Electrochemistry
advertisement

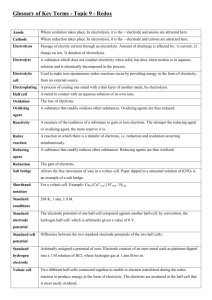
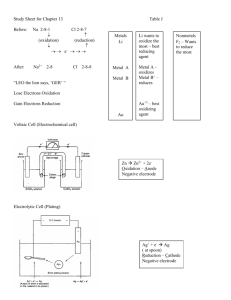
CHAPTER 21 NOTES ELECTRODE POTENTIALS AND THEIR MEASUREMENTS In this chapter we will examine spontaneity with respect to redox reactions. One of the most common redox reactions is that which occurs when metals are immersed into solutions of dissimilar metal ions – the single replacement reaction. In other cases, mixing ions (in the presence of acid) leads to more complicated redox reactions (recall the iodometric titration in the fall). Regardless of compelexity, some metals and ions are more easily reduced than others. This difference in affinity for electrons drives chemical reactions. In previous chapters we discussed related concepts such as electronegativity, electron affinity and ionization energy. In this chapter, the ease of oxidation, or the more accepted ease of reduction, is known as the Standard Electrode Potential. It is measured in volts which have the units, Joules/coulomb. As generally depicted above, if you place a metal like Cu in a solution of Cu2+ ions (at 1.0 M) an equilibrium is quickly established: Cu (s) Cu2+ (aq) + 2e- This particular situation creates what is known as a half-cell. It is more a theoretical idea than anything functional – a half-cell represents the "half reaction" from balancing redox reactions last fall and "half" of a reaction cannot occur. Remind yourself that if the reaction above is read left to right it is called oxidation. Each metal will have a unique equilibrium with its ions that will result in a slight positive or negative charge build-up on the electrode. This tendency cannot be measured directly but can be measured by connecting different half-cells to each other. Electrons will flow from the electrode of higher e- density to the electrode of lower e- density (akin to electrons "diffusing"). To measure the difference in potential we connect two half-cells by joining the electrodes with a wire and the cells to each other with a salt bridge to maintain electro-neutrality. As ions related to the electrode enter or leave the solution, spectator ions in the salt bridge migrate into the solutions to maintain charge balance. The two half- cells, external electron circuit and the salt bridge is an Electrochemical Cell. The voltage is the potential difference between the two half-cells and is called the electromotive force (emf) or the cell potential. A cell diagram symbolically represents an electrochemical cell. It has the following characteristics. 1. The anode (oxidation) is placed on the left side of the diagram. 2. The cathode (reduction) is placed on the right side of the diagram. 3. The boundary between different phases (solid to aq) is a single line. 4. The boundary between the half-cells (salt bridge) is a double line. An electrochemical cell that spontaneously produces electricity is called a voltaic or galvanic cell. The following cell diagram represents the electrochemical cell on the top of the next page. Although not labeled as such, the wire on the cathode side of the cell is commonly platinum. Zn|Zn2+(1M)||H+(1M),H2(1atm)|Pt It would be convenient to assign a potential to each half reaction. Problem: They only occur in pairs – so how would we know what voltage contribution is provided by each half reaction? Solution: Define one half reaction as having 0.00 volts. Introducing the Standard Hydrogen Electrode Define the S.H.E. Where 2H+ + [H+] = 1.00 M 2e- & H2 E°red = 0.00 V PH2 = 1.00 atm. A standard electrode potential, E°, measures the tendency for a reduction process to occur at an electrode. A standard cell potential, E°cell, is the total potential formed from two standard electrodes, one reduction and one oxidation. Examine the electrochemical cell reaction between Zn and H2. Since the E°cell is +0.76 V, the reaction is not only spontaneous as depicted but it tells us that Zn metal is more easily oxidized than H2 by a potential of +0.76 V. Inverting our thinking, it also means (more importantly for convention sake) that H+ is more easily reduced than Zn2+. This leads us to conclude that the half reaction representing the reduction of Zn2+ ions should be listed below the reduction half reaction of H+ on a standard reduction table. Since the tendency for reduction of H+ is defined as 0.00 V, the reduction potential for Zn2+ must