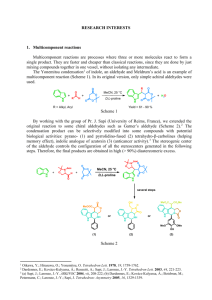
The thesis entitled “Synthesis of β
advertisement

Abstract The thesis entitled “Synthesis of mixed β-peptides, Imidazoles and Base Catalysed Condensation Reactions” is divided into two chapters. First chapter is dealt with the synthesis of (R) and (S)--homoserine, oxetane -amino acid, enatiomeric C-linked carbo--amino acids from D-and L-arabinose and their use in the synthesis of peptides. Second chapter describes the synthesis of imidazoles and Knovenagel condensations. Chapter I: Synthesis of new β-amino acids and mixed β-peptides This chapter is dealt with synthesis of (S)- and (R)--hSer (from L- and D-phenyl alanine), oxetane-β-amino acid and new enantiomeric C-linked carbo-β-amino acids and their use in the design and synthesis of mixed carbo-β-peptides. Glycolipids and glycoproteins play a major role in inflammation, immune response, metastasis, fertilization and many other biomedically important processes.1-4 Majority of the natural proteins contain (oligo)-saccharide side chains and the saccharide residues are covalently linked to the protein backbone either N- (via asparagines) or Oglycosidically (via serine, threonine, or tyrosine).5 The incorporation of C-glycosyl amino acids in glycopeptides may serve in preparing chemically and metabolically resistant analogues that display inhibitor activity towards O- or N-glycosidases.6 β-Amino acids are part structures of several natural products and very important components. Due to the importance of such unusual amino acids and their nonavailability from ‘chiral pool’ work was undertaken towards their synthesis and development of non-natural peptides with new conformations. Seebach et al.7a and Gellman et al.7b designed β-peptides with 14- and 12-helical structures by using aliphatic and constrained cyclic β-amino acids8 respectively. Even though sugar amino acids have been used for the peptide synthesis, synthesis of βpeptides having carbohydrates as side chains is not reported. For the synthesis of such peptides using C-linked carbo-β- and -amino acids (/-Caa), with carbohydrate side chains, TBAF9 was used an efficient base in promoting the aza-Michael addition of benzylamine on sugar-based γ-alkoxy α,β-unsaturated ester10 A to result in B (Scheme 1). The continued interest in developing new bases for efficient aza-Michael addition of 1 Abstract amine an α,β-unsaturated ester, revealed DBU as an efficient base to give B while reaction of A with BnNH2 gave B and C. Scheme 1 O O H3CO D-Glucose H3CO A TBAF or DBU BnNH2 O O H3CO 68% O O NHBn O H3CO C O H3CO + O NHBn O H3CO B Our earlier work on the carbo-β-peptides,11 prepared from (S)- and (R)- β-Caas alternatingly resulted in 10/12- and 12/10-mixed helices, wherein conformational constraints were observed with (S)-β-Caa. Based on the above observation, synthesis of a new set of monomers such as homoserines (S)-1 and (R)-2 (Figure 1) and small ring oxetane β-amino acids 3 were envisaged to relieve the constraints. It was assumed that, such side chains in the new -amino acids would help in the formation of well-defined helices in the thus made