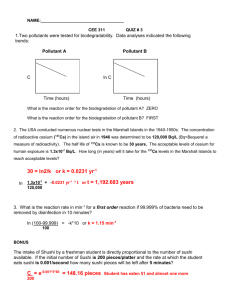
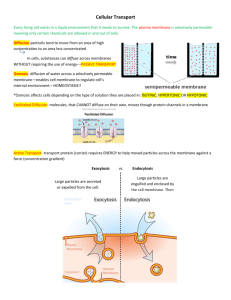
Systematic identification of proteins regulated by the TOR Complex
advertisement

Systematic identification of proteins regulated by the TOR Complex 2-dependent kinase Ypk1 in Saccharomyces cerevisiae By Alexander Muir A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Molecular and Cell Biology in the Graduation Division of the University of California, Berkeley Committee in charge: Professor Jeremy W. Thorner, Chair Professor David G. Drubin Professor John Kuriyan Professor Kevan M. Shokat Spring 2015 Abstract Systematic identification of proteins regulated by the TOR Complex 2-dependent kinase Ypk1 in Saccharomyces cerevisiae by Alexander Muir Doctor of Philosophy in Molecular and Cell Biology University of California, Berkeley Professor Jeremy W. Thorner, Chair Abstract: Eukaryotic plasma membranes are highly complex structures, both with respect to molecular composition and spatial organization. This composition and organization is essential for cellular function and must be maintained during dramatic shifts in extracellular environment that challenge membrane homeostasis. In the model eukaryote Saccharomyces cerevisiae, signaling by TOR Complex 2 (TORC2) and its effector kinases Ypk1 and Ypk2 is modulated in response to various membrane stresses and cells deficient in TORC2-Ypk signaling have membrane defects, suggesting that this signaling pathway is important for maintaining membrane homeostasis. To understand the molecular mechanisms by which TORC2-Ypk1 signaling regulates membrane homeostasis, I devised a three-tiered genome-wide screen to identify substrates of Ypk kinases. This screen identified 12 novel Ypk substrates in a variety of processes linking TORC2-Ypk signaling to membrane homeostasis including: lipid metabolism, glycerol metabolism, peroxisome function and autophagy. I then performed detailed studies of the regulation of identified substrates Lac1 and Lag1, components of the ceramide synthase complex, a lipid metabolism enzyme. In response to various membrane stresses, TORC2-Ypk1 activates ceramide synthase. This activation is essential in allowing cells to survive these stresses. Interestingly, it appears that activation of ceramide synthase promotes stress survival not by increasing ceramide levels, but rather by preventing the accumulation of ceramide precursors. Lastly, I characterized the TORC2-Ypk regulation of identified substrates Fps1, Gpt2 and Smp1, proteins involved in glycerol metabolism and the cellular response to hyperosmotic shock. I show that TORC2-Ypk is a novel hyperosmotic shock responsive pathway and that TORC2-Ypk coordinately regulates glycerol metabolism substrates to promote accumulation of glycerol, balance osmolarity and promote cellular survival. Thus, this work has identified at least some of the molecular mechanisms by which TORC2-Ypk signaling restores membrane homeostasis in the face of environmental stress. 1 Table of Contents Acknowledgements iii List of tables iv List of figures v List of abbreviations vii Chapter 1 - Introduction Molecular composition of the eukaryotic plasma membrane Molecular organization of the plasma membrane Role of plasma membrane organization and composition in cellular physiology TORC2-Ypk signaling and maintenance of membrane homeostasis Outstanding questions and outline of this thesis Figures 1 1 2 Chapter 2 - Materials and methods Construction of yeast strains Plasmids and recombinant DNA methods Bioinformatic prediction of Ypk1 substrates Yeast growth assays and synthetic dosage lethality screening Purification of Ypk1-as purification of putative Ypk substrates Ypk1-as in vitro kinase assays Preparation of cell extracts and immunoblotting Analysis of sphingolipid species In vitro ceramide synthase assay Fluorescence microscopy of Smp1-GFP and Fps1-GFP Measurement of STL1 promoter transcription by fluorescence activated cell sorting (FACS) Measurement of intracellular glycerol accumulation Co-immunoprecipitation of Fps1 and Rgc2 Tables 12 12 12 12 13 13 14 14 15 16 17 Chapter 3 - A three-tiered screen identifies novel Ypk substrates Introduction Results A three-tiered screen to identify new Ypk1 substrates Bioinfomatic prediction of Ypk substrates A synthetic dosage lethality screen to identify Ypk interacting proteins Biochemical screening of bioinformatically and genetically predicted Ypk substrates Discussion 29 29 29 29 29 i 3 4 7 9 17 17 17 19 30 31 32 Figures Tables 36 39 Chapter 4 - Ypk1 phosphorylates ceramide synthase to stimulate synthesis of comples sphingolipids Introduction Results Global screen indicates Lac1 and Lag1are Ypk1 substrates Lac1 and Lag1 are phosphorylated by Ypk1 in vivo Lac1 and Lag1 phosphorylation increases under stress and is required for cell survival Calcineurin down-regulates Lac1 and Lag1 phosphorylation Ypk1-mediated phosphorylation of Lac and Lag1 stimulate ceramide synthase activity Ypk1-mediated stimulation of Lac1 and Lag1 prevents autophagy induction during TORC2-driven up-regulation of sphingolipid biosynthesis Is Atg21 a physiologically-relevant Ypk1 substrate? Discussion Figures Chapter 5 - TOR Complex 2-Ypk is a MAPK-independent osmolarity sensing pathway that promotes glycerol accumulation and stress survival Introduction Results Smp1 is phosphorylated by Ypk kinases and this modification influences Smp1 function and localization Fps1 is phosphorylated by Ypk kinases and this modification is blocked when cells are subjected to hyperosmotic stress TORC2-Ypk regulation of Fps1 is independent of the known Ypk-mediated phosphorylation of Fps1 promotes channel opening and glycerol efflux TORC2-Ypk regulation of Fps1 is independent of the known Fps1 regulators Hog1 and Rgc1/2 and allows cells to survive hyperosmotic stress in the absence of Hog1 Discussion Figures Tables 45 45 46 46 46 47 48 49 50 51 52 56 69 69 71 71 72 74 75 76 78 90 Chapter 6 - Concluding remarks Conclusions Future directions Final remarks 91 91 92 94 Literature cited 96 Appendix – A study of places to walk around Berkeley and the Bay Area 118 ii Acknowledgements I grew up watching Total Request Live on MTV. No, no, don’t worry. I am not going to thank Carson Daly for raising me or anything like that. I just liked the part where he’d ask audience members to give a “shout out” to someone special. I am very happy to shout out the many people to whom I am indebted for making my life in Berkeley happy, fulfilling and productive. First of all, I have to thank the members of the Thorner lab for creating such an exciting place to work. I owe much of my scientific training to Françoise Roelants, an amazing scientist whom I am tremendously lucky to count as a friend. As a researcher, I would be pretty hopeless without her training, and her goodwill helped me tremendously through my difficult early years in the lab. Additionally, I have to thank the Thorner lab post-doctorates, in particular, Subu Ramachandran, for their input into my work over the years. My fellow graduate students also contributed tremendously to my scientific growth. In particular, Dr. Jesse Patterson’s doggedness, technical skill, and willingness to help out were a source of inspiration. I would also like to thank Chris Alvaro for being thoughtful, being razor sharp, and singing beautifully in the lab late at night. What more could you ask of a colleague? I am also incredibly fortunate to have worked with two undergraduate students, Jeff Liu and Garrett Timmons. Jeff, Garrett and I collaborated on much of the work described here and we made an excellent team. I am also profoundly indebted to Garrett for helping me cajole the lab into a skydiving trip. I am grateful to the scientific mentors I have had throughout my training. My advisor Jeremy Thorner taught me much, but two lessons stand out. First, science is not merely a body of facts, but a community of people who collect the facts and interpret them. Jeremy taught by example that teaching and being a good member of this community is of utmost importance. I remember once a student came well over an hour late to an appointment with him. He was in the middle of something else, but he stopped and made time to answer her questions. I aspire to be so caring. Jeremy also taught me, however, that the facts do matter. The usefulness of his encyclopedic knowledge continues to inspire me to be as well read as possible. I am also grateful to my thesis committee members, David Drubin, John Kuriyan and Kevan Shokat. They always had interesting ideas for other directions I could take my projects. I regret that I didn’t always have the time to try all their suggestions out. Lastly, I am very thankful to my undergraduate advisor Ilaria Rebay, who took a chance, asked me to work in her lab, and introduced me to research. I have to say thank you to my amazing friends. Jeremy Amon, Alex Wu and I spent many hours bowling, eating salads at the Sizzler, and discussing everything from contemporary music videos to new frontiers in salads. I would not have had 1/10th the fun or be 1/10th the person I am today (for better or worse) without them. I certainly would have never experienced children’s day at the Chabot Gun Club. I am also extremely grateful to have found kindred adventuring spirits in Eric Estrin, Crystal Pierce, Erik Lokensgard, Debbie Lehmann and Sarah Price. I’ll never forget camping in the snow and rain, road trips and wanderings with all of you. If I could still request a music video for y’all, it would be Kelly Clarkson’s “My Life Would Suck Without You”. Finally, I thank Mom, Dad, Faith, Alyssa, Caelin, Jeremy, Grandma, Ben, Christine, Emerson and my favorite aunt Dixie. It is impossible to thank you enough for all that you have done for me. I won’t try to put anything in words, other than to offer the following work as thanks to you. iii List of tables Table 2.1 Yeast strains used in this study 19 Table 2.2 Plasmids used in this study 24 Table 3.1 Known Ypk1 substrates and potential substrates predicted by MOTIPS listed under GO Slim terms 39 Synthetic dosage lethality of various Fps1 mutants in ypk1-as ypk2Δ cells 90 Table 5.1 iv List of figures Figure 1.1 Sequence alignment of Ypk1 and Ypk2 9 Figure 1.2 TORC2-Ypk signaling pathway 10 Figure 1.3 Initially identified Ypk substrates give insight into TORC2-Ypk regulation of membrane homeostasis 11 Figure 3.1 A three-part screen to identify likely Ypk1 substrates 36 Figure 3.2 Identified Ypk substrates 38 Figure 4.1 Lac1 and Lag1 are components of the ceramide synthase enzyme complex 56 Genetic and biochemical screening indicates Lac1 and Lag1 are Ypk1 substrates 57 Figure 4.3 Ypk1 phosphorylates Lac1 and Lag1 at S23 and S24 in vivo 58 Figure 4.4 Enhanced Ypk1 phosphorylation of Lac1 and Lag1 under sphingolipid and heat 59 Activation of calcineurin leads to rapid dephosphorylation of Lac1 and Lag1 without affecting Ypk1 function 60 Ypk1 phosphorylation of Lac1 and Lag1 stimulates ceramide synthase activity 61 Failure of Ypk1 to upregulate ceramide synthase causes LCBP accumulation that triggers autophagy 63 Figure 4.2 Figure 4.5 Figure 4.6 Figure 4.7 Figure 4.8 TORC2-Ypk1 signaling globally activates sphingolipid synthesis, selectively directs flux toward ceramide metabolites, and preventsLCBP cross-talk to the autophagy pathway 65 Figure 4.9 Loss of Ypk activity or putative Ypk phosphorylation of Atg21 does not appear to prevent CVT or amino acid starvation induced autophagy 67 Figure 5.1 Gpt2 is not phosphorylated by Ypk or other basophilic kinases 78 Figure 5.2 Smp1 is phosphorylated at predicted Ypk sites and phosphorylation of these residues is important for Smp1 function 79 v Figure 5.3 Figure 5.4 Figure 5.5 Figure 5.6 Figure 5.7 Figure 5.8 Changes in Fps1 dosage does not affect TORC2 of Pkh phosphorylation of Ypk1 Ypk in response to TORC2 signaling phosphorylates Fps1 at three sites in vivo 81 82 TORC2 phosphorylation of Ypk1 decreases rapidly in response to hyperosmotic shock resulting in Fps1 dephosphorylation 84 Phosphorylation of Fps1 by TORC2-Ypk increases channel transport activity 85 TORC2-Ypk regulates Fps1 independently of known regulators Hog1 and Rgc1/2 87 Saccharomyces cerevisiae utilizes two independent osmosensing systems to rapidly increase intracellular glycerol upon hyperosmolarity 89 vi List of abbreviations 1-NM-PP1 3-MB-PP1 ATP BSA CoA CVT DAPI DHAP DHS DHS-1-P DMSO DNA DTT EDTA EGTA GFP GST IPC IPTG LC LCB LCB-1-P MAPK MIPC MOTIPS MS/MS OD600 PCR PHS PHS-1-P PQD SC SEM SDL SDS-PAGE SPOTS TAP TCA TLC TOR TORC1 TORC2 YP 1-(1,1-dimethylethyl)-3-(1-naphthalenylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4amine 1-(tert-Butyl)-3-(3-methylbenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine Adenosine triphosphate Bovine serum albumin Co-enzyme A Cytoplasmic to vacuole pathway 4',6-diamidino-2-phenylindole Dihydroyacetone phosphate Dihydrosphingosine Dihydrosphingosine-1-phosphate Dimethylsulfoxide Deoxyribonucleic acid Dithiothreitol Ethylenediaminetetraacetic acid Ethylene glycol tetraacetic acid Green fluorescent protein Glutathione S-transferase Inositol phosphorylceramide Isopropyl β-D-1-thiogalactopyranoside Liquid chromatography Long chain base Long chain base-1-phosphate Mitogen activated protein kinase Mannosylinositol phosphorylceramide Motif analysis pipeline Tandem mass spectrometry Optical density at 600 nm Polymerase chain reaction Phytosphingosone Phytosphingosone -1-phosphate Pulsed-Q dissociation Synthetic complete medium Standard error of the mean Synthetic dosage lethality Sodium dodecyl sulfate-polyacrylamide gel electrophoresis Serine palmitoyl-CoA transferase complex Tandem affinity purification Trichloroacetic acid Thin layer chromatography Target of rapamycin Target of rapamycin complex 1 Target of rapamycin complex 2 Yeas extract peptone medium vii Chapter 1: Introduction Molecular composition of the eukaryotic plasma membrane All cells are bound by a ~30 Å thick hydrophobic membrane that defines the cell’s boundary, ensures cellular integrity and mediates interactions between the individual cell and its environment. In eukaryotic cells, this plasma membrane is a complex supramolecular assembly composed of thousands of different chemically diverse lipid and protein species (Guan and Wenk, 2006; Shevchenko and Simons, 2010). It is clear that the plasma membrane and its compositional complexity is important to eukaryotic organisms, as ~5% of the protein coding capacity of the human genome is dedicated to lipid metabolism (van Meer et al., 2008) and ~26% of the genome codes for membrane proteins (Fagerberg et al., 2010). Below I will outline the major molecular classes that compose the plasma membrane, before moving to an overview of how these species are spatially organized in biological membranes. Lipids are the major structural component of biological membranes and they represent ~50% of eukaryotic membrane mass (Cooper and Hausman, 2013). Membrane lipids are of three major classes: glycerophospholipids, sphingolipids and sterols (van Meer et al., 2008). The main lipid class of eukaryotic membranes is glycerophospholipids; ~70-80% of membrane phospholipids are of this type. These lipids consist of a hydrophobic diacylglycerol moiety, containing fatty acyl chains of various lengths and degrees of unsaturation. Various hydrophilic head groups can be attached to diacylglycerol giving rise to the large variety of glycerophospholipids observed in eukaryotic membranes. The most common glycerophospholipids observed in cellular membranes are phosphatidylcholine (~40% membrane phospholipid by mole), phosphatidylethanolamine (~20%), phosphatidylserine (~10%) and various phosphatidylinositols (~2-5%) (van Meer et al., 2008). Sphingolipids are the second major membrane lipid class and are structurally distinct from glycerophospholipids. The hydrophobic sphingolipid backbone is ceramide, a molecule composed of a long chain (sphingoid) base that is amide linked to a very long chain saturated fatty acid. As with glycerophospholipids, various polar head groups (typically composed of carbohydrates) can be appended to ceramide to form the full complement of cellular sphingolipids (Futerman and Hannun, 2004). Sphingolipids typically represent ~25% of all plasma membrane lipids (van Meer et al., 2008). Lastly, sterols are a third structurally different class of lipid. Sterols are planar molecules composed of multiple carbon rings resulting from cyclization of squalene derived from the mevalonate biosynthetic pathway (Buhaescu and Izzedine, 2007). They are, with the exception of a hydroxyl group, non-polar molecules and they are the major non-polar lipid species in eukaryotic membranes (van Meer et al., 2008). In addition to lipid species, thousands of various membrane proteins also reside in the membrane (Fagerberg et al., 2010). These proteins play diverse biological roles, primarily acting as receptors, transporters and enzymes (Sachs and Engelman, 2006; Almén et al., 2009). Thus, proteins are also a major component and source of compositional complexity in membranes. It is also becoming clear that these molecules are not merely passengers in the lipid membrane where 1 they perform functions, but many of these proteins also play important roles in generating and maintaining membrane organization, as discussed below. Molecular organization of the eukaryotic plasma membrane In recent decades, it has become increasingly clear that the thousands of lipid and protein species that compose the plasma membrane are not homogenously mixed, as often illustrated in textbook models of the membrane, for example, as when depicting the fluid mosaic model (Singer and Nicolson, 1972). Rather, the plasma membrane is a highly structured organelle. Localization and organization of specific lipid species has been observed along two axes: laterally, with respect to the plane of the bilayer; and, trans-bilayer, with respect to each of the two leaflets. In the lateral axis, the physical properties of sphingolipid and sterol species allows them to cluster away from glycerophospholipids and form local phase separations termed “lipid rafts” (Simons and van Meer, 1988). Lipid rafts were initially defined as small (10-200 nm diameter) domains of the membrane enriched in sphingolipids and sterols. The very long saturated acyl chains of membrane sphingolipids was presumed to lead to thickening of membrane in these domains (Gandhavadi et al., 2002), which could in turn lead to recruitment of certain membraneembedded and membrane-associated proteins, especially GPI-anchored protein or Spalmitoylated proteins (Brown and Rose, 1992; Zacharias et al., 2002; Foster et al., 2003). The function and even the existence of lipid-mediated membrane domain organization in live cells has been controversial (Simons and Sampaio, 2011). However recent advances in resolution and dynamics of live cell microscopy has revealed the presence of such structures in cells (Eggeling et al., 2009). In addition to lipid-mediated lateral membrane organization, cells use other protein-driven processes to generate membrane organization. Local restriction of lipids and membrane proteins by cortical cytoskeleton “fences” has been observed to form membrane domains (Chichili and Rodgers, 2009). Membrane-associated proteins also generate lateral membrane organization. For example, cytokinetic septin proteins alter lipid diffusion, generating regions of distinct lipid composition (Beise and Trimble, 2011; Clay et al., 2014) and BAR domain-containing proteins bend membranes forming stable domains (Ziółkowska et al., 2011; Zhao et al., 2013). Integral membrane proteins themselves generate lateral membrane organization. Caveolin proteins form sphingolipid- and sterol-enriched domains, termed caveolae (Monier et al., 1996). In the context of integral membrane proteins, both protein-protein and protein-lipid interactions have been observed to generate domains (Douglass and Vale, 2005). Lastly, cells also use directed vesicular transport to form stable lateral membrane domains (Dustin and Groves, 2012). In addition to lateral membrane organization, bilayer asymmetry is also a well established property of all biological membranes and has been demonstrated in many cell types (Devaux, 1991). Sphingolipids and phosphatidylcholine predominate in the extracellular leaflet of the membrane bilayer, whereas other glycerophospholipids are enriched in the inner leaflet (van Meer et al., 2008). Lipids are able to spontaneously “flip” from one leaflet to the other, although kinetically this is a slow process (Kornberg and McConnell, 1971). Therefore, to maintain bilayer asymmetry, cells use a variety of membrane-embedded enzymatic machines (e.g. "flippases, floppases and scramblases") to translocate lipids from one bilayer leaflet to the other 2 (Sharom, 2011). Lastly, beyond the structuring and ordering of lipid species in the plasma membrane, there is also dramatic ordering of the protein content of the plasma membrane. Oligomeric membrane-binding proteins have been observed to form microdomains (Monier et al., 1995; Ziółkowska et al., 2011) and represent one such class of membrane protein organization. Additionally, signaling proteins have been observed to cluster at “signaling synapses” and play important role in cellular signaling events (Dustin and Groves, 2012). Recently, high-throughput microscopy experiments have found that membrane proteins in general are not diffusely localized in the membrane. Rather they are found in multiple non-overlapping domains (Spira et al., 2012). Thus, distinct clustering of lipids and proteins leads to a patchwork organization of the membrane - a "mosaic tile" model (Engelman, 2005) rather than the rapid diffusion and homogeneity of the previously proposed "fluid mosaic" model (Singer & Nicolson, 1972). Role of plasma membrane organization and composition in cellular physiology It is increasingly being appreciated that the above described membrane organization is crucial for determining the physical properties of biological membranes and thus their function. Studies have found that loss of bilayer asymmetry reduces the mechanical stability of cellular membranes (Manno et al., 2002) and leads to dramatic defects on the ability of membranes to bend and bud off vesicles (Devaux, 1991; Williamson and Schlegel, 1994; Devaux et al., 2008). Accordingly, flippase function and regulation has been found to be important for aspects of cellular physiology including intracellular membrane trafficking (Hua et al., 2002; Fairn et al., 2011), polarized cell growth (Saito et al., 2007; Das et al., 2012), cell signaling (Sartorel et al., 2015), and cytokinesis (Roelants et al., 2015). Recent work demonstrates that absence of a flippase causes mislocalization of a Drosophila olfactory receptor (Ha et al., 2014). At the organismal level, loss of flippases and membrane asymmetry results in a number of pathologies (Ikeda et al., 2006), including profound defects in lymphocyte development (Clark, 2011). Thus, asymmetric lipid organization in the bilayer axis is of functional importance. In addition to the functional roles ascribed to bilayer asymmetry, lateral membrane asymmetry is increasingly understood to have functional roles in cellular physiology as well. This lateral compartmentation of the plasma membrane has been well documented in fungi and plants (Malinsky et al., 2010; Malinsky et al., 2013). In animal cells, lateral membrane microdomains are important for propagation of extracellular signals across membranes (Dustin and Groves, 2012), examined particularly thoroughly in the case of Ras (Schmick et al. 2015; Zhou & Hancock, 2015), and membrane trafficking processes (Simons and Sampaio, 2011). Recent studies have found that just displacing membrane proteins from their appropriate microdomains, even while leaving them intact in the plasma membrane can drastically affect membrane protein function (Berchtold et al., 2012; Spira et al., 2012). Thus, lateral membrane organization does indeed seem to play important roles in various cellular processes. Lastly, membrane composition is also incredibly important for determining function. Classically, in order to understand the functional role of certain lipids in cellular function, cells defective in biosynthesis of certain lipids (either by genetic manipulation or pharmacological inhibition) were phenotypically compared to appropriate control cells. For example, by studying mutants 3 defective in sphingolipid metabolism, it was found that sphingolipids are essential for endocytosis (Zanolari et al., 2000). This experimental modality has successfully identified functional roles for certain lipid classes and by this approach composition has been shown to affect membrane fluidity, the function of membrane proteins and membrane trafficking (Spector and Yorek, 1985). Recent advances in mass spectrometry based analytical chemistries have made it possible to quantitatively examine all membrane lipids, termed the lipidome (Ejsing et al., 2009; Brügger, 2014). These techniques have been used to identify and quantify dynamic changes to the cellular lipidome in response to various stimuli, during transit through the cell division cycle, and in various mutant backgrounds (Atilla-Gokcumen et al., 2014; da Silveira Dos Santos et al., 2014; Casanovas et al., 2015). Excitingly, these unbiased lipidomics approaches have identified dynamic changes in lipid composition that are required for various cellular processes, such as a requirement for glycosphingolipid biosynthesis in cytokinesis (Atilla-Gokcumen et al., 2011; Atilla-Gokcumen et al., 2014) and a requirement for increased cardiolipin synthesis during the diauxic shift to mitochondrial respiration in the yeast Saccharomyces cerevisiae (Casanovas et al., 2015). These recent findings from lipidomic analysis suggest that dynamic changes in membrane composition are an important component of the cellular response to many stimuli and during developmental processes, and further that many new functional roles for lipids in these processes remain to be discovered. TORC2-Ypk signaling and maintenance of membrane homeostasis Given that membrane composition and organization (collectively, I will refer to these properties as 'membrane status') is of vital importance to cellular function, and that cells appear to dynamically alter their membranes in response to environmental stimuli, it is important to understand what mechanisms cells use to sense membrane status and take action to restore functional homeostasis. The model eukaryote S. cerevisiae has proven useful in identifying a number of signaling pathways that respond to various types of membrane stress. Using this organism, mitogenactivated protein kinase (MAPK) pathways have been shown to be important in eliciting the responses required by the cell to survive a number of different stressful conditions. Signaling pathways that activate the MAPK Hog1 are important for viability under hyperosmotic conditions, a stress that leads to cell shrinkage and membrane compression (Hohmann, 2015; Saito and Posas, 2012). The MAPK Slt2/Mpk1 downstream of Pkc1 signaling is important for survival under hypotonic conditions and after physical wounding of the cell wall (Levin, 2011; Kono et al., 2012). These signaling pathways are conserved across eukaryotes [mammalian ortholog of Hog1 is MAPK p38 and the mammalian ortholog of Slt2 is MAPK Erk5], and mammalian cells similarly use MAPKs (Zarubin and Han, 2005) and PKC (Togo et al., 2000; Liu et al., 2003; Hermoso et al., 2004; Bullard et al., 2007) to respond to osmolarity change or membrane stress. Another less well characterized, yet important, membrane stress-responsive signaling pathway controls two highly related effectors: protein kinases Ypk1 and Ypk2 (formerly Ykr2). As assessed by genetic analysis, these two enzymes appear to carry out vital functions in membrane homeostasis. Cells