be -0.76 V. Note the sign and remember that a negative reduction potential means that the reverse reaction (oxidation of Zn) must be +0.76V – the diagram above depicts this! E°Cell AND SPONTANEOUS CHANGE. When a voltaic cell operates, it does work. The work, w, done by a cell is given by the product of three terms: w nFEcell where n is the moles of electrons from the net equation, F is Faraday’s constant (the charge per mole of electrons: 96485 coulombs) and E° is the volts (Joules/coulomb) of the cell. You can see by unit analysis, that work is measured in joules! The amount of energy to do work is also known as the free energy, ∆G! Therefore we can say that G nFEcell The negative sign identifies that a positive cell voltage and a ∆G° both mean spontaneity. Most metals react with acids such as HCl. How can you tell which will? Any metal with a negative reduction potential will react with mineral acids (HCl, HBr, HI). Remember, if the reduction of H+ to H2 is 0.00 Volts, it has a greater tendency to reduce than any metal with a lower reduction potential. This means the metal will oxidize in the presence of the acid! Silver lies above the H+ reduction tendency. Silver will not dissolve in HCl – but it will react in HNO3. Look right above the silver ion reduction reaction. The nitrate ion, rich in oxygen and in the presence of H+, oxidizes the silver while it is reduced to NO. If we can relate ∆G° to an equilibrium constant as in the last chapter, we must be able to do so with E°cell. Start with the simple substitutions of the following equations: ∆G° = -RTlnKeq & ∆G° = -nFE°cell -nFE°cell = -RTlnKeq Now rearrange the last equation above into the more functional versions. The left equation is standard form but can be simplified by merging constants R, T and F to give the right equation. Be advised that the equation sheet on the national exam does not supply you with the right equation. It gives the analog using base 10 logs. E cell RT ln Keq nF E cell 0.0257 ln K eq n The following graphic presents the glorious relationships between all that we have been studying. It is worth your time. Nonstandard Conditions Suppose we are working with electrochemical cells with concentrations other than 1.0 M ions? In these nonstandard situations, we can also define a cell potential. It is the Nernst Equation. You should check the text for the derivation Ecell E cell 0.0257 ln Q n but please note the beautiful symmetry to the nonstandard ∆G equation. ∆G = ∆G° + RTlnQ A concentration cell is a special electrochemical cell where the two electrodes are identical; only the concentration of the two solutions is different. The Nernst equation is your tool. Using this equation you can see that if the two half-cells are identical, the standard cell potential is 0.00 V. What makes the voltage deviate from zero is the fact that the two half-cells have different concentrations of ions. These values create a voltage in the "lnQ" term. If you are interested in the voltage qualitatively, think and use LeChatelier's principle. Believe it or not but this concept relates to how a pH meter reads the H+ concentration in a solution. A reference [H+] solution inside the electrode sets up a small potential with the solution’s [H+]. Prior to this, the pH electrode is immersed in a standard buffer solution and calibrated to the measured potential (in the millivolt range). A second buffer with a different pH is then calibrated to its voltage. A calibration curve is set and the device measures millivolts but displays pH. This idea has evolved into other sealed electrodes with standard concentrations of ions. Imagine placing an electrode containing a sealed solution of standard Ag+ ions into a saturated solution of silver chloride (relatively insoluble). AgCl (s) Ag+ (?M) + Cl- (?M) A very small potential is set up and that value, inserted into the Nernst equation, defines the [Ag+] in the saturated solution. If you know this, you also know the [Cl-] and you can solve for the Ksp. If you know the Ksp, you also know the ∆G° for the reaction. Wow! BATTERIES Primary batteries: nonreversible Secondary batteries: reversible -- they can be recharged. Fuel Cells: These are equivalent to a combustion process (redox) where chemical energy is directly converted to electrical energy. It is not really a battery but an energy converter with amazing