The work carried out during my research tenure has been compiled
advertisement

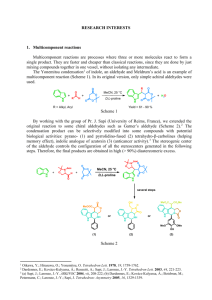
SYNOPSIS The work carried in my research tenure has been compiled in the form of a thesis entitled “Synthesis c][1,4]benzodiazepine of Hybrids DNA-interactive and C2/C8-Linked Combinatorial Pyrrolo[2,1- Synthesis of its Dimers/Hybrids and Bioactive Fused [2,1-b]quinazolinones” The main aim of this work is to design and synthesize biologically active molecules like pyrrolobenzodiazepines, which are known for their DNA-binding ability and potent anticancer activity. The biologically active pyrrolobenzodiazepine dimers/hybrids and bioactive fused [2,1-b]quinazolinones namely vasicinone and deoxyvasicinone have been synthesized by solid-phase combinatorial methods. The thesis has been divided into four chapters. CHAPTER I: This chapter gives the general introduction about cancer chemotherapy, covalent interactions of drug-DNA, particularly of pyrrolo[2,1c][1,4]benzodiazepine (PBD) antitumour antibiotics, combinatorial chemistry, the aim and objectives of the present work. CHAPTER II: This chapter has been divided into two sections. SECTION-A: Consists of the synthesis and DNA-binding affinity of novel 6chloropurine linked pyrrolo[2,1-c][1,4]benzodiazepine hybrids and their antitumour activity against eight human tumour cell lines. SECTION-B: Consists of the design, synthesis and DNA-binding affinity of novel C2/C8-linked 1,2,3-triazene-pyrrolo[2,1-c][1,4]benzodiazepine conjugates. CHAPTER III: This chapter describes the development of solid-phase synthetic strategies for pyrrolo[2,1-c][1,4]benzodiazepine dimers, its hybrids and this chapter has been divided in to three sections. SECTION-A: This section consists of the selective reduction of aromatic azides to amines in solution as well as on solid-phase and resin cleavage employing BF3. OEt2/EtSH. This reagent system has been utilized for the synthesis of pyrrolo[2,1-c][1,4]benzodiazepines and fused [2,1- b]quinazolinones. SECTION-B: This section describes the combinatorial synthesis of I SYNOPSIS C8-linked chalcone-pyrrolobenzodiazepine congugates and chalcone-napthalimide hybrids have been synthesized for the first time on solid-support, which are potential DNA-binding agents. SECTION-C: The biologically important DNAinteractive pyrrolo[2,1-c][1,4]benzodiazepine dimers (DSB-120) and their C2fluorinated analogues have been synthesized on solid-support. CHAPTER IV: This chapter deals with the synthesis of fused [2,1-b]quinazolinones namely vasicinone and deoxyvasicinone also for the first time on solid-phase. One of the methods is resin attached to amino functionality while the other is an aza-Wittig reductive cyclization approach. This methodology is amenable to the generation of combinatorial libraries with diversity in both A- and C-rings to afford a variety of fused [2,1-b]quinazolinones. CHAPTER-I INTRODUCTION This chapter describes the general introduction about pyrrolobenzodiazepines and combinatorial chemistry.. PYRROLO[2,1-c][1,4]BENZODIAZEPINES Cancer is a diseases characterized by uncontrolled growth or spread of abnormal cells. It involves the conversion of any normal cell to a cancerous cell showing tandem replication and cell division at much faster rate in comparison to the normal cells and thus provides a potential target area for the development of chemotherapeutic agents. It is now clear that chemotherapy’s most effective role in solid tumours is as an adjuvant to initial therapy by surgical or radiotherapeutic procedures. Chemotherapy becomes critical to effective treatment because only systemic therapy can attack micrometastases. These agents can be categorized into functional subgroups like alkylating agents, antimetabolites, antibiotics, and antimitotics. The pyrrolo[2,1-c][1,4]benzodiazepines (PBDs) belonging to the class of DNA-interactive antitumour antibiotics have the potential as regulators of gene II SYNOPSIS expression with possible therapeutic application in the treatment of genetic disorders including cancer. The first PBD antitumour antibiotic anthramycin has been described by Leimgruber et. al., in 1963, and since then a number of compounds have been developed on PBD ring system leading to DNA binding ligands. H3C 8 OH H 9 N OCH 3 H 11 10 7 N 5 6 N HO H 1 11a 2 4 3 O Anthramycin N H O N N H3CO CONH 2 O Tomaymycin O N N OCH 3 H3CO O SJG-136 H O Figure 1 Pyrrolo[2,1-c][1,4]benzodiazepines (PBDs) are a family of potent naturally occurring low molecular weight antitumour antibiotics originally isolated from various Streptomyces species. Their common interaction with DNA has been extensively investigated and it is considered unique since they bind within the minor groove of DNA forming a covalent aminal bond between the C11-position of the central B-ring and the N2 amino group of guanine base. A number of naturally occurring and synthetic compounds based on PBD ring system, such as anthramycin, tomaymycin, DC-81 and its dimers (presently, SJG-136 is under clinical evaluation), have shown varying degrees of DNA binding affinity and anticancer