lacking both Ypk1 and Ypk2/Ykr2 are inviable (Chen et al., 1993; Roelants et al., 2002). Phenotypic characterization of cells deficient in Ypk activity revealed that they have 4 dramatic defects in endocytosis, fail to couple cell wall growth to membrane growth, and are dramatically sensitive to inhibitors of sphingolipid biosynthesis (Sun et al., 2000; deHart et al., 2002; Roelants et al., 2002; Schmelzle et al., 2002; Roelants et al., 2004; Roelants et al., 2010). All of these phenotypes suggested that Ypk1 and Ypk2 play an important role in maintaining membrane homeostasis. Ypk1 and Ypk2 are very similar proteins (especially in their kinase domain) (see Fig. 1.1) and are functionally semi-redundant (Chen et al., 1993; Roelants et al., 2002; Roelants et al., 2004). These paralogs likely arose from a wholesale genome duplication that occurred during the evolutionary path that led to modern-day S. cerevisiae (Byrne and Wolfe, 2005). Ypk1 and Ypk2 are members of the AGC family of protein kinases, one of the conserved groups in this enzyme super-family (Pearce et al., 2010). Mammalian SGK1/SGK (serum-and glucocorticoid-induced protein kinase) is the sequence and functional ortholog of yeast Ypk1 (Casamayor et al., 1999). Activation of the catalytic activity of AGC kinases requires phosphorylation of a conserved Thr in their activation loop ("T-loop"). This modification causes conformational changes that make the enzyme competent to catalyze phosphoryl transfer from ATP to a target protein (Pearce et al., 2010). The upstream activating protein kinases that mediate T-loop phosphorylation of Ypk1 and Ypk2 are the protein kinases Pkh1 and Pkh2. Mammalian PDK1 (2-phosphoinositide-dependent protein kinase) is the sequence and functional ortholog of Pkh1 and Pkh2 (Casamayor et al., 1999; Roelants et al., 2002; Roelants et al, 2004). Pkh1 and Pkh2 localize in cortical puncta at the cell periphery (Roelants et al., 2002) in tight association with two PtdIns4,5P2-binding proteins (Pil1 and Lsp1) that are major components of plasma membrane-bound complexes, referred to as eisosomes (Walther et al., 2006; Walther et al., 2007). Thus, Pkh1 and Pkh2 could potentially serve as sensors of some aspect of membrane status. In addition to T-loop phosphorylation, AGC kinases can be further activated by phosphorylation of conserved sites - the so-called turn and hydrophobic motifs sites - in the carboxy-terminal extension downstream of their catalytic domain (Pearce et al., 2010). Complexes containing the atypical protein kinase TOR (target of rapamycin) are responsible for phosphorylation of AGC kinases at these C-terminal sites (Pearce et al., 2010). In yeast, there are two TOR paralogs, Tor1 and Tor2 (Heitman et al. 1991), whereas in mammalian cells there is a single gene product (mTOR/RAFT/RAPT/FRAP) (Sabatini et al., 1994). One of the TOR-containing complexes in yeast, TORC1, contains either Tor1 or Tor2, as well as other subunits diagnostic of this protein ensemble that ensure its localization to the cytosolic face of the lysosome and dictate its substrate specificity (Loewith et al., 2002; Wedaman et al., 2003; Reinke et al., 2004; Loewith & Hall, 2011). The other TOR-containing complex in yeast, TORC2, contains only Tor2, as well as other subunits diagnostic for this assembly that direct its localization to the plasma membrane and impose its substrate specificity (Loewith et al., 2002; Wedaman et al., 2003; Reinke et al., 2004; Loewith & Hall, 2011). Mammalian cells have highly homologous TORC1 and TORC2 complexes that contain mTOR (Kim et al., 2002; Sarbassov et al., 2004; Huang & Manning, 2009; Betz & Hall, 2013). TORC1 and TORC2 regulate different aspects of cell physiology. TORC1, but not TORC2, is specifically sensitive to inhibition by the macrolide rapamycin and its structural relatives ("rapalogs") (Benjamin et al., 2011; Chiarini et al., 2015), which has allowed for extensive characterization of the cellular functions of TORC1 (Loewith et al., 2002; Loewith & Hall, 2011; 5 Albert & Hall, 2015). TORC1 is most active when nutrients, especially amino acids, and in particular Leu and Gln (Jewell et al. 2015), are available in both yeast (Panchaud et al., 2013; Chantranupong et al., 2015) and animal cells (Bar-Peled & Sabatini, 2014; Efeyan et al., 2015) and, in animal cells, when cells have been concomitantly stimulated with a growth factor (e.g. EGF, PDGF, insulin, etc.) that activates PtdIns 3-kinase (Huang & Manning, 2009; Chiarini et al., 2015). Active TORC1 phosphorylates targets that promote anabolic processes, such as ribosome biogenesis, protein synthesis, pyrimidine biosynthesis and lipogenesis, while inhibiting catabolic processes, such as autophagy and lysosome biogenesis (Laplante and Sabatini, 2012; Howell et al. 2013; Albert & Hall, 2015). In contrast, there has not been (until recently, see Kliegman et al., 2013; Rispal et al., 2015), a specific inhibitor of TORC2; hence, our understanding of the physiological functions of this complex is more rudimentary. What is known is that TORC2 mediates phosphorylation of AGC kinases distinct from those phosphorylated by TORC1 (Pearce et al., 2010). In yeast, phosphorylation of Ypk1 and Ypk2 at their C-terminal turn and hydrophobic motifs is mediated by TORC2 and phosphorylation at these sites does considerably increase their catalytic activity (Kamada et al., 2005; Roelants et al., 2011). In yeast, tor1∆ mutants are viable, whereas tor2∆ are inviable, indicating that TORC2 function is essential. Strikingly, the absence of TORC2 activity can be rescued by alleles of Ypk1 or Ypk2 that make these enzymes constitutively hyperactive, suggesting that the sole function of TORC2 that is essential for cell viability is activation of Ypk1 and Ypk2 (Kamada et al., 2005; Roelants et al., 2011. Because Ypk1 and Ypk2 are the effectors that execute the essential function(s) of TORC2, its stands to reason, therefore, that characterization of the substrates of Ypk1 and Ypk2 should shed considerable light on those aspects of cellular physiology that are under TORC2 control. In this regard, and given the phenotypes of Ypk-deficient cells, initial research on the roles of Ypk1 and Ypk2 centered on attempts to understand how membrane status is sensed and relayed to the Ypk kinases. Based on in vitro biochemical assays, two groups (Friant et al., 2001; Liu et al., 2005) reported that the activity of Pkh1 and Pkh2 could be stimulated by addition of long chain sphingoid base (mainly, phytosphingosine in yeast), suggesting that the Pkh kinases might serve as sensors of plasma membrane composition that could, in turn, modulate Ypk kinase activity appropriately to adjust the rates of the reactions necessary to maintain homeostasis. However, more recent results demonstrated that, in vivo, T-loop phosphorylation of the Ypk kinases by the Pkh kinases is not subject to control by sphingolipids or their precursors (Roelants et al., 2010). Therefore, it appears that other Ypk kinase regulators must be the sensors of membrane status, and TORC2 clearly represents one of the most likely candidates for that role. Indeed, more recent work has found that TORC2 activity is sensitive to a variety of membrane stresses. Three types of membrane stress - inhibition of sphingolipid synthesis (Roelants et al., 2011), heat shock (Sun et al., 2012), and hypotonic conditions (or mechanical stretch) (Berchtold et al., 2012) - clearly elevate TORC2 function. In these cases, change in TORC2 activity was measured indirectly by monitored the status of Ypk phosphorylation at its hydrophobic motif, by assessing the specific activity of immunoprecipitated Ypk1, and/or by evaluating the phosphorylation state of downstream Ypk1 substrates (e.g., Orm2). In the case of hypotonic stress, it appears that the increase in TORC2 activity is due to dissociation of two TORC2 components (Slm1 and Slm2) from the rest of the TORC2 complex and their sequestration into a separate plasma membrane microdomain (Berchtold et al., 2012; Niles et al., 2012). Conversely, 6 upon exposure of cells to hyperosmotic conditions, our laboratory has found, in collaborative studies, that TORC2 activity dramatically decreases (Lee et al., 2012b), although the precise mechanism by which this rapid and marked reduction in apparent activity occurs has not yet been determined. In summary, although T-loop phosphorylation by the Pkh kinases is necessary to license Ypk activity, it appears that TORC2 senses and responds to membrane stress and conveys this information to Ypk1. Furthermore, the downstream outputs under the control of TORC2-Ypk1 are important for maintaining membrane homeostasis, as judged by the fact that cells defective in TORC2-Ypk signaling exhibit dramatic membrane-related phenotypes. Pkh- and TORC2depndent Ypk signaling and its membrane phenotypes are schematized in Figure 1.2. Outstanding questions and outline of this thesis These findings raised an obvious and important question - what processes does TORC2-Ypk signaling modulate in order to maintain membrane homeostasis in response to stressful stimuli? In contrast to the hyperosmotic stress responsive Hog1 MAPK pathway and the hyposmotic stress responsive Rho1-dependent Pkc1 pathway (sequence ortholog of mammalian RHOactivated PKN2/PRK2), where the downstream functional outputs are largely known (Levin, 2011; Saito and Posas, 2012; Hohmann, 2015), relatively little was understood about the downstream responses of TORC2-Ypk to membrane stress. However, two approaches provided initial insight about the downstream effectors of TORC2-Ypk stress-induced signaling. The first tactic was a genetic approach. To find factors whose full activity requires Ypk-mediated phosphorylation, a selection for dosage suppressors of the temperature sensitivity of a ypk1-ts ypk2∆ strain was conducted (Roelants et al., 2002), which yielded plasmids containing the SMP1 gene, which encodes a MADS box-family transcription factor that I have now demonstrated (see Chapter 5) is an authentic in vivo substrate of Ypk1. Conversely, to find factors whose activity is inhibited by Ypk-mediated phosphorylation, a selection for transposon insertion mutations that suppressed the temperature sensitivity of ypk1-ts ypk2∆ cells was conducted (Roelants et al., 2002). This screen recovered Orm2, which is an inhibitor of the first enzyme unique to sphingolipid biosynthesis, L-serine:palmitoyl-CoA acyltransferase comples (SPOTS) (Breslow et al., 2010). Subsequently, it was found that Ypk1 phosphorylates and inhibits the Orm proteins, and thus TORC2-Ypk1 signaling increases the rate of formation of the long-chain base precursor (phytoshingosine) to yeast sphingolipids (Berchtold et al., 2012; Roelants et al., 2011). Thus, increased TORC2-Ypk1 signaling in response to sphingolipid depletion or membrane stretch allows cells to increase sphingolipid production to manage these stresses (Berchtold et al., 2012; Roelants et al., 2011). A more recent chemical genetic screen in which a small molecule inhibitable tor2-as allele was systematically introduced to a collection of yeast mutants deleted for single genes similarly found genetic interactions between TORC2 and sphingolipid biosynthetic genes (Kliegman et al., 2013). This screen also found intriguingly that TORC2 function is required for the pentose phosphate pathway that generates high energy metabolites 7 needed for nucleotide and lipid biosynthesis. Thus, genetic screening has proven useful in identifying functionally important Ypk substrates and TORC2 regulated processes. Another approach for identifying downstream effectors of TORC2-Ypk1 signaling was biochemical. Use of synthetic peptides indicated that Ypk1 has a rather stringent phosphoacceptor site sequence preference (Casamayor et al., 1999), which was fully confirmed wben much more extensive combinatorial peptide libraries were employed (Mok et al., 2010). By scanning the yeast proteome for proteins with occurrences of this motif that also had some connection to plasma membrane physiology, a candidate substrate emerged, namely Fpk1 (and its paralog Fpk2), a protein kinase responsible for activation of plasma membrane-localized aminophospholipid flippases (Nakano et al. 2008). Indeed, it was then demonstrated that Ypk1 phosphorylates and negatively regulates Fpk1 both in vitro and in vivo (Roelants et al., 2010). Thus, in addition to stimulating production of sphingolipid precursor, TORC2-Ypk fine-tunes the glycerophospholipid composition of the inner leaflet of the membrane bilayer. Similarly, based in its phosphoacceptor motif, it was found that Ypk1 also phosphorylates and inhibits one of the two isoforms (Gpd1) of glycerol-3-phosphate dehydrogenase, a primary source of glycerol-3-P for production of glycerophospholipids (Lee et al., 2012b), but also, when dephosphorylated, a source of the glycerol that yeast cells produce as a innocuous osmo-protectant when subjected to hyperosmotic conditions (Hohmann, 2009). As discussed above, we have found that TORC2 activity is markedly decreased during hyperosmotic shock, which prevents Ypk1-mediated inhibition of Gpd1, thereby increasing synthesis of glycerol-3-P, which can be dephosphorylated to produce the glycerol osmolyte that yeast cells accumulate as a means to counteract the increase in external osmotic pressure (Lee et al., 2012b). Thus, by identifying only a handful of Ypk substrates (Fpk1, Fpk2, Orm1, Orm2 and Gpd1), a great deal was learned at the mechanistic level about how Ypk function maintains membrane homeostasis downstream of stimuli that both increase and decrease TORC2 activity. This degree of progress is outlined in Figure 1.3. Nonetheless, an important outstanding question remains what other substrates in the cell are regulated by Ypk action? I felt that identification of previously uncharacterized substrates would undoubtedly give further insight not only about how TORC2-Ypk signaling is connected to membrane biology, but about those essential aspects of cell physiology that are under the control of TORC2. Thus, the main aim of my doctoral dissertation research was to develop a genome-wide strategy to systematically identify and characterize new Ypk kinase substrates. As described in this thesis, I devised a novel approach towards matching substrates with cognate kinases and applied it to identify Ypk1 substrates (Chapter 3). I then proceeded to characterize in depth a number of the candidate Ypk1 substrates that I pinpointed by applying my methodology. Chapter 4 documents TORC2-Ypk-mediated regulation of Lac1 and Lag1, conserved catalytic subunits of the ceramide synthase complex, an enzyme central for the production of sphingolipids. Finally, in Chapter 5, I present studies showing that TORC2-Ypk regulates Fps1 (an aquaglyeroporin) and Smp1 (a transcription factor), two substrates involved in the cellular response to hyperosmotic shock, which led me to the discovery that TORC2-Ypk signaling is a novel MAPK-independent pathway that cells use to survive the challenge of hyperosmotic stress. 8 Figures Fig. 1.1: Sequence alignment of Ypk1 and Ypk2. Primary sequence diagrams of Ypk1 and Ypk2 are shown with regions of significant sequence similarity shown. Darker coloring indicates higher sequence similarity. The percent identity between each region is indicated. 9 Fig. 1.2: TORC2-Ypk signaling pathway. Diagram outlines known relationships between TORC2 [complex members and interactions adopted from (Loewith et al., 2002; Wullschleger et al., 2005)], Pkh and Ypk kinases. Downstream of Ypk, membrane related phenotypes that are observed in Ypk mutants are shown. 10 Fig. 1.3: Initially identified Ypk substrates give insight into TORC2-Ypk regulation of membrane homeostasis. Diagram of the TORC2-Ypk signaling pathway from Fig. 1.1 is shown. However, the known substrates of Ypk kinases are now shown. Arrows indicate interactions that increase activity, while blunted lines indicate interactions that decrease activity of the indicated protein. Identification of these 5 substrates gives mechanistic insight into the membrane phenotypes associated with loss of TORC2-Ypk signaling. Identification of the flippase activating kinases Fpk1/2 as Ypk1 substrates mechanistically links TORC2-Ypk signaling to membrane asymmetry. Identification of TORC2-Ypk regulation of the Orm1/2 proteins , negative regulation of the SPOTS complex, implicates TORC2-Ypk as an activator of sphingolipid metabolism. This explains why Ypk deficient strains are sensitive to sphingolipid biosynthetic inhibitors. Lastly, identification of Gpd1 as a Ypk substrate links TORC2-Ypk signaling to osmotic induced glycerol metabolism, an essential feature of the hyperosmotic shock response in S. cerevisiae. This provides mechanistic insight to the osmotic shock sensitivity observed in Ypk deficient strains. 11 Chapter 2: Materials and Methods Construction of yeast strains All S. cerevisiae strains used in this study are listed in Table 2.1. Strains were constructed using standard yeast genetic manipulations (Burke et al., 2005). For all strains constructed, integration of the desired DNA fragment into the correct genomic loci was confirmed by PCR using an oligonucleotide complementary to the integrated DNA fragment and an oligonucleotide complementary to genomic sequence at least 150 bases away from the integration site. After confirmation that the DNA fragment was integrated at the correct locus, additional PCRs were performed to generate fragments covering the integrated DNA fragment. These reaction products were sequenced to confirm the DNA fragment introduced into the genome had the correct sequence. Plasmids and recombinant DNA methods All plasmids used in this study (except the library of PGAL1-based overexpression plasmids for synthetic dosage lethality (SDL) screening) are listed in Table 2.2. All plasmids were constructed and progated in E. coli using standard laboratory methods (Green and Sambrook, 2012) or by Gibson assembly (Gibson et al., 2009) using the Gibson Assembly Master Mix Kit according to manufacturers specifications (New England Biolabs). For SDL screening, the entire open reading frame of each predicted and known Ypk1 substrate was amplified by PCR from BY4741 genomic DNA and ligated into the multiple cloning site of YCpLG (CEN, PGAL1, LEU2), generating a vector allowing galactose-inducible overexpression of each substrate. All constructs generated in this study were confirmed by sequence analysis covering all promoter and coding regions in the construct. Bioinformatic prediction of Ypk1 substrates A Nx20 position weight matrix defining Ypk1 phosphoacceptor site specificity was made merging previously published data sets defining Ypk1 peptide substrate selectivity (Casamayor et al., 1999; Mok et al., 2010). This position weight matrix was then used with MOTIPS (Lam et al., 2010) to identify proteins with likely phosphorylated occurrences of this motif. Yeastmine (Balakrishnan et al., 2012) was used to identify all S. cerevisiae genes that showed genetic interactions with Ypk1, Ypk2, any component of the sphingolipid biosynthetic pathway or any component of known Ypk1 regulators (TORC2 and protein phosphatase 2 A). Genes that showed significant growth phenotypes on myriocin, aureobasidin A and caspifungin (all compounds that cause severe growth phenotypes in ypk1Δ strains) were identified from the literature (Hillenmeyer et al., 2008). Lastly, MOTIPS predicted Ypk1 phosphorylation sites were compared to Phosphogrid (Sadowski et al., 2013) to identify those sites known to be phosphorylated in vivo. To be considered a potential Ypk1 substrate in this study, a protein had to have a myriocin, aureobasidin or caspofungin phenotype or a Yeastmine identified genetic interaction and: (a) 4 or more MOTIPS predicted sites, (b) 3 sites with at least one above a MOTIPS likelihood score of 0.7 or a Phosphogrid identified site or (c) 1-2 sites with a MOTIPS likelihood score of 0.7 and a Phosphogrid identified site. I also considered for further analysis a limited number of proteins that did not meet these criteria, but which still contained Ypk1 motifs. 12 These are indicated in Table 3.1. Yeast growth assays and synthetic dosage lethality screening For SDL screening, yAM135-A and BY4741 strains were both transformed with each SDL plasmid. Transformants were then cultured overnight in SC medium containing 2% raffinose and 0.2% sucrose. Ten-fold serial dilutions of overnight cultures starting from A600 nm = 1.0 were then made in sterile water and spotted onto SC solid medium with 2% galactose (to induce protein expression) or 2% dextrose (no protein expression). These plates also contained 1:1000 DMSO, 1 μM 3-MB-PP1 or 2 μM 3-MB-PP1 to inhibit to varying degrees Ypk1-as kinase activity in yAM135-A. Serially spotted cultures were allowed to grow in the dark at 30˚C for 3 days. Plates were then scanned on a flatbed scanner and growth phenotypes were assessed and scored. For all SDL overexpression constructs that caused toxicity upon overexpression in WT BY4741 strains, I also performed Ypk1 dosage rescue growth assays. PMET25-Ypk1 or PMET25Ypk1(K367A) (kinase-dead) plasmids were co-transformed into BY4741 with the toxic SDL plasmid. Transformants were then cultured overnight in SC medium containing 2% raffinose and 0.2% sucrose. Ten-fold serial dilutions of overnight cultures starting from A600 nm = 1.0 were then made in sterile water and spotted onto SC solid medium with 2% galactose (to induce protein expression) or 2% dextrose (no protein expression), in the presence of methionine to repress Ypk1 expression or in the absence of methionine to derepress Ypk1 expression. Serially spotted cultures were allowed to grow in the dark at 30˚C for 3 days. Plates were then scanned on a flatbed scanner and growth phenotypes were assessed to determine if Ypk1 overexpression could rescue toxicity of overexpression of the SDL protein. For broth growth assays, cultures in exponential phase in rich (YP) medium (Burke et al., 2005) with 2% dextrose were diluted to A600 nm = 0.1. A portion (100 μL) of each culture was placed in a well in a 96-well plate with vehicle or drug at the indicated concentration. Cultures were grown with orbital shaking at 30˚C in a Tecan Infinite M-1000 PRO plate reader (Tecan Systems Inc.) for 24 h. Absorbance measurements were taken every 15 min Absorbance values were converted to A600 nm values using a standard curve of absorbance values of cultures at known A600 nm taken on the same plate reader. Purification of Ypk1-as and purification of putative Ypk substrates To purify Ypk1-as kinase, pAX50 (2μ, PGAL1-Ypk1(L424A)-TAP, URA3) transformed yAM135A yeast were diluted to A600 nm = 0.125 in 3L of SC with 2% raffinose+0.2% sucrose and grown with shaking at 30˚C to mid-exponential phase. Expression was induced for ~18 h by the addition of 2% galactose. Cells were harvested by centrifugation and frozen in liquid nitrogen. The cells were then lysed cryogenically using Mixer Mill MM301 (Retsch). The lysate was resuspended at 2 mL/g in TAP-B [50 mM Tris-Cl pH 7.5, 200 mM NaCl, 1.5 mM MgOAc, 1 mM DTT, 2 mM NaVO4, 10 mM NaF, 10 mM Na-PPi, 10 mM β-glycerol phosphate, 1x cOmplete protease inhibitors™ (Roche)]. The lysate was clarified by centrifugation at 15,000xg for 20 min Clarified lysate was then further centrifuged at 100,000xg for 1 h and then brought to 0.15% NP-40 using 10% NP-40 detergent stock. Ypk1-as-TAP fusion was then affinity purified from the lysate using IgG-agarose resin (GE Healthcare). The resin was extensively washed with Protease 3C Buffer (50 mM Tris-Cl pH 7.5, 200 mM NaCl, 1.5 mM MgOAc, 1 mM DTT, 0.01% NP-40, 10% Glycerol, 2 mM NaVO4, 10 mM NaF, 10 mM Na-PPi, 10 mM β-glycerol 13 phosphate) and then resuspended in 1 mL Protease 3C Buffer. Ypk1-as was eluted by the addition of 80 U Prescission Protease™ (human rhinovirus 3C protease) (GE Healthcare) for 5h at 4˚C. Protease 3C was removed by the addition of glutathione-agarose (GE Healthcare). Each putative Ypk1 substrate (or a large fragment thereof) was fused to the C-terminus of GST in the vector pGEX-6P-1 and the resulting GST-substrate fusion proteins expressed in and purified from E. coli strain BL21. Cultures (1L) were grown at 37˚C to mid-exponential phase and then induced at room temperature for 4 h with 0.5 mM IPTG. Cells were harvested by centrifugation and the fusion proteins were purified by affinity chromatography using glutathione-agarose and standard procedures. Ypk1-as in vitro kinase assays For kinase assays, 0.25 μg of purified Ypk1-as was incubated with a purified GST-substrate in Kinase Assay Buffer (50 mM Tris-Cl, pH 7.5, 200 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA) with 2 μCi [γ-32P]ATP at 30˚C in the presence of absence of 10 μM 3-MB-PP1 for 30 min Reactions were terminated by the addition of SDS-PAGE sample buffer containing 6% SDS followed by boiling for 5 min Labeled proteins were resolved by SDS-PAGE and analyzed by Coomassie blue staining and autoradiography with Phosphorimager plates (Molecular Dynamics) on a Typhoon imaging system (GE Healthcare). Preparation of cell extracts and immunoblotting To extract proteins from yeast cells, an alkaline lysis and trichloroacetic acid (TCA) precipitation method were used, as previously described (Westfall et al., 2008). Cell extracts were then resolved by various SDS-PAGE methods described below prior to immunobotting. To resolve Lac1, Lag1, Fps1, Smp1(1-274) and Ypk1 phosphorylated species, 15 μL of TCA extract was resolved by SDS-PAGE [8% acrylamide, 35 µM Phos-tag™ (Wako Chemicals USA, Inc.), 35 µM MnCl2 at 160 V]. Inclusion of the Phos-tag™ reagent retards the mobility of and improves the resolution of phosphorylated species (Kinoshita et al., 2009). To resolve Gpt2 phosphorylated species, 15 μL of the TCA extract was resolved by SDS-PAGE [8% 75:1 acrylamide:bisacrylamide), at 120 V]. Reducing the degree of gel crosslinking by increasing the ratio of acrylamide monomer to bisacrylamide is a strategy also known to improve the separation between phosphorylated and unphosphorylated species (Huang et al., 1997). To resolve full length Smp1 phosphorylated species 15 μL of TCA extract was resolved by SDS-PAGE [8% acrylamide, 45 µM Phos-tag (Wako Chemicals USA, Inc.), 45 µM MnCl2 at 160 V]. After separation by SDS-PAGE, the resolved proteins were transferred to nitrocellulose and incubated with primary antibody in Odyssey buffer (Licor Biosciences), washed, and incubated with IRDye680LT-conjugated anti-mouse IgG (Licor Biosciences) in Odyssey buffer with 0.1% Tween-20 and 0.02% SDS. Blots were imaged using an Odyssey infrared scanner (Licor Biosciences). Primary antibodies and dilutions used in this study were: 1:1000 rabbit anti-HA (Covance Inc.), 1:5000 mouse anti-FLAG (Sigma-Aldrich), 1:5000 rabbit anti-FLAG (SigmaAldrich), 1:100 tissue culture medium containing mouse anti-c-myc mAb 9E10 (Monoclonal Antibody Facility, Cancer Research Laboratory, Univ. of California, Berkeley; 1:100), 1:500 14 mouse anti-GFP (Roche), 1:500 rabbit anti-pSGK (T256) (to recognize Ypk1 phosphorylated at T504) (Santa Cruz Biotechnology), 1:20000 rabbit anti-Ypk1(pT662) (a generous gift from Ted Powers), 1:10000 rabbit anti-Ape1 (a generous gift from Daniel Klionsky), and 1:10000 rabbit anti-PGK1 (our laboratory). Analysis of sphingolipid species Complex sphingolipids were analyzed by thin layer chromatography. Logarithmically growing cultures of strains were adjusted to A60o nm = 1.0 and 2 ml cultures were labeled with 100 µCi of [32P] H3PO4 and cells allowed to grow for 3 h. Lipids were extracted and resolved as previously described (Hanson and Lester, 1980; Momoi et al., 2004) with minor modifications. The cell pellet was washed twice with 2 ml water and treated with 5% trichloroacetic acid for 20 min on ice. Pellets were extracted twice with 0.75 ml of ethanol/water/diethyl-ether/pyridine/NH4OH (15:15:5:1:0.018) at 