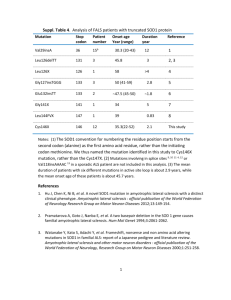
qui - Istituto di Ricerche Farmacologiche Mario Negri
advertisement

Mutant SOD1 and familial ALS: from the molecule to man Milan (Italy) September 13-16, 2007 MARIO NEGRI Institute Organizers Caterina Bendotti; bendotti@marionegri.it Maria Teresa Carrì; carri@Bio.uniroma2.it Alberto Ferri; a.ferri@hsantalucia.it Lavinia Cantoni; cantoni@marionegri.it Scientific Committee Caterina Bendotti Maria Teresa Carrì Stefan L. Marklund Giuseppe Rotilio Pamela Shaw Joan S. Valentine Secretariat Annalisa Volpe; a.volpe@hsantalucia.it Dear Colleagues and Friends, It is a great pleasure for us to welcome you to this first meeting on mutant SOD1 and familial ALS in Milano, Italy. The conference is focussed on the selective neurotoxic properties of mutant SOD1, from biochemical and biophysical data in vitro to results in vivo from studies in animal models and patients. Hopefully, this meeting will offer the opportunity to review and discuss existing evidence and new hypotheses on the mechanisms of mutant SOD1-linked ALS, and encourage scientific collaboration. We are certain that this will be of help both to all the neuroscientists already working in the field and to those that are now joining in the attempt to understand the cause of this rather obscure disease. Since the conference is running in Milano, we are also pleased to be able to offer you the exclusive opportunity to visit the famous “Last Supper” painted by Leonardo da Vinci from 1495 to 1498. We wish to thank all participants, all the people who have assisted us in the organization of the meeting and our sponsors that made it possible. Last, but not least, many thanks are due to Prof. Silvio Garattini, director of the Mario Negri Institute, for kindly hosting the meeting. Caterina Bendotti Maria Teresa Carrì 2 Scientific Program Thursday 13 September Registration 3.30 pm Welcome Ceremony 5.30 pm Caterina Bendotti, Maria Teresa Carrì Silvio Garattini ( Director of Mario Negri Institute) Mario Melazzini ( President of AISLA) Opening lecture 6.00 pm S1 - Giuseppe Rotilio (Italy) From erythrocuprein to SOD1: memories of a Biochemist. Welcome party 7.00 pm Friday 14 September Session I 8.30 am – 10.45 am Biochemistry of Mutant SOD1s Chairpersons: J.S. Valentine – G. Rotilio S2 - Joan S. Valentine (USA) How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? S3 - Mikael Oliveberg (Sweden) ALS-associated SOD1 mutations preferentially reduce the proteins repulsive charge S4 - Lucia Banci (Italy) Interplay between structural and dynamical properties and protein aggregation in SOD1 S5 - Joseph S. Beckman (USA) Direct measurement of zinc-deficient SOD localized the ventral gray matter in G93A SOD rats Selected oral communication SO1 - Albrecht Clement (Germany) Dismutase-inactive mutant SOD1 has different functional properties when heterodimerized with wild type SOD1 Coffee break (10.45 am -11.15 am) 3 Session II 11.15 am – 1.00 pm Mutant SOD1s toxicity in cell models Chairpersons: S.L. Marklund – M.T. Carrì S6 - Pamela Shaw (UK) Mutant SOD1 in motoneuronal cells: mechanisms from the genomic and proteomic profile S7 - Gen Sobue (Japan) Mutant SOD1s and protein aggregation/degradation in motoneuronal cells S8 - Maria Teresa Carrì (Italy) Mutant SOD1s and mitochondria in motoneuronal cells Selected oral communication SO2 - Mauro Cozzolino (Italy) Cysteine 111 affects aggregation of mutant SOD1 in NSC34 cells Lunch (1.00 pm – 2.00 pm) Session III 2.00 pm – 4.15 pm Mutant SOD1s toxicity in animal models Chairpersons: Z.S. Xu – W. Robberecht S9 - Stefan L. Marklund (Sweden) Soluble misfolded SOD1 species in ALS S10 - Jean-Pierre Julien (Canada) From secretion of SOD1 mutants to immunotherapeutic approaches S11 - Jasna Kriz (Canada) Live imaging of disease progression in SOD1 mutant mice S12 - Valentina Bonetto (Italy) Proteomics of spinal cord aggregates in G93A SOD1 transgenic mouse Selected oral communication SO3 - Avi Chakrabartty (Canada) Detection of misfolded SOD1 in sporadic and familial ALS Coffee break (4.15 pm – 4.45 pm) 4 Session IV 4.45 pm – 6.45 pm Genetics of Familial ALS Chairpersons: C.E. Shaw – A. Chiò S13 - Peter M. Andersen (Sweden) Genetics of SOD1-linked fALS S14 - Christopher E. Shaw (UK) Genetics of non-SOD1-linked fALS S15 - Stefania Battistini (Italy) SOD1 in Italian ALS patients S16 - Wim Robberecht The pathogenesis of sporadic amyotrophic lateral sclerosis Selected oral communication SO4 - Richard Wroe (UK) The Evolution of the Amyotrophic Lateral Sclerosis Database (ALSOD) Social Event - Visit to Leonardo’s “Last Supper” 7.30 pm Social Dinner – 8.30 pm Saturday 15 September Session V 8.30 am – 10.15 am Motor neuron death in SOD1-linked fALS: intra-cellular mechanisms Chairpersons: L. Cantoni – P. Pasinelli S17 - Piera Pasinelli (USA) The double life of two pro-survival proteins: when SOD1 and Bcl-2 get mutated, wild and pro-death S18 - Claudia Crosio (Italy) Role of Bcl2a1 in SOD1-linked fALS S19 - Angelo Poletti (Italy) The nucleus as target for mSOD1 toxicity S20 - Massimo Tortarolo (Italy) Role of MAP kinases cascade in SOD1-linked fALS Selected oral communication SO5 - Haining Zhu (USA) Interaction between familial ALS-linked SOD1 mutants and the dynein complex 5 Coffee break (10.15 am – 10.45 am) Session VI 10.45 am – 12.30 noon Motor neuron death in SOD1-linked fALS: inter-cellular mechanisms Chairpersons: M. Bentivoglio – L. Barbeito S21 - Luis Barbeito (Uruguay) SOD1 mutations and astrocyte dysfunction in ALS S22 - Marina Bentivoglio (Italy) Neuroinflammatory signalling in the motor circuit in SOD1-linked fALS S23 - Davide Trotti (USA) Post-translational processing of the glial glutamate transporter EAAT2 in ALS Selected oral communication SO7 - Giambattista Bonanno (Italy) Excessive exocytotic release of glutamate in the spinal cord of SOD1 G93A mutant mice Lunch (12.30 noon – 1:30 pm) Session VII 1:30 pm – 3.15 pm Motor neuron death in SOD1-linked fALS: remote contributions Chairpersons: J.P.Loeffler – A.Musarò S24 - Antonio Musarò (Italy) Muscle control of motor neuron survival and activity S25 - Linda Greensmith (UK) Axonal Transport deficits in models of motor neuron disease S26 - Jean-Philippe Loeffler (France) Energy homeostasis and denervation processes at the neuro-muscular junction in ALS Selected oral communication SO8 - Valeria Padovano (Italy) Effects of physical exercise and the steroid nandrolone on neuromuscular junctions in G93A -SOD1 transgenic mice Coffee break (3.15 pm – 3.45 pm) Session VIII 3.45 pm – 5.45 pm Poster Session 6 Sunday 16 September Session IX 8.30 am – 12.30 noon Therapeutics in SOD1 mutant mice Chairpersons: V. Silani – C. Raoul S27 - Cedric Raoul (Switzerland) Lentiviral and adeno-associated viral vectors for motoneuron gene therapy S28 - Zuoshang Xu (USA) An RNAi approach to understanding the mechanism and therapy of ALS caused by mutant SOD1 S29 - Vincenzo Silani (Italy) Translational research: possible explanations for the failure in ALS Coffee break (10.00 am -10.30 am) S30 - Jonathan Glass (USA) SOD1-People: It's time for a trial S31 - Lucie Bruijn (USA) Drug discovery and development for ALS Selected oral communication SO9 - Rita Perlingeiro (USA) The potential of endothelial progenitors to recover the stem cell niche in ALS Closing remarks Caterina Bendotti (Italy) ________________________________________________________________ Individual departure (Sandwiches available) 7 Invited Lectures 8 S1 From erythrocuprein to SOD-1: memories of a Biochemist Giuseppe Rotilio Department of Biology, “Tor Vergata” University of Rome. Italy Recollecting steps of his lifelong experience over decades of biochemical research on Cu,Zn SOD, the author will touch upon some findings obtained in the Rome laboratory, which scan the evergreen primacy of this green protein on different stages of biochemistry, even with changing pen – names. Born as a humble vehicle for copper in blood and tissues (erythrocuprein), grown up unexpectedly to an unrivalled leading role as superfast enzyme, top model for metal–binding protein centres and primary defence against ROS (superoxide dismutase), then rejuvenated in the play of metal traffic in the cell (cytocuprein), it is presently recognized as an invaluable key to understand biochemical mechanisms of life, death and disease (SOD 1). 9 S2 How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Joan Selverstone Valentine Department of Chemistry and Biochemistry, UCLA, Los Angeles, California 90095-1569, USA. More than 100 different mutations in the gene encoding copper-zinc superoxide dismutase (SOD1) cause familial forms of amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease in which aggregation of the SOD1 protein is widely believed to be the primary mode of pathogenesis. We have studied the mutant SOD1 proteins using two different approaches: characterization of expressed and purified protein and analysis of SOD1 in transgenic mouse spinal cords. In vitro, mutant SOD1 proteins show remarkably diverse structures, activities and native state stabilities. Some of the mutations cause enormous changes while others seem to have no measurable effect on its biophysical and biochemical properties. To address the question of how such widely differing proteins can all cause the same disease, we have turned to analysis of the SOD1 aggregates isolated from the spinal cords of transgenic ALS mice. Analysis of spinal cords from three different strains of ALS-SOD1 mutant mice by mass spectrometric and shotgun-proteomic methods revealed that the SOD1 proteins that are recovered from the isolated aggregates are predominantly the fulllength SOD1 polypeptide. The recovered SOD1 proteins did not bear post-translational modifications such as ubiquitination, carboxymethylation, or extensive oxidation. The aggregates were composed almost entirely of SOD1, although various proteins that are highly abundant in spinal tissue were found in very low amounts, along with SOD1. The significance of these findings for ALS will be discussed. 10 S3 ALS-associated SOD1 mutations preferentially reduce the proteins repulsive charge Mikael Oliveberg Department of Biochemistry and Biophysics, Stockholm University, Sweden. We provide bioinformatical evidence that protein charge plays a key role in the disease mechanism of amyotrophic lateral sclerosis (ALS). Analysis of 100 ALS-associated mutations in Cu/Zn superoxide dismutase (SOD1) shows that these are site-selective with a preference to decrease the proteins net repulsive charge. For each SOD1 monomer this charge is normally –6. As biomolecules as a rule maintain net negative charge to assure solubility in the cellular interior, the result lends support to the hypothesis of protein aggregation as an initiating event in the ALS pathogenesis. The strength of this preferential reduction of repulsive charge is higher in SOD1-associated ALS than in other inherited protein disorders. 11 S4 Interplay between structural and dynamic properties and aggregation in SOD1 Lucia Banci CERM & Department of Chemistry, University of Florence, Italy SOD1, in its mature form, is characterized by several post-translational modifications, such as metal binding, disulfide bond formation, and quaternary structure, i.e. a dimeric state, all of which are essential for a fully active enzyme. These features are interlinked and affect the conformational and dynamic properties of SOD1 as well as dramatically affect SOD1 tendency to aggregate. SOD1 has been suggested to be related to the onset of Familial Amyotrophic Lateral Sclerosis (FALS) as a very large number of single point mutations have been found in FALS patients. It has been also suggested that the pathogenic action of SOD1 mutants resides in their gain of a toxic function, the latter being related to their tendency to form protein aggregates. One of the striking features of these mutants is that they are spread all over the protein sequence without any apparent general correlation between their nature and location and the potential effects on SOD1 properties. We have characterized the structural and dynamic features of various SOD1 states, both for WT and mutants, in terms of metallation and oxidation, and we have investigated the interlinks/correlations among these properties(1, 2). We have identified some of the key factors which induce protein aggregation in SOD1. They are independent of the mutation and operative also in WT SOD1. This knowledge allowed us to suggest a general mechanism for SOD1 aggregation, which can reconcile the common behavior of such a diverse set of mutations (1). 1 Banci, L.; Bertini, I.; Girotto, S.; Martinelli, M.; Vieru, M.; Whitelegge, J.P.; Durazo, A.; Valentine, J.S., submitted. 2 Banci, L.; Bertini, I.; Boca, M., Girotto, S.; Martinelli, M.; Vieru, M.; unpublished data. 12 S5 Direct measurement of zinc-deficient SOD1 localized in the ventral gray matter in G93A SOD rats Beckman JS, Nylin K, Morré J and Arbogast B Linus Pauling Institute, Environmental Health Sciences Center, Oregon State University The loss of zinc from both wild type and mutant SOD offers a compelling hypothesis to explain how SOD causes the selective degeneration of motor neurons in ALS. Using 0.5 mm punches of frozen spinal cord and carefully optimized methods using electrospray-ToF mass spectrometry, we can quantify SOD with its metals fully retained by a rapid procedure. We found that zinc-deficient SOD accumulates to be nearly equamolar with Cu,Zn SOD (each was approximately 15 µM) specifically in ventral gray matter of G93A SOD transgenic rats and mice at the time when disease symptoms first develop (around 60 days). There was remarkably little evidence of protein aggregation until the animals are terminal. Zinc-deficient SOD is not present in dorsal spinal cord or in the brain of the same animals. X-ray structures show the loss of zinc causes both the electrostatic loop and the zinc binding loop become disordered, causing the copper to become more exposed as well as affecting the dimer interface. The increased redox activity of copper in zinc-deficient SOD depletes motor neurons of ascorbate and glutathione and increases formation of peroxynitrite and tyrosine nitration. The increased oxidative and nitrative stress catalyzed by zinc-deficient SOD is exceeding toxic to motor neurons. In addition, zinc-deficient SOD makes astrocytes reactive, which would contribute to the progressive death of motor neurons. 13 S6 Mutant SOD1 in motoneuronal cells: gene expression profiling to unravel mechanisms of cellular injury. Janine Kirby, Laura Ferraiuolo, Paul Heath, Hazel Holden, Pamela Shaw. Sheffield Care and Research Centre for Motor Neuron disorders, Academic Neurology Unit, Section of Neuroscience, University of Sheffield, UK Despite intensive research effort, the causes of motor neuron injury in the presence of mutant SOD1 remain incompletely understood. Microarray analysis has been applied to the problem of motor neuron degeneration. Early studies employed tissue analysis of whole spinal cord, in which changes in the motor neuron transcriptome are masked by changes in other cell types. Recently, laser capture microdissection has allowed the identification of gene expression changes in specific cell types. The initial aims of the functional genomics programme at the University of Sheffield are to: 1. Identify the cell specific properties of motor neurons and differences in gene expression between groups of MN vulnerable and spared in the disease process in ALS; 2. To understand the cellular pathways of motor neuron injury in the presence of mutant SOD1 at different time points; 3. To identify targets for drug therapies. A cellular model (NSC34 cell line stably transfected to express mutant or normal human SOD1) and the G93A mSOD1 mouse model have been employed. Gene expression changes are verified in human ALS using optimally processed CNS tissue from patients. Motor neurons are isolated from spinal cord sections using the Arcturus Pixcell laser capture microdissector. RNA is extracted using Picopure kit (Arcturus), amplified (where necessary) using the RiboAmp Amplification kit (Arcturus) and labelled using the BioArray High Yield RNA g cRNA is applied to the r Transcript Labelling Kit (Enzo). 10elevant Affymetrix GeneChip, and data analysis is performed using ArrayAssist System (Iobion); Pathway Architect and GenMAPP1.1. New insights arising from 2 key studies will be discussed: 1. Transcriptome changes in the NSC34 cell line in the presence of mutant SOD1. 268 genes were differentially expressed and key insights were the overall transcriptional repression and the downregulation of the Nrf2 anti-oxidant response element genes. The development of this pathway as a target for neuroprotective therapy with Nrf2 inducing drugs will be illustrated. 2. Gene expression changes in motor neurons at 3 disease time points in the G93A mouse model have been investigated. Key changes at the pre-symptomatic stage (252 genes differentially expressed) include a marked upregulation of lipid and carbohydrate metabolism and mitochondrial function, as well as genes involved in transcription and translational functions. At the late disease stage (120days) 167 genes are significantly altered, with the development of marked transcriptional repression, but up-regulation of complement system components and key cyclins involved in cell cycle regulation. Gene expression profiling of motor neurons in health and disease generates greater understanding of the cell specific features of motor neurons and the cellular pathways which become dysfunctional during motor neuron injury. These pathways represent rational targets for neuroprotective therapy development. 14 S7 Mutant SOD1 and protein aggregation/ degradation in motorneuronal cells Niwa J, Yamada S, Ishigaki S, Sone J, Takahashi M, Katsuno M, Tanaka F, Doyu M, Sobue G Department of Neurology, Nagoya University Graduate School of Medicine, Nagoya, Japan Mutations in the Cu/Zn-superoxide dismutase (SOD1) gene cause familial amyotrophic lateral sclerosis (ALS) through the gain of a toxic function. Ubiquitylated aggregates of mutant SOD1 proteins in affected lesions are pathological hallmarks of the disease and are involved in motor neuron death. Ubiquityl ligases, Dorfin has been reported to ubiquitylate mutant SOD1. Dorfin is a RING-finger/IBR (in-between ring-finger) domain-containing ubiquityl ligase, which we previously identified from human spinal cord. Dorfin physically binds and ubiquitylates various familial ALSlinked SOD1 mutants and subsequently targets them for proteasomal degradation. We have investigated the effect of overexpression of Dorfin and found that it protects neuronal cells against the toxic effects of mutant SOD1 and reduces the number of aggregates composed of mutant SOD1 in cell culture model system. Furthermore, we crossed Dorfin- overexpressing mice onto G93A SOD1 mice and found that double transgenic mice showed longer survival and reduced number of aggregates formation in spinal cord as compared to controls, although motor performance was not affected. In order to get more direct and powerful effect of proteasomal degradation of mutant SOD1, we next used 20 S proteasomes of archaebacteria (archaea). Archaeal 20 S proteasomes are structurally simple and proteolytically powerful and thought to be an evolutionary precursor to eukaryotic proteasomes. We successfully reproduced the archaeal proteasome in a functional state in mammalian cells, and we show that the archaeal proteasome effectively accelerated degradation of mutant SOD1 and reduced the cellular toxicities. Further, we demonstrated that archaeal proteasome can also degrade other neurodegenerative disease- associated proteins such as mutant polyglutamine tract extended androgen receptor, α-synuclein and tau. Meanwhile, recent studies suggest that mutant SOD1 readily forms an incorrect disulfide bond upon mild oxidative stress in vitro and the insoluble SOD1 aggregates in spinal cord of ALS model mice contain multimers cross-linked via intermolecular disulfide bonds. Here we show that a non-physiological intermolecular disulfide bond between cysteines at positions 6 and 111 of mutant SOD1 is important for high molecular weight aggregate formation, ubiquitylation, and neurotoxicity; all of which were dramatically reduced when the pertinent cysteines were replaced in mutant SOD1 expressed in Neuro-2a cells. Also we found that Dorfin ubiquitylated mutant SOD1 by recognizing the Cys6, 111- disulfide cross-linked form and targeted it for proteasomal degradation. From a perspective of protein aggregation/ degradation in motorneuronal cells, our results presented here open an avenue to develop a new therapeutic approach for ALS. (1) Yamada S, Niwa J, Ishigaki S, Takahashi M, Ito T, Sone J, Doyu M, and Sobue G. (2006) Archaeal proteasomes effectively degrade aggregation-prone proteins and reduce cellular toxicities in mammalian cells. J Biol Chem. 281, 23842-23851. (2) Niwa J, Ishigaki S, Hishikawa N, Yamamoto M, Doyu M, Murata S, Tanaka K, Taniguchi N, and Sobue G. (2002) Dorfin ubiquitylates mutant SOD1 and prevents mutant SOD1-mediated neurotoxicity. J Biol Chem. 277, 36793-36798. 15 S8 Mutant SOD1s and mitochondria in motoneuronal cells Ferri A (1,2), Cozzolino M (2), Crosio C (2,3), Nencini M (2), Casciati A (2), Gralla EB (4), Rotilio G (5), Valentine JS (4) and Carrì MT (2,5). (1) Inst. of Neuroscience CNR, Rome, Italy. (2) Lab. of Neurochemistry, Fondazione S.Lucia, Rome, Italy. (3) Department of Physiological, Biochemical and Cell Sciences, University of Sassari, Italy. (4) Department of Chemistry and Biochemistry, UCLA, Los Angeles, California 90095-1569, USA. (5) Department of Biology, University of Rome “Tor Vergata”, Rome, Italy Recent studies suggest that the toxicity of familial amyotrophic lateral sclerosis (fALS) mutant Cu,Zn superoxide dismutase (SOD1) arises also from its selective recruitment to mitochondria (1). We have demonstrated that each of twelve different fALS-mutant SOD1s with widely differing biophysical properties are associated with mitochondria of motoneuronal cells to a much greater extent than wild type SOD1 and that this effect may depend on the oxidation of Cys residues. We demonstrated further that mutant SOD1 proteins associated with the mitochondria tend to form cross-linked oligomers and that their presence causes a shift in the redox state of these organelles and results in impairment of respiratory complexes. The observation that such a diverse set of mutant SOD1 proteins behave so similarly in mitochondria of motoneuronal cells and so differently from wild type SOD1 suggests that this behavior may explain the toxicity of ALS-mutant SOD1 proteins that causes motor neurons to die (2). Considering that ALS is not only a multifactorial, but also a multi-system disease, and that signals from non-neuronal cells contribute in determining the progression of the disease, we have extended our observations by studying the effect of inflammatory cytokines on the localization of mutant SOD1 proteins. Our data indicate that the amount of SOD1 associated with mitochondria and the impairment of mitochondria functionality are increased in motoneuronal cells treated with cytokines, thus supporting the view that death of motor neurons is a non-cell autonomous event (3). (1) Manfredi G. and Xu Z. (2005) Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 5, 77-87. (2) Ferri A., Cozzolino M., Crosio C., Nencini M., Casciati A., Gralla E.B., Rotilio G., Valentine J.S. and Carri M.T. (2006) Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 103, 13860-13865. (3) Boillee S., Vande Velde C. and Cleveland D.W. (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39-59. 16 S9 Soluble misfolded SOD1 species in ALS Marklund SL (1), Zetterström P (1), Bergemalm D (1), Stewart H (2,3), Graffmo KS (1), Andersen P (2), Brännström T (1), and Oliveberg M (4). (1) Department of Medical Biosciences, and (2) Pharmacology and Clinical Neuroscience, Umeå University, Sweden. (3) Brain Research Center, University of British Columbia, Canada. (4) Department of Biochemistry and Biophysics, Stockholm University, Sweden. Mutants of superoxide dismutase-1 (SOD1) cause amyotrophic lateral sclerosis (ALS) by an unidentified cytotoxic mechanism. We have previously shown that the stable human (h) SOD1 mutants D90A and G93A are abundant and show highest levels in liver and kidney in transgenic murine ALS models, while the unstable G85R and G127insTGGG (G127X) mutants are scarce but enriched in the CNS. These data indicated that minute amounts of misfolded SOD1 enriched in the motor areas might exert the ALS-causing cytotoxicity. The objective of this study was to determine amounts and analyse the structure of soluble misfolded hSOD1 species in tissues of murine transgenic ALS models. For the analyses we used antibodies specific for misfolded hSOD1 generated with peptides in the sequence of the enzyme. These were used in an immunocapture protocol for analysis of misfolded hSOD1 in 20.000 g supernatants of murine tissue homogenates. A hydrophobic interaction chromatography protocol was also developed and used for that purpose. All G127X and the major part of the G85R hSOD1s bound in the assays, but only minute subfractions of the G93A and D90A mutants. Wild-type hSOD1 bound even less. The absolute levels of misfolded hSOD1 were, however, similar in the murine ALS models and they were enriched in the susceptible spinal cord. This enrichment was seen from birth until death, and the levels of soluble misfolded hSOD1 were comparable to the amounts of hSOD1 that become sequestered in aggregates in the terminal stage. The misfolded hSOD1 was composed of disulfide-reduced subunits lacking metal ions, and also subunits that apparently carried non-native intrasubunit disulfide bonds. Misfolded hSOD1, released from the antibodies by the peptides used for immunization, was shown by gel chromatography to be composed of monomers, trimers and oligomers. The soluble misfolded hSOD1 species expose sticky hydrophobic internal structures which might interact with essential cellular factors in ways that cause cytotoxicity. They form a least common denominator amongst hSOD1 mutants with widely different molecular characteristics, and are thus potential culprits for the cytotoxicity that causes ALS. The susceptibility of the motor areas of the CNS may be caused by an inadequate ability to recognize and degrade misfolded SOD1 species. 17 S10 From secretion of SOD1 mutants to immunotherapeutic approaches Urushitani M (1,2), Abou Ezzi (3), Gros-Louis F and Julien JP (1-3) Research Centre of CHUL, Laval University, Quebec, Canada Despite a decade of investigation on familial ALS caused by missense mutations in the superoxide dismutase (SOD1) gene, the mechanism of toxicity to motor neurons has remained elusive. Although it is well known that SOD1 is a cytosolic protein without specific translocation sequence, a yeast twohybrid screen led us to discover that chromogranins, components of neurosecretory vesicles, are interacting partners of SOD1 mutants linked to ALS, but not of wild-type SOD1. The existence of such interactions was confirmed by co-immunoprecipitation assays using either lysates from Neuro2a cells co-transfected with chromogranins and SOD1 mutants or from spinal cord of ALS mice. Cell culture studies showed that chromogranins may act as chaperones to promote the secretion of mutant SOD1. Cell-free translocation assay using recombinant SOD1 and microsomes showed that apo-form SOD1 of both wild and mutant types translocated into microsomes in ATP-dependent fashion. Furthermore, we discovered that extracellular SOD1 mutant can trigger microgliosis and death of motor neurons in culture suggesting a pathogenic mechanism based on toxicity of secreted SOD1 mutant proteins. This led us to test immunization protocols aiming to reduce the burden of extracellular SOD1 mutant in nervous tissue of mice models of ALS, using recombinant SOD1 mutant protein as an immunogen. At first, a vaccination protocol was tested on a G37R SOD1 mouse strain with late-onset disease exhibiting moderate levels of mutant SOD1. The repeated injections of adjuvant-SOD1 mutant with a last boost injection before symptoms at 6 month-old was effective in delaying disease onset and extending life span of G37R SOD1 mice by over 4 weeks. Western blot analysis using a monoclonal antibody specific to mutant SOD1 forms provided evidence of clearance of SOD1 species in the spinal cord of vaccinated G37R SOD1 mice. In addition, a passive immunization through intraventricular infusion of purified anti-human SOD1 antibody with osmotic minipump succeeded in alleviating disease symptoms and prolonging the lifespan of G93A SOD1 mice. From these results, we propose that immunization should be considered as potential therapeutic strategy for familial ALS caused by SOD1 mutations. (1) Urushitani, M, Sik A, Sakurai T, Nukina N, Takahashi, R. and Julien, J.-P. (2006) Chromograninmediated secretion of mutant superoxide dismutase proteins linked to ALS. Nature Neuroscience 9, 108-118. (2) Urushitani, M., Abou Ezzi S. and Julien, J.-P. (2007) Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc.Natl. Acad. Sci, 104, 2495-2500. (3) Abou Ezzi, S., Urushitani, M. and Julien, J.-P. (2007) Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem., on line. 18 S11 Live imaging of disease onset and progression in SOD1 mutant mice Jasna Kriz Dept. Anatomy and Physiology, Laval University, Centre de Recherche CHUL (CHUQ), Québec, QC, Canada Transgenic mice expressing a mutant sodium dismutase 1 (SOD1) develop phenotype with many pathological features resembling human disease. Although major clinical symptoms in amyotrophic lateral sclerosis (ALS) arise from neurodegeneration and death of motoneurons, recent studies suggest that non neuronal cells, such as astrocytes and microglia, play a role in the toxicity to motor neurons. Their precise role in onset and progression of the disease remain however unknown. To further investigate the role of non-neuronal cells in the disease onset and progression, we developed a mouse model for live imaging of astrocyte activation in ALS. Glial fibrilary acidic protein (GFAP) is strongly up-regulated in ALS. It is a known marker of astrocyte activation, and GFAP up-regulation has been associated with the disease progression. To generate a mouse model for live imaging of astrogliosis, we took advantage of reporter mice carrying the firefly luciferase gene under the transcriptional control of GFAP promoter (GFAP-luc, Xenogen, CA) and crossed them with SOD1G93A mutant mice. Live imaging of astrocyte activation was performed weekly starting as early as 3-4 weeks of age till the end stage of the disease using a high resolution CCD camera for small animal optical imaging. Data collected by in vivo imaging showed that photon emission/GFAP signal was first detected at the lumbar spinal cord area. The signal first arose from small multiple areas of astrocytes activation which then converged into a larger signal around 80100days of age. Moreover the peak signals arising from the spinal cord at approx. 100 days correlated with the abrupt onset of sensori-motor deficits and paralysis. GFAP-luc/SOD1G93A mice will provide unique tools for understanding disease pathology and longitudinal responses to drug testing. 19 S12 Proteomics of spinal cord aggregates in G93A SOD1 transgenic mouse Valentina Bonetto Dulbecco Telethon Institute and Mario Negri Institute for Pharmacological Research. Proteinaceous inclusions rich in mutant Cu, Zn superoxide dismutase (SOD1) and ubiquitin have been found in tissues of amyotrophic lateral sclerosis (ALS) patients and mutant SOD1 animals, even before disease onset. However, very little is known on the aggregate protein constituents and therefore on the actual role and mechanism of aggregation in ALS pathogenesis. We have recently shown that mutant SOD1 progressively accumulates in a Triton X-100-insoluble fraction from the spinal cord of the G93A SOD1 mouse model of ALS, and that part of insoluble SOD1 is oligoubiquitinated, therefore not targeted to the proteasome. To investigate the impact and role of aggregation in disease pathogenesis we attempted a comprehensive characterization of the proteins and their post-translational modifications of the spinal cord Triton-insoluble fraction from G93A SOD1 mice at different stages of the disease. Proteins were separated by two-dimensional gel electrophoresis and identified by MALDI-TOF mass spectrometry. The identified proteins are cytoskeletal proteins, mainly intermediate filaments, several mitochondrial proteins, chaperones, proteins of the endoplasmic reticulum, proteins involved in metabolic pathways and signaling. Now we are performing the characterization of the post-translational modifications, including nitration, carbonylation and phosphorylation. Interestingly, we could see that the majority of the aggregated proteins are oxidized suggesting a possible link between oxidative stress and aggregation pathways in ALS pathogenesis. 20 S13 Genetics of SOD1-linked ALS Peter M. Andersen Pharmacology and Clinical Neuroscience, Umeå University, Sweden. In 1993 a consortium reported the finding of 11 missense mutations in the gene encoding the enzyme CuZn-superoxide dismutase (EC 1.15.1.1, superoxide:superoxide oxidoreductase, SOD1) in 13 of 18 FALS pedigrees (Rosen et al., 1993). The human SOD1 gene spans 11 kb of genomic DNA on 21q21-22 with 5 exons separated by 4 introns. The 5 exons codes for 153 highly-conserved aminoacids which together with a catalytic Cu ion and a stabilizing Zn ion forms a subunit. Each subunit is also stabilized by an disulfid bridge spanning C57 and C146. Two identical subunits combine through non-covalent binding to form the homodimeric SOD1enzyme. The only known function of SOD1 is to catalyze the reduction of superoxide anion O2.- to molecular oxygen O2 and hydrogenperoxide H2O2, which in turn is reduced to H2O by gluthation peroxidases and catalase. SOD1 is ubiquitously expressed in all cells in all organisms above the bacteria, and constitutes about 0.5-0.8% of the soluble protein in the human brain. At the cellular level, it is found in the cytosol, nucleus, and between the two mitochondrial membranes. There are some evidence that it may be secreted through a chromogranin-mediated mechanism. In the mitochondrial matrix is the isoenzyme manganese containing SOD (Mn-SOD, SOD2), and extracellularly body fluids the tetrameric CuZnSOD (SOD3). The discovery of mutations in SOD1 in FALS sparked world-wide search for mutations in the gene. Since 1993 , a total of 153 mutations have been found:137 exonic mutations alters the amino acid sequence of the CuZn-SOD and are assumed to be disease causative. Also, eight silent mutations and six intronic variants have been reported and are presumably non-causative. Two more intronic variants close to the border between intron 4 and exon 5 have been predicted to either introduce 3 novel amino acids FLQ between exon 4 and exon 5 (creating a CuZn-SOD monomer of 156 amino acids) or introduce five novel amino acids FFTGP after exon 4 truncating the mutated polypeptide after amino acid number 123 (Andersen et al., 2003). Of the 139 disease-associated mutations, 123 are missense mutations causing a change of one amino acid to another but keeping the polypeptide lenght of 153 aminoacids. The 123 missense mutations are in 72 different codons throughout the 5 exons, including eight in exon 3 which codes for the catalytic site. The remaining 16 mutations are non-sense and deletion mutations that either introduces new nucleotides or removes nucleotides in the DNA sequence in exons 2, 4 or 5, or intron 4 as described. The result is a change in the lenght of the final polypeptide. The shortest of reported mutant polypeptides is 121 and the longest 156 amino acids long (Andersen et al., 2003) Though most missense mutations are in exon 4 and the non-sense mostly in the beginning of exon 5, there is no obvious correlation of the mutated sites and conserved residues through evolution, enzymic stability or enzymatic function. Little clinical data have been published for patients with SOD1 mutations. A 1996-review concluded that with few exceptions there are no clinical correlate between a specific mutation and phenotype. However, there are no blindedstudy comparing patients with with patients without a SOD1 gene mutation to support this statement. Now more data are available and some of the mutants can be grouped according to survival time (Table 1), variability in site of onset (Table 2), complete or incomplete disease penetrance (Table 3) with reservations for small numbers for some mutants. Inexplainable some mutants (i.e. A4V, G41S, D83G, G93A) are consistently associated with a very rapid disease independently of site of onset, while other mutants (G41D, H46R, A89V, homozygosity for D90A, E100K) are always associated with onset in the lower limbs and very slowly ascending paresis. It is the authors clinical experience that patients carrying any of the five latter mutations are remarkably similar in every aspect. In a third group of mutants, some individuals have very short survival while others in the same family survive for a decade or more. This is best illustrated by the G37R (survival range 2 to 36 years), I104F (3 to 38 years) and I113T (2-20 years). Without any obvious correlation to survival time, some mutants consistently have onset in the lower limbs (G37R, H46R, D76V, L84F, D90A homozygous, E100K) or upper limbs (L84V). Variable site of onset is the rule for many mutations. For most mutants, there appears to be both intrafamilial and interfamilial variation in sites of onset with somewhat more frequent onset in the lower limbs than in ALS not associated with SOD1 mutations. Bulbar onset has been claimed to be rare among patients with SOD1 mutations but has been reported (Table 2). The pedigrees used to find linkage to the SOD1 gene were pedigrees with high penetrance. Unfortunately, only few detailed pedigrees have been published and at present it is only safe to state 21 that 24 mutations are associated with high if not complete penetrance (Table 3). Likewise, some mutations appears to regularly be inherited with reduced penetrance sometimes obscuring the heredity of the disease and making genetic counseling difficult. This is particular the case for the widespread I113T, which is associated with a higher mean age of onset (58 years) than is most commonly reported for SOD1 mutations (47 years) (Jones et al., 1994). It is documented that I113T can pass asymptomatic from a patient down through to the grand-children or even great grandchildren before becoming manifest again (Suthers et al., 1994). The grouping listed in Tables 1- 5 are based on available published data and may change as data are published. For many SOD1 mutants no clinical data are available or the mutation has only been found in singular patients making characterisation impossible at present. The mean age at onset of first symptom is 47 years for ALS patients with a SOD1 mutation, 50.5 years for FALS and 56-58 years for SALS both without SOD mutation (Cudkowicz et al., 1997). The youngest onset of ALS in a patient with a SOD1 mutation was 6 years (I104F) and 18 years (G16S), and the oldest 84 years (A4V; I113T) and 94 years (D90A homozygous), respectively. The shortest documented survival time was 14 weeks (N86S homozygous) and the longest 36 years (G37R), 38 years (I104F) and 44 years (L144F), respectively. There is surprisingly little variability in the mean age of onset for nearly all mutants, with the exception of L37R, V38L and G114A with somewhat lower mean age of onset (perhaps biased by small numbers) and I113T with somewhat higher as mentioned earlier. That the mutants have the same mean age of onset but very different disease progression rates, uniformity in site of onset or disease penetrance implies, that onset of the disease and expression may be different processes (Andersen et al., 1997; Cudkowicz et al., 1997). While many patients with SOD1 mutations reportedly are clinically identical to patients without SOD1 mutations, a predominantly lower motor neuron pattern is the rule for patients with a SOD1 mutation. No case with predominantly upper motor neuron (UMN) features have been reported. A feature that may be unique to ALS cases with SOD1 mutations is very prolonged central motor conduction times upon transcranial magnetic stimulation of the motor cortex (MEP). Delayed central latency has been found in many patients with slowly progressing ALS heterozygous for D76Y or homozygous for D90A as well as in fast progressing cases heterozygous for G12R, G41S and N139H (Weber et al., 2000). Some mutants may show features of involvement of other parts of the nervous system. Autonomic failure has been reported in cases with G93S and V118L. Sensory symptoms, paresthesies, lancinating pain in the back, localized neuralgic pain in the buttocks, hips or knees, ataxia, or bladder disturbance (in some cases progressing to incontinence) have been reported for some mutants (Table 4). These atypical features may precede the onset of paresis and may cause difficulty in setting the ALS diagnosis. In H46R and in D90A homozygous patients, this preparetic disease phase may last for a few months to several years (Andersen et al., 1996). Patients with SOD1 gene mutations have frequently been claimed to be cognitively intact but no formal studies of this have been published. On the contrary, Masé et al. (2001) reported a patient heterozygous for the L144F SOD1 with ALS with severe frontallobe-type dementia and Battistini et al. (2005) reported two patients heterozygous for the G41S SOD1 gene mutation with aggressive ALS and frontallobe-type dementia. The most common SOD1 mutation globally is the D90A followed by the A4V (accounting for about half of all cases in the U.S.A.), and I113T. Patients with the D90A SOD1 mutation has been found in almost every country where patients have been screened for SOD1 mutation. The D90A is also the only one of the 139 mutations to show recessive inheritance and has as such been found in many apparently SALS cases. Homozygosity for the D90A gives rise to an easily identifiable phenotype with initially a preparetic phase followed months to years later by slowly ascending creeping paresis and wasting always beginning asymmetrically in the lower limbs. After a mean of some 4 years from onset in the legs, paresis appears in the upper limbs and a year and a half later bulbar symptoms appears. With time bulbar symptoms have progressed to anarthria and aphagia, and some patients have shown pseudo-bulbar palsy. A few patients have shown ataxia in the earlier stages of the diseases but it later disappears. At end stage, the patient is completely tetraparalytic with generalized wasting, cachectic and in some cases with bladder incontinence. Dementia has not been observed in D90A-homozygous ALS patients surviving for more than two decades. The mean survival time is 14 years (Andersen et al., 1996). Presently, only the D90A has been proven to show recessive inheritance but homozygosity for three other mutations (L84F, N86S, L126S) have been found in single individuals in heavily inbred families (Boukaftane et al., 1998). In these families heterozygous individuals also develops ALS and the inheritance is therefore not recessive. Interestingly, the phenotype in two of these homozygous cases appears to be far more aggressive than in the heterozygous cases suggesting a dose-effect. Recently, two siblings with ALS were found to be carriers of both a D90A and a D96N SOD1 alleles and the phenotype was rather similar to the D90A homozygous cases. This is the only instance of compound heterozygosity in ALS, but the published pedigree is also consistend with dominant inheritance of D96N with incomplete penetrance. 22 The prevalence of patients with SOD1 mutations varies greatly from country to country: No patient with a SOD1 mutation has been found in Poland, and only 9 mutations in a few patients in Germany contrasting with four mutations in Scotland, seven in Sweden and 26 in Japan. In Scotland, the I113T has been found in several FALS and apparently SALS cases (all shown to have the same common ancestor), while in northern Scandinavia the D90A is very common in particular in SALS cases. Other mutations reported in apparently SALS are listed in table 5. For only one of these, the H80R, has it been possible through paternity studies to shown that it is a de novo mutation in one of the parents. The others are likely to be FALS with low penetrance though this has only been proven for N19S, D76Y, L84F, D90A and I113T. The high prevalence of the A4V in the caucasian population in North America has in a haplotype study been shown to be unrelated to the three A4V families found in Italy and Sweden and is probably of Asian-Indian origin. I113T is most commonly found in the United Kingdom and its former colonies, L144F is widespread on the Balkan and in northern Italy, while L84F is the most frequent mutation in central Italy. The most common mutation in Germany appears to be R115G and in Japan H46R though large epidmiological studies have not been performed. Some mutations have been found in very different ethnic groups (H46R in caucasians in eastern Norway, Germany and the U.S.A., as well as in Pakistan and Japan; E100K in Afro-Americans as well as in caucasians in eastern Germany) and are probably separate mutational events in the past. While 26 mutations have been reported in the 128-million Japanese population, only recently were the first four mutations reported from Korea and China. The global distribution of different mutations may change radically when mass screening is performed in all of Asia, Africa and South America. The present knowledge is based mainly on screening a small proportion of the ALS patients in the western countries. The national differences in the reported prevalences of SOD1 mutations is not only explained by the different ethnic backgrounds but also whether only FALS cases were studied, the number of studied cases (in some countries as few as 30-40 cases have been studied) and the laboratory technique used to analyze for mutation. Screening for SOD1 mutations has revealed that SOD1 mutations are not found in other diseases than ALS, that the only SOD1 polymorphism is the D90A and that about a fifth-a sixth of all diagnosed FALS cases and a few percentage of apparently SALS cases carries a SOD1 mutation (Table 6). The 139 mutations that causes a change in the CuZn-SOD polypeptide are probably all disease causative though for only a small part statistical analysis have shown linkage to ALS or have been shown to cause a motor neuron disease when expressed in transgenic mice (G37R, G85R, G93A, G93D, D90A, G127X) or transgenic rats (H46R, G93A). The possibility that some mutants found in single patients (i.e. V14G, G16S, S134N) are co-incidental findings can not be excluded untill further studies have been done. TABLES Table 1: Disease survival time in ALS associated with SOD1 mutations (without artificial ventilation. Het = heterozygous, hom = homozygous) Fast (< 3 years) A4T A4V C6F C6G V7E L8Q G10V G41S H43R H48Q L84V D90V G93A D101H L106V I112M I112T R115G G127X A145T V148G V148I Medium (3-10 years) Slow (>10 years) Variable G85R G41D E21G G93R H46R G37R G93V D76V L38V E100G A89V D76Y D101G D90A hom L84F D101N G93D N86S G108V G93S D90A het L126X E100K G93C G141E G93R I104F I113T L144F L144S 23 Table 2: Site of onset in ALS associated with a SOD1 mutation Uniform Variable Bulbar onset G37R H46R D76V L84F L84V D90A hom E100K E100G A4V C6G G41S N86S D90A het I113T L144F V148I A4V C6G L8Q D76Y D90A het V148I I151T Table 3: Disease penetrance in SOD1 gene mutations associated with ALS: Complete (>90% by age 70) Incomplete A4V A4T G37R L8Q L38V N19S G41S E21G H43R N65S H46R G72S D76V D76Y L84F N86S L84V A89V N86K D90A hom D90A het E100G A95T D101H G93S I104F I113T G108V L126S C111Y S105L I112M N139H G114A L126GQRWKX G127GGQRWKX G141E L144F V148G V148I Table 4: SOD1 mutationes associated with atypical features: A4V (neuralgic pain), V14G, E21G, H46R, H48Q, L84F, D90A, G93S, E100G, I104F, V118L (only after having been placed on IV), L144F, I151T. Sexual disinhibition: G41S Cognitive disturbance: L144F, G41S Table 5: SOD1 mutations reported in cases with apparently sporadic ALS (SALS): A4V, L8V, G12R, V14G, G16S, N19S, E21K, V29A, H48Q, C57R, N65S, G72S, D76Y, H80R (only de novo mutation confirmed), L84F (of unknown father), N86I, N86S, N86D, A89V, D90A,G93S, A95T, V97L, D101N, S105delSL, L106P, D109Y, I113T, T116R, V118L, V118KTGPX, D124G, E133E, K136X, L144F Table 6: Frequency of SOD1 mutations In SALS: : 7.3% (3/41) in Italy (Corrado L et al., Neuromuscul Disord 2006;16:800-804) 7% (4/56) in Scotland (Jones CT et al., J Med Genet 1995;32:290-292) 6% (3/48) in Italy (Gellera C et al., ALS & other MND’s 2001;2(suppl 1):543-546) 4% (14/355) in Scandinavia (Andersen PM et al., Brain 1997;10:1723-1737) 3% (5/175) in the UK (Shaw CE et al., Ann Neurol 1998;43:390-394) 3% (5/155) in England (Jackson M et al., Ann Neurol 1997;42:803-807) 24 1.2% (1/87) in Spain (García-Redondo A et al., Muscle & Nerve 2002;26:274-278) 0% (0/225) in Italy (Battistini S et al., J Neurol 2005;252:782-788) In FALS: 23.5% (12/51) in Scandinavia (Andersen PM et al., Brain 1997;10:1723-1737) 23.5% (68/290) in the USA (Cudkowicz ME et al., Ann Neurol 1997;2:210-221) 21% (8/38) in the UK (Shaw CE et al., Ann Neurol 1998;43:390-394) 19.7% (14/71) in the UK (Orrell R et al., Neurology 1997;48:746-751) 18% (2/11) in Spain (García-Redondo A et al., Muscle & Nerve 2002;26:274-278) 18% (7/39) in Italy (Battistini S et al., J Neurol 2005;252:782-788) 14.3% (10/70) in France (Boukaftane Y et al., Can J Neurol Sci 1998;25:192-196) 12% (9/75) in Germany (Niemann S et al., JNNP 2004;75:1186-1188) Without classification to hereditary disposition: 7.2% (148/2045) in North America (Andersen PM et al., ALS & Other MND’s 2003;4:62-73). 25 S14 Genetics of non-SOD1-linked fALS Christopher E. Shaw Department of Clinical Neuroscience, King’s College London, Box 43 Institute of Psychiatry, De Crespigny Park, London SE5 8AF, UK. chris.shaw@iop.kcl.ac.uk Traditional gene hunting strategies in autosomal dominant (AD) conditions rely on ascertaining DNA and detailed clinical information from multiple affected individuals in large kindreds. Because classical amyotrophic lateral sclerosis (ALS) is a rapidly progressive and fatal illness the identification of a robust locus has only been achieved in a handful of kindreds. The identification of SOD1 on Chromosome 21q21 was followed by linkage to 18q21 (ALS3), 16q21 (ALS6) and 20p13 (ALS7). Disappointingly the genetic mutation responsible for ALS in these kindreds has been elusive despite exhaustive gene sequencing. Confirmation of linkage in multiple kindreds to these loci has only been achieved for ALS 6 and genome-wide scans of the linked kindreds using microarrays has challenged our confidence that these are the only potentially linked regions. Relatively recently it has been recognised that ALS and fronto-temporal dementia exist in a phenotypic spectrum and can occur within the same AD kindred as a single gene disorder. Five families with AD ALS-FTD were previously linked to 9q21. More recently we have identified a locus on chromosome 9p13-21, (maximal multipoint LOD score of 3.02 (=0) at D9S1878). Four other kindreds have now been linked to the same overlapping locus. Recombination has narrowed the conserved haplotype in our kindred to 12cM (11Mb) at 9p13.2-21.3 (flanking markers D9S2154 and D9S1874). Bioinformatic analysis of the region has identified 103 known genes but no pathogenic mutation has been identified to date. New genomic research platforms are being developed which we anticipate will advance our ability to identify new loci and gene mutations. 26 S15 SOD1 in Italian ALS patients Stefania Battistini Department of Neuroscience, Neurology Section, University of Siena, Siena, Italy Presently, more than 100 different mutations located in the five exons of the SOD1 gene have been identified worldwide in ALS patients with an autosomal dominant transmission or rarely, recessive transmission. In surveys of different populations in Europe and North America, the frequency of SOD1 mutations in familial ALS (FALS) was reported to be 12% to 23.5% and in sporadic ALS (SALS) 1.2% to 7% (1). We and others have shown that, in Italy, the frequency of SOD1 gene mutations in FALS cases is 18% to 22% and in SALS cases 1% to 4.5% (2-4), a figure consistent with those of other surveys. Among the more than 100 SOD1 mutations associated with ALS, some occur as recurrent mutations while others as founder mutations and a geographic distribution of SOD1 mutations is beginning to emerge. The A4V mutation results the most prevalent mutation in the United States while the D90A mutation the most prevalent in Europe and a founder effect has been demonstrated for both. In Italy, at the present time, 18 different SOD1 gene mutations have been reported in ALS patients (2-5). Overall, the D90A results the most common mutation among SOD1 mutated Italian ALS cases. Recently, we first reported two Italian ALS patients heterozygous for the D90A mutation, providing evidence of the coexistence in Italy, like in other countries, of ALS cases with heterozygous and homozygous D90A mutation. The G41S, together with the L84F, results the most prevalent SOD1 gene mutation found in SOD1 mutated Italian FALS patients (3). These mutations seem to be responsible for ALS clustering in Central Italy. Indeed, three of the L84F families originated from the same mountainous area of the Marche region, while all the seven ALS patients carrying the G41S mutation identified by us, belonged to five unrelated FALS pedigrees originating from the same restricted area of northwest of Tuscany. Interestingly, the G41S mutation has only been found so far in Italian families. This mutation is associated with a quite uniform clinical phenotype with early UMN and LMN involvement in one or both lower limbs and a rapidly progressive disease course with appearance of bulbar signs within 1 year and death few months later. Genotyping with markers from the SOD1 locus showed that the seven Italian G41S FALS patients share a common haplotype (6). Our findings strongly suggest that the G41S mutation in the Italian population originates from a common ancient founder. (1) Andersen PM. (2006) Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Current Neurology and Neuroscience Reports 6, 37-46. (2)Gellera C, Testa D, Passariello P, et al. (2003) SOD1 gene mutations in amyotrophic lateral sclerosis Italian patients. Amyotroph Lateral Scler Other Motor Neuron Disord. 4 (Suppl 1), 98. (3) Battistini S, Giannini F, Greco G, et al. (2005) SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study. J Neurol. 252, 782-8. (4) Corrado L, D’Alfonso S, Bergamaschi L, et al. (2006) SOD1 mutations in Italian patients with sporadic amyotrophic lateral sclerosis (ALS). Neuromuscul Disord. 16,800-4. (5) ValentinoP, Conforti FL, Pirritano D, et al. (2005) Brachial amyotrophic diplegia associated with a novel SOD1 mutation (L106P). Neurology. 64, 1477-1478. (6) F. Giannini, S. Battistini, C. Ricci, et al. (2005) Phenotypic-genotypic study of amyotrophic lateral sclerosis Italian families with the G41S SOD1 gene mutation. Amyotroph Lateral Scler Other Motor Neuron Disord. 6 (Suppl 1),79. 27 S16 The pathogenesis of sporadic amyotrophic lateral sclerosis Wim Robberecht Dept. of Neurology and Section of Experimental Neurology,University Hospital Leuven, Leuven Belgium. The pathogenesis of sporadic amyotrophic lateral sclerosis (ALS) remains unknown. It is generally accepted that a genetic component and/or an environmental factor on the background of an aging nervous system are involved. Several attempts to identify an environmental factor have been unsuccessful. Genetic susceptibility factors or modifying genes can be identified in a candidate gene approach based on molecular studies of models of motor neuron degeneration in vivo or in vitro, based upon genome wide association studies in ALS patients, or through the genetic screening of small animal models for the disease. These different approaches are likely to be complementary as it is hypothesized that many factors are likely to be involved. Excitotoxicity, the vascular endothelial growth factor (VEGF) system and abnormalities of RNA metabolism have been identified as pathogenic contributors to the mechanism of motor neuron degeneration. In the present presentation, data supporting these hypotheses will be highlightened, and new supportive evidence will be presented. In addition, a zebrafish model generated to identify new modifying genes will be presented. 28 S17 The double life of two pro-survival proteins: when SOD1 and Bcl-2 get mutated, wild and pro-death Piera Pasinelli Farber Institute for Neurosciences, Thomas Jefferson University 900 Walnut Street, Philedelphia, PA 19107 USA Mitochondrial abnormalities and activation of the mitochondrial apoptotic pathway are both characteristic features of mutant SOD1-mediated motor neuron death. Contrary to WT SOD1 (a well known anti-apoptotic protein), mutant SOD1 is pro-apoptotic. The mechanisms by which WT SOD1 protects and mutant SOD1 kills the cells are unknown. Because SOD1 represents one of the major anti-oxidant defenses in the cells, and an increased production of reactive oxygen species (ROS) and oxidative stress have been implicated in many paradigms of cell death, the anti-apoptotic and pro-survival properties of SOD1 have mainly been attributed to its anti-oxidant activity. We found that Sod1 interacts with Bcl-2 raising the intriguing question of whether the anti-apoptotic properties of SOD1 are a consequence of both its dismutase/anti-oxidant function and, in parallel but perhaps independently, its interactions with Bcl-2 and the anti-apoptotic machinery. The molecular switches that confer toxic properties to mutated SOD1 are unknown. Similarly to WT SOD1, mutant SOD1 binds Bcl-2. However, the nature of mutant SOD1 binding to Bcl-2 differs from that of WT SOD1. Contrary to WT SOD1, mutant SOD1 specifically localizes to spinal cord mitochondria were it forms SDS-resistant high molecular weight aggregates that bind and entrap Bcl2 Thus, opposed to the WT SOD1/Bcl-2 complex, mutSOD1/Bcl-2 aggregates could be a molecular determinant of cell death in ALS. In the present study, we began a series of in vitro and cell-based studies to determine whether indeed both WT and mutant SOD1, at least partially, depend on Bcl-2. We will show our unpublished, preliminary evidence indicating that both WT and mutant SOD1 may require Bcl-2 to exert their protective and toxic function respectively. Consequences for development of therapies against mutant SOD1-mediated cell death will be discussed. 29 S18 Role of Bcl2a1 in SOD1-linked fALS Crosio C (1,2), Casciati A (2), Iaccarino C (1,2), Rotilio G (3) and Carrì MT (2,3) (1) Department of Physiological, Biochemical and Cell Sciences, University of Sassari, Italy. (2) Lab. of Neurochemistry, Fondazione S.Lucia, Rome, Italy. (3) Dept. of Biology, University of Rome “Tor Vergata” Motor neurons are the only cells in both animal models and ALS patients that undergo degeneration upon mutant SOD1 over-expression. Up to date, the molecular bases of mutant SOD1-induced cell death are still debated. A crucial step in filling this gap is the identification of genes whose expression is altered by mutant SOD1 in motor neurons. We provide evidence of the selective induction of Bcl2a1 in motor neurons of a mouse model of ALS and its functional switch upon Tumor Necrosis Factor alpha (TNFalpha) treatment in neuronal cell lines. Bcl2a1 up-regulation may represent an early event aimed to protect cells from oxidative stress induced by mutant SOD1, but at later stages, when neuroinflammation occurring in ALS is maximal, Bcl2a1 expressed early in motor neurons, may switch from anti-apoptotic to pro-apoptotic. In these conditions increase in Bcl2a1 constitutes a reinforcing event in the progressive loss of motor neurons occurring in ALS. 30 S19 The nucleus as target for mSOD1 toxicity Sau D (1,4), Crippa C (1,4), De Biasi S (2), Vitellaro-Zuccarello L (2), Simonini F (1,4), Onesto E (1,4), Rusmini P (1,4), Bolzoni E (1,4), Bendotti C (3), Poletti A (1,4). (1) Institute of Endocrinology, Center of Excellence on Neurodegenerative Diseases, University of Milan (Italy). (2) Department of Biomolecular Sciences and Biotechnologies, University of Milan (Italy). (3) Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri, Milan (Italy). (4) InterUniversity Center on Neurodegenerative Diseases, Universities of Florence, Rome and Milan (Italy). Cu/Zn Superoxide-Dismutase type-1 (SOD1) is one of the major intracellular antioxidant enzyme responsible for the removal of toxic free radical species. The enzyme is a homodimer of about 32 kDa, which is mainly localized in the cytoplasm, but smaller amounts are present in the nucleus, in the peroxisomes and lysosomes, as well as in mitochondria (1, 2). It is well know that point mutations in the gene coding for SOD1 are linked to almost 15% of cases of familial amyotrophic lateral sclerosis (fASL), but in the majority of the mutations, the SOD1 enzymatic activity is preserved, suggesting that motor neuron death is due to a gain of neurotoxic function(s) of the aberrant SOD1. However, we recently found that the two mutant SOD1s (G93A and A4V) misfold generating insoluble species and aggregates, that alter the nuclear diffusion of the enzyme. The lack of the antioxidant defense mechanisms in the nucleus correlates with an increased sensitivity to DNA damage, induced by the free radical species formed after treatment with high doses of hydrogen peroxide. Thus, although, we confirmed that the formation of mutant misfolded SOD1 aggregates is associated to the impairment of the ubiquitin-proteasome pathway (UPP), and possibly the trigger of the disease, the alteration of the genomic DNA in postmitotic motor neuronal cells may be a contributing factor in the pathogenesis and progression of fALS. In order to counteract the primary cause of mutant SOD1 toxicity linked to protein misfolding, we have then analysed the role of a small chaperone protein on the solubility and degradation of this antioxidant enzyme, and shown that the clearance of the mutant SOD1 is greatly enhanced by the HspB8 protein, without affecting the UPP. This suggests that the modulation of the levels of this small Hsp protein may provide novel potential therapeutical approaches for fALS. 1) Fridovich I., (1975) Superoxide dismutases. Annu. Rev. Biochem. 1975, 44, 147-159. 2) Valentine, JS, and Hart PJ (2003) Misfolded CuZnSOD and amyotrophic lateral sclerosis. PNAS. 100, 3617-3122. GRANTS: Telethon - Italy (#GGP06063), MIUR-FIRB (#RBAU01NXFP), MIUR-Cofin (2005057598_002), University of Milan-FIRST, FONDAZIONE CARIPLO 31 S20 Role of MAP kinases cascade in SOD1-linked fALS M. Tortarolo, G. Spaltro, D. Lidonnici, P.Veglianese, D. Lococo and C. Bendotti Dept. Neuroscience, Institute for Pharmacological Research” Mario Negri”, Milano, Italy Activation of mitogen activated protein kinases (MAPKs), which consists of three main kinase subfamilies, JNK, p38MAPK and ERK, represents an important step in the signaling transduction cascades leading to neuronal death in response to a variety of extracellular stimuli including excitotoxicity, oxidative stress and inflammatory processes. These pathways are involved in several neurodegenerative diseases and have been implicated in the mechanisms underlying of motor neuron degeneration in amyotrophic lateral sclerosis (ALS). In particular we have found that p38MAPK cascade, including MKK3-6, MKK4 and ASK1, is selectively activated in the motor neurons of transgenic mice overexpressing SOD1 mutants before the symptoms onset in concomitance with the accumulation of phosphorylated neurofilaments in the perikarya, an early sign of axonal dysfunction and neuronal degeneration. Increased activated p38MAPK and ASK1 immunoreactivity was also found in the spinal motor neurons from sporadic ALS patients indicating that this phenomenon is likely to play a relevant role in the pathogenesis of ALS. No changes of JNK activation was found in the motor neurons of mouse models and ALS patients. We have also found that activation of p38MAPK pathway in the motor neurons is associated with a concomitant upregulation of the TNFalpha but not IL1 beta, receptors. With the progression of the disease p38MAPK activation becomes prominent also in the reactive astrocytes and microglia of SOD1 mutant mice although this effect was not accompanied by the activation of the upstream cascade, suggesting different regulatory mechanisms of this kinase in motor neurons and glial cells. JNK is also activated in the glial cells of mouse models and patients with ALS. Overactivation of these kinases in the glial cells may induce the over production of inflammatory cytokines and nitric oxide and this may strongly contribute to propagate the damage to adjacent healthy cells. Studies using spinal neurons-astrocytes co-culture system expressing or not the mutant SOD1,are underway to verify this hypothesis. p38MAPK has been also implicated in a specific FAS-linked death pathway occurring in motor neurons. Recently, a specific inhibition of FAS ligand has demonstrated a delay in the onset and increased survival in SOD1 mutant mice mediated by the inhibition of p38MAPK. However a direct inhibitor of p38MAPK function was shown to protects motor neurons but did not prevent disease progression in SOD1 mutant mice. Thus, the direct involvement of p38MAPK as primary event or epiphenomenon in the degeneration of motor neurons is still controversial and will be discussed. Supported by Telethon. 32 S21 SOD1 mutations and astrocyte dysfuction in ALS Luis Barbeito Institut Pasteur de Montevideo, Uruguay. Glial cells surrounding motor neurons can greatly influence neurodegeneration in mice expressing the mutations of the superoxide dismutase-1 (SOD1) linked to familiar ALS. We analyzed mitochondrial activity in astrocytes expressing the SOD1G93A mutation and determined whether such astrocytes influenced survival of neighboring motor neuron. Mitochondria from astrocyte cultures obtained from transgenic rats expressing the SOD1G93A displayed a defective respiration, decreased oxygen consumption, lack of ADP-dependent respiratory control and reduced membrane potential. Similar defects were found in mitochondria isolated from the spinal cord of SOD1 G93A rats. Furthermore, protein 3-nitrotyrosine was detected immunochemically in mitochondrial proteins from SOD1G93A astrocytes revealing that mitochondrial defects were associated to nitro-oxidative damage. Pre-treatment with the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) restored mitochondrial function, forming adducts with mitochondrial proteins in vivo. Remarkably, SOD1G93A astrocytes induced death of motor neurons cultured on the top of the astrocyte monolayers, as compared to non-transgenic ones. Motor neuron loss was prevented by pre-incubation of SOD1G93A astrocytes with antioxidants and NOS inhibitors. We also tested recently designed mitochondrially-targeted antioxidants, ubiquinone or carboxy proxyl nitroxide covalently coupled to a triphenylphosphonium cation (MitoQ and Mito-CP, respectively), since they could act as multimodal drugs, having multiple cellular synergistic effects. In our hands, both MitoQ and Mito-CP completely restored mitochondrial respiration in SOD1G93A-expressing astrocytes and stimulated the ability of astrocytes to support motor neuron survival. Remarkably, neuroprotection was accomplished at low nanomolar concentrations of the oxidants, which make them much more effective than therapies with conventional antioxidants. We hypothesize that the systemic administration of these drugs to SOD1G93A mice will down-regulate glial-mediated neurotoxicity. Taken together, our results indicate that mitochondrial dysfunction in astrocytes critically influence motor neuron survival, which may explain key features of ALS progression. 33 S22 Neuroinflammatory signalling in the motor circuit in SOD1-linked fALS Bentivoglio M (1), Kassa R.M. (1), Cupidi C. (1,2), Mariotti R. (1) (1) Department of Morphological and Biomedical Sciences, University of Verona, Verona, Italy, (2) Department of Experimental Medicine, University of Palermo, Italy. A wealth of data obtained in the murine model of SOD1-linked familial amyotrophic lateral sclerosis (fALS), as well as experimental findings on motoneuron degeneration after peripheral nerve damage, recall attention on the role of neuroinflammatory signalling in motoneuron death, but this role is still unclear. In our laboratory, we investigated alterations of central motor circuits in SOD1-mutant mice, with special reference to inflammatory parameters and to the response to peripheral challenges. The role of retrograde inflammatory signals in motoneuron degeneration was studied in presymptomatic SOD1(G93A) mice, applying a “double hit” paradigm. This sequential insult consisted of inflammation elicited in the target muscles by injections of the endotoxin lipopolysaccharide (LPS), followed by facial axotomy. The data obtained with these manipulations revealed an enhancement of motoneuron loss and microglial activation in the facial nucleus of SOD1-mutant mice after nerve transection with respect to wild-type (wt) mice, and inflammatory priming further enhanced the degeneration of axotomized SOD1-mutant facial motoneurons. In addition, recruitment of T lymphocytes in the injured facial nucleus was affected by inflammatory priming in a different manner in SOD1-mutant and wt mice, since the transgenic animals, at variance with wt ones, showed a low degree or no T cell infiltration in the axotomized facial nucleus after LPS pretreatment. This abnormal lymphocytic response was not due to impairment of cellular adhesion mechanism or to alternative recruitment of neutrophils. The data point out the interesting findings of i) an effect of retrograde inflammatory signalling on the enhanced susceptibility of SOD1-mutant motoneurons to death-inducing axonal injury, and ii) an effect of mutant SOD1 on immune-related responses to peripheral inflammatory challenges. Furthermore, investigation of the motor cortex of SOD1(G93A) mice revealed alterations of corticospinal neurons and surrounding glia. Shrinkage of the cell bodies of corticospinal neurons, characterized with retrograde tract tracing from the spinal cord, as well as marked activation of the surrounding astrocytes and microglia were found in the presymptomatic stage of disease and worsened in the terminal stage. Upregulation of mRNAs of pro-inflammatory mediators (interleukin, IL,-1α and IL-1β, inducible nitric oxide synthase and NF-κB) was documented in the deep layers of the motor cortex, where corticospinal neurons reside, of SOD1-mutant mice. Altogether the findings point to damage of the entire central motor circuit in murine SOD1-linked fALS, and highlight that such alterations are accompanied by inflammatory events and implicate alterations of immunerelated responses. (Supported by FIRB RBNE01B5WW and MIUR PRIN 2005057598) 34 S23 Post-translational processing of the glial glutamate transporter EAAT2 in ALS Davide Trotti Farber Institute for Neurosciences, Thomas Jefferson University 900 Walnut Street, Philedelphia, PA 19107 USA EAAT2, also known as GLT-1 in rodents, is a high affinity, Na+-dependent glutamate transporter of glial origin that is essential for the clearance of synaptically released glutamate and prevention of excitotoxicity. During the course of human amyotrophic lateral sclerosis (ALS) and in transgenic mutant SOD1 mice models of the disease, expression and activity of EAAT2 is remarkably reduced. We previously showed that some of the mutant SOD1 proteins exposed to oxidative stress inhibit EAAT2 and the impairment was largely brought about by caspase-3 cleavage at a single defined locus, giving rise to two fragments that we termed truncated EAAT2 (Tr-EAAT2) and C-terminus of EAAT2 (CTE). In this study we report that analysis of spinal cord homogenates prepared from mutant G93A-SOD1 mice reveals CTE to be of a higher molecular weight than expected due to conjugation with SUMO-1. The sumoylated CTE fragment (CTE-SUMO-1) accumulates in the spinal cords of these mice as early as pre-symptomatic stage (70 days of age) and not in other central nervous system areas which are unaffected by the disease. CTE-SUMO-1 appearance is specific to the ALS mice, as it does not occur in the R6/2 mouse model for Huntington’s disease. Using an astroglial cell line, primary culture of astrocytes and tissue samples from G93A-SOD1 mice, we show that CTE-SUMO-1 is targeted to promyelocytic leukemia nuclear bodies (PML-NBs). As one of the proposed functions of PML-NBs is regulation of gene transcription we suggest a possible novel mechanism by which the glial glutamate transporter EAAT2 could contribute to the pathology of ALS. 35 S24 Muscle control of motor neuron survival and activity Dobrowolny G (1), Aucello M (1), Fanò G (2), Protasi F (2), Molinaro M(1), Rosenthal N (3), Sandri M (4), and Musarò A (1) (1) Department of Histology and Medical Embryology, Sapienza University of Rome, Italy. (2) CeSI, Centro Scienze dell'Invecchiamento, Universita G. d'Annunzio, Chieti, Italy. (3) EMBL Mouse Biology Program, Monterotondo, Italy; (4) Venetian Institute of Molecular Medicine, Univ Padova, Italy. One of the crucial systems severely affected in several neuromuscular diseases is the loss of effective connection between muscle and nerve, leading to a pathological non-communication between the two tissues. The best examples of impaired interplay between the two tissues is the disease Amyotrophic Lateral Sclerosis (ALS), due to mutation in the SOD1 gene. Motor neurons degeneration and muscle atrophy are the major pathological processes associated with ALS suggesting that nerve activity plays an important role in muscle homeostasis and remodeling. However, other cells may be involved in the pathogenesis of ALS and open the possibility that alteration in skeletal muscle homeostasis represents one of the principal mediators of motor neurons degeneration. In this context, skeletal muscle can play a direct role in the pathogenesis of ALS or, alternatively, provide the appropriate factors to sustain neuron survival. In this presentation I will discuss the contribution of skeletal muscle to motor neuron survival and activity and the potential muscle signals, including IGF-1, that influence neuron survival, axonal growth and maintenance of synaptic connections. 36 S25 Axonal transport deficits in models of motor neuron disease Linda Greensmith Institute of Neurology, University College London, Queen Square, London, UK Increasing evidence suggests that deficits in axon transport pathways play an important role in MND pathogenesis. In SOD1 mice, defects in both retrograde and anterograde transport have been observed prior to disease onset. Indeed, we previously observed deficits in retrograde axonal transport in SOD1 motoneurons as early in development as embryonic day 13, which represents one of the earliest pathological changes reported in this model1. Results from other mouse models of MND have also shown a link between axonal transport deficits and motoneuron degeneration. For example, Loa mice homozygous for mutations in the molecular motor dynein, show an impairment of axonal retrograde transport in addition to significant motoneuron degeneration and postnatal overexpression of the dynein subunit dynamitin, also induces motoneuron degeneration in mice. Furthermore, human disorders involving degeneration of motoneurons have been linked to mutations in motor proteins e.g. the anterograde motor Kinesin 1 and dynactin which is an essential component of the dynein motor complex. Surprising results from our laboratory provided further evidence linking transport deficits to motoneuron degeneration, when we found that the deficit in retrograde transport in SOD1 motoneurons is completely rescued when SOD1 mice are crossed with Loa mice. Moreover, Loa/SOD1 mice show amelioration in disease symptoms and a significant extension in lifespan. It has been recently been shown that mutant SOD1 interacts and co-localizes with dynein, suggesting a direct gain of function of mutant SOD1 which may result in impaired axonal transport. However, the mechanism by which a mutation in a motor protein ameliorates symptoms and axon transport defects in SOD1 motoneurons remains unclear. Nevertheless, our unexpected findings not only indicate that defective transport may play a significant role in MND pathogenesis but also suggest that manipulation of axonal transport pathways may have therapeutic value. We are currently investigating the mechanism by which axonal transport deficits may cause motoneuron degeneration for example by investigating mitochondrial transport in models of MND. We are also studying axonal transport in a number of alternative mouse models of MND, for example a new mouse strain called Abnormal rear legs (Arl), which has a different point mutation in the cytoplasmic dynein heavy chain to that described for the Loa mouse. Finally, we are also studying axonal transport in motoneurons of a mouse model of Spinal Bulbar Muscular Atrophy (SBMA), in which motoneurons die as a consequence of an expansion of CAG trinucleotide repeats in the androgen receptor gene. 37 S26 Energy homeostasis and denervation processes at the neuro-muscular junction in ALS Loeffler JP, Dupuis L, Gonzalez de Aguilar JL, René F and Meininger V. Laboratoire INSERM U692 “Signalisations Moléculaires et Neurodégénérescence”, Université Louis Pasteur, Strasbourg France and (MV) Centre SLA Salpêtrière Fédération de Neurologie, Paris France. Despite the traditional view of ALS as a pure motor neuron (MN) disease, growing evidence suggests that the disease is, in fact, a multisystem disorder with additional extramotor neurological manifestations. Beyond the nervous system, intriguing metabolic alterations have also been observed in asssociation with the course of the disease. In particular, recent studies revealed that two thirds of ALS patients present with a stable hypermetabolism that correlates with survival (Desport et al., 2005). The cause of the hypermetabolism in ALS is still unknown but our research on genetic animal models has shed some light into this question. Very early, transgenic mice with mutated SOD1 genes present with reduced adiposity and increased rates of energy expenditure, which reveals the presence of a metabolic deficit before any apparent sign of motor impairment. The burden of these metabolic alterations exerts some influence on the neurodegenerative process, since increasing the lipid content of the diet offers neuroprotection and extends survival (Dupuis et al., 2004). We have also shown that the gastroinstestinal absorption of lipids as well as the peripheral clearance of triglyceriderich lipoproteins are markedly increased in the mutant SOD1 mice, which strongly suggests an increase in the consumption of lipids by muscles, a situation that could account for the protective effect of the high fat regimen in these animals (Fergani et al., 2007). Our recent studies demonstrates that hyperlipidemia is a typical feature of ALS patients and, most importantly, that bearing an abnormally elevated LDL/HDL ratio significantly increased survival by more than 12 months (Dupuis et al., 2007). An early hallmark of ALS is the destruction of the neuromuscular junction (NMJ), an event that might suggest that the anatomical target of MNs is somehow involved in the disease initiation. To test whether increased energy expenditure in skeletal muscle is sufficient to initiate denervation, we analyzed transgenic mice overexpressing uncoupling protein 1 (UCP1) under the control of the muscle specific creatine kinase (MCK) promoter. We show that these mice present progressive denervation (as shown by electromyographic recording), loss of functional NMJ, loss of MN and muscle atrophy. These results show that an increased energy demand in the muscle fibre is sufficient to initiate major disorders in the motor unit. The metabolic disorder associated with ALS may represent a physiopathological mechanism whose importance in disease initiation and progression has been largely underestimated. References Desport et al., 2005 Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis 2:202-207. Dupuis et al., 2004 Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA 101:1115911164. Dupuis et al., 2007 Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology (in press). Fergani et al., 2007 Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J Lipid Res 48:1571-1580. 38 S27 Lentiviral Vectors and Adeno-associated Vectors for Motoneuron Gene Therapy Raoul C 1,2, Towne C 1 and Aebischer P1 1 Integrative Biosciences Institute, SV LEN, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland. 2 Present address: Institut National de la Santé et de la Recherche Médicale (INSERM) Avenir team, INMED, campus de Luminy, Marseille, France. Approximately 20% of familial ALS cases are caused by mutations in the Cu/Zn superoxide dismutase (SOD1) gene that result in a deleterious gain of function for this enzyme. The posttranscriptional gene silencing mechanism of RNA interference (RNAi) thus represents a promising therapeutic strategy for this disease. We have previously described the proof of principle for RNAibased knockdown of SOD1 mutant in a fALS mouse model. Lentiviral vectors expressing short hairpin RNA (shRNA) targeting mutated SOD1 were injected into the lumbar spinal cord of presymptomatic SOD1 mice retarding disease onset and progression in the hindlimbs. The next challenge is therefore to enhance the delivery of these silencing instructions to reach more cells involved in disease pathogenesis. Towards this goal, we are evaluating the use of systemic injections of adeno-associated viral vectors (AAV), which have been shown to infect large quantities of skeletal muscle following intravenous delivery. Since AAV has been shown to transduce motoneurons through retrograde transport following local intramuscular delivery, we hypothesised that this systemic approach would facilitate widespread transduction of motoneurons. Accordingly, we have generated an AAV serotype 6 vector driving expression of shRNA against SOD1 (AAV6-shSOD1) with an EGFP reporter to trace transduced cells. We first observed efficient retrograde transport of AAV6-shSOD1 to the motoneuron soma following intramuscular injection and have also demonstrated that this vector was capable of silencing SOD1 in muscle at the mRNA and protein level. We have thus performed systemic administration of the vector by tail vein injection. Intravenous AAV6-shSOD1 injections resulted in high levels of transduction in skeletal muscles, including gastrocnemius, biceps, tongue, masseter, intercostals and diaphragm. Strikingly, anaylsis of the transduction pattern of AAV6 in the central nervous system revealed transduced motoneurons at lumbar, thoracic and cervical levels of the spinal cord and also within the motoneuron nuclei of the brain stem. We are currently examining the effect of this delivery technique on disease onset, progression and ultimately, survival, by injecting presymptomatic adult mice with the AAV6-shSOD1 vector. 39 S28 An RNAi approach to understanding the mechanism and therapy of ALS caused by mutant SOD1 Zuoshang Xu Department of Biochemistry and Molecular Pharmacology,University of Massachusetts Medical School, Worcester, Massachusetts, USA. RNAi is a conserved cellular mechanism in metazoan. Triggered by double stranded RNA, RNAi causes degradation of RNA molecules that share homology to the double stranded RNA. Therefore, RNAi can be used to target specific mRNA for degradation, leading to gene silencing. My lab tries to harness this mechanism to accomplish two goals: establishing transgenic RNAi as a reverse genetics tool for silencing specific genes in mice and developing RNAi as a therapeutic means for treating neurodegenerative diseases. Currently, the main method for reverse genetics in mammals is gene knockout by homologous recombination. Although highly effective, this method is complex, expensive, time-consuming and limited to mouse. These limitations hinder rapid and wide application of reverse genetics in mammalian species. We have developed a transgenic RNAi method to silencing specific genes in transgenic mice and demonstrated that this method can reproduce phenotypes of gene knockout with lower cost and shorter time. Recently we have developed an inducible silencing method with which one can induce gene silencing in specific cell types. We are applying this technology to study genes that are relevant to neurodegenerative diseases including ALS. Because of its specificity, RNAi has a great potential as a therapeutic method for a variety of diseases. We proposed to develop RNAi as a therapy for ALS caused by mutations in SOD1 and other neurodegenerative diseases that are caused by dominant, gain-of-function type of genetic mutations. We demonstrated that RNAi is effective in knock down the expression levels of mutant SOD1 in vitro, in cultured cells and in vivo. We are testing delivery of RNAi in animals by several different ways, including gene therapy using recombinant adenovirus and adeno-associated virus, and direct infusion of chemically stabilized siRNA. Our preliminary results suggest that both delivery methods can be effective in extending the survival of mice expressing mutant SOD1G93A. These results hold promise that RNAi can be applied to treat ALS caused by mutations in SOD1 gene as well as other neurodegenerative diseases caused by gain-of-function type of genetic mutations. 40 S29 Translational research: possible explanations for the failure in ALS Vincenzo Silani Department of Neurology and Laboratory of Neuroscience, University of Milan Medical School - IRCCS Istituto Auxologico Italiano, Milano, Italy The most popular animal model in ALS is the SOD1 G93A mutant mouse. The use of this transgenic in understanding the pathogenesis of the disease has become invaluable since 1994 (Gurney et al, 1994). As natural extension of neurobiological studies, any treatment to ALS patients has been pretested in the SOD1 mouse. Today obligate informations in pre-clinical settings indicate drug trials in this animal model as a prerequirement for any therapeutical approach in humans. The number of treatments tested in the SOD1 mutant mouse has been endless but the applicability to ALS patients on the basis of the efficacy in the model really poor. Success in human clinical trials have been extremely limited, calling into question the utility of preclinical data for identifying therapeutical agents that are worthy of further studies in humans. There are several possible explanations for the failure of the successful animal studies to translate into effective therapies in humans. The main possibility is that the SOD1 mouse really represents an animal model for familial rather than sporadic ALS. A second possible explanation is that therapies in the mouse are most commonly initiated prior to the clinical onset of the disease. These considerations and many others have been recently reviewed (Benatar, 2007). The scientific community has reached a large awareness of the scarce therapeutical translation from mice to humans in a desperate disease as ALS. A preliminary document has been produced and guidelines for the preclinical in vivo evaluation of pharmacological active drugs assembled (Ludolph et al., 2007). We need animal models mimicking motoneuronal selective vulnerability such as SOD1 transgenic mouse that, compared to transgenics for other neurodegenerative disease, represents an elegant study model to define the pathobiology of ALS. However, we need to further set accepted standards for drug testing. Translation of the SOD1 transgenic data to humans needs to be perfected but the ALS scientific community has the skill and commitment to achieve the goal. References Benatar. Neurobiol Dis. 2007 Apr;26(1):1-13. Epub 2007 Jan 3; Gurney et al. Science 1994, 264, 1772-1775; Ludolph et al. Amyotrophic Lateral Sclerosis First published on 05 April 2007; DOI: 10.1080/17482960701292837. 41 S30 SOD1-People: It's time for a trial Jonathan Glass Emory Center for Neurodegenerative Disease Woodruff Memoriall Research Building, Atlanta GA 30322, USA A small minority of people with ALS carry pathogenic mutations in the SOD1 protein. This discovery led to the development of transgenic models of SOD1-related ALS in mice and rats, as well as in non-mammals such as fish and worms. The biological consequences of SOD1 mutations are many, and there are a number of hypotheses regarding how SOD1 mutant proteins lead to motor neuron dysfunction and death. Even so, it is still unclear how mutant SOD1 leads to clinical disease in animals, and there is precious little data on the mechanism(s) of disease in SOD1 people. There have been more than 150 reports of therapeutic interventions in animal models of SOD1 related ALS (Benatar, 2007), many of which demonstrate the "effectiveness" of treatments directed at one or several of the hypothetical mechanisms of disease. Several of these promising animal studies have led to human clinical trials. Unfortunately, none of these trials has shown beneficial effects in humans with ALS. These failures in human clinical trials must force us to re-evaluate how we evaluate preclinical data from SOD1 models as tools for translation to humans. There are several potential problems with this approach: 1. The overwhelming majority of participants in ALS clinical trials have sporadic ALS, and not familial ALS. Very few would be expected to have SOD1-related ALS. 2. There are no data supporting the idea that the mechanism(s) of disease in sporadic and familial ALS or SOD1-related ALS are the same. 3. Many of the therapeutic "successes" in animal models were demonstrated in experimental paradigms requiring presymptomatic treatment of animals. 4. The animal models of SOD1-related ALS require that the mutant protein be massively overexpressed in order to generate a clinical or pathological phenotype. This is not the case in humans with SOD1-related ALS. A logical and reasonable approach to better understanding the pathogenesis of ALS and developing new treatments is to focus more attention on people and families with SOD1-related ALS. These people represent the true human counterpart to the mutant SOD1 that have been studied so intensively. We have found this to be highly motivated to participate in epidemiologic and genetic surveys, and many have expressed their willingness to participate in a preventive clinical trial. Much can be learned from this population: What is the true risk of carrying an SOD1 mutation? What factors other than genetics influence the onset of disease? Are there proteomic, physiologic, or other biomarkers that can be used to predict disease onset? Are there similarities or differences in disease mechanisms in SOD1-related ALS and sporadic ALS? Will a therapeutic trial in people carrying SOD1 mutations show positive results that correspond to similar trials in animals? SOD1 people are an experimental resource that are waiting to be studied. Its time. This work is presented as a collaborative effort with Michael Benatar, MD, and Meraida Polak, RN, along with Samantha Kaplan and Crystal Richards, MPH. Funding for this project is from Emory University's Woodruff Health Science Center and a fellowship grant to MB from the American Academy of Neurology and the ALS Association. 42 S31 Drug discovery and development for ALS Lucie Bruijn The ALS Association, 4338 Lavender Drive, Palm Harbor FL34685; email: lucie@alsa-national.org Currently there is only one FDA approved drug for ALS, Riluzole, which has a modest effect on patient survival. There is a real need for the development of effective drugs for the disease. There is no single cell based assay that best represents the disease mechanism in ALS (it is likely that multiple mechanisms are involved in disease progression). In partnership with The National Institute of Neurological Disorders (NINDS), The ALS Association tested 1000 FDA approved compounds in 10 different assays modeling aspects of the disease. The most promising lead from this screen has moved forward into clinical trials for ALS. Expanding on these efforts and taking a more directed and focused approach to ensure that projects once initiated and promising will be taken through the necessary steps to move better chemical entities into model systems and finally the clinic, The ALS Association launched its new program Translational Research Advancing Therapies for ALS (TREAT ALS) in 2005. The intent of this new program is to develop novel compounds and take advantage of drugs in development that have potential for ALS from the academic and biotech sector. This presentation will provide an overview of ongoing efforts and future directions for Drug Discovery and Development in ALS. 43 Selected oral communications 44 SO1 Dismutase-inactive mutant SOD1 has different functional properties when heterodimerized with wild type SOD1 Heidrun Witan (1), Ingrid Koziollek-Drechsler (1), Rebecca Wade (2), Christian Behl (1) and Albrecht M. Clement (1) Institute for Physiological Chemistry and Pathobiochemistry, University of Mainz, Medical School, 55099 Mainz; (2) EML Research, Heidelberg. The toxic properties of amyotrophic lateral sclerosis (ALS)-causing mutant Cu/Zn superoxide dismutase (SOD1) proteins are still elusive. Among the proposed, but recently challenged, toxic qualities are the increased aggregation capacity and an aberrant enzymatic activity. Furthermore, contradictory reports exist to which extend the presence of wild type SOD1 ( WTSOD1) contributes to disease onset and progression. In order to investigate the role of hWTSOD1 in ALS, we initially tested if mutant monomers can form functional dimers with hWTSOD1 in HEK-293 cells. The wild type-like mutants A4VSOD1, G37RSOD1 and G93ASOD1 but also copper-free G85RSOD1 form dismutase active SOD1 dimers after transient transfection. In order to study the biochemical and functional properties of heterodimers, we generated SOD1 dimer constructs allowing the expression of a fusion protein of two covalently linked SOD1 monomers with an EGFP tag. Although the two SOD1 monomers are in close proximity, they do not form a static functional dimer as others have reported by introducing intermolecular disulfide bonds. We demonstrated that hWTSOD1, A4VSOD1, G37RSOD1 and G93ASOD1 homodimers as well as mutant-WT heterodimers display dismutase activity, suggesting that the dimer formation and 3Dstructure remain unaffected by the linker and the EGFP-tag. Even G85RSOD1 heterodimers showed activity, whereas G85RSOD1 homodimers are inactive as previously published. Homo- and heterodimers show different aggregation behaviour. When expressed transiently, mutant homodimers form visible aggregates, whereas heterodimers have a largely reduced aggregation potential. These results have been confirmed by centrifugation assays. Mutant homodimers are enriched in the pellet fractions, whereas heterodimers remain largely in the cytosolic fractions. Based on their differential activity and aggregation behaviour, we propose that studying the functional properties of homo- and heterodimeric SOD1 proteins may give new insights into the role of hWTSOD1 in ALS pathogenesis and possibly will shed more light on the toxic properties of mutant SOD1 This work was supported by the MAIFOR Program and the IFZN of the University of Mainz. H.W. was supported by the “Graduiertenkolleg Neuroscience” of the University of Mainz. 45 SO2 Cysteine 111 affects aggregation of mutant SOD1 in NSC34 cells Cozzolino M. (1), Amori I. (1), Pesaresi M.G. (1), Ferri A. (1,3), Nencini M. (1), Carrì M.T. (1,2). (1) Lab. of Neurochemistry, Fondazione S. Lucia IRCCS, Rome, Italy; (2) Department of Biology, University of Rome “Tor Vergata”, Rome, Italy; (3) Inst. of Neuroscience CNR, Dept. of Psychobiology and Psycopharmacology, Rome, Italy Converging evidence indicates that aberrant aggregation of mutant SOD1 is strongly implicated in FALS. Mutant SOD1 forms high molecular weight oligomers which disappear under reducing condition, both in neural tissues of FALS transgenic mice and in cells overexpressing mutSOD1, indicating a role of aberrant intermolecular disulfide crosslinking in the oligomerization and aggregation process 1, 2. To study the specific contribution of cysteines in the mechanism of aggregation, we mutated human SOD1 in each of its cysteine residues in position 6, 57, 111 and 146. Using a cell transfection assay for SOD1 solubility and aggregation, we analysed the behaviour of those SOD1s. Preliminary results showed that cysteines affected hSOD1 solubility to different extents, with mutation of Cys6 the more effective in promoting SOD1 aggregation and mutation of Cys111 having no effect. A mutated cysteine background was also used to test the solubility of G93A mutSOD1. Interestingly, the removal of Cys111 strongly reduced the ability of G93A SOD1 to form aggregates in cultured NSC34 cells. Accordingly, insoluble G93A mutSOD1 showed an altered reactivity of Cys11 to modification by maleimide, suggesting the occurrence of cysteine oxidation. Moreover, treatments that depleted the cellular pool of reduced glutathione (GSH) exacerbated mutSOD1 insolubility, while an overload of intracellular GSH significantly rescued mutSOD1 solubility. These data are consistent with the view that the redox environment influences the oligomerization/aggregation pathway of mutant SOD1 and point to Cys111 as a key mediator of this process. 1. Ferri, A. et al. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A 103, 13860-5 (2006). 2. Deng, H. X. et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A 103, 7142-7 (2006). 46 SO3 Detection of misfolded SOD1 in sporadic and familial ALS Rakhit R (1), Robertson J (1), Vande Velde C (2), Horne P (1), Ruth DM (1), Griffin J (1), Cleveland DW (2), Cashman NR (3), Chakrabartty A (1) (1) Dept Med Biophysics/CRND, University of Toronto, Toronto, Canada. (2) Ludwig Inst., UCSD, La Jolla, USA. (3) Dept Medicine, UBC, Vancouver, Canada. Protein misfolding diseases result from the toxicity associated with conversion of the native state of a protein into a pathologically misfolded conformation. In 20% of familial ALS, mutations in SOD1 cause the protein to misfold and form intracellular inclusions. Toxicity of these cytoplasmic aggregates is thought to arise from aberrant interactions with the protein-folding chaperones or from inhibition of proteasomes. Toxicity has also been proposed to result from aberrant interactions with mitochondrial proteins (Tom20/Bcl-2) because SOD1 has been detected in mitochondria from spinal cord, and mitochondrial vacuolization is an early event in ALS models. The ability to detect misfolded proteins in vivo would be very beneficial for diagnosis and for furthering research into the molecular basis of the disease. Our approach to developing in vivo conformational probes involves generation of antibodies that detect non-native states of a protein, yet do not bind natively folded molecules. We have developed an antibody that specifically recognizes monomer/misfolded forms of SOD1. This antibody was raised to an epitope within the SOD1 dimer interface, which is normally buried within the native obligate homodimer. This SOD1 exposed dimer interface (SEDI) antibody, only recognizes SOD1 conformations where the native dimer is disrupted/misfolded, exposing the hydrophobic dimer interface. We have used this antibody to detect misfolded SOD1 in transgenic rodent ALS models and in human tissue. Using the SEDI antibody we establish the presence of monomer/misfolded SOD1 in three ALS mouse models, with G37R, G85R or G93A -SOD1 mutations, and in a human individual with an A4V SOD1 mutation. Despite ubiquitous expression, misfolded SOD1 is found primarily within degenerating motor neurons and is preferentially enriched in mitochondrial and membrane fractions from affected spinal cords. Misfolded SOD1 appears before symptom onset and decreases at disease end-stage, concomitant with motor neuron loss. Extracellular misfolded SOD1 was also observed by exclusion of double staining with markers of CNS cell types. Misfolded SOD1 was present in brain and spinal cord, but absent in tissues that are unaffected by disease: liver, heart, muscle, and kidney. Spinal cord sections from human individuals with sporadic ALS and familial ALS with and without SOD1 mutations are being examined. 47 SO4 The Evolution of the Amyotrophic Lateral Sclerosis Database (ALSOD) Wroe RJ, Butler AW, Powell JF and Al-Chalabi A MRC Centre for Neurodegeneration Research, Department of Neuroscience, Institute of Psychiatry, King’s College London, UK. To date, more than 100 different point mutations spanning across the 153 amino acid SOD1 sequence have been identified causing ALS. In 1999 the ALSOD database was generated to store these point mutations along with ALS patient information, in an attempt to correlate ALS genotype with ALS phenotype (1). The aim of the database was to further understand the disease producing allelic variations and genetic diversity associated with ALS. Here we report our ongoing development and redesign of the ALSOD database and its automated procedures. The significant new features have improved ALSOD helping to link the genetic mutations of the SOD1 protein to the hypothetical 3D structural rearrangements caused by the mutation and the resulting patient phenotype. In addition to the ALS genotype to phenotype relationship, the new version of ALSOD now provides a more comprehensive knowledgebase for ALS, detailing genetic, proteomic, and bioinformatics information associated with the disease. The redeveloped site also enables ALSOD to be searched for a specific ALS subject based on their demographic and disease information. We believe that ALSOD provides an ideal vehicle so that ALS data can not only be recorded but also searched. (1) Radunovic A, Leigh PN. ALSODatabase: database of SOD1 (and other) gene mutations in ALS on the Internet. European FALS Group and ALSOD Consortium. Amyotroph Lateral Scler Other Motor Neuron Disord. 1999 Dec;1(1):45-9. 48 SO5 Interaction between familial ALS-linked SOD1 mutants and the dynein complex Fujian Zhang (1), Anna-Lena Ström (2), Kei Fukada (2), Sangmook Lee (3), Lawrence J. Hayward (3) and Haining Zhu (1,2) (1) Graduate Center for Nutritional Sciences, (2) Department of Molecular and Cellular Biochemistry, College of Medicine, Lexington, KY 40536, (3) Department of Neurology, University of Massachusetts Medical School, Worcester, MA 01655 Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive motor neuron death. More than 90 mutations in the copper-zinc superoxide dismutase (SOD1) gene cause a subset of familial ALS. Toxic properties have been proposed for the ALS-linked SOD1 mutants, but the nature of the toxicity has not been clearly specified. Cytoplasmic inclusion bodies containing mutant SOD1 and a number of other proteins are a pathological hallmark of mutant SOD1 mediated familial ALS, but whether such aggregates are toxic to motor neurons remains unclear. In this study, we identified a dynein subunit as a component of the mutant SOD1-containing highmolecular-weight complexes using proteomic techniques. We further demonstrated interaction and colocalization between dynein and mutant SOD1, but not normal SOD1, in cultured cells and also in G93A and G85R transgenic rodent tissues. Moreover, the interaction occurred early, prior to the onset of symptoms in the ALS animal models and increased over the disease progression. Motor neurons with long axons are particularly susceptible to defects in axonal transport. Our results demonstrate a direct “gain-of-interaction” between mutant SOD1 and dynein, which may provide insights into the mechanism by which mutant SOD1 could contribute to a defect in retrograde axonal transport or other dynein functions. The aberrant interaction is potentially critical to the formation of mutant SOD1 aggregates as well as the toxic cascades leading to motor neuron degeneration in ALS. 49 SO7 Excessive exocytotic release of glutamate in the spinal cord of SOD1 G93A mutant mice Milanese M. (1), Zappettini S. (1), Raiteri L. (1), Popoli M (2), Barbiero V.S. (2), Bonanno G. (1) (1) Department of Experimental Medicine, Pharmacology and Toxicology Section, and Centre of Excellence for Biomedical Research, University of Genoa, Italy (2) Department of Pharmacological Sciences, Center of Neuropharmacology, and Center of Excellence on Neurodegenerative Diseases, University of Milan, Italy Among the different hypotheses to explain motor neurons death in amyotrophic lateral sclerosis (ALS), glutamate-mediated excitotoxicity may play a mayor role. Abnormalities in glutamate transport, mainly a reduced expression and function of GLT-1, were observed in synaptic preparations of motor cortex and spinal cord in ALS. It has been suggested that this reduction in GLT-1 activity could explain the higher levels of glutamate in ALS patients and in animal models of the disease Since GLT1 is mainly localized in astroglial processes, it was hypothesized that glial cells play a role in the development of the disease. Alternatively, elevated extra-cellular concentrations of glutamate may well be due to augmentation of neuronal glutamate release rather than to the astrocyte-localized inhibition of reuptake. We have previously shown that the heterotransporter-mediated glycine- ad GABA-induced release of [3H]D-aspartate ([3H]D-Asp) from mouse spinal cord synaptosomes was more elevated in mice expressing human SOD1 with the Gly93A substitution [SOD1(+)/G93A(+)], a transgenic model of ALS, respect to mice expressing the unmodified human SOD1 [SOD1(+)] and in non transgenic littermates [SOD1(-)/G93A(-)] (1, 2). We have here studied the release of 3HD-Asp and of endogenous glutamate induced by depolarizing and non depolarizing stimuli known to induce exocytotic neurotransmitter release. The spontanoeus outflow of the excitatory amino acid was higher in SOD1/G93A(+) than in SOD1(+) or SOD1(-)/G93A(-) mice. Exposure to 15 or 25 mM KCl or to 0.3 µM ionomycin provoked an almost complete Ca2+-dependent release of [3H]D-Asp from spinal cord synaptosomes. The exocytotic release of 3HD-Asp and glutamate induced by KCl or ionomycin was dramatically increased in symptomatic SOD1(+)/G93A(+) mice than in controls. The higher glutamate release in mutant animals was already present in pre-symptomatic 70-90 and 30-40 day-old mice. Noticeably, both the stimulus-evoked release of GABA in spinal cord and of [3H]D-Asp in motor cortex of SOD1(+)/G93A(+) mice did not differ from controls As to the molecular determinants of this increased glutamate exocytosis, we have studied the SNARE-complex proteins expression, the SNARE-complex formation, CaM kinasi II phosphorylation, its interaction with syntaxin-1. The results indicate that spinal cord glutamatergic nerve terminals undergoes to some presynaptic modifications which may sustain the increased glutamate exocytosis. Our in vitro results show that the exocytotic release of glutamate is enhanced mutant SOD1 mice. If it occurs in vivo, the different modulation of GLU and GABA release here reported could induce an unbalance between spinal inhibitory and excitatory transmission in ALS. (1) Raiteri L., Paolucci E., Prisco S., Raiteri M. and Bonanno G. (2003) Activation of a glycine transporter on spinal cord neurons causes enhanced glutamate release in a mouse model of amyotrophic lateral sclerosis Br. J. Pharmacol. 138, 1021-1025. (2) Raiteri L., Stigliani S., Zappettini S., Mercuri N.B., Raiteri M. and Bonanno G. (2004) Excessive and precocious glutamate release in a mouse model of amyotrophic lateral sclerosis Neuropharmacology 46, 782-792. 50 SO8 Effects of physical exercise and the steroid nandrolone on neuromuscular junctions in G93A -SOD1 transgenic mice Francolini M.(1), Cappello V.(1), Matteo Fossati (1), Mariotti R.(2), Kassa R. M.(2), Padovano V. (1), Bentivoglio M (2), and Pietrini G. (1) (1) Department of Pharmacology, School of Medicine, Università degli Studi di Milano; CEND- Center of neurodegenerative diseases; CNR-Institute of Neuroscience , Milan, Italy. (2) Department of Morphological and Biomedical Sciences, University of Verona, Verona, Italy. Evidence in mice and man indicates that ALS is a distal axonopathy, with pathological changes of motoneurons beginning at the distal axons (with early mitochondrial damages and selective loss of neuromuscolar synapses) and proceeding in a "dying back" pattern (1). Quantitative analysis demonstrated denervation at the neuromuscular junction (NMJ) by day 47 in SOD1(G93A) mice in which signs of clinical disease appeared at about 80 days. A confirmation of ALS as a distal axonopathy also in the human pathology derived from autoptic findings in a patient, demonstrating denervation and reinnervation changes in muscle but normal appearing motoneurons (2). The molecular mechanisms underlying these defects are not known, but they suggest an altered plasticity of NMJs. SOD1(G93A) transgenic mice have been used as a model to study the effects of strenuous exercise and/or the anabolic steroid nandrolone on NMJs by immunofluorescence and electron microscopy. Transgenic SOD1-G93A mice and their wild type littermates were sacrificed at disease onset (P90) and diaphragm and triceps has been dissected, fixed by perfusion and subsequent immersion in 4% paraformaldehyde for 2 hours and extensively washed with phosphate buffer. Teased muscles bundles composed of 20-30 fibers were excised from diaphragms and stained with TRITC-conjugated alpha-bungarotoxin (Molecular Probes) to label the nAchR in the postsynaptic membrane. These bundles were then immunolabeled with anti-synapsin in order to evaluate the percentage on innervated/denervated fibres in the different experimental models to evaluate the protective or detrimental effect of the treatments in this muscle. For the ultrastructural analyses bundles of fibers were dissected, fixed with 2% glutaraldehyde and further processed for transmission electron microscopy. Our preliminary results indicate that strenuous exercise decreases the number of innervated fibres in both transgenic and wild type littermates, with a remarkable effect in transgenic mice. On the contrary, the chronic steroid treatment appears beneficial, although insufficient to prevent the negative effect of physical exercise in G93A transgenic mice. It has been recently proposed that mutant SOD1 accumulated in the presynaptic terminal may contribute directly to denervation or to failure of successful reinnervation of neuromuscular junctions (3), we are therefore looking for the presence and the localization of mutant SOD1 in the NMJs of transgenic mice diaphragms. (1) Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases.J Neurosci.20, 2534-2542. (2) Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man.Exp Neurol. 185, 232-240. (3) Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. (2006) Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 26, 8774-8786. 51 SO9 The potential of endothelial progenitors to recover the stem cell niche in ALS Akbarloo N., Darabi R. and Perlingeiro R.C.R. Department of Developmental Biology, UTSouthwestern Medical Center, Dallas-TX, USA Amyotrophic Lateral Sclerosis (ALS) is a progressive neurodegenerative disorder in which motor neurons die beginning in mid-adult life. The loss of large projection neurons results in severe movement dysfunction. Transgenic mice that express mutant SOD1 develop a progressive motor neuron disease that resembles human ALS, representing a valuable animal model to study novel therapies for this disease. To date, ALS is still incurable and current cellular approaches to treat ALS have attempted to replace damaged or deteriorating neurons by stem cell transplantation. Although some improvement has been observed, regeneration of large projection neurons capable of establishing appropriate connections to their targets is still out of reach. An alternate approach to treat ALS would be to improve the microenvironment, which in turn would stimulate cell regeneration. Endothelial progenitors mediate revascularization, which promotes improved blood flow in ischemic tissues, a fundamental requirement for tissue regeneration. In this regard, human umbilical cord blood (UCB) has been pointed out as a rich potential supply of endothelial progenitors, which are known to express the surface marker CD133. To test the hypothesis that endothelial progenitors isolated from human cord blood are able to repair the neuronal microenvironment in ALS, we have generated an immunodeficient SOD1 mouse model, SOD1;Rag2-/-;gammaC-/-. These mice are devoid of B, T, and NK cells and make excellent xeno-transplant recipients, allowing us to perform human to mouse transplantations without immunosuppression. This immunodeficient mouse model shows similar disease progression to conventional SOD1 mice. By week 18, untreated immunodeficient SOD1 mice as well as the ones that had been transplanted with unfractionated mononuclear cells (MNCs) presented the usual symptoms of disease, including significant weight loss, marked kyphosis, and hind limb muscle wasting, while mice that had received transplantation of UCB-derived CD133+ cells presented no signs of disease by this time. Overall, an extension of 4 weeks was observed in the lifespan of SOD1;Rag2-/-;gammaC-/- infused with endothelial progenitors. Rotarod analyses further demonstrated a delay in the impairment of motor function in mice that received CD133+ cell transplantation when compared to untreated mice (an approximate 4 week latency in motor performance deficits: week 20 vs week 16, respectively). Analyses of spinal cord cryosections showed that transplanted mice had twice as many surviving motoneurons as did nontransplanted mice. These findings corroborate our hypothesis that early endothelial progenitors can preserve or repair the neurogenic niche and provide scientific rational for the therapeutic application of UCB transplantation in ALS. 52 Posters Posters Posters 53 P1 The Molecular Mechanism of Superoxide Dismutase Aggregation in Lou Gehrig's Disease Sagar D. Khare, Michael Caplow, Kyle Wilcox and Nikolay V. Dokholyan Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, School of Medicine, Campus Box 7260 Chapel Hill, NC 27599 USA Mutation-induced aggregation of the dimeric enzyme Cu, Zn superoxide dismutase (SOD1) has been implicated in the familial form of the amyotrophic lateral sclerosis (ALS) or Lou Gehrig's disease, but the mechanism of aggregation is not known. We studied the folding thermodynamics and kinetics of SOD1 using a hybrid molecular dynamics approach. We found that the residues which contribute the most to SOD1 thermal stability are also crucial for apparent two-state folding kinetics. Surprisingly, we found that these residues are located on the surface of the protein and not in the hydrophobic core. Mutations in some of the identified residues are found in patients with the disease. We argue that the identified residues may play an important role in aggregation (1,2). We further investigated in silico the sequence and structural determinants of SOD1 aggregation: (a) we identified sequence fragments in SOD1 that have a high aggregation propensity, using only the sequence of SOD1, and (b) we performed molecular dynamics simulations of the SOD1 dimer folding and misfolding. In both cases, we identified identical regions of the protein as having high propensity to form intermolecular interactions. These regions correspond to the N- and C-termini, and two crossover loops and two -strands in the Greek-key native fold of SOD1. Our results suggest that the high aggregation propensity of mutant SOD1 may result from a synergy of two factors: the presence of highly amyloidogenic sequence fragments ("hot-spots"), and the presence of these fragments in regions of the protein that are structurally most likely to form inter-molecular contacts under destabilizing conditions. Therefore, we postulated that the balance between the self-association of aggregation-prone sequences and the specific structural context of these sequences in the native state determines the aggregation propensity of proteins (3,4). We further experimentally measured dissociation rate and equilibrium constants for human SOD1 dimer dissociation. We have also determined that loss of Zn is required for SOD1 aggregation. Using computation and experiments, we determined the rates of Zn loss and further SOD1 aggregation. Thus, we have identified a minimal aggregation sequence of SOD1 and measured the rate and equilibrium constants for each step of this sequence. Our findings suggest the role of mutations that are associated with familial ALS (5). (1) S. D. Khare, F. Ding, and N. V. Dokholyan, "Folding of Cu, Zn superoxide dismutase and Familial Amyotrophic Lateral Sclerosis" Journal of Molecular Biology, 334: 515-525 (2003) (2) S. D. Khare and N. V. Dokholyan, "Common dynamical signatures of FALS-associated structurallydiverse Cu, Zn superoxide dismutase mutants" Proceedings of the National Academy of Sciences USA, 103: 3147-3152 (2006) (3) S. D. Khare, K. C. Wilcox, P. Gong, and N. V. Dokholyan, "Sequence and structural determinants of Cu, Zn superoxide dismutase aggregation" Proteins: Structure, Function, and Bioinformatics, 61: 617632 (2005) (4) S. D. Khare, M. Caplow, and N. V. Dokholyan, "FALS mutations in Cu, Zn superoxide dismutase destabilize the dimer and increase dimer dissociation propensity: a large-scale thermodynamic analysis" Amyloid: the Journal of Protein Folding Disorders, 13: 226 - 235 (2006) (5) S. D. Khare, M. Caplow, and N. V. Dokholyan, "The rate and equilibrium constants for a multi-step reaction sequence for the aggregation of superoxide dismutase in ALS" Proceedings of the National Academy of Sciences USA, 101: 15094-15099 (2004) 54 P2 Early misfolding events in human Cu-Zn superoxide dismutase apo-enzyme are revealed by molecular dynamics Richard W. Strange*, Chin W. Yong*, William Smith & S. Samar Hasnain CCLRC Daresbury Laboratory, Warrington, Cheshire WA4 4AD Mutations of the gene encoding Cu-Zn superoxide dismutase (SOD1), a vital antioxidant enzyme, cause 20 % of the familial cases of the progressive neurodegenerative disease amyotrophic lateral sclerosis (FALS). A growing body of evidence suggests that it is the molecular behaviour of the dimeric molecule in the absence of metals that leads to gain of toxic properties via misfolding, unfolding and aggregation. Structural studies have provided static snapshots on the behaviour of the molecule in the absence of the metals. Using atomic resolution structure of fully metallated human SOD1 and highly parallelised MD code on a high performance capability computer, we have employed molecular dynamics to reveal the first stages of misfolding due to metal-deletion. These calculations reveal the first steps in the formation of experimentally observed aggregations of FALS mutant SOD1. This result could have implications for the role of de-metallated wild-type SOD1 in sporadic cases of ALS, for which the molecular cause still remains undiscovered. 55 P3 Oxidized wild type SOD1 acquires toxic properties reminiscent of mutant SOD1 species Abou Ezzi S. (1), Urushitani M. (2), Julien J.-P. (1) (1) CHUL research center, Laval University, Quebec, QC, Canada; (2) Molecular Neuroscience research center, Shiga university, Shiga, Japan Sporadic and familial ALS cases are clinically indistinguishable, suggesting that common pathogenic mechanisms are involved. Oxidative stress is implicated in the pathogenesis of neurodegenerative diseases including Parkinson’s, Alzheimer’s and ALS (1). Recent studies suggest that superoxide dismutase (SOD1) may represent a major target of oxidative damage in neurodegenerative diseases. To test the possibility that oxidized species of wild type (WT) SOD1 might be involved in pathogenic processes, we analyzed the properties of the WT human SOD1 protein after its oxidation in vivo or in vitro by hydrogen peroxide (H2O2) treatment. Using transfected Neuro2a cells expressing WT or ALSlinked SOD1 species, we show that exposure to H2O2 modifies the properties of WT SOD1. Oxidized WT SOD1 can be conjugated with poly-ubiquitin and can interact with Hsp70. Chromogranin B, a neurosecretory protein that interacts with mutant SOD1 but not with WT SOD1(, was coimmunoprecipitated with oxidized WT SOD1 from lysates of Neuro2a cells treated with H 2O2. Treatment of microglial cells (line BV2) with either oxidized WT SOD1 or mutant SOD1 recombinant proteins induced tumor necrosis factor-α and inducible nitric oxide synthase. Furthermore, exposure of cultured motor neurons to oxidized WT SOD1 caused cell death like mutant SOD1 proteins. These results suggest that WT SOD1 may acquire binding and toxic properties of mutant forms of SOD1 through oxidative damage. Hence, the possibility that sporadic and familial ALS may share a common pathogenic pathway involving abnormal SOD1 species must be considered. (1) Shaw P. J. (2005) Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry 76, 1046-1057. (2) Urushitani M., Sik A., Sakurai T., Nukina N., Takahashi R. and Julien J. P. (2006) Chromograninmediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 9, 108-118. 56 P4 SOD1 intracellular localization in copper-depleted NSC-34 motoneuronal cells Arciello M (1), Capo CR (1), Ferri A (2), Cozzolino M (2), Carrì MT (1,2), Rossi L (1) (1) Department of Biology, “Tor Vergata” University of Rome. Italy; (2) Laboratory of Neurochemistry, Fondazione S.Lucia IRCCS, Rome, Italy. A small fraction of SOD1 locates in the intermembrane space of mitochondria, where it enters as apo-protein (1); it has been postulated that the accumulation of mutant SOD1 in mitochondria may be a possible cause of motoneuron death in familial amyotrophic lateral sclerosis (fALS) (2). We have previously demonstrated that the treatment of neuroblastoma cells with the copper chelator Trientine depletes SOD1 activity (3); this might increase the amount of copper-devoid SOD1 entering mitochondria. Contradictory evidence has been reported on the correlation between copper-depletion and ALS: treatment of fALS model mice with Trientine prolongs survival (4), while copper-deficient patients show evidence of lower motor neuron disease, similar to ALS patients (5). In order to clarify the role of copper deficiency in SOD1 sub-cellular localization and motor neuron degeneration, we treated murine motoneuronal NSC-34 cells, transfected with human wild-type SOD1, with Trientine for 72 hours. Intracellular copper level was measured by atomic absorption spectrometry, showing a decrease of about 50%. The activity of transfected SOD1 (assessed by specific staining of gels, after electrophoresis under non-denaturing conditions) decreased, as well as its protein content, evaluated by Western blotting. Mitochondria were isolated from transfected NSC34 cells following copper depletion and showed accumulation of wild-type SOD1. Cell viability as well as cell cycle, analysed by flow cytometry, were not affected, indicating that accumulation in mitochondria of wild-type SOD1 is not detrimental to cells. The same experimental design, but carried out on NSC-34 expressing mutants SOD1, will elucidate whether these proteins behave differently than the wild-type form under conditions of copper-depletion, thus affecting motorneuron viability. (1) Sturz L.A., Diekert K., Jensen L.T., Lill R. and Culotta V.C. (2001). A fraction of Cu,Znsuperoxide dismutase and its metallochaperone , CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276, 38084-38089. (2) Ferri A., Cozzolino M., Crosio C., Nencini M., Casciati A., Gralla E.B., Rotilio G., Valentine J.S. and Carrì M.T. (2006). Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. 103, 13860-13865. (3) Rossi L., Marchese E., Lombardo M.F., Rotilio G. and Ciriolo M.R. (2001). Increased susceptibility of copper-deficient neuroblastoma cells to oxidative stress-mediated apoptosis. Free Radic. Biol. Med. 30,1177-1187. (4) Nagano S., Fujii Y., Yamamoto T., Taniyama M., Fukada K., Yanagihara T. and Sakoda S. (2003)The efficacy of trientine or ascorbate alone compared to that of the combined treatment with these two agents in familial amyotrophic lateral sclerosis model mice. Exp. Neurol. 179, 176-180. (5) Weihl C.C. and Lopate G. (2006). Motor neuron disease associated with copper deficiency. Muscle nerve. 34, 789-793. 57 P5 Doubling expression of SOD1 in SH-SY5Y cells leads to altered SOD1 forms that are increased on G93A mutant expression Cerqueira FM (1), Medinas DB (1), Demasi M (2), Carrí MT (3), Augusto O (1). (1) Departmento de Bioquímica, Instituto de Química, Universidade de São Paulo, São Paulo, Brazil. (2) Departmento de Biologia, Instituto Butantan, São Paulo, Brazil. (3) University of Roma “Tor Vergata”, Rome, Italy. About 20% of familial ALS (fALS) cases are due to mutations in the Cu,Zn-superoxide dismutase (SOD1) gene. It was thought that the mutated enzymes would have impaired SOD activity, but this has not been corroborated so far. Presently, it is more accepted that the mutated enzymes acquire a new toxic function. What this new toxic function is and how it relates to the degeneration of motor neurons remains debatable. Here, we compared human neuroblastoma cells transfected with fALS mutant G93A (SH-SY5YG93A) or wild-type SOD1 (SH-SY5YWT) with parent cells (SH-SY5Y) (1) in regard to growth, viability, basal oxidant production, SOD activity and SOD forms. Transfected cells presented increased growth rate and higher basal oxidant production. SH-SY5YWT and SH-SY5YG93A cells in early culture stages showed the expected 2-fold increase in SOD expression and activity. After 4 weeks of culture, SH-SY5YG93A maintained SOD1 expression levels but dismutase activity decreased to parent cell levels. Cells transfected with SOD1 presented increased levels of altered SOD1 forms such as the reduced enzyme, disulfide multimers and anionic detergent-insoluble forms, particularly in SH-SY5YG93A cells. Among the insoluble forms, a thiol-resistant SOD dimer was detected by SDS-PAGE. A similar dimer has been previously detected during the bicarbonatedependent peroxidase activity of human SOD1 (2). Altered SOD forms are probably responsible for the increase in the chymotrypsin-like activity of the proteasome, verified in transfected cells. In conclusion, doubling SOD1 expression was sufficient to increase altered SOD1 forms and the G93A mutation enhanced their yields. To establish SOD1 dimer structure appears to be particularly relevant because dimeric SOD forms have been detected in ALS animal models and patients, and shown to shadow the onset and progression of motor neuron degeneration (3). Supported by: FAPESP and CNPq. (1) Carri, M. T.; Ferri, A.; Battistoni, A.; Famhy, L.; Gabbianelli, R.; Poccia, F.; Rotilio, G. (1997) Expression of a Cu,Zn-superoxide dismutase typical of familial sclerosis induces mitochondrial alteration and increased of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett., 414, 365-368. (2) Zhang H, Andrekopoulos C, Joseph J, Crow J, Kalyanaraman B. (2004) The carbonate radical anion-induced covalent aggregation of human copper, zinc superoxide dismutase, and alphasynuclein: intermediacy of tryptophan- and tyrosine-derived oxidation products. Free Radic Biol Med. 36, 1355-1365. (3) Shaw BF, Valentine JS. (2007) How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem. Sci. 32, 78-85. 58 P6 Effect of the of small heat shock protein HspB8 in a cellular model of familial Amyotrophic Lateral Sclerosis Crippa V (1), Simonini F (1), Bolzoni E (1), Onesto E (1), Rusmini P (1), Sau D (1), Carra S (2), Landry J (2), Poletti A (1) (1) Institute of Endocrinology, Centre of Excellence on Neurodegenerative Diseases of the University of Milan and InterUniversity Centre on Neurodegenerative Diseases (University of Florence, Rome and Milan) Via Balzaretti 9, 20133 Milan. tel 02-5031.8215; fax 02-5031.8204; E-mail: angelo.poletti@unimi.it (2) Centre de recherche en cancerologie de l'Universite Laval. L'Hotel-Dieu de Quebec, 9, rue McMahon, Quebec, QC, CANADA G1R 2J6. tel: (418) 691-5281/fax: (418) 691-5439 About 10% of cases of amyotrophic lateral sclerosis (ALS) are familial (fALS) and 20% of them are linked to mutations in Cu/Zn Superoxide Dismutase 1 (SOD1), a soluble enzyme present in cytosol, nucleus, peroxisomes, lysosomes, and mitochondria (1,2). Compelling data support the idea that fALS should be considered a protein misfolding disorder, in which a non native, toxic, oligomeric conformation is generated in the mutant protein. To study fALS we have produced a model based on immortalized motoneuron transfected with plasmids coding for wild type (wt) or mutant SOD1 (wtSOD1/G93A-SOD1). While, as expected, wtSOD1 was widely diffused in the cell, G93A-SOD1 was mainly present in the perinuclear region forming citoplasmic and nuclear aggregates. High molecular weight (MW) species of G93A-SOD1 oligomers and SDS-resistant forms were detectable only in G93A-SOD1 samples. Filter retardation assay revealed that wt protein was not retained by the cellulose acetate membranes, instead significant amounts of insoluble G93A-SOD1 were detectable, suggesting that G93A-SOD1 generates misfolded species capable to produce aggregates. Using the YFPu, a reporter protein targeted to degradation by the proteasome, we have shown a proteasome impairment when G93ASOD1 was expressed. We have studied the effects of overexpression of HspB8 protein, a small heat shock protein, that has already shown its ability on reducing aggregate formation of mutant polyglutamine proteins. Overexpression of HspB8 reduced the levels of monomeric form of either wt and G93A-SOD1 and increased the clearence of G93A-SOD1 oligomeric high MW species. Similar results were obtained in presence of the proteasome inhibitor MG132. In fact, robust decrease of G93A-SOD1 insolubile species was observed in presence of HspB8 both in basal condition and in presence of MG132. Moreover, HspB8 reduced YFPu levels, suggesting a desaturation of the proteasome system. By immunoprecipitation analysis we found no interaction between HspB8 and SOD1 proteins, indicating that, probably, HspB8 does not need a direct interaction to exert its chaperone function. The ability of HspB8 to reduce G93A-SOD1 levels even in presence of MG132 and to desaturate the proteasome suggests that this chaperone might be involved in an alternative degradative pathway. (1) Fridovich, I. Annu. Rev. Biochem. 1975, 44, 147. (2) Valentine, JS, and Hart PJ. PNAS 2003. 100, 3617 Grants Telethon - Italy (#GGP06063), MIUR-FIRB (#RBAU01NXFP); (2005057598_002), University of Milan-FIRST, FONDAZIONE CARIPLO. MIUR-Cofin 59 P7 Altered redox environment in a motor neuron-like cell model for SOD1-related forms of amyotrophic lateral sclerosis D’Alessandro G (1), Tartari S (1), Babetto E (2), Rizzardini M (1), Conforti L (2) and Cantoni L (1). (1) Laboratory of Molecular Pathology, Dept. of Molecular Biochemistry and Pharmacology, Istituto di Ricerche Farmacologiche “Mario Negri”, Milan, Italy.(2) Babraham Institute, Cambridge, United Kingdom. About 20% of familial ALS cases are associated to mutations in the Cu/Zn superoxide dismutase gene (SOD1). The toxic functions acquired by these mutant SOD1 forms are still unclear. The presence of oxidative damage was documented in ALS patients and in experimental models. Glutathione is the main low-molecular weight thiol in mammalian cells. The GSSG/2GSH couple provides a very large pool of reducing equivalents and is considered the most important cellular redox buffer. However, data on the levels of GSH or GSSG in tissues of ALS patients and in SOD1 transgenic mice are scanty. Even in the SOD1-mouse model it has not been studied whether a different disease stage might differently affect this parameter. Furthermore, when analyzing a whole tissue, these investigations are made difficult by the heterogeneous nature of cell populations in the sample. This study investigated how a prolonged exposure to the mutant G93ASOD1 affects the pool of reduced glutathione (GSH) and glutathione disulfide (GSSG) in a cell model. Motor neuron-like cell lines expressing a high level of wtSOD1 and a high or low level of G93ASOD1 were cultured for four, seven and ten weekly passages. GSH and GSSG were determined spectrophotometrically in the total cell lysates. In parallel, an untransfected control cell line was studied, that showed constant levels of GSH and GSSG in a ratio of 100:1. At the fourth passage, GSH and GSSG were significantly higher than in controls in all the transfected cells; however, the largest increase (about three-fold) in GSH was in the high-G93ASOD1 cells; while GSSG increased more (about 2.5-fold) in the wtSOD1 cells. At the tenth passage, the GSH increases were no longer evident, while GSSG remained higher than in control cells. These changes increased the GSH/GSSG ratios in the G93ASOD1 cells expressing a high level of the mutant protein at the early passages. They also shifted the half-cell reduction potential values (Ehc) (calculated by the Nernst equation) towards a more negative value only in the G93ASOD1 cells. In conclusion, G93ASOD1 altered not only the reducing capacity but also the redox environment differently from the wtSOD1; exposure time influenced this effect. This might modify key cellular processes such as signal transduction, DNA and RNA synthesis, protein synthesis, enzyme activation and even regulation of the cell cycle and be relevant to G93ASOD1 toxicity to motor neurons. Financial support was provided by MIUR, FIRB, Protocol RBIN04J58W_000 --- 60 P8 Induction of oxidative DNA damage in cells expressing mutant Superoxide Dismutase 1 (SOD1) Sau D(1), De Biasi S (4), Vitellaro L (4), Crippa V (1), Bolzoni E (1), Onesto E (1), Simonini F (1), Riso P (2), Bendotti C (3), Poletti A (1) (1) Inst. of Endocrinology, Centre of Excellence on Neurodegenerative Diseases, University of Milan, Via Balzaretti 9, 20133 Milan, Italy. Tel/Fax 02-5031.8215/04 - angelo.poletti@unimi.it Inter-University Research Centre on the molecular basis of neurodegenerative diseases (2) Department of Food Science and Microbiology (DiSTAM), Human Nutrition Unit, University of Milan; (3) Institute of Pharmacological Research Mario Negri, Via Eritrea 62- 20157 Milan, Italy. (4) Department of Biomolecular Sciences and Biotechnology, University of Milan,ViaCeloria 2620133 Milan, Italy. Amyotrophic Lateral Sclerosis (ALS) is characterized by a selective loss of both upper and lower motorneurons. About 10% are inherited cases (familial ALS or fALS) and, among them, about 20% are linked to Superoxide Dismutase 1 (SOD1) mutations. SOD1, one of the major cellular antioxidant enzyme is localized in the cytosol, nucleus, peroxisomes, lysosomes, and mitochondrial intermembrane space (1, 2); the active form is composed of a soluble homodimer of ~32 kDa., whose enzymatic activity is preserved in most mutant forms. Immunocytochemical analysis, performed on spinal cord sections from transgenic mice, expressing either wild type (wt), or mutant G93A-SOD1 (Gly to Ala in 93), showed that human wtSOD1 was detectable both in cytoplasm and, at lower levels, in the nuclei of motorneurons, while nuclear levels of mutant SOD1 were reduced. We have confirmed this peculiar distribution of the mutant SOD1 in immortalized motor neurons (NSC34), transfected with wt and mutant SOD1. In these cells, we also observed the formation of cytoplasmic and nuclear inclusions. Moreover, western blot and filter retardation assay showed that G93A-SOD1 aggregates formed insoluble species. We have analyzed the Ubiquitin-Proteasome-Pathway (UPP), using reporter plasmids expressing Yellow Fluorescent Protein (YFP), fused with a short degron which direct s YFP to proteasome degradation, and also carrying either a Nuclear Localization Signal (YFP-NLS) or a Nuclear Exporting Signal (YFP-NES); we found proteasome impairment only in the cytoplasm. The effect of mutant SOD1 exclusion from nuclei on genomic DNA integrity was analyzed using a COMET assay (Single Cell Gel Electrophoresis), on NSC34 expressing SOD1s, after induction of oxidative stress with H2O2. Cells expressing G93A-SOD1 showed a higher DNA damage compared to those expressing wtSOD1, possibly because of a loss of nuclear protection. Therefore, an increased nuclear concentration of free radical species may results from their lower clearance due to the sequestration of the SOD1 enzyme into insoluble inactive forms. The data obtained suggest that the nucleus may be a target of G93A-SOD1 neurotoxicity in fALS which might arise from an initial misfolding (gain-of-function), generating nuclear deprivation of the active enzyme (loss-offunction in the nuclei), a process that may be involved in ALS pathogenesis. 1) Fridovich, I. Annu. Rev. Biochem. 1975, 44, 147. 2) Valentine, JS, and Hart PJ PNAS 2003. 100, 3617 Grants Telethon Italy (#GGP06063), MIUR-FIRB (#RBAU01NXFP); (2005057598_002), University of Milan-FIRST, FONDAZIONE CARIPLO. MIUR-Cofin 61 P9 Glutathione and protein-glutathione mixed disulfides compartmentalization in intact motor neuron-like cells Tartari S (1), D’Alessandro G (1), Veglianese P (1), Rizzardini M (1), Ottersen OP (2) and Cantoni L (1). (1) Istituto di Ricerche Farmacologiche “Mario Negri”, Milan, Italy. (2) Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway Mutations in the Cu/Zn superoxide dismutase gene (SOD1) are associated with 20% of familial amyotrophic lateral sclerosis (fALS) cases, oxidative stress being one of the mechanisms possibly involved in the toxicity of the mutant protein(s). Glutathione is an ubiquitous tripeptide. Its reduced form (GSH) has important antioxidant properties and is in equilibrium with oxidized species, including protein-GSH mixed disulfides (Pr-SSG). This equilibrium is altered in the presence of oxidative/nitrosative stress. Usually, levels of intracellular GSH and PrSSG are measured using total cell lysates or subcellular fractions obtained by differential centrifugation, but very poor information and methodology are available to determine the compartmentalization of GSH and Pr-SSG in the intact cell. This could be relevant to understand the pathogenic mechanisms of mutant forms of SOD1, since it was suggested that toxicity could be associated with depletion of the mitochondrial pool of GSH in motor neurons, the cell population preferentially affected in ALS. The aim of this study was to assess the localization and the relative distribution of GSH and PrSSG in a motor neuron-like cell line (NSC-34-tTA40) established in our laboratory. Cells were fixed and stained with a polyclonal GSH-specific antibody coupled to an ALEXA-Fluor 594 secondary antibody and analyzed by laser confocal microscopy. GSH immunostaining was clearly evident in the cell body and in the neurites while the central area of the cell, likely corresponding to the nucleus, was almost completely devoid of staining. Treatment with buthionine sulfoximine (20 M for 24 hr), an inhibitor of GSH synthesis, drastically diminished the intensity of the labelling confirming the antibody selectivity. In another experiment, NSC-34-tTA40 cells were exposed to the thiol oxidant diamide (1mM for 5 minutes), fixed and treated with a specific washing procedure to remove unbound GSH. Marked immunoreactivity for Pr-SSG (virtually absent in the controls) became evident and it was localized mainly on the cellular surface and within plasma membrane blebs. To investigate the presence of GSH in mitochondria, cells were double stained with the anti-GSH and an anti-HSP60 (as a marker for mitochondria). Immunoreactivity for HSP60 showed only partial overlap with that for GSH, suggesting that mitochondria might be heterogeneous with respect to GSH content. In conclusion, this confocal microscopy analysis appears suitable to study the compartmentalization of GSH and PrSSG. Experiments are in progress to apply this technique to study the effect(s) of the mutant G93ASOD1 in a conditional cell model that utilizes the NSC-34-tTA40 cell line developed in our laboratory. Financial support was provided by MIUR, FIRB, Protocol RBIN04J58W_000 62 P10 Interaction between SOD1 and cystatin b in “in vitro” models of ALS Ulbrich L (1), Cozzolino M (2), Melli M (3), Carrì MT (2,4), Augusti-Tocco G (1) (1) Dept. of Cell and Developmental Biology, University of Rome “La Sapienza”, Italy (2) Fondazione Santa Lucia IRCCS, Rome, Italy (3) Department of Biology, Bologna University, Italy. (4) Dept. of Biology, University of Rome “Tor Vergata”, Italy Cystatin B (CSTB) is an inhibitor of the cysteine proteases of the cathepsin family (1). Furthermore, CSTB, invitro, is used as model to study the formation of amyloid fibrils (4-6). The cstb gene is frequently mutated in a rare neurodegenerative disease called Progressive Myoclonus Epilepsy (2, 3). The interaction of CSTB with a number of proteins involved in the cytoskeletal function, is also reported (7, 9). Recently, Wootz et al. (8) have shown increased levels of CSTB and C in the spinal cord of amyotrophic lateral sclerosis (ALS) transgenic mice. This may correlate with the influence of the redox environment on aggregate formation of cystatin B and its mutants (9) Western blot analysis of neuroblastoma SH-SY5Y cells transfected with wild type or mutant copper/zinc superoxide dismutase (SOD1) shows that overexpression of SOD1 correlates with incresed levels of CSTB, as compared to non transfected cells. Using anti-CSTB abs, co-immuneprecipitation of SOD1 and CSTB from WT SH-SY5Y protein extract was also observed. Similar results were obtained by the immuneprecipitation of CSTB from the NSC34 hybrid cell line described as a model for motor neurons, transfected with both Cystatin b and SOD1. Furthermore, in agreement with Cipollini et al. (9) overexpression of WT CSTB in WT and parental SH-SY5Y cells triggers aggregation of the protein in the cytoplasm. Altogether, these results suggest interaction between SOD1 and Cystatin b and their possible cooperation in the functional alteration of motoneurons in ALS. 1 Turk V. and Bode W. (1991) The cystatins: Proteins inhibitors of cysteine proteinases. FEBS Lett. 285, 213-219 2 Lalioti M., Mirotsou M, et al. (1997) Identification of mutation in cystatin b, the gene responsabile for the Unverricht-Lundeborg type of progressive myoclonus epilepsy (EPM1).Am.J.Hum.Genet. 60, 342-351 3 Pennacchio L.A., Lehesjoki A.E. et al (1996) Mutations in the gene encoding cystatin b in progressive myoclonus epilepsy (EPM1). Science 271, 1731-1733. 4 Staniforth RA, Giannini S, Higgins LD, Conroy MJ, Hounslow AM, Jerala R, Craven CJ, Waltho JP (2001) Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J 20: 4774-4781 5 Zerovnik E., Pompe-Novak M., et al. (2002) Human stefin B readily forms amyloid fibrils in vitro, Biochim. Biophys 1594,1-5. 6 Zerovnik E., Zavasnik-Bergant V., et al (2002) Amyloid fibril formation by human stefin B in vitro: immunogold labelling and comparison to stefin A- Biol. Chem 383, 859-863. 7 Di Giaimo R., Riccio M., et al. (2002) New insights into the molecular basis of progressive myoclonus epilepsy: a multiprotein complex with cystatin B. Human Molecular Genetics 11, 29412950. 8 Wootz H., Weber E., Korhonen L, and Lindholm D., (2006) Altered distribution and levels of cathepsin D and cystatins in amyotrophic lateral sclerosis transgenic mice: possible roles in motor neuron survival. Neuroscience 143, 419-130. 9 Cipollini E., Riccio M., et al Wild type cystatin B and its EPM1 mutants are polymeric and generate amyloid-like aggregates in vivo (submitted). 63 P11 Studies on zinc and metallothioneins in the G93A SOD1 rat mutant Averill SA, Yee J, Ismajli M., Malaspina A. and Michael-Titus AT. Neuroscience Centre, ICMS, Bart’s and the London School of Medicine, Queen Mary, University of London, 4 Newark Street, London E1 2AT, UK. Zinc is a divalent cation with essential structural and functional roles in the central nervous system (1). In the context of acute neurological injury, it has been suggested that excess zinc is associated with neurotoxicity. Metallothioneins are small proteins which bind zinc and have been shown to be associated with neuronal injury (2). An increase in the expression of these proteins has been reported in the mouse G93A SOD1 model of amyotrophic lateral sclerosis (ALS) (3) and in ALS patients. This study explored the distribution of zinc and of metallothioneins in the G93A SOD1 rat model of ALS. Adult transgenic G93A SOD1 rats and matched wild-type animals were used throughout. Zinc was detected using an autometallographic technique, following the injection of sodium selenite (10 mg/kg, i.p.) at various times before sacrifice, and metallothionein I/II (MT) expression was detected by immunocytochemistry. In wild-type rats zinc was present both at spinal and supraspinal level and also in the dorsal root ganglia (DRG). In the spinal cord, the staining appeared in the neuropil at 2.5 h after the injection of sodium selenite, and became more intense at 6 h after injection. At 24 h, zinc was also detected in neuronal cell bodies. In the DRG, the zinc-labelled cells were mainly small and medium sized IB4-positive cells. In ALS rats at late stage disease (20-25 weeks), there was a marked decrease in zinc in the spinal cord. In contrast, in the same animals there was a marked increase in MT immunoreactivity, in particular in the ventral horn and white matter tracts. However, motorneurones did not appear to express MT. Most of the MT immunoreactivity was present in astrocytes, confirmed by double labeling with GFAP. The observations on MT confirm previous observations on MT up-regulation in a murine ALS model and in ALS patients, and suggest that pathogenetic events associated with the disease (e.g. an increased oxidative stress) may trigger an increased MT expression, which may have a neuroprotective role. This is the first report of changes in zinc in the spinal cord of an ALS animal model at an advanced stage of the disease. It remains to be seen if these changes are linked with the pathogenesis of the disease. (1) Frederickson C.J., Suh S.W. Silva D., Frederickson C.J. and Thompson R.B. (2000) Importance of zinc in the central nervous system: the zinc-containing neuron. J. Nutr. 130, 1471S-1483S (2) Stankovic R.K., Chung R.S. and Penkova (2007) Metallothioneins I and II; neuroprotective significance during CNS pathology. Int. J. Biochem. Cell Biol. 39, 484-489. (3) Gong Y.H. and Elliott J.L. (2000) Metallothionein expression is altered in a transgenic murine model of familial amyotrophic lateral sclerosis. Exp. Neurol., 162, 27-36. 64 P12 Force plate for measuring G93A SOD1 mice forces by digital speckle pattern interferometry Calvo AC (1), Arroyo MP (3), Bea JA (2), Manzano R (1), Andres N (3), Grasa J (2), Zaragoza P (1), Doblare M (2) and Osta R (1). (1) LAGENBIO-INGEN.I3A, Facultad de Veterinaria, Universidad de Zaragoza, Zaragoza, Spain. (2) GEMM.I3A, Centro Politécnico Superior, Universidad de Zaragoza, Zaragoza, Spain. (3) Gnomo, Instituto de Investigación en Ingeniería de Aragón (I3A), Universidad de Zaragoza, Zaragoza, Spain. Digital Speckle Pattern Interferometry (DSPI) is a well established technique for measuring small displacements of scattering surfaces (1). This technique can be very useful when an objective test for measuring the muscle strength is necessary. In particular, we studied the state of development of the amyotrophic lateral sclerosis (ALS) in G93A transgenic mice, in terms of muscle strength as a parameter to measure the forces exerted by the animal during locomotion over a sensing plate. The experimental apparatus for obtaining surface displacements consists of two acrylic (PMMA) walls inserted in a box and attached to an aluminium plate which has a central hole where the sensing force plate is placed. The walls define a central corridor where the animal is allowed to walk freely. A mirror placed at 45º under the box and a camera above it are used to take photos of the animal while it walks on the sensing plate. A diode pumped solid state (DPSS) laser was used as a source of illumination. The scattered light from the plate was collected onto a charge-couple device (CCD) sensor where a focused plate image was formed. The calculation of applied forces magnitudes and position involves a first step that includes the calculation of the phase for each specklegram, using a Fourier transform method (2), and its difference with the reference specklegram phase. Then, the maps are filtered, unwrapped and transformed into displacement fields. At first stage, we carried out calibration experiments for estimating the sensing plate elastic parameters. We observed that 3 m is the maximum displacement produced by the 10 grams static weight and occurred when the weight is placed on the plate centre. We then challenged experiments from wild type and G93A SOD1 mice varying in age and weight. Manual triggering is used to record time sequences of 500 specklegrams every time the mouse walks on the sensing plate. One of the specklegrams, corresponding to no force on the plate, is taken as the reference specklegram. We have obtained force traces for singlelimb contacts in mice with only 20 grams weight body. The maximum force magnitude is around 50 % of the animal weight, which is within the range of values previously reported form mice (3). We report that DSPI can be used as a tool to objective measure the force plate displacement fields produced by G93A SOD1 mice, shedding light on the knowledge of ALS. (1) Rastogi P.K. (2001) Measurement of static surface displacements, derivatives of displacements and three-dimensional surface shapes-examples of applications to non-destructive testing. Digital speckle pattern interferometry and related techniques, John Wiley & Sons, Chichester. (2) Lobera J., Andrés N. and Arroyo M.P. (2004) Digital speckle pattern interferometry as a holographic velocimetry technique. Meas. Sci. Technol. 15, 718-724. (3) Zumwalt A.C., Hamrick M. and Schmitt D. (2006) Force plate for measuring the ground reaction forces in small animal locomotion. J. Biomechanics 3, 2877-2881. 65 P13 Characterization of SOD1G93A mouse muscle by a combined genomic and proteomic approach D. Capitanio (1), C. Fallini (2), M. Vasso (1,3), A. Ratti (2), G. Grignaschi (4), M. Volta (2), C. Daleno (4), S. Calza (5), C. Bendotti (4), C. Gelfi (1,3), V. Silani (2) (1) Department of Biomedical Sciences and Technologies, University of Milan, Segrate, Italy. (2) Dept. Neuroscience, University of Milan Medical School – IRCCS Istituto Auxologico Italiano, Milano, Italy. (3) Institute of Molecular Bioimaging and Physiology, CNR, Segrate, Milano. (4) Lab. Molecular Neurobiology, Dept. Neuroscience, Mario Negri Institute for Pharmacological Research, Milano. (5) Department of Biomedical Statistics, University of Brescia, Brescia, Italy. ALS is a fatal, progressive paralysis arising from the premature death of motoneurons. Alterations in the morphology and metabolism of skeletal muscle have also been identified both in animal models and in patients affected by the disease. Furthermore, muscular modulation of IGF-1 and GDNF have been proved to delay the onset and prolong survival of mutant SOD1 mice. On the contrary, recent evidence suggests that SOD1mut-mediated damage within muscle is not a significant contributor to non-cell-autonomous pathogenesis of ALS. Despite these contrasting results, muscle tissue may represent a valuable source for identification of disease biomarkers and for the understanding of the pathogenetic mechanisms of ALS. We have characterized the gene expression profile of triceps and gastrocnemius muscles from transgenic SOD1G93A and control mice at presymptomatic (7 weeks) and symptomatic (14 weeks) stages of the disease by the use of Affymetrix GeneChip technology. This analysis was combined with the proteomic profiling of gastrocnemius muscles using a two-dimensional difference in gel electrophoresis (2D-DIGE) approach coupled to mass spectrometry. To evaluate and to exclude from our data gene and protein expression changes associated to physiological denervation processes and muscular atrophy, we have also analysed gastrocnemius muscle after crush of the sciatic nerve. At presymptomatic stage of the disease, few changes in gene expression profile were observed in gastrocnemius and triceps muscles. On the contrary, at a symptomatic stage, Gene Ontology analysis of both muscles revealed their different involvement in the disease. In particular, triceps seemed to be still engaged in regenerative processes, while gastrocnemius was already atrophic with a predominant activation of the apoptotic pathway. Proteomic profiling of SOD1G93A versus control mice revealed the differential expression of 82 spots at presymptomatic stage and 153 spots at 14 weeks of age. Interestingly, 55 out of 82 spots (67%) were common to the two disease stages, and 16 of them were identified. Moreover, we identified 8 additional proteins specifically changed only at 7 weeks of age. These proteins were all associated to mitochondrial metabolism and architecture. Our data support the importance of skeletal muscle in the study of ALS since it well recapitulates the pathological processes that lead to the degeneration of motor neurons. The use of comparative genomic and proteomic technologies may represent a valid approach for the identification of clinically relevant biomarkers for neurological disorders. 66 P14 Effect of phrenic nerve degeneration on diaphragm contraction in a murine transgenic model of ALS Severini C. (1), Carunchio I. (2,3), Pieri M. (2,3), Curcio L. (2,3), Panico M.B. (2), Pisani V. (2) , Massa R. (2), Zona C. (2,3). (1) Institute of Neurobiology and Molecular Medicine, CNR, Rome, Italy, (2) Department of Neuroscience, University of Rome ‘‘Tor Vergata’’, Rome, Italy, and (3) IRCCS Fondazione S. Lucia, Rome, Italy. Amyotrophic lateral sclerosis (ALS) is a late-onset progressive neurodegenerative disease affecting motor neurons. The etiology of most ALS cases remains unknown, but 2% of instances are due to mutations in Cu/Zn superoxide dismutase enzyme (SOD1). Transgenic mice overexpressing human mutant SOD1 genes (G93A) have been obtained and are the most widely used animal model for the study of ALS. They develop the disease and die at 130-140 days. In this work we studied electrophysiological and histopathological features of phrenic nerve and diaphragm muscle obtained from control and G93A mice at the age of 90 and 130 days. We examined the contractile activity by physiological assessment of muscle force of isolated phrenic nerve-diaphragm in vitro preparations. By 90 days of age the muscular tension, measured through direct and indirect electric stimulation, was similar in control and G93A mice. By 130 days muscular contractile characteristics remained unvaried in control and G93A mice following a direct stimulation, whereas the indirect stimulation of the muscle through the phrenic nerve was significantly less efficient in G93A mice compared to control mice. Samples of the phrenic nerve and diaphragm muscle were analysed from mice at the same age for histopathological analysis. At 90 days, initial axonal degerneration was observed in the phrenic nerve. At 120 days widespread axonal degeneration characterized the phrenic nerve whereas there was a mild myofiber atrophy in diaphragm. The present results show that the muscle contractility is not significantly impaired also at the endstage of the disease, whereas at this stage an axonal degeneration rapidly involves the majority of motor nerve fibers, inducing diaphragm paralysis. 67 P15 Role of the ubiquitin proteasome pathway dysfunction in a mouse model of amyotrophic lateral sclerosis Cheroni C (1), Marino M. (1), Dantuma N (2), Bendotti C (1) (1) Department of Neuroscience, M. Negri Institute for Pharmacological Research, Milan, Italy (2) Department of Cell and Molecular Biology, Karolinske Institute, Stockholm, Sweden Accumulation of aggregated and ubiquitinated proteins are pathological hallmarks of both familial and sporadic Amyotrophic Lateral Sclerosis (ALS)and are found in the spinal cord of transgenic mice carrying human mutant SOD1, a model of the disease. The ubiquitin-proteasome pathway (UPP) is the main proteolytic system in eukaryotic cells; increasing evidence suggest a link between UPP impairment and the aggregate formation in neurodegenerative disease; however, it is still unclear whether an impairment of the UPP occurs in the motor neurons and/or in glial cells of ALS mouse models and if it is related with accumulation of protein aggregates. Real time PCR experiments on lumbar spinal cord of SOD1G93A mice revealed no changes in the mRNA levels of the catalytic constitutive 20S proteasome subunits prior to symptom onset while both 19S and 11S resulted significantly decreased. At the symptomatic stage, the mRNA levels of 20S subunit still remained unchanged between G93A and NTg littermates. On the contrary, LMP7, MECL1 were significantly increased in G93A lumbar spinal cord, while the nonresulted decreased. At the end stage of disease progression a decrease of the mRNA levels for all the constitutive subunits of proteasome was found in the lumbar spinal cord of SOD1G93A mice The phenomena resulted restricted in the pathological area, since no changes were detected in the hippocampus (Beta5, LMP7, alpha and 19S tested). In order to evaluate the UPP function at the cellular levels, SOD1G93A mice were cross-bred with the UbG76V-GFP transgenic model, that expresses a reporter protein for UPP functionality. Immunohistochemical analyses of symptomatic double transgenic mice revealed the presence of rare GFP-positive cells in the spinal cord; at the end stage the accumulation of the reporter protein was more extended in the spinal cord and involved also the motor nuclei of the brainstem. Co-localization experiments in the spinal cord of end stage double transgenic mice revealed that Ub-GFP accumulation was rarely observed in the glial population. In fact, the majority of GFP-positive cells showed a neuronal morphology. At the symptomatic stage, about one third of GFP positive neurons showed perikaryal staining of phosphorylated neurofilaments, while two third of all GFP positive cells were also ubiquitin positive.These data suggest that early changes of transcript levels for 19S and 11S in ventral spinal cord may be the first sign of UPP change in G93A mice. Progressive decrease of constitutive proteasome subunits observed at the later stages may contribute to accumulation of protein aggregates found in neurons and neurites. Supported by Telethon 68 P16 Spinal cord degeneration in amyotrophic lateral sclerosis: the role of retinoid signaling Natasa Jokic, Yong Yong Ling, Rachael E. Ward, Adina T. Michael-Titus, John V. Priestley, Andrea Malaspina Neuroscience Centre, Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, United Kingdom Retinoid signaling (ReS), a complex of molecular pathways that mediate the effects of vitamin A and its derivatives, is increasingly recognized as a component of the repair capacity that could be activated to induce protection and regeneration in the mature nervous tissue. Changes in distribution and expression of retinoid receptors have been described as part of a spinal cord protective response to acute injury and to chronic degeneration. We have recently reviewed the expression of ReS gene candidates and functionally synergic nuclear receptors in human spinal cord from healthy adults and from patients with amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disorder causing extensive motor neurons loss and death from respiratory failure in 3 to 5 years from disease onset (1). This tentative ReS expression profile included data generated from in-house gene expression analysis and information retrieved from independent datasets, obtained through serial analysis of gene expression and array investigations. In the present study (2), we have combined RNA and protein expression analysis to characterize the expression profile of retinoid receptor isotypes in the lumbar spinal cord of the superoxide dismutase 1 G93A transgenic rat model of ALS throughout the disease development and in wild type littermates. We show that key pathogenic features associated to disease progression, including gliosis and motor neuron loss appear to develop in parallel with a specific pattern of retinoid receptors expression change, involving RXRbeta up-regulation in astrocytes and a selective gamma motor neuron RARalpha staining at end-stage disease. Our work provides experimental evidence supporting the role of retinoid signaling in neurodegeneration of the spinal cord and suggests that future studies could explore new treatment strategies based on retinoid-modulating agents. (1) Malaspina A, Turkheimer F. A review of the functional role and of the expression profile of retinoid signaling and of nuclear receptors in human spinal cord. Brain Res Bull. 2007; 71: 437-46. (2) Jokic N., Ling Y., Ward R., Michael-Titus A.T., Priestley J.V., Malaspina A. Retinoid receptors in chronic degeneration of the spinal cord: observations in a rat model of amyotrophic lateral sclerosis. Submitted to J. Neurochem. 69 P17 ATF-3 Expression in the SOD1 Rat Model of Amyotrophic Lateral Sclerosis Ngoh SFA, Ward RE, Hall JCE, Jones C, Jokic N, Averill SA, Michael-Titus AT, Malaspina A, Priestley JV. Neuroscience Centre, Bart’s and the London School of Medicine and Dentistry, Queen Mary University of London, Institute of Cell & Molecular Sciences, 4 Newark Street, London E1 2AT Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that leads to progressive paralysis. The molecular pathogenesis of this ultimately fatal disease remains poorly understood. It has previously been reported that expression of the stress-activated transcription factor ATF-3 precedes the death of motoneurones in mice that express G93A mutant superoxide dismutase SOD1 (1). We carried out this study to characterize the expression of ATF-3 in the SOD1 rat model of ALS. Transgenic Sprague Dawley rats expressing the G93A SOD1 gene mutation, along with age and sexmatched wild-type littermates, were subject to behavioural analysis to monitor disease progression. The rats were anaesthetized and perfusion fixed for histological analysis at either a presymptomatic (10 week) or end-stage (20 - 25 weeks) of the disease. Tissue obtained from the spinal cord and dorsal root ganglia (DRG) of these animals was processed for ATF-3 immunoreactivity and double labeled with a range of antisera used to identify motoneurones, glia, and sub-populations of DRG neurones. Motoneurones in the spinal cord of the end-stage SOD1 transgenic rats exhibited ATF-3 immunoreactivity, in contrast with motoneurones in wild-type controls, which displayed no ATF-3. Most of the ATF-3 positive cells were also immunoreactive for choline acetyl transferase (ChAT), and some of the ATF-3 positive cells had low ChAT expression. Consistent with degeneration of motoneurones, strong ATF-3 immunoreactivity was also observed in Schwann cells in the ventral roots and spinal nerves. However, ATF-3 immunoreactivity was also present in a few Schwann cells in the dorsal roots and in a sub-population of DRG neurones. ATF-3 immunoreactive DRG neurones were predominantly large diameter, and immunoreactive for heavy-chain neurofilament. ATF-3 immunoreactivity was rarely found in small diameter calcitonin gene related peptide (CGRP)-positive neurones, or in the DRG of wild-type animals. Our results in SOD1 mutant rats mirror earlier results obtained in SOD1 mice (1) and in studies of peripheral nerve injury (2). ATF-3 is a marker of cellular injury and its upregulation in motoneurones and their associated Schwann cells may play a role in the pathogenesis of ALS. An upregulation of ATF3 in sensory systems in ALS has not been reported previously, and ongoing studies are investigating whether this is a secondary response to motoneuron loss or a parallel primary event. We gratefully acknowledge support from St. Bartholomew and the London Charitable Foundation. (1) Vlug, A.S., Teuling, E., Haasdijk, E.D., French, P., Hoogenraad, C.C., and Jaarsma, D. (2005) ATF3 expression precedes death of spinal motoneurons in amyotrophic lateral sclerosis- SOD1 transgenic mice and correlates with c-Jun phosphorylation, CHOP expression, somato- dendritic ubiquitination and Golgi fragmentation. European Journal of Neuroscience. 22: p1881- 1894 (2) Hunt, D., Hossain-Ibrahim, K., Mason, M.R., Coffin, R.S., Lieberman, A.R., Winterbottom, J., Anderson, P.N. (2004) ATF3 upregulation in glia during Wallerian degeneration: differential expression in peripheral nerves and CNS white matter. BMC Neuroscience 5: 9 70 P18 Inclusions containing superoxide dismutase-1 are regularly present in amyotrophic lateral sclerosis patients lacking mutations in the enzyme Nilsson K (1), Jonsson, P.A.(2), Graffmo K(1), Andersen P.M.(3), Marklund S (2) and Brännström T (1) (1) Dep. of Medical Bioscience, Umeå University, Sweden. (2) Dep. of Clinical Chemistry, Umeå University, Sweden. (3) Dep. of Pharmacology and Clinical Neuroscience, Umeå University, Sweden Six percent of amyotrophic lateral sclerosis (ALS) is linked to mutations in the ubiquitously expressed enzyme superoxide dismutase-1 (SOD1), making it the most common cause of the disease. The cause of the remaining cases is largely unknown. Histopathologycally, three types of inclusions are seen in neurons of the brain and spinal cord – Bunina bodies, Lewybody-like-hyaline inclusions and skein inclusions. In addition, in familial ALS (FALS) caused by mutated SOD1 another type of inclusions are observed, which contain aggregated units of the misfolded enzyme. Misfolded and aggregation-prone forms of mutant SOD1s are thought to trigger the disease. The aim of this study was to investigate if SOD1 is involved in ALS patients lacking mutations in the enzyme and to find the subcellular localization of the SOD1 inclusions. CNS tissue was collected from 30 patients with sporadic ALS (SALS), 7 patients with FALS, and from 46 control patients with and without other neurodegenerative diseases. Paraffin embedded spinal cord sections were immunostained using immunohistochemistry and immunofluorescence for detection and co-localization of SOD1 with lysosomes, endoplasmatic reticulum (ER) and mitochondria. An antibody recognizing the human TAR DNA-binding protein (TDP-43) was used to study a possible co-lozalisation with SOD1 inclusions. Confocal microscopy was used for analysis of the samples. Numerous small somal round inclusions in motoneurons, immunoreactive for SOD1, were a feature of both SALS and FALS. They were relatively homogenous in size and measured 0.5-3µm. All investigated SALS and FALS patients had SOD1 containing inclusions. In contrast none or very few controls had inclusions. Double immunofluorescence staining showed a partial co-localization with the lysosomal marker. No co-localization was found between SOD1 inclusions and ER, mitochondria or TDP-43. Our results suggest that inclusions of misfolded, aggregated SOD1 are a general histopathological feature, regularly present in the motoneurons of ALS patients lacking mutations in the enzyme.The inclusions are situated in the cytoplasm of the neurons and do not co-localize with TDP-43, mitochondria or ER and only partly with lysosomes. Our results suggest that SOD1 is involved in the pathogenesis of all forms of ALS. 71 P19 Direct Detection and Quantitation of Metals Bound to Cu,Zn Superoxide Dismutase from discrete regions of the central nervous system of ALS animals models Keith E. Nylin (1,2), Brian Arbogast (3), Nathan Lopez (1), Mark Levy (2), Jeff Morre (3), Joseph S. Beckman (1,2,3) (1) Department of Biochemistry & Biophysics, Oregon State University, Oregon, USA, (2) Linus Pauling Institute, Oregon State University, Oregon, USA, (3) Environmental Health Science Center, Oregon State University, Oregon, USA We have hypothesized the loss of zinc from Cu,Zn superoxide dismutase (SOD1) plays a direct role in the disease mechanism for amyotrophic lateral sclerosis (ALS). Several lines of evidence support this hypothesis, however, because the affected tissue regions of ALS are so minute, it has previously been impossible to quantify metals bound to SOD from tissue, and therefore Zn-deficient SOD has never been measured directly from ALS tissue. Consequently, we have developed a sensitive method to measure the metal content of SOD directly from discrete regions of the spinal cord using electrospray mass spectrometry. We collect small necropsy punches (250 g) from flash frozen central nervous tissue then thaw in 5 L of buffer. The supernatant from the thawed tissue is collected after centrifugation and loaded onto a C4 ZipTip from Millipore and eluted into the mass spectrometer with 40% acetonitrile/water. In addition to SOD, we have identified several proteins which elute from the ZipTip including ubiquitin, CCS, and -synuclein. Furthermore, by analyzing blood with this new assay, we are able to genotype an animal in less than two minutes. By extensive testing of controls we show the metal status of SOD is preserved throughout the assay and the results obtained from the mass spectrometer accurately reflect the metal status in vivo. By using Cu,Zn bovine SOD as an internal standard, we find nearly 15 M of SOD in the ventral grey matter of a rat model of ALS is missing zinc, equal to the amount of Cu,Zn SOD, while over 20 M is apo. Strikingly, we find the dorsal grey matter is almost void of Zn-deficient SOD, but still maintains 20 M apo SOD. Furthermore, we do not see Zn-deficient SOD in 30 days old transgenic rats, long before pathology of the disease begins. By using this new sensitive assay we developed we have found a spatial and temporal correlation between the presence of Zn-deficient SOD and affected tissue in ALS, further supporting the hypothesis that Zn-deficient SOD plays a direct role in the ALS disease mechanism. 72 P20 Flavoproteins and ALS: expression of Fmo gene family in SOD1 mutated mice Ogliari P. (1), Corato M (1), Cova E. (1), Cereda C. (1), Gagliardi S. (1), Daleno C. (2), Bendotti C. (2) and Ceroni M (1,3,4). (1) Lab. of Experimental Neurobiology, Neurological Institute IRCCS C. Mondino, Pavia, Italy, (2) Dept. of Neuroscience, “Mario Negri” Institute for Pharmacological Research, Milano, Italy (3) Dept. of Neurosciences, Policlinico di Monza, Monza, Italy (4) Dept. of Neurology, University of Pavia, Pavia, Italy. Amyotrophic Lateral Sclerosis (ALS) is an adult-onset, progressive and fatal neurodegenerative disease, whose pathogenesis is unknown. Motor neuronal degeneration represents the main pathological feature in ALS, for which two major pathogenetic hypotheses have been proposed: oxidative stress and excitotoxicity. Flavin-containing monooxigenases (FMO) represent a gene family coding for microsomal enzymes which are involved in the oxidative metabolism of a variety of xenobiotics. This family contains five genes, FMO1, FMO2, FMO3, FMO4, FMO5, and at least six pseudogenes in humans. Function of FMO genes is largely studied in liver, kidney and lung but not in nervous system. Recently, the observation of a 80% reduction in FMO1 mRNA expression in spinal cord of sporadic ALS patients pointed to a relation between ALS and FMO genes. In addition, in a previous study, we found an association between two 3’-UTR single nucleotide polymorphisms of FMO1 gene and the development of sporadic ALS. To investigate the mRNA levels of FMO1, FMO2, FMO3, FMO4, FMO5 in murine nervous system in normal mice and in a transgenic murine model of ALS.The C57BL/6J mouse strain dealing the human-SOD1 G93A mutation has been used as disease model; C57BL/6J normal mice have been used as controls. Four brain areas have been examined in all animals: cerebellum, cerebral hemispheres, brainstem and spinal cord. A Real-Time PCR technique with TaqMan probes has been used for expression analysis. Normalization was optimized using Hprt as housekeeping gene.Our study demonstrates that all FMO genes, except for Fmo3, are expressed in the murine nervous system. The FMO expression is sex-dependent and varies over different brain areas. Moreover, alterations in FMO expression have been found in G93A mutated mice. Particularly, in G93A males, we observed in different cerebral areas a lower expression of Fmo2 and Fmo4 gene compared to WT mice. Conversely, G93A female mice expressed significantly greater amounts of Fmo2 and Fmo5 genes in cerebellum and cerebral hemispheres.Our study represents the first attempt to analyse Fmo gene expression in different areas of the nervous system and particularly in spinal cord. We confirmed the sex-dependent expression of Fmo genes in nervous system, as already found in other tissues. The finding of altered Fmo expression in a model of motor neuron degeneration might suggest a role for Fmo genes in the development of ALS, even if the pathogenic link is presently unknown. 73 P21 Valosin-Containing Protein Mutation Accelerates Paralysis and Death in SOD1G93A Transgenic Mice Yan J , Fu R , Deng HX , Caliendo J , Zhai H , Chen W , Liu E , Siddique T, Department Neurology, Northwestern Medical University, Chicago, IL USA SOD1G93A transgenic mouse is an established animal model of human familial ALS. A subgroup of ALS is associated with frontotemporal dementia (FTD). Mutations in the VCP gene cause autosomal dominant FTD with inclusion body myopathy and Paget disease of bone (IBMPFD). VCP performs a variety of essential cellular functions. Mutant SOD1 can be degradated via Dorfin, a ubiquitin ligase (E3). VCP directly interacts with Dorfin and both colocalize in Lewy body-like inclusions in the spinal cord motor neurons of ALS(1). We found a VCP R155P mutation in a IBMPFD family. To explore the effect of mutant VCP on the onset and progression of SOD1 G93A transgenic mice, we established VCPR155P mouse using a 17.9kb human genomic DNA fragment containing the entire VCP gene with site-directed mutation. VCPR155P mouse were crossbred with SOD1G93A to generate SOD1G93A /VCPR155P double transgenic mice. Transgenes were identified by PCR, DNA sequencing and Southern blot analysis. Mice were observed weekly for changing weight and tested on the Rotarod system. We generated four SOD1G93A /VCPR155P double transgenic mice, two littermates with VCPR155P, two littermate with SOD1G93A and two nontransgenic littermates. SOD1G93A /VCPR155P double transgenic mice stopped gaining weight after 61 days and the onset of hind legs paralysis was at 77.7±3.9 days and end-stage was at 84.3±4.6 days. The SOD1G93A and VCPR155P littermates were not affected by 112 days. The average onset of paralysis in SOD1G93A mice (n=273) was 120.4±8.2 days and end-stage time of SOD1G93A mice (n=397) was 128.5±10.7 days. The SOD1G93A /VCPR155P vastus lateralis muscle showed remarkable muscle fiber atrophy and fiber size variation, but without obvious rimmed vacuoles. Muscle morphology of SOD1G93A / VCPR155P littermates were normal at the time SOD1G93A /VCPR155P reached end stage. It appears that VCP mutation significantly accelerates the onset and progression of disease in SOD1G93A mice. VCP immunohistochemic analysis and characterization of the muscles, brain and spinal cord is currently underway. (1) Ishigaki S, Hishikawa N, Niwa J, et al. Physical and functional interaction between Dorfin and Valosin-containing protein that are colocalized in ubiquitylated inclusions in neurodegenerative disorders. J Biol Chem. 2004; 279(49):51376-85. 74 P22 p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis Jozsef Gal, Anna-Lena Ström, Renee Kilty, Fujian Zhang and Haining Zhu Department of Molecular and Cellular Biochemistry, University of Kentucky, 741 South Limestone Street, Lexington, Kentucky 40536 Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by motor neuron death. A hallmark of the disease is the appearance of protein aggregates in the affected motor neurons. We have found that p62, a protein implicated in protein aggregate formation, accumulated progressively in the G93A mouse spinal cord. The accumulation of p62 was in parallel to the increase of polyubiquitinated proteins and mutant SOD1 aggregates. Immunostaining studies showed that p62, ubiquitin and mutant SOD1 co-localized in the protein aggregates in affected cells in G93A mouse spinal cord. The p62 protein selectively interacted with familial ALS mutants, but not WT SOD1. When p62 was co-expressed with SOD1 in NSC34 cells, it greatly enhanced the formation of aggregates of the ALS-linked SOD1 mutants, but not wild-type SOD1. Cell viability was measured in the presence and absence of overexpressed p62 and the results suggest that the large aggregates facilitated by p62 were not directly toxic to cells under the conditions in this study. Deletion of the ubiquitin-association (UBA) domain of p62 significantly decreased the p62-facilitated aggregate formation, but did not completely inhibit it. Further protein interaction experiments also showed that the truncated p62 with the UBA domain deletion remained capable of interacting with mutant SOD1. The findings of this study show that p62 plays a critical role in forming protein aggregates in familial ALS, likely by linking misfolded mutant SOD1 molecules and other cellular proteins together. 75 P23 Identification of a novel splice-site mutation in the Cu/Zn superoxide dismutase (SOD1) gene associated with amyotrophic lateral sclerosis Corrado L (1), Negri G (1), Testa L (2), Oggioni GD (2), Momigliano-Richiardi P (1), Mazzini L (2), D’Alfonso S (1) (1) Human Genetics Laboratory, Dept. of Medical Sciences, Eastern Piedmont University , (2) Dept of Neurology, Azienda Ospedaliera Maggiore della Carità, Novara, Italy We report a novel mutation of the Cu/Zn superoxide dismutase (SOD1) gene localized in intron 4 and found in an ALS patient with a family history for amyotrophic lateral sclerosis (FALS). The patient is a 56 years old woman, who presented at our outpatients’ department with a two-year history of speech disturbances and progressive weakness in her left arm. At the time of our visit (June 2006) neurological and neurophysiological examinations confirmed upper and lower motoneuron involvement both at bulbar and spinal level. The patient was finally diagnosed as ALS with bulbar onset. Her maternal grandmother died of motoneuron disease whereas her mother is 84 years old and does not manifest any sign of motoneuron disease. The mutation is a T nucleotide insertion at +3 downstream the exon4-intron4 junction Although the invariant GT donor site was not apparently affected, bioinformatics analysis (http//: l25.itba.mi.cnr.it/~webgene/wwwspliceview.html) predicted that a T at position +3 completely abolished the exon 4 donor site suggesting a possible alternatively spliced mRNA. To determine whether this mutation could affect splicing mechanisms, reverse trascription PCR (RT PCR) was performed on polyA-RNA isolated from peripheral blood lymphocytes of the patient. The analysis identified two different aberrant transcripts together with the normal isoform. One of these presented the retention of intron 4 sequence. We predict that the addition of intronic extrabases would result in an introduction of four aa (V-K-F-S- end) before a premature stop codon at aa 122. A second aberrant transcript was generated using a different donor site localized at the 3’ end of exon 4 (three nucleotides upstream the natural donor site) leading to an in frame 3bp deletion. We predict for this transcript the production of a SOD1 protein lacking a single amino acid (val 118) in a conserved region close to the active site loop (aa 121-144). We have no data about the quantitative ratio among the three mRNA isoforms and about the pathogenic effect of the two aberrant transcripts. We can hypothesize a main pathogenic role for the transcript with the intron 4 retention since at least two similarly truncated proteins at residues 124 and 121 were reported in two patients with a familial and a sporadic disease respectively. In conclusion we report a likely pathogenic new SOD1 intronic mutation with reduced penetrance. 76 P24 Novel SOD1 Q23R mutation associated with muscle mitochondrial dysfunction in familial ALS Stefania Corti, Andreina Bordoni, Dario Ronchi, Domenico Santoro, Sabrina Salani, Chiara Donadoni, Costanza Lamperti, Valeria Lucchini, Veronica Crugnola, Maurizio Moggio, Nereo Bresolin, Giacomo P. Comi Dino Ferrari Center, Department of Neurological Sciences. Fondazione I.R.C.C.S. Ospedale Maggiore, Policlinico, Mangiagalli and Regina Elena, University of Milan, Milan Italy Superoxide dismutase 1 (SOD1) gene mutations are responsible for approximately 20% of all familial amyotrophic lateral sclerosis (ALS) cases. Substantial evidence suggests that mitochondrial dysfunction is involved in ALS pathogenesis. In fact, two mitochondrial DNA mutations have been described in motor neuron disease patients, and recent studies suggest that the toxicity of mutant SOD1 arises from its selective recruitment to motoneuron (MN) mitochondria. Here we describe the clinical feature of a 40-year-old man from Bangladesh that presents progressive proximal lower limb muscle weakness by the age of 37, involving after one year also the arms. At age 40 years, neurological examination shows proximal and distal limb muscle weakness with distal atrophy, characterized by lower rather than upper MN impairment. Diffuse fasciculations were present, also in the tongue. The patient s mother died at 35 age for pneumonia, while a maternal uncle presented a referred muscle impairment at the lower limbs. EMG showed acute and chronic denervation findings, while brain and spinal MRI was normal. Direct sequencing revealed a heterozygous mutation CAG to CGG in codon 23 substituting glutamine to arginine in the SOD1 gene (Q23R). A muscle biopsy showed neurogenic pattern associated with cytochrome c oxidase (COX) deficieny in several muscle fibers. No apoptotic nuclei were found with TUNEL reaction. The novel SOD1 Q23R mutation affects a highly conserved aminoacidic position and results in a early-onset ALS phenotype. The mutation is likely to confer to the mutant SOD1 protein a mitochondrial toxicity also in muscle tissue, as demonstrated by partial cytochrome c oxidase dysfunction. 77 P25 Gender difference in levels of Cu, Zn-Superoxide Dismutase (SOD1) in cerebrospinal fluid of patients with amyotrophic lateral sclerosis Katrin Frutiger (1), Thomas J. Lukas (2), George Gorrie (3), Senda Ajroud-Driss (1), Teepu Siddique (1) (1) Davee Department of Neurology and Clinical Neurosciences, (2) Department of Molecular Pharmacology and Biological Chemistry, Northwestern University Feinberg School of Medicine, Tarry building, Room 13-715, 303 East Chicago Avenue, Chicago, IL 60611 USA, (3) Southern General Hospital, Department of Neurology, Glasgow, UK BACKGROUND: Currently the best studied mechanism for amyotrophic lateral sclerosis (ALS) is the one caused by mutations in the gene for cytosolic Cu/Zn-binding superoxide dismutase (SOD1). Mutant SOD1-protein causes motor neuron degeneration due to the gain of a novel toxic function. OBJECTIVES AND METHODS: To evaluate the relevance of SOD1 levels in cerebrospinal fluid (CSF) in ALS patients, the SOD1 concentration was immunoassayed in the CSF of 11 patients with ALS and 19 neurological controls. RESULTS: The mean level of SOD1 in CSF from all samples was 45.5 +/-11.3 ng/ml. There was no statistically significant difference between the levels of SOD1 in CSF of ALS patients and neurological control subjects. Here we show that the SOD1 concentration in the CSF is significantly higher in male ALS patients (54.0 +/-9.0 ng/ml) compared to female ALS patients (38.1 +/-6.4 ng/ml) (p=0.007). This gender difference is not observed in the CSF of neurological controls. CONCLUSION: The observation of a gender difference in levels of SOD1 in CSF of ALS patients needs further investigation to determine whether it is relevant to the observed gender related difference in disease incidence. 78 P26 SOD1 Gene Mutations in Italian Amyotrophic Lateral Sclerosis Patients Gellera C (1), Ticozzi N (2), Passariello P (1), Ratti A (2), Plumari M (1), Colombrita C (2), Castellotti B (1), Lauria G (1), Fallini C (2), Testa D (1), Corbo M (2), Taroni F (1), Silani V (2). (1) Fondazione IRCCS – Istituto Neurologico C. Besta – Milano – Italy. (2) Dipartimento Neuroscienze, Università degli Studi di Milano - IRCCS Istituto Auxologico Italiano –– Milano – Italy. Mutations in the SOD1 gene are present in 10-20% of ALS familial (FALS) and in 3-6% sporadic (SALS) cases with more than one hundred different mutations described so far. Disease progression and duration often correlate with specific SOD1 mutations, although a great variability has been observed among and within families carrying the same mutation. A great variability in SOD1 gene mutations has also been observed in populations with different ethnic backgrounds. Our work has focused on the identification of mutations in SOD1 gene in a large cohort of Italian ALS patients from two Neurological referral Centers. We have screened 762 Italian patients including 149 FALS and 613 SALS cases. We have found mutations in 18/149 (12%) unrelated FALS and in 7/613 (1%) SALS patients. 10/18 (55%) FALS carried mutations frequently described in different ethnic backgrounds (A4V, L84F, G93D and L144F). Interestingly, we identified 4 new SOD1 mutations. In an attempt to speculate about genotype-phenotype correlations, we suggest that, excluding A4V cases that associate with a severe and rapid course of the disease, FALS SOD1 cases show a slower and less aggressive phenotype than idiopathic cases. It is possible that susceptibility genes may be involved not only in ALS variants with unknown aetiology, but could also explain the incomplete penetrance and variable expression observed in pedigrees with the same SOD1 gene mutation. Other candidate genes have also been screened in a subset of our SOD1-negative patients: alsin gene in 75 patients with a juvenile onset and SETX gene in 44 patients with a dominant inheritance. No mutations were identified in exons where mutations were previously described to occur. We have analyzed Angiogenin gene as a risk factor for ALS pathogenesis in our cohort of patients. We identified 7 different mutations in 13 patients and one mutation (I46V) in a control sample (1.9% vs 0.2%, p 0,014). Six mutations were novel and were found both in familial (4.5%) and sporadic (1.3%) patients (p 0.035). A subset of sporadic patients (n=95) were also screened for deletions in SMN2 gene, another candidate ALS susceptibility gene. We did not find a significant association of SMN2 deletions in ALS patients respect to controls. Our results indicate that SOD1 gene mutations still remain the main determinant in the pathogenesis of hereditary ALS and ANG gene represents a strong susceptibility factor for both SALS and FALS cases in the Italian population. 79 P27 S-adenosylmethionine-dependent methylation in amyotrophic lateral sclerosis: preliminary results and pathogenetic hypotheses Monsurrò MR (1), Trojsi F (1), Salvatore A (2), Piccirillo G (1), Capone F (1), Maiorano P (1), De Bonis ML (2), D’Angelo S (2), Salemme S (2), Galletti P (2), Tedeschi G (1) (1) Department of Neurological Sciences and (2) Department of Biochemistry and Biophysics “Francesco Cedrangolo”, Second University of Naples, Naples, Italy Amyotrophic lateral sclerosis (ALS) is a progressive degenerative disorder of upper and lower motor neurons. It is sporadic in 90% of cases, while familiar ALS (fALS) is mainly caused by missense mutation of superoxide dismutase 1 (SOD1) gene, responsible of SOD activity decrease and reactive oxygen species increase (1). The aim of this study was to evaluate levels of methylation in erythrocyte membrane proteins of ALS patients, as a possible marker of oxidative stress. Blood samples of 35 ALS patients (20 men, 15 women, 54±10 years) and 32 healthy age- and sexmatched controls were processed to assess methylation of intact erythrocytes membrane proteins by electrophoretic analysis (as index of abnormal levels of L-isoaspartyl residues) and intracellular concentrations of methyl donor S-adenosylmethionine (AdoMet) and AdoMet demethylated product S-adenosylhomocysteine (AdoHcy) by high performance liquid chromatography. Abnormal L-isoaspartyl residues were significantly higher in ALS patients erythrocyte membrane proteins with an increased methyl accepting capability (P<0.05). AdoMet concentration in ALS patients erythrocytes was about 50% lower, while AdoHcy levels were equivalent, measuring a low AdoMet/AdoHcy ratio in ALS patients. Several evidences underlined the role of reactive oxygen species and nitric oxide in neurodegenerative mechanism (1). Furthermore, previous studies demonstrated that increased formation of L-isoaspartyl residues is one of the major structural alterations occurring in erythrocyte membrane proteins as a result of oxidative stress (2). These abnormal residues are physiologically converted into normal L-aspartyl residues by protein-L-isoaspartyl methyltransferase (PIMT), which methylates the alpha-carboxyl group of atypical L-isoaspartyl residues. PIMT is ubiquitous and its repair function is particularly crucial in erythrocytes, as a consequence of molecular ageing and oxidative damage (2). Our preliminary results suggest the presence of an injured action of PIMT in ALS patients erythrocytes, with accumulation of L-isoaspartyl residues, notably responsible of abnormal protein conformation. Thus, we hypothesize a concomitant injured mechanism of DNA methylation, dependent on AdoMet/AdoHcy ratio, which probably causes a deregulated gene expression. DNA hypomethylation in ALS could damage epigenetic control of long term silencing genes expression, such as imprinted or X-linked genes, with possible involvement in ALS pathogenesis, as described in some human genetic disorders. Further studies might be planned to evaluate methylation and gene expression in motor neurons of ALS animal models. (1) Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344:1688-1700 (2) Ingrosso D, D'Angelo S, di Carlo E, Perna AF, Zappia V, Galletti P (2000) Increased methyl esterification of altered aspartyl residues in erythrocyte membrane proteins in response to oxidative stress. Eur J Biochem 267:4397-4405 80 P28 SOD1 mutations in ALS patients: an epidemiological study in Piemonte, Italy Restagno G (1), Lombardo F (1), Sbaiz L (1), Fimognari M (1), Calvo A (2), Ghiglione P (2), Cammarosano S (2), Cavallo E (2), Mutani R (2), Chiò A (2) (1) (2) Laboratorio di Genetica Molecolare, ASO OIRM-Sant’Anna, Torino, Italy Dipartimento di Neuroscienze, Università di Torino, Italy According to series from ALS referral centres, superoxide dismutase 1 (SOD1) mutations may account for 20% of FALS and 2-4% of apparently sporadic cases (1). However, no studies have evaluated the frequency of SOD1 mutations in population-based series of cases. We assessed SOD1 mutations in familial and sporadic ALS in an epidemiological setting. From 1995 all ALS cases resident in Piemonte are actively enrolled in a population register. After 2000, most patients from Torino province (population, 2,214,934) have been evaluated for SOD1 mutations. Genomic DNA from patients and normal controls was isolated and purified from 150 µl of peripheral blood on ABI PrismTM 6100 Nucleic Acid Prep Station. DHPLC was performed using the WAVE system (Trasgenomic, Inc). Direct sequence of the entire coding region (five exons plus flanking introns) of the SOD1 gene was performed using dideoxinucleotides method with the Big Dye kit (Applied Biosystems) on ABIPrism 3100 automated sequencer. PCR products were band-purified on lowmelting-point agarose gel using QIAquick (QIAGEN) purification columns. Products were sequenced directly from both ends using Big Dyes Terminator and then purified with Centrisep columns. The products were run on an ABI PRISM 3100 DNA sequencer and analysed with softwares “Factura” and “Sequence Navigator.” From 2000 to 2005 a total of 386 ALS patients (223 men and 163 women) were diagnosed in the Province of Torino (mean annual incidence rate, 2.91/100,000 [95% CI, 2.63-3.21]). Twenty-two patients (5.7%) had a positive family history for ALS. FALS were younger than SALS (FALS 57.4 [SD 12.2]; SALS 65,6 [SD 10.5]; p=0.00001) and had a lower gender ratio (1:1 vs.1.4:1). SOD1 was analyzed in 325 patients (84.2%), including all FALS. Five patients had a SOD1 exon mutation: 3 FALS (13.6% of FALS), and two apparently sporadic cases (0.7% of SALS). The following mutations were found: G41S (2 FALS), G93D (1 FALS), N19S (1 SALS), and a deletion of a glutamic acid at codon 133, E133ΔE (1 SALS). In our epidemiologic study, the frequency of FALS was lower than that reported by clinical series, whereas the frequency of SOD1 mutations among FALS was in the range of expected values. Only less than 1% of SALS cases had a SOD1 mutation. These discrepancies indicate that referral series overestimate both the frequency of FALS and the frequency of SOD1 mutations in SALS patients, probably because they preferentially perform SOD1 analysis in younger subjects, who have a higher risk of carrying SOD1 mutations. (1) Andersen P. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6:37-46 81 P29 DNA damage, p53 activation and apoptosis in a cellular model of ALS Barbosa LF (1), Cerqueira F (1), Macedo A (1), Sogayar MC (1), Augusto O (1), Carrì MT (2), Di Mascio P (1) and Medeiros MHG (1). (1)Departamento de Bioquímica, Instituto de Química, Universidade de São Paulo, Sao Paulo, Brazil. (2)Dipartimento di Biologia, Università di Roma “Tor Vergata”, Rome, Italy. Mutations in Cu,Zn-Superoxide Dismutase (SOD) have been linked to familial amyotrophic lateral sclerosis (FALS) (1) leading to a gain of a “toxic function”, not completely understood. It has been suggested that FALS-mutant SOD induces motor neuron apoptosis (2). Oxidative damage in DNA (3) and p53 upregulation (4) have been detected in ALS neuronal tissues. It is known that accumulation of DNA damage triggers apoptosis by p53 activation (5). We examined DNA damage, p53 activity and apoptosis in human neuroblastoma SH-SY5Y cells transfected with SOD wild-type (SOD-WT) and a mutant SOD (SOD-G93A) typical of FALS. The levels of 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodGuo) and 1,N2-etheno-2´-deoxyguanosine (1,N2-dGuo) were determined by HPLC coupled with eletrochemical detection and mass espectrometry, respectively. DNA strand breaks were analyzed by the comet assay. The activity of p53 was quantified by reporter gene assay and apoptosis by DNA fragmentation followed by flow cytometry. Significantly increased levels of DNA damage (3 fold), increased p53 activity (3 fold) and a higher level of apoptosis (4 fold) were observed in SH-SY5Y cells transfected with SOD-G93A, compared to cells transfected with SOD-WT and parental cells. Moreover, western blot analyses showed that SOD-G93A accumulates in the nucleus and is more associated to DNA than SOD-WT. These results indicate that this mutant SOD has a pro-oxidative and pro-apoptotic activity, and its accumulation in the nucleus and association to DNA may induce DNA damage and trigger apoptosis by p53 activation. Significantly increased levels of DNA damage (3 fold), increased p53 activity (3 fold) and a higher level of apoptosis (4 fold) were observed in SH-SY5Y cells transfected with SOD-G93A, compared to cells transfected with SOD-WT and parental cells. Moreover, western blot analyses showed that SOD-G93A accumulates in the nucleus and is more associated to DNA than SOD-WT. These results indicate that this mutant SOD has a pro-oxidative and pro-apoptotic activity, and its accumulation in the nucleus and association to DNA may induce DNA damage and trigger apoptosis by p53 activation. Acknowledgements: FAPESP, CNPq, FINEP, Instituto do Milênio Redoxoma, USP. (1) Rosen D.R. et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 362, 59-62. (2) Cozzolino M. et al. (2006) Apaf1 mediates apoptosis and mitochondrial damage induced by mutant human SOD1s typical of familial amyotrophic lateral sclerosis. Neurobiol Dis, 21(1), 69-79. (3) Ferrante R.J. et al (1997) Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 69(5), 2064-2074. (4) Martin L.J. (2000) p53 is abnormally elevated and active in the CNS of patients with Amyotrophic Lateral Sclerosis. Neurobiol Dis 7, 613-622. (5) Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell 88, 323-331. 82 P30 The slow Wallerian degeneration gene (WldS) blocks early formation of focal axonal dystrophy in CNS axons undergoing degeneration Bogdan Beirowski (1), Elisabetta Babetto (1), Guillermo Garcia Alias (2), Keith R.G. Martin (2), Michael P. Coleman (2) (1) The Babraham Institute and, (2) Centre for Brain Repair, Cambridge, UK Degeneration of axons is a neuropathological hallmark in various neurodegenerative diseases such as ALS. Generally, axons respond with diverse morphology, topology and speed to various lesion paradigms and noxious effects and recent studies involving the slow Wallerian degeneration (Wld S) phenotype suggest a common mechanism underlying these responses. Previously we have shown in the PNS that individual wild-type and WldS axons following transection and crush injury degenerate progressively displaying different morphological, topological and temporal degeneration patterns. Here we extend these studies to the CNS and investigate the spatiotemporal patterns of axonal degeneration in wild-type and WldS optic nerve and spinal cord following physical lesion. Assessment of axonal survival on transverse optic nerve and spinal cord dorsal column sections suggested a progressive nature of axon degeneration in the CNS following different lesion paradigms. Surprisingly, in contrast to cut or crushed PNS axons, where no major morphological changes are detectable within the ca. 36 h latent phase, optic nerve and dorsal column axons showed focal hypertrophic swellings as early as 10 h, long before axonal fragmentation. Ultrastructural analysis revealed that these swellings were either filled with diverse vesicular bodies, disorganized neurofilaments and degenerating organelles or appeared empty. Interestingly, these swellings showed a gradient along the lesioned nerve tracts with more swellings at proximal sites. Lesioned optic nerves and dorsal columns of WldS rats contained significantly fewer hypertrophic swellings, especially at sites far distal to the lesion. Observations of cultured YFP-H optic nerves showed that hypertrophic axonal swellings arose on uninterrupted nerve fibres after a few hours. In summary, we found that the morphology of axons undergoing Wallerian degeneration in the CNS differs from that in the PNS and resembles focal axonal dystrophy that occurs in many neurodegenerative disorders. We propose a novel working model for CNS axon degeneration where lesioned optic nerve axons become dystrophic long before they fragment, possibly due to early focal axonal transport defects. The WldS phenotype delays this focal axonal dystrophy. 83 P31 The Arl Mouse - A New Mouse Strain With A Mutation In the Cytoplasmic Dynein Heavy Chain Bros V (1), Golding M (2), Chia R (1), Schiavo G (2), Flenniken A (3), Adamson SL (3), Rossant J (3). Fisher EMC (1), Greensmith L (1), Hafezparast M (4), (1) Institute of Neurology, London, UK; (2) Cancer Research UK, London, UK (3) Centre For Modeling Human Disease, Toronto, Canada; (4) Department of Biochemistry, University of Sussex, Brighton, UK Mutations in cytoplasmic dynein have been implicated in the pathogenesis of amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disorder characterized by motor neuron degeneration and muscle paralysis. For example, we have previously shown that Loa mice with a mutation in the cytoplasmic dynein heavy chain show defects in retrograde axonal transport and reduced motoneuron survival. Furthermore, postnatal over-expression of the dynein subunit dynamitin, also induces motoneuron degeneration in mice. In addition, mutations in motor proteins such as the anterograde motor Kinesin 1 or dynactin, an essential component of the dynein motor complex, have been linked with motoneuron degeneration in human disorders. Here we present a new mouse strain called the Abnormal rear legs (Arl), which has a different point mutation in the cytoplasmic dynein heavy chain to that described for the Loa mouse. Our initial examination suggests that the ‘Arl’ mouse may have a more severe phenotype than the Loa mouse. For example, from a young age, the Arl mice show a distinctive “amphibian-like” gait. Their hind limbs are splayed laterally so that they have difficulty in supporting their pelvis, which hinders forward movement. We are therefore carrying out a full phenotypic characterisation of the Arl mice in terms of axonal transport, physiological analysis of muscle force and motor unit survival as well as a morphological assessment of motoneuron survival. Our initial results indicate that there is a clear pathology in the sciatic nerves of these mice, resulting in a significant reduction in nerve diameter compared to their wildtype (WT) littermates. The underlying pathology of these nerves is under investigation. 84 P32 The nucleocytoplasmic transport system is altered in Motor Neurons of a mouse model of familial ALS Peviani M., Spano G., Bendotti C . Lab. Molecular Neurobiology, Istituto di Ricerche Farmacologiche “Mario Negri”, Milan (Italy). Amyotrophic Lateral Sclerosis (ALS) is a rare neurodegenerative disease that affects upper and lower motor neurons. It determines progressive muscle atrophy, leading to ultimate paralysis and death. It has recently been suggested that alteration of distal axons may take part in the early steps leading motor neurons to death in ALS (1). Moreover, evidence is emerging that a dysfunction of nucleocytoplasmic transport system may also be involved in the mechanisms underlying motor neuron degeneration in ALS (2). Importins are recruited for important steps of nucleocytoplasmic transport system, and upregulation of importin beta has been found to be specifically triggered in peripheral nerves to modulate regeneration of injured neurons after crush injury (3). These evidences prompted us to investigate whether importin-mediated nucleocytoplasmic transport system is modified in neuronal perikaria or at the periphery during progressive degeneration of neuronal cells, in a mouse model of ALS. We observed a reduction of importin beta levels in homogenates from ventral horn spinal cord concomitantly with the appearance of first symptoms of the pathology. Moreover using confocal miscroscopy we detected an altered distribution of the importins in motor neurons with impaired retrograde axonal transport, and in neurons showing accumulation of phosphorylated neurofilaments, an hallmark of neural degeneration. We suggest that alterations of nucleocytoplasmic transport-related proteins may represent an additional mechanism involved in the degeneration of motor neurons in ALS pathology. (1) Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 185(2):232-40. (2) Zhang J, Ito H, Wate R, Ohnishi S, Nakano S, Kusaka H. (2006) Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol (Berl). 112(6):673-80. (3) Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, vanMinnen J, Twiss JL, Fainzilber M. (2003) Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 18;40(6):1095-104. 85 P33 Proteomic analysis of astrocytic secretion in a mouse model of familial amyotrophic lateral sclerosis Basso M (1,2), Tortarolo M (2), Gabriella Spaltro (2), Dario Lidonnici (2), Salmona M (2), Bendotti C (2), Bonetto V (1) (2) (1) Dulbecco Telethon Institute, (2) Institute for Pharmacological Research “Mario Negri”, Milan, Italy Amyotrophic lateral sclerosis (ALS) is a progressive, fatal neurological disease characterized by the specific degeneration of motor neurons in the cortex, brainstem and spinal cord. The mechanisms involved in this selective degeneration are still unidentified. In recent years, a clear interplay between motor neurons and other cellular population have been proven (1-2). In particular, the role of astrocytes seems to be extremely important for their contribution to spinal motor neuron degeneration (3). It seems that the toxicity is caused by factors and proteins released into the extracellular space by astrocytes and motor neurons (3). The aim of this work is to investigate the role of astrocytes in the pathogenesis of familial ALS. For this purpose we analyzed the secretome from primary culture of astrocytes expressing the pathogenic mutated SOD1 or wild-type SOD1. Secreted proteins were separated by two-dimensional electrophoresis, and the gel maps were compared by computerized image analysis. The most abundant proteins secreted were identified by MALDI mass spectrometry. About forty proteins are differentially secreted by astrocytes expressing the mutated SOD1 in comparison with controls. We are currently identifying these proteins. These preliminary results suggest that the only expression of mutated SOD1 has an impact on astrocyte secretion pathways. We are now comparing astrocyte secretome with the secretome of motor neuron/astrocytes co-cultures carrying or not the mutation with a special focus on SOD1. At the same time we are investigating the expression of astrocytic proteins when the mutated SOD1 is present or not, in order to understand which are the intracellular molecular pathways which determine alteration in the secretome and may be important in mediating SOD1 toxicity. (1) Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillee, S., Rule, M., McMahon, A. P., Doucette, W., Siwek, D., Ferrante, R. J., Brown, R. H., Jr., Julien, J. P., Goldstein, L. S., and Cleveland, D. W. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302(5642), 113-7. (2) Boillee, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., Kollias, G., and Cleveland, D. W. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312(5778), 1389-92. (3) Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., and Przedborski, S. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10(5), 615-622. 86 P34 Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity Bogaert E. (1), Van Damme P. (1), Callewaert G. (2), Herijgers P. (3), Van Den Bosch L. (1), Robberecht W. (1) (1) Laboratory of Neurobiology, (2) Laboratory of Physiology, (3) Laboratory of Experimental Surgery and Anaesthesiology, University of Leuven, Belgium. Influx of Ca2+ through AMPA receptors contributes to the neuronal damage in stroke, epilepsy and neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS). The Ca 2+ permeability of AMPA receptors is determined by the GluR2 subunit, receptors lacking GluR2 being permeable to Ca2+. We identified a difference in GluR2 expression in motor neurons between two different rat strains (Wistar and Holtzman), which correlated with a difference in vulnerability to AMPA receptormediated excitotoxicity. In an in vitro model Wistar and Holtzman motor neurons, were cultured on a pre-established feeder layer of astrocytes. Compared to Holtzman motor neurons, Wistar motor neurons had lower GluR2 expression levels, and were more vulnerable to AMPA receptor-mediated excitotoxicity. Astrocytes were found to determine the neuronal GluR2 expression levels. In order to demonstrate an increased vulnerability of Wistar motor neurons to AMPA receptor-mediated excitotoxicity in vivo, transient spinal cord ischemia was studied. We found that the extent of paresis and motor neuron degeneration induced by ischemia was more pronounced in Wistar compared to Holtzman rats. We also studied survival of Wistar and Holtzman rats overexpressing mutant SOD1, a reliable model for human ALS. However, no difference was found, in spite of the known contribution of AMPA receptor-mediated excitotoxicity to mutant SOD1-induced motor neuron degeneration. The lack of protection was found to be due to the presence of mutant SOD1 in Holtzman astrocytes. Holtzman astrocytes overexpressing mutant SOD1 did no longer protect motor neurons against excitotoxicity and abolished their ability to stimulate GluR2 expression. All together, these results reveal a new mechanism through which astrocytes influence neuronal functioning in health and disease. 87 P35 Glutamate release induced by glycine and GABA is enhanced in the spinal cord of SOD1 G93A mutant mice. Zappettini S. (1), Raiteri L. (1), Milanese M. (1), Raiteri M. (1), Bonanno G. (1) (1) Department of Experimental Medicine, Pharmacology and Toxicology Section, and Centre of Excellence for Biomedical Research, University of Genoa, Italy. There is increasing evidence that the extra-cellular levels of the excitatory neurotransmitter glutamate are enhanced in amyotrophic lateral sclerosis (ALS) and that glutamate-mediated excitotoxicity is implicated in the disease. As a cause of excessive glutamate, abnormalities of the glutamate transport were observed, which were subsequently ascribed to a selective loss of the GLT1 glutamate transporter. Since GLT1 is mainly localized at astroglial processes, the hypothesis that glial cells play a role in the development of the disease appeared to be likely. An alternative way to produce elevated extra-cellular concentrations of glutamate may well consists of the increase of neuronal glutamate release rather than of astrocyte-localized inhibition of reuptake. We have previously shown that glycine evokes glutamate and GABA release from mouse spinal cord synaptosomes by penetrating in the glutamate- or GABA-releasing nerve terminals through transporters (heterotransporters) of the GLYT1 and GLYT2 type (1, 2) and that GABA evokes glutamate, but not glycine, release via GAT1 heterotransporters sited at glutamate-releasing nerve terminals (3). We here compared the glycine-evoked release of glutamate or GABA and the GABAevoked release of glutamate in mice expressing a mutant form of human SOD1 with a Gly93Ala substitution [SOD1(+)/G93A(+)], a transgenic model of familiar ALS; in mice, expressing the unmodified human SOD1 [SOD1(+)] and in non-transgenic littermates [SOD1(-)/G93A(-)]. In [SOD1(+)] control animals, the basal efflux of 3HD-aspartate, a marker of the glutamate intraterminal pools, was concentration-dependently increased by GLY (EC50=68.3 M; Emax=186% of potentiation) and by GABA (EC50=3.6 M; Emax=113%) through a heterotransporter-mediated process. Similar results were observed in SOD1(-)/G93A(-) mice. The effects of GLY and GABA were significantly higher in symptomatic mutant SOD1(+)/G93A(+) mice than in controls (E max= 260% vs 186% and 159% vs 113%, respectively, with no changes in EC 50 values). GLY and GABA increased also the release of endogenous glutamate more efficiently in SOD1(+)/G93A(+) animals respect to controls. On the contrary, the release of tritiated or endogenous GABA, induced by glycine, was not modified in ALS mice. The higher release of 3HD-aspartate release in mutant animals was already present in pre-symptomatic 70-90 and 30-40 day-old mice. Our results show an excessive release of glutamate in an animal model of familiar ALS. The understanding of the mechanisms of this excessive release requires detailed investigation. If it occurs also in vivo, the different modulation of glutamate and GABA release here reported could induce an unbalance between inhibitory and excitatory transmission in ALS. (1) Raiteri L., Raiteri M. and Bonanno G. (2001) Coexistence and function of different neurotransmitter transporters in the plasma membrane of CNS neurons. J. Neurochem. 76, 18231832,. (2) Raiteri L., Stigliani S., Siri A., Passalacqua M., Melloni E., Raiteri M. and Bonanno G. (2005) Glycine taken up through GLYT1 and GLYT2 heterotransporters into glutamatergic axon terminals of mouse spinal cord elicits release of glutamate by homotransporter reversal and through anion channels. Biochemical Pharmacology, 69, 159-168. (3) Raiteri L., Stigliani S., Patti L., Usai C., Bucci G., Diaspro A., Raiteri M. and Bonanno G. (2005) Activation of GABA GAT-1transporters on glutamatergic terminals of mouse spinal cord mediates glutamate release through anion channels and by transporter reversal. J. Neurosci. Res. 80, 424-433. 88 P36 Glutamate uptake by presynaptic compartment is decreased in transgenic SOD1G93A mice, an animal model of amyotrophic lateral sclerosis Elena Fumagalli, Simona Colleoni, Caterina Bendotti and Tiziana Mennini Istituto di Ricerche Farmacologiche Mario Negri, Milan, Italy Excitatory amino acid transporters guarantee glutamate homeostasis in the CNS, and both glial and neuronal transporter dysfunction could be of relevance in pathological conditions. While the role played by glial cells in glutamate clearance in vivo is well documented, the involvement of the presynaptic compartment is still unclear, also due to the difficulties in isolating pure functional cellular fractions from the whole tissue. A decreased glial GLT1 expression with reduced glutamate uptake has been reported in CNS tissues of amyotrophic lateral sclerosis (ALS) patients and in transgenic SOD1G93A mice, one of the most used animal model of familiar ALS. However, studies were done using crude synaptosomal fractions, that are a mixed preparation containing both neuronal and glial elements. We evaluated the glutamate uptake in purified gliosomes and synaptosomes obtained from spinal cord of SOD1G93A mice and non transgenic C57 mice, and compared the results with those obtained in the classical crude synaptosomal preparation. Western blot analysis showed 90% enrichment of synaptophysin in the purified synaptosomes, with about 30% contamination from glial cells (GFAP). Two time points were considered, the early symptomatic stage (16 weeks) and at the late stage of the disease (24 weeks). The different preparations showed similar Km and different capacities of glutamate uptake (Vmax), with higher Vmax in the purified synaptosomes, both in transgenic and control mice. We found no changes in Km and Vmax of glutamate uptake between transgenic and non transgenic mice at the early stage of disease, while a significant reduction of Vmax (-30%) was evident at the late stage in transgenic SODG93A mice, selectively in the purified synaptosomes. A slight reduction of the Vmax was also found in crude synaptosomes (-12%) while value was unchanged in gliosomes. Pharmacological properties of these preparations were also investigated, in order to try to identify the glutamate transporter located at the nerve terminals. However, no differences were found in terms of sensitivity to different glutamate transporters inhibitors (DHK, SOS, TBOA) between transgenic and non transgenic mice and among the different purified preparations. We confirmed a decrease in the expression of the glutamate transporter GLT1 and found a reduction of immunoreactivity for the neuronal marker synaptophysin, likely a consequence of the degeneration of nerve terminals of the corticospinal tract. Thus we conclude that the loss of presynaptic glutamatergic neurons is responsible for reduced glutamate uptake in the spinal cord of 24 week-old SOD1G93A mice. 89 P37 Longitudinal study of the onset and progression of Amyotrophic Lateral Sclerosis disease in SOD1 mutant mice by non invasive imaging KELLER A.Florence & KRIZ Jasna Dept. Anatomy and Physiology, Laval University, Centre de Recherche CHUL(CHUQ), Québec, QC, Canada Amyotrophic lateral sclerosis (ALS) is a late onset neurological disease characterized by progressive spinal motor neurons degeneration associated with paralysis and eventually death. Transgenic mice expressing a mutant sodium dismutase 1 (SOD1) develop phenotype with many pathological features resembling human familial and sporadic ALS. Although major clinical symptoms in ALS arise from neurodegeneration and death of motoneurons, recent studies suggest that non neuronal cells could play a role in the toxicity to motor neurons. Their precise role in onset and progression of the disease remain however unknown. To further investigate the role of non-neuronal cells in the disease onset and progression, we developed mouse model for live imaging of astrogliosis in ALS. As a glial fibrilary acidic protein (GFAP) is strongly up-regulated in ALS, we used it as a hallmark of astrogliosis to create our model. We thus crossed mice carrying the firefly luciferase gene under the transcriptional control of mouse GFAP promoter (GFAP-luc, Xenogen, CA) with mice carrying the SOD1G93A mutation. The double transgenic GFAP-luc/SOD1G93A mice were used in the study as well as GFAP (wt)-SOD1G93A and GFAP-luc as controls. Live imaging was performed weekly starting from postnatal weeks 3-4 till the end stage of the disease. Loss of extension reflex, weight-loss and motor deficits were used as indicators of clinical symptoms. The results were obtained from 19 female/male mice matched-age littermates and were compared to adequate controls (n=7). Data collected by in vivo imaging showed that photon emission/GFAP signal was first detected at the lumbar spinal cord area. The signal first arose from small multiple areas of astrocytes activation which then converged into a larger signal around 80-100days of age. The correlation analysis between live imaging and behaviour data revealed that increase in GFAP signal in the spinal cord at 70-80 days correlated with the initial disease onset (loss of extension reflex). Moreover the peak signals arising from the spinal cord at approx. 100 days correlated with the abrupt onset of sensorimotor deficit and paralysis. The end-stage of the disease (approx 133d) was characterized by an increase of bioluminescent signals arising form the different brain structures that coincided with the loss of body weight. GFAP-luc//SOD1G93A mice will provide unique tools for understanding disease pathology and longitudinal responses to drug testing. Acknowledgement: CIHR, FRSQ, RRTQ 90 P38 Effects of mutant FALS-SOD1 on microglia secretion Padovano V., Massari S., Righi M. and Pietrini G. Department of Pharmacology, School of Medicine, Università degli Studi di Milano; CEND- Center of neurodegenerative diseases; CNR-Institute of Neuroscience , Milan, Italy. Increasing evidence indicates a participation of the microglia to motor neuron degeneration in human and murine ALS. Microglia represents the resident immune competent cells of the CNS, and noxious stimulus may elicit microglia activation and its subsequent release of toxic factors that accelerate neuronal degeneration and death. To unravel the contribution of microglia to the pathogenesis of ALS, we are testing the hypothesis of altered pathways of secretion in microglia induced by the expression of the G93A mutant of SOD1. To this purpose we have established microglia cell culture models expressing constitutive or inducible human SOD1 (WT and G93A SOD1). Our data indicate that the expression of mutant SOD1 alters the basal and LPS-stimulated release of potentially toxic molecules including the G93A SOD1, whereas the expression of even higher amount of the WT SOD1 decrease the release of these molecules. Moreover, our data suggest that these molecules are released via unconventional pathways of secretion not involving the endoplasmic reticulum-Golgi complex route. We are now performing experiments aimed at identifying the secretory pathways followed by these molecules, which is a crucial step in order to develop appropriate therapeutic strategies to prevent toxicity mediated by microglia. 91 P39 Microglial activation and MCP-1 staining pattern develop differently with disease progression in the spinal cord of SOD1 mutant mouse and rat Siklos L (1), Paizs M (1), Henkel J (2), Beers DR (2) and Appel SH (2). (1) Institute of Biophysics, Biological Research Center, Szeged, Hungary (2) Department of Neurology, Methodist Neurological Institute, Houston, USA Neuropathological findings and experimental data support the decisive role of microglia in the pathogenesis of amyotrophic lateral sclerosis (ALS) (1,2). Additionally, the involvement of monocyte chemoattractant protein-1 (MCP-1) in neuron-microglia interaction has been suggested (3), which chemokine is critical for the recruitment of inflammatory cells of monocytic lineage after injury in the central nervous system. For modeling the disease, transgenic mice (4) as well as transgenic rats (5), overexpressing mutant (G93A) Cu2+/Zn2+ superoxide dismutase (mSOD1) have been developed which animals show signs of progressive motor neuron disease resembling clinical and pathological features of human ALS. In both transgenic animals, we characterized the change of microglial and MCP-1 immunostaining in the lumbar segment of the spinal cords as the disease progressed. One set of animals was used from each species: rats were sacrificed at age of 18, 40, 50, 70, 90, 102 days and at endstage (day 122); mice were killed at age of 16, 38, 80, 111 days and at endstage (day 141). After perfusion, the spinal cords were dissected, frozen sections were cut (30 m) from the lumbar segment and immunostaining was made on free-floating sections with the avidin-biotin technique. In transgenic rats, MCP1-staining started to develop at age of 40 days, reached its maximum at day 70 and disappeared at day 102. Only the motor neurons were stained. According to the change in their appearance, microglia activation commenced at days 50-70, then their proliferation progressed until endstage, although activated microglia were mostly confined to the ventro-lateral region of the spinal cord. In mSOD1 transgenic mice, MCP-1 staining could be seen at every time point, which staining was not restricted to motor neurons. The staining intensity decreased from a high level (at age of 16 day) to a low level (at age of 38-80 days), then started to increase again and reached a high level for a second time at endstage. This unexpected U-shaped time dependence of the MCP-1 staining intensity was supported by data from RT-PCR measurements (6). Microglia activation in mice was evident at age of 80 days then their proliferation continuously increased until endstage with a homogeneous distribution in the whole grey matter. The contrasting time dependence of MCP-1 staining in mice and rats as well as the different distribution pattern of the activated microglia at close to endstage suggest that the same mutant enzyme may lead to different pathological course even in relatively close species. (1) Kawamata T., Akiyama H., Yamada T. and McGeer P.L. (1992) Immunologic reactions in amyotrophic lateral sclerosis, brain and spinal cord. Am.J.Pathol. 140, 691-707. (2) Beers D.R., Henkel J.S., Xiao Q., Zhao W., Wang J., Yen A.A., Siklos L., McKercher S.R. and Appel S.H. (2006) Wild-type microglia extend survival in Pu.1 knockout mice with familial amyotrophic lateral sclerosis. Proc.Natl.Acad.Sci.USA. 103, 16021-16026. (3) Flügel A., Hager G., Horvat A., Spitzer C., Singer G.M., Graeber M.B., Kreutzberg G.W. and Schwaiger F.W. (2001) Neuronal MCP-1 expression in response to remote nerve injury. J.Cereb.Blood Flow Metab. 21, 69-76. (4) Gurney M.E., Pu H., Chiu A.Y., et al. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772-1775. (5) Howland D.S., Liu J., She Y., Goad B., Maragakis N.J., Kim B., Erickson J., Kulik J., DeVito L., Psaltis G., DeGennaro L.J., Cleveland D.W. and Rothstein J.D. (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc.Natl.Acad.Sci.USA. 99, 1604-1609. (6)Henkel J.S., Beers D.R., Siklos L. and Appel S.H. (2006) The chemokine MCP-1 and the dendritic and myeloid cells are increased in the mSOD1 mouse model of ALS. Mol.Cell Neurosci. 31, 427-437. 92 P40 Ivermectin inhibits AMPA receptor-mediated excitotoxicity in vitro and extends the life span of an ALS mouse model Andries M (1), Van Damme P (2), Robberecht W (2), and Van Den Bosch L (2) (1) Department of Molecular Cell Biology, Faculty of Medicine, K.U.Leuven, Campus Gasthuisberg, Leuven, Belgium. (2) Laboratory of Neurobiology, Faculty of Medicine, K.U. Leuven, Campus Gasthuisberg, Leuven, Belgium We have shown before that α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptormediated excitotoxicity contributes to the selective motor neuron death in amyotrophic lateral sclerosis (ALS) (1). In this study, we investigated the effect of P2 receptor-influencing substances on kainate-induced motor neuron death in an in vitro model for AMPA receptor-mediated excitotoxicity. Complete protection was found after preincubation of the motor neurons with ivermectin or Cibacron Blue 3G-A. Preincubation with both P2X4 modulators did not influence the number or Ca2+ permeability of the AMPA receptors and addition during kainate stimulation alone had no effect. Preincubation with a low concentration of ATP, the natural agonist of the P2X 4 receptor, also protected the motor neurons against a subsequent excitotoxic stimulation, while high concentrations of ATP were toxic. Moreover, ivermectin increased the toxicity of low ATP concentrations, indicating that ivermectin can potentiate the effect of ATP on its receptor. Ivermectin and ATP also protected against hypoxia/hypoglycemia. To further investigate the relevance of these findings for ALS, we treated mutant SOD1G93A-mice with ivermectin. This resulted in an extension of the life span of these mice with almost 10%. We conclude that ivermectin induces a mechanism in motor neurons, in vivo and in vitro, that protects against subsequent excitotoxic insults. Our in vitro data indicate that this protective mechanism is due to the potentiation by ivermectin of an effect of ATP mediated by the P2X4 receptor. (1) Van Damme P., Leyssen M., Callewaert G., Robberecht W. and Van Den Bosch L. (2003) The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 343, 81–84. 93 P41 Mesenchymal stem cell treatment induces survival prolonging and symptom amelioration in mutant SOD1 G93A mice Milanese M. (1), Zappettini S. (1), Casazza S. (2), Caponetto C. (2), Bonanno G. (1), Uccelli A. (2). (1) Department of Experimental Medicine, Pharmacology and Toxicology Section, and Centre of Excellence for Biomedical Research, University of Genoa, Italy (2) Department of Neuroscience, Neurology Section, University of Genoa, Italy Amyotrophic lateral sclerosis (ALS) is a chronic neuromuscular disorder clinically characterized by muscle wasting, weakness and spasticity reflecting a progressive degeneration of upper and lower motor neurons. To date hardly any treatments prolong survival in ALS patients of some extent. Development of more effective neuroprotective therapies is impeded by lack of understanding of the mechanisms of neuronal death and how the disease propagates. Mesenchymal stem cells (MSC), a subset of adult stem cells derived from the bone marrow stroma, have generated much enthusiasm as possible cell source for tissue repair including the nervous system. Recent studies have shown that MSC can also modulate immune responses and exert an anti-apoptotic effect on different cells including neurons. In addition, MSC can migrate into the central nervous system when i.v. injected. We studied here the effects of MSC administration in mice expressing mutant human super oxide dismutase (SOD1) with a G93A substitution [SOD1(+)/G93A(+)], a transgenic animal model of ALS. Mesenchymal cells (106 cells/animal, i.v.) were injected at day 90, well after the onset of the first disease symptoms, that can be recorded at about day 60. Saline injected SOD1(+)/G93A(+) were used as controls. Control mice survived about 120 days while the MSC-treated mice exhibited a statistically significant prolonged survival time compared to saline injected controls. Such effect was associated with a significant amelioration in the performance of behavioral motor tests in the MSCtreated animals. Studying neurotransmitter release, we have found that glutamate exocytosis is enhanced in the spinal cord of SOD1(+)-G93A(+) mice, respect to controls. Interestingly, MSC treatment almost abolished this extra-release of the excitatory amino acid. Upon i.v injection, a few luciferase-labeled MSCs were detected inside the mice spinal cord. Amelioration of some histological parameters was observed irrespective neural trans-differentiation. We can conclude that the treatment with MSC may be considered as an appealing therapeutic opportunity for ALS although the effect does not seem to rely on tissue repair. 94 List of Participants 95 Chiara Abrescia Bioindustry Park Canavese spa Colleretto Giacosa (TO), Italy abrescia@bipcamail.it Ilaria Amori Lab. Of Neurochemistry Fondazione Santa Lucia IRCCS Roma, Italy ilariaamori@hotmail.com Peter Andersen Department of Neurology Institute of Clinical Neuroscience Umeå University SE-901 85 Umeå, Sweden peter.andersen@neuro.umu.se Svetlana Antonyuk Molecular Biophysics Group Daresbury Laboratory Warrington, U.K. s.antonyuk@dl.ac.uk Mario Arciello Dept. of Biology University of Rome "Tor Vergata" Rome, Italy arciello.mario@libero.it Gabriella Augusti-Tocco Dept. of Cell and Developmental Biology University of Rome “La Sapienza” Rome, Italy gabriella.tocco@uniroma1.it Elisabetta Babetto The Babraham Institute Cambridge, UK elisabetta.babetto@bbsrc.ac.uk Lucia Banci Department of Chemistry University of Florence Sesto Fiorentino (FI), Italy banci@cerm.unifi.it Luis Barbeito Istituto Clemente Estable Montevideo, Uruguay barbeito@pasteur.edu.uy Livea Barbosa Departamento de Bioquímica Instituto de Química Universidade de São Paulo São Paulo, Brazil livea@iq.usp.br Manuela Basso Department of Molecular Biochemistry and Pharmacology Istituto di Ricerche Farmacologiche “Mario Negri” Milano, Italy basso@marionegri.it Stefania Battistini Azienda Osp. Univ. Senese U.O.C. Neurologia Policlinico “Le Scotte” Siena, Italy battistinis@unisi.it Andrea Battistoni Dept. of Biology University of Rome "Tor Vergata" Rome, Italy andrea.battistoni@uniroma2.it Joseph S. Beckman Linus Pauling Institute Department of Biochemistry and Biophysics, Oregon State University Corvallis OR 97331USA joe.beckman@oregonstate.edu Bogdan Beirowski The Babraham Institute Cambridge, UK bogdan.beirowski@bbsrc.ac.uk Caterina Bendotti Dept. Neuroscience Istituto di Ricerche Farmacologiche "Mario Negri" Milano, Italy bendotti@marionegri.it Marina Bentivoglio Department of Morphological and Biomedical Sciences University of Verona Verona, Italy marina.bentivoglio@univr.it 96 Elke Bogaert Laboratory of Neurobiology University of Leuven Leuven, Belgium elke.bogaert@med.kuleuven.be Irene Carunchio Dept. of Neuroscience University of Rome “Tor Vergata” Rome, Italy neurolab@virgilio Giambattista Bonanno Department of Experimental Medicine University of Genoa Genoa, Italy bonanno@pharmatox.unige.it Cristina Cereda IRCCS Ist. Neurologico “Mondino” Pavia, Italy cristina.cereda@mondino.it Valentina Bonetto Istituto di Ricerche Farmacologiche "Mario Negri" Milano, Italy bonetto@marionegri.it Fernanda Cerqueira Departmento de Bioquímica Instituto de Química Universidade de São Paulo São Paulo, Brazil fernandamcerqueira@yahoo.com.br Virginie Bros-Facer Institute of Neurology University College London, UK v.bros@ion.ucl.ac.uk Avi Chakrabartty Dept Med Biophysics/CRND University of Toronto Toronto, Canada chakrab@uhnres.utoronto.ca Lucie Bruijn The ALS Association Palm Harbor, FL 34685, USA. lucie@alsa-national.org Albrecht Clement Institute for Physiological Chemistry and Pathobiochemistry University of Mainz Medical school Mainz, Germany clement@uni-mainz.de Lavinia Cantoni Istituto di Ricerche Farmacologiche "Mario Negri" Milano, Italy cantoni@marionegri.it Daniele Capitanio Dept. of Biomedical Sciences and Technologies University of Milan Segrate (MI), Italy daniele.capitanio@unimi.it Concetta Capo Dept. of Biology University of Rome "Tor Vergata" Rome, Italy capo@uniroma2.it Maria Teresa Carrì Dept. of Biology University of Rome "Tor Vergata" Rome, Italy carri@bio.uniroma2.it Claudia Colombrita Dip. Scienze Neurologiche Università di Milano Milano, Italy claudia.colombrita@studenti.unimi.it Mauro Cozzolino Lab. of Neurochemistry Fondazione S. Lucia IRCCS Rome, Italy m.cozzolino@hsantalucia.it Valeria Crippa Institute of Endocrinology Centre of Excellence on Neurodegenerative Diseases of the University of Milan Milan, Italy valeria.crippa@unimi.it 97 Claudia Crosio DiFBC Università degli Studi di Sassari Sassari, Italy ccrosio@uniss.it Elena Fumagalli Istituto di Ricerche Farmacologiche "Mario Negri" Milano, Italy fumagalli@marionegri.it Giuseppina D’ Alessandro Istituto di Ricerche Farmacologiche "Mario Negri" Milano, Italy dalessandro@marionegri.it Stella Gagliardi Università degli studi di Pavia Pavia, Italy stellagag@libero.it Ruth Danzeisen International Copper Association New York NY 10016 USA rdanzaisen@copper.org Paolo Di Mascio Instituto de Química Universidade de São Paulo São Paulo, Brazil pdmascio@iq.usp.br Gabriella Dobrowolny Dip. Istologia ed Embrionologia Medica Università “La Sapienza” Roma, Italy gabriella.dobrowolny@uniroma1.it Jacques Durand Lab. de la Plasticitè et Physio-Pathologie de la Motricité UMR 6196 CNRS Aix-Marseille Université Marseille, France durand@dpm.cnrs-mrs.fr Samer Abou Ezzi CHUL Research Center Laval University Quebec, Canada samer_ezzi@yahoo.ca Claudia Fallini Dipartimento di Scienze Neurologiche Università di Milano Milano, Italy claudia.fallini@unimi.it Valentina Gallo Imperial College of London London, UK v.gallo@imperial.ac.uk Jonathan Glass Emory Center for Neurodegenerative Disease Whitehead Biomedical Research Building Atlanta, GA 30322 USA jglas03@emory.edu Genevieve Gowing Laval University Quebec, Canada genevieve.gowing@crchul.ulaval.ca Linda Greensmith Sobell Department of Motor Neuroscience and Movement Disorders Institute of Neurology London, UK l.greensmith@ion.ucl.ac.uk Francois Gros-Louis Dept.of Oncology and Molecular Endocrinology CHUL Research Center Laval University Quebec, Canada f_groslouis@hotmail.com Jean-Pierre Julien Dept.of Oncology and Molecular Endocrinology CHUL Research Center Laval University Quebec, Canada jean-pierre.julien@crchul.ulaval.ca Alberto Ferri Lab. of Neurochemistry Fondazione S. Lucia IRCCS Rome, Italy a.ferri@hsantalucia.it 98 Ali Murat Kaya Institute for Physiological Chemistry and Pathobiochemistry University of Mainz Medical school Mainz, Germany kaya@uni-mainz.de Antonio Musarò Department of Histology and Medical Embryology University of Rome “La Sapienza” Rome, Italy antonio.musaro@uniroma1.it Jasna Kriz Dept.of Oncology and Molecular Endocrinology CHUL Research Center Laval University Quebec, Canada jasna.kriz@crchul.ulaval.ca Monica Nencini Lab. of Neurochemistry Fondazione S. Lucia IRCCS Rome, Italy monicanencini@hotmail.com Michael Kyba Dept.of Developmental Biology University of Texas Georgetown, Texas, USA michael.kyba@utsouthwestern.edu Jean-Philippe Loeffler Lab. de Signalisations Mol. et Neurodeg, Universite Louis Pasteur Strasbourg, France loeffler@neurochem.u-strasbg.fr Andrea Malaspina Institute of Cell and Molecular Science Queen Mary University of London London E1 2AT, UK a.malaspina@qmul.ac.uk Stefan Marklund Department of Medical Biosciences Umea University SE-901 85 Umea, Sweden stefan.marklund@medbio.umu.se Marco Milanese Department of Experimental Medicine, Pharmacology and Toxicology Section University of Genoa Genoa, Italy milanese@pharmatox.unige.it Maria Rosaria Monsurrò Department of Neurological Sciences Second University of Naples Naples, Italy mrmonsurro@hotmail.com Keith Nylin Department of Biochemistry & Biophysics Oregon State University Oregon, USA keith.nylin@oregonstate.edu Paolo Ogliari IRCCS Ist. Neurologico “Mondino” Pavia, Italy paolo_ogliari@yahoo.com Mikael Oliveberg Department of Biochemistry and Clinical Neuroscience Umea University SE-901 85 Umea, Sweden mikael.oliveberg@dbb.su.se Valeria Padovano Department of Pharmacology School of Medicine Università degli Studi di Milano Milano, Italy valeria.padovano@unimi.it Piera Pasinelli Farber Institute for Neurosciences Thomas Jefferson University Philadelphia, PA 19107 USA piera.pasinelli@jefferson.edu Rita Perlingeiro Department of Developmental Biology UTSouthwestern Medical Center Dallas, TX USA rita.perlingeiro@utsouthwestern.edu Maria Grazia Pesaresi Lab. of Neurochemistry Fondazione S. Lucia IRCCS Rome, Italy lorpesaresi@libero.it 99 Grazia Pietrini Department of Pharmacology School of Medicine Università degli Studi di Milano Milan, Italy grazia.pietrini@unimi.it Daniela Sau Inst. of Endocrinology Centre of Excellence on Neurodegenerative Diseases University of Milan, Milan, Italy daniela.sau@unimi.it Angelo Poletti Institute of Endocrinology Center of Excellence on Neurodegenerative Diseases University of Milan Milan, Italy angelo.poletti@unimi.it Christopher E. Shaw Department of Neurology King's College London School of Medicine London, UK chris.shaw@iop.kcl.ac.uk Cédric Raoul INSERM-AVENIR Team Campus de Luminy Parc Scientifique de Luminy 13200 Marseille cedex 09 France cedric.raoul@ibdm.univ-mrs.fr Antonia Ratti Dipartimento di Scienze Neurologiche Università di Milano Milano, Italy antonia.ratti@unimi.it Claudia Ricci Università di Siena Siena, Italia ricci6@unisi.it Wim Robberecht Department of Neurology and Experimental Neurology University Hospital Gasthuisberg University of Leuven Leuven, Belgium wim.robberecht@uz.kuleuven.ac.be Luisa Rossi Dept. of Biology University of Rome "Tor Vergata" Rome, Italy luisa.rossi@uniroma2.it Giuseppe Rotilio Dept. of Biology University of Rome "Tor Vergata" Rome, Italy rotilo@bio.uniroma2.it Pamela J. Shaw Academic Neurology Unit University of Sheffield Sheffield, UK pamela.shaw@shef.ac.uk Laszlo Siklos Institute of Biophysics Biological Research Center Szeged, Hungary siklos@brc.hu Vincenzo Silani Dep.of Neurology and Lab.of Neuroscience IRCCS Istituto Auxologico Italiano Milan, Italy vincenzo@silani.com Gen Sobue Department of Neurology Nagoya University Graduate School of Medicine Nagoya 466-8500, Japan sobueg@med.nagoya-u.ac.ip Richard Strange CCLRC Daresbury Laboratory Warrington Cheshire WA4 4AD, UK r.w.strange@dl.ac.uk Michael Swash Royal London Hospital London, Uk mswash@btinternet.com Silvia Tartari Istituto di Ricerche Farmacologiche "Mario Negri" Milano, Italy tartari@marionegri.it 100 Nicola Ticozzi IRCCS-Istituto Auxologico Italiano Università di Milano Scuola Specializzazione Neurologia Milano, Italy n.ticozzi@fastwebnet.it Massimo Tortarolo Dept.of Neuroscience Istituto di Ricerche Farmacologiche "Mario Negri", Milano, Italy tortarolo@marionegri.it Davide Trotti Farber Institute for Neurosciences Thomas Jefferson University Philadelphia Philadelphia PA 19107, USA davide.trotti@jefferson.edu Joan S. Valentine Department of Chemistry and Biochemistry University of California Los Angeles CA 90095, USA jsv@chem.ucla.edu Ludo Van den Bosch Laboratory for Neurobiology Experimental Neurology University of Leuven Leuven, Belgium ludo.vandenbosch@med.kuleuven.be Monika Vestling Molecular Biology Umezi University Umea, Sweden monika.vestling@molbiol.umu.se Zuoshang Xu Department of Biochemistry and Molecular Pharmacology University of Massachusetts Medical School Worcester, MA 01605 USA Zuoshang.xu@umassmed.edu Simona Zappettini Farmacologia Università degli studi di Genova Genova, Italia simonazappettini@libero.it Haining Zhu Department of Molecular and Cellular Biochemistry College of Medicine University of Kentucky Lexington, KY 40536 USA haining@uky.edu 101 Alphabetical list of contributors 102 A Adamson Aebischer Ajroud-Driss Akbarloo Al-Chalabi Amori Andersen Andres Andries Appel Arbogast Arciello Arroyo Aucello Augusti-Tocco Augusto Averill P31 S27 P25 SO9 SO4 SO2 S9, S13, P18 P12 P40 P39 S5, P19 P4 P12 S24 P10 P5, P29 P11, P17 B Babetto Banci Barbeito Barbiero Barbosa Basso Battistini Bea Beckman Beers Behl Beirowski Bendotti Bentivoglio Bergemalm Bogaert Bolzoni Bonanno Bonetto Bordoni Brännström Bresolin Bros Bruijn Butler P7, P30 S4 S21 SO7 P29 P33 S15 P12 S5, P19 P39 SO1 P30 S19, S20, P8, P13, P15, P20, P32, P33, P36 S22, SO8 S9 P34 S19, P6, P8 SO7, P35, P41 S12, P33 P24 S9, P18 P24 P31 S31 SO4 C Caliendo Callewaert Calvo Calza Cammarosano Cantoni Capitanio P21 P34 P12, P28 P13 P28 P7, P9 P13 Caplow Capo Capone Caponnetto Cappello Carra Carrì Carunchio Casazza Casciati Cashman Castellotti Cavallo Cereda Cerqueira Ceroni Chakrabartty Chen Cheroni Chia Chiò Clement Cleveland Coleman Colleoni Colombrita Comi Conforti Corato Corbo Corrado Corti Cova Cozzolino Crippa C. Crippa V. Crosio Crugnola Cupidi Curcio P1 P4 P27 P41 SO8 P6 S8, S18, SO2, P4, P5, P10, P29 P14 P41 S8, S18 SO3 P26 P28 P20 P5, P29 P20 SO3 P21 P15 P31 P28 SO1 SO3 P30 P36 P26 P24 P7 P20 P26 P23 P24 P20 S8, SO2, P4, P10 S19 P6, P8 S8, S18 P24 S22 P14 D Daleno D’Alessandro D’Alfonso D’Angelo Dantuma Darabi De Biasi De Bonis Demasi Deng Di Mascio Doblare Dobrowolny Dokholyan Donadoni P13, P20 P7, P9 P23 P27 P15 SO9 S19, P8 P27 P5 P21 P29 P12 S24 P1 P24 103 Doyu Dupuis S7 S26 E Ezzi P13, P26 S24 S8, SO2, P4 P31 P28 P31 SO8 SO8 P25 P21 SO5 P36 P20 P22 P27 P30 P13 P26 P28 S30 P31 S26 P25 S9, P18 S8 P12 S25, P31 SO3 P13 S10 H Hafezparast Hall Hasnain Hayward Henkel Herijgers Horne P31 P17 P2 SO5 P39 P34 SO3 I Iaccarino Ishigaki Ismaili P16, P17 P17 P18 S10, P3 K G Gagliardi Gal Galletti Garcia Alias Gelfi Gellera Ghiglione Glass Golding Gonzales De Aguilar Gorrie Graffmo Gralla Grasa Greensmith Griffin Grignaschi Gros-Louis Jokic Jones Jonsson Julien S10, P3 F Fallini Fanò Ferri Fisher Fimognari Flenniken Fossati Francolini Frutiger Fu Fukada Fumagalli J S18 S7 P11 Kassa Katsuno Keller Khare Kilty Koziollek-Drechsler Kriz S22, SO8 S7 P37 P1 P22 SO1 S11, P37 L Lamperti Landry Lauria Lee Levy Lidonnici Ling Liu Lococo Loeffler Lombardo Lopez Lucchini Lukas P24 P6 P26 SO5 P19 S20, P33 P16 P21 S20 S26 P28 P19 P24 P25 M Macedo Maiorano Malaspina Manzano Marino Mariotti Marklund Martin Massa Massari Mazzini Medeiros Medinas Melli Meininger Mennini Michael-Titus Milanese Moggio Molinaro Momigliano-Richiardi Morré Monsurrò Musarò Mutani P29 P27 P11, P16, P17 P12 P15 S22, SO8 S9, P18 P30 P14 P38 P23 P29 P5 P10 S26 P36 P11, P16, P17 SO7, P35, P41 P24 S24 P23 S5, P19 P27 S24 P28 104 N Negri Nencini Ngoh Nilsson Niwa Nylin S P23 S8, SO2 P17 P18 S7 S5, P19 O Oggioni Ogliari Oliveberg Onesto Osta Pinzalos Ottersen P23 P20 S3, S9 S19, P6, P8 P12 P9 P Padovano Paizs Panico Pasinelli Passariello Pesaresi Perlingeiro Peviani Piccirillo Pieri Pietrini Pisani Plumari Poletti Popoli Powell Priestley Protasi SO8, P38 P39 P14 S17 P26 SO2 SO9 P32 P27 P14 SO8, P38 P14 P26 S19, P6, P8 SO7 SO4 P16, P17 S24 R Raoul Raiteri L. Raiteri M. Rakhit Ratti René Restagno Righi Riso Rizzardini Robberecht Robertson Ronchi Rosenthal Rossant Rossi Rotilio Rusmini Ruth S27 SO7, P35 P35 SO3 P13, P26 S26 P28 P38 P8 P7, P9 S16, P34, P40 SO3 P24 S24 P31 P4 S1, S8, S18 S19, P6 SO3 Salani Salemme Salmona Salvatore Sandri Santoro Sau Sbaiz Schiavo Severini Shaw C. Shaw P. Siddique Siklos Silani Simonini Smith Sobue Sogayar Sone Spaltro Spano Stewart Strange Stroem P24 P27 P33 P27 S24 P24 S19, P6, P8 P28 P31 P14 S14 S6 P21, P25 P39 S29, P13, P26 S19, P6, P8 P2 S7 P29 S7 S20, P33 P32 S9 P2 SO5, P22 T Takahashi Tanaka Taroni Tartari Tedeschi Testa Ticozzi Tortarolo Towne Trojsi Trotti S7 S7 P26 P7, P9 P27 P23, P26 P26 S20, P33 S27 P27 S23 U Uccelli Ulbrich Urushitani P41 P10 S10, P3 V Valentine Van Damme Van Den Bosch Vande Velde Vasso Veglianese Vitellaro-Zuccarello Volta S2, S8 P34, P40 P34, P40 SO3 P13 S20, P9 S19, P8 P13 105 W Wade Ward Wilcox Witan Wroe SO1 P16, P17 P1 SO1 SO4 X Xu S28 Y Yamada Yan Yee Yong P21 P11 P2 Z Zappettini Zaragoza Zetterstroem Zhai Zhang Zhu Zona SO7, P35, P41 P12 S9 P21 SO5, P22 SO5, P22 P14 S7 106 This Meeting has been made possible by the generous contribution of our sponsors (in alphabetical order): Associazione Italiana per la Sclerosi Laterale Amiotrofica (AISLA) Bancaintesa San Paolo Bio-Rad Fondazione Cariplo GE Healthcare Gilson Harlan International Brain Research Organization (IBRO) International Copper Association (ICA) International Society for Neurochemistry (ISN) Italian Society for Biochemistry (SIB) M-MEDICAL Motor Neuron Disease Association (MND Association) Olympus Space Import-Export s.r.l. We thank for their patronage: EALSC (European ALS Consortium) SIN (Società Italiana di Neurologia) SINS (Società Italiana di Neuroscienze) The abstract book is a generous gift of Punzo&Colombo Canon Business Center, Bergamo 107