Rare inherited disorders of fibrinogen
advertisement

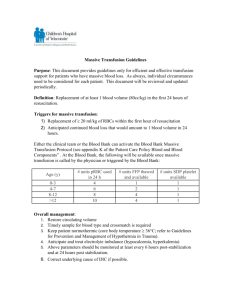
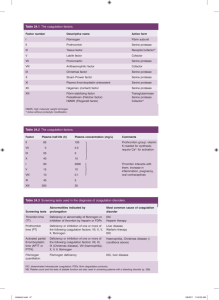
Haemophilia (2008), 14, 1151–1158 DOI: 10.1111/j.1365-2516.2008.01831.x ORIGINAL ARTICLE Rare inherited disorders of fibrinogen S. S. ACHARYA and D. M. DIMICHELE Department of Pediatrics, Weill Medical College of Cornell University, New York, USA Summary. Fibrinogen, a hexameric glycoprotein encoded by three genes – FGA, FGB, FGG – clustered on chromosome 4q is involved in the final steps of coagulation as a precursor of fibrin monomers required for the formation of the haemostatic plug. Inherited disorders of fibrinogen abnormalities are rare and not as well clinically characterized as some other inherited bleeding disorders. To characterize the clinical manifestations, molecular defects and treatment modalities of these rare disorders, a Medline search from January 1966 to September 2007 for these disorders reported in large studies and registries was undertaken. Inherited fibrinogen disorders can manifest as quantitative defects (afibrinogenemia and hypofibrinogenemia) or qualitative defects (dysfibrinogenemia). Quantitative fibrinogen deficiencies may result from mutations affecting fibrinogen synthesis, or processing while qualitative defects are caused by mutations causing abnormal polymerization, defec- Introduction Fibrinogen is a 340-kDa glycoprotein synthesized in the liver, with a multitude of functions including fibrin clot formation, non-substrate thrombin binding, platelet aggregation and fibrinolysis [1]. The first clinical report of congenital afibrinogenemia dates back to 1920 when a 9-year-old boy suffering from recurrent bleeding episodes since birth and lacking fibrinogen in blood was described and shown subsequently to be autosomal recessive in inheritance with variable penetrance [2,3]. The estimated prevalence of afibrinogenemia which is the most severe form of the disorder is around 1 in 1 000 000 [4] and Correspondence: Suchitra S. Acharya, MD, Weill Medical College of Cornell University, Pediatric Hematology/Oncology, 525 East 68th Street, P-695, New York, NY 10021, USA. Tel.: 212-746-3418; fax: 212-746-8986; e-mail: ssa2001@med.cornell.edu Accepted after revision 5 July 2008 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd tive cross-linking or defective assembly of the fibrinolytic system. Clinical manifestations vary from being asymptomatic to developing catastrophic life-threatening bleeds or thromboembolic events. Management of bleeds includes use of purified plasma-derived concentrates, cryoprecipitate or fresh frozen plasma. Use of some of these products carries risks of viral transmission, antibody development and thromboembolic events. Establishment of registries in Iran, Italy and North America has fostered a better understanding of these disorders with an attempt to explore molecular defects. Rare Bleeding Disorder Registries developed through the United States and international efforts hopefully will encourage development and licensure of safer, effective products. Keywords: bleeding, congenital afibrinogenemia, dysfibrinogenemia, fibrinogen genes, hypofibrinogenemia, thrombosis recent registries from Italy, Iran and North America have greatly improved understanding of the clinical spectrum of presentation [5–7]. However, knowledge on the incidence of these disorders has been confounded by publication bias. In populations where consanguinity is high, as noted in the Iranian Registry, the prevalence may be similar to other autosomal recessive disorders [5,8]. In fact, a sevenfold higher incidence of fibrinogen disorders was observed in the Iranian Registry for Rare Bleeding Disorders in comparison with similar registries in Italy and United Kingdom [8]. Conversion of fibrinogen to insoluble fibrin plays a pivotal role in haemostatic balance and exposure of its non-substrate thrombin-binding sites after fibrin clot formation promotes anti-thrombotic properties [9]. Thus, because of its multi-faceted roles in coagulation, quantitative and qualitative modifications of this molecule can result in bleeding or thrombotic phenotypes. Consequently, these fibrinogen disorders can present as afibrinogenemia or 1151 1152 S. S. ACHARYA and D. M. DIMICHELE hypofibrinogenemia (quantitative defects) or dysfibrinogenemia (qualitative defects) and hence, clinical manifestations