60°C for 1 h. Glycerophospholipids in the extract were hydrolyzed by treating with 0.1 M monomethylamine at 50°C for 1 h after which the base was neutralized by addition of 12 µl of glacial acetic acid. Lipids were extracted with 1 ml chloroform, 0.5 ml methanol and phases separated with addition of 1 ml water. For some samples 1 ml of 4 N NaCl was used, in order to facilitate the separation of the aqueous and organic phases. The organic layer was dried under vacuum and resuspended in 50 µl of chloroform/methanol/water (16:16:5) and resolved on a silica gel TLC plate with chloform/methanol/4.2 N NH4OH (9:7:2). Radioactivity on the TLC plate was visualized with a Phosphorimager screen and Typhoon imaging system. LCBs and LCB-1-Ps levels were monitored by liquid chromatography-mass spectrometryOvernight cultures were adjusted to A60o nm = 0.2 and allowed to grow to an A60o nm = 1.0. Cell pellets from 10 ml of this culture equivalent to 10 A600 nm units was used for analysis. C17sphingosine (Avanti Polar Lipids), (5 nmol) was added to all samples as an internal standard. Lipids were extracted as described above and the final dried lipid extract was dissolved in methanol/water/formic acid (79:20:1), centrifuged to remove insoluble material and an aliquot of this material was injected into the HPLC for analysis. Lipid extracts were analyzed using an Agilent 1200 liquid chromatograph (LC; Santa Clara, CA) that was connected in-line with an LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source (ESI; Thermo Fisher Scientific, Waltham, MA). The LC was equipped with a C4 analytical column (Viva C4, 150 mm length × 1.0 mm inner diameter, 5 µm particles, 300 Å pores, Restek, Bellefonte, PA) and a 100 µL sample loop. Solvent A was 99.8% water/0.2% formic acid and solvent B was 99.8% methanol/0.2% formic acid (v/v). Solvents A and B both contained 5 mM ammonium formate. The sample injection volume was 85 µL (partial loop). The elution program consisted of isocratic flow at 30% B for 2 min, a linear gradient to 65% B over 0.1 min, a linear gradient to 100% B over 4.9 min, isocratic flow at 100% B for 4 min, a linear gradient to 30% B over 0.1 min, and isocratic flow at 30% B for 18.9 min, at a flow rate of 170 µL/min The column and sample compartments were maintained at 40°C and 4°C, respectively. The injection needle was rinsed with a 1:1 methanol/water (v/v) solution after each injection. The column exit was connected to the ESI probe of the mass spectrometer using PEEK tubing 15 (0.005” inner diameter × 1/16” outer diameter, Agilent). Mass spectra were acquired in the positive ion mode over the range m/z = 250 to 1200 using the Orbitrap mass analyzer, in profile format, with a mass resolution setting of 100,000 (at m/z = 400, measured at full width at halfmaximum peak height). In the data-dependent mode, the five most intense ions exceeding an intensity threshold of 30,000 counts were selected from each full-scan mass spectrum for tandem mass spectrometry (MS/MS) analysis using pulsed-Q dissociation (PQD). MS/MS spectra were acquired in the positive ion mode using the linear ion trap, in centroid format, with the following parameters: isolation width 3 m/z units, normalized collision energy 40%, and default charge state 1+. A parent mass list was used to preferentially select ions of interest for targeted MS/MS analysis. To avoid the occurrence of redundant MS/MS measurements, real-time dynamic exclusion was enabled to preclude re-selection of previously analyzed precursor ions, using the following parameters: repeat count 2, repeat duration 30 s, exclusion list size 500, exclusion duration 180 s, and exclusion width 0.1 m/z unit. Data were analyzed using Xcalibur software (version 2.0.7 SP1, Thermo) and LIPID MAPS Online Tools (Fahy et al., 2007). Exact masses of precursor ions in positive ion (M+H+) state obtained from LIPID MAPS Online Tools were as follows- phytosphingosine- 318.3003, dihydrosphingosine-302.3053, phytosphingosine-1 phosphate-398.2666 and dihydrosphingosine-1 phosphate- 382.2717. The ions were further confirmed by tandem MS and identification of the fragments; product ions for the phytosphingosine headgroup was 282.3 and dihydrosphigosine headgroup was 266.4. In vitro ceramide synthase assay Yeast expressing 3xFLAG-Lag1 (wild-type and phospho-site mutations) were grown at 30°C to mid-exponential phase and microsomes were prepared as described previously (Schorling et al., 2001) and resuspended in B88 buffer (20 mM HEPES-KOH pH 6.8, 150 mM KAc, 5 mM MgOAc, and 250 mM sorbitol). Ceramide synthase was then immunopurified from these microsomes using anti-FLAG agarose (Sigma) as described previously (Vallée and Riezman, 2005). Prior to assembling ceramide synthase reactions, a small alioquot of each (15 μL) immunoprecipitate was taken and proteins were resolved by SDS-PAGE and immunoblotted with anti-FLAG to determine the relative amount of immunopurified ceramide synthase in each reaction. Recovered ceramide levels for each reaction were normalized to the amount of ceramide synthase determined by this procedure. Ceramide synthesis reaction was assembled in B88 buffer with 5 mg/ml BSA, in a total reaction volume of 0.1 ml containing 47.5 µL anti-FLAG agarose immunoprecipitates, 50 µM phytosphingosine and 0.1 mM of steroyl (C18)- CoA. Reactions were incubated at 30 °C for 1 h. Reactions were terminated by addition of 0.4 ml methanol/CHCl3 (2:1) and further extracted with 0.4 ml CHCl3 and 0.4 ml water. The CHCl3 phase was separated by centrifugation and removed and dried under vacuum. The dried lipids resuspended in methanol/water/formic acid (79/20/1) and an aliquot analyzed by LC-MS as described above. The product from the reaction, C18-phytoceramide was identified by the exact mass of its precursor ion at 584.5612 and its product ion upon fragmentation at 282.3. Additionally, authentic C18-phytoceramide (Matreya, LLC.) standard was used to obtain a standard curve in order to establish that values from ceramide synthase reaction were in the linear range of estimation. 16 Fluorescence microscopy of Smp1-GFP and Fps1-GFP Cells transformed with plasmids that express GFP-tagged proteins were grown in selective medium to mid-exponential phase (A60o nm ~0.6) or stationary phase. For cells stained with DAPI to visualize nuclear DNA, 2.5 μg/mL of DAPI was added to medium for 30 min Stained cells were then washed in fresh medium. Cells were viewed directly with an epifluorescence microscope (model BH-2; Olympus America, Inc.) using a 100X objective fitted with appropriate band-pass filters (Chroma Technology Corp.). Images were collected using a CoolSNAP MYO charge-coupled device camera (Photometrics). Measurement of STL1 promoter transcription by fluorescence activated cell sorting (FACS) Cells with PSTL1-GFP integrated at the HIS3 locus were grown to early exponential phase in SCD medium lacking tryptophan. Cells with the ypk1-as were continuously grown in the presence of 10 μM 3-MB-PP1 to inhibit Ypk1 activity. Cells were then switched into fresh medium with or without 0.4 M NaCl to trigger hyperosmotic stress. Cells were grown under these conditions for 90 min Cells were then pelleted and 100 μL of paraformaldehyde solution (4% w/v paraformaldehyde, 3.4% sucrose) was added to cells. Cells were incubated for 15 min at room temperature. Cells were then pelleted again and washed in wash solution (1.2 M sorbitol, 100 mM potassium phosphate, pH 7.5). The cell pellet was then resuspended in the wash solution. Cells were sonicated in a sonicator bath and then cellular GFP fluorescence was then measured by FACS using a Guava easyCyte (Merck Millipore). For each sample, fluorescence in 10 5 cells was measured. The average cellular fluorescence was then plotted. Measurement of intracellular glycerol accumulation Measurement of intracellular glycerol was conducted as described in Albertyn et al. (1994). Briefly, cells from ~40 mL of exponentially-growing culture were harvested by centrifugation and washed with 1 mL of medium. The resulting cell pellets were frozen in liquid N2 and stored at -80˚C prior to analysis. Each cell pellet was then boiled at 100˚C for 10 min in 1 mL of 50 mM Tris-Cl, pH 7.0. The lysate was then clarified by centrifugation at 16,000xg for 15 min. Glycerol concentration in the resulting supernatant solution was measured using an enzymatic assay kit (Sigma Aldrich) and was normalized to extract protein concentration, as measured by Bradford assay (Bradford, 1976). Co-immunoprecipitation of Fps1 and Rgc2 Co-immunoprecipitation experiments were performed as previously described (Lee et al., 2013), with only minor modifications. Cells expressing Fps1-3xFLAG (yAM271 – A), Fps13A-3xFLAG (yAM272 – A) or untagged Fps1 (BY4742) were transformed with a plasmid expressing Rgc23xHA under the control of the MET25 promoter (Lee et al., 2013). Cultures of each were grown to mid-exponential phase in SCD -uracil medium. Cultures were then diluted to A60o nm = 0.2 in 1 L of SCD-Met-Ura medium to induce expression of Rgc2-3xHA. Cultures were grown at 30˚C for 5 h. Cells were harvested by centrifugation and resuspended in 5 mL of TNE+Triton+NP-40 [50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 4 mM NaVO4, 50 mM NaF, 20 mM Na-PPi, 5 mM 17 EDTA, 5 mM EGTA, 0.5% Triton-X100, 1.0% NP-40 1x cOmplete protease inhibitor™ (Roche)]. The cells were then lysed cryogenically using Mixer Mill MM301 (Retsch). The lysate was thawed on ice and then clarified twice by centrifugation at 13,000xg for 20 min. Protein concentration of the clarified lysate was measured by BCA assay (Thermo Scientific) and normalized between the lysates. Fps1-3xFLAG was then isolated from ~10 mg of lysate using 50 uL of mouse anti-FLAG antibody coupled agarose resin (Sigma Aldrich) equilibriated in TNE+Triton. Binding was allowed to occur for 2 h at 4˚C. The resin was then extensively washed with TNE+Triton and precipitates were then processed for SDS-PAGE resolution and immunoblot detection of Fps1-3xFLAG and Rgc2-3xHA. 18 Tables Table 2.1 Yeast strains used in this study Strain Relevant Genotype Source/reference BY4741 MATa his3∆1 leu2∆0 met15∆0 ura3∆0 Research Genetics, Inc. BY4742 MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 Research Genetics, Inc. CGA65 BY4741 snf1∆::KanMX Christopher Alvaro, this lab DL3188 BY4742 rgc1∆::KanMX rgc2∆::KanMX (Beese et al., 2009) JTY5173 W303-1B MATα ade2-1 can1-100 his3-11,15 leu2- (Zaman et al., 2009) 3,112 ura3∆0 trp1-1 tpk1-as tpk2-as tpk3-as JTY5468 BY4741 tor2-29::KanMX (Li et al., 2011) JTY5537 BY4742 pbs2∆::KanMX Research Genetics, Inc. JTY5538 BY4742 ssk2∆::KanMX Research Genetics, Inc. JTY5539 BY4742 ssk22∆::KanMX Research Genetics, Inc. JTY5540 BY4742 sho1∆::KanMX Research Genetics, Inc. JTY5541 BY4743 ssk1∆::KanMX/ssk1∆::KanMX Research Genetics, Inc. JTY5574 BY4741 cna1∆::KanMX4 cna2∆::KanMX4 M.S. Cyert, Stanford Univ. JTY6142 BY4741 ypk1∆::KanMX4 Research Genetics, Inc. JTY6322 BY4743 fps1∆::KanMX/fps1∆::KanMX Research Genetics, Inc. JTY6328 BY4741 gpd1∆::KanMX4 gpd2∆::HIS3 (Lee et al., 2012) yAM120-A BY4741 ypk2∆::KanMX4 This study yAM134-A BY4741 Ypk1(L424A)::URA3- This study 19 yAM135-A BY4741 Ypk1(L424A)::URA3- ypk2∆:: KanMX4 This study yAM159-A BY4741 3xFLAG-Lag1::LEU2 This study yAM163-A BY4741 3xFLAG-Lag1(S23A S24A)::LEU2 This study yAM165-A BY4742 3xHA-Lac1::HIS3 This study yAM166-A BY4742 3xHA-Lac1 (S23A S24A)::HIS3 This study yAM168 BY4741 3xHA-Lac1::HIS3 3xFLAG-Lag1::LEU2 This study yAM181 - A BY4742 fps1∆::natNT2 This study yAM184 BY4741 3xHA-Lac1(S23A S24A)::HIS3 3xFLAG- This study Lag1(S23A S24A)::LEU2 yAM192-A BY4741 MET15+ 3xHA-Lac1(S23E S24E)::HIS3 This study 3xFLAG-Lag1(S23E S24E)::LEU2 yAM197 BY4742 smp1∆::LEU2 This study yAM205-A BY4742 Lac1::LEU2 Lag1::LEU2 This study yAM207-B BY4742 Lac1(S23A S24A)::LEU2 Lag1(S23A This study S24A)::LEU2 yAM210 BY4742 Lac1(S23E S24E)::LEU2 Lag1(S23E This study S24E)::LEU2 yAM219 – A BY4741 gpt2∆:: Hygr This study yAM221 - A BY4741 atg21∆::Hygr This study yAM231 BY4741 gpt2∆:: Hygr sct1∆::natNT2 [pRS315 This study PGPT2-Gpt2-3xFLAG; pAX240] yAM232 BY4741 gpt2∆:: Hygr sct1∆::natNT2 [pRS315 PGPT2-Gpt23A-3xFLAG; pAX248] 20 This study yAM233 BY4741 gpt2∆:: Hygr sct1∆::natNT2 [pRS315 This study PGPT2-Gpt23E-3xFLAG; pAX245] yAM234 - B BY4741 PTPI1-GFP-Atg8::HIS3 This study yAM235 - A BY4741 PTPI1-GFP-Atg8::HIS3 atg21∆::Hygr This study yAM236 - B BY4741 PTPI1-GFP-Atg8::HIS3 This study Ypk1(L424A)::URA3- ypk2∆:: KanMX4 yAM241 - A BY4741 PSTL1-GFP::HIS3 This study yAM242 - A BY4741 PSTL1-GFP::HIS3 ypk1-as::URA3- This study yAM243 BY4741 PSTL1-GFP::HIS3 smp1∆::LEU2 This study yAM244 – A BY4741 PSTL1-GFP::HIS3 ypk1-as::URA3- This study smp1∆::LEU2 yAM246 - A BY4741 Smp1-3xFLAG::URA3 This study yAM247 - A BY4741 Smp1(T20A T107A T221A)- This study 3xFLAG::URA3 yAM248 - A BY4741 Smp1(T20E T107E T221E)- This study 3xFLAG::URA3 yAM275 BY4742 LYS2+ Fps1-3xFLAG::URA3 This study hog1∆::KanMX yAM278 BY4742 Fps1(S181A S185A S570A)- This study 3xFLAG::URA3 hog1∆::KanMX yAM281 BY4742 Ypk1(L424A)::URA3- ypk2∆:: KanMX4 This study Fps1-3xFLAG::URA3 yAM284 - A BY4742 Ypk1(L424A)::URA3- ypk2∆:: KanMX4 21 This study Fps1(S181A S185A S570A)-3xFLAG::URA3 yAM291 - A BY4742 Fps1(S570A)-3xFLAG::URA3 This study hog1∆::KanMX yAM301 - A BY4742 Fps1(S181A S185A)-3xFLAG::URA3 This study yAM307 - A BY4742 Fps1(Δ544-581)-3xFLAG::URA3 This study yAM308 - A BY4742 Fps1(I218A V220A)-3xFLAG::URA3 This study yAM309 - A BY4742 Fps1(S181A S185A I218A V220A This study S570A)-3xFLAG::URA3 yAM310 - A BY4742 Fps1(T147A)-3xFLAG::URA3 This study yAM315 BY4741 Rgc2(S344A T808A S948A S75A S827A This study S1021A S1035A)-3xHA:: Hygr yAM318 BY4741 Fps1(S181A S185A S570A)- This study 3xFLAG::URA3 Rgc2(S344A T808A S948A S75A S827A S1021A S1035A)-3xHA:: Hygr YDB379 BY4741 Ypk1-3xFLAG::natNT2 J.S. Weissman, Univ. of California, San Francisco yGT12 BY4742 LYS2+ Lac1::LEU2 Lag1::LEU2 This study lcb3∆::natNT2 yGT13 BY4742 LYS2+ Lac1(S23A S24A)::LEU2 This study Lag1(S23A S24A)::LEU2 lcb3∆::natNT2 yGT14 BY4742 LYS2+ Lac1(S23E S24E)::LEU2 This study Lag1(S23E S24E)::LEU2 lcb3∆::natNT2 yGT21 BY4742 Fps1-3xFLAG::URA3 This study 22 yGT22 BY4742 Fps1(S181A S185A S570A)- This study 3xFLAG::URA3 yGT24 BY4742 Fps1(S570A)-3xFLAG::URA3 This study YJP544 BY4741 hog1∆::KanMX Jesse Patterson, this lab yKL4 BY4741 TOR2+::Hygr Kristin Leskoske, this lab yKL5 BY4741 Tor2(L2178A)::Hygr Kristin Leskoske, this lab 23 Table 2.2 Plasmids used in this study Plasmid Description Source/reference pGEX6P-1 GST tag, bacterial expression vector GE Healthcare, Inc. pGEX4T-1 GST tag, bacterial expression vector GE Healthcare, Inc. YCpLG CEN, LEU2, PGAL1 vector (Bardwell et al., 1998) BG1805 2µm, URA3, PGAL1, C-terminal tandem affinity Open Biosystems, Inc. (TAP) tag vector pRS313 CEN, HIS3, vector (Sikorski and Hieter, 1989) pRS316 CEN, URA3, vector (Sikorski and Hieter, 1989) pRS416 CEN, URA3, vector (Sikorski and Hieter, 1989) pRS426 2µm, URA3, vector (Sikorski and Hieter, 1989) pBC111 CEN, LEU2, vector (Iida et al., 2007) CHp282 pRS416 PMET25-GFP Chris Huynh, this laboratory p3151 pRS316 PMET25-Rgc2-3xHA (Lee et al., 2013) pAX131 pGEX4T-1 Lac1(1-76) This study pAX132 pGEX4T-1 Lac1(1-76)(S23A S24A) This study pAX133 pGEX4T-1 Lag1(1-80)(S23A S24A) This study pAX134 pGEX6P-1 Muk1(1-305) This study pAX135 pGEX6P-1 Fps1(531-669)(S570A) This study pAX136 pRS313 PLAC1-3xHA-Lac1 This study pAX215 pGEX6P-1 Cyk3 This study pAX223 pGEX6P-1 Gpt2(1-35) This study pAX224 pGEX6P-1 Gpt2(570-743) This study 24 pAX225 pGEX6P-1 Bre5 This study pAX226 pGEX6P-1 Npr1(1-437) This study pAX227 pGEX6P-1 Pal1 This study pAX228 pGEX6P-1 Ysp2(97-665) This study pAX229 pGEX6P-1 Ysp2(1072-1282) This study pAX230 pGEX6P-1 Atg21 This study pAX231 pGEX6P-1 Pex31(250-462) This study pAX238 pRS316 PGPT2-Gpt2-3xFLAG This study pAX239 CEN, LEU2, PTPI1-GFP-Atg8 This study pAX240 pRS315 PGPT2-Gpt2-3xFLAG This study pAX241 pRS316 PATG21-Atg21-3xFLAG This study pAX244 pRS316 PGPT2-Gpt2(S649A S650A S651A)- This study 3xFLAG pAX245 pRS315 PGPT2-Gpt2(S649E S650E S651E)- This study 3xFLAG pAX246 pRS316 PATG21-Atg21(S237E)-3xFLAG This study pAX247 pRS316 PATG21-Atg21(S237A)-3xFLAG This study pAX248 pRS315 PGPT2-Gpt2(S649A S650A S651A)- This study 3xFLAG pAX250 pRS313 PTPI1-GFP-Atg8 This study pAX253 pRS316 PSMP1-Smp1-3xFLAG This study pAX260 pRS316 PSMP1-Smp1(T20A T107A T221A)- This study 3xFLAG 25 pAX263 pRS316 PSMP1-Smp1(T20E T107E T221E)- This study 3xFLAG pAX270 pRS316 PSMP1-Smp1-GFP This study pAX274 pRS316 PFPS1-Fps1-3xFLAG This study pAX275 pRS316 PFPS1-Fps1(S181A S185A S570A)- This study 3xFLAG pAX286 pRS316 PSMP1-Smp1(1-274)-3xFLAG This study pAX287 pRS316 PSMP1-Smp1(1-274)(T20A T107A This study T221A)-3xFLAG pAX290 pRS316 PFPS1-Fps1-GFP This study pAX293 pRS316 PFPS1-Fps1(S570A)-GFP This study pAX294 pRS316 PFPS1-Fps1(S181A S185A)-GFP This study pAX295 pRS316 PFPS1-Fps1(S181A S185A S570A)-GFP This study pAX50 BG1805 Ypk1(L424A) This study pAX53 pRS416 PMET25-Ypk1(K376A) This study pAX55 pGEX6P-1 Lcb3(1-79) This study pAX56 pGEX6P-1 Cdc1(1-41) This study pAX58 pGEX6P-1 Her1(1-224) This study pAX59 pGEX6P-1 Rts3 This study pAX62 pGEX6P-1 Fkh1 This study pAX63 pGEX6P-1 Yhp1 This study pAX66 pGEX6P-1 YNR014W This study pAX67 pGEX6P-1 YHR097C This study 26 pAX94 pGEX6P-1 Mds3(545-1016) This study pAX96 YCpLG Fps1 This study pBCT- pBC111 PTDH3-Cch1 (Iida et al., 2007) pBT12 pGEX6P-1 Smp1 This study pBT6 pGEX6P-1 Fps1(1-255) This study pBT7 pGEX6P-1 Fps1(531-669) This study pFR203 pGEX4T-1 Orm1(1-85) (Roelants et al., 2011) pFR252 pRS315 PYPK1-Ypk1(S51A S57A S71A T504A This study CCH1H S644A S653A T662A)-myc pFR273 pRS316 PYPK1-Ypk1(D242A) (Roelants et al., 2011) pFR291 pGEX4T-1 Lag1(1-80) This study pGT02 pRS426 PFPS1-Fps1-3xFLAG This study pGT05 pRS426 PFPS1-Fps1(Δ12-231) -3xFLAG This study pGT06 pRS426 PFPS1-Fps1(Δ534-650)-3xFLAG This study pGT11 YCpLG Fps1(Δ12-231) This study pGT12 YCpLG Fps1(Δ534-650) This study pGT13 YCpLG Fps1(Δ544-581) This study pGT14 YCpLG Fps1(S181A S185A S570A) This study pGT15 YCpLG Fps1(S181E S185E S570E) This study pGT17 pRS426 PFPS1- Fps1(S181A S185A S570A)- This study 3xFLAG pGT18 pRS426 PFPS1- Fps1(S181A S185A S570A)- 27 This study 3xFLAG pLB215 pRS416 PMET25-Ypk1 (Niles et al., 2012) 28 Chapter 3: A three-tiered screen identifies novel Ypk substrates This chapter is based on work published in Muir A et al., eLife, 2014. I devised the threetiered screen for Ypk substrates with Prof. Jeremy Thorner. I was responsible for all bioinformatic and biochemical screening. I constructed the substrate overexpression library and performed all synthetic dosage lethality genetic screening with the assistance of undergraduate Garrett Timmons. Introduction To gain further insight into how the TORC2-Ypk1 signaling axis contributes to plasma membrane homeostasis and potentially other cellular processes, I aimed to identify novel Ypk substrates. A variety of mass spectrometry-based approaches for identifying kinase substrates are available (Koch and Hauf, 2010). In general, these approaches involve inhibiting the kinase of interest in vivo, extracting phosphopeptides from this sample and comparing levels of phosphopeptides to an appropriate control. Those peptides that change upon inhibition are candidate substrates for the kinase of interest. However, these mass spectrometry-based approaches have the drawback that they are not sensitive enough to detect low-copy number signaling proteins and can also fail to detect membrane proteins (Koch and Hauf, 2010). As nearly all of the previously identified Ypk substrates fall into these categories, use of mass spectrometry based approaches seemed ill advised. Instead, I devised a three-step procedure that would avoid the drawbacks of mass spectrometrybased approaches to screen in an unbiased and genome-wide manner for additional candidate Ypk1 substrates whose physiological relevance I could then evaluate. I first used bioinformatics to search the yeast proteome for presumptive targets, then applied a genetic method to narrow down the hits to likely, functionally important substrates, and then used biochemical analysis both in vitro and in vivo to validate the best prospects. In this chapter, I will describe in detail each step of the screen and report here the general outcomes of this screen. Chapters 4 and 5 describe characterization of the functional role of Ypk phosphorylation of substrates involved in sphingolipid metabolism and substrates involved in glycerol metabolism, respectively. Results A three-tiered screen to identify new Ypk1 substrates I devised a three-step strategy (Fig. 3.1A) to pinpoint bona fide cellular targets of Ypk1, utilizing bioinformatics to predict potential Ypk1 substrates, then an in vivo genetic test involving a novel variation of the synthetic dosage lethality method to winnow the list to likely candidates, and finally biochemical analysis in vitro to confirm whether the identified gene product serves as a direct substrate of Ypk1. The physiological relevance of Ypk1-dependent modification of each protein on the resulting final list could then been evaluated. Bioinfomatic prediction of Ypk substrates For initial bioinformatic search of the yeast proteome, I developed a position-weighted 29 consensus sequence logo (Fig. 3.1B) for the preferred Ypk1 phospho-acceptor site based on two primary criteria: (a) the known Ypk1 sites in five, validated in vivo targets (Fpk1, Fpk2, Orm1, Orm2, and Gpd1) (Roelants et al., 2010; Roelants et al., 2011; Lee et al., 2012b; Niles et al., 2012; Sun et al., 2012); and, (b) the sequence preference displayed by Ypk1 for phosphorylation of synthetic peptides in vitro (Casamayor et al., 1999; Mok et al., 2010). All demonstrated substrates either in vivo or in vitro contain Arg at positions -5 and -3 with respect to the phosphorylated Ser (or Thr); thus, these positions were invariant in the search motif. Given that nearly all the verified sites within known in vivo targets possess a hydrophobic residue (V, I, F) at position +1, the search motif gave preference to sites with a hydrophobic residue at the +1 position. I then took advantage of the existing MOTIPS motif analysis package (Lam et al., 2010) to identify those S. cerevisiae gene products that contain occurrences of the search logo. Several authentic Ypk1 substrates (e.g., Fpk1, Orm1, and Orm2) contain multiple Ypk1 phosphorylation sites. Thus, I filtered our search further by prioritizing candidates containing multiple matches to the search logo, as predicted by MOTIPS. However, to avoid disregarding potential Ypk1 substrates with a single predicted match to the sequence logo, I also considered MOTIPS hits wherein there was existing evidence in the PhosphoGRID database (Sadowski et al., 2013) indicating that a predicted site is phosphorylated in vivo. To narrow down the list of potential substrates further, I chose to pursue those gene products containing matches to the sequence logo for which there was existing information in the literature suggesting a phenotypic relationship to known Ypk1-dependent processes: (a) a loss-of-function mutation in the candidate gene exhibits elevated sensitivity to agents (aureobasidin A, caspofungin and/or myriocin) toward which a ypk1∆ mutant is also sensitive (Hillenmeyer et al., 2008); (b) the candidate gene product is reported to be involved in a genetic or biochemical interaction with Ypk1, as curated in YeastMine (Balakrishnan et al., 2012); (c) the candidate gene product is connected in some way to known Ypk1 regulators (e.g., TORC2, PP2A); and/or, (d) the candidate gene product is involved in a known Ypk1-regulated process (e.g., sphingolipid metabolism) (for further details, see Chapter 2). Reassuringly, our approach identified three known Ypk1 substrates (Fpk1, Orm1 and Orm2); absence of Fpk2 and Gpd1 from the list generated solely by the MOTIPS search criteria arose from the fact that these substrates contains only a single predicted site that is not presently recorded in PhosphoGRID. For this reason, I also restored for consideration additional gene products that contain a single match to the consensus sequence logo that YeastMine indicated are involved in processes in which Ypk1 is implicated. The resulting candidates, grouped via cellular process on the basis of current GO Slim terminology (http://www.geneontology.org/GO.slims.shtml), are cataloged in Table 3.1, and represent fewer than 100 gene products out of the approximately 6,000 protein-coding genes in the yeast genome (Lin et al., 2013) [although the number of authentic open-reading-frames undergoes constant revision (http://www.yeastgenome.org/cache/genomeSnapshot.html)]. A synthetic dosage lethality screen to identify Ypk interacting proteins As the secondary filter for the candidates recognized bioinformatically, I developed an in vivo approach to identify those gene products that manifested an expected hallmark of proteinsubstrate interaction. I reasoned that under conditions where the level of activity of an essential kinase, like Ypk1, is near-limiting for normal growth, high-level over-expression of an authentic substrate might tie up the available pool of active enzyme and prevent efficient phosphorylation of other cellular substrates necessary for growth and/or viability (Fig. 3.1C). This scheme is a 30 novel variation on a genetic approach referred to as synthetic dosage lethality (SDL) (Sopko et al., 2006; Sharifpoor et al., 2012). To limit Ypk1 activity, I used ypk1-as ypk2∆ cells, which express from the YPK1 locus an analog-sensitive allele, Ypk1(L424A), and titrated down its activity by addition of a low concentration of an efficacious inhibitor,1-(tert-butyl)-3-(3methylbenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3-MB-PP1) (Burkard et al., 2007) that has no effect on wild-type cells. To achieve high-level over-expression, each bioinformatic hit was expressed from the galactose-inducible GAL1 promoter on a CEN plasmid. As proof of concept, I used two known Ypk1 substrates, Orm1 and Orm2, as positive controls and GFP, which is not a Ypk1 substrate (data not shown), as a negative control. In the absence of limiting the activity of Ypk1(L424A) with inhibitor, overexpression of neither Orm proteins nor GFP was deleterious to cell growth (Fig, 3.1D, left panels, compare lower to upper). However, in the presence of a low dose of 3-MB-PP1, overexpression of Orm1 and Orm2 on galactose medium prevented cell growth, whereas overexpression of GFP did not (Fig. 3.1D, middle and right panels, compare lower to upper). I was able to test the majority of the candidates (90/96) that arose in the bioinformatic search in this same fashion [however, 10/90 caused toxicity upon over-expression even in wild-type cells and, hence, could not be scored]. Those candidates that, like Orm1 and Orm2, exhibited toxicity only on galactose medium and only when Ypk1(L424A) activity was limited in the presence of 3-MB-PP1, but not when inhibitor was absent, were designated SDL hits (Table 3.1, column 4). Moreover, use of a series of 3-MB-PP1 concentrations allowed for quantification of the strength of the SDL effect (from + to ++++). In one case (Fps1), a marked SDL effect was observed upon overexpression in the ypk1-as ypk2Δ cells in the absence of chemical inhibition; I considered this a valid SDL hit because GAL promoter-driven over-expression of Fps1 was not growth inhibitory in wild-type (YPK1+ YPK2+) cells. Thus, as summarized in Table 3.1 (column 4), a significant fraction (20/90) of the candidates identified bioinformatically that were tested in this fashion, but far from all, displayed an SDL phenotype. In this regard, it is important to note that all known Ypk1 substrates tested (Gpd1, Fpk1, Orm1 and Orm2) displayed an SDL phenotype, whereas nearly 80% of the bioinformatic hits, like GFP, did not. Consistent with the view that the SDL phenotype could arise from the over-expressed target serving as a decoy substrate that titers a