efficiency. You should study this section for the descriptive aspects of batteries. CORROSION This section is largely a descriptive section. through is advised. A quick read- Cathodic protection - a special idea worth highlighting. To protect a metal such as iron, a chunk of some other metal (e.g. Mg) that is more easily oxidized is connected to it. This other metal, the sacrificial anode, effectively protects the Fe from rusting. ELECTROLYSIS: NONSPONTANEOUS CHANGE If one takes a voltaic cell that has a potential of 1.5 V and applies an external voltage greater than 1.5 V in the nonspontaneous direction, the reaction can be made to run backwards. In this situation, the cell is defined as an electrolytic cell. In practice the means of making an electrolytic cell is more difficult than the above statement implies. Sometimes a greater amount of voltage is needed to force the reaction backwards. This extra voltage is called the overpotential. Secondly, competing reactions, often undesirable, may take place. Quantitative aspects of electrolysis remind us of stoichiometry. Consider the reduction of Al3+ to Aluminum metal and these facts: 1. 1.00 mol e- = 96485 coulombs 2. charge (C) = current in amperes (C/s) x time (s) 3. Al3+(aq) + 3e- Al(s) What mass of Al will be plated out in 1.00 hr by 1.62 A? 60min 60sec 1.62C 1mol e - 1mol Al 26.98g 1hr 0.544g hr min sec 96485C 3mol e 1mol ELECTROCHEMISTRY: THE STUDY OF THE INTERCHANGE OF CHEMICAL AND ELECTRICAL ENERGY. Section 1. Consider this redox reaction: MnO4- + 8H+ + 5Fe2+ Galvanic Cells Mn2+ + 5Fe3+ + 4H2O To balance, break it into half reactions. The Fe2+ to Fe3+ oxidation has to occur 5x for every permanganate reduction. When these ions are in the same solution, the electrons are transferred directly. This is a very important analytical method but when it comes to tapping the electrons -- Result: NO USEFUL WORK IS OBTAINED -just waste heat - ugh! The key is to separate the oxidizing agent from the reducing agent; require the electron transfer to occur through a wire. The current can be directed through a device to do useful things. To complete the circuit, a salt bridge provides spectator ions to both half-cells. The following is the line notation for the permanganate and iron(II) reaction above. Pt | Fe2+ (aq) , Fe3+ (aq) || MnO4- (aq) , Mn2+ (aq) | Pt(s) Section 2. Standard Reduction Potentials Standard Reduction Potentials: voltages relating the tendency for reduction to occur when ions are 1.00 M and gases are 1.00 atm. Large positive voltages indicate a favored reduction. Large negative voltages indicate an unfavored reduction BUT if reversed, a favored oxidation. When a half reaction is an oxidation, E°oxid = -E°red. CONSIDER THE FOLLOWING REDOX EQUATION AT 25°C. Al + Ag+ Al3+ + Ag The half-reactions and their standard potentials are: anode: oxidation = Al Al3+ + 3eE°oxid = 1.66 V cathode: reduction = 1e- + Ag+ Ag E°red = 0.80 V (The reduction is multiplied by 3 to balance electrons produced in the oxidation. Voltage remains fixed – it is the potential for the reaction and independent of quantity.) When you add the half reactions, you add the voltages. Al + 3Ag+ Al3+ + 3Ag Therefore E°cell = 2.46 V Line Notation: For the above galvanic cell, it would be: Al(s) | Al3+ (aq) || Ag+ (aq) | Ag(s) We also know the relationship to thermodynamics: ∆G° = -nFE° Solving for ∆G°: G 3mol e 96485coul mol e 2.46 J coul - - G 712kJ And we know the relationship to the equilibrium constant: G RT ln K K OVERFLOW IN OTHER WORDS, K IS REALLY BIG! Thus, the maximum cell potential is directly related to the free energy difference between reactants and products in the galvanic cell. When E° is positive, ∆G must be negative -- the condition for spontaneity and K must be greater than 1! Those calculations were all based on standard cell potentials Consider the same voltaic cell but under nonstandard conditions where [Al3+] is at 4.0 M and the [Ag+] is at 0.001 M. What is the voltage under these conditions? According to our friend "LeChat" the voltage should drop because I have boosted the product concentration and reduced the reactant concentration. Let's check out the math! Remember the Q expression with initial concentrations raised to their coefficients. The Nernst equation is the tool of choice. E 2.46V 0.0257 4 ln 3 0.0013 Ecell 2.27V "Le Chat" is all over this stuff – He rules!