include the potential for either mucocutaneous and/or deep tissue bleeding tendencies or thrombotic tendencies. This review will focus on characterizing these rare inherited fibrinogen disorders with respect to clinical presentations, diagnostic criteria including molecular defects and the possibility of genotype–phenotype correlations and current products used in their management. Material and methods This report consists of available data on these inherited fibrinogen disorders including the estimated incidence, identified gene defects, clinical manifestations, diagnostic considerations and current treatment strategies. This was achieved through a Medline search conducted for the English language literature published from January 1966 to September 2007. The key terms fibrinogen, bleeding, clotting, thrombosis, fibrinogen genes were used, supplemented with additional cross-references through bibliographies. In order to avoid selection bias we included data from larger studies and published registries. Structure–function relationship Fibrinogen is secreted into the bloodstream as a disulphide-linked hexamer composed of two identical heterotrimers, each consisting of one Aa, one Bb and one c chain with molecular masses of 67 (610 residues), 57 (461 residues) and 47 kDa (411 resi- dues), respectively [9]. The hexamer is characterized by a symmetrical structure with a central E domain, connected to two peripheral D domains (Fig. 1). The three chains are encoded by paralogous genes (FGA, FGB and FGG coding for Aa, Bb and c chains, respectively), clustered in a 50-kb region on chromosome 4 (4q31.3–4q32.1) [10]. Conversion of fibrinogen to fibrin occurs after removal of fibrinopeptides A (FPA) and B (FPB) by thrombin from the N-termini of the Aa and Bb chains at the Arg16–Gly17 and the Arg14–Gly15 bonds, respectively. FPA release takes place faster and earlier than FPB release and is sufficient to induce clot formation. Abnormalities at the thrombin cleavage site can cause impaired release of FPA, inhibiting conversion of fibrinogen to fibrin leading to bleeding. FPB (Bb 1–14) cleavage occurs more slowly and contributes to lateral fibril and fibre association. Absent or slow FPB release with delayed polymerization of the fibrin monomers can cause a bleeding phenotype while impaired FPB release results in abnormalities of polymerization that are associated with thrombotic events. Finally, the soluble fibrin clot is stabilized by the amidolytic action of activated factor XIII (FXIII) to form gamma– gamma dimers and alpha polymers. Plasmin cleavage sites include regions between D and E domains in all the three chains producing fragments Y, D and E (Fig. 1). Abnormal fibrinogens that exhibit defective cross-linking by activated FXIII have been associated with abnormal wound healing while abnormalities that interfere with plasminogen binding or activation on the fibrin clot result in clinical thrombosis (9). (a) D domain D domain (b) D domain Haemophilia (2008), 14, 1151–1158 D domain Fig. 1. Schematic representation of the fibrinogen molecule. Fibrinogen consists of three pairs of polypeptide chains, Aa, Bb and c, joined by disulphide bonds to form a symmetric dimeric structure (a). The NH2 terminals of all six chains form the central domain (E domain) of the molecule containing fibrinopeptides A and B (FPA and FPB) sequences which are cleaved by thrombin during enzymatic conversion to fibrin. Enzymatic conversion of fibrinogen to fibrin (b) by thrombin cleavage results in release of FPA and FPB. Binding sites for IIa, tissue plasminogen activator and factor XIII are indicated on the fibrinogen or fibrin molecule. 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd INHERITED FIBRINOGEN DISORDERS Types of fibrinogen disorders The normal plasma fibrinogen concentration is approximately 1.5–3.5 g L)1 with a half-life of about 4 days. Inherited fibrinogen disorders are traditionally categorized on the basis of plasma concentration as follows: quantitative or type 1 deficiencies (including afibrinogenemia and hypofibrinogenemia) with reduced levels of antigen and functional activity and qualitative or type II deficiencies (including dysfibrinogenemias and hypodysfibrinogenemias) with normal or reduced antigen levels associated with abnormal functional activity. Functional activity assay measures clottable protein on the basis of either coagulation