limited pool of active Ypk1 away from acting on its essential substrates, I observed that overexpressed catalytically-inactive Fpk1 caused an SDL phenotype equivalent to or stronger than wild-type Fpk1 (Table 3.1). If such SDL phenotypes reflect occlusion of a limited pool of enzyme by over-expressed substrate, then, conversely, co-overexpression of Ypk1 or even of a kinase-dead allele Ypk1(K376A) (driven from the MET25 promoter) might rescue the toxicity. Indeed, the deleterious effect of Smp1 over-expression was rescued by co-over-expression of either Ypk1 or Ypk1(K376A) (Table 3.1), suggesting that the SDL phenotype of over-expressed Smp1 also arises from titration of a limited amount of Ypk1 away from essential substrates. Biochemical screening of bioinformatically and genetically predicted Ypk substrates Lastly, to determine whether the gene products that displayed an SDL phenotype are indeed substrates for Ypk1, I incubated those (17/20) that I was able to successfully express and purify as recombinant proteins or protein fragments (as GST fusions) from E. coli with [-32P]ATP and Ypk1(L424A), which was highly purified from yeast cells as described in Chapter 2. I chose to use the analog-sensitive allele, even though it is only about 50% as active as wild-type Ypk1 31 (Roelants et al., 2011), because ablation of activity in the presence of 3-MB-PP1 allowed us to confirm that any 32P incorporation into substrate observed was due to phosphorylation by Ypk1(L424A) itself (and not due to some other protein kinase that might be present in the preparation). All known in vivo substrates of Ypk1 (Fpk1, Fpk2, Gpd1, Orm1 and Orm2) display robust incorporation (as judged by autoradiography) in this in vitro assay (Lee et al., 2012b; Roelants et al., 2010; Roelants et al., 2011). Therefore, in testing each candidate, an appropriate positive control, Orm1(1-85) was included (Fig. 3.1E), which also allowed comparison between independent assays. Gratifyingly, 12/17 (70%) of the SDL hits tested in this manner displayed readily detectable and Ypk1-specific phosphorylation (Table 3.1, right column). Moreover, the results of such assays clearly demonstrated that Ypk1 is not a "promiscuous" enzyme. For example, Fps1 has three possible N-terminal Ypk1 sites (RPRGQT147T, RRRSRS181R and RSRATS185N) and one C-terminal site. The C-terminal site is a Ypk1 target (see Chapter 5); however, none of the N-terminal motifs serves as an efficient Ypk1 phospho-acceptor site (Fig. 3.1E), perhaps because each lacks a hydrophobic residue at +1. Thus, I considered it very likely that the dozen candidates (shown schematically in Fig. 3.2 and highlighted in bold in Table 3.1) identified bioinformatically that also displayed an SDL phenotype and served as Ypk1 substrates in vitro would be functionally important Ypk1 substrates in vivo. To validate this conclusion and confirm that these candidates are indeed physiologically relevant Ypk1 targets, I chose to characterize Lac1 and Lag1, two of the dozen candidates (Table 3.1), because they are the catalytic subunits of the ceramide synthase complex and might further our understanding about how sphingolipid production is regulated by the TORC2-Ypk1 signaling axis (see Chapter 4). Discussion Identification of new TORC2-Ypk1 substrates Our approach identified 12 new Ypk substrates. I was able to generated reagents to examine the physiological relevance of five of these twelve candidates. In Chapters 4 and 5, I report that four of the five tested substrates are indeed bona fide targets of Ypk in vivo. These findings validate the ability of our methods for discovery of physiologically relevant protein kinase substrates. Phospho-acceptor site motif pattern-matching alone, although useful in identifying kinase substrates in some cases (Gwinn et al., 2008; Holt et al., 2007; Hutti et al., 2009; Linding et al., 2007; Mah et al., 2005; Manning et al., 2002; Rennefahrt et al., 2007; Yaffe et al., 2001), can yield a large number of false positives. This possibility was a significant concern for us because certain features of the basophilic Ypk1 motif are shared with other protein kinases (Mok et al., 2010). As a means to avoid this problem, I devised a novel SDL-based genetic approach to apply as a secondary filter to parse the bioinformatically selected candidates further. Although not the only possible explanation for detecting an SDL hit, one mechanism our genetic method should be able to assess is whether the candidate gene product displays a primary characteristic of a true substrate, namely the ability to compete with other substrates for association with Ypk1. A genuine Ypk1 substrate should, when highly over-expressed, sequester a large fraction of the available pool of active kinase, and thus impede its actions on its essential cellular targets. In the absence of Ypk2, over-expression of an authentic Ypk1 substrate should 32 therefore be deleterious for growth when the activity of an analog-sensitive Ypk1 allele is reduced with a selective inhibitor. The fact that 70% of the SDL hits were indeed substrates for Ypk1-mediated phosphorylation in vitro verified that our use of this genetic method as a secondary filter to pick out true substrates from the list of bioinformatically identified candidates was well justified. In this regard, overexpression of Fpk1(KD), a catalytically-inactive nonessential Ypk1 substrate, yielded a readily detectable SDL phenotype. Thus, using the protocol I devised, SDL may be a generally useful way to identify substrates and perhaps other binding partners of protein kinases, not just those directly connected to any output phenotype being measured. Other genetic approaches have also been useful in identifying physiologically relevant targets of Ypk1. For example, a transposon insertion that suppressed the growth defect of a ypk1-ts ypk2Δ strain at an otherwise non-permissive temperature initially identifed Orm2 as a potential Ypk1 substrate (Roelants et al., 2002), a finding later corroborated by us (Roelants et al., 2011) and others (Berchtold et al., 2012; Liu et al., 2012; Niles et al., 2012; Sun et al., 2012). Similarly, Smp1, which I confirmed here is a likely Ypk1 substrate (see also Chapter 5), was first identified as a potential Ypk1 target because it was isolated as a dosage suppressor of the temperaturesensitive phenotype of ypk1-ts ypk2Δ cells (Roelants et al., 2002). Smp1 is a transcription factor (Dodou & Treisman, 1997; de Nadal et al., 2003) that mediates iron toxicity (Lee et al., 2012a) and, similarly, Ypk1 is required for iron toxicity (Lee et al., 2012a). Thus, TORC2-Ypk1 signaling might be mechanistically coupled to iron metabolism by modulation of Smp1 transcriptional output (see Chapter 5). Likewise, other situations where chemical genetics can be applied have proven useful in gleaning what aspects of cell function are controlled by a protein kinase. For example, mutagenesis of yeast Tor2 to confer susceptibility to a chemical inhibitor and thereby selectively inhibit TORC2 action has been achieved (Kliegman et al., 2013). This tool was combined with a collection of deletion mutants to identify what processes, when eliminated, are especially deleterious to cell growth and survival when TORC2 action (and presumably Ypk1 activity) is limiting. This analysis suggested some connection between TORC2 action and the pentose-phosphate pathway (Kliegman et al., 2013), in keeping with the growth-promoting roles of both TORC1 and TORC2 and the demand for NADPH in many cellular anabolic reactions. Similarly, use of TOR inhibitors has implicated TORC2-Ypk1 signaling in regulation of actin filament formation that is somehow required for yeast cell survival in response to low levels of DNA damage (Shimada et al., 2013). By contrast, the genetic approach in our SDL method is quite different, in that it scores the deleterious effect arising from overexpression of a gene product (increased protein dosage) rather than from the total absence of a gene product, when the activity of the kinase of interest is limited by the presence of a moderate concentration of a specific inhibitor. Theoretically, under our conditions, I should also have been able to observe synthetic dosage rescue ("SDR"); however, I found no such examples. In any event, our method independently identified nearly all of the previously known in vivo substrates of Ypk1, as well as nearly a dozen genuine Ypk1 targets, including Lac1 and Lag1 (Chapter 4), that have only been pinpointed by our three-tiered method. Thus, our SDL screening technique provides a complementary approach for identifying substrates of a protein kinase above and beyond those accessible through genetic interactions 33 between the kinase and single-gene deletions or other genetic schemes. Many of the gene products identified by our screen are involved in processes that the TORC2Ypk1 signaling axis is already known to regulate. For example, Ypk1 regulates glycerol-3phosphate production via phosphorylation and inhibition of glycerol-3-phosphate dehydrogenase Gpd1 (Lee et al., 2012b). Interesting, a potential Ypk1 target that met all of the criteria in our screen is Gpt2, sn-glycerol-3-phosphate 1-acyltransferase (Zheng and Zou, 2001), an enzyme that esterifies glycerol-3-phosphate as the first step in glycerolipid formation. In this same regard, as another very likely Ypk1 target I also identified Fps1, a membrane channel (aquaglyceroporin) that regulates efflux of glycerol (a glycerol-3-phosphate-derived metabolite) (Luyten et al., 1995). These results strengthen the conclusion that TORC2-Ypk1 signaling is intimately involved in modulating the level of the precursor to both glycerophospholipids and the osmolyte glycerol (Lee et al., 2012b) (see Chapter 5). Ypk1 action has been implicated in regulation of both fluid phase and receptor-mediated endocytosis (deHart et al., 2002). In this regard, I identified as a very likely Ypk1 target an endocytic adaptor, the α-arrestin Rod1, which is necessary for ubiquitinylation-triggered internalization of nutrient permeases (Becuwe et al., 2012; Lin et al., 2008) and the pheromone receptor Ste2 (Alvaro et al., 2014). Thus, as it does for plasma membrane lipids, TORC2-Ypk1 signaling may modulate plasma membrane protein composition via this α-arrestin, a possibility being pursued by my fellow graduate student Christopher G. Alvaro and Ann Aindow, an undergraduate working with him. Ypk1 function has also been implicated in regulating production of reactive oxygen species (ROS) (Niles et al., 2014), but an as yet undefined mechanism. In our screen, I found Ysp2, a protein that regulates mitochondrial morphology and ROS levels (Sokolov et al., 2006), as a very likely Ypk1 substrate, possibly providing insight into the molecular basis of the connection between TORC2-Ypk1 signaling and ROS levels. Similarly, several other prospects that were identified by our screen as very likely Ypk1 substrates remain to be validated. Such candidates include Muk1, a guanine nucleotide exchange factor (GEF) for yeast Rab 5-type small GTPases (Vps21, Ypt52, and Ypt53) involved in vesicle-mediated Golgi body-to-endosome trafficking (Paulsel et al., 2013), suggesting that Ypk1 may also control switches that direct the flow of lipids. Muk1 is also intriguing for another potential reason. In S. pombe, a Rab 5-like GTPase (Ryh1) and its Muk1-like GEF were identified in a screen for TORC2 activators (Tatebe et al., 2010). Thus, if Ypk1-mediated phosphorylation inhibits Muk1 function, it could represent a negative feedback mechanism exerted on TORC2; conversely, if Ypk1-mediated phosphorylation stimulates Muk1 function, it could represent a mechanism for self-reinforcing maintenance of TORC2 activity and, thus, a high level of activated Ypk1. Clearly, by further investigating the physiological relevance of these and other remaining candidates much new biology may be learned. Indeed, several gene products of totally unknown function, such as Yhr097c and Ynr014w, as well as gene products (e.g., Atg21 and Pex31) not previous linked to either TORC2 or Ypk1, if validated, may provide new mechanistic insight into additional cellular processes regulated by TORC2-Ypk1 signaling. In conclusion, our screening approach has uncovered a significant number of candidate 34 substrates. These appear to be high-quality candidates because the majority that have been tested, to date, are functionally relevant in vivo substrates of Ypk1. Characterization of the remaining substrates will very likely shed further light on aspects of cellular function that are regulated by TORC2-Ypk1 signaling. 35 Figures 36 Fig. 3.1: A three-part screen to identify likely Ypk1 substrates. (A) The three-part screening strategy to identify Ypk1 substrates is shown schematically as a flow chart. Numbers indicate the number of hits/considered genes at each step in the screen. (B) The bioinformatic approach towards identifying Ypk1 substrates is schematized as a flowchart with each filter as a box. Genes were first filtered by MOTIPS on the basis of having likely phosphorylatable Ypk1 motifs. Subsequently, substrates were filtered by having many Ypk1 motifs or having a Ypk1 site known to be phosphorylated in published data sets. Lastly, genes were filtered by requiring the gene to have a published chemical sensitivity like Ypk1 does, or a published interaction with Ypk1, Ypk1 regulators (TORC2 or PP2A) or sphingolipid biosynthetic machinery.(C) A possible explanation for Ypk1 synthetic dosage lethality interactions is shown. Normally, the cell has enough kinase activity to buffer overexpression of a substrate (Substrate 2), so that essential substrates are regulated and normal growth is unperturbed. However, concurrent decrease in kinase activity coupled with substrate overexpression causes loss of regulation of essential substrate(s) (Substrate 1) leading to observable growth defects. (D) ypk1-as ypk2Δ (yAM135 – A) cells were transformed with PGAL1-GFP (negative control), PGAL1-Orm1 or PGAL1-Orm2 (known Ypk1 substrates, positive SDL controls) plasmids. Overnight cultures were then serially diluted onto either dextrose (to repress substrate overexpression) or galatose (to induce substrate overexpression) containing media with increasing concentrations of the Ypk1-as inhibitor 3-MBPP1. (E) GST-Orm1(1-85) (pFR203) and GST-Fps1(1-255) (pBT6) were purified from E. coli and incubated with [γ-32P]ATP and Ypk1-as, purified from S. cerevisiae, in the absence or presence of 3-MB-PP1. The products were then resolved by SDS/PAGE and analyzed as described in chapter 2. 37 Fig. 3.2: Identified Ypk kinases substrates. Diagram of the TORC2-Ypk pathway from chapter 1. Known Ypk substrates and newly predicted substrates are grouped by the biological processes (if known) to which the substrates contribute. Known substrates are shown in black whereas newly predicted substrates are shown in red. 38 Tables Table 3.1 Known Ypk1 substrates and potential substrates predicted by MOTIPS listed under GO Slim terms.a Gene GPD1/YDL022W* FPK1/YNR047W FPK1(D621A) [Kinase-dead mutant] ORM1/YGR038W ORM2/YLR350W AVO1/YOL078W AVO2/YMR068W BEM2/YER155C BNI1/YNL271C CDH1/YGL003C ENT1/YDL161W GIC2/YDR309C LSB3/YFR024C-A PAL1/YDR348C SLA1/YBL007C TSC11/YER093C YHR097C YSC84/YHR016C MOTIPS Sites 24P 37, 200, 244, 436, 481 37, 200, 244, 436, 481 52, 53 47, 48 552P,597, 1078 273P, 305, 407 83, 168, 1810, 1813 119, 1138P, 1533 51, 195, 213P 160P 90, 312, 345P 262P 49P, 391, 436 445, 447P, 449P, 477 19P, 97, 188 58, 288P, 294P 274, 374P Chemical Sensitivity/YeastMine Interaction(s) Known Ypk1 Substrates YeastMine SDL Score Ypk1 Dosage Rescue In vitro substrate +++ N/A + YeastMine + N/A + YeastMine ++ N/A + YeastMine YeastMine Cytoskeleton Organization +++ ++++ N/A N/A + + YeastMine - N/A N/A YeastMine - N/A N/A Myr, YeastMine - N/A N/A AbA, Casp, YeastMine - N/A N/A AbA, YeastMine TOXIC - N/A YeastMine - N/A N/A Myr, AbA - N/A N/A YeastMine - N/A N/A AbA, Casp ++++ N/A - YeastMine - N/A N/A YeastMine N/A N/A N/A Myr +++ N/A +/- Myr, YeastMine - N/A N/A 39 Biological Process Unknown COM2/YER130C ECM3/YOR092W* ICS2/YBR157C JIP4/YDR475C KKQ8/YKL168C RTS3/YGR161C* SEG1/YMR086W YDR186C YHR080C YNR014W YPK3/YBR028C EPL1/YFL024C FKH1/YIL131C GAL11/YOL051W HCM1/YCR065W* HOT1/YMR172W* RLM1/YPL089C* SMP1/YBR182C* SSN2/YDR443C YHP1/YDR451C* BCK2/YER167W ESC2/YDR363W 251, 370, 380 312, 350 14, 95, 136, 172P 348, 352, 592, 649 83, 113, 144, 146, 212, 293 30, 238P 56, 118P, 217, 634, 752P 334P, 540P, 542P, 620, 715P 401, 513, 667 54, 115, 156, 197 72, 73, 90P 24, 28, 61 404 1003P 80 387, 520, 586 20 20, 107 608P 180, 182 12, 38, 373, 430 114, 143, 145 Myr, YeastMine - N/A N/A Myr, AbA, YeastMine - N/A N/A Myr - N/A N/A Myr, AbA N/A N/A N/A YeastMine - N/A N/A YeastMine - N/A N/A Myr + N/A N/A YeastMine - N/A N/A YeastMine - N/A N/A YeastMine +++ N/A + Myr, Casp - N/A N/A Transcription from RNA Polymerase II Promoter YeastMine Myr, YeastMine Myr, YeastMine AbA, YeastMine TOXIC - N/A N/A N/A N/A N/A N/A Myr - N/A N/A Myr YeastMine Myr Myr Mitotic Cell Cycle TOXIC TOXIC - + N/A N/A N/A + N/A N/A YeastMine TOXIC - N/A YeastMine - N/A N/A 40 PTK2/YJR059W SET3/YKR029C SWI4/YER111C VHS2/YIL135C ZDS1/YMR273C* ZDS2/YML109W 59P, 82, 91, 171, 275 236, 405, 428 816P 316, 318, 325P 78, 370 183, 267, 345 Myr, YeastMine + N/A N/A YeastMine - N/A N/A Myr, YeastMine - N/A N/A Myr, Casp - N/A N/A AbA, YeastMine - N/A N/A YeastMine - N/A N/A YeastMine - N/A N/A YeastMine + N/A N/A Myr - N/A N/A Myr, AbA, YeastMine TOXIC - N/A YeastMine ++++ N/A - Myr, YeastMine - N/A N/A Myr, Casp, YeastMine - N/A N/A YeastMine - N/A N/A Myr, AbA - N/A N/A N/A - N/A + Myr, AbA, YeastMine - N/A N/A Myr Myr, YeastMine +++ +++ N/A N/A + + 24P Myr, YeastMine +++ N/A + 16P Myr, YeastMine Cellular Ion Homeostasis and Transport - N/A + Myr, AbA, YeastMine - N/A N/A YeastMine - N/A + Protein Phosphorylation P HAL5/YJL165C KIN1/YDR122W KIN2/YLR096W KSP1/YHR082C NPR1/YNL183C SKY1/YMR216C YAK1/YJL141C YPL150W P 17 , 217 , 233 652, 791P, 879, 986P 665P, 818, 1020 594, 827P, 884P 125P, 255P, 257P, 317P 383P 128P, 206, 240 371P, 452, 890 Lipid Metabolic Process ADR1/YDR216W CDC1/YDR182W* CKI1/YLR133W GPT2/YKR067W LAC1/YKL008C* LAG1/YHL003C LCB3/YJL134W AVT3/YKL146W b CCH1/YGR217W 180, 230P, 756 9 P 14 , 25P, 30P 27, 652 23, 24 55, 59P, 172, 174 146, 148, 347 41 FPS1/YLL043W MNR2/YKL064W NHA1/YLR138W* PPZ1/YML016C DED1/YOR204W HCR1/YLR192C* HEF3/YNL014W* RPL3/YOR063W SUI2/YJR007W* TEF1/YPR080W* BPH1/YCR032W CSR2/YPR030W ROM2/YLR371W SSD1/YDR293C BRE5/YNR051C EXO84/YBR102C MUK1/YPL070W RGP1/YDR137W 147, 181, 185, 570P 165, 620, 621, 826 544, 830 122, 203, 250P P 84, 576 223 898 24P, 337 58 72P 1334, 1336, 1963 61, 103, 525, 987 76P, 193p, 396 164P, 482P, 503 398P 76, 313, 494, 554 173, 184P, 185P 220, 364P, 450, 452 Myr, YeastMine +++++ N/A + AbA - N/A N/A Myr, YeastMine - N/A N/A Myr, YeastMine TOXIC - N/A - N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A Casp - N/A N/A Myr - N/A N/A YeastMine - N/A N/A Myr, AbA, YeastMine TOXIC - N/A Golgi Vesicle Transport Myr, YeastMine +++ N/A - YeastMine - N/A N/A Myr +++ N/A +/- YeastMine - N/A N/A YeastMine N/A N/A N/A Myr, AbA - N/A N/A Myr, AbA, YeastMine +++ N/A - YeastMine N/A N/A N/A Translation YeastMine Myr, AbA, YeastMine Myr, AbA, YeastMine Myr, AbA, YeastMine Myr Myr Cell Wall Organization or Biogenesis Signaling IRA2/YOL081W GIS3/YLR094C* MDS3/YGL197W SYT1/YPR095C 882, 884, 1578, 1745, 3069 249, 333 757, 824, 842, 851, 1204 277P, 410, 728 DNA Replication 42 CDC13/YDL220C CTI6/YPL181W RIM4/YHL024W 314, 333P 155, 216P 93, 429, 525, 607 YeastMine Myr, YeastMine TOXIC - N/A N/A N/A YeastMine - N/A N/A Myr - N/A N/A Myr +++ N/A +/- YeastMine - N/A N/A Myr, YeastMine - N/A N/A Myr, AbA TOXIC - + Myr, YeastMine +++ N/A + YeastMine - N/A N/A Myr N/A N/A N/A +++ N/A - - N/A N/A ++ N/A + - N/A N/A +++ N/A + Endocytosis ALY2/YJL084C ROD1/YOR018W ROG3/YFR022W 166P, 201, 225, 803 563, 617, 807, 823 425, 584, 718 Other FRT1/YOR324C HER1/YOR227W YSP2/YDR326C 167, 201, 203, 228P, 385 28P, 102p, 157P 326, 518, 1237 RNA Catabolic Process P JSN1/YJR091C PUF2/YPR042C 174, 275 , 600 55, 143, 246, 902 Cytokinesis CYK3/YDL117W 159, 207P, 746 DSN1/YIR010W 240, 250P PEX31/YGR004W 432P PAM1/YDR251W 471, 553P, 625 ATG21/YPL100W 237P AbA, YeastMine Chromosome Segregation YeastMine Peroxisome Organization YeastMine Pseudohyphal Growth Myr, AbA, Casp, YeastMine Response to Starvation Myr a Genes that are not bioinformatically predicted Ypk1 substrates, but contain Ypk1 motifs and were included in this study are marked with an asterisk. SDL assay results are listed for each bioinformatically predicted Ypk1 substrate. The scoring system reports growth phenotypes of the ypk1-as ypk2Δ strain transformed with the indicated PGAL1-SUBSTRATE plasmid upon overexpression on galactose with varying levels of 3-MB-PP1-imposed Ypk1-as inhibition. A growth phenotype is defined as at least 1 serial dilution spot less growth than YCpLG-GFP control at the given 3-MB-PP1 concentration. A strong growth phenotype is defined as no growth at the given 3-MB-PP1 concentration. (+++++) indicates a growth phenotype with no 3MB-PP1. (++++) is a strong growth phenotype on 1 μM 3-MB-PP1. (+++) indicates a growth 43 phenotype on 1 μM 3-MB-PP1. (++) is defined as no phenotype on 1 μM 3-MB-PP1, but a strong growth phenotype on 2 μM 3-MB-PP1. (+) indicates no phenotype on 1 μM 3-MB-PP1, but a detectable growth phenotype on 2 μM 3-MB-PP1. (-) indicates no growth phenotype at any concentration of 3-MB-PP1 tested. TOXIC indicates overexpression of the putative substrate on galactose-containing media was deleterious to growth even in the wild-type control strain (BY4741). These toxic genes were then tested for Ypk1 dosage rescue (for details, see Materials and Methods); here, (+) indicates that Ypk1 overexpression could at least partially rescue the overexpression toxicity of the indicated gene and (-) indicates that Ypk1 overexpression could not rescue the overexpressiion toxicity. Lastly, the results of testing the indicated purified predicted Ypk1 target as a substrtate in the in vitro protein kinase assay with purified Ypk1-as; here, (+) indicates that Ypk1-as- dependent (3-MB-PP1 inhibitable) incorporation was detectable for the substrate at a level comparable seen for incorporation into the positive control (the known Ypk1 substrate, GST-Orm1(1-85); (+/-) indicates that readily detectable Ypk1-as-dependent incorporation was found, but at a level lower than that seen for an equivalent amount of GSTOrm1(1-85) protein. (N/A) indicates that the indicated gene product was not tested in the indicated assay. Genes listed in bold are those that are both SDL interactors and in vitro Ypk1 substrates and are considered hits in our screen. bThe Cch1 SDL assay was performed with a plasmid constitutively overexpressing Cch1 under the TDH3 promoter (pBCT-CCH1H, (Iida et al., 2007)), as efforts to generate a PGAL1-Cch1 vector failed. 44 Chapter 4: Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids This chapter is based on work published in Muir et al., 2014, eLife. I was responsible for all in vivo experiments examining Ypk1 regulation of ceramide synthase and with Research Specialist Françoise Roelants, Ph.D., I performed experiments examining Ypk1 phosphorylation of ceramide synthase in vitro. Postdoctoral researcher Subramaniam Ramachandran, Ph.D., and I performed all the lipid analyses and in vitro ceramide synthase experiments. Unpublished results on Ypk1 regulation of Atg21, in which I performed all of the studies, are also reported here. Introduction The proteins Lac1 and Lag1 were identified as likely Ypk substrates from the screen described in Chapter 3. Lac1 (418 residues) and Lag1 (411 residues) are apparent paralogs at the primary sequence level (69% identity, 77% similarity) (Byrne and Wolfe, 2005) and are polytopic integral proteins in the ER membrane with the predicted Ypk1 site in each protein residing near its N-terminus (Fig. 4.1A). It has been shown that the N-termini of these proteins are exposed to the cytosol (Kageyama-Yahara and Riezman, 2006). Along with a small accessory subunit Lip1 (150 residues), Lac1 and Lag1 are demonstrated constituents of the ceramide synthase complex (Schorling et al., 2001; Vallée and Riezman, 2005), which catalyzes N-acylation of the free amino group on the long-chain base (LCB), mainly phytosphingosine in yeast, with a preference for very long chain fatty acyl-CoAs as the acyl donor, thereby forming phytoceramide (Fig. 4.1B). Genetically, Lac1 and Lag1 appear to play overlapping functional roles; lac1Δ or lag1Δ single mutants are viable, whereas a lac1Δ lag1Δ double mutant is reportedly either inviable (Jiang et al., 1998) or extremely slow growing (Barz and Walter, 1999; Schorling et al., 2001; Vallée and Riezman, 2005). The ceramide synthase reaction lies at an important branch point in the sphingolipid metabolic network (Fig. 4.1B) because de novo synthesis of ceramides both consumes LCBs and prevents conversion of LCBs to their 1-phosphorylated derivatives (LCBPs). Thus, the rate of ceramide synthesis is tightly coupled to the levels of both LCBs and LCBPs (Kobayashi and Nagiec, 2003); and, moreover, the balance between ceramides and total LCBs and LCBPs affects growth rate in both fungi (Kobayashi and Nagiec, 2003) and mammalian cells (Spiegel and Milstien, 2003). By virtue of their position in the pathway, Lac1 and Lag1 are situated to be important regulators of this balance. TORC2-Ypk signaling is already known to regulate the first step of sphingolipid biosynthesis via Ypk1-mediated phosphorylation of Orm1 and Om2, alleviating their inhibition of the LCBproducing SPOTS complex (Roelants et al., 2011), and this regulation is important for surviving sphingolipid depletion (Roelants et al., 2011) and membrane stretch under hypotonic stress (Berchtold et al., 2012). Therefore, I sought to understand what type of control TORC2-Ypk1mediated phosphorylation exerts on Lac1 and Lag1 and what role their regulation of ceramide synthase plays in sphingolipid homeostasis and metabolism. 