F HP 2
advertisement

Phase problem:
Detectors can only measure amplitudes (intensities), but no phases which yield the bulk of structural
information.
- We cannot measure the phase of a photon.
- We cannot measure the phase difference between two photons.
We measure the intensities of the diffracted beams I(hkl) proportional to |F(hkl)|^2 (the square of
the structure factor amplitude).
1
−2𝜋𝑖(ℎ𝑥+𝑘𝑦+𝑙𝑧)+ 𝑖𝛼(ℎ𝑘𝑙)
ρ(xyz) = 𝑉 ∑ℎ𝑘𝑙 |𝐹(ℎ𝑘𝑙)|𝑒
We have to know the phases α of all reflections (h k l) -> high speed computers.
|F(hkl)|
? α(hkl)
But although we are free to fix the absolute phase we can determine the relative phase of different
diffracted rays:
- By convention a wave scattered from the origin of the crystal coordinate system has a phase of 0.
- By convention any other wave (scattered from other points in the crystal) is phase-shifted by the
difference in position.
Intensity of scattered X-rays are measured by a
Phases could be calculated from the coordinates
detector:
of all atoms
proportional to |F|^2
=> Amplitude |F| is derived from the experiment
ρ(xyz) = (1/V)ΣF(hkl)exp[-2πi(hx + ky + lz)]
with F(hkl) = |F|exp(2πi) = Σfiexp[-Bi(sin/)]exp[2πi(hx + ky + lz)]
MIR – Multiple isomorphous replacement
-
Solve the phase problems for macromolecules like DNA, RNA, proteins
Binding of heavy atoms to the macromolecule changes diffraction pattern
Requirements:
The crystal of the native macromolecule and the crystal of its heavy atom derivative should be
isomorphous (of the same crystal form):
=> Differences in diffraction patterns only due to contribution of the heavy atoms.
The unit cell dimensions have to be the same:
Binding of the heavy atom should not change the crystal packing or the conformation of
macromolecule.
=> The only difference between the native macromolecule and its heavy atom derivative is the
presence of one or more heavy atoms.
Heavy atoms:
The perturbation of diffraction pattern has to be large enough -> high atomic number.
-
Platinum, Gold, Mercury, Lead, Uranium, Thorium, Rhenium, Iridium, Osmium
Small macromolecules: Silver, Palladium (of lower atomic number)
Bromine, Iodine: iodinated tryrosin or nucleic bases
Lanthanides: replacing Calcium or Magnesium
Nobel gases like Xenon, Krypton
Derivatisation:
Soak the crystal of the native macromolecule with a solution of heavy atoms (salts or
coordination compounds) for minutes, weeks or months.
Try different concentrations (0.05mM – 100mM) and different pH values.
Channels in the crystal (about 50% are water) allow the heavy atoms to reach the
macromolecules and bind to functional groups.
Generate a heavy atom derivative and then crystallize this derivative.
This method could avoid non-isomorphism.
It is useful if ligands, substrates, inhibitors are bound to the heavy atom.
Diffraction pattern:
Compare the diffraction pattern of the native and the heavy atom crystal:
You will find deviations in the intensities of corresponding reflections.
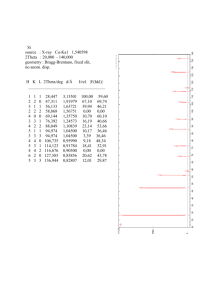
The picture below shows the diffraction patterns of the native and derivative crystals of a
nitrogenase.
- Pairs of reflections are underlined.
- Their relative intensities are reversed in the second pattern.
(native: right = darker; derivative: left = darker).
Determining the phase of the structure factor of the macromolecule FP:
The following equations have to be solved for all reflections F(hkl).
FHP = FP + FH => FP = FHP - FH
|FHP|2 = |FP|2 + |FH|2 + 2|FP||FH|cos(p + H)
=> p = H + arcos [(|FHP|2 - |FP|2 - |FH|2 ) / (2|FP||FH|)]
|FHP| and |FP|
can be read out from the diffraction pattern.
|FH| = |FHP| - |FP| and H
can be determined if we know the position of the atom.
