Microsoft Word
advertisement

SYNOPSIS
The thesis entitled “Synthesis of C1-C11 Fragment of Borrelidin via Desymmetrization
Strategy, Enzymatic Resolution of 4-Tetrahydropyranols and Application in Total
Synthesis of Diospongin-A” is divided into three chapters.
CHAPTER I: This chapter is further divided into two sections (Section A and Section B).
Section A: This section deals with the introduction and previous synthetic approaches of
Borrelidin.
Section B: This section describes the present synthesis of C1-C11 fragment of Borrelidin.
CHAPTER II: This chapter is further divided into two sections (Section A and Section B).
Section A: This section deals with the enzymatic resolution of 4-tetrahydropyranols by Candida
rugosa lipase.
Section B: This section deals with the total synthesis of Diospongin-A.
CHAPTER III: This chapter is further divided into two sections (Section A and
Section B).
Section A: This section deals with the InCl3-catalyzed tandem Michael/Friedel-Crafts cyclization
for the synthesis of chiral 2, 4-disubstituted tetrahydroquinolines.
Section B: This section describes the CeCl3·H2O/NaI-promoted stereoselective synthesis of 2, 4disubstituted chiral tetrahydroquinolines.
CHAPTER I
SECTION A:
Introduction and previous synthetic approaches of Borrelidin:
This chapter describes about some important antibiotics, antivirals and introduction of
Borrelidin.
9
HO
10
11
12
NC
8
13
7
5
6
O
14
16
15
OH
4 3
2
1
O
17
H
18
23
CO2H
Borrelidin
1
Borrelidin 1 is a structurally unique 18-membered macrolide, structural and functional
features of borrelidin include a reduced polypropionate moiety with the 4, 6, 8, 10-methyl groups
(borrelidin numbering) possessing a distinctive syn/syn/anti relationship, a Z/E cyanodiene unit
at C12-C15, and a cyclopentane carboxylic acid subunit at C17.
Borrelidin possesses interesting biological activity including antibacterial activity, which
involves selective inhibition of threonyl tRNA synthetase, anti-Borrelia activity, antiviral
activity, antiangiogenesis activity, and inhibitory activity towards cyclindependent kinase
Cdc28/Cln2 of Saccharomyces cerevisiae.
It was first isolated from Streptomyces rochei in 1949 by Berger et al. The planar
structure of borrelidin was elucidated by Keller-Schierlein in 1967, and its absolute configuration
was determined by Anderson et al. by X-ray crystallography of a chiral solvate. This chapter also
deals with the previous synthetic approaches of Borrelidin.
SECTION B
Present synthesis of C1-C11 fragment of Borrelidin
During the course of the on going program on the synthesis of different bioactive natural
products by desymmetrization strategy. Borrelidin, a novel antibacterial, antiviral with complex
structural beauty, attracted the interest to initiate a program on the synthesis of C 1-C11 fragment
of Borrelidin.
The retrosynthetic strategy revealed that the synthetic plan for the Borrelidin involves an
intramolecular Reformatsky-type reaction of α-bromo- α, β/γ, δ-unsaturated nitrile compound 2
with macrocyclization at C11-C12 after esterification between acid 3 and alcohol 4. Acid
compound 3 would be synthesized from compound 5 by simple reduction and oxidation
reactions, which in turn could be obtained from compound 6 by extending two carbons using
Wittig reaction and diastereo selective Evans alkylation. This could be obtained from compound
7 by diastereoselective opening of epoxide, which in turn could be obtained from compound 8.
This could be obtained from the known precursor 9 which could be synthesized from compound
10 which in turn could be obtained from furan 11 and 2,4-dibromo-3-pentanone 12 (Scheme 1).
OTBS
OH
HO
O
OHC
O
O
Br
O
NC
H
NC
H
CO2H
OPMB
2
1
+
CO2H
3
OTHP
OH
Br
OTBS
H
NC
OPMB
4
Ph
O
N
O
OBn
O
5
OTBS
O
HO
OBn
OH
TBDPSO
OTBS
6
7
O
OTBDPS
BnO
8
O
O
OH
9
O
Br
Br
O
10
OBn
O
12
11
Scheme 1
Synthesis of lactone 9
OBn
The synthesis was started by employing (3+4) cycloaddition of oxyallyl cation and furan
as reported by Hoffman and co-workers. The acid catalyzed bromination of 3-pentanone with
two equivalents of bromine furnished the known 2, 4-dibromo-3- pentanone 12 in 80% yield.