45 Results Global screen indicates Lac1 and Lag1are Ypk1 substrates Among the bioinformatically predicted substrates, both Lac1 and Lag1 displayed a readily detectable SDL phenotype (Fig. 4.2A) and both GST-Lac1(1-76) and GST-Lag1(1-80) served as in vitro substrates for Ypk1, albeit with the phosphorylation of Lac1 being reproducibly much more robust than Lag1 (Fig. 4.2B). Site-directed mutagenesis confirmed that the Ypk1-mediated phosphorylation of both substrates occurred exclusively at the predicted phospho-acceptor site(s), specifically Ser23 and Ser24 in both proteins (Fig. 4.2B). Lac1 and Lag1 are phosphorylated by Ypk1 in vivo To determine whether both Lac1 and Lag1 are phosphorylated in vivo in a Ypk1-dependent manner and at their Ypk1 consensus sites, an integrated 3xHA-tagged version of each protein and its corresponding S23A S24A mutant was expressed in yeast and extracts of the cells were analyzed by phosphate-affinity SDS-PAGE (Phos-tag gels) (Kinoshita et al., 2009). In this separation technique, the more highly phosphorylated the protein, the more its migration is retarded. Both Lac1 (Fig. 4.3A, left) and Lag1 (Fig. 4.3A, right) migrated as two species, and the slower of the two could be attributed to phosphorylation because it was eliminated if the sample was pre-treated with calf intestinal phosphatase. This slower mobility species represented phosphorylation at S23 and S24 because the band was also eliminated in Lac1(S23A S24A) and Lag1(S23A S24A) mutants (Fig. 4.3A). I noted that phosphatase treatment, even of the Lac1(S23A S24A) and Lag1(S23A S24A) mutants, resulted in appearance of a third, even faster migrating species, presumably due to removal of a phosphorylation(s) elsewhere in these proteins, consistent with indirect evidence that Lac1 and Lag1 might be subject to casein kinase II (yeast Cka2)-dependent modification (Kobayashi and Nagiec, 2003). In agreement with their relative efficacies as in vitro substrates (Fig. 4.2B), I found that, reproducibly, the majority of Lac1 was present in the cell as the slower mobility isoform, whereas the opposite was true for Lag1 (Fig. 4.3A). Because the behavior of Lac1 gave us greater sensitivity of detection, and for the sake of conciseness, some of our subsequent findings are illustrated with data for Lac1 only. However, all experiments were repeated with both proteins with virtually identical results and conclusions. To further confirm that Ypk1 is the protein kinase responsible for phosphorylation at Ser23 Ser24 in vivo, plasmids encoding 3xHA-tagged tagged Lac1 and Lag1 were introduced by DNAmediated transformation into wild-type, ypk1Δ and ypk2Δ strains. Cultures of the resulting cells were grown in the absence or presence of a sub-lethal dose of myriocin, a condition which several previous studies have shown activates TORC2- and Ypk1-mediated signaling (Roelants et al., 2011; Berchtold et al., 2012; Sun et al., 2012), and the resulting extracts were analyzed on Phos-tag gels. As observed previously for two other bona fide substrates, Orm1 and Orm2 (Roelants et al., 2011), absence of Ypk1 totally abrogated the appearance of phosphorylated Lac1 (Fig. 4.3B) and phosphorylated Lag1 (data not shown), whereas elimination of Ypk2 had no effect. Thus, Ypk1 is the paralog solely responsible for the observed in vivo phosphorylation of Lac1 and Lag1 at Ser23 and Ser24. Furthermore, under conditions that stimulate TORC246 Ypk1 signaling, nearly all of the Lac1 (Fig. 4.3B) and much more of the Lag1 (data not shown) were converted to the phosphorylated isoform indicating that TORC2 activation is relayed to ceramide synthase via Ypk1. To further confirm that TORC2 signaling is essential for Ypk1 mediated phosphorylation of ceramide synthase, Lac1 phosphorylation was monitored in TOR2 and tor2-as strains after the addition of the tor2-as specific inhibitor BEZ-235 (Kliegman et al., 2013). TORC2 inhibition led to rapid and sustained dephosphorylation of Lac1 (Fig. 4.3C), confirming that TORC2 activity is necessary for Ypk1-mediated ceramide synthase phosphorylation. Thus, our screening approach was successful in revealing two, previously uncharacterized, cellular targets of the TORC2-Ypk1 signaling axis. Lac1 and Lag1 phosphorylation increases under stress and is required for cell survival The fact that impeding LCB production with a sub-lethal dose of the SPOTS inhibitor myriocin stimulated Ypk1-mediated Lac1 and Lag1 phosphorylation suggested that this modification is important for their physiological function. As one means to confirm that reduction in sphingolipid biosynthesis capacity results in up-regulation of Lac1 and Lag1 phosphorylation, we subjected the pathway to blockade near its end by treating the cells expressing integrated 3xHA-tagged Lac1 or Lag1 with aureobasidin A (Heidler and Radding, 1995), an antibiotic that prevents formation of complex sphingolipids in yeast by inhibiting Aur1 (phosphatidylinositol: ceramide phosphoinositol transferase) (Nagiec et al., 1997). As observed for treatment with myriocin, the amount of phosphorylated Lac1 (Fig. 4.4A) and Lag1 (data not shown) was markedly increased in response to treatment with aureobasidin A. Another perturbation that has been shown to transiently up-regulate TORC2-Ypk1-mediated signaling is heat shock (Sun et al., 2012). Consistent with that response, it has been shown previously that heat shock leads to a transient increase in sphingolipid production and that sphingolipid production is important for heat shock survival (Jenkins et al., 1997; Cowart et al., 2003). Moreover, measurement of pathway intermediates and mathematical modeling also suggested that a sharp spike of increased ceramide synthase activity may occur after heat shock (Chen et al., 2013). Therefore, I subjected the same cells to heat shock and monitored Lac1 and Lag1 phosphorylation at various times thereafter. Within 5 min, the amount of phosphorylated Lac1 (Fig. 4.4B) and Lag1 (data not shown) increased markedly, but was back to the resting level by 30 min If these changes in phosphorylation state at Ser23 and Ser24 in Lac1 and Lag1 are important for the metabolic adjustments that the cell needs to adapt appropriately, then preventing phosphorylation at these sites should impair cell survival. To test this prediction, I integrated as the sole source of ceramide synthase, mutant versions of Lac1 and Lag1 in which Ser23 and Ser24 were mutated to Ala and, hence, cannot be phosphorylated under any circumstances. As a control, I also generated integrated versions of Lac1 and Lag1 in which Ser23 and Ser24 were mutated to Glu, to mimic conversion of the entire population to the phosphorylated state. Indeed, I found that the cells co-expressing Lac1(S23A S24A) and Lag1(23A S24A) grew detectably less well when challenged with either myriocin or aureobasidin A than either otherwise isogenic wild-type cells or cells co-expressing Lac1(S23E S24E) and Lag1(S23E S24E) (Fig. 4.4C). This growth difference could not be attributed to differences in protein levels between the ceramide synthase alleles, as immunoblot analysis indicates similar steady state levels of ceramide 47 synthase components under these various stresses (Fig. 4.4D). Thus, Ypk1-dependent phosphorylation of these sites in Lac1 and Lag1 is functionally important for cell survival in response to the stress of limiting sphingolipid biosynthesis. Furthermore, the fact that the Lac1(S23E S24E) Lag1(S23E S24E) strain phenocopied a LAC1+ LAG1+ strain under conditions that promote phosphorylation of Lac1 and Lag1 indicates that these mutations generated effective phosphomimetic alleles. Calcineurin down-regulates Lac1 and Lag1 phosphorylation As a means to delineate what cellular phosphatase is responsible for counteracting the Ypk1mediated phospho-regulation of Lac1 and Lag1, plasmid-borne 3xHA-tagged Lac1 was expressed in a collection of deletion strains lacking each of the non-essential protein phosphatase genes or their associated factors, and the phosphorylation state of Lac1 was assessed using Phostag gels. By this approach, I was unable to find any phosphatase-deficient mutant that exhibited a significant increase in the amount of phosphorylated Lac1 compared to control cells (data not shown). However, considerable genetic evidence indicates a strong connection between Ca2+ signaling and sphingolipid biosynthesis (Beeler et al., 1998). Moreover, it has been reported previously that TORC2-Ypk-dependent regulation of sphinglipid biosynthesis is antagonized by the action of the Ca2+/calmodulin-dependent protein phosphatase calcineurin (also known as phosphoprotein phosphatase 2B), although the level at which the phosphatase acted was unknown (Aronova et al., 2008). Hence, I conducted the converse experiment by stimulating the cells expressing 3xHA-tagged Lac1 acutely with 0.2 M CaCl2, a condition known to robustly activate calcineurin in yeast (Stathopoulos-Gerontides et al., 1999). Within <10 min, I found total abrogation of phospho-Lac1 (Fig. 4.5A, left) and total abrogation of phospho-Lag1 (data not shown) in wild-type cells, whereas in otherwise isogenic cna1Δ cna2Δ mutants (which lack both calcineurin catalytic subunit paralogs) a substantial portion of the phosphorylated species remained (Fig. 4.5A, right). These results suggested that calcineurin may directly reverse the phoshorylations introduced into Lac1 and Lag1 by Ypk1. However, it is also the case that two of the ancillary subunits associated with TORC2, Slm1 and Slm2, are demonstrated calcineurin-binding proteins (Bultynck et al., 2006; Tabuchi et al., 2006) and that calcineurin action appears to oppose TORC2-dependent signaling (Mulet et al., 2006; Daquinag et al., 2007; Berchtold et al., 2012). Hence, it was possible, therefore, that the observed loss of phospho-Lac1 and -Lag1 might be due to a Ca2+stimulated and calcineurin-mediated reduction in TORC2 and/or Ypk1 activity, rather than to direct action of calcineurin on phospho-Lac1 and -Lag1. To rule out the former possibility, I examined Lac1 and Lag1 phosphorylation in cells expressing constitutively-active Ypk1(D242A), which I have demonstrated bypasses the need for its TORC2-dependent activation (Roelants et al., 2011). I found that Ca2+ addition still led to nearly complete Lac1 and Lag1 dephosphorylation in these cells (Fig. 4.5B). Furthermore, as judged by immunoblotting with a phospho-site specific antibody, Pkh1- and Pkh2-dependent phosphorylation of the activation loop in Ypk1 (Fig. 4.5C) [or in Ypk1(D242A) (data not shown)] was not diminished in cells stimulated with Ca2+, confirming that there was no calcineurin-mediated decrease in the amount of active Ypk1 present. Therefore, it seems clear that calcineurin dephosphorylates the sites in Lac1 and Lag1 phosphorylated by Ypk1 directly, rather than through down-regulation of Ypk1 function. 48 Ypk1-mediated phosphorylation of Lac and Lag1 stimulates ceramide synthase activity Given the phenotypic evidence that TORC2-Ypk1-dependent modulation of Lac1 and Lag1 is physiologically important (Fig. 4.4C), I next conducted biochemical analysis to determine how Ypk1-mediated phosphorylation affects the function of Lac1 and Lag1 in ceramide synthesis. Toward that end, I first used LC-MS to monitor the levels of LCBs and LCBPs extracted from equivalent numbers of cells from exponentially-growing cultures of wild-type cells or otherwise isogenic cells expressing as the sole source of Lac1 and Lag1 either the non-phosphorylatable Lac1(S23A S24A) and Lag1(S23A S24A) mutants or the phosphomimetic Lac1(S23E S24E) and Lag1(S23E S24E) mutants. Relative to wild-type cells, the Lac1(S23A S24A) Lag1(S23A S24A) cells accumulated significantly more PHS (Fig. 4.6A, top left), as well as more dihydrosphingosine (DHS) (Fig. 6A, top right), which is a more minor LCB in yeast (note the difference in scale of the ordinate), whereas the Lac1(S23E S24E) Lag1(S23E S24E) cells displayed a level of both PHS and DHS that was somewhat lower, and a similar trend was observed even for PHS-1-P, an even less abundant metabolite (Fig. 4.6A, bottom left). The level of DHS-1-P (Fig. 4.6A, bottom right) was so low as to make reliable measurement difficult, but no differences between strains could be detected. Given that the ceramide synthase complex is responsible for the conversion of LCBs into ceramides, these findings indicate that, in the absence of Ypk1-mediated phosphorylation, the rate of LCB utilization is significantly reduced, consistent with the conclusion that Ypk1-dependent modification of Lac1 and Lag1 promotes ceramide synthase function. As an independent means to measure flux through the sphingolipid pathway, and because all complex sphingolipids in yeast contain inositol-phosphate, an equivalent number of cells of the same three strains were pulse-labeled, in triplicate, with [32P]PO4-3, and the acidic sphingolipids extracted and analyzed by thin-layer chromatography. Strikingly, the amount of complex sphingolipids generated during the pulse was higher in the Lac1(S23E S24E) Lag1(S23E S24E) cells than in the wild-type controls, while Lac1(S23A S24A) Lag1(S23A S24A) cells generated lower levels of complex sphingolipids (Fig. 4.6B). This finding is again consistent with the conclusion that Ypk1-mediated phosphorylation of Lac1 and Lag1 stimulates the production of the ceramide precursors to complex sphingolipids. Ypk1-mediated phosphorylation could stimulate the ceramide synthase reaction in vivo by stabilizing Lac1 and Lag1 thereby increasing their steady-state level, by enhancing their association with the small ancillary subunit Lip1, and/or by direct activation. Immunoblotting of exponentially-growing cultures expressing the 3xHA-tagged versions of wild-type Lac1 and Lag1 and the Lac1(S23A S24A) Lag1(S23A S24A) and Lac1(S23E S24E) Lag1(S23E S24E) indicated no discernible difference in their steady-state level (see Fig. 4.4D). Likewise, in cells co-expressing the same proteins and FLAG-tagged Lip1 (gift of Howard Riezman, Univ. of Geneva), I observed no difference in the efficiency of Lip1 co-immunoprecipitation between wild-type Lac1 and Lag1 and either the Lac1(S23A S24A) Lag1(S23A S24A) or Lac1(S23E S24E) Lag1(S23E S24E) mutants (data not shown). These results suggested that Ypk1-mediated phosphorylation may directly enhance the catalytic efficiency of Lac1 and Lag1. To test this possibility directly, 3xFLAG-tagged versions of Lac1 and Lag1 were immuno-purified from detergent-solubilized microsomes isolated from exponentially-growing cells and the resulting protein assayed in vitro, monitoring the formation of ceramide from PHS and steroyl-CoA by 49 LC-MS. No product was observed in the absence of added steroyl-CoA (data not shown), and product formation was reduced by 85% in the presence of PHS and steroyl-CoA if 1 μM australifungin, a demonstrated and specific ceramide synthase inhibitor (Mandala and Harris, 2000), was added (data not shown), confirming that the reaction measured was catalyzed by ceramide synthase. I found that the specific activity of the Lac1(S23E S24E) Lag1(S23E S24E) complex was reproducibly ~2 higher than either the Lac1(S23A S24A) Lag1(S23A S24A) mutant or the wild-type complex (Fig. 4.6C upper panel), similar to the degree of difference in sphingolipid metabolites between these same strains that I measured by other means (Fig. 4.6A and 4.6B). In two independent trials (each performed in triplicate), the identical trend was found when microsomes from these same cells were assayed directly (i.e. without detergent solubilization and enzyme enrichment by immunoprecipitation (data not shown). Additionally, I found that immunopurified ceramide synthase from wild-type cultures treated with myriocin had higher activity than ceramide synthase prepared from untreated cells (Fig. 4.6C, lower panel). Furthermore, this increase in ceramide synthase activity in response to myriocin treatment was not observed in Lac1 (S23A S24A) Lag1 (S23A S24A) cells, consistent with TORC2-Ypk1 signaling increasing ceramide synthase activity by phosphorylation at these residues. These findings are also in agreement with a reported ~2 fold decrease in the rate of ceramide production by ceramide synthase complex isolated from TORC2-deficient yeast (Aronova et al., 2008). Hence, I conclude that Ypk1 phosphorylation directly increases the catalytic activity of ceramide synthase. Ypk1-mediated stimulation of Lac1 and Lag1 prevents autophagy induction during TORC2-driven up-regulation of sphingolipid biosynthesis The observed increase in ceramide synthase activity in response to Ypk1-mediated phosphorylation could serve two roles that are not mutually exclusive: (i) to produce more ceramide and the derived complex sphingolipids; and, (ii) to prevent inadvertent accumulation of LCBs and the derived LCBPs when TORC2-driven Ypk1 activation stimulates metabolic flow into the sphingolipid pathway by alleviating Orm1- and Orm2-imposed inhibition of SPOTS (see Fig. 4.8). I reasoned that if the latter were one of the important physiological functions of Ypk1dependent stimulation of ceramide synthase, then TORC2 activation would be detrimental to cells in which the Lac1(S23A S24A) Lag1(S23A S24A) mutant is the sole source of this enzyme. To mimic TORC2-stimulated elevation of Ypk1 activity, the constitutively-active Ypk1(D242A) allele (hereafter referred to as Ypk1*) was expressed from the YPK1 promoter on a CEN plasmid in either wild-type cells or the Lac1(S23A S24A) Lag1(S23A S24A) mutant. Indeed, compared to the empty vector control, expression of Ypk1* was well tolerated by cells containing wild-type Lac1 and Lag1, but deleterious to the growth of the Lac1(S23A S24A) Lag1(S23A S24A) mutant cells, whether measured on agar plates (Fig. 4.7A, top) or in liquid culture (Fig. 4.7A, bottom). The most likely metabolic perturbation responsible for the observed decrease in growth in the Lac1(S23A S24A) Lag1(S23A S24A) expressing Ypk1* is the accumulation of either LCBs or LCBPs. I asked if levels of these metabolites were altered in Ypk1* expressing cells. As previously noted (see Fig. 4.6A) Lac1(S23A S24A) Lag1(S23A S24A) cells had higher PHS levels than wild-type controls, but the addition of the Ypk1* plasmid caused no further increase (Fig. 4.7B upper panel). This indicates even in the absence of TORC2-Ypk1 mediated ceramide 50 synthase regulation, cells are able to buffer the levels of LCBs even with active TORC2 signaling. However, the addition of Ypk1* did cause an increase in PHS-1-P levels in Lac1(S23A S24A) Lag1(S23A S24A) but not wild-type cells (Fig. 4.7B lower panel). Thus, I reasoned that abnormally high LCBP levels might cause the growth defect observed in Ypk1* expressing Lac1(S23A S24A) Lag1(S23A S24A) cells. If accumulation of LCBPs in Lac1(S23A S24A) Lag1(S23A S24A) co-expressing Ypk1* is indeed responsible, then reduction in LCBP synthesis by elimination of the gene LCB4, which encodes the major LCB kinase (Nagiec et al., 1998), should alleviate the growth inhibition. As expected, introduction of an lcb4∆ null mutation into the Lac1(S23A S24A) Lag1(S23A S24A) mutant suppressed the growth inhibitory effect of Ypk1* (Fig. 4.7C). Conversely, and consistent with toxicity arising from accumulation of LCBPs when Lac1 and Lag1 cannot be stimulated by Ypk1, I found that the poor growth phenotype of cells lacking the gene (LCB3) encoding the major LCBP phosphatase (Mandala et al., 1998), was markedly exacerbated by introduction of the Lac1(S23A S24A) Lag1(S23A S24A) alleles, but not by introduction of the Lac1(S23E S24E) Lag1(S23E S24E) alleles (Fig. 4.7D). In fact, and strikingly, presence of the Lac1(S23E S24E) Lag1(S23E S24E) allele afforded nearly complete rescue of the slow-growth phenotype of the lcb3∆ mutation (Fig. 4.7D). Consistent with the toxicity of LCBPs when LCB utilization by ceramide synthase is inefficient, an elo3 mutation, which prevents efficient formation of C26-CoA (an acyl chain found in yeast ceramides), was synthetically lethal with lcb3∆ (Kobayashi and Nagiec, 2003). It has been reported that aberrant increases in LCBP level impede growth by triggering autophagy even under nutrient-rich conditions (Zimmermann et al., 2013). In agreement with the poor growth arising from LCBP-evoked induction of autophagy, I found that preventing autophagy by deletion of ATG1, which encodes a protein kinase necessary for induction of autophagophore formation and its elongation (Papinski and Kraft, 2014), provided substantial rescue of the growth debilitating effect of Ypk1* expression in Lac1(S23A S24A) Lag1(S23A S24A) mutant cells (Fig. 4.7F). This rescue was not a result of ATG1 mutation indirectly affecting LCBP levels as LCBPs did not significantly change in atg1Δ strains and still accumulated in Ypk1* expressing atg1Δ Lac1(S23A S24A) Lag1(S23A S24A) cells (Fig. 4.7G). Collectively, these results indicate that, aside from stimulating ceramide synthesis per se, a physiologically important role of Ypk1-dependent ceramide synthase activation is also to prevent accumulation of LCBPs and thereby avoid inadvertent induction of autophagy under nutrientsufficient conditions (Fig. 4.8). Is Atg21 a physiologically-relevant Ypk1 substrate? Given that TORC2-Ypk signaling appears to play an important role in preventing autophagy induction due to LCBP accumulation, I was also interested in the fact that Atg21, a PtdIns3Pbinding -propeller (WD-40 repeat) protein required for vesicle formation in the cytoplasm-tovacuole targeting (Cvt) pathway (Krick et al., 2008; Nair et al., 2010), was another potential Ypk1 substrate that I identified in my screen (see Fig. 3.2). CVT is a form of selective autophagy that occurs under basal conditions and is used to deliver certain enzymes into the vacuole (Lynch-Day & Klionsky, 2010; Umekawa & Klionsky, 2012). Therefore, I asked whether I could detect any alteration of CVT pathway function either in cells with diminished Ypk activity or in cells expressing mutant alleles of Atg21 where the predicted Ypk sites were mutated either to Ala to mimic the absence of Ypk1-mediated phosphorylation or to Glu to mimic permanent 51 Ypk1-mediated phosphorylation. To examine CVT pathway activity, I monitored the status of GFP-Atg8 and the processing of Ape1 under nutrient rich (basal autophagy) conditions. Under these circumstances, proteolytic maturation of both these proteins requires CVT pathway activity and Atg21 function (Umekawa and Klionsky, 2012). However, I did not observe any significant differences in these two biochemical read-outs of CVT pathway activity, compared to control cells, when Ypk activity was inhibited (Fig. 4.9A) or for cells expressing either type of Atg21 phospho-site mutant (Fig. 4.9B). Recently, it has been reported that Ypk1 activity is important for autophagy initiated in response to amino acid starvation, but not in response to general nitrogen source starvation (Vlahakis et al., 2014). Processing of GFP-Atg8 provides a robust read-out for amino acid starvation evoked autophagy (Guimaraes et al., 2015). Hence, I monitored the status of GFP-Atg8 in response to leucine limitation in strains expressing my Atg21 phospho-site mutants. Again, however, I observed no difference between control cells and cells expressing either class of Atg21 alleles in the predicted Ypk1 sites (Fig. 4.9C). Thus, I have not yet uncovered any evidence that Ypk1-mediated phosphorylation of Atg21 plays any role in controlling its function. It is possible that pulse-chase experiments might provide a better way to observe whether the kinetics of the CVT and autophagy pathway are affected than the steady-state analysis I conducted. Furthermore, I have not yet conclusively shown that Atg21 is phosphorylated at its predicted Ypk sites in vivo. Thus, future experiments examining Ypk1 phosphorylation of Atg21 in vivo, and more careful phenotypic analysis of the consequences of this phosphorylation, will be necessary to determine if Atg21 is an authentic Ypk1 substrate or a false positive from my screen. Discussion Mechanism of phosphoregulation of ceramide synthase As I have demonstrated here, TORC2-dependent Ypk1-mediated phosphorylation of Lac1 and Lag1 stimulates the function of the ceramide synthase complex. Consistent with our findings, a previous study found a consistent decrease in ceramide synthase activity in microsomal fractions isolated from cells in which TORC2 had been inactivated (and thus Ypk1 activity was presumably reduced) (Aronova et al., 2008), although indirect effects of the loss of TORC2 function on ceramide synthase activity could not be ruled out. Our findings make it clear that the role of TORC2 is to promote the Ypk1-dependent phosphorylation of Lac1 and Lag1 subunits of this enzyme. However, the precise molecular mechanism by which this post-translational modification stimulates this enzyme is still not completely clear. In this regard, it has been shown that mammalian ceramide synthase activity increases upon heterodimerization of different catalytic subunit isoforms (Laviad et al., 2012). However, as judged by co-immunoprecipitation, I found no difference in the state of Lac1-Lag1 association between the wild-type proteins and either our Lac1(S23A S24A) Lag1(S23A S24A) or Lac1(S23E S24E) Lag1(S23E S24E) mutants (data not shown). As mentioned in Results, I found no difference in the steady-state level of these same complexes or in their content of Lip1, a non-catalytic component of the complexes also essential for ceramide synthase activity (Vallée and Riezman, 2005). Thus, understanding of how phosphorylation of Ser23 and Ser24 in Lac1 and Lag1 stimulate ceramide synthase activity may require detail structural information, which will be challenging to obtain for these polytopic integral membrane proteins. 