Since cos(x) = cos(-x) arcos has two solutions. Therefore we need a second heavy atom derivative
with the heavy atom at another position yielding new |FHP|, |FH| and H: multiple instead of single
isomorphos replacement.
|FP|
|FHP1|
|FH2|
|FH1|
|FHP2|
Procedure:
1) Characterize and collect the diffraction pattern of the native macromolecule.
=> |FP|
2) Prepare two heavy atom derivatives.
3) Characterize and collect the diffraction patterns of the heavy atom derivatives.
=> |FHP|
4) Determine the position of the heavy atoms in the unit cells with the help of Patterson
function.
=> |FH| and H
5) Calculate the phase of the structure factor of the protein.
=> p = H + arcos [(|FHP|2 - |FP|2 - |FH|2 ) / (2|FP||FH|)]
6) Calculate the electron density.
ρ(r) = 1/V Σ |F(S)|exp(-2πirS + iP)
7) Build the 3D model of electron density and refine it.
MAD –Multi-wavelength anomalous dispersion
-
Anomalous scatteres change the diffraction pattern.
A method related to MIR: It uses one type of crystal and different wavelength instead of
different crystals and one wavelength. => no problems with non-isomorphism
Requirements:
- Tunable radiation of third generation synchrotrons
- Careful measurements of intensities because differences caused by anomalous scatterers are
small:
o exposure times as function of expected anomalous signal.
o High completeness
o High reduncdancy
o Friedel pairs are measured nearly at the same time
o Flash freezing for constant crystal conditions
Anomalous scatterers:
- Heavy atoms introduced in the crystal (see MIR: Bromine, Iodine, Krypton, Xenon, …)
i.e. Bromine-Uridine instead of Thymine in DNA
i.e. Seleno-Methionine instead of Methionine (expressed in auxotroph E.coli strains) – This
method is extensively used and has a high rate of success.
- Heavy atoms already part of the native macromolecule: Iron, Copper, Zinc, etc.
<= Absorption edges of atoms normally found in macromolecules (light atoms like carbon, oxygen,
nitrogen) are not near the wavelength of X-rays used in crystallography.
MIR with anomalous scattering:
The structure factor of the heavy atom derivative:
F2 = F1 + ΔFr + ΔFi
fa = fo + Δf’ + iΔf’’ = f’ + iΔf’’
Be aware that Friedel’s law is true for λ1 but not for λ2.
F1 (hkl)= F2(hkl) - ΔFr(hkl) - ΔFi(hkl)
F1 (hkl)= F2(-h-k-l) - ΔFr(hkl) – [-ΔFi(hkl)]
because upon reflection: Fr1 (-h-k-l)= F1 (hkl), ΔFrr(-h-k-l) = ΔFr(hkl)
but ΔFri(-h-k-l) = [-ΔFi(hkl)]
There is only one solution fulfilling this two equations.
ΔFr and ΔFi:
- looked up in tables
- phases depend upon the position of the heavy atom in the unti cell (Patterson function).
|Fλ1(hkl)|
|Fλ2(hkl)| and |Fλ2(-h –k –l)|
|Fλ2(-h-k-l)|
-ΔFi(hkl))
ΔFr(hkl)
|Fλ1(hkl)|
ΔFi(hkl)
)
|Fλ2(hkl)|
Now we know FHP = | Fλ1(hkl)|exp(iαλ1):
intensities in anomalous data set -> phase in normal (nonanomalous) data set
FP = FHP - FH can be solved.
amplitude and phase of FHP
see above
amplitude and phase of FH
Patterson function
Procedure of SIR with anomalous scattering:
1) Diffraction pattern of native crystal at λ1
=>| FP|
2) Diffraction pattern of derivative crystal at λ1
=> | FHP|
3) Determination of the heavy atom
4) Diffraction pattern of derivative crystal at λ2
=> αHP
Hand problem:
The added phase information from anomalous scattering can make hand selection possible.
Multi wavelength anomalous dispersion:
Data sets from heavy atom derivative at different wavelength are like those from distinct heavy atom
derivatives due to the dependence of Δf’ and Δf’’ on the wavelength.
=> Measurements at several wavelength close to an absorption edge yield data sets with distinct
values for the real and the imaginary contribution of anomalous scattering
- Reflection intensities between Friedel mates vary at the same wavelength (= Bijvoet difference)
- Reflection intensities vary slightly with wavelength (= dispersive difference).
=> This differences contain phase information. You only have to solve equations which resemble
those for MIR.
FH, FH’’ and –FH’’: amplitudes as well as phases are known (Patterson map)
|FPH|
|FP|
Bijvoet difference:
ΔF = |Fλ(hkl)| - |Fλ(-h –k –l)| at a given wavelength λ, Friedel mate and its opposite
Dispersive difference:
ΔF = |Fλ1(hkl)| - |Fλ2(hkl)| at different wavelength λ1 and λ2, one reflection
High precision is easier than for Bijvoet signal. Systematic errors of datasets at two wavelength
cancel out.
Bijvoet signal:
is derived from the differences in the intensities of Friedel mates collected at a wavelength which
causes anomalous scattering.
<|ΔF(+/-h)|>/<|F(h)|> = [(0.5)^0.5][(N(A)/N(P)^0.5]2(f’’/Z(eff))
- N(A) = number of anomalous scatterers
- N(P) = number of protein atoms
- Z(eff) = average number of non-H-atoms
Karle formulation:
The structure factor for an individual reflection h at wavelength λ:
λF(h) = °F (h) + λF (h) = °F (h) + °F (h) + λF ’(h) + iλF ’’(h) = °F (h) + λF ’(h) + iλF ’’(h)
N
A
N
A
A
A
T
A
A
-
°FN(h) = normal part
λ
FA(h) = anomalous part
Simplest case: only one scattering atom:
Intensity is proportional to | λF(-h)|2
| λF(-h)|2 =
|°FT|2
+ a(λ) |°FA|2
+ b(λ) |°FT| |°FA| cos(°φT-°φA)
+/-c(λ) |°FT| |°FA| sin(°φT-°φA)
a(λ) = (f’2+ f’’2)/f°2
b(λ) = 2(f’/ f°)
c(λ) = 2(f’’/ f°)
(All of the wavelength-dependence has been collected into the coefficients a(λ), b(λ), and c(λ). All
amplitudes are of wavelength-independent structur factors °F.)
Procedure:
1) Preparing the derivative.
2) Characterize and collect diffraction patterns at three different wavelength:
PI = point of inflection of f’ (lowest possible value)
PK = peak of f’’ = absorption edge
RE = a point remote of the absorption edge
3)
4)
5)
6)
PI crystal of native molecule
PK and RE crystals of derivates
Determine the position of the anomalous scatterers. (Patterson function).
Calculate the phase of the structure factor of macromolecule.
Calculate the electron density and build the 3D model.
Refine the model.
Patterson function: Determination of the position of heavy
atom/anomalous scatterers
P(u v w) = 1/V * Σhkl e2πi(hu + kv + lw)
= 1/V * Σhkl |F(h k l)|2 cos[2π(hu + kv + lw)]
= ∫r1 ρ(r1) * ρ(r1 + u)dv
This function is a Fourier transform without phases (we do not know) whose amplitude is the square
of the norm of one structure factor (proportional to the measured intensity).
Like ρ(x y z) has peaks (areas of high denity) at locations of the atoms P(u v w) has peaks at
locations corresponding to vectors between atoms.
- x y z are the coordinates in the electron denity map
- u v w are the coordinates in the Patterson map with dimension identical to the real cell
X
1) A structure of three atoms
- N atoms
2) All possible vectors between the atoms: red, violet and blue (not shown in b).
i.e. from 1 to 3 but also from 3 to one (opposite direction) and also from 1 to 1
3) Vectors transferred to the origin of the Patterson map with empty unit cells around it
- N2 vectors including the ones between an atom and itself (concentrated at the origin
causing a high peak at the origin)
- N(N-1) vectors excluding the ones between an atom and itself
The head of a vector is the location of a Peak in Patterson map: Patterson atom
The coordinates of this atom represent vectors between the real atoms:
(u v w) = (x1 – x2 y1 – y2 z1 – z2)
4) Patterson atoms from one unit cell added to the other cells so that each cell contains N(N-1)
Patterson atoms.
=> This is what you see when you compute a Patterson map:
- N(N-1) maxima
- distance between maximum and origin = length of an interatomic vector
- Intensities of maxima are proportional to ZiZj (atomic numbers of two atoms i and j)
Just another demonstration:
P(u v w) = ∫r1 ρ(r1) * ρ(r1 + u)dv
- ρ(r1) = electron density at the beginning of u
- ρ(r1 + u) = electron density at the end of u
=> Only if ρ(r1) as well as ρ(r1 + u) are nonzero will P(u v w) be nonzero.
!! We know u but not r1.
Integrating ρ(r1) * ρ(r1 + u) with r1 over the entire unit cell includes that r1 assumes different length
and different directions so that the calculated cosine argument assumes angles between 0 and 2π
and the integral lead to zero except all phase angles are zero.
P(u v w) = 1/V * Σhkl |F(h k l)|2 cos[2π(hu + kv + lw)]
Because of centrosymmetry the complex sine terms disappear.
The Patterson function is a convolution of a structure and its inverse:
1
Convolution: C(x) =∫𝜂=0 𝑓(𝜂)𝑔(𝑥 − 𝜂)𝑑𝜂 = f*g
We can change Patterson function into the form P(u) = ∫r’’ρ(r’’)ρinv(u-r’’)dv
with r’’ = r1 + u
and ρinv(u-r’’) = electron density distribution of the inverse structure compared to ρ(r’’-u)
Patterson map -> structure:
1) Determine the number of atoms: N(N-1) is number of peaks of the Patterson function.
2) A number of Patterson atoms form the original structure with the origin (gray triangle).
3) Trial and error: Searching Patterson atoms forming the original structure.
I.e. green x falsifies a vector linking a light violet and an orange dot.
Simplify the search by unit cell symmetry:
- 21 axis (twofold screw) on edge b:
each atom (x y z) -> counterpart atom (-x, y+1/2, -z)
connecting vectors (u v w) = (2x, ½, 2z) all lying in a plane cutting the Patterson unit cell at v = ½
called the u(1/2)w plane (a so called Harker section – analogous there are also Harker lines.)
Such a case you will find if heavy atoms bind at equivalent positions. Their Patterson map peaks will
be found on Harker sections.
- Determine u v and w of the peaks and calculate x, y and z.
-> y not specified = loss of information
The hand problem:
If the arrangement of heavy atoms in a protein unit cell is enantiomeric you have a problem. I.e. a
threefold screw axis can be left or right handed. But Patterson map does not distinguish between
inverted images because you can not tell whether a Patterson vector ab is a-b or b-a.
Patterson map in MIR and MAD:
Since there are more vectors between atoms than are atoms in the unit cell we only want to
solve the Patterson function for the heavy atom/anomalous scatterer. Therefore we
construct a difference Patterson function:
Δ|F|2 = (|FPH| - |FP|)2
We are extracting the simple diffraction pattern of the heavy atom from the complicate
diffraction pattern of the heavy atom derivative.
To determine the position of anomalous scatterers in single heavy atom derivatives
we need anomalous Patterson and dispersive Patterson maps
with Δ|F|2 = |FPK(hkl)-FPK(-h-k-l)|2 and Δ|F|2 = |FRE(hkl)-FPI(hkl)|2 respectively
MR – Molecular Replacement:
-
Using phases from a known protein with similar structure
Known protein = phasing/search model
-> placed in the unit cell of the new (unknown) protein
Used for:
- mutants and related proteins
- complexes with ligands, co-factors etc.
- Multidomain proteins and Macromolecular complexes (domains/subunits are known)
- molecule in another space group
Requirements:
- sequence homology
- differences limited (rmsd between Cα of search model and of target < 1Å)
- quality and completeness of diffraction data
- dimension of phasing model with respect to the target protein
- criteria for discriminating the correct solution
Adjust the model:
- Deletion of mobile, flexible, non conserved loops
- Deletion of ligands, cofactors, inhibitors, metals waters…
- Ala truncation of non-identical residues
- Poly-Ala truncation of all side chains
- Average structure (after superposition of many similar structures of
individual search models)
weighting of atoms by B-factors derived from rms distance between
identical atoms
Obtaining phases:
The phase contains the bulk of information but intensities provide enough information about the
differences between the two proteins.
! Phases of the molecular structure factor F(hkl) depend on the location of the atoms in the unit cell.
=> The phase of the phasing model can only be used if its orientation and position are the same as of
the new protein.
To satisfy this condition we have to rotate and position the phasing model in the unit cell and then
calculate the phase of this properly arranged model.
x1 = Rx2 + T
- x1 and x2 arrangements of the target and the model respectively
- R and T rotation function and translation function
Because we have to know how to arrange the phasing model we determine the unit cell dimension
and the symmetry of the new protein with the help of its native data.
Trial and error: Compare |F(hkl)calculated| of the model with |F(hkl)observed| of the target protein and
find the best match (R = 0 for perfect agreement, Rmax = 1 for total non-superimposition)
R = (Σhkl||F(hkl)observed| - |F(hkl)calculated||)/Σhkl|F(hkl)observed|
Since the number of trial orientations and positions is enormous the search for superimposition is
split into
- search for the best orientation (rotation about three axis x, y, z – three parameters) and
- search for the best position (with three coordinates x, y, z – further three parameters).
If the number of the possible values of each parameter is N then we have to investigate N3 + N3
possible arrangements instead of N6.
Vectors in Patterson map:
- Intramolecular vectors = self vector set
between atoms of the same molecule (not longer than rmax)
convolution between the electron density for one molecule and the density for the same
molecule
-
depend on the structure and orientation of the molecule (-> determination of rotation)
Intermolecular vectors = cross vector set
between atoms of different (symmetry related) molecules (average > rmax)
convolution between the electron density for one molecule and the density for another
(symmetry related) molecule
depend on the structure and orientation and position (crystal packaging) of the
molecule
(-> determination of translation after rotation)
Patterson map of
intermolecular vectors
between a molecule and a
symmetry related molecule
(twofold symmetry = rotation
about 180°.
Upon moving the molecule the
symmetry related molecule
moves in the opposite
direction relative to the
twofold axis.
Therefore intermolecular
vectors length shifts by double
that amount.
Procedure:
1) Patterson function <- diffraction data from the target
2) Correlation with intramolecular vectors of the search model <- rotation
=> determination of orientation
3) Correlation with intermolecular vectors of the search model <- translation
=> determination of position
4) Structure of our target = Search Model * R(r) + T(r)
=> Coordinates of atoms => phase α(hkl)
Rotation function:
It evaluates the correlation of the Patterson map for the target and for the phasing model in
various orientations:
- maximum value at maximum overlap
R(Φ φ χ) = ∫u v w Ptarget(uvw)Pmodel{(uvw) * [Φ φ χ]}du dv dw
When either Patterson function is zero the product is zero.
-> Maximum value at maximum overlap.
Translation function:
It compares sets of intermolecular vector with observed Patterson map and is a product
function like rotation function is.
Maximum value at maximum overlap.
- search by Patterson function, correlation coefficient between |Fo| and |Fc| or R factor between
|Fo| and |Fc|
Calculating electron density:
1
−2𝜋𝑖(ℎ𝑥+𝑘𝑦+𝑙𝑧)+ 𝑖𝛼(ℎ𝑘𝑙)
ρ(xyz) = 𝑉 ∑ℎ𝑘𝑙 |𝐹(ℎ𝑘𝑙)|𝑒
- small molecules: |𝐹(ℎ𝑘𝑙)| = |Fobserved|
- macromolecules: |𝐹(ℎ𝑘𝑙)| = 2|Fobserved| - |Fcalculated|
- difference density: |𝐹(ℎ𝑘𝑙)| = |Fobserved| - |Fcalculated|