories of solvents tested: 1) polar, protic solvent (acetic acid); this tended to smear both
samples equally, though with some minor difference (< 1 cm) in the distance from the starting point
between red and blue. The reason for smearing may have to do with hydrogen exchange, which readily
carries and drops solute molecules with little selectivity. 2) nonpolar solvents (hexane, cyclohexane,
petroleum distillates {especially branched pentane and butane}); these did not pick up the samples,
particularly because the samples used were polar and ionic, thus opposed to spontaneous strong interaction
with nonpolar species. In addition, the solvent could not bind to the plate as effectively, hence as quickly,
as polar solvents. 3) moderately polar solvents (in order of polarity: methylene chloride, trichloromethane,
50/50 methylene chloride/isopropanol, and the originally mentioned 50/50 mixture, methylene
chloride/methanol); these solvents carried the samples without smearing and on the extreme ends of the list,
did not move the samples more than a centimeter in the less polar, while in the given solvent, samples ran
at a relatively fast rate without great selectivity. The ideal solvent in the case of these charged azobenzene
dyes was the methylene chloride/isopropanol solvent, with considerable separation of about 3.5 cm
between the final streaks, even on the plate running the mixed sample.
Dammer, 6
Discussion
Caffeine and salicylic acid structures are given respectively in figures 3 and 4. In order to separate
a mixture of these two compounds in solution, given the experience garnered with methyl red and
methylene blue, it is apparent that certain methods would be more appropriate to achieve certain ends.
Before employing any of the above methods, a solvent would have to be found to dissolve both
compounds. It would appear that both species would readily dissolve in protic compounds, due to
protonation of the sp2-bonded nitrogens in caffeine and proton exchange or donation by the carboxyl in the
acid. Caffeine, in fact, is stabilized if protons bond to three filled pi antibonding orbitals corresponding to
the amine heteroatoms. Otherwise, the rings in caffeine have an even number of pairs of pi electrons,
making the compound antiaromatic. In a polar aprotic solvent, the solubilities of both compounds would
decrease, but remain sufficient for the purposes of separation, because each has carbonyl substituents
contributing to substantial dipole moments in either of these molecules.
Once relegated to a good streaking solvent with polar solute interaction like that seen like the
50/50 methylene chloride/isopropanol mixture, thin layer chromatography would effectively separate trace
samples of the two compounds from mixture in the solvent. There is a difference in the number of
electrostatic “handles” on either of the molecules; caffeine has 3 filled pi antibonding orbitals, one per
nitrogen—and two polar carbonyl groups. Compared to the two polar aryl substituents in ortho positions
relative to each other, the streaking solvent would pull caffeine to the top of the plate, while the remaining
acid would be considerably lower down, because the polar solvent will have carried each up at sufficiently
different rates, assuming the plate contains silica.
The complication with TLC in this case not yet mentioned is that both compounds are colorless.
Therefore, dry samples from the separated streaks on the plate ideally should be analyzed using IR, NMR,
or mass spectrometry techniques to verify the purity of the compounds and the accuracy of this prediction.
Another combination of moderately polar solvents maximizing regioselective electrostatic interaction based
on steric hindrance at the key molecular “handles” of one molecule only.
Fig. 4: Salicylic Acid
In extraction, larger quantities of the
(Oblique View)
compounds could be separated practically. In a
basic polar solvent, such as water pH adjusted
Figure 3: Caffeine
with ammonia gas, caffeine would initially
dissolve, along with salicylic acid, which would
immediately become deprotonated. Introducing
a second immiscible solvent phase to the
extraction environment would begin the
separation. A nonprotic, nonpolar solvent in
this second phase would invite some caffeine into the new phase, as its
hydrocarbon branches and neutral net charge will permit and encourage
this. In addition, Le Châtelier’s principle would explain the transfer of
some fraction of the total caffeine concentration to the added solvent so
that concentration in both solvents could reach equilibrium. Even better transfer equilibrium for the
purpose of separation could be forced, had the ammonia gas been bubbled through the apparatus already
containing both solvents and the mixture, because there would be a decreased solubility in the water as the
pH is driven up, while the ammonia consumes water in the process of becoming aqueous ammonium
hydroxide. The result would be partial separation in either case, but the nonpolar solvent, such as
cyclohexanol, could be removed from the apparatus and then forced to deposit caffeine by distillation,
whereupon it could be recycled back into the extraction apparatus and the process could be repeated until
no caffeine remained available for phase transfer.
The purity of the caffeine obtained in this manner might be fractional, because salicylic acid can
dissolve in (or through) nonpolar substances like the lipid bilayer of cell membranes, but it is less likely to
do so when it necessarily has a charge. It would seem likely that the caffeine obtained might be quite pure,
while it is less clear that the process here mentioned would completely remove residual caffeine from the
water, as it remains a molecule in its uncharged form, like protonated salicylic acid, capable of dissolving
in either nonpolar hydrophobic layers and hydrophilic layers—the reason it can find its way from soda or
coffee to effectively passing the blood-brain barrier and causing vasodilation within a half hour, or faster.
Dammer, 7
In chemical terms, the caffeine remains soluble in either phase because the ring substituents of like
character can suspend the molecule in either phase.
An improvement to the extraction procedure, were it imperative to increase the purity of salicylic
acid, could involve a reversal of the pH in the aqueous phase, below the pKa of salicylic acid, particularly
that pKa in the nonpolar phase—thanks to solvation effects, in fact higher than the aqueous pKa. At this
point, caffeine entering the aqueous phase would be captured as its nitrogens became protonated, and thus
positively charged. The protonated salicylic acid would likely be at least as good of a solute as methyl red
in the methylene chloride solvent, because it is no more polar, and also a smaller molecule, so that it can
interact with more of the small solvent molecules, maximizing solvation effects. The methylene chloride
would be decanted from the base of the apparatus, and the distillation of the solvent should thus produce a
high-grade purity of the salicylic acid.
Another option in the above examples employing simple extraction technique would be to keep
the liquid phase buffered slightly below the pKa of the (at least) partially protonated caffeine conjugate
acid, which should be higher than the pKa of the acid. Keeping the solute dilute relative to the saturation
point of either compound, detergent could be added to selectively remove either of the charged species—
SDS would transfer caffeine while CTAB would transfer salicylic acid. An excess of nonpolar reagent
could be added to the nonpolar phase so that the transferred compound is consumed, in which case, the
detergents would eventually shuttle all of one compound out of the aqueous solution, and this solution
could be decanted and evaporated to give the desired product. While one of the original mixture
components is lost, purity and efficiency should be considerably higher than other methods mentioned.
This conclusion fits with this researcher’s finding that phase transfer catalysis methods are of use in
industry because of their efficiency and ability to target reagents to particular phases so accurately.
Selected Reading
University of Maryland Physical Chemistry Research Abstracts.
<http://www.chem.umd.edu/physical/walker/research.html>.