52 TORC2-Ypk1 control of ceramide synthesis is conserved Although the enzymic steps that carry out sphingolipid biosynthesis have been largely elucidated, much less was known, until recently, about regulation of these enzymes (Breslow & Weissman, 2010; Breslow, 2013). The first insight came when it was demonstrated (Roelants et al., 2011; Berchtold et al., 2012; Sun et al., 2012) that, in response to membrane stresses, including treatment with myriocin and aureobasidin A, TORC2-Ypk1 signaling is activated and alleviates inhibition of the SPOTS complex by phosphorylating the negative regulatory proteins Orm1 and Orm2 (Fig. 8). As a direct consequence, the rate of de novo production of the LCB precursor to sphingolipids is increased. Although TORC2 signaling had been implicated in promoting synthesis of ceramide, the product of LCB N-acylation (Aronova et al., 2008), it was unknown whether that role was simply the result of TORC2-Ypk1-dependent stimulation of SPOTS function and the resulting increase in LCB supply. As I demonstrated here, Ypk1mediated phosphorylation of Lac1 and Lag1 also increases in response to both myriocin and aureobasidin A, suggesting that ceramide synthesis per se, and not simply general elevation of LCB levels, is important for allowing the cells to cope with the effects of these antibiotics. Indeed, collectively, the findings I describe here demonstrate unequivocally that, in addition to up-regulation of SPOTS, TORC2-Ypk1 exerts direct control on the ceramide synthase step of the sphingolipid biosynthetic pathway by phosphorylating and stimulating the function of the Lac1 and Lag1 subunits of the ceramide synthase complex. Moreover, as I also demonstrated, the ceramide synthase reaction represents an important branch point in sphingolipid biosynthesis (Figs. 2B and 8). LCBs produced by the SPOTS reaction can either be converted to ceramides or become phosphorylated by LCB kinase Lcb4 (and its paralog Lcb5) to form LCBPs (Nagiec et al., 1998). Accumulation of LCBPs has been shown to be toxic to yeast cell growth (Kim et al., 2000), as I have also confirmed here, at least in large part because, as is now known, these metabolites trigger inappropriate induction of autophagy (Zimmermann et al., 2013). Thus, the rate of ceramide production must be properly adjusted to maintain the pool of LCBs and derived LCBPs at a non-deleterious level, in agreement with evidence in yeast and other organisms that ceramides and LCBPs generally play antagonist roles and must be maintained in the proper dynamic balance (Kobayashi and Nagiec, 2003; Spiegel and Milstien, 2003; Kihara et al., 2007; Dickson, 2008; Breslow and Weissman, 2010; Bikman and Summers, 2011). Hence, the function I have discovered and described here for TORC2-Ypk1 in stimulating ceramide synthase promotes utilization of the increased LCB generated upon TORC2-Ypk1-mediated up-regulation of SPOTS. This metabolic control has multiple clear-cut physiological benefits to the cell: (a) directing flow in the sphingolipid pathway toward complex sphingolipids to populate the plasma membrane barrier; (b) reduction of the level of potentially toxic LCBPs; and, (c) avoidance of inappropriate induction of autophagy under nutrient-sufficient conditions (Fig. 8). Indeed, our results indicate that a significant role for the coordination exerted by TORC2-Ypk1 between the level of SPOTS activity and the level of ceramide synthase activity is to prevent metabolic "cross-talk" to the autophagy pathway. If this function of TORC2-Ypk1 signaling is important, then it should be conserved. Indeed, alignments of the primary structures of Lac1 and Lag1 homologs predicted from sequenced fungal genomes, from Saccharomyces sensu stricto species to the very distantly related Ustilago maydis, nearly all contain a basophilic sequence that matches the Ypk1 phospho-acceptor site consensus and is located, as in S. cerevisiae Lac1 and Lag1, in their N-terminal cytosolic 53 extensions, suggests that Ypk1-related protein kinases may regulate ceramide synthase across essentially all fungal species. Consistent with this suggestion, a genetic interaction between a Ypk1 homolog (YpkA) and a Lac1/Lag1-like ceramide synthase component (BarA1) has been reported in Aspergillus nidulans (Colabardini et al., 2013). However, this mode of regulation may not extend to multicellular eukaryotes; of the six distinct ceramide synthase catalytic subunts encoded in the human genome, only CerS6 has a sequence that resembles a Ypk1 site (43-R-x-R-x-x-x-S-Hpo-50), and it serves as only a weak phospho-acceptor for SGK1, the human ortholog of Ypk1 (S. Ramachandran and J. Thorner, unpublished results). Orm1 and Orm2, when phosphorylated at unique sites by protein kinase Npr1, appear to promotes steps in the sphingolipid pathway that lead to more complex sphingolipids (Shimobayashi et al., 2013). In contrast to Ypk1, which is activated by TORC2, Npr1 is inhibited by TORC1 (MacGurn et al., 2011). Thus, this control mechanism will only be exerted under conditions that inactivate TORC1, such as amino acid starvation (Loewith and Hall, 2011), a condition that presumably requires adjustment of both plasma membrane lipid and protein composition to maximize the cell's ability to scavenge and assimilate nutrients. Under the same condition, autophagy is induced because, like Npr1, TORC1 also negatively regulates the autophagy-inducing protein kinase Atg1-Atg13 complex (Alers et al., 2014). Conversely, under nutrient sufficient conditions, TORC1 is active, and phosphorylates and stimulates protein kinase Sch9. Interestingly, Sch9 action should act in concert with TORC2-Ypk1 signaling to help keep the levels of LCBs and LCBPs low and ceramides high. This is likely because Sch9 promotes transcriptional repression of genes (YDC1 and YPC1) that encode ceramidases and inhibits a phosphosphingolipid phospholipase C (Isc1) that hydrolyzes complex sphingolipids (Swinnen et al., 2014). Such complex multi-component controls may be a general feature of signaling modalities that interface with biosynthetic pathways that have intermediates, like LCBPs, that are not innocuous, but are themselves bioactive metabolites. Calcineurin negatively regulates ceramide synthase Prior studies had suggested that calcineurin negatively regulates sphingolipid production via effects on the function of the ancillary TORC2 subunits, Slm1 and Slm2 (Bultynck et al., 2006; Mulet et al., 2006; Tabuchi et al., 2006; Daquinag et al., 2007), although the molecular connection between Slm1 and Slm2 and sphingolipid biosynthesis was unclear. Subsequently, it was observed that, in cells lacking the regulatory subunit (Cnb1), there was an increase in C26containing ceramides, suggesting that calcineurin somehow antagonizes TORC2-dependent signaling (Aronova et al., 2008). I found that calcineurin negatively regulates ceramide synthesis by directing the dephosphorylation of Lac1 and Lag1. This Ca2+-activated calcineurin-dependent dephosphorylation occurred even in cells expressing a TORC2-independent constitutively-active Ypk1 allele. Moreover, I showed here that calcineurin does not affect Pkh1- (and Pkh2-) mediated phosphorylation of the activation loop of Ypk1, and I demonstrated previously that presence or absence of calcineurin does not alter TORC2-mediated phosphorylation of Ypk1 or cause any substantial change in Ypk1 specific activity (Roelants et al., 2011). Thus, direct downmodulation of either TORC2 or Ypk1 by calcineurin cannot account for the negative regulation it exerts on sphingolipid biosynthesis. What our findings now make clear is that calcineurin negatively regulates the sphingolipid pathway at the level of ceramide synthesis, at least in large part, by direct dephosphorylation of the stimulatory phosphorylations in the ceramide synthase 54 subunits Lac1 and Lag1 that are installed by TORC2-Ypk1 signaling. Calcineurin recognizes substrates via a docking motif (PxIxIT or variants thereof), typically also accompanied quite a distance upstream by a secondary docking site (LxVP) (Roy and Cyert, 2009). However, these sites can be quite degenerate; for example, the more hydrophobic variant PVIVIT is much more potent in recruiting calcineurin when it is used to replace the native "PxIxIT" sequences in either the transcription factor Crz1 (PIISIQ) (Roy et al., 2007) or the endocytic adaptor Aly1 (PILKIN) (O'Donnell et al., 2013). In both Lac1 and Lag1, there is a similar hydrophobic sequence located at the identical position in both proteins (355PIVFVL360). Whether this or any other degenerate match represents a calcineurin-binding site remains to be determined. 55 Figures Fig. 4.1: Lac1 and Lag1 are components of the ceramide synthase enzyme complex. (A) A diagram of the membrane topology of Lac1 and Lag1 derived from (Kageyama-Yahara and Riezman, 2006). Lac1 and Lag1 are experimentally determined to have 6 transmembrane domains. The N terminus of these proteins is cytosolic and therefore accessible for Ypk1 phosphorylation. (B) Diagram of yeast de novo sphingolipid biosynthesis derived from (Dickson, 2008). Skeleton structures of metabolites are shown and enzymes are indicated as ovals. Metabolites shaded in green are those directly produced or derived from ceramide synthase while those in red are alternative products at the ceramide synthase branch point. 56 Fig. 4.2: Genetic and biochemical screening indicates Lac1 and Lag1 are Ypk1 substrates. (A) SDL results for Lac1 and Lag1. ypk1-as ypk2Δ (yAM135 – A) or wild-type (BY4741) cells were transformed with PGAL1-GFP (negative control), PGAL1-Lac1 or PGAL1-Lag1 plasmids. The SDL assay was then performed as described in Materials and Methods. (B) GST-Lac1(1-76) (pAX131), GST-Lag1(1-80)(pFR29), GST-Lac1(1-76)(S23A S24A)(pAX132) and GST-Lag1(180)(S23A S24A)(pAX133) were purified from E. coli and Ypk1-as kinase assays were performed as described in chapter 2. 57 Fig. 4.3: Ypk1 phosphorylates Lac1 and Lag1 at S23 and S24 in vivo. (A) 3xHA-Lac1 (yAM165-A) 3xHA-Lac1(S23A S24A) (yAM166-A), 3xFLAG-Lag1 (yAM159-A) and 3xFLAG-Lag1(S23A S24A) (yAM163-A) strains were grown exponentially in YPD. Cells were then harvested and whole-cell extracts were prepared. Extracts were split and one fraction was then treated with calf intestinal phosphatase. The extracts resolved by Phos-tag SDS-PAGE and immunoblotted with HA or FLAG and Pgk1 (loading control) antibodies. P-Lac1 and P-Lag1 indicate the band corresponding to S23 S24 phosphorylation. * indicates a non-specific band that appears in HA blots of yeast whole cell extracts. (B) Wild-type (BY4741), ypk1Δ (JTY6142) and ypk2Δ (yAM120 – A) strains were transformed with a plasmid centromeric plasmid encoding 3xHA-Lac1 expressed under control of its endogenous promoter (pAX136). Cells were grown to mid-exponential phase and then treated with a sublethal dose of myriocin (0.625 μM) or methanol (vehicle) for 2 h. Cell extracts were then prepared, resolved by Phos-tag SDS-PAGE and blotted as above in (A). (C) TOR2 (yKL04) or tor2-as (yKL05) strains were transformed with a 3xHA-Lac1 expressing plasmid (pAX136). Cells were grown to mid-exponential phase and treated with 1 uM BEZ-235 for the indicated times. Cell extracts were then prepared, resolved by Phos-tag SDS-PAGE and blotted as above in (A). 58 Fig. 4.4: Enhanced Ypk1 phosphorylation of Lac1 and Lag1 under sphingolipid and heat stress. (A) 3xHA-Lac1 (yAM165-A) cells were grown to early exponential phase in YPD. Cultures were then treated with sublethal doses of myriocin (0.625 μM) or methanol (vehicle) or aureobasidin A (1.8 μM) of ethanol (vehicle) for 2 h. (B) 3xHA-Lac1 (yAM165-A) cells were grown to exponential phase in YPD at 30˚C. A sample of this culture was then harvested. The remaining culture was then moved to 42˚C to initiate heat shock and samples were harvested at the indicated time points. Whole cell extracts were prepared from each sample, resolved by Phos-tag SDS-PAGE, and immunoblotted as in Figure 4.3. P-Lac1 indicates the band corresponding to S23 S24 phosphorylation. * indicates a non-specific band that appears in HA blots of yeast whole cell extracts. (C) LAC1 LAG1 (yAM205 - A), Lac1SSAA Lag1SSAA (yAM207 - B) and Lac1SSEE Lag1SSEE (yAM210) were grown to exponential phase in YPD. Serial dilutions of each culture were made and spotted on YPD plates containing vehicle or the indicated concentration of myriocin or aureobasidin A. Cells were allowed to grow for 3 days at 30˚C prior to imaging. (D) 3xHA-Lac1::HIS3 3xFLAG-Lag1::LEU2 (yAM168) 3xHA-Lac1(S23A S24A)::HIS3 3xFLAG-Lag1(S23A S24A)::LEU2 (Lac1SSAA Lag1SSAA) (yAM184) and 3xHALac1(S23E S24E)::HIS3 3xFLAG-Lag(S23E S24E)1::LEU2 (Lac1SSEE Lag1SSEE) (yAM192 – A) cells were treated with 1.0 uM myriocin or 18.2 nM aureobasidin A for 2h. Extracts were prepared from each sample, resolved by SDS-PAGE, and immunoblotted as indicated. 59 Fig. 4.5: Activation of calcineurin leads to rapid dephosphorylation of Lac1 and Lag1 without affecting Ypk1 function. (A) Wild-type (BY4741) or cna1Δ cna2Δ (JTY5574) strains were transformed with a centromeric plasmid encoding 3xHA-Lac1 expressed under control of its endogenous promoter (pAX136). Cultures were grown to mid-exponential phase in selective media and then treated with 200 mM CaCl2 for 10 minutes to activate calcineurin. Cultures were then harvested and whole cell extracts were prepared from each sample, resolved by Phos-tag SDS-PAGE, and immunoblotted as in Figure 4.3. (B) 3xHA-Lac1 (yAM165-A) cells were transformed with a centromeric plasmid encoding the hyperactive TORC2 independent Ypk1D242A allele expressed under its own promoter (pFR273) or vector (pRS316). Cultures were grown in selective media to mid-exponential phase before treatment with 200 mM CaCl2 for 10 min Cultures were then harvested and whole cell extracts were prepared from each sample, resolved by Phos-tag SDS-PAGE, and immunoblotted as above. (C) 3xFLAG-Ypk1 (YDB379) cells were grown to midexponential phase in YPD. Cultures were treated with or without 200 mM CaCl2 for 10 min Cells were then harvested and whole extracts prepared. Extracts were resolved by SDS-PAGE and blotted with anti-pSGK(T256), which recognizes Ypk1 T504 activation loop phosphorylation and anti-FLAG antibody (Ypk1 loading control). The blot is representative of triplicate samples and the quantitation of the ratio of pT504/FLAG from these replicates is shown below the blot. 60 61 Fig. 4.6: Ypk1 phosphorylation of Lac1 and Lag1 stimulates ceramide synthase activity. (A) Cultures of LAC1 LAG1 (yAM205 - A), Lac1SSAA Lag1SSAA (yAM207 - B) and Lac1SSEE Lag1SSEE (yAM210) strains were grown to mid-exponential phase and then harvested. Sphingolipids were extracted and analyzed as described in chapter 2. Values represent the mean of three independent experiments (each performed in triplicate) and error bars represent SEM. (B) Triplicate exponentially growing cultures of LAC1 LAG1 (yAM205 - A), Lac1SSAA Lag1SSAA (yAM207 - B) and Lac1SSEE Lag1SSEE (yAM210) were adjusted to OD600 = 1.0. Complex sphingolipids were labeled and analyzed by thin layer chromatography (TLC) as in chapter 2. A representative TLC plate is shown with the origin at the bottom of the image. The assigned identity of species as IPCs and MIPCs was confirmed by pharmacological or genetic inhibition of the production of these species in control cultures (data not shown). Quantification of total complex sphingolipids was performed in ImageJ by integrating the phosphorimager screen intensity across the lane for each sample and normalized to 1 for wild-type ceramide synthase samples. (C) (upper) Ceramide synthase was purified by FLAG immunopurification from 3xHALac1::HIS3 3xFLAG-Lag1::LEU2 (yAM168) 3xHA-Lac1(S23A S24A)::HIS3 3xFLAGLag1(S23A S24A)::LEU2 (Lac1SSAA Lag1SSAA) (yAM184) and 3xHA-Lac1(S23E S24E)::HIS3 3xFLAG-Lag(S23E S24E)1::LEU2 (Lac1SSEE Lag1SSEE) (yAM192 – A) cells. Immunoprecipitates were then split into 3 fractions and in vitro ceramide synthase assays (60 min reactions) were performed in triplicate. A small sample of each ceramide synthase assay was resolved by SDS-PAGE and immunoblotted. The signal intensity quantified from the immunoblot was then used to normalize ceramide synthase activity in each sample. (lower) Ceramide synthase was immunopurified from 3xHA-Lac1::HIS3 3xFLAG-Lag1::LEU2 (yAM168) 3xHA-Lac1(S23A S24A)::HIS3 or 3xFLAG-Lag1(S23A S24A)::LEU2 (Lac1SSAA Lag1SSAA) (yAM184) cells as above except cultures were treated with 1.0 uM myriocin or methanol vehicle prior to harvesting. Values represent the mean of three independent experiments (each performed in triplicate) and error bars represent SEM. Statistical significance of values (Student's t-test): *, p = <0.05, **, p = <0.009; and, ***, p <0.0009. 62 63 Fig. 4.7: Failure of Ypk1 to upregulate ceramide synthase causes LCBP accumulation that triggers autophagy. (A) LAC1 LAG1 (yAM205 - A) or Lac1SSAA Lag1SSAA (yAM207 - B) were transformed with PYPK1-Ypk1D242 (shown as Ypk1*) (pFR273) or pRS316 (E.V.). Transformants were grown strains were grown to exponential phase in synthetic complete media and then diluted to OD600=0.1 and grown in microtiter plates (lower) or on agar plates (upper). For liquid cultures, each was grown in at least quadruplicate and the error bars indicate the SEM of replicates at each time point. (B) cells from (A) were grown to mid-exponential phase in selective synthetic complete media and then harvested. Sphingolipids were extracted and analyzed as described in chapter 2. Values represent the mean of three independent experiments (each performed in triplicate) and error bars represent SEM. (C) LAC1 LAG1 lcb4Δ (yAM237) or Lac1SSAA Lag1SSAA lcb4Δ (yAM238 – A) were transformed with PYPK1-Ypk1D242 (shown as Ypk1*) (pFR273) and growth experiments performed as in (A). (D) Liquid growth assays were performed as in (A) for LAC1 LAG1 (yAM205 - A), Lac1SSAA Lag1SSAA (yAM207 - B), Lac1SSEE Lag1SSEE (yAM210), LAC1 LAG1 lcb3Δ (yGT12), Lac1SSAA Lag1SSAA lcb3Δ (yGT13) and Lac1SSEE Lag1SSEE lcb3Δ (yGT14) strains. (E) LAC1 LAG1 (yAM205 - A) or Lac1SSAA Lag1SSAA (yAM207 - B) or LAC1 LAG1 lcb3Δ (yGT12) strains were transformed with PYPK1-Ypk1D242 (shown as Ypk1*) (pFR273) or pRS316 (E.V.) and additionally PTPI1-GFP-Atg8. Growing cultures treated with vehicle or 2 μg/mL rapamycin for 2h and then harvested and whole extracts prepared. Extracts were resolved by SDS-PAGE and blotted with anti-GFP to detect GFP-Atg8 and free GFP arising from GFP-Atg8 autophagic processing and anti-Pgk1 antibody. The blot is representative of triplicate samples and the quantitation of the ratio of free GFP/Pgk1 from these replicates is shown below the blot. (F) LAC1 LAG1 atg1Δ (yAM239 - A) or Lac1SSAA Lag1SSAA atg1Δ (yAM240 – A) sensitivity to PYPK1-Ypk1D242 was measured as in (A). (G) Cells from (F) were grown to mid-exponential phase in selective synthetic complete media and then harvested. Sphingolipids were extracted and analyzed as described in (B). 64 65 Fig. 4.8: TORC2-Ypk1 signaling globally activates sphingolipid synthesis, selectively directs flux toward ceramide metabolites, and prevents LCBP cross-talk to the autophagy pathway. (A) Diagram of yeast de novo sphingolipid biosynthesis shown is derived from (Dickson, 2008). Enzymes are in ovals. Skeletal structures of metabolites are shown. Increased color intensity indicates level of metabolite increase in response TORC2-Ypk1 activation. TORC2-Ypk1 signaling globally activates de novo sphingolipid biosynthesis via derepression of the SPOTS complex (Berchtold et al., 2012; Roelants et al., 2011; Sun et al., 2012), potentially increasing levels of all metabolites. However, Ypk1 also upregulates ceramide synthesis via phosphorylation of Lac1 and Lag1, thus primarily directing this increased flux towards ceramides and away from LCBs and LCBPs. (B) In the absence of Ypk1 mediated ceramide biosynthesis regulation, increased sphingolipid flux raises LCB and LCBP levels. This slows cell growth by activating autophagy. Thus, TORC2-Ypk1 signaling not only activates sphingolipid biosynthesis in response to stress, but also insulates this flux towards ceramides to prevent metabolite mediated crosstalk to the autophagy machinery. 66 67 Fig. 4.9: Loss of Ypk activity or putative Ypk phosphorylation of Atg21 does not appear to prevent CVT or amino acid starvation induced autophagy. (A) Wild-type (yAM234 - B), atg21Δ (yAM235-A), or ypk1-as ypk2Δ (yAM236-B) strains expressing GFP-Atg8 from a constitutive promoter (TPI1) integrated at the HIS3 locus were grown with the indicated concentrations of 3-MB-PP1 into mid-exponential phase in rich (YPD) medium. Based on doubling time of ypk1-as ypk2Δ (yAM135–A) cells in YPD, 0.5 μM 3-MB-PP1 leads to ~30% reduction in Ypk1 activity, 1.0 μM 3-MB-PP1 leads to ~50% reduction in Ypk1 activity, and 2.0 μM 3-MB-PP1 leads to ~90% reduction in Ypk1 activity (data not shown). Cells were harvested, and then whole extracts were prepared and blotted as in Fig. 4.7E. (B) atg21Δ (yAM221-A) cells were co-transformed with a centromeric PTPI1-GFP-Atg8 plasmid (pAX239) and empty vector (pRS316), Atg21-3xFLAG (pAX241), Atg21 (S237A)-3xFLAG (pAX247) or Atg21(S237E)3xFLAG (pAX246) plasmids. Cells were grown in selective SCD medium into mid-exponential or stationary phase. Cells were harvested and blotted for GFP-Atg8 processing as in (A). Additionally, blotting was performed for Ape1. (C) atg21Δ (yAM221-A) cells were cotransformed with a centromeric PTPI1-GFP-Atg8 plasmids (pAX250) and empty vector (pRS316), Atg21-3xFLAG (pAX241), Atg21 (S237A)-3xFLAG (pAX247) or Atg21(S237E)-3xFLAG (pAX246) plasmids. Cells were grown to mid-exponential phase in selective SCD medium. Cells were then washed and put into SCD medium without leucine (yAM221–A is a leucine auxotroph) to trigger amino acid starvation. Cells were harvested and blotted for GFP-Atg8 processing as in (A). 68 Chapter 5: TORC2-Ypk1 signaling is a MAPK-independent mechanism for promoting glycerol accumulation and survival in response to hyperosmotic stress Introduction Cells routinely encounter many diverse environmental and developmental stresses that physically challenge the plasma membrane. For example, mechanical stretching of skin keratinocytes or environmental osmotic shocks in microbes generate forces that threaten membrane integrity and ultimately, if unresolved, cell viability. Understanding how cells cope with commonly occurring membrane stresses is essential for understanding how membrane homeostasis is maintained in changing environments. How S. cerevisiae cells respond to hyperosmotic shock has served as a model system for understanding how the cell copes with reduced turgor pressure and relaxed membrane tension. In yeast, two pathways have been identified, to date, that are triggered when cells are subjected to hypertonic stress. In response to such a challenge, plasma membrane-embedded osmosensors (Sln1 and Sho1) associated with heavily O-glycosylated cell wall-embedded mucins (Msb2 and Hkr1) (Cullen, 2007; Tanaka et al., 2014; Tatebayashi et al., 2015) initiate pathways that activate a MAPK family member, Hog1 (Brewster, 1993; Brewster & Gustin, 2014). The sequence and functional ortholog of Hog1 in mammals is p38/SAPK/MAPK14 (Han et al., 1994; Cuenda & Rousseau, 2007; Cuadrado & Nebreda, 2010). Activated Hog1 drives a transcriptional and posttranscriptional program that increases glycerol production and retention (Hohmann, 2009; Saito and Posas, 2012; Hohmann, 2015). Intercellular glycerol acts as a counter-balancing osmolyte critical for optimal survival when cells are placed in hyperosmotic medium. In addition, hyperosmotic shock increases cytosolic Ca2+ concentration (Denis and Cyert, 2002) to a level sufficient to activate phosphoprotein phosphatase 2B/calcineurin (CN), which then specifically associates with and dephosphorylates the PtdIns4,5P2 5-phosphatase Inp53/Sjl3 to stimulate retrieval of excess plasma membrane via clathrin-mediated endocytosis (Guiney et al., 2015). Conversely, when yeast cells are placed in hypotonic conditions, different mechanosensors (Wsc1, Wsc2, Wsc3, Mid2 and Mtl1) that span the plasma membrane and the cell wall (Kock et al. 2015) activate GEFs for the small GTPase Rho1, which in its GTP-bound state stimulate a protein kinase, Pkc1, that initiates a signaling cascade that activates another MAPK family member, Mpk1/Slt2 (de Nobel et al., 2000; Levin, 2011). Activated Slt2/Mpk1 drives the expression of factors that strengthen the cell wall layers (Orlean, 2012; Free, 2013) to combat the increased turgor pressure and, hence, is a response referred to as the cell wall integrity pathway (Levin, 2011; Engelberg et al., 2014). The sequence and functional ortholog of yeast Pkc1 in mammals is the RHO- (and RAC-) activated PKC-related enzyme PRK2/PKN2 (Mukai, 2003; Thumkeo et al., 2013) and the sequence and functional ortholog of yeast Slt2/Mpk1 in mammals is ERK5 (Piper et al., 2006; Truman et al., 2006). Collectively, therefore, these studies of the response to stress in yeast have helped to illuminate how mammalian cells respond to osmolarity changes and/or the degree of membrane stretch. Despite this progress in understanding the cellular response to extracellular osmolarity and membrane stress, regulation of even well studied cellular survival mechanisms still remains incomplete. For example, an important part of the yeast response to hyperosmolarity is 69 prevention of glycerol efflux from cells that is mediated by the aquaglyceroporin Fps1 (Luyten et al., 1995; Tamás et al., 1999; Ahmadpour, et al., 2014). Although Hog1 can negatively regulate Fps1 by displacing the Fps1-activating protein Rgc2 in response to hyperosmotic shock (Lee et al., 2013), Fps1 still closes in response to hyperosmotic shock in the absence of Hog1 (Tamás et al., 1999; Babazadeh et al., 2014). Thus, there must be at least one additional osmosensing mechanism that exists that is able to transduce the hyperosmolarity signal to Fps1 and prevent efflux of glycerol and, thus, promote adaptation to hypertonic stress. Recently, we and others have found that the TORC2 complex in S. cerevisiae, which was known to respond to membrane stresses such as depletion of sphingolipids and heat shock (Roelants et al., 2011; Berchtold et al., 2012; Sun et al., 2012) also responds to changes in osmotic conditions and/or membrane stretch. As judged by phosphorylation of the TORC2 effector Ypk1, TORC2 activity apparently increases under hypo-osmotic stress (or by mechanical stretch) and the increase in TORC2-Ypk1 signaling elevates the level of membrane sphingolipids to manage this stress (Berchtold et al., 2012). Conversely, as judged by the same criterion, TORC2 activity seems to decrease dramatically during hyperosmotic shock and the loss of TORC2-Ypk1 signaling helps attenuate the hyperosmotic shock, at least in part, through alleviating Ypk1dependent inhibition of glycerol-3-phosphate dehydrogenase Gpd1, leading to the production of more glycerol-3-P, which, after dephosphorylation, increases intracellular glycerol (Lee et al., 2012b). As with the other osmosensing machinery in yeast, mammalian TORC2 activity is also responsive to changes in osmotic conditions (Sengupta et al., 2013) and membrane stretch (Kippenberger et al., 2005). Thus, TORC2 is a conserved, membrane stress-responsive signaling module whose output modulates both membrane composition and intracellular osmolyte level. To understand how TORC2-Ypk1 signaling contributes to cellular osmotic and membrane stress response, I recently performed a genome-wide screen to identify novel Ypk1 substrates. In this way, I demonstrated that activated Ypk1 stimulates the ceramide synthase complex to to promote the formation of sphingolipids (Muir et al., 2014). In this screen, I also identified two enzymes involved in glycerol metabolism. The aquaglyceroporin Fps1 and Gpt2/Gat1, a glycerol-3phosphate/dihydroxyacetone phosphate acyltransferase that consumes glycerol-3-phosphate to generate phosphatidic acid, a precursor to phospholipids or triacylglycerol (Zheng and Zou, 2001; Bratschi et al., 2009; Henry et al., 2012). Additionally, the screen uncovered a transcription factor Smp1, that has been previously implicated in the response to hyperosmotic shock (de Nadal et al., 2003) and also in Ypk1 physiology (Roelants et al., 2002). In this chapter, I document that, although Gpt2 does not appear to be under TORC2-Ypk1 regulation, Fps1 and Smp1 are bona fide substrates of Ypk1 in vivo and their Ypk1-dependent phosphorylation is physiologically important. I found that, under normal growth conditions, Ypk1 phosphorylates Fps1 and that this phosphorylation is essential for Fps1 channel activity. Thus, in the absence of stress, TORC2-Ypk1 signaling licenses glycerol efflux. However, upon hyperosmotic shock, TORC2 activity and, in turn, Ypk1 activity are rapidly lost, resulting in closure of the Fps1 channel. Therefore, the reduction in TORC2-Ypk1 signaling that occurs under hyperosmotic conditions allows cells to cope with this stress by promoting accumulation of intracellular glycerol not only by increasing glycerol production (Lee et al., 2012b), but also by ensuring glycerol retention. Lastly, I show that abrogation of TORC2-Ypk1 phosphorylation of Fps1 occurs independently of the MAPK Hog1 and other components of known hyperosmotic 70 sensing pathways. Thus, my work has uncovered a previously uncharacterized mechanism that contributes to cellular adaptation to high osmolarity by driving a coordinated program of glycerol production and retention. Results Smp1 is phosphorylated by Ypk kinases and this modification influences Smp1 function and localization To examine the role of TORC2-Ypk1 signaling in glycerol metabolism, I examined, first, phosphorylation of a candidate Ypk1 substrate that emerged from my screen (Muir et al. 2014) that is connected to utilization of glycerol-3-P, namely the glycerol-3-P/dihydroxyacetone-P sn-1 O-acyltransferase Gpt2/Gat1. There is one perfect match to the Ypk1 consensus phosphoacceptor sequence (646RSRSSSI652) in this 743-residue enzyme. Based on past experience, we have found that at such sites containing multiple Ser residues more than just the canonical one (in red) can be phosphorylated in a Ypk1-dependent manner (Roelants et al., 2011). For that reason, I prepared a Gpt2(S649A S650A S651A) mutant as well as a phosphomimetic Gpt2(S649E S650E S651E) mutant. One or more of these three Ser residues is phosphorylated in vivo because, compared to wild-type Gpt2, the Gpt23A mutant exhibited a distinctly faster mobility upon SDSPAGE, a hallmark of decreased phosphorylation (Fig. 5.1A). However, this phosphorylation did not appear to be dependent on Ypk1 activity because there was no change in Gpt2 mobility when a ypk1-as ypk2Δ strain was treated with inhibitor 3-MB-PP1 (Fig. 5.1A). Other AGC kinase family members with prominent roles in metabolic regulation have, like Ypk1, basophilic consensus phosphoacceptor motifs, including yeast AMPK (Snf1) and yeast PKA (Tpk1, Tpk2 and Tpk3). However, like loss of Ypk1 function, absence of these kinases did not prevent Gpt2 phosphorylation (Fig. 5.1A). Furthermore, based on a phenotypic criterion, it appears that phosphorylation at the predicted Ypk1 consensus site(s) has no physiological role. In the absence of Gpt2, the growth of yeast cells is inhibited by high concentrations of the fatty acid oleate, whereas wild-type cells grow well under this condition (Zaremberg & McMaster, 2002). Moreover, in the absence of both Gpt2 and its paralog Sct1/Gat2, cells are inviable (Zaremberg & McMaster, 2002). I found that gpt2∆ sct1∆ cells expressing either the Gpt23A or Gpt23E alleles grew just as well as cells expressing wild-type Gpt2 (Fig. 5.1B). Thus, in the absence of any evidence for Ypk1-mediated phosphorylation of Gpt2 or any evidence for an effect of phosphorylation at the putative Ypk1 site, I turned my