peptides. Similarly, enantiomeric -Caas 4 and 5 (Figure 1) also were prepared to study the synthesis with alternating chirality. Figure 1 O H3CO NHBo c OAC O O NHBo c H3CO OAC H3CO O NHBo c O H3CO H3CO O SOaa (3) (R)--hSer (2) (S)--hSer (1) NHBo c O O H3CO O NHBo C O O O H3CO (R)-Caa (4) (S)-Caa (5) Synthesis of β-amino acids 1, 2 and 3 The requisite -amino acids 1 and 2 were prepared from commercially available L- and D-phenyl alanine respectively. Accordingly, L-phenyl alanine 6 (Scheme 2) was treated with I2 and NaBH4 in dry THF to afford phenyl alaninol 7, which on reaction with (Boc)2O in THF gave 8. Acetylation of 8 with Ac2O and oxidative cleavage of phenyl ring in 9 afforded (S)-β-homoserine 1. 2 O O Abstract Scheme 2 NH2 NH2 a Ph 7 d OAc Ph 8 NHBoc O NHBoc c OH Ph OH Ph COOH 6 NHBoc b OAc HO 1 9 a) NaBH4, I2, THF, 18 h, 0 C-reflux; b) (Boc)2O, Et3N, THF, 4 h, rt; c) Ac2O, Et3N, CH2Cl2, DMAP, 30 min, rt; d) RuCl3, NaIO4, CCl4: CH3CN : H2O, 48 h, rt Similar sequence of reactions on D-phenyl alanine 10 (Scheme 2) gave (R)-βhomoserine 2. Scheme 3 NH2 NHBoc NH2 a Ph 11 O NHBoc c OAc Ph OH Ph OH Ph COOH 10 b 12 NHBoc d OAc HO 13 2 a) NaBH4, I2, THF, 18 h, 0 oC-reflux; b) (Boc)2O, Et3N, THF, 4 h, rt; c) Ac2O, Et3N, CH2Cl2, DMAP, 30 min, rt; d) RuCl3, NaIO4, CCl4: CH3CN : H2O, 48 h, rt The oxetane--amino acid (3, -Oaa) was prepared from D-manitol. Accordingly, mannitol diacetonide, prepared from D-mannitol, on oxidative cleavage gave (R)glyceraldehye 14 (Scheme 4). Reaction of 14 with allyl bromide and zinc dust12 in THF at room temperature for 4 h afforded homoallylic alcohol 15, which on osmylation with catalytic amount of OsO4 and NMO in acetone:water (4:1) at room temperature for 24 h afforded 16. Further, cleavage of 1,2-diol 16 using NaIO4 in CH2Cl2 and catalytic sat. NaHCO3 at 0 °C to room temperature for 5 h afforded aldehyde 17, which was used as such for the next reaction. Aldehyde 17 was treated with NaBH4 in MeOH at 0 °C for 30 min to furnish alcohol 18. Primary alcohol 18 on tosylation with p-TsCl and Et3N in CH2Cl2 and catalytic amount of DMAP at room temperature for 12 h afforded 19, which was reacted with NaH in DMF at room temperature for 12 h to give 20 in. Acetonide deprotection in compound 20, on reaction with 60% aq. AcOH at room temperature for 12 h afforded the diol 21. 3 Abstract Scheme 4 O a O O O CHO 14 O 15 O OH OH e b O O 16 OH O c O f 17 O g O d OH 21 O j i H3CO h O 20 OH CHO OH HO O O OHC O O OH OH OH OTs 19 18 O NHBn O k,l 3 24 H3CO O 23 22 a) Allyl bromide, Zn, THF, sat. NH4Cl, 4 h, rt; b) OsO4, NMO, 4:1, Acetone: water, 24 h, rt; c) NaIO4, CH2Cl2, sat. NaHCO3, 5 h, rt; d) NaBH4, MeOH, 30 min, 0 oC; e) p-TsCl, Et3N, DMAP, CH2Cl2, 12 h, rt; f) NaH, DMF, 12 h, rt; g) 60% aq. Acetic acid, 12 h, rt; h) NaIO4, MeOH-H2O, 1 h, rt; i) Ph3P=CHCO2Me, MeOH, 2 h, rt; j) BnNH2, 12 h, rt; k) 10% Pd/C, H2, MeOH, 12 h, rt; l) (Boc)2O, Et3N, THF, 4 h, rt Further, cleavage of 1,2-diol in 21 using NaIO4 in MeOH-H2O at room temperature for 1 h gave aldehyde 22, which was immediately treated with (methoxycarbonylmethylene)triphenyl phosphorane in MeOH at room temperature for 2 h to give 23. Reaction of Michael acceptor 23 with benzyl amine (2.5 equivalents)13 at room temperature gave oxetane-β-amino acid ester 24. Ester 24 was subjected to hydrogenation with 10% Pd-C in methanol under hydrogen atmosphere and the amine was treated with (Boc)2O and Et3N in THF to result in 3. In our reported studies on the carbo--peptides, the epimeric -Caas were utilized with ‘alternating chirality’. It was planned to synthesise enantiomeric -Caas and use them in the peptide design with alternating chirality. In this direction, enantiomeric Caas were prepared from D-and L-arabinose. Accordingly, the new C-linked carbo-βamino acid 4 was prepared from L-arabinose as shown in Scheme 5. Readily available L(+)-arabinose was selectively silylated using TBDPSCl in DMF at 0 C to room temperature for 18 h to afford 25, which was treated with dry acetone, CuSO4 and catalytic amount of conc. H2SO4 at room temperature for 5 h to furnish 26. Compound 26 was subjected to alkylation using MeI and NaH in THF at 0 °C to room temperature for 12 h to give 27, which on desilylation using TBAF at 0 °C to room temperature for 14 h 4 Abstract gave 28. Oxidation of 28 with IBX in DMSO gave aldehyde 29, which on Witting reaction in C6H6 at reflux for 5 h gave 30. Reaction of 30 with benzyl amine (2.5 equivalents) at room temperature for instance gave a separable mixture of C-linked carbo-β-amino acid esters 31 and 32. Ester 31 was subjected to hydrogenolysis with 10% Pd-C in methanol under hydrogen atmosphere to give the amine, which on treatment with (Boc)2O and Et3N in CH2Cl2 afforded 4. Scheme 5 O O d OR TBDPSO c TBDPSO O TBDPSO O a b L-(+)-Arabinose O MeO O HO HO OH 27 26 25 R = H 25a R = TBDPS O O O O g O HO O f e H3CO O H O O O H3CO H3CO 28 30 H3CO 29 O O NHBn O O NHBn O H3CO O + h, i O 4 H CO 3 H3CO 31 O O H3CO 32 a) TBDPSCl, Imidazole, DMF, 18 h, 0 oC-rt ; b) Acetone, H+, CuSO4, 5 h, rt, ; c) NaH, MeI, THF, 12 h, 0 0C-rt ; d) TBAF, CH2Cl2, 14 h, 0 oC-rt ; e) IBX, DMSO, 6 h, rt; f) Ph3P=CHCO2Me, C6H6, 5 h, reflux; g) BnNH2, 12 h, rt; h) Pd/C, H2, MeOH, 12 h, rt ; i) (Boc)2O, Et3N, CH2Cl2, 4 h, rt O In a similar way, as described for 4, readily available D-(+)-arabinose was selectively silylated using TBDPSCl in DMF to afford 33 (Scheme 6). Compound 33 was treated with dry acetone, CuSO4 and H2SO4 (cat) to furnish 34, which was subjected to alkylation with MeI and NaH in THF to give 35. Deprotection of 35 using TBAF gave 36, which on oxidation with IBX in DMSO gave aldehyde 37; Witting olefination on 37 in C6H6 at reflux temperature for 5 h gave 38. Reaction of 38 with benzyl amine gave a separable mixture of Caas 39 and 40. Compound 39 was subjected hydrogenation with 10% Pd/C and resultant amine was treated with (Boc)2O and Et3N in CH2Cl2 to result in 5. 5 Abstract Scheme 6 O a TBDPSO D-(+)-Arabinose O O O H3CO O H3CO b H3CO 36 NHBnO O O O + H3CO H3CO 40 OO O TBDPSO H3CO 34 O O O H CO Ff 3 35 g H3CO 38 37 NHBn O O O H3CO c O O O O H e OO O TBDPSO HO HO OH 33 R = H 33a R = TBDPS O HO d OR h, i 5 39 a) TBDPSCl, imidazole, DMF, 18 h, 0 C-rt ; b) Acetone, H+, CuSO4, 5 h, rt, ; c) NaH, MeI, THF, 12 h, 0 C-rt ; d) TBAF, CH2Cl2, 14 h, 0 C-rt ; e) IBX, DMSO, 6 h, 0 C - rt; f) Ph3P=CHCO2Me, C6H6, 5 h, reflux; g) BnNH2, 12 h, rt; h) Pd/C, H2, MeOH, 12 h, rt ; i) (Boc)2O, Et3N, CH2Cl2, 4 h, rt Synthesis of mixed β-peptides with epimeric -Caas and (R)/(S)--hSer/(S)--Oaa Having successfully prepared new -amino acids 1-5, it was firstly proposed to synthesise mixed -peptides using 41 and 41a alternatingly with 1 and 2 (Figure 2). The main intention of the work was to understand the impact of a small side chain on the helix forming capability. Peptides were prepared by using standard peptide coupling conditions15 in the presence of water soluble 1-hydroxy-1H-benzotriazole (HOBt), 1-[3(dimethylamino) propyl]-3-ethylcarbodiimide hydrochloride (EDCI) and DIPEA as a base in dry CH2Cl2. Figure 2 O H3CO NHBoc O C C O C4 C1 C3 H3CO O H3CO C HO C C4 C1 O C3 C2 O H3CO (R)--Caa (41) O NHBoc O C2 O (S)--Caa (41 a) NHBoc O OAc HO (S)--hSer (1) NHBoc OAc (R)--hSer (2) 6 Abstract In the synthesis of mixed C-linked carbo-β-peptides di-, tri-, tetra- and pentapeptides 42-45 respectively were prepared with (S)-C-linked carbo-β-amino acid, 41a and (S)-β-hSer 1, alternatingly by adopting segment condensation method (Scheme 7). Dipeptide 42 was prepared by condensation of two monomers, (S)-Caa 41a and (S)--hSer 1. Tripeptide 43 was prepared by coupling of dipeptide amine and acid of (S)-βCaa, while tetrapeptide 44 was prepared by coupling of tripeptide amine and (S)--hSer acid. The pentapeptide 45 was prepared from tetrapeptide amine 44 and (S)-β-Caa acid. Scheme 7 H N O H N S H N S O S OCH3 O O O 42 O OAc H3CO H N O O H3CO O O O H N H N O OAc H3CO S H N S O O O H3CO O O H N S O O 44 O O O 43 O S O OAc H3CO O O H N S H N S O H N S O OCH3 O H N O O O O O O OAc H3CO O O O OCH3 O O O O OAc H3CO S O OAc H3CO H N S H N S S OCH3 O O O 45 In a further study, it was proposed to use the β-Caa monomer and β-h-Ser with alternating chirality for the synthesis of new β-peptides. Accordingly, the dipeptide 46, tripeptide 47, tetrapeptide 48 and pentapeptide 49 (Scheme 8) were prepared from (S)-Clinked carbo-β3-amino acid (Caa) 41a and (R)--h-Ser 2 by using standard peptide coupling conditions. NMR, CD and MD studies clearly indicated that mixed-β-peptides 48 and 49 folded into 12/10/12- and 10/12/10/12-mixed helical pattern (Figure 3). 7 Abstract Scheme 8 H N O H N R O S O OAc H3CO 46 OCH3 O H N R O O O H N S O O O H3CO O O AcO H N S O O H3CO 47 O OCH3 O O O 12 10 H N R O OAcO H N S O H3CO H N R O O OAcO 48 O H N S O H3CO OCH3 O O O O 12 10 10 H N O O H3CO O O O O AcO H N H N H N O H3CO O O O AcO H N O H3CO O O OCH3 O O O 49 Figure 3: a) Circular dichroism spectrum of tetrapeptide 48 in CH3OH at concentration of 0.1 mM; b) MD structure (side view) of 48 (bundle of 20 conformers lowest in energy determined by restrained molecular dynamics). In a further study, (R)--Caa 41 and (S)--hSer 1 were successfully utilized for the synthesis of new β-peptides. Accordingly, the dipeptide 50, tripeptide 51, tetrapeptide 52, and pentapeptide 53 (Scheme 9) were prepared. NMR, CD and MD studies clearly indicated that, these mixed-β-peptides folded in a 12/10- mixed helical pattern. Peptides 51 and 53 have shown 12/10- and 12/10/12/10- mixed helices, while 52 indicated the presence of a 10/12/10-mixed helix. 8 Abstract 12 Scheme 9 H N O H N S O R O OAc H3CO H3CO O O H N S O AcO R H N R H N S O O O H3CO O H3CO O O H N O OAc H3CO O R OCH3 O O O 10 AcO H N R OCH3 O O H3CO 52 O O O O 12 12 H N 51 O H N 12 O O O O 10 O H N S O O O O 50 H N R O OCH3 10 10 H N R S O AcO 10 H N S O H3CO O O O O AcO H N R O O H3CO O O OCH3 O O 53 Figure 4: CD spectrum of 52 in CH3OH at concentration of 0.1 mM NMR solution structure; (b) MD structure side view of 52 (bundle of 20 conformers lowest in energy determined by restrained molecular dynamics). Similarly, di-, tri- and tetrapeptides 54-56 (Scheme 10) were prepared from (R)Caa 41 and (R)--hSer 2. In 1 H NMR spectra of peptides 46-48, characteristic information on well-defined secondary structures was not observed. These peptides have not shown the presence of any secondary structure. 9 Abstract Scheme 10 H N O H N R O R O OAc H3CO H N R O H3CO O O 55 O H N R O O O O O H N O OCH3 O OAc H3CO O R OCH3 O O O 54 H N O H N R O H N R O OAc H3CO O R O OAc H3CO O O O H N R O 56 OCH3 O O O Thus study on the synthesis of mixed -peptides using (R/S)--Caa and β-hSer (R/S) amply indicated that the peptides with a CH2OAc side chain also behaved like the earlier carbo--peptides prepared from alternatingl (R)- and (S)--Caas. Like the homooligomeric peptides derived from (R)-β-Caa and (S)-β-Caa, the mixed peptides derived from (R)-β-Caa/(R)--hSer and (S)-β-Caa/(S)--hSer do not present any secondary structures. In a further new design, it was planned to synthesize the mixed β-peptides using the (R)-β-Caa 41 and (S)--Oaa 3 alternatingly. Accordingly, 41 and 3 were successfully utilized for the synthesis of new mixed-β-peptide 57 (Scheme 11). Scheme 11 H N O H N O OCH3 O H3CO O O O O O 57 Similarly, dipeptide 58 was prepared from 41 and 3, having 3 at N-terminus (Scheme 11). 10 Abstract Scheme 12 H N O H N O O O OCH3 O O H3CO O O 58 However, attempted conversion of the above dipeptides 57 and 58 into the corresponding higher homologues gave unexpected problems in the synthesis. Hence, work could not be extended in this direction. Thus, even though new -Caas were prepared with a CH2OAc and oxetane side chains and peptides were made, only the peptides with alternating -Caa and β-hSer have shown well-defined 12/10-mixed helical patterns. However, for reasons not known enantiomeric -Caas derived from L-and D-arabinose did present difficulties in their synthesis of enantiomeric β-peptides. Chapter II described ZrCl4 catalysed synthesis of tri- and tetrasubstituted imidazoles and Knoevenagel condensation reactions by modified Ni-Al layered double hydroxides Section A: ZrCl4 Catalysed synthesis of tri- and tetrasubstituted imidazoles A general protocol has been developed for the rapid synthesis of 2,4,5trisubstituted and 1,2,4,5-tetra-substituted imidazoles in good yields using ZrCl4 as a catalyst at room temperature (Scheme 13). The synthetic strategy is based on the condensation of 1,2-diarylethanedienones with aldehydes or aldehydes and amines resulting in 2,4,5-tri-substituted and 1,2,4,5tetra-substituted imidazoles respectively using 20 mol% of ZrCl4 as a catalyst. Of the several methods reported in the literature for the synthesis of imidazoles, the four component one-pot condensation of aryl glyoxals, aldehydes, amines and ammonium 11 Abstract acetate in refluxing acetic acid is the most desirable and convenient method (Scheme 13). Scheme 13 Ph Ph R-CHO O + NH4OAc, ZrCl4 N CH3CN, rt or Ph O R1NH2, NH4OAc, ZrCl4, CH3CN, rt Ph R N R1 R = Aliphatic, Aromatic, sugars R1= Differnt amines Initially, benzaldehyde (Table 1) with an equimolar quantity of benzil and excess of ammonium acetate in acetonitrile was treated with 20 mol% ZrCl4 and stirred at room temperature for 0.75 h to furnish 2,4,5-triphenyl imidazole. The reaction of benzil with a range of other aromatic aldehydes carrying electron withdrawing or electron releasing substitutes afforded high yields of products (Table 1). Noteworthy to mention that reaction of benzil with p-anisaldehyde and excess of ammonium acetate, the reaction was only 50% complete. Aliphatic aldehydes (entry 6, 7), heterocyclic aldehyde (entry 8a) and sugar aldehydes (9, 10, 11, 12) too gave the products in high yields. Similarly, terenaldehyde (entry 13) with two equivalents of benzil and excess of ammonium acetate gave bis imidazolyl benzene. This approach was used for the preparation of tetrasubstituted imidazoles too successfully in good yields. Thus, benzil on reaction with equimolar quantity of aldehyde, amines (14-16) along with excess of ammonium acetate in acetonitrile at room temperature furnished 1,2,4,5-tetrasubstituted imidazoles. The reaction was carried out with aliphatic amine (entry 14), aromatic amine (entry 15) and heterocyclic amine (entry 16) successfully to give 14a, 15a and 16a respectively. 12 Abstract Table 1: ZrCl4 catalysed synthesis of tri/tetrasubstituted imidazoles Starting material Entry Time (h) Product Ph OHC Yield (%) N 1 N H Ph R R 2 1 1a R = H 0.75 95 2 2a R = Cl 10 93 96 89 3 3 3a R = F 5 4 4 4a R = NO2 7 5 OHC Ph O N Ph OHC O N H Ph 5 N 6 7 CH 3 OHC CH3 CH 3 7 8 N H 6a Ph 6 Ph CH3 N N H Ph Ph 6 95 2 91 4 84 5a CH3 CH3 7a N OHC N H Ph 4.5 84 6 84 1.25 88 1.5 87 8a 8 9 9 O OHC O 10 13 O 9a N OCH3 Ph O O O O 11a O OO N H O NH2 14 Ph-CHO H N N 13a 15 + 15 NH2 16 N 16 Ph 2 87 Ph N Ph N 14a CH3-(CH2)5-NH2 Ph-CHO Ph 85 O OO O CHO 1.5 O 12a 12 O + O N Ph Ph OCH3 N H 13 14 O 10a N Ph O N H O O Ph Ph Ph O O O OO O O N N H H3CO Ph 11 OHC N H Ph Ph 10 O OHC O 12 N Ph O H3CO 11 Ph CHO OHC Ph 1 86 0.75 88 1.25 89 Ph N Ph N Ph N Ph 15a Ph Ph N + Ph-CHO N 16a 13 Abstract Section B: Knovenagel condensation by modified Ni-Al layered double hydroxides. Calcined Ni-Al hydrotalcite is an efficient and environmentally attractive solid base catalyst for the selective synthesis of -nitroalkanols, ,- unsaturated nitriles or esters by Henry or Knoevenagel reactions of the activated nitro alkanes, carboxylic esters or nitriles with various aldehydes respectively (Scheme 14). The main advantages with this catalytic system are simple work-up procedure and reusability with quantitative yields. Scheme 14 R O H R1 CN Calcined Ni-Al HT R CN room temperature H R1 A R = Aryl, R1 = CN / CO2Et As is seen from Table 2, all the aldehydes 1-8 underwent smooth condensation selectively to give dehydrated products 1a-8a, 1b-4b, 6b-8b without any self-condensed or hydrated products of Knoevenagel adducts. The reaction using calcined Ni-Al hydrotalcite catalyst between benzaldehyde (1) and ethyl cyanoacetate (entry 1, Table 2) gave selectively Knoevenagel adduct 1b whereas alkali metal containing MCM-41 gives a mixture of hydrated and dehydrated products. The rate of reaction is very impressive and comparable with recently reported results under microwave irradiation employing phosphorous pentoxide. The condensation of furfural 8 with malononitrile proceeds at a faster rate under the present reaction conditions (entry 8, Table 2), unlike in the earlier reports. Further, the calcined Ni-Al hydrotalcite was reused without loss of its activity and selectivity. No reaction occurred, when the reaction was conducted with the filtrate of 14 Abstract solid catalyst after reaction, indicating that the active ingredient was not leached out of the solid catalyst. Thus, calcined Ni-Al hydrotalcite is used as an excellent solid base catalyst for Knoevenagel condensations. This new solid base catalyst becomes a potential alternate to soluble bases with the following advantages; a) high catalytic activity under very mild liquid phase conditions with 100% selectivity for -nitroalkanols, b) easy separation of the catalyst by simple filtration. c) reaction involves non-toxic and inexpensive materials d) No side reactions hence waste minimization and e) recycling of the catalyst. 15 Abstract Table 2: Knoevenagel condensation catalysed by calcined Ni-Al hydrotalcite. S.No Starting material Product CHO CN H R 1a R = CN 1b R = CO2Et 1 1 CHO CN H R 2a R = CN 2b R = CO2Et Cl 2 CHO O2N 3 NO 2 3 CN H R 3a R = CN 3b R = CO2Et H R 4a R = CN 4b R = CO2Et 4 CHO 5 CHO CN MeO 5 H 97 91 83-84 47-48 3.0 2.0 92 91 161-162 91-92 1.5 2.5 93 95 160-161 169-170 2.0 3.0 90 92 77-79 67-69 2.0 81 ___ 2.0 2.0 89 86 2.0 90 ___ 1.5 93 ___ 2.0 96 93-94 5a CN CN H R 6a R = CN 6b R = CO2Et OMe 6 CHO MeO MeO OMe OMe CN MeO H H CHO O 8 114-115 79-80 OMe O 7 8 2.5 4.0 MeO 6 7 m.p. OMe CN CHO OMe OMe Yield (%) Cl 2 4 Time (h) 7a CN CN R 8a R = CN 8b R = CO2Et 16 Abstract References 1. Reviews: a) Seebach, D.; Matthews, J. L. Chem. Commun. 1997, 2015; b) Gellman, S, H. Acc. Chem. Res. 1998, 31, 173; c) DeGrado, W, F.; Schneider, J. P.; Hamuro, Y. J. Pept. Res. 1999, 54, 206; d) Gademann, K.; Hintermann, T.; Schreiber, J. V. Curr. Med. Chem. 1999, 6, 905; e) Cheng, R. P.; Gellman, S. H.; DeGrado, W. F. Chem. Rev. 2001, 101, 3219; f) Steer, D. L.; Lew, R. A.; Perlmutter, P.; Smith, A. I.; Aguilar, M-I. Curr. Med. Chem. 2002, 9, 811. 2. Banerjee, A.; Balaram, P. Curr. Science 1997, 73, 1067. 3. Varki, A. Glycobiology 1993, 3, 97. 4. a) Phillips, M. L.; Nudelman, E.; Geeta, F. C. A.; Perez, M.; Singhal, A. K.; Hakomori, S.; Paulson, J. C. Science 1990, 250, 1130; b) Lasky, L. A. Science 1992, 258, 964; c) Miller, D. J.; Macek, M. B.; Schur, B. D. Nature 1992, 357, 589; d) Schulze, I. T.; Manger, I. D. Glycoconjugate J. 1992, 9, 63; e) Yaki, T.; Hirabayashi, Y.; Ishikawa, H.; Kon, S.; Tanaka, Y.; Matsumoto, M. J. Biol. Chem. 1986, 261, 3075; f) Spohr, U.; Lemieux, R, U. Carbohydr. Res. 1988, 174, 211; g) Tulsiani, D. R. P.; Yoshida-Komiya, H.; Araki, Y. Biol. Reprod. 1997, 57, 487. 5. a) Arsequell, G.; Valencia, G. Tetrahedron: Asymmetry 1997, 8, 2839; b) Lis, H.; Sharon, N. Eur. J. Biochem. 1993, 218, 1. 6. a) Marcaurelle, L. A.; Bertozzi, C. R. Chem. Eur. J. 1999, 5, 1384; b) Sears, P.; Wong, C.-H. Angew. Chem., Int. Ed. Engl. 1999, 38, 2300. 7. a) Seebach, D.; Ciceri, P.; Overhand, M.; Juan, B.; Rigo, D.; Oberer, L.; Hommel, U.; Amstutz, R.; Widmer, H. Helv. Chim. Acta 1996, 79, 2043; b) Applequist, J.; Bode, K. A.; Appella, D. H.; Christianson, L. A.; Gellman, S. H. J. Am. Chem. Soc. 1998, 120, 4891. 8. Reviews: a) Cole, D. C. Tetrahedron 1994, 50, 9517; b) Juaristi, E.; Quintana, D.; Escalante, J. Aldrichim. Acta 1994, 27, 3; c) Cardillo, G.; Tomasini, C. Chem. Soc. Rev. 1996, 117; d) Liu, M.; Sibi, P. Tetrahedron 2002, 58, 7991. 9. Sharma, G. V. M.; Goverdhan Reddy, V.; Subhash Chander, A.; Ravinder Reddy, K. Tettrahedron: Asymmetry 2002, 13, 21. 10. Costa, J. S.; Dias, A. G.; Anholeto, A. L.; Monteiro, M. D.; Patrocinio, V. L.; Costa, P. R. R. J. Org. Chem. 1997, 62, 4002. 17 Abstract 11. Sharma, G. V. M.; Ravinder Reddy, K.; Radha Krishna, P.; Ravi Sankar, A.; Narasimulu, K.; Kiran Kumar, S.; Jayaprakash, P.; Jagannadh, B.; Kunwar. A. C. J. Am. Chem. Soc. 2003, 125, 13670. 12. Abushnab, E.; Venishetti, P.; Leiby, R. W.; Singh, H. K.; Mikkilineni, A. B.; Wu, D. C.-J.; Saibaba, R.; Panzica, P. J. Org. Chem. 1988, 53, 2598. 13. Trochet, J. M. J.; Bachler, B. J.; Eder, H.; Hong, F. L.; Perret, N.; Poncet, J.; Zumbwald, B. J. Helv. Chim. Acta. 1973, 56, 1310. 14. Patil, N. T.; Tilekar, J. N.; Dhavale, D. D. J. Org. Chem. 2001, 66, 1065. 15. Bodanszky, M. Peptide Chemistry – A Practical Textbook, Springer Verlag, Berlin 1988. 16. Krieg, B.; Mancke, G. Z. Naturforschg 1967, 22b, 132. 17. (a) Lash, T. D.; Bellettini, J. R.; Bastian, J. A.; Couch, K. B. Synthesis, 1994, 170; (b) Solladiecavallo, A.; Quazzotti, S. Tetrahedron Asymm. 1992, 3, 39. 18. (a) Gompper, Chem Ber. 1957, 90, 374; (b) Beccalli, E.; Majori, L.; Marchensini, A.; Jorricelli, C. Chem. lett. 1980, 657; (c) Alper, H.; Amaratunga, S. J. Org. Chem. 1982, 47, 3595. 19. Otterill, I. C.; Usytinsky, A; Ya.; Arnold, J. M.; Clark, D. S.; Dordick, J. S.; Michels, P.C.; Khmelnitsky, Yu, L. Tetrahedron Lett. 1998, 39, 1117. 20. Siddiqui, S, A.; Narkhede, U. C.; Palimkar, S, S.; Daniel, T.; Lahoti, R, J.; Srinivasan, K. V. Tetrahedron 2005, 61, 3539. 21. Knovenagel, E. Chem. Ber. 1894, 27, 2345. 22. House, H. O in ‘Modern Synthetic Reactions’, 2nd Edn.Benjamin, Menlo Park, 1972, 646. 23. Reeves, R. L. In ‘The Chemistry of the Carbonyl Group’ Ed. S Patai, Wiley Inter Science, Newyork, 1996, Vol. 1, 567. 24. Tietze L. F.; Eicher, T. ‘Reactions and Synthesis’, 1st Edn. University Science Books, Mill Vally, 1989. 25. Subbarao, Y. V.; Choudary, B. M. Synth. Commun. 1991, 21, 1163. 18