activity. O O N HN H2N N H N HN H N HH N N H N DNA N N O 11R/S aminal O N10-C11 imine Figure 2. PBD-DNA interaction III N N DNA SYNOPSIS COMBINATORIAL CHEMISTRY Combinatorial chemistry is a new methodology developed by researchers in the pharmaceutical industry to reduce the time and costs associated with producing effective and competitive new drugs. By accelerating the process of chemical synthesis, this method is having a profound effect on all branches of chemistry, especially on drug discovery. Through the rapidly evolving technology of combinatorial chemistry, it is now possible to produce libraries of small molecules to screen for novel bioactivities. This powerful new technology has begun to help pharmaceutical companies to find new drug candidates quickly, save significant money in preclinical development costs and ultimately change their fundamental approach to drug discovery. Combinatorial chemistry is used to synthesize large number of chemical compounds by combining sets of building blocks. Each newly synthesized compound’s composition is slightly different from the previous one. In this way the bench chemists can single handedly prepare many hundreds or thousands of compounds in the time usually taken to prepare only a few by routine methodologies. Over the last few years, the combinatorial chemistry has emerged as an exciting new paradigm for the drug discovery. In a very short time the topic has become the focus of considerable scientific interest and research efforts. CHAPTER-II (SECTION-A) DESIGN, SYNTHESIS OF NOVEL C2 AND C8-LINKED PURINE-PYRROLOBENZODIAZEPINE HYBRIDS AS ANTITUMOUR ANTIBIOTICS In the past few years design and synthesis of symmetrical cross-linking agents, particularly based on pyrrolobenzodiazepines (PBDs) has been of considerable interest. The PBDs are of current interest due to their ability to recognize and subsequently form covalent bonds to specific base sequences of double stranded DNA. These antitumour antibiotics bind covalently to the N2 of IV SYNOPSIS guanine at Pu-Gu-Pu sites in the minor groove of DNA. All cancers are characterized by an abnormal control of cell proliferation. This is caused by mutation or mis-regulation of cell-cycle regulatory genes and proteins. Cyclindependent kinases (CDKs) are a family of serine/threonine kinases that become active only when associated with regulatory partners called cyclins. The design and development of selective CDK inhibitors is however, a serious challenge for medicinal chemists for the target of anticancer agents. Further, chloro-purine nucleoside ring systems are reported to have shown a variety of biological activities particularly CDK inhibitors and selective adenosine receptors. The objective of the present work is to combine the feature of CDK inhibitor and adenosine receptor property in the purine nucleoside moiety. Therefore, it has been of considerable interest to couple this moiety to the C2 and C8-position of PBDs. In the present part of the chapter (Section-A) the design, synthesis of novel C2 and C8-linked purine-pyrrolobenzodiazepine hybrids as antitumour antibiotics has been described. SYNTHESIS OF C8-LINKED PURINE-PBD HYBRIDS Synthesis of C8-linked 6-chloropurine-PBD (11a,b) hybrids has been carried out by employing the commercially available vanillin. Oxidation of vanillin followed by benzylation and nitration by employing the literature methods provide 4-benzyloxy-5-methoxy-2-nitrobenzoic acid (1). This has been further coupled to Lproline methylester to afford the compound 2, which upon reduction with DIBAL-H produces the corresponding aldehyde (3). The aldehyde group of this product has been protected with EtSH/TMSCl to give 4. Compound 4 upon debenzylation affords (2S)-N-(4-hydroxy-5-methoxy-2-nitrobenzoyl)pyrrolidine-2-carboxaldehyde diethylthioacetal (5) which upon etherification by Boc-protected bromoalkanes followed by deprotection provides 7a,b. These compounds have been coupled with V SYNOPSIS 2-amino-1,3-dichloropyrimidine in the presence of Et3N to produce the corresponding 8a,b. The compounds 8a,b upon cyclization with CH(OEt)3 affordsScheme 1 BnO NO 2 NO 2 CHO BnO ii) OH H3CO NO 2 COOCH 3 BnO i) N H3CO O 1 N H3CO O O 2 3 iii) Boc H N NO 2 CH(SEt)2 O n v) N H3CO NO 2 CH(SEt)2 HO N H3CO O 6a; n = 2 6b; n = 3 vi) O 4 vii) N N H3CO N H3CO H N Cl NO 2 CH(SEt)2 O n NO 2 CH(SEt)2 BnO O 5 NH 2 H2N iv) N O n NO 2 CH(SEt)2 N H3CO O 8a,b; n = 2,3 O 7a,b; n = 2,3 viii) Cl Cl N N ix) 10a,b; n = 2,3 N O n NO 2 CH(SEt)2 H3CO N H3CO N N NH 2 CH(SEt)2 O n N N N N O 9a,b; n = 2,3 O x) Cl N N N N O n N H3CO H N 11a; n = 2 O 11b; n = 3 Reagents and conditions: (i) (a) SOCl2, C6H6, 1-3-drops DMF, rt, 2 h; (b) Et3N, L-proline methylester hydrochloride, THF-H2O, 0 oC-rt, 2 h, 85%; (ii) DIBAL-H, CH2Cl2, -78 oC, 1 h, 71%; (iii) EtSH/TMSCl, CH2Cl2, rt, 8 h, 72%; (iv) BF3. OEt2, EtSH, CH2Cl2, rt, 12 h, 75%; (v) K2CO3, DMF, Boc-protected aminobromoalkanes, rt, 48 h, 65-75%; (vi) TFA, CH2Cl2, rt, 6 h; (vii) 2-amino-1,3-dichloropyrimidine, Et3N, n-BuOH, reflux, 48 h, 58%; (viii) CH(OEt)3, HCl (10%), rt, 16 h, 62%; (ix) SnCl2. 2H2O, MeOH, reflux, 85-87%; (x) HgCl2-CaCO3, CH3CN-H2O (4:1), 68-71%. VI SYNOPSIS -9a,b. These purine-substituted nitrothioacetal intermediates 9a,b upon reduction with SnCl2. 2H2O in methanol gives the aminothioacetal precursors 10a,b and these on deprotection by HgCl2/CaCO3 affords the desired purine-PBD hybrids (11a,b) as shown in Scheme 1. SYNTHESIS OF C2-LINKED PURINE-PBD HYBRIDS Synthesis of C2-linked 6-chloropurine-PBD hybrid (22a) has been carried out by employing the commercially available 4,5-dimethoxy-2-nitrobenzoic acid (12a). This has been further coupled to 4-hydroxy L-proline methylester to afford the compound 13a. Mesylation of C2-hydroxy group followed by azidation (bimolecular nucleophilic substitution reaction SN2) with NaN3 produces 15a. This upon reduction with DIBAL-H produces the corresponding aldehyde 16a. The aldehyde group of this product has been protected with EtSH/TMSCl to give 17a. The compound 17a has been reduced with TPP followed by coupling with 2-amino-1,3dichloropyrimidine in the presence of Et3N produces the corresponding 19a. This upon cyclization with CH(OEt)3 affords the cyclized product 20a. Further, the purine-substituted nitrothioacetal intermediate 20a upon reduction with SnCl2. 2H2O in methanol gives the aminothioacetal precursors 21a and this on deprotection by HgCl2/CaCO3 affords the desired purine-PBD hybrid (22a). Compound 22b has also been obtained in the same manner by employing commercially available vanillin. Oxidation of vanillin followed by benzylation and nitration by employing the literature methods provides 4-benzyloxy-5-methoxy-2-nitrobenzoic acid (12b) as shown in Scheme 2. The DNA binding ability of these compounds have also been investigated by thermal denaturation studies using calf thymus (CT) DNA at pH 7.0, incubated at 37 °C. It is observed that compound 11a,b elevates the helix melting temperature (∆Tm) of the CT-DNA to 2.1 and 2.2 °C after incubation for 18 h while compounds 22a and 22b have not exhibited any significant ∆Tm value. In the same experiment the naturally occurring DC-81 exhibits a ∆Tm of 0.7 °C. VII SYNOPSIS Scheme 2 R1 NO 2 R1 i) OH R2 NO 2 COOCH 3 R2 N O 1 N R1 v) O S O CH 3 O N O 14a,b NO 2 CHO R2 N3 O 17a,b NO 2 COOCH 3 iii) NO 2 CH(SEt)2 R2 R1 R2 OH O 13a,b 2 12a; R , R = OCH3 12b; R1 = OBn, R2 = OCH3 R1 ii) N iv) N3 O 16a,b R1 NO 2 COOCH 3 R2 N N3 O 15a,b vi) R1 NO 2 CH(SEt)2 R2 N O 18a,b R1 vii) NO 2 CH(SEt)2 R2 NH 2 N Cl viii) R1 NH 2 CH(SEt)2 R2 ix) Cl N N N O 21a,b R1 N Cl N N O 20a,b N N NO 2 CH(SEt)2 R2 N NH 2 NH O 19a,b N N N x) R1 N H Cl N R2 N N O 1 2 N N 22a; R , R = OCH3 22b; R1 = OBn, R2 = OCH3 Reagents and conditions: (i) SOCl2, C6H6, 4-hydroxy L-proline methylester hydrochloride, THF-H2O, oC-rt, 2 h, 85%; (ii) mesyl chloride, Et3N, CH2Cl2, rt, 6 h, 85%; (iii) NaN3, DMF, 50-60 o C, 6 h; 80%; (iv) DIBAL-H, CH2Cl2, -78 oC, 1 h, 71%; (v) EtSH/TMSCl, CH2Cl2, rt, 8 h, 72%; (vi) TPP, THF-H2O, rt, 24 h, 75%; (vii) 2-amino-1,3-dichloropyrimidine, Et3N, n-BuOH, reflux, 48 h, 58%; (viii) CH(OEt)3, HCl (10%), rt,16 h, 62%; (ix) SnCl2, 2H2O, MeOH, reflux, 6 h, 85-87%; (x) HgCl2-CaCO3, CH3CN-H2O (4:1), 12 h, 68-72%. The C8-linked purine-PBD hybrids (11a,b) have been evaluated against eight human tumour cell lines derived from eight cancer types that are MCF7 (breast VIII SYNOPSIS cancer), Hop62 (non-small cell lung cancer), KB, Colo205, PC3 (prostate cancer), Gurav, Zr-75-1 and A549. These compounds exhibited in vitro anticancer activity in selected human cancer cell lines. CHAPTER-II (SECTION-B) SYNTHESIS OF NOVEL C2/C8-LINKED 1,2,3-TRIAZENE SUBSTITUTED PYRROLO[2,1C][1,4]BENZODIAZEPINES AS ANTICANCER AGENTS The development of hybrid molecules comprising of two types of cytotoxic moieties represent a new approach in the discovery of new antitumour agents. DNA is the major cause of cytostatic activity of antitumour agents such as the methyltriazenes, temozolomide and dacarbazine in guanosine residues of Omethylation. In light of the high mortality rates associated with these cancers, the role of cytotoxic chemotherapeutic drugs continues to be investigated. One such drug which in recent years has emerged as arguably the most effective anticancer agent for the treatment of malignant brain methylimidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one tumours is (temozolomide). 8-carbamoyl-3Several DNA alkylating and cross-linking agents are currently in clinical trials in combination with temozolomide. Further, in literature a number of anticancer agents have been developed in which triazene-alkylating agents have played a key role in their biological activity. The objective of the present work is to combine the feature of DNA alkylating and cross-linking properties in the 1,2,3-triazene molecules. Therefore, it has been considered of interest to couple this moiety at C2, C8 and C2/C8-position of the PBD ring system. In the present part of the chapter (Section-B), the synthesis of novel C2/C8-linked 1,2,3-triazene substituted pyrrolo[2,1-c][1,4]benzodiazepines potential anticancer agents have been described. IX as SYNOPSIS SYNTHESIS OF C8-LINKED TRIAZENE-PBD HYBRIDS Synthesis of C8-linked triazene-PBD hybrids (27a-c) has been carried out by employing the commercially available vanillin. Oxidation of vanillin followed by benzylation and nitration by employing the literature method provides 4-benzyloxy5-methoxy-2-nitrobenzoic acid. The (2S)-N-(4-hydroxy-5-methoxy-2- nitrobenzoyl)pyrrolidine-2-carboxaldehyde diethylthioacetal (5) has been carried out by the same procedure as mentioned in Section-A (Scheme 1). Compound 5 upon etherification with dibromoalkanes followed by azidation with NaN3 provides 24a-c. These compounds have been cyclized with dimethyl acetylene dicarboxylate to produce the corresponding 25a-c that is called a “click reaction” as the biologically diverse molecules are obtained in one step. These triazene ester substituted nitrothioacetal intermediates 25a-c upon reduction with SnCl2. 2H2O in methanol gave the aminothioacetal precursors 26a-c which upon deprotection by HgCl2/CaCO3 afford the desired C8-linked triazene-PBD hybrids (27a-c) as shown in Scheme 3. X SYNOPSIS Scheme 3 NO 2 CH(SEt)2 HO Br i) N H3CO NO 2 CH(SEt)2 O n N H3CO O O 23a; n = 3 ii) 23b; n = 4 23c; n = 5 5 H3CO 2C N N N H3CO 2C n NO 2 CH(SEt)2 O N3 iii) N H3CO NO 2 CH(SEt)2 O n N H3CO O O 24a-c; n = 3-5 25a-c; n = 3-5 iv) H3CO 2C N N N H3CO 2C O n NH 2 CH(SEt)2 v) N N N H3CO 2C N H3CO H3CO 2C O 26a-c; n = 3-5 n O H3CO 27a; n = 3 27b; n = 4 27c; n = 5 N H N O Reagents and conditions: (i) dibromoalkane specers, K2CO3, dry DMF, 12 h, rt, 78-82%; (ii) NaN3 (0.5M) in DMSO, 80 oC, 6 h, 85-88%; (iii) dimethyl acetylenedicarboxylate, dry benzene, reflux, 6 h, 87-92%; (iv) SnCl2.2H2O, MeOH, reflux, 5 h, 75-83%; (v) HgCl2-CaCO3, CH3CN-H2O (4:1), rt, 6 h, 58-62%. SYNTHESIS OF C2-LINKED TRIAZENE-PBD HYBRIDS (2S,4S)-N-[2-nitrobenzoyl]-4-azidopyrrolidine-2-carboxaldehyde diethylthio-acetal (9a-c) has been prepared by employing the same procedure described in earlier Section-A (Scheme 2). These compounds (28a-c) have been cyclized with dimethyl acetylene dicarboxylate to provide the corresponding 29a-c. The triazene ester substituted nitrothioacetal intermediates 29a-c by reduction with SnCl2. 2H2O in methanol gave the aminothioacetal precursors 30a-c and these upon deprotection by HgCl2/CaCO3 afford the desired C2-linked triazene-PBD hybrids (31a-c) (Scheme 4). XI SYNOPSIS Scheme 4 R1 NO 2 CH(SEt)2 i) R2 N N R2 H N O H3CO 2C N N N N N CO 2CH 3 H3CO 2C ii) iii) N N 29a-c O 28a; R1 = R2 = H 28b; R1 = R2 = OCH3 28c; R1 = OBn, R2 = OCH3 R1 NO 2 CH(SEt)2 R2 N3 O R1 R1 NH 2 CH(SEt)2 R2 N O 30a-c CO 2CH 3 N N H3CO 2C N CO 2CH 3 31a; R1 = R2 = H 31b; R1 = R2 = OCH3 31c; R1 = OBn, R2 = OCH3 Reagents and conditions: (i) dimethylacetylene dicarboxylate, dry benzene, reflux, 6 h, 88-90%; (ii) SnCl2.2H2O, MeOH, reflux, 5 h, 80-85%; (iii) HgCl2-CaCO3, CH3CN-H2O (4:1), rt, 6 h, 58-61%. SYNTHESIS OF C2/C8-LINKED TRIAZENE-PBD HYBRIDS Compound 29 has been prepared by employing the same procedure described in earlier Scheme 4 (29c). This compound 29 upon debenzylation affords the corresponding debenzylated product 32. This upon etherification with dibromoalkanes followed by azidation with NaN3 provides the precursors 34a-c. These compounds have been cyclized with dimethyl acetylene dicarboxylate to produce the corresponding 35a-c. This triazene ester substituted nitrothioacetal intermediates 35a-c by reduction with SnCl2. 2H2O in methanol gave the aminothioacetal precursors 36a-c and these upon deprotection by HgCl2/CaCO3 afford the desired C2/C8-linked triazene-PBD hybrids (37a-c) as shown in Scheme 5. XII SYNOPSIS Scheme 5 BnO NO 2 CH(SEt)2 N N H3CO N H3CO N N NO 2 CH(SEt)2 HO i) O N N N O CO 2CH 3 H3CO 2C 29 32 CO 2CH 3 H3CO 2C ii) N3 O n Br NO 2 CH(SEt)2 N N H3CO N NO 2 CH(SEt)2 O n iii) N H3CO N N O O CO 2CH 3 H3CO 2C N N 33a; n = 3 33b; n = 4 33c; n = 5 34a-c; n = 3-5 H3CO 2C CO 2CH 3 iv) H3CO 2C N N N H3CO 2C NO 2 CH(SEt)2 O n H3CO 2C NH 2 CH(SEt)2 O n H3CO 2C O n H3CO N N O CO 2CH 3 H3CO 2C vi) N N N N N H3CO 36a-c; n = 3-5 H3CO 2C N CO 2CH 3 H3CO 2C v) H3CO 2C N O 35a-c; n = 3-5 N N N N N H3CO N H N N 37a; n = 3 O 37b; n = 4 H3CO 2C 37c; n = 5 N N CO 2CH 3 Reagents and conditions: (i) BF3. OEt2, EtSH, CH2Cl2, rt, 12 h, 75%; (ii) dibromoalkane spacers, K2CO3, dry DMF, 12 h, rt, 78-80%; (ii) NaN3 (0.5M) in DMSO, 80 oC, 6 h, 89-92%; (iii) dimethyl acetylenedicarboxylate, dry benzene, reflux, 6 h, 88-90%; (iv) SnCl2.2H2O, MeOH, reflux, 5 h, 78-85%; (v) HgCl2-CaCO3, CH3CN-H2O (4:1), rt, 6 h, 58-62%. XIII SYNOPSIS The DNA binding ability of these compounds has also been investigated by thermal denaturation studies using calf thymus (CT) DNA at pH 7.0, incubated at 37 °C. It is observed that compound 27a,c; 31a-c and 37a,c elevates the helix melting temperature (∆Tm) of the CT-DNA to 2.0, 1.0, 1.0; 1.0, 1.1, 2.0 and 0.9, 5.9 °C incubation of 0 oC and after 18 h while compounds 27b and 37b have not exhibited any significant ∆Tm value. In the same experiment the naturally occurring DC-81 exhibits a ∆Tm of 0.7 °C. CHAPTER-III SYNTHESIS OF PYRROLOBENZODIAZEPINES, ITS DIMERS AND HYBRIDS ON SOLID-PHASE The pyrrolo[2,1-c][1,4]benzodiazepines (PBDs) are a group of potent, naturally occurring, antitumour antibiotics produced by various Streptomyces species. A number of naturally occurring and synthetic compounds based on this PBD ring system, such as anthramycin, chicamycin, abbeymycin, DC-81 and its dimers (DSB-120) have shown varying degrees of DNA binding affinity and anticancer activity. Moreover, pyrrolo[2,1-c][1,4]benzodiazepine-5,11-diones (PBD5,11-diones) are known as non-covalent interactive minor groove binders. These are also intermediates for the synthesis of structurally modified PBD-imines via oxidation of secondary amines. There are many methods known for the solution phase synthesis of PBD imines. However, there are only few reports on the solidphase synthesis of these PBD antitumour antibiotics and this chapter has been divided into three sections. CHAPTER-III (SECTION-A) SELECTIVE REDUCTION OF AROMATIC AZIDES IN SOLUTION/SOLID-PHASE AND RESIN CLEAVAGE EMPLOYING BF3. OEt2/EtSH: PREPARATION OF DC-81 The selective reduction of aromatic azido functionalities to the corresponding amines is an important transformation in synthetic organic chemistry, used to construct a variety of biologically active molecules, especially in heterocyclic and XIV SYNOPSIS medicinal chemistry. In recent literature, the reduction of aromatic nitro/azido functionalities is readily accomplished with a wide variety of reagents. Many of these methods, however, require heavy metal catalysts, acid conditions, high temperatures or pressure, which renders most of them not suitable for application to synthetic organic chemistry. In recent years, Lewis acid based reagents have found wide applications in synthetic chemistry because of their ready availability, easy handling and low costs. In the present work in search of solution-phase and solid-phase compatible method a process employing BF3. OEt2/EtSH reagent system has been developed for the reduction of aromatic azides (1a-g) to the amines (2a-g) as illustrated in Scheme 1. Scheme 1 N3 R BF3. OEt2/EtSH CH2Cl2, r,t. 1.5-2.5 h. 1a-g NH 2 R 2a-g PREPARATION OF PYRROLO[2,1-c][1,4]BENZODIAZEPINE-5,11-DIONES AND FUSED [2,1b]QUINAZOLINONES BY REDUCTIVE CYCLIZATION APPROACH EMPLOYING BF3. OEt2/EtSH This methodology has also been applied to the synthesis of pyrrolo[2,1c][1,4]benzodiazepin-5,11-diones as intermediates in the synthesis of naturally occurring and synthetically modified PBD imines such as tomaymycin and chicamycin. With a view to develop new efficient solution- and solid-phase procedures, BF3. OEt2/EtSH reagent system has been employed for the azido reductive cyclization process. The substrates 3a-f have been taken in CH2Cl2, BF3. OEt2 and EtSH is added to provide the PBD-5,11-diones (4a-f) Similarly, it has been considered of interest to synthesize fused [2,1-b]quinazolinones (e.g., deoxyvasacinone) by using this reagent system for the azido reductive cyclization protocol for the substrates 3g-i to afford the fused [2,1-b]quinazolinones (4g-i) as shown in Scheme 2. XV SYNOPSIS Scheme 2 R N3 COOCH 3 N O 3a-f R N3 O N R1 BF3. OEt2/EtSH R H N CH2Cl2, rt, 1.5-2.5 h. O H N O 4a-f BF3. OEt2/EtSH R R1 N N CH2Cl2, rt, 1.5-2.5 h. O O 3g-i 4g-i PREPARATION OF NATURALLY OCCURRING DC-81 This approach has been further applied to the preparation of the naturally occurring PBD antibiotic DC-81 (9). This compound exerts its biological activity by covalently binding to the N2 of guanine in the minor groove of DNA. Various approaches towards compounds of this type have been investigated over the past few years; most of these methods have met with varying degrees of success and have different limitations. Herein, we report an efficient and simple method for the synthesis of naturally occurring DC-81 by employing BF3. OEt2/EtSH. 4-Benzyloxy-5-methoxy-2-azidobenzoic acid (5) has been converted to acid chloride by using oxalyl chloride. This azidobenzoyl chloride has been coupled with L-proline methylester in presence of Et3N to give the coupled azido benzoyl proline methylester (6). This upon reduction with DIBAL-H gave the aldehyde 7. The selective reduction of the azido group of 7 with varying amounts of BF3. OEt2/EtSH (10:20 eq or 2.5:5 eq) to give the ethanethiol protected intermediates 8 and 10. Then deprotection with HgCl2-CaCO3 in CH3CN:H2O (4:1) produces the naturally occurring DC-81 (9) and benzylated DC-81 (11) as shown in Scheme 3. XVI SYNOPSIS Scheme 3 N3 BnO OH H3CO O iii) O BnO 7 NH 2 CH(SEt)2 v) N H3CO N3 CHO HO O 7 iv) N H3CO O 10 H N O DC-81 9 NH 2 CH(SEt)2 BnO N H3CO O 8 N H3CO N H3CO 6 HO N3 CHO BnO ii) N H3CO O 5 N3 COOCH 3 BnO i) v) BnO H3CO N H N O Benzylated DC-81 11 Reagents and Conditions: (i) (a) (COCl)2 , CH2Cl2, O oC-rt, 2 h; (b) Et3N, L-Proline methylester hydrochloride, THF, O oC-rt, 6 h; ii) DIBAL-H, CH2Cl2, -78 oC, 1 h; (iii) BF3. OEt2/EtSH (10:20 eq), CH2Cl2, rt, 6 h; (iv) BF3. OEt2/EtSH (2.5:5 eq), CH2Cl2, rt, 6 h; (v) HgCl2-CaCO3, CH3CN:H2O (4:1), rt, 12 h. Interestingly, when this procedure has been used for the reduction of resinlinked aryl azides to the corresponding aminophenols have been obtained. There are some reports in the literature on the Lewis acid assisted cleavage of resins have been employed, however; to the best our knowledge this is the first report on cleavage of a resin employing BF3. OEt2/EtSH. SOLID-PHASE SYNTHESIS REDUCTIVE CYCLIZATION OF PBD-5,11-DIONES EMPLOYING BF3. OEt2/EtSH BY During the course of our studies in this laboratory on the development of solid-phase synthetic methods for the PBD ring system, a number of procedures mainly based on reductive-cyclization approaches have been studied. In this connection, the boron trifluoride diethyletherate reagent system has been reacted with resin-linked aryl azido proline methylesters (16a-e) to afford PBD-5,11-diones (17a-e) by the simultaneous reduction of the azido functionality and cleavage of the resin as shown in Scheme 4. This method is advantageous as the reductive XVII SYNOPSIS cyclization and the resin cleavage takes place in the same step, unlike in the previous procedures. Scheme 4 N3 N3 COOCH 3 i) R R N COOH OH O 13a-c 12a-c NH ii) OH O Wang resin 15 14 N3 COOCH 3 H N N3 COOCH 3 15 iii) R CCl 3 iv) R N R N OH O 13a-c O O 17a-c N3 COOCH 3 iv) N H3CO O 16d,e N O O 16a-c O H HO H3CO R H N OH O H N R O 17d,e R = H, OH Reagents and conditions: (i) (a) (COCl)2, CH2Cl2, 0 oC-rt, 2 h; (b) Et3N, hydroxy-L-proline methylester hydrochloride,THF, 0 oC-rt, 1 h; (ii) trichloroacetonitrile, DBU, CH2Cl2, 40 min, 0 oC; (ii) trifluoromethane sulphonic acid, CH2Cl2, 40 min; (iii) CH2Cl2:BF3. OEt2:EtSH (2:1:0.25), 2 h. The DBU has been added to a suspension of Wang resin (14) and trichloroacetonitrile to give TCA resin (15), which has been coupled with azidoesters (13a-c) in presence of trifluoromethane sulphonic acid to provide the precursors 16ac. The compounds 13a-c has been prepared from substituted 2-azidobenzoic acids through their acid chlorides on coupling with hydroxy-L-proline methylester hydrochloride. Similarly, 16d,e has been prepared by the coupling of bromomethyl Wang resin and hydroxy azidobenzoyl proline methylester in presence of Cs2CO3NaI. Reductive cyclization and resin cleavage of 16a-e employing BF3. OEt2/EtSH in CH2Cl2 to gave PBD-5,11-diones (17a-e) as illustrated in Scheme 4 (Tetrahedron Lett. 2006, 47, 4253-4257). XVIII SYNOPSIS CHAPTER-III (SECTION-B) SOLID-PHASE SYNTHESIS AND DNA BINDING AFFINITY OF C8-LINKED CHALCONEPYRROLOBENZODIAZEPINE CONJUGATES AND CHALCONE-NAPTHALIMIDE HYBRIDS Chalcones are a class of anticancer agents that have shown promising therapeutic efficacy for the management of human cancers. Chalcones, considered as the precursor of flavonoids and isoflavonoids, are abundant in edible plants. Chemically they consist of open-chain flavonoids in which the two aromatic rings are joined by a three-carbon α,β-unsaturated carbonyl system. The objective of the present work is to combine the feature of anticancer property in the chalcone molecule. Therefore, it has been considered of interest to couple to C8-position of PBD through alkyl spacer. In the present part of chapter (Section-B), the solid-phase synthesis, DNA binding affinity of C8-linked chalcone-pyrrolobenzodiazepine conjugates and chalcone-napthalimide hybrids have been described. SOLID-PHASE SYNTHESIS OF A CHALCONE LIBRARY 2-Hydroxyacetophenone (18) has been coupled to commercially available Merrifield resin (19, 2.00 mmol/g) with Cs2CO3 and NaI to give the resin-bound 2hydroxyacetophenone (20). This reaction has been confirmed by IR spectrum, which showed the keto group absorption at 1739 cm-1. The resin-bound 2- hydroxyacetophenone (20) has been condensed with vanillin (21) employing NaOMe (as a 0.5M solution in MeOH) to give the resin-bound enone product 22. This reaction has also been monitored by FT-IR, which showed a strong peak of the carbonyl absorption at 1747 cm-1 (Scheme 5). Scheme 5 OH Cl OH CH 3 18 O OHC O 19 i) 21 CH 3 OCH 3 O OH ii) OCH 3 O O 20 22 Reagents and conditions: (i) Cs2CO3, NaI, DMF, rt, 24 h; (ii) NaOMe/THF/MeOH, rt, 4 days. XIX SYNOPSIS SOLID-PHASE SYNTHESIS OF A C8-LINKED CHALCONE-PBD LIBRARY The derivatized resin (22) has been coupled with bromo-substituted azidobenzoyl proline methyl esters (23a-c) in presence of K2CO3 to afford the resin 24a-c as indicated by IR spectra that shows strong azide stretching vibrations between 2106 and 2110 cm-1. After that the derivatized resins (24a-c) have been reduced selectively with DIBAL-H to give the corresponding resin-bound azido aldehyde 25a-c. These azido aldehyde chalcone-resins (25a-c) have been reduced and cyclized via Staudinger reaction by employing TPP to produce 26a-c. This step has been confirmed by IR analysis (the azido peak disappeared). Finally, the resins 26a-c have been cleaved by employing TFA/CH2Cl2 (1:1) to afford the products 27ac. The resin-bound chalcone PBD dilactams (28a-c) have been obtained by the reduction of 24a-c employing TPP and followed by cleavage with TFA to afford 29ac (62-73%) as shown in Scheme 6. The DNA binding ability of these compounds has also been investigated by thermal denaturation studies using calf thymus (CT) DNA at pH 7.0, incubated at 37 °C. It is observed that compounds 27a-c elevates the helix melting temperature of the CT-DNA to 7.9, 2.1 and 5.0 °C after incubation of 18 h. In the same experiment the naturally occurring DC-81 exhibits a ∆Tm of 0.7 °C. XX SYNOPSIS Scheme 6 O OH Br + N O 23a-c; n = 1-3 O 22 i) O n N3 O 24a-c; n = 1-3 COOCH 3 N OCH 3 H3CO O N3 COOCH 3 O H3CO OCH 3 O n O ii) iii) O O n O N3 N OCH 3 H3CO O O O n O O O 25a-c; n = 1-3 O H H N N OCH 3 H3CO iii) O 28a-c; n = 1-3 O O iv) n O N O n O 26a-c; n = 1-3 H N OCH 3 H3CO O OH CHO O O H H N iv) N OCH 3 H3CO O 29a-c; n = 1-3 O OH O n O N N OCH 3 H3CO O 27a-c; n = 1-3 H O Reagents and conditions: (i) K2CO3/DMF, 50 oC, 48 h; (ii) DIBAL-H, CH2Cl2, -78 oC, 2 h; (iii) TPP/toluene, rt, 16 h; (iv) TFA/CH2Cl2 (1:1), rt, 2 h; XXI SYNOPSIS SOLID-PHASE SYNTHESIS OF A CHALCONE-NAPTHALIMIDE LIBRARY The resin-coupled chalcone-napthalimides (32a-c) have been prepared by a similar procedure as shown in Scheme 6. Further, the resins have been cleaved with TFA/CH2Cl2 (1:1) to afford the compounds 32a-c as shown in Scheme 7. Scheme 7 O Br O OH n O O 30a-c; n = 1-3 OCH 3 O N O O N n O OCH 3 i) 22 O 31a-c; n = 1-3 ii) O OH O n N O OCH 3 O 32a-c; n = 1-3 Reagents and conditions: (i) K2CO3/DMF, 50 oC, 48 h; (ii) TFA/CH2Cl2 (1:1), rt, 2 h. CHAPTER-III (SECTION-C) SOLID-PHASE SYNTHESIS OF BIOLOGICALLY IMPORTANT DNA-INTERACTIVE PYRROLO[2,1-c][1,4]BENZODIAZEPINE DIMERS (DSB-120) AND THEIR C2-FLUORINATED ANALOGUES As part of our continuing effort to develop new heterocyclic solid-phase strategies for synthesizing nitrogen-rich heterocyclic compounds based on pyrrolobenzodiazepines, we required libraries of the dimers of pyrrolobenzodiazepines for lead generation against certain disease targets, particularly cancer. In continuation of earlier efforts in this laboratory on the structural modifications of PBDs and their dimers we were interest to develop solidphase methodologies, particularly for the PBD dimers. The solid-phase synthesis of PBD dimers such as DSB-120 and its C2-fluoro substituted analogues has been carried out for the first time. It has been observed that in most solution-phase XXII SYNOPSIS procedures for the preparation of PBD dimers there are problems relating to solubility. Therefore, the development of solid-phase protocols for PBD dimers is of immense importance and this could address the solubility difficulties encountered during work-up. The 4-hydroxy-3-methoxybenzaldehyde (vanillin) (21) has been oxidized with sulphamic acid and sodium chlorite in ice-cold water to give 33. Further, esterification followed by dimerization with dibromo alkanes in presence of K 2CO3 gave 35a-c. These 35a-c upon nitration with freshly prepared mixture of SnCl4/HNO3, followed by subsequent reduction with SnCl2. 2H2O produce the starting materials (37a-c) as shown in Scheme 8. Scheme 8 HO HO i) H3CO OH H3CO CHO 21 33 HO ii) OCH 3 H3CO O 34 O iii) O2N H3CO O n O NO 2 OCH 3 OCH 3 H3CO O 36a-c; n = 1-3 O iv) H3CO O n O OCH 3 OCH 3 H3CO O 35a-c; n = 1-3 O v) H2N H3CO O n O NH 2 OCH 3 OCH 3 H3CO O 37a-c; n = 1-3 O Reagents and conditions: i) NaClO2, sulphamic acid, H2O, 1 h, rt; ii) MeOH, H2SO4, reflux, 2 h; iii) dibromo alkanes, K2CO3, DMF, 12 h, rt; iv) SnCl4/HNO3, CH2Cl2, -35 oC, 10 min; v) SnCl2. 2H2O, MeOH, reflux, 5 h. The starting materials (37a-c) have been treated with p-nitrophenyl carbonate Wang resin (38) using 1-hydroxybenzotriazole (HOBt) and diisopropylethylamine (DIPEA) in CH2Cl2:DMF (2:1) to give 39a-c. Hydrolysis of the methyl esters (39a-c) afforded the corresponding acids in presence of 1N NaOH to give 40a-c. These have XXIII SYNOPSIS O Scheme 9 37a-c; n = 1-3 O O 38 i NO 2 O O HN O H3CO O O n O NH OCH 3 OCH 3 H3CO O O 39a-c; n = 1-3 O ii O HN O HO O O O n NH OH OCH 3 H3CO O O HO 40a-c; n = 1-3 iii 41 R = H NH 42 R = F R O HO O HN O N R n N OCH 3 H3CO N R iv O O N R O 43a-f; n = 1-3 O OH NH O O HO H O O O n O O N OCH 3 H3CO O OH H N R O 44a-f; n = 1-3 v N H N R O n O N N OCH 3 H3CO O 45a-c, R = H 45d-f, R = F H O R Reactions and conditions: i) HOBt, DIPEA, CH2Cl2:DMF (2:1) 6 h, rt; ii) 1N NaOH, 1,4-dioxane, 80 o C,12 h; iii) EDCI, HOBt,15-24 h, rt; iv) DMSO, (COCl)2, Et3N, CH2Cl2, -78 oC, 2 h; v) TFA/CH2Cl2 (1:1), 2 h, rt. XXIV SYNOPSIS -been coupled with 2-pyrrolidinemethanol (41) in the presence of EDCI and HOBt to provide 43a-c. Similarly, 4-fluoro-2-pyrrolidinemethanol (42) has also been coupled same as mentioned above procedure to give 43d-f. Swern oxidation of 43a-f then gave 44a-f via an oxidative cyclization process in presence of COCl2 and Et3N. Finally, the resins (44a-f) have been cleaved by using TFA/CH2Cl2 (1:1) to afford the target products 45a-f via loss of the hydroxyl groups as shown in Scheme 9 (Tetrahedron Lett. 2006, 47, 6553-6556). CHAPTER-IV SOLID-PHASE COMBINATORIAL SYNTHESIS OF ALKALOIDS (VASICINONE AND DEOXYVASICINONE) FUSED [2,1-b]QUINAZOLINONE Combinatorial chemistry has become an extremely powerful technique for the generation of drug-like small organic molecule libraries in medicinal chemistry programmes. Solid-phase organic synthesis (SPOS) is especially useful in creating large numbers of hit and lead compounds in combinatorial libraries for use in highthroughput screening. As a result, an increasing range and number of pharmaceutically useful heterocyclic compounds have been prepared. In this investigation, we have developed new solid-phase strategies for synthesizing nitrogen-containing heterocyclic compounds based on fused [2,1-b]quinazolinones, employing Wang resin and polymer-supported (PS) 4-nitrophenyl carbonate linkers. Vasicinone is a fused [2,1-b]quinazolinone alkaloid isolated from the aerial parts of Adathoda vasica Nees (family: Acanthacea; Sanskrit-Vasaka), which is an evergreen subherbaceous bush used extensively in indigenous medicine for treatment of colds, coughs, bronchitis and asthma. The leaves of this plant are rich in essential oil and quinazoline alkaloids viz,. (–)-vasicinone, (–)-vasicine, vasicinolone, deoxyvasicinone, vasicoline, adathaodine, luotonin A and anisotine. Vasicinone and related alkaloids have also been reported to be present in other plants namely, Peganum harmala and Sida cordifolia. Fitzgerald et al., isolated mackinazolinone and XXV SYNOPSIS 6,7,8,9-tetrahydro-11H-pyrido[2,1-b]quinazoline, analogues of vasicinone and vasicine analogues respectively from Mackinlaya macrosciadia and Mackinlaya subulata of the family Araliaceae. The present work describes the solid-phase combinatorial synthesis of fused [2,1-b]quinazolinones including vasicinone for the first time on solid-phase, starting from different anthranilic acids, substituted 2-azidobenzoic acid and coupling with various lactams. The lactams have been synthesized starting from γ-butyrolactone (1a) and δvelarolactone (1b). The synthesis involves use of red phosphorus and bromine at 80 C to obtain the corresponding tribromo compounds (2a,b) which upon treatment o with ammonia followed by cyclization using NaH afforded the 3-bromo-lactams (4a,b) as shown in Scheme 1. Scheme 1 O O i) n O Br n Br Br Br n = 1; 1a n = 2; 1b n iii) NH 2 Br n = 1; 2a n = 2; 2b Br O ii) n = 1; 3a n = 2; 3b n O N H n = 1; 4a n = 2; 4b Reagents and conditions: i) Red P/Br2, 0-80 oC, 3 h; ii) NH3, H2O/CHCl3, 10-30 oC, 1 h; iii) NaH, THF, 0-10 oC-rt, 1 h. A stirred solution of commercially available substituted anthranilic acids (6a-c) have been coupled to the activated polymer-supported p-nitrophenyl carbonate (5) (1.00 g, 0.93 mmol/g) by using 1-hydroxybenzotriazole (HOBt) and diisopropylethylamine (DIPEA) in DMF-CH2Cl2 (1:2) to give 7a-c. Then 7a-c has been coupled to different lactams (8a,b and 4a,b) using dicyclohexylcarbodiimide (DCC) and catalytic amount of 4-dimethylaminopyridine (DMAP) in CH2Cl2 to afford 9a,b; 10a,b and 11a-c; 12a-c. The solid-supported bromides have been reacted with potassium acetate and catalytic amount of 18-crown-6-ether to afford acetates (13a-c) and (14a-c). These upon deacetylation with K2CO3 in THF-MeOH (1:1) gave XXVI SYNOPSIS 15a-c and 16a-c. Finally, 9a,b; 10a,b and 15a-c; 16a-c have been treated with TFACH2Cl2 (1:1) for the effective cleavage from the resin followed by cyclization to produce 17; 18a-b; 19a-c and 20, 21; 22 and 23 as shown in Scheme 2. Scheme 2 O O O R1 O R2 O X OH O R1 NH 2 i) R2 6a; R1 = H, R2 = H 6b; R1 = H, R2 = Cl 6c; R1 = OMe, R2 = OBn NO 2 5 O HN R1 n ii) OH 8a; n = 1 8b; n = 2 NH O R2 X=H 7a-c O 1 n N 20; n 21; n 22; n 23; n iii) v) O N n R2 =1 =1 =2 =2 O N N OH R2 17 18a,b; n = 1 19a-c; n = 2 O N R2 R1 X 11a-c; n = 1 12a-c; n = 2 X = Br O O n 9a,b; n = 1 10a,b; n = 2 X = H v) R1 NH O 4a; n = 1 4b; n = 2 X = Br O R N NH O n iv) OH O O 15a-c; n = 1 16a-c; n = 2 R1 N R2 NH O n OAc O O 13a-c; n = 1 14a-c; n = 2 Reagents and conditions: i) HOBt, DIPEA, CH2Cl2:DMF (2:1), 6 h, rt; ii) DCC, DMAP, CH2Cl2, 0 oC, 12 h, rt; iii) KOAc, 18-crown-6-ether, CH3CN, 80 oC, 6 h or DMF, 8 h, rt; iv) K2CO3, THF:MeOH (1:1), rt, 6 h; v) TFA, CH2Cl2 (1:1), 2 h, rt. Similarly, in Scheme 3 the compound 24 has been attached to bromomethyl Wang resin (25) (1.0 g, 0.8-1.0 mmol/g) with Cs2CO3 and NaI. This upon hydrolysis with 1N NaOH and coupling with 4a,b gave 27a,b as indicated by IR spectrum that showed a strong azide peak in the range 2110 cm-1. Azides 27a,b have been cyclized by an aza-Wittig mediated reductive-cyclization approach by using triphenylphosphine (TPP) in dry toluene to give 28a,b. Intermediates 28a,b have XXVII SYNOPSIS been converted to 31a,b by employing reactions similar to those described in Scheme 2. These transformations have been monitored by FT-IR spectroscopy of the resin beads (Tetrahedron Lett. 2006, 47, 9025-9028). Scheme 3 O Br H3CO OCH 3 HO O O H3CO OH 25 O i) N3 HN H3CO O n N n ii) 4a; n = 1 4b; n = 2 N3 26 24 O Br N3 O Br 27a; n = 1 27b; n = 2 iii) O H3CO O O O N v) H3CO N N n n O 30a; n = 1 30b; n = 2 iv) N n H3CO O N OH 29a; n = 1 29b; n = 2 OAc N Br 28a; n = 1 28b; n = 2 vi) O H3CO HO N n N 31a; n = 1 OH 31b; n = 2 Reagents and conditions: i) (a) Cs2CO3, NaI, DMF, 24 h, rt; (b) 1N NaOH, 1,4-dioxane, 100 oC, 12 h; ii) DCC, DMAP, CH2Cl2, 12 h, 0 oC-rt; iii) TPP, toluene, 3 h, rt; iv) KOAc, 18-crown-6-ether, CH3CN, 80 oC, 6 h or DMF, 8 h, rt; v) K2CO3, THF:MeOH (1:1), 6 h, rt; vi) TFA, CH2Cl2, (1:1) 1 h, rt. XXVIII