attention to another candidate Ypk1 substrate that emerged from our screen (Muir et al., 2014), namely the MADS-box transcription factor Smp1 (Shore & Sharrocks, 1995; Dodou & Treisman, 1997), which had been connected previously to both Ypk1 function (Roelants et al, 2002) and in the response of yeast cells to hyperosmotic stress (de Nadel et al., 2003). The mammalian orthologs of Smp1 (and its paralog Rlm1) are the MEF2 family of transcription factors (Potthoff & Olson, 2007). There are three predicted Ypk1 phosphorylation sites (15RNRTVTF21, 102RRRRATV108 and 216RIRPDTH222) in the 452-residue Smp1 protein. To enhance the ability to detect phosphorylation-dependent mobility shifts, I generated a C-terminal truncation, Smp1(∆275-452), because the resulting Smp1(1-274) fragment contains all three predicted Ypk1 sites, but lacks sequences that contain predicted Hog1 (MAPK) phosphorylation sites (de Nadel et al., 2003). Using Smp1(1-274) produced a readily interpretable banding pattern upon SDS-PAGE containing Phos-tag™, a separation method designed to resolve phospho71 isoforms (2009). I found that a slower mobility species of Smp1(1-274) was detectable in wildtype cells that became markedly more prominent when cells were treated with a stimulus (addition of the antibiotic myriocin) known to lead to Ypk1 activation (Roelants et al. 2011) (Fig. 5.2A). Moreover, this species was completely eliminated when a Smp1(1-273; T20A T107A T221A) mutant was examined (Fig. 5.2A). Resolution of full-length Smp1 by Phos-tag SDS-PAGE yielded a complicated banding pattern; yet, absence of the three predicted Ypk1 sites caused a readily detectable decrease in the mobility of many of these species (Fig. 5.2B). Collectively, these observations indicate that Smp1 is phosphorylated in cells in a Ypk1dependent manner and at its predicted Ypk1 sites. Furthermore, based on a phenotypic criterion, it appears that phosphorylation at the predicted Ypk1 consensus site(s) has an important physiological role. Unlike wild-type cells, cells lacking Smp1 are resistant to high concentrations of ferric (Fe3+) ions (Lee et al., 2012a). I found that both an Smp1(T20A T107A T221A) allele and a Smp1(T20E T107E T221E) allele were just as resistant to the toxic effects of Fe3+ as a complete null mutant (smp1∆) (Fig. 5.2C), even though wild-type Smp1 and the two mutants were expressed at comparable levels (Fig. 5.2D). Thus, authentic phosphorylation at its Ypk1 sites is necessary for Smp1 function (and, in the case of this protein, Glu residues at the same positions cannot replace the presence of actual phosphate groups). Ypk1 activity also appears to affect the subcellular localization of Smp1. Using an Smp1-GFP fusion, which has been shown previously to be functional (de Nadal et al., 2003), I found that Smp1 localizes to the cytoplasm during exponential growth, but translocates to the nucleus during stationary phase, in agreement with what others have observed before (de Nadal et al., 2003). However, in cells expressing the ypk1-as allele, when 10 μM 3-MB-PP1 was added to completely inhibit Ypk1 activity (while leaving Ypk2 activity, and thus cell viability unaffected), I observed that Smp1 was present in the nucleus even during exponential growth (Fig. 5.2E). Thus, one role for Ypk1-mediated phosphorylation of Smp1 is to prevent its nuclear import. Lastly, Smp1 mediates part of the transcriptional response to hyperosmotic shock (de Nadal et al., 2011), including contributing to full transcriptional upregulation of the STL1 gene (de Nadal et al., 2003), which encodes a glycerol/H+ symporter responsible for glycerol import (Duskova et al., 2015). Using a STL1promoter-GFP gene fusion as a transcriptional reporter, I measured STL1promoter-driven expression in smp1Δ, ypk1-as or double mutant cells both basally and after exposure to hyperosmotic shock (0.4 M NaCl). I found that cells lacking Smp1 had only a modest diminution in its salt stress-induced expression, similar to what has been described previously (de Nadal et al., 2003) (Fig. 5.2F). Strikingly, however, in cells in which Ypk1-as had been inhibited with 3MB-PP1, salt stress induction of STL1 expression was almost completely prevented. The requirement for Ypk1 activity for salt stress-evoked STL1 expression was observed even in cells lacking Smp1. Hence, Ypk1 function clearly modulates Smp1 localization and function, but also influences gene expression independently of its modification of Smp1. It would be worthwhile to construct and examine additional reporter plasmids containing other predicted Smp1-responsive and hyperosmotic shock-regulated promoters to try to characterize how Ypk1 modulates transcription. Whether TORC2-Ypk1 signaling has a role in regulating other types of membrane stress-induced transcription is also of significant interest. Fps1 is phosphorylated by Ypk kinases and this modification is blocked when cells are subjected to hyperosmotic stress A third candidate Ypk1 substrate that emerged from my screen (Muir et al. 2014) that is directly 72 connected to control of intracellular glycerol concentration is the aquaglyceroporin Fps1, which regulates glycerol efflux (Karlgren et al., 2004; Lee et al., 2013; Duskova et al. 2015). I observed before that GAL promoter-driven overexpression of Fps1 in ypk1-as ypk2Δ cells was growth inhibitory, whereas Fps1 overexpression had no effect on wild-type (YPK1+ YPK2+) cells (Muir et al. 2014). Moreover, in a global screen for interactors with all yeast protein kinases and phosphatases, it has been reported that the paralog of Ypk1, Ypk2, is able to physically associate with Fps1 (Breitkreutz et al., 2010). For these reasons, I initially entertained the possibility that the reason for the observed toxicity of Fps1 overexpression in cells with only limiting Ypk1 activity might be that the elevated level of Fps1 sequestered the available pool of Ypk1 away from the membrane microdomains that harbor the essential Ypk1 activators Pkh1 (and Pkh2) and TORC2, resulting in a lack of activation of Ypk1, a kinase that is essential for cell viability (Chen et al. 1993; Roelants et al., 2002). Therefore, I examined Ypk1 phosphorylation at T504 (the T-loop/activation loop residue that is the target of Pkh1 and Pkh2) and T662 (a residue in the so-called hydrophobic motif that is a target of TORC2) in strains lacking or overexpressing Fps1. I found that the level of Fps1 expression had no significant effect on Ypk1 phosphorylation at either of these sites (Fig. 5.3). Therefore, it did not appear that Fps1 was identified in our screen because it interferes with Ypk1 activation. Hence, it seemed more likely that Fps1 was recovered in our screen because it is, in fact, a bona fide Ypk1 substrate. Despite the technical difficulties in examining an integral membrane protein, I confirmed this conclusion, as follows. In Fps1, there are four, predicted, Ypk1 consensus phosphorylation sites (142RPRGQTT148, 176RRRSRSR182 , 180RSRATSN186 and 565RARRTSD148) (Fig. 5.4A). A C-terminal fragment of Fps1 (and its proteolysis products) containing the single Ypk1 site Ser570 was phosphorylated in a Ypk1-specific manner in vitro and mutation of Ser570 to Ala demonstrated that the observed phosphorylation was specific to the predicted Ypk site (Fig. 5.4B). Using Phos-tag SDS-PAGE analysis, I confirmed that Ser570 is phosphorylated in vivo because a corresponding S570A mutation caused a dramatic shift to a faster mobility species (Fig. 5.4C, rightmost lanes). An Nterminal fragment of Fps1 containing the three other predicted Ypk1 sites was not detectably phosphorylated by Ypk1 in vitro (Fig. 5.4B); however, when expressed in vivo, and based on the effect on mobility in Phos-tag SDS-PAGE of mutating Ser181 and Ser185 to Ala, these two sites are clearly phosphorylated in the cell (Fig. 5.4C). Likewise, the mobility of Fps1 underwent the same dramatic shift to faster mobility in ypk1-as ypk2Δ cells treated with 3-MB-PP1, but not in wild-type cells (Fig. 5.4D). Moreover, the mobility of the resulting species was not different from that of an Fps1(S181A S185A S570A) mutant, Fps13A hereafter, and inhibition of Ypk1 activity did not further increase its mobility. One likely possibility to explain the difference between our in vitro results (performed with N- and C-terminal fragments of Fps1) and our in vivo results (examining intact Fps1) is that Ypk1 docks at a binding site in the C-terminal domain in order to phosphorylate sites in the N-terminal domain. Furthermore, as expected for a Ypk1dependent modification, phosphorylation of these same sites in Fps1 in vivo was also TORC2dependent, as judged by the fact that inhibition of TORC2 activity with a pharmacological agent (NVP-BEZ235) (Maira et al., 2008; Kliegman et al., 2013) also drastically reduced Fps1 phosphorylation (Fig. 5.4E). I conclude that Fps1 is an authentic in vivo substrate of Ypk1. Recently, we documented that TORC2-mediated phosphorylation of Ypk1 is greatly decreased when cells are subjected to hyperosmotic shock (Lee et al., 2012b). Using a Ypk17A mutant, which allows for facile detection of mobility shifts arising from its TORC2-specific 73 phosphorylation, I found that loss of TORC2-mediated phosphorylation of Ypk1 after hyperosmotic shock occurs very rapidly (within 1 min) (Fig. 5.5A). This loss of TORC2mediated phosphorylation of Ypk1 is transient, however. By 20 min after hyperosmotic shock, TORC2-mediated phosphorylation of Ypk1 is again readily detectable and is back to its prestress level by 75 min after application of the stress (Fig. 5.5A). The reduction in TORC2mediated phosphorylation of Ypk1 in response to hypertonic stress was still observed in mutants that abrograte signal propagation in either the Sho1- or Sln1-dependent branches of the HOG pathway, or in the absence of their common terminal effector, Hog1 MAPK (Fig. 5.5B). Given that TORC2-Ypk1 action results in direct Fps1 phosphorylation and given that TORC2-mediated phosphorylation of Ypk1 is greatly decreased when cells are subjected to hyperosmotic shock, I examined whether phosphorylation of Fps1 was affected when cells were subjected to hyperosmotic shock. Indeed, as expected, hyperosmotic shock led to loss of phosphorylation of Fps1 that mirrored the kinetics of the loss of TORC2-mediated phosphorylation of Ypk1 in response to hypertonic stress and yielded a species with a mobility indistinguishable from that of the Fps13A mutant lacking Ypk1 phosphorylation sites (Fig. 5.5C). Taken together, these results demonstrate that loss of TORC2-mediated phosphorylation of Ypk1 in response to hyperosmotic stress dramatically down-modulates Fps1 phosphorylation in a HOG pathway-independent manner. Ypk-mediated phosphorylation of Fps1 promotes channel opening and glycerol efflux To determine whether this newly revealed mode of Fps1 regulation serves any function in promoting cell survival in the face of hypertonic stress, I next investigated how Ypk1-mediated phosphorylation may affect Fps1 function. First, I repeated the SDL assay (Muir et al., 2014) to assess the phenotype caused by overexpression of wild-type Fps1, Fps13A, or previously characterized mutants of Fps1 that are known to cause the channel to be constitutively open (Tamás et al., 2003; Hedfalk et al., 2004) or constitutively closed (Lee et al., 2013). I found that overexpression of wild-type, Fps13A, and a known closed channel mutant gave an SDL phenotype, whereas known open channel mutants did not (Table 5.1). Based on this criterion, the behavior of Fps13A resembles that of a closed channel, suggesting that Ypk1-mediated phosphorylation promotes channel opening. Fps1 can mediate the influx of xylitol (not a physiological carbon source for Saccharomyces cerevisiae; Fraenkel, 2011) and expression of constitutively open channel mutants allows cells defective in glycerol production (gpd1Δ gpd2Δ mutants) to survive when challenged with a high concentration of exogenous xylitol by permitting the xylitol to equilibrate across the membrane, thereby restoring osmotic balance (Hedfalk et al., 2004). Expression of an N-terminal truncation mutant, Fps1(∆12-231), that is constitutively open allowed cells to survive on 1M xylitol, whereas Fps13A did not (Fig. 5.6A). Thus, by this second criterion, the behavior of Fps13A resembles that of a closed channel. Fps1 can also mediate the entry of a toxic metalloid, arsenite, and thus arsenite sensitivity is a read-out of Fps1 function (Thorsen et al., 2006). Unlike wild-type Fps1, all Fps1 mutants predicted to lack transport activity, such as a null mutant (fps1∆), a cell lacking known activators that promote Fps1 channel opening (rgc1Δ rgc2Δ double mutant) (Beese et al., 2009), and a mutant (fps1ΔPHD) unable to bind Rgc1 or Rgc2 (Lee et al., 2013), exhibited resistant to arsenite 74 toxicity (Fig. 5.6B). Strikingly, cells expressing Fps13A as the sole source of this channel were equally if not more arsenite resistant (Fig. 5.6B). Hence, by this third criterion, the behavior of Fps13A resembles that of a closed channel. If Ypk1-mediated phosphorylation at multiple sites promotes Fps1 channel opening in a graded manner, then one might expect that phosphorylation at individual Ypk1 sites would contribute additively to channel opening and, conversely, that absence of phosphorylation at individual Ypk1 sites would contribute additively to arsenite resistance, which is exactly what I observed (Fig. 5.6C). Collectively, these results indicate that TORC2-stimulated Ypk1-mediated phosphorylation of Fps1 promotes opening of this channel and its transport activity. It has been shown that loss of Fps1 function leads to retention of intracellular glycerol (Tamás et al., 1999). Consistent with Ypk1-dependent phosphorylation favoring the open state of Fps1, I found that cells expressing Fps13A accumulated ~2 fold as much glycerol as wild-type cells (Fig. 5.6D). In agreement with this finding, we previously documented that cells deficient in Ypk activity have a higher intracellular glycerol level (Lee et al., 2012b). For this reason, I also examined the arsenite resistance of cells in which I attempted to reduce Ypk activity because loss of the kinase should mimic Fps13A and, perhaps, allow enhanced resistant to the toxic effect of arsenite. However, it is not possible to completely inhibit Ypk function and maintain viability. However, even at a sub-lethal dose of 3-MB-PP1, I did not detect any arsenite resistance in either ypk1-as or ypk1-as ypk2Δ cells (Fig. 5.6E). Presumably, as with maintenance of viability, the residual Ypk activity present is still sufficient to phosphorylate Fps1 and keep the channel open. I also cannot rule out the possibility that reduction of Ypk activity may have additional consequences that might increase the arsenite sensitivity of cells, even if there is some down-modulation of Fps1 function. Nevertheless, taken together, all of our other results show conclusively that Ypk1 phosphorylates and thereby promotes opening of Fps1. I considered that TORC2-dependent Ypk1-mediated phosphorylation of Fps1 might also promote its channel function in other ways, such as stabilizing the protein or increasing its dwell time and, hence, its concentration, in the plasma membrane. However, immunoblotting (Fig. 5.6F) and fluorescence microscopy (Fig. 5.6G) indicate that steady-state level and localization of wild-type Fps1 and Fps13A are indistinguishable. Therefore, the primary effect of Ypk1dependent phosphorylation of Fps1 is to favor the open state of this channel. TORC2-Ypk regulation of Fps1 is independent of the known Fps1 regulators Hog1 and Rgc1/2 and allows cells to survive hyperosmotic stress in the absence of Hog1 Fps1 is negatively regulated by Hog1 MAPK in two ways: (i) in response to challenge with acetic acid or arsenite (but, curiously, not hyperosmotic stress), direct phosphorylation of Fps1 by Hog1 stimulates channel internalization and degradation (Thorsen et al., 2006; Mollapour and Piper, 2007; and (ii) in response to hyperosmotic stress, direct phosphorylation of Fps1 by Hog1 causes dissociation of its positive activators Rgc1 and Rgc2 (Beese et al., 2009; Lee et al., 2013). I found that Fps13A was still arsenite resistant even in a hog1Δ mutant (Fig. 5.7A), quite similar to the behavior of a strain that expresses an Fps1 mutant unable to bind Hog1 (Lee et al., 2013) 75 (Fig. 5.7B). Thus, mechanistically, TORC2-Ypk1 regulation of Fps1 occurs in a manner independent of any role for Hog1. In addition, as judged by their co-immunoprecipitation, I found that the lack of Ypk1-mediated phosphorylation does not cause dissociation of the positive Fps1 activator Rgc2 because as much or more Rgc2 is associated with Fps13A as with wild-type Fps1 (Fig. 5.7C). Thus, although the exact mechanism by which TORC2-Ypk1 phosphorylation favors Fps1 channel opening is not yet known, it is clear that it is not achieved through modulation of the Hog1-Rgc1/2 system. Our results suggest that loss of TORC2-dependent phosphorylation of Ypk1 that occurs when cells are subjected to hypertonic conditions and the ensuing reduction of Ypk1-mediated phosphorylation of Fps1 provides a novel means, independent from the HOG pathway, for both sensing high osmolarity and responding to it because decreased Fps1 channel function should permit intracellular glycerol accumulation. Indeed, tellingly, I found that a Fps13A hog1Δ double mutant cells is more resistant to hyperosmotic stress than hog1Δ cells, exhibiting growth at a near wild-type level at 0.5 and 0.75 M sorbitol concentrations (Fig. 5.7D). This striking rescue confirms that loss of Ypk1-mediated Fps1 channel opening is sufficient for cells to accumulate an adequate amount of glycerol to physiologically cope with hyperosmotic stress, even in the absence of Hog1. Discussion Our screen for new substrates (Muir et al., 2014) reinforces the conclusion that glycerol metabolism as an important process regulated by the TORC2-Ypk1 axis. Here I have documented that the loss of TORC2 stimulation of Ypk1 that occurs in response to hyperosmotic shock reduces Ypk1-mediated phosphorylation of the glycerol efflux channel Fps1. All evidence supports the conclusion that Ypk1 phosphorylation of Fps1 favors its open state and, thus, absence of Ypk1 phosphorylation closes the channel. By this means, in response to hyperosmotic shock, Fps1-mediated glycerol efflux is prevented allowing intracellular glycerol to accumulate. We demonstrated previously that Gpd1, the glycerol-3-P dehydrogenase that is the rate-limiting enzyme in glycerol biosynthesis (Remize et al., 2001), is as bona fide in vivo substrate for Ypk1 and that Ypk1-mediated phosphorylation inhibits this enzyme (Lee et al., 2012b). This step is rate-limiting for glycerol production because glycerol is formed directly by dephosphorylation of the glycerol-3-P generated from DHAP by Gpd1 action (Fraenkel, 2011). Thus, the inactivation of TORC2-Ypk1 signaling that occurs upon hyperosmotic shock has at least two coordinated consequences that work synergistically to cause glycerol accumulation and promote cell survival (Fig. 5.8). First, loss of TORC2-Ypk1 signaling alleviates inhibition of Gpd1, thereby increasing glycerol production. Second, loss of TORC2-Ypk1 signaling results in closing of the Fps1 channel allowing for retention of the glycerol produced. In addition, my preliminary evidence suggests that loss of TORC2-Ypk1 signaling will permit the nuclear entry of the Smp1 transcription factor whose function appears to contribute to survival under hypertonic conditions (de Nadal et al., 2003). Thus, like the HOG response, the TORC2-Ypk1 signaling axis seems to modulate both transcriptional and post-transcriptional processes that help the cell cope with hyperosmotic stress. Future studies will be required to understand how Ypk1 (and Ypk2) regulates Smp1 function and what roles Smp1 has in rewiring of cellular physiology in response to hyperosmotic shock and other stresses that perturb plasma membrane structure and function. 76 Despite the similarities in stimuli sensed and pathway outputs, I have shown here that the TORC2-Ypk1 response is independent of any connection to Hog1. Why S. cerevisiae has two independent mechanisms for regulating glycerol levels in response to hyperosmotic stress is an interesting question. The presence of two systems might allow cells to adjust their response optimally to stresses that occur with different intensity, duration, or frequency. It has been shown that Hog1 primarily responds to stresses occurring no more frequently than every 200 seconds (Hersen et al., 2008; McClean et al., 2009). Perhaps TORC2-Ypk1 signaling allows response to occur in environments that fluctuate even more rapidly. Another possibility is that TORC2-Ypk1 signaling drives adaptive glycerol accumulation in response to stimuli not sensed by the Sln1- or Sho1-branches of the HOG pathway, or vice-versa. How does TORC2 sense high osmolarity? A recent report provided some evidence that Sho1, an integral plasma membrane protein that is thought to be a component of one of the osmosensors in the Hog1 pathway, can interact directly with Tor2, the catalytic subunit of TORC2 (Lam et al., 2015). This observation suggested the possibility that TORC2 activity could be subject to direct negative regulation via the osmolarity sensors of the Hog1 pathway itself. However, as I have shown here, in sho1Δ cells and mutants in other components of both the Sho1 and Sln1 branches of the Hog1 pathway, TORC2 inactivation in response to hyperosmolarity is unaffected. My results demonstrate that TORC2-Ypk1 is a novel osmosensing pathway independent of the known MAPK response. Identification of mechanism by which TORC2-mediated phosphorylation of Ypk1 is rapidly inactivated in response to high osmolarity is an important goal for future work. Osmosensing by TORC2 does not appear to be a phenomenon confined to S. cerevisiae. mTORC2 activity has been found to be altered in response to mechanical stretch (Kippenberger et al., 2005) and to osmotic shock (Sengupta et al., 2013) in human cell lines. Additionally, the downstream mTORC2 effector kinase SGK1, which is the mammalian ortholog of Ypk1 (Casamayor et al., 1999), has been implicated in the response to increased salt (osmolyte) levels in murine T cells (Wu et al., 2013). Furthermore, mTORC2 signaling is important for regulating levels of ion transporters (Sengupta et al., 2013; Gleason et al., 2015). Collectively, these results suggest that mTORC2 might respond to osmotic pressure by regulating ion transporters to balance osmolarity, much like TORC2 in yeast responds to osmolarity and regulates Fps1 to balance osmolarity. Thus, our screen has given us insight into a novel potentially conserved signaling system by which eukaryotes respond to changes in osmotic pressure. 77 Figures Fig. 5.1: Gpt2 is not phosphorylated by Ypk or other basophilic kinases. (A) Wild-type (BY4741), ypk1-as ypk2Δ (yAM135 - A), snf1Δ (CGA65) or tpk1-as tpk2-as tpk3-as (JTY5173) cells were transformed with Gpt2-3xFLAG (pAX238) or Gpt23A-3xFLAG (pAX244) plasmids. Cells were grown to mid-exponential phase and then treated with 10 μM 3-MB-PP1 (ypk1-as ypk2Δ), 5 μM 1NMPP1 (tpk1-as tpk2-as tpk3-as) (Zaman et al., 2009) or vehicle for 90 minutes. Cells were harvested, extracts prepared, resolved by SDS-PAGE, and blotted as described in chapter 2. (B) Wild-type (BY4741), gpt2Δ (yAM219 - A) or gpt2Δ sct1Δ strains covered with Gpt2-3xFLAG (yAM231), Gpt23A-3xFLAG (yAM232) or Gpt23E-3xFLAG (yAM233) plasmids were spotted and grown on YPD or YPD supplemented with 3.5 mM oleate for 3 days at 30C. gpt2Δ sct1Δ mutants are not viable (Zaremberg and McMaster, 2002). Therefore, Gpt23A or Gpt23E mutants do not completely abrogate Gpt2 activity as these plasmids would not be able to sustain cell growth on YPD. Furthermore, these mutations do not appear to decrease the ability of Gpt2 to allow cells to survive oleate stress. Cells with Gpt23A or Gpt23E as the sole source of glycerol-3-phosphate/dihydroxyacetone phosphate acyltransferase do not exhibit sensitivity to oleate as gpt2Δ cells do and are no more sensitive than wild-type cells. 78 79 Fig. 5.2: Smp1 is phosphorylated at predicted Ypk sites and phosphorylation of these residues is important for Smp1 function. (A) Wild-type (BY4741) cells were transformed with Smp1(1-274)-3xFLAG (pAX286) or Smp1(1-274)3A-3xFLAG (pAX287) plasmids. Cells were grown to mid-exponential phase. Cells were harvested, extracts prepared, resolved by SDSPAGE, and blotted as described in chapter 2. (B) Wild-type (BY4741) cells were transformed with Smp1-3xFLAG (pAX253) or Smp13A-3xFLAG (pAX260) plasmids. Cells were grown to mid-exponential phase. Cells were harvested, extracts prepared, resolved by SDS-PAGE, and blotted as described in chapter 2. (C) Upper panel Wild-type (BY4741), smp1Δ (yAM197), Smp1-3xFLAG (yAM246 - A), Smp13A-3xFLAG (yAM247 - A) and Smp13E-3xFLAG (yAM248 - A) cells were serially diluted, spotted and grown on SCD or SCD supplemented with indicated ferric iron chloride concentrations [prepared as described in (Lee et al., 2012a)] for 3 days at 30C. Lower panel smp1Δ (yAM197) cells were transformed with Smp1-3xFLAG (pAX253), Smp13A-3xFLAG (pAX260) and Smp13E-3xFLAG (pAX263) plasmids. Transformants were serially diluted, spotted and grown on SCD medium lacking uracil (to select for the plasmid) or SCD without uracil supplemented with indicated ferric iron chloride concentrations for 3 days at 30C. (D) smp1Δ (yAM197), Smp1-3xFLAG (yAM246 - A), Smp13A-3xFLAG (yAM247 - A) and Smp13E-3xFLAG (yAM248 - A) strains from (C Upper) were grown to mid-exponential phase, harvested, extracts prepared, resolved by SDS-PAGE, and blotted as described in chapter 2. (E) Wild-type (BY4741) or ypk1-as (yAM134 - A) cells were transformed with a centromeric plasmids Smp1-GFP from its own promoter (pAX270). Cells were then grown to mid-exponential phase or stationary phase as indicated and microscopy was performed as described in chapter 2. ypk1-as cells were treated with 10 μM 3-MB-PP1 to inhibit Ypk1 activity. Arrows indicate cell nuclei as determined by DAPI staining. (F) Wild-type (yAM241 - A), smp1Δ (yAM243), ypk1-as (yAM242 - A) or smp1Δ ypk1-as (yAM244 - A) cells with PSTL1-GFP integrated at the HIS3 locus were grown to mid-log phase in SCD without tryptophan with 10 μM 3-MB-PP1 in ypk1-as strains to inhibit Ypk1 activity. Cells were then transferred to SCD without tryptophan with or without 0.4 M NaCl to cause hyperosmotic shock. Cells were then harvested, fixed and intracellular GFP was measured by FACS as described in chapter 2. 80 Fig. 5.3: Changes in Fps1 dosage does not affect either TORC2- or Pkh1-mediated phosphorylation of Ypk1. Haploid wild-type (BY4742) and fps1Δ (yAM181 - A) cells or diploid wild-type (BY4743) and fps1Δ/ fps1Δ (JTY6322) cells were co-transformed with plasmids expressing Fps1 under the control of the GAL1/10 (pAX96) promoter or empty vector control (YCpLG) and a plasmid expressing Ypk1-HA under the control of the MET25 promoter (pLB215). Cells were grown to mid-exponential phase SC media without methionine to stimulate Ypk1-HA expression with 2% raffinose before addition of 2% galactose to induce Fps1 expression for 3.5 hours. Cells were harvested, extracts prepared, resolved by SDS-PAGE, and blotted as described in chapter 2. The upper hand corresponds to Ypk1-HA as judged by comigration between bands staining for HA and pT504. The faster migrating species seen in pT662 and pT504 blots result from these antibodies recognizing phosphorylation of endogenous Ypk1 and Ypk2. 