time or velocity (Clauss or thrombin clotting method). The antigen level is measured by most labs using a radioimmunodiffusion assay. Quantitative fibrinogen deficiencies may result from mutations affecting fibrinogen synthesis, assembly, intracellular processing (with or without endoplasmic retention), domain stability and protein secretion. Qualitative defects are caused by mutations in any of the three fibrinogen genes affecting any one of the functional properties of fibrinogen, including absence or delayed release of FPA and FPB, delayed or enhanced polymerization, defective cross-linking, decreased thrombin binding and defective assembly of the fibrinolytic system. 1153 (7%), menometrorrhagia, first-trimester abortions, ante-partum and postpartum haemorrhage, hemoperitoneum after corpus luteum rupture have been reported [12,13]. In one series of 13 pregnancies in six women with afibrinogenemia, seven (54%) ended in spontaneous abortion at 6–7 weeks gestation supporting requirement of fibrinogen for embryo implantation [14]. Further, impaired wound healing and wound dehiscence postsurgery has been reported because of the non-tensile clot and inadequate deposition of healing proteins delaying wound healing. Paradoxical arterial and venous thromboembolic events can occur in afibrinogenemia in the presence of co-inherited thrombophilia, after replacement therapy and without any of these risk factors [15,16]. It is hypothesized that afibrinogenemia patients can generate thrombin in the initial and secondary burst of thrombin generation promoting enhanced thrombin generation [17]. Fibrin can also act as antithrombin I by sequestering and downregulating thrombin activity. Hence, thrombin not trapped by the clot is available for platelet activation in the arterial wall [17]. In fibrinogen knock-out mice the number of embolized thrombi was sixfold higher than the wild-type leading to vessel occlusion supporting the anti-thrombotic properties of fibrinogen [18]. Hypofibrinogenemia Clinical manifestations Afibrinogenemia is often diagnosed in the newborn period because of umbilical cord bleeding. Hypofibrinogenemia is associated with fewer bleeding episodes and may not be diagnosed until a traumatic or surgical challenge occurs. Dysfibrinogenemias are commonly diagnosed in adulthood [11]. Afibrinogenemia Afibrinogenemia is associated with a bleeding tendency, which is highly variable including life-threatening and spontaneous/trauma-related bleeds. Bleeding can start in the neonatal period with 85% presenting with umbilical cord bleeding [5] or bleeding into the skin, gastrointestinal (GI) tract, genitourinary tract or central nervous system (CNS) bleeding (5%) [7,8]. Musculoskeletal bleeding occurs less frequently with haemarthroses reported in 54% of the patients [6–8]. Unusual manifestations such as spontaneous rupture of the spleen and presence of bone cysts in afibrinogenemia [12] have also been reported. In females, normal menses, menorrhagia 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd Hypofibrinogenemia the patients have similar bleeding patterns but have a milder course and may follow invasive procedures or major trauma largely exceeding spontaneous events (80% vs. 20%) [7]. In a questionnaire report on 100 patients with inherited fibrinogen disorders from 10 different countries from North America and Europe the most frequent haemorrhagic symptom was observed to be menorrhagia (46%) followed by muscle haematoma (12%), GI bleeding (5%) with no cases of CNS, intraperitoneal bleeding, miscarriage or thrombosis reported in this group [19]. A familial hypofibrinogenemia with hepatic storage disease associated with different point mutations in the fibrinogen gamma chain have been described [20,21]. Dysfibrinogenemia Dysfibrinogenemia the patients have an unpredictable clinical phenotype. A compilation of 250 patients revealed asymptomatic patients (53%), bleeders (26%) and clotters (21%) some of whom also had bleeding [22]. Postpartum thrombosis, Haemophilia (2008), 14, 1151–1158 1154 S. S. ACHARYA and D. M. DIMICHELE spontaneous abortions, stillbirths are described in the ISTH database [23]. Pregnant women with dysfibrinogenemia are at particular risk for bleeding following vaginal delivery, C-section and regional analgesia. Patients may also experience delayed wound healing and dehiscence. Data from the United Kingdom Health Centre DoctorsÕ Organisation reported an association between deep vein thrombosis, thrombophlebitis and pulmonary embolus in 26 index patients with dysfibrinogenemia at a young age (mean age: 32 years). The prevalence of dysfibrinogenemia in patients with a history of venous thrombosis is low, i.e. 0.8% as deduced from nine studies on 2376 patients [23]. The thrombotic risk increases with acquired risk factors and coinheritance of other thrombophilia [24]. Skin necrosis has been described with certain dysfibrinogenemias (fibrinogen Marburg) and less commonly arterial thromboses [23]. Laboratory diagnosis Afibrinogenemia (homozygous/compound heterozygous state) is characterized by the complete absence or reduced amounts of immunoreactive fibrinogen as measured by antigenic and functional assays (less than 0.1 g L)1). All coagulation tests which depend on fibrin as the endpoint, i.e. prothrombin time (PT), activated partial thromboplastin time (aPTT), thrombin time (TT) and reptilase time are infinitely prolonged. Fibrinogen is undetectable by both functional (Clauss) and antigenic assays. With more sensitive enzyme-linked immunosorbent assays (ELISA), afibrinogenemia could be defined as unmeasurable level of functional fibrinogen associated with trace amounts of immunoreactive fibrinogen. Hypofibrinogenemia (heterozygous state) is defined by a decreased level of normal fibrinogen (activity and antigen between 0.5 g L)1 and lower limit of normal range for local laboratory). As hypofibrinogenemia is a proportional decrease of functional and immunoreactive fibrinogen, most fibrin-based coagulation tests are variably prolonged, the most sensitive test being thrombin time (usually prolonged at fibrinogen activity < 1 g L)1). The total clot-based and immunogenic fibrinogen levels are both reduced to a similar level as the functional fibrinogen. Tests should be interpreted with regard to possibility of acquired hypofibrinogenemia – consumptive coagulopathy, hepatic failure, l-asparaginase therapy and family studies may be helpful in differentiating from the congenital variety. Dysfibrinogenemia is characterized by a structural abnormality of the fibrinogen molecule resulting in altered functional properties. Classically, the Haemophilia (2008), 14, 1151–1158 functional assay of fibrinogen yields low levels compared with immunological assays but levels may be concordant and functional levels may be normal. Therefore, a discrepancy between clottable protein and immunologically measured fibrinogen is characteristic of dysfibrinogenemia. A prolonged reptilase time in the presence of a normal functional fibrinogen provides strong evidence of dysfibrinogenemia. A normal or increased antigen with a lower functional level resulting in a low functional antigen ratio (most commonly 1:2) is usually diagnostic [25]. The sensitivity of coagulation tests to dysfibrinogenemia depends on the specific mutation, reagents and techniques [26]. A definitive diagnosis can be established by demonstrating the molecular defect. However, as these disorders are dominantly inherited, family studies may be helpful to differentiate from acquired dysfibrinogenemia. In the small percentage that presents with thrombosis, a thrombophilia work up to exclude co-existing prothrombotic defects may be useful. Hypodysfibrinogenemia which is defined by both quantitative and qualitative defects in fibrinogen result in levels ranging from 0.5 to 1.2 g L)1. Molecular diagnosis Afibrinogenemia and hypofibrinogenemia mutations Mutations causing afibrinogenemia have been detected in all three genes; the majority found to date are in FGA which are mainly deletions, frameshift, nonsense or splicing mutations. These can lead to deficiency of fibrinogen by several mechanisms: these can act at the DNA level, RNA level by affecting mRNA splicing or stability or at the protein level by affecting protein synthesis, assembly or secretion [11]. In a database compiled by Hans and Biot these mutations have been associated with clinical bleeding and thromboses there being no genotype–phenotype correlation which could possibly be related to modifier genes or common variant thrombophilic genes [22]. There seems to be considerable overlap between the causative mutations accounting for afibrinogenemia and hypofibrinogenemia. In many cases asymptomatic patients are in fact heterozygous for null mutations which in homozygosity or heterozygosity would cause afibrinogenemia. Mutations of the FGA gene which include null mutations, large deletions (11 kb, 1238 bp and 15 kb in the FGA gene), frameshift (IVS4+A>G and )1138C>T in the FGA gene), splice-site and early truncating nonsense mutations are the most frequently identified mutations irrespective of geographic 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd INHERITED FIBRINOGEN DISORDERS locations in individuals of European and nonEuropean descent [27]. In contrast, the spectrum of mutations in FGG and FGB includes an excess of missense mutations in the C-terminal globular domains [28]. It is important to identify these mutations for designing efficient strategies for mutation detection in new cases. In the majority of patients with afibrinogenemia or hypofibrinogenemia for whom the causative mutation has been identified there is no intracellular accumulation of the mutant fibrinogen chain. However, missense mutations identified in the FGG can cause fibrinogen deficiency in the heterozygous state owing to intracellular retention of the mutant gamma chain and progressive liver disease with hepatocellular intracytoplasmic inclusions [20,21]. Dysfibrinogenemia mutations Consistent with dominant transmission of dysfibrinogenemia, the majority of patients with dysfibrinogenemia are heterozygous for missense mutations in one of the three fibrinogen genes leading to delayed or absent FPA release or defective fibrin polymerization. Mutations at these sites are estimated to account for approximately 45% of dysfibrinogenemia mutations [22]. The majority of these mutations are in FGA. Thus, dysfibrinogenemia can cause either a bleeding disorder or thrombophilia; some mutations can cause both. Not all are symptomatic; many are in fact discovered following routine laboratory tests before surgery, by prolonged TT. Individuals (65%) with FGG mutations are asymptomatic with 5% having bleeding symptoms and 30% thrombosis [29]. A defective binding of thrombin to abnormal fibrin which leads to increased thrombin levels has been implicated in thrombosis (fibrinogens Malmo, Naples, New York I, Pamplona II and Poitiers) and a defective stimulatory function of abnormal fibrin in the tissue plasminogen activator-mediated fibrinolysis has also been implicated in thrombotic events (fibrinogens Argenteuil, Chapel Hill III, Date, New York I, Nijmegen, Pamplona II and Paris V) [23]. These ÔthrombophilicÕ mutations have been found predominantly in the C-terminal domain of the Aa chain and the thrombin cleavage site of the Bb chain [30]. Interestingly, dysfibrinogens Marburg and Bern V both a chain mutation form clots which are fragile with failure to form normal fibrin aggregates leading to bleeding symptoms. However, there is also impaired fibrinolysis leading to thrombotic complications [29]. Therefore, with dysfibrinogenemias there may be a potential for genotype–phenotype correlations. A database is 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd 1155 available on the Study Group on Hemostasis and Thrombosis: http://www.geht.org/databseang/fibrinogen [22], which lists all fibrinogen variants identified to date in patients with dys-, hypo- and afibrinogenemia. Thus, mutation analysis can provide a valuable tool for diagnosis confirmation, identification of potential carriers and prenatal diagnosis. Prenatal diagnosis Afibrinogenemia and hypofibrinogenemia are autosomal recessive in inheritance while dysfibrinogenemia is autosomal dominant. Hence, pregnant females with a family history, especially those with a history of consanguinity should be appropriately counselled with regard to risks of producing a child with the disease. If the mutation is known, genetic testing should be planned early in pregnancy by chorionic villous sampling in order to aid appropriate and safe delivery of the affected child. In babies born to known/suspected carrier parents, cord blood testing for genetic defects may be offered. Indirect prenatal diagnosis by the use of linkage studies could also be performed in these disorders [11]. Avoidance of arterial sticks, intramuscular injections, traumatic interventions and screening for intracranial bleeds by head ultrasound at birth is recommended. Management Afibrinogenemia and hypofibrinogenemia Replacement therapy is generally effective in treating bleeding episodes in congenital afibrinogenemia. PatientÕs personal and family history of bleeding or thrombosis can help guide therapy. Options for replacement include plasma-derived fibrinogen concentrate (used in Europe), cryoprecipitate (used in the United States) and fresh frozen plasma [31,32]. Clinical situations include spontaneous bleeding and surgery as postoperative bleeding is reported in 40% in the Iranian series [6]. Antifibrinolytic agents may be used especially for dental procedures. Caution needs to be exerted in those with a history of thrombosis and other risk factors such as pregnancy, surgery and immobilization when they may be at increased risk for thrombosis. During pregnancy, treatment is recommended as soon as possible to prevent foetal loss (as early as 6–7 weeks to aid in implantation) and continued throughout pregnancy and postpartum with the Haemophilia (2008), 14, 1151–1158 1156 S. S. ACHARYA and D. M. DIMICHELE aim of maintaining levels above 0.5 g L)1. Oestrogen–progesterone preparations may be helpful in menorrhagia in the absence of a thrombotic tendency [33]. Dosage. In general, level of fibrinogen to be achieved is 1.0 g L)1 and should be maintained until haemostasis is achieved and wound healing is complete. In the United States, cryoprecipitate in a dose of 1 bag per 5 kg body weight followed by 1 bag per 15 kg body weight is used. It could be continued daily or every other day in the absence of consumption with close monitoring of fibrinogen activity depending on the clinical indication and response [31]. Dosage calculation. Dose (g) = desired increment in g L)1 · plasma volume (plasma volume is 0.07 · (1– haematocrit) · weight (kg). replacement products will depend on the clinical response and laboratory monitoring. Topical fibrin glue or antifibrinolytic agents may be advocated for superficial bleeds. In a pregnant woman with a bleeding phenotype, recommendations for afibrinogenemia/hypofibrinogenemia can be followed. During delivery, it should be assumed that the baby has dysfibrinogenemia and invasive monitoring should be avoided. Caution should be exercised when interpreting dysfibrinogenemia tests at birth because of physiological or acquired dysfibrinogenemia in the newborn. Thromboprophylaxis in women with a personal or family history of thrombosis should include compression stockings with low molecular weight heparin and/or replacement products reserved strictly for thrombotic events. For those with a bleeding phenotype vaginal delivery can be conducted by observation and replacement therapy only if bleeding occurs and maintained until wound healing is complete. Dysfibrinogenemia The guidelines for dysfibrinogenemia are not standardized with lack of sufficient data in bleed management. As the dysfunctional fibrinogen can interfere with the function of the infused fibrinogen and assays used for monitoring, functional level of fibrinogen should be raised above 1.0 g L)1 depending on the clinical response and maintained until wound healing is complete [31]. Repeat doses of Complications of disease and treatment Acquired inhibitors have been reported after replacement therapy [34]. In addition, treatment associated thromboembolic complications could be averted by administering small doses of heparin along with fibrinogen concentrate [16]. Successful use of direct thrombin inhibitors such as Lepirudin in individuals who suffered recurrent thrombosis despite adequate Table 1. Clinical, laboratory characterization and treatment recommendations for rare inherited disorders of fibrinogen. Transmission Impact Fibrinogen level Symptoms Laboratory Treatment Afibrinogenemia Hypofibrinogenemia Dysfibrinogenemia Autosomal recessive (both parents are carriers) One in 1 million <0.1 g L)1 plasma Autosomal dominant* and recessive** Less than afibrinogenemia Between 0.1 g L)1 and lower limit of normal for lab Umbilical cord bleeding Cutaneous bleeding Gastrointestinal haemorrhage Intracranial bleeding (infrequent) Articular bleeding aPTT is normal; increased PT-N, TT mildly prolonged, fibrin assay between 0.1 g L)1 and lower limit for lab Autosomal dominant* and recessive** 1 in 1 million £1.5 and 3.5 g L)1 of plasma No symptoms Haemorrhage Thrombosis Umbilical cord bleeding Cutaneous bleeding Gastrointestinal haemorrhage Intracranial bleeding (infrequent) Articular bleeding PTT, aPTT, TT markedly prolonged; all fibrin assays zero or trace Cryoprecipitate/fibrinogen concentrate/antifibrinolytic agents Cryoprecipitate/fibrinogen concentrate/antifibrinolytic agents PT, aPTT are normal. increased TT and reptilase time; fibrin assay is normal; higher fibrinogen for immunogenic than thrombin clotting method Cryoprecipitate/fibrinogen concentrate with anticoagulants in thrombosis aPTT, activated partial thromboplastin time; PT, prothrombin time; TT, thrombin time; PT-N, prothrombin time-normal. *Only one parent is a carrier. **Both parents are carriers. Haemophilia (2008), 14, 1151–1158 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd INHERITED FIBRINOGEN DISORDERS heparin and aspirin [35] has also been explored. Finally, routine surveillance for disease- and treatment-related complications and immunization against hepatitis in a comprehensive care setting is highly recommended. 1157 Acknowledgements The authors thank Susan Parker for secretarial assistance. This project was partly funded by the ChildrenÕs Cancer and Blood Foundation. Prophylaxis Disclosures Prophylactic infusions of cryoprecipitate have been employed after life-threatening bleeds such as intracranial bleeds. However, this strategy needs to be judicious given the possibility of transmission of infectious agents, allergic reactions, venous access problems, development of inhibitors and the risk of thrombo-embolic complications. Moreover, the prophylaxis schedule may need to be tailored based on individual pharmacokinetics, which is highly variable on replacement therapy [36]. In a retrospective analyses, the mean number of bleeding episodes on demand and prophylaxis were not significantly different (0.7 vs. 0.5) [19]. Hence, caution needs to be exercised before routinely recommending prophylaxis. The clinical, laboratory and recommended treatments are summarized in Table 1. The authors stated that they had no interests which might be perceived as posing a conflict or bias. Clinical trials None of the fibrinogen concentrates are available in the United States at the present time. A prelicensure phase II pharmacokinetic study sponsored by CSLBehring with a plasma- derived concentrate – Haemocomplettan – which enrolled 15 subjects was completed in April 2008. The results of this trial will be presented to the Food and Drug Administration for licensing approval. Individuals doing research in pathophysiology/ molecular basis 1. M. Neerman-Arbez, Geneva, Switzerland; Marguerite.Arbez@medicine.unige.ch 2. D. Galanakis, New York, USA; dgalanak@path. som.sunysb.edu 3. D.H. Farrell, Portland, USA; farrelld@ohsu.edu 4. M.W. Mosesson, Milwaukee, USA; mwmosesson @bcsew.edu 5. P.M. George, Christchurch, New Zealand; peter. george@chmeds.ac.nz 6. S. Duga, Milan, Italy; Stefano.duga@unimi.it Links to organizations – professional, lay: National Hemophilia Foundation Orientation manual for Healthcare workers: http://www.hemophilia. org/NHFWeb/Resource/ 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd References (* indicates key references) *1 Mosesson MW, Siebenlist KR, Meh DA. The structure and biological features of fibrinogen and fibrin. Ann N Y Acad Sci 2001; 936: 11–30. 2 Rabe F, Salomon E. Uber-faserstoffmangel in Blute bei einem Falle von Haemophilie. Arch Int Med 1920; 95: 2. 3 Werder E. Congenital afibrinogenemia. Helv Paediatr Acta 1963; 18: 208–29. 4 Martinez J, Palascak J, Peters C. Functional and metabolic properties of human asialofibrinogen. J Lab Clin Med 1977; 89: 367–77. 5 Mannucci PM, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood 2004; 104: 1243–52. *6 Lak M, Keihani M, Elahi F, Peyvandi F, Mannucci PM. Bleeding and thrombosis in 55 patients with inherited afibrinogenaemia. Br J Haematol 1999; 107: 204–6. *7 Acharya SS, Coughlin A, Dimichele DM, North American Rare Bleeding Disorder Study Group. Rare Bleeding Disorder Registry: deficiencies of factors II, V, VII, X, XIII, fibrinogen and dysfibrinogenemias. J Thromb Haemost 2004; 2: 248–56. *8 Peyvandi F, Duga S, Akhavan S, Mannucci PM. Rare coagulation deficiencies. Haemophilia 2002; 8: 308– 21. *9 Asselta R, Duga S, Tenchini ML. The molecular basis of quantitative fibrinogen disorders. J Thromb Haemost 2006; 4: 2115–29. 10 Kent WJ, Sugnet CW, Furey TS, et al. The Human Genome Browser at UCSC. Genome Res 2002; 12: 996–1006. *11 Neerman-Arbaz M. Molecular basis of fibrinogen deficiency. Pathophysiol Haemost Thromb 2006; 35: 187–98. 12 al-Mondhiry H, Ehmann WC. Congenital afibrinogenemia. Am J Hematol 1994; 46: 343–7. 13 Kobayashi T, Asahina T, Maehara K, Itoh M, Kanayama N, Terao T. Congenital afibrinogenemia with successful delivery. Gynecol Obstet Invest 1996; 42: 66–9. *14 Suh TT, Holmback K, Jensen NJ, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev 1995; 9: 2020–33. *15 Girolami A, Ruzzon E, Tezza F, Scandellari R, Vettore S, Girolami B. Arterial and venous thrombosis in rare Haemophilia (2008), 14, 1151–1158 1158 16 17 18 *19 20 21 *22 *23 24 *25 26 S. S. ACHARYA and D. M. DIMICHELE congenital bleeding disorders: a critical review. Haemophilia 2006; 12: 345–51. Dupuy E, Soria C, Molho P, et al. Embolized ischemic lesions of toes in an afibrinogenemic patient: possible relevance to in vivo circulating thrombin. Thromb Res 2001; 102: 211–9. Mosesson MW. Antithrombin I. Inhibition of thrombin generation in plasma by fibrin formation. Thromb Haemost 2003; 89: 9–12. Ni H, Denis CV, Subbarao S, et al. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest 2000; 106: 385–92. Peyvandi F, Haertel S, Knaub S, Mannucci PM. Incidence of bleeding symptoms in 100 patients with inherited afibrinogenemia or hypofibrinogenemia. J Thromb Haemost 2006; 4: 1634–7. Rubbia-Brandt L, Neerman-Arbez M, Rougemont AL, Malé PJ, Spahr L. Fibrinogen gamma375 arg–>trp mutation (fibrinogen Aguadilla) causes hereditary hypofibrinogenemia, hepatic endoplasmic reticulum storage disease and cirrhosis. Am J Surg Pathol 2006; 30: 906–11. Medicina D, Fabbretti G, Brennan SO, George PM, Kudryk B, Callea F. Genetic and immunological characterization of fibrinogen inclusion bodies in patients with hepatic fibrinogen storage and liver disease. Ann NY Acad Sci 2001; 936: 522–5. Hans M, Biot F. A database for human fibrinogen variants. Ann N Y Acad Sci 2001; 936: 89–90. Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost 1995; 73: 151–61. Siebenlist KR, Mosesson MW, Meh DA, DiOrio JP, Albrecht RM, Olson JD. Coexisting dysfibrinogenemia (gammaR275C) and factor V Leiden deficiency associated with thromboembolic disease (fibrinogen Cedar rapids). Blood Coagul Fibrinolysis 2000; 11: 293–304. Hayes T. Dysfibrinogenemia and thrombosis. Arch Pathol Lab Med 2002; 126: 1387–90. Lefkowitz JB, DeBoom T, Weller A, Clarke S, Lavrinets D. Fibrinogen Longmont: a dysfibrinogenemia Haemophilia (2008), 14, 1151–1158 *27 28 *29 *30 *31 *32 33 *34 35 *36 that causes prolonged clot-based test results only when using an optical detection method. Am J Hematol 2000; 63: 149–55. Neerman-Arbez M, de Moerloose P, Bridel C, et al. Mutations in the fibrinogen A alpha gene account for the majority of cases of congenital afibrinogenemia. Blood 2000; 96: 149–52. Vu D, de Moerloose P, Batorova A, Lazur J, Palumbo L, Neerman-Arbez M. Hypofibrinogenaemia caused by a novel FGG missense mutation (W253C) in the gamma chain globular domain impairing fibrinogen secretion. J Med Genet 2005; 42: e57. Roberts HR, Stinchcombe TE, Gabriel DA. The dysfibrinogenaemias. Br J Haematol 2001; 114: 249– 57. McDonagh J. Dysfibrinogenemia and other disorders of fibrinogen structure or function. In: Colman R, Hirsh J, Marder V, Clowes A, George J, eds. Hemostasis and Thrombosis, 4th edn. Philadelphia, PA: Lippincott Williams & Wilkins, 2001: 855–92. Kreuz W, Meili E, Peter-Salonen K, et al. Efficacy and tolerability of a pasteurised human fibrinogen concentrate in patients with congenital fibrinogen deficiency. Transfus Apher Sci 2005; 32: 247–53. Santagostino E, Mancuso ME, Morfini M, et al. Solvent/detergent plasma for prevention of bleeding in recessively inherited coagulation disorders: dosing, pharmacokinetics and clinical efficacy. Haematologica 2006; 91: 634–9. Rizk DE, Kumar RM. Congenital afibrinogenemia: treatment of excessive menstrual bleeding with continuous oral contraceptive. Am J Hematol 1996; 52: 237–8. RaÕanani P, Levi Y, Varon D, Gitel S, Martinowitz U. Congenital afibrinogenemia with bleeding, bone cysts and antibodies to fibrinogen. Harefuah 1991; 121: 291–3. Schuepbach RA, Meili EO, Schneider E, Peter U, Bachli EB. Lepirudin therapy for thrombotic complications in congenital afibrinogenaemia. Thromb Haemost 2004; 91: 1044–6. Kreuz W, Meili E, Peter-Salonen K, et al. Pharmacokinetic properties of a pasteurised fibrinogen concentrate. Transfus Apher Sci 2005; 32: 239–46. 2008 The Authors Journal compilation 2008 Blackwell Publishing Ltd