The oxyallyl cation (generated from 2, 4-dibromo-3-pentanone with zinc copper couple at -10
o
C) when treated with furan underwent (3+4) cycloaddition to afford 2, 4-dimethyl-8-
oxabicyclo(3.2.1)oct-6-en-3-one (13, 14, 15). The stereoselective reduction of these ketones
using DIBAL-H afforded mixture of alcohols, which on purification gave the major alcohol 16.
This was further protected as its benzylether using sodium hydride and benzylbromide in
refluxing tetrahydrofuran to afford compound 10. Asymmetric hydroboration of olefin 10 using
(-)-IPC2BH in THF afforded alcohol 17. This was then converted to the keto compound 18 by
PCC oxidation, which was further subjected to Bayer-Villiger oxidation with m-CPBA to give
lactone 9 (Scheme 2).
Br
Br2, AcOH, Water
O
rt, 12 h, 80%
Br
O
Zn-Cu couple
DME, -10 oC, 80%
O
12
O
O
O
DiBAL-H
+
+
O
14
13
O
THF, -10 oC, 70%
O
15
O
O
dry THF, reflux
6 h, 90%
OH
16
O
H2O2, NaOH, 95%
10
OBn
O
O
PCC, CH2Cl2
rt, 3 h, 90%
HO
(-)-Ipc2BH
NaH / BnBr
+ mixture of isomers
OBn
17
m-CPBA, NaHCO3
O
OBn
18
Scheme 2
Synthesis of compound 8
CH2Cl2, 25 oC
10 h, 90%
O
O
9
OBn
Our next goal was to introduce α-methyl at C2 position of compound 9, which has been
achieved by introducing olefin followed by hydrogenation. Treatment of compound 9 with LDA,
paraformaldehyde resulted alcohol 19, which was treated with MsCl, Et3N to yield O-mesylated
product 20 and then it was reacted with DBU in dichloromethane to obtain olefinic compound
21. This was subjected to hydrogenation by using Pd-C, Na2CO3 in ethylacetate to furnish
compound 22, which was subjected to reductive opening by using DIBAL-H to give triol
compound 23. This was protected as its acetonide 24 using 2, 2-dimethoxypropane and cat.
PTSA in dry DCM, further benzyl group was removed by using Li, Naphthalene to obtain diol
25, and then primary alcohol was selectively protected as its benzylether 26 by using standard
conditions. The secondary alcohol was protected as its xanthate ester 27 and it was treated with
Bu3SnH and catalytic amount of AIBN to obtain compound 28. Acetonide was deprotected by
using standard conditions to obtain diol 29, selective protection of primary alcohol by using
TBDPS-Cl, imidazole yielded compound 8 (Scheme 3).
O
O
HO
LDA, paraformaldehyde,
O
THF, -78 oC, 80%
O
MsCl, Et3N, DCM, 0-6 oC
O
12 -16h, 84%
O
9 BnO
19 BnO
O
O
MsO
DBU, DCM, rt
O
H2, 10% Pd-C, Na2CO3
O
10h,90%
O
EtOAc, rt, 10 -12h, 95%
O
BnO
21
20
OBn
O
DIBAL-H, DCM, rt
O
HO
OH
4-5h,85%
O
BnO
22
OBn OH
23
HO
2,2-DMP, acetone, PTSA, rt
OH
Li, Napthalene, -23 oC
HO
OBn O
30min, 90%
O
3-4h, 78%
OBn OH
24
23
NaH, BnBr, THF, 0 oC
HO
OH
O
O
BnO
3h, 90%
25
Bu3SnH, cat. AIBN
O
O
BnO
95%
cat. PTSA, MeOH, rt
Toluene, reflux, 3h, 90%
O
O
O
2h, 88%
28
27
BnO
TBDPS-Cl, imidazole
29
O
26
BnO
MeS2CO
NaH, CS2, MeI, THF
OH
OH
OH
OTBDPS
BnO
DCM, rt, 2h, 90%
8
OH
Scheme 3
Synthesis of compound 7
Compound 8 was protected as its xanthate ester 30, by using earlier procedure and then
reduced with Bu3SnH and catalytic amount of AIBN to obtain compound 31. Benzyl group of
compound 31 was deprotected by using Li, Naphthalene to give alcohol 32, which was oxidized
to aldehyde 33 using IBX, DMSO. This was treated with C2- Wittig reagent in benzene to yield
compound 34. This was selectively reduced to allylic alcohol 35 by using DIBAL-H, followed
by Sharpless asymmetric epoxidation using (-)-DIPT to give epoxide 7 (Scheme 4).
OTBDPS
BnO
95%
OH
8
Bu3SnH, cat. AIBN
30
3-4h, 80%
31
OTBDPS
IBX, DMSO, THF, rt
OTBDPS
H
30min, 95%
33
O
32
Ph3PCHCOOEt, benzene, rt
OCS2Me
Li, Napthalene, -23 oC
OTBDPS
BnO
Toluene, reflux, 3h, 90%
HO
OTBDPS
BnO
NaH, CS2, MeI, THF
OEt DIBAL-H, DCM, rt
TBDPSO
1h, 92%
40min, 91%
O
34
TBDPSO
OH
(-)-DIPT, TBHP, Titanium isopropoxide
DCM, 88%
35
O
TBDPSO
OH
7
Scheme 4
Synthesis of compound 5
Compound 7 was regieoselectively reduced by using Red-Al to yield 1,3-diol 36, which
was selectively protected as its mono benzylether 37 by using standard conditions and the
secondary hydroxyl group was protected as its TBS ether compound 38. On selective
deprotection of TBDPS by using NH4F, yielded compound 6. This was oxidized to aldehyde 40
by using IBX, DMSO and immediately used for the next reaction. It was treated with C2 Wittig
reagent in benzene to obtain trans-α,β-unsaturated ester 40. Selective reduction of the olefin by
using NaBH4, NiCl4 yielded compound 41, this ester was then hydrolyzed to acid 42 by using
LiOH. Compound 42 was activated by forming mixed anhydride using Piv-Cl, Et3N and in situ
treated
with
(S)-4-benzyl-2-oxazolidinone
to
obtain
compound
43,
which
was
diastereoselectively methylated by using NaHMDS and MeI to give compound 5 (Scheme 5).
O
TBDPSO
7
NaH, BnBr, THF, 0 oC
o
OH Red-Al, THF, 0 C
5h, 81%
TBDPSO
HO
EtO
Ph3PCHCOOEt, benzene, rt
1h, 92%
OTBS
OBn NaBH4, NiCl4, MeOH
40
EtO
OBn
O
1h, 92%
OTBS
HO
LiOH, MeOH, H2O, THF (1:1:1), 0 oC
OTBS
6
OBn
OHC
39
O
OBn
2d, 78%
OTBS
IBX, DMSO, THF, rt
30min, 95%
DCM, 0 oC, 1h, 92%
OH
NH4F, MeOH, rt
OBn
OH
TBSOTf, 2,6-Lutidine
OBn
37
OH
36
TBDPSO
3h, 90%
38
TBDPSO
41
OBn
O
4.5h, 85%
42
OTBS
Ph
Et3N, Piv-Cl, THF
O
(S)-4-Benzyl-2-oxazolidinone, LiCl
84%
O
N
OBn
O
OTBS
43
Ph
NaHMDS, MeI, THF, -78 oC
3h, 70%
O
O
N
OBn
O
5
Scheme 5
Synthesis of C1-C11 fragment of Borrelidin 3:
OTBS
OTBS
Compound 5 was reduced to alcohol 44 by using NaBH4 and it has been protected as its
THP ether 45 by using standard conditions, and then benzyl group was deprotected by treating
with Li, naphthalene to obtain alcohol 46, which was oxidized using mild oxidizing agent
TEMPO and BAIB to yield the desired carboxylic acid 3 (Scheme 6).
Ph
O
N
O
NaBH4, MeOH
OBn
O
5
44
THPO
OTBS
45
THPO
OH
3-4h, 76%
OTBS
46
BAIB, TEMPO, Acetone, Water (8:2)
OTBS
OBn
2h, 90%
Li, Napthalene, THF, -23 oC
OBn
1h, 80%
OTBS
3,4-Dihydropyran, cat. PTSA, DCM
HO
THPO
45min, 72%
COOH
3
OTBS
Scheme 6
CHAPTER II
Section A:
Enzymatic resolution of 4-tetrahydropyranols by Candida rugosa lipase.
This chapter includes the introduction, previous approaches of the Prins cyclization and
introduction to enzymatic kinetic resolution.
Present work
Our ongoing research for the synthesis of biologically active molecules, we were
interested to construct the enantiomerically pure 4-hydroxytetrahydropyran units and to utilize in
the synthesis of complex molecules. However, there have been no reports on the kinetic
resolution of racemic tetrahydropyranols via lipase-mediated transesterification. Racemic 4hydroxy tetrahydropyrans were easily prepared by the condensation of homoallylic alcohols with
aldehydes via Prins-cyclization.
Kinetic resolution of the racemic 4-tetrahydropyranol via Candida rugosa lipase
furnished the corresponding (2S, 4R)-tetrahydropyranyl acetate and (2R, 4S)-tetrahydropyranol.
Then alcohol was treated with acetic anhydride, pyridine to form its corresponding acetate.
Product chirality was confirmed by reductive opening of the resolved hydroxypyranol by using
10% Pd-C in ethanol. Analytical data was compared and found matching with the reported
compound {(Specific rotation for the compound 52:
[α] D25 = (+)18.1 (c 0.4, CHCl3)} (Scheme 7).
OAc
OH
OH
Candida rugosa
Ar
+
vinyl acetate,cyclo hexane, rt
O
Ar
O
48
47
OH
Ar
O
49
OAc
Acetic anhydride
pyridine
Ar
O
Ar
49
50
OH
OH
10% Pd/C
Ph
O
O
EtOH, rt
Ph
HO
(S)-5-phenylpentane-1,3-diol
52
51
Scheme 7
Similarly, various 4-hydroxytetrahydropyranols were resoluted in the same conditions to yield
corresponding enantiomerically pure acetate and alcohol. The alcohol was treated with acetic
anhydride, pyridine to form corresponding acetate and compared (Table 1).
Entry
Acetate 48
Substrate 47
25b
[a]
ee(%)c
D
21.2
O
Cl
11.3
O
OH
OAc
O
O
PhO
88
F
MeO
i
Me
80
-27.9
89
9
85
-16.7
83
12
86
-62.1
90
8
81
O
OMe
OAc
-4.3
87
6
79
O
-7.5
87
9
82
-12.3
88
Br
OAc
98
PhO
O
O
OMe
O
5.8
92
8.9
90
MeO
F
F
OAc
OAc
O
O
OAc
OAc
OH
O
10
O
OAc
OH
h
87
O
OAc
OH
O
-18.3
O
94
Br
O
OMe
79
OAc
70.8
MeO
6.5
OAc
O
OH
g
87
OAc
O
O
-9.8
MeO
19.6
f
90
O
29.8
PhO
75
Cl
MeO
Br
6
O
OAc
O
d
95
OAc
OH
MeO
92
Cl
OH
e
-21.3
OAc
21.7
c
Yield (%)e
O
OAc
OH
b
98
O
O
25b
c
[a]
D ee(%) Time (h)
OAc
OAc
OH
a
Acetate 50
13.9
92
Me
O
Me
10
86
a
All products were characterized by IR, NMR and mass spectroscopy.
Optical rotations were recorded in CHCl3 (c = 1.0)
c
Enantiomeric excess of acetates were achieved by using chiral HPLC.
d
Alcohols resulting after resolution were converted to their respective acetates so as to match the
optical rotation.
e
Isolated and unoptimized yields.
b
Table 1
Section B: Total synthesis of Diospongin-A.
This chapter describes about introduction and previous synthetic approaches of
Diospongin-A.
Present synthesis:
Retrosynthetic analysis of 53 illustrates that it can be obtained from compound 54 by
Mitsunobu inversion followed by Wacker oxidation, this in turn can be envisaged from
tetrahydropyranol 55 by means of an enzymatic kinetic resolution using PPL. Compound 55
could be easily prepared from allylation product of benzaldehyde and cinnamaldehyde which is
further proceeded to the Prins-cyclization (Scheme 8).
OH
OH
O
O
O
54
53
OH
OH
CHO
O
55
+
1-phenylbut-3-en-1-ol
Cinnamaldehde
Scheme 8
The synthesis of Diospongin A began with cinnamaldehyde and 1-phenylbut-3-en-1-ol,
which could be easily obtained by Barbier allylation of benzaldehyde. This was subjected to
Prins-cyclization with cinnamaldehyde to produce tetrahydropyranol 55, subsequent resolution
of product 55 by using Porcine pancreatic lipase and vinyl acetate in the presence of
cyclohexane afforded desired acetate 56 and alcohol 57 in approximately 1:1 ratio, the desired
acetate 56 was then deprotected by using K2CO3 in MeOH to furnish alcohol 54. Inversion of the
alcohol 54 by employing Mitsunobu reaction conditions afford compound 58. This was oxidized
in Wacker oxidation conditions using PdCl2 and CuCl in DMF/H2O to afforded compound 59, it
was subjected to hydrolysis by using K2CO3 in MeOH to furnish the target molecule, Diospongin
A 53 (Scheme 9).
OH
OH
CHO
1. TFA, DCM, 3h
+
1-phenylbut-3-en-1-ol
O
2. K2CO3, MeOH
78%
Cinnamaldehyde
55
OAc
PPL, Vinylacetate
OH
O
Cyclohexane, rt, 5d
O
+
57
56
OAc
OH
K2CO3, MeOH
O
TPP, DIAD,
O
p-nitrobenzoic acid, Toluene
3h, 90%
15min, 92%
56
54
NO2
NO2
O
PdCl2, CuCl, O2
O
O
O
DMF, Water (1:7), 55 oC, 3d, 89%
O
O
59
58
OH
O
K2CO3, MeOH
O
O
15min, 90%
53
Scheme 9
CHAPTER III
SECTION A
InCl3-catalyzed tandem Michael/Friedel-Crafts cyclization for the synthesis of chiral 2, 4disubstituted tetrahydroquinolines
In continuous efforts towards the development of new methodologies a novel route for
the synthesis of optically active tetrahydroquinolines in a one-pot operation we have also studied
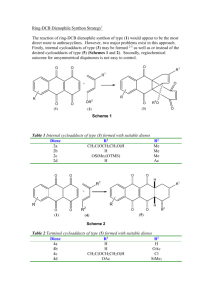
the condensation of aniline (60a, R = R1 = R2= H) with optically active 4,6-di-O-acetyl-2,3dideoxyaldehydo-D-erythro-trans hex-2-enose 61 as a model reaction using 10 mol% of InCl3.
The reaction went to completion within 3 hours at room temperature and the product 62a was
obtained with high selectivity (entry a, Table 2, Scheme 10).
1
NH2
R
+
2
R
OAc
R
AcO
R
CHO
60
10 mol% InCl3
CH3CN
OH
R
H H
N
1
R2
H
O
OAc
OAc
62
61
Scheme 10
Encouraged by the results obtained with aniline, we turned our attention towards various
arylamines. Interestingly, arylamines such as mono-, di- and trisubstituted anilines reacted
efficiently with 61 under similar conditions to give the corresponding benzo-fused heterobicycles
in good yields. However, ortho-hydroxy substituted trans-cinnamaldehyde and aniline did not
give the expected cyclized product under the reaction conditions. Furthermore, simple α, βunsaturated aldehydes without δ-hydroxyl group did not afford the bicyclic adduct. The reaction
was successful only with δ-hydroxy-α,β-unsaturated aldehydes.
The method is highly stereoselective affording novel benzo fused heterobicycles under
mild reaction conditions. The scope and generality of this process is illustrated with respect to
various substituted arylamines and the results are presented in Table 2.
Entry
a ,ß -Unsaturated
Aldehyde
61
Aryl amine
60
Producta
62
H H
N
OAc
NH 2
60a
AcO
H
Br
Me
H H
N
H
60c
Me
60d
NH 2
Cl
H
Cl
4.5
Cl
H
Cl
NH 2
60f
OAc
5.0
Cl
Cl
Cl
H
Br
OAc
H
5.0
83
3.0
95
OAc
O
H H
N
Br
90
OAc
O
NH 2
60i
OAc
H H
N
Me
Me
4.5
OAc
O
H
NH 2
60h
OAc
H H
N
NH 2
60g
O
H
Me
83
OAc
H H
N
Me
80c
OAc
O
60e
Cl
93
3.0
OAc
H H
N
Cl
88
OAc
O
H
NH 2
OAc
H H
N
Cl
5.0
OAc
H H
N
NH 2
91
3.5
OAc
O
Me
Yield(%)b
OAc
O
Br
NH 2
Me
OAc
CHO
OH
60b
Reaction time(h)
O
OAc
OAc
3.5
87
Entry
a ,ß -Unsaturated
Aldehyde
61
Aryl amine
60
Producta
62
OAc
NH 2
60j
AcO
H H
N
CHO
OH
Cl
Cl
H
F
F
OAc
H
OAc
OAc
60l
Ph
Ph
H
H2N
60m
H
4.0
82
5.0
90
OAc
O
H H
N
85
OAc
O
H H
N
NH 2
5.0
Yield(%)b
OAc
O
H H
N
NH 2
60k
Reaction time(h)
OAc
O
4.5
92
OAc
a
Products were characterized by 1H NMR, 13C NMR, IR and mass spectroscopy.
Yield refers to pure products after chromatography.
c
Contains 15% of the other regioisomer as determined by 1H NMR spectroscopy.
b
Table 2
CHAPTER III
SECTION B
CeCl3·H2O/NaI-promoted
stereoselective
synthesis
of
2,4-disubstituted
chiral
tetrahydroquinolines
In continuation of our interest in the synthesis of C- and O-glycosides, we have
accomplished a novel approach for the synthesis of sugar derived chiral tetrahydroquinolines
from D-glucal and aryl amines. Thus, treatment of 3,4,6-tri-O-acetyl- D-glucal 64 with aniline
63a in the presence of an equimolar ratio of CeCl3·7H2O and NaI in water afforded sugar fused
tetrahydroquinoline 65a in 82% yield (Scheme 11).
R1
R
OAc
R
OAc
NH2
+
R2
63
CeCl3.7H2O/NaI
OAc
O
H H
N
1
o
H2O, 80 C
R
R2
H
O
OAc
OAc
65
64
Scheme 11
Interestingly, a variety of aryl amines including mono-, and di-substituted anilines
reacted smoothly with glucal triacetate under similar conditions to afford the corresponding
benzo-fused heterobicycles in good yields. However, 3,4,6- tri-O-methyl-D-glucal or 3,4,6-tri-Obenzyl-D-glucal did not react with aryl amines under identical reaction conditions (entry o, Table
3). The reaction was successful only with glucal triacetate.
Furthermore, the reaction did not proceed with 2,6-disubstituted anilines such as 2,6dichloroaniline and 2,6-dimethylaniline under the reaction conditions (entry n, Table 3). These
results clearly indicated that one of the ortho positions of aniline should be free from substitution
for the success of the reaction.
The scope and generality of this process is illustrated with respect to various aryl amines
and D-glucal (Table 3).
Entry
Aryl amine
63
Producta
65
D-Glucal
64
H H
N
OAc
NH2
OAc
O
NH2
H
O
H H
N
OAc
Cl
OAc
O
NH2
Cl
H
OAc
H H
N
OAc
OAc
63c
Me
OAc
O
Me
H
63d
F
NH2
Br
OAc
Me
H
H H
N
OAc
Br
H
OAc
O
OAc
OAc
NH2
O
OAc
H
H H
N
OAc
63g
MeO
O
OAc
OAc
Cl
O
H
O
H H
N
Cl
OAc
85
9.0
75
8.5
72
7.5
83c
OAc
8.0
MeO
OAc
NH2
7.5
OAc
O
OAc
80
OAc
H H
N
Me
8.0
OAc
O
OAc
O
NH2
F
OAc
OAc
63f
OAc
OAc
O
82
OAc
O
H H
N
OAc
NH2
7.0
OAc
O
OAc
H
Yield(%)b
OAc
OAc
63b
63h
OAc
OAc
63a
63e
Reaction time(h)
O
70
OAc
OAc
OAc
9.0
80c
Entry
Aryl amine
D-Glucal
63
64
Cl
63i
Producta
65
Cl
OAc
NH2
Me
H
NH2
63j
OAc
O
H
Me
OAc
NH2
OAc
O
Br
NH2
63l
Me
OAc
O
Me
H
H H
N
H
7.5
82
8.0
86
9.5
65
8.5
70
OAc
OAc
O
Cl
OAc
O
OAc
63m
79
OAc
O
OAc
NH2
OAc
H H
N
OAc
8.5
OAc
O
H
OAc
OAc
H H
N
OAc
63k
Br
H H
N
OAc
O
OAc
OAc
OAc
NH2
OAc
63n
Cl
OAc
O
No reaction
8.0
No reaction
9.0
OR1
OR1
NH2
63o
O
OR1
R1=Me or Bn
a
Products were characterized by 1H NMR, 13C NMR, IR and mass spectroscopy.
Yield refers to pure products after chromatography.
c
5-10% other regioisomer was obtained.
b
A
Yield(%)b
OAc
O
Cl
OAc
Me
OAc
OAc
O
Cl
H H
N
Me
OAc
Reaction time(h)