81 82 Fig. 5.4: Ypk in response to TORC2 signaling phosphorylates Fps1 at three sites in vivo. (A) Diagram of Fps1 [adapted from (Lee et al., 2013)] with predicted Ypk sites indicated as red circles. Primary sequence context of predicted sites are shown at left. (B) Upper GST-Fps1(1255) (pBT6), GST-Fps1(531-669)(pBT7) were purified from E. coli and Ypk1-as kinase assays were performed as described in chapter 2. Lower GST-Fps1(531-669)(pBT7) and GSTFps1(531-669)(S570A) (pAX135) were purified from E. coli and Ypk1-as kinase assays were performed as above. (C) Extracts from Fps1-3xFLAG (yGT21), Fps1(T147A)-3xFLAG (yAM310 - A), Fps1(S181A S185A)-3xFLAG (yAM301 - A), Fps1(S570A)-3xFLAG (yGT24) or Fps13A-3xFLAG (yGT22) expressing strains were prepared, resolved by Phos-tag SDS-PAGE and immunoblotted as per chapter 2. Red asterisks indicate the appearance of higher mobility non-phosphorylated Fps1 species. (D) Fps1-3xFLAG (yGT21), Fps13A-3xFLAG (yGT22), ypk1as ypk2Δ Fps1-3xFLAG (yAM281) and ypk1-as ypk2Δ Fps13A-3xFLAG (yAM284 - A) strains were grown to mid-exponential phase and treated as indicated with 10 μM 3-MB-PP1 or vehicle for 60 minutes. Fps1-3xFLAG was resolved by Phos-tag SDS-PAGE as in (C), and blotted as in (C). Red asterisks indicate the appearance of higher mobility non-phosphorylated Fps1 species. (E) tor2-as (yKL5) cells expressing Fps1-3xFLAG (pAX274) or Fps13A-3xFLAG (pAX275) were grown to mid-exponential phase and then treated with 2μM BEZ-235 or vehicle for 30 min Extracts were resolved and immunoblotted as in (C). 83 Fig. 5.5: TORC2 phosphorylation of Ypk1 decreases rapidly in response to hyperosmotic shock resulting in Fps1 dephosphorylation. (A) Wild-type (BY4741) or tor2-29 (JTY5468) cells expressing Ypk17A-myc (pFR252) were grown at 26˚C to midexponential phase before switching their media to include 1 M sorbitol for the indicated time periods. Extracts were prepared, resolved and blotted as described in chapter 2. (B) As in (A) except extracts came from hog1Δ (YJP544), sho1Δ (JTY5540), ssk1Δ (JTY5541), ssk22Δ (JTY5539), ssk2Δ (JTY5538) or pbs2Δ (JTY5537) strains expressing Ypk17A-myc treated with 1 M sorbitol for 1 min (C) Fps13xFLAG (yGT21) or Fps13A-3xFLAG (yGT22) expressing cells were treated with 1 M sorbitol for indicated time points and Fps1-3xFLAG was resolved and blotted as in (Fig. 5.4). 84 85 Fig. 5.6: Phosphorylation of Fps1 by TORC2-Ypk increases channel transport activity. (A) gpd1Δ gpd2Δ cells (JT76328) that cannot balance osmolarity by glycerol production were transformed with high copy number plasmids expressing Fps1-3xFLAG (pGT02), Fps1(Δ12231) -3xFLAG (pGT05), Fps1(Δ534-650)-3xFLAG (pGT06), Fps13A-3xFLAG (pGT17), Fps13E-3xFLAG (pGT18) or empty vector (pRS426). Cells were then serially diluted and plated on SCD lacking uracil to select for plasmid or SCD without uracil supplemented with 1 M xylitol. Growth was allowed for 3 days at 30 ˚C prior to imaging. Constitutively open Fps1(Δ12231) allows cells to live. Interestingly, previous reports suggest that Fps1(Δ534-650) is constitutively open as well (Hedfalk et al., 2004). However, this mutant does not allow cells to survive this stress. (B) Cultures of Fps1-3xFLAG (yGT21), Fps13A-3xFLAG (yGT22), Fps1ΔPHD-3xFLAG (yAM307 - A), rgc1Δ rgc2Δ (DL3188) and fps1Δ (yAM181 - A) were adjusted to OD600=1.0 and serial dilutions were then spotted onto YPD plates containing the indicated concentration of arsenite. Cells were allowed to grow for 4 days at 30˚C prior to imaging. (C) As in (A) except Fps1-3xFLAG (yGT21), Fps1(T147A)-3xFLAG (yAM310 - A), Fps1(S181A S185A)-3xFLAG (yAM301 - A), Fps1(S570A)-3xFLAG (yGT24) or Fps13A3xFLAG (yGT22) cultures were used and cells were grown for 2 days at 30˚C prior to imaging. (D) Extracts were made from triplicate exponentially growing cultures of wild-type (BY4742), fps1Δ (yAM181 - A), Fps1-3xFLAG (yGT21) and Fps13A-3xFLAG (yGT22) strains. Concentrations of glycerol and protein in these extracts were then determined (see Materials and Methods). These measurements were used to calculate the ratio of glycerol to protein mass in each extract. (E) Wild-type (BY4741), fps1Δ (yAM181 - A), ypk1-as (yAM134 - A) and ypk1-as ypk2Δ (yAM135 - A) cells were serially diluted and plated on YPD media containing 3-MB-PP1 to inhibit Ypk1as and/or arsenite as indicated. Growth was allowed for 3 days at 30 ˚C prior to imaging. (F) Extracts were made from strains in (C) and resolved by standard SDS-PAGE using 8% acrylamide gels. (G) fps1Δ (yAM181 - A) cells expressing Fps1-GFP (pAX290), Fps1(S181A S185A)-GFP, (pAX294), Fps1(S570A)-GFP (pAX293) or Fps13A-GFP (pAX295) were imaged as described in chapter 2. Representative images are shown. 86 87 Fig. 5.7: TORC2-Ypk regulates Fps1 independently of known regulators Hog1 and Rgc1/2. (A) Cultures of Fps1-3xFLAG (yGT21), Fps1570A-3xFLAG (yGT24), Fps13A-3xFLAG (yGT22), Fps1-3xFLAG hog1Δ (yAM275), Fps1570A-3xFLAG hog1Δ (yAM291 - A) and Fps13A-3xFLAG hog1Δ (yAM278) strains were adjusted to OD600=1.0 and serial dilutions were then spotted onto YPD plates containing the indicated concentration of arsenite. Cells were allowed to grow for 2 days at 30˚C prior to imaging. (B) As in (A) except Fps1IVAA-3xFLAG (yAM308 - A), Fps1(3A)IVAA-3xFLAG (yAM309 - A), Rgc27A (yAM315) and Rgc27A Fps13A-3xFLAG (yAM318) strains were tested. Fps1IVAA mutation prevents Hog1 binding to and regulation of Fps1 while the Rgc27A cannot be phosphorylated and displaced from Fps1 by Hog1. These mutations render the channel constitutively open and arsenite sensitive (Lee et al., 2013). (C) Fps1-3xFLAG (yGT21) or Fps13A-3xFLAG (yGT22) strains were transformed with a PMET25Rgc2-HA plasmid (p3151). After Rgc2-HA expression, Fps1 was immunopurified (see chapter 2). Immunoprecipitates were resolved by SDS-PAGE and Fps1-Rgc2 interaction was determined by blotting for Rgc2-HA in the precipitates. (D) Wild-type (BY4741), hog1Δ (YJP544) or Fps13A-3xFLAG hog1Δ (yAM278) strains were plated onto synthetic complete medium lacking tryptophan with 2% dextrose and the indicated concentration of sorbitol. Cells were grown for 3 days prior to imaging. Serial dilutions are shown from the same plate. 88 Fig. 5.8: Saccharomyces cerevisiae utilizes two independent osmosensing systems to rapidly increase intracellular glycerol upon hyperosmolarity. (A) Diagram of the Hog1 MAPK mediated response to acute hyperosmotic stress [adapted from (Hohmann, 2015)]. Left shows the unstressed condition while right denotes cells under hyperosmotic stress. Hog1 is activated downstream of multiple osmosensing pathways. Hog1 increases glycolytic flux through phosphorylation of Pkf26 allowing for more glycerol production and also prevents glycerol efflux by phosphorylation and displacement of the Fps1 activators Rgc1/2 (Lee et al., 2013). On longer time scales, Hog1 also translocates to the nucleus where it drives transcription of glycerol production and uptake genes (de Nadal et al., 2011) (not depicted). (B) The TORC2-Ypk response to hyperosmolarity similarly increases glycerol production and accumulation. Basal TORC2-Ypk activity normally represses Gpd1 (Lee et al., 2012b) and is important for Fps1 channel activity. Hyperosmotic inactivation of TORC2 thus modulates the two rate limiting steps of glycerol production (Remize et al., 2001) to promote glycerol accumulation. Diagram of TORC2 adapted from (Liao and Chen, 2012; Wullschleger et al., 2005). 89 Tables Table 5.1 Synthetic dosage lethality of various Fps1 mutants in ypk1-as ypk2Δ cellsa Fps1 Mutant Wild-type Fps1(Δ12-231) Fps1(Δ534-650) Fps1(Δ544-581) Fps13A Fps13E SDL Phenotype +++++ +++++ +++++ +++++ Fps1 channel activity Unknown Constitutively open (Tamás et al., 2003) Constitutively open (Hedfalk et al., 2004) Constitutively closed (Lee et al., 2013) Unknown (but phenotype indicates closed) Unknown a SDL assays were performed as described in Chapter 2 and Table 3.1, except that the following plasmids were used for overexpressing Fps1 (pAX96), Fps1(Δ12-231) (pGT11), Fps1(Δ534650) (pGT12), Fps1(Δ544-581) (pGT13), Fps13A (pGT14) and Fps13E (pGT15). 90 Chapter 6: Concluding Remarks Conclusions If a cell is to survive, it must adjust its plasma membrane lipid and protein composition properly during growth and in response to environmental insults. Understanding the mechanisms that a cell uses to maintain its plasma membrane homeostasis is a fundamental question in cell biology. Many years of research using S. cerevisiae and its plasma membrane (van der Rest et al., 1995) as a model has given tremendous insight into conserved molecular mechanisms and pathways that a eukaryotic cell uses to cope with various membrane stresses (Ding et al., 2009; Shima & Takagi, 2009; Bravim et al., 2010; Jendretzki et al., 2011; Levin, 2011; Saito and Posas, 2012; Hohmann, 2015). Despite this progress, the pathways already described do not represent the complete set of molecular mechanisms that have evolved to cope with such stresses. Recently, our lab (Roelants et al., 2011) and others (Berchtold et al., 2012; Sun et al., 2012) demonstrated that signaling from TORC2 and the downstream protein kinases that it stimulates, Ypk1 (and its paralog Ypk2), is important for cell survival in response to stresses that perturb membrane lipid composition and organization. Surprisingly, despite intense interest in TORC1, especially in animal cells, because of its central role in growth control (Betz & Hall, 2013; Lemming & Sabatini, 2013; Bar-Peled & Sabatini, 2014; Shimobayashi & Hall, 2014; Albert & Hall, 2015; Efetan et al., 2015), relatively little was known about how TORC2 activity is regulated or how its effectors influence cell physiology (Huang & Manning, 2009; Masui et el., 2014). Thus, mechanistically how TORC2 action allows cells to cope with stress was unknown. The studies I conducted for my doctoral research and that are presented in this dissertation describe my discoveries about the mechanisms by which TORC2-Ypk1 signaling in yeast influences cellular processes to allow cells to maintain plasma membrane homeostasis in the face of stresses that otherwise would compromise cell viability. In Chapter 3, I described a productive approach - combining bioinformatics, a novel genetic screen that exploited synthetic dosage lethality, and in vitro biochemistry - to generate a high quality list of previously uncharacterized Ypk1 substrates. The dozen substrates identified in this manner include targets in processes in which TORC2 signaling was already implicated, such as in control of sphingolipid metabolism, suggesting more complex and nuanced roles for TORC2 signaling in these processes. Other identified substrates indicate that TORC2 has roles in modulating other areas of cell physiology in which it was not previously implicated. Thus, my search for new Ypk1 substrates increased the breadth and depth of our understanding of the regulation of cellular processes by TORC2. Of the five candidates that I explored in detail (Lac1, Lag1, Gpt2, Smp1, Fps1), all except Gpt2 were demonstrably Ypk1 substrates in vivo and their Ypk1-mediated phosphorylation had readily detectable physiological consequences. Thus, my approach for identifying protein kinase substrates was very successful and might be very useful in identifying substrates for protein kinases situated in other pathways. Mass spectrometry-based methods for identifying protein kinase substrates have been very successful (McLachlin & Chait, 2001; Graves & Haystead, 2003; Corthals et al., 2005; Cohen & Knebel, 2006; Nita-Lazar et al., 2008; Macek et al., 2009; Yates et al., 2009; Dephoure et al., 2013; Jünger & Aebersold, 2013) and have been applied usefully to yeast (Ficarro et al., 2002; Chi et al., 2007; Bodenmiller & Aebersold, 2011). However, it is acknowledged by its 91 practioners that, for technical reasons, mass spectrometry typically does not query easily either membrane proteins or proteins present in low abundance, which is the case for many proteins involved in signaling (Koch and Hauf, 2010). In this regard, it is striking that my approach yielded three authentic Ypk1 targets that are polytopic integral membrane proteins (Fps1 in the plasma membrane, and Lac1 and Lag1 in the ER membrane), as well as a low-abundance transcription factor (Smp1) that shuttles between the cytosol and the nucleus. Thus, the approach I developed for protein kinase substrate identification may be especially useful for matching protein kinases to their cognate substrates in those circumstances where the targets are anticipated to include membrane proteins and/or low-abundance proteins. In Chapter 4, I presented detailed characterization of the role that Ypk1 phosphorylation plays in regulation of Lac1 and Lag1, the two catalytic subunits of the ceramide synthase complex (Muir et al., 2014). These proteins and their function have been conserved throughout eukaryotic evolution. I found that Ypk1-mediated phosphorylation of Lac1 and Lag1 stimulated the activity of the ceramide synthase complex and that this upregulation was essential for survival in response to certain types of membrane stress. We had shown previously that TORC2-Ypk1 signaling increased the activity of the first step of sphingolipid biosynthesis (Roelants et al., 2011). My new findings documented the further fact that TORC2-Ypk1 not only increases flux into the sphingolipid pathway, but concomitantly channels the intermediates into end-product (complex sphingolipids). In addition, my results also indicate that upregulation of ceramide synthase also helps prevent accumulation of long chain base precursors and their phosphorylated derivatives, which, when elevated, can inadvertently trigger autophagy under non-starvation conditions (Zimmermann et al., 2013). This coordinate regulation may be a general feature of signaling mechanisms that interface with biosynthetic pathways in order to prevent ‘metabolic crosstalk’ by pathway intermediates that have multiple bioactive functions. Lastly, in Chapter 5, I described characterization of TORC2-Ypk1 regulation of the aquaglyceroporin Fps1 and a stress-responsive transcription factor, Smp1. I found that Fps1 is phosphorylated in a TORC2- and Ypk1-dependent manner and that phosphorylation is required for glycerol transport across the plasma membrane. Similarly, I found that Smp1 is also phosphorylated by TORC2-Ypk1 and these phosphorylations are required both to impede the nuclear entry of Smp1, but also for its function. Both of these substrates are involved in the cellular response to hyperosmotic shock. This led me to examine the role of TORC2 signaling during this stress. I confirmed that TORC2-mediated phosphorylation of Ypk1 is rapidly, but transiently, down-regulated in response to hyperosmotic shock. This apparent inhibition does not require any of the known high osmolarity sensing pathways in yeast. I found that the drop in TORC2-Ypk1 signaling simultaneously increases glycerol production by alleviating Ypk1mediated inhibiton of the glycerol-3-P dehydrogenase Gpd1 (Lee et al., 2012b) and promote glycerol retention by preventing Ypk1-dependent opening of the Fps1 channel. As a result, intracellular glycerol accumulates to add the cell in coping with hypertonic conditions. Thus, my screen for novel substrates revealed that TORC2-Ypk1 signaling provides a novel mechanism for rapid response to hyperosmotic stress. Future directions The work presented here provides some insight into the mechanisms by which TORC2-Ypk1 92 signaling promotes cellular survival during membrane stress. I believe future characterization of additional substrates identified in Chapter 3 will continue to provide important new insights into TORC2 signaling. For example, another candidate substrate I identified is Rod1, a member of the family of endocytic adaptors known as -arrestins (O'Donnell et al., 2013; Alvaro et al., 2014). Characterizing how TORC2-Ypk1 signaling regulates the α-arrestin Rod1 might explain whether and how plasma membrane protein composition is regulated in concert with plasma membrane lipid composition in response to stress. Additionally, after bioinformatic identification, only the top ~100 hits were subjected to the secondary, more laborious SDL screen described in Chapter 3. It should be possible to increase the throughput of the SDL screen to permit more presumptive candidate substrates to be tested in an unbiased manner. If additional candidate Ypk1 substrates are found, it would seem likely that their detailed characterization would yield further insights into the mechanisms by which TORC2 signaling regulates cellular physiology. Identification of the substrates Fps1 and Smp1 has revealed that TORC2-Ypk1 signaling is a novel hyperosmotic shock responsive pathway. TORC2-Ypk1 ouput is rapidly diminished upon hyperosmotic shock; and, although in the converse direction from HOG pathway signaling with respect to activation state, TORC2-Ypk1 function responds to the same environmental stress and with the same kinetics and with similar output (accumulation of intracellular glycerol) as the HOG pathway. This situation prompts an obvious question: why does the cell have two pathways that trigger the same response to the same stress? I look forward to future studies that may shed additional light on the nuances between these two stress-responsive pathways and that may explain, hopefully, or at least in part, why yeast have evolved such seemingly redundant systems. Additionally, identification of Smp1 prompted us to examine the role of TORC2-Ypk1 signaling in hyperosmotic stress-evoked transcriptional changes. Interestingly, cells lacking Ypk1 activity failed to activate transcription from the STL1 promoter, a classical hyperosmotic stress-induced promoter, regardless of whether Smp1 was present or not. This observation suggests that the nuclear entry and/or function of other transcription factors, or alleviation of repression by certain transcriptional repressors, or the function of other components of the RNA polymerase II transcription machinery, requires Ypk1-dependent phosphorylation, either directly or indirectly. Indeed, among the candidate Ypk1 substrates identified by my approach (see Chapter 3) are Gal11 and Ssn2, two subunits of the mediator complex that acts as a bridge between site-specific DNA-binding transcriptional activators and the RNA polymerase II holoenzyme (Kornberg, 2005) and Epl1, a subunit of the essential NuA4 histone acetyl-transferase complex that also promotes gene expression (Doyon & Cóté, 2004). Moreover, in addition to Smp1 itself, a number of other DNA-binding transactivators were identified in my screen, including Rlm1 (another MADS-box family transcription factor and paralog of Smp1), Fkh1 and Hcm1 (two Forkhead family transcription factors), and Hot1. Hot1 (for "high osmolarity-induced transcription") is especially intriguing with regard to the necessity for Ypk1 action in STL1 induction in response to hyperosmotic shock because Hot1 has been shown to be required for the induction of GPD1 (and its paralog GDP2) under hypertonic conditions (Alepuz et al., 2003; Gomar-Alba et al., 2013). In contrast to all of the above factors that act positively on transcription, among candidate Ypk1 targets identified by my methods only a single negative regulator was identified, Yhp1, a transcriptional repressor of the Homeobox family. In any event, my findings indicate that TORC2-Ypk1 signaling may play an important role in the induction of 93 the expression of gene products that aid the cell in coping with membrane stress. Therefore, I am also looking forward to future studies that will address this aspect of TORC2-Ypk1 function. Although the work described in this thesis addresses downstream aspects of TORC2-Ypk1 signaling, it is important to consider how TORC2 "senses" the status of the plasma membrane. We know very little about how TORC2 function (state of assembly, localization, activity state, stability) may be influenced by membrane lipid and protein composition during normal growth and by stresses (organic solvent, heat shock, lipid synthesis inhibition, hypertonic and hypotonic conditions, etc.) that perturb plasma membrane composition, structure and/or function. How TORC2 activity is controlled is an important unanswered question in TOR signaling. By comparison, significantly greater headway has been made in understanding how TORC1 activity is regulated (Loewith & Hall, 2011; Laplante & Sabatini, 2012). Importantly, TORC2 activity appears to be poised at an intermediate level. Some stresses, e.g., hypo-osmotic shock, appear to upregulate TORC2 activity (Berchtold et al., 2012; Niles et al., 2012), whereas other stresses, e.g., hyperosmotic shock, appear to inactivate TORC2 function (Lee et al., 2012b). Perhaps each type of stress influences TORC2 by a unique and separate mechanism, or perhaps TORC activity is coupled to a single property of the membrane, e.g., membrane tension, that can be differentially affected by each type of stress. Either model can explain the effects of stress observed to date. I am particularly excited for future studies that will hopefully define the molecular mechanisms by which membrane status modulates TORC2 action. TORC2 signaling is conserved in other eukaryotes, including humans (Cybulski & Hall, 2009; Huang & Manning, 2009). TORC2 signaling in mammalian cells appears to be sensitive to membrane stretch and osmolarity much like TORC2 is in yeast (Kippenberger et al., 2005; Sengupta et al., 2013). Future studies examining regulation of TORC2 and how TORC2 then regulates cell physiology in other organisms will be interesting as they identify common themes and species-specific adaptive differences in signaling from this conserved pathway. Lastly, I am looking forward to future experiments that will continue to give us new understanding of the varied roles lipids play in cell physiology. My studies of TORC2 found that strict regulation of LCB-1-P levels were necessary to prevent autophagy. However, due to the relative difficulties of studying lipids compared to other biomolecules, it is still unknown how LCB-1-P triggers autophagy. I look forward to studies that will address the role that intracellular LCB-1-P plays in autophagy induction. In this regard, I am excited by the new lipidomics techniques and in vivo chemistries to analyze, perturb and visualize lipids that are becoming available (Atilla-Gokcumen et al., 2014; Muro et al., 2014; Casanovas et al., 2015). Recent advances have made it possible to even determine the spatial distribution of lipid species on the micron scale in situ (Soltwisch et al., 2015). It will be exciting to see these new tools being used to study dynamic changes in the membrane composition of cells undergoing physiological changes. These new techniques will hopefully give us new understanding of the role that changing lipid composition plays in membrane function and cellular adaptation, and the mechanisms that cells use to achieve these outcomes. Final remarks The cell membrane is the point of contact between the cell and the outside world. Through this 94 border the cell experiences and is assaulted by the environment. It is fascinating to learn the mechanisms by which the cell keeps this essential organelle in working shape. TORC2 signaling is one such conserved mechanism by which eukaryotes accomplish this task. I am very pleased that my doctoral dissertation research has provided significant new insight and additional understanding about the molecular mechanisms by which TORC2 action maintains plasma membrane homeostasis. 95 Literature Cited Albert V, Hall MN (2015) mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 33: 55-66. Albertyn, J., Hohmann, S., and Prior, B.A. (1994). Characterization of the osmotic-stress response in Saccharomyces cerevisiae: osmotic stress and glucose repression regulate glycerol-3phosphate dehydrogenase independently. Curr Genet 25, 12-18. Alers, S., Wesselborg, S., and Stork, B. (2014). ATG13: Just a companion, or an executor of the autophagic program? Autophagy 10, 944-956. Almén, M.S., Nordström, K.J., Fredriksson, R., and Schiöth, H.B. (2009). Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol 7, 50. Alepuz PM, de Nadal E, Zapater M, Ammerer G, Posas F (2003) Osmostress-induced transcription by Hot1 depends on a Hog1-mediated recruitment of the RNA Pol II. EMBO J. 22: 2433-2442. Alvaro CG, O'Donnell AF, Prosser DC, Augustine AA, Goldman AR, Brodsky JL, Cyert MS, Wendland B, Thorner J (2014) Specific -arrestins negatively regulate Saccharomyces cerevisiae pheromone response by down-modulating the G-protein coupled receptor Ste2. Mol. Cell. Biol. 34: 2660-2681. Ahmadpour D, Geijer C, Tamás MJ, Lindkvist-Petersson K, Hohmann S (2014) Yeast reveals unexpected roles and regulatory features of aquaporins and aquaglyceroporins. Biochim. Biophys. Acta 1840: 1482-1491. Aronova, S., Wedaman, K., Aronov, P.A., Fontes, K., Ramos, K., Hammock, B.D., and Powers, T. (2008). Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab 7, 148-158. Atilla-Gokcumen, G.E., Bedigian, A.V., Sasse, S., and Eggert, U.S. (2011). Inhibition of glycosphingolipid biosynthesis induces cytokinesis failure. J Am Chem Soc 133, 10010-10013. Atilla-Gokcumen, G.E., Muro, E., Relat-Goberna, J., Sasse, S., Bedigian, A., Coughlin, M.L., Garcia-Manyes, S., and Eggert, U.S. (2014). Dividing cells regulate their lipid composition and localization. Cell 156, 428-439. Babazadeh, R., Furukawa, T., Hohmann, S., and Furukawa, K. (2014). Rewiring yeast osmostress signalling through the MAPK network reveals essential and non-essential roles of Hog1 in osmoadaptation. Sci Rep 4, 4697. Balakrishnan, R., Park, J., Karra, K., Hitz, B.C., Binkley, G., Hong, E.L., Sullivan, J., Micklem, G., and Cherry, J.M. (2012). YeastMine--an integrated data warehouse for Saccharomyces cerevisiae data as a multipurpose tool-kit. Database (Oxford) 2012, bar062. 96 Bardwell, L., Cook, J.G., Zhu-Shimoni, J.X., Voora, D., and Thorner, J. (1998). Differential regulation of transcription: repression by unactivated mitogen-activated protein kinase Kss1 requires the Dig1 and Dig2 proteins. Proc Natl Acad Sci U S A 95, 15400-15405. Bar-Peled L, Sabatini DM (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol. 24: 400-406. Barz, W.P., and Walter, P. (1999). Two endoplasmic reticulum (ER) membrane proteins that facilitate ER-to-Golgi transport of glycosylphosphatidylinositol-anchored proteins. Mol Biol Cell 10, 1043-1059. Becuwe, M., N., V., Lara, D., Gomes-Rezende, J., Soares-Cunha, C., Casal, M., HaguenauerTsapis, R., Vincent, O., Paiva, S., and Léon, S. (2012). A molecular switch on an arrestin-like protein relays glucose signaling to transporter endocytosis. J Cell Biol 196, 247-259. Beeler, T., Bacikova, D., Gable, K., Hopkins, L., Johnson, C., Slife, H., and Dunn, T. (1998). The Saccharomyces cerevisiae TSC10/YBR265w gene encoding 3-ketosphinganine reductase is identified in a screen for temperature-sensitive suppressors of the Ca2+-sensitive csg2∆ mutant. J Biol Chem 273, 30688-30694. Beese, S.E., Negishi, T., and Levin, D.E. (2009). Identification of positive regulators of the yeast fps1 glycerol channel. PLoS Genet 5, e1000738. Beise, N., and Trimble, W. (2011). Septins at a glance. J Cell Sci 124, 4141-4146. Benjamin D, Colombi M, Moroni C, Hall MN (2011) Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 10: 868-880. Berchtold, D., Piccolis, M., Chiaruttini, N., Riezman, I., Riezman, H., Roux, A., Walther, T.C., and Loewith, R. (2012). Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol 14, 542-547. Betz C, Hall MN (2013) Where is mTOR and what is it doing there? J. Cell Biol. 203: 563-574. Bikman, B.T., and Summers, S.A. (2011). Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest 121, 4222-4230. Bodenmiller B, Aebersold R (2011) Phosphoproteome resource for systems biology research. Methods Mol. Biol. 694: 307-322. Bradford, M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248-254. Bratschi MW, Burrowes DP, Kulaga A, Cheung JF, Alvarez AL, Kearley J, Zaremberg V (2009) Glycerol-3-phosphate acyltransferases Gat1 and Gat2 are microsomal phosphoproteins with 97 differential contributions to polarized cell growth. Eukaryot. Cell 8: 1184-1196. Bravim F, de Freitas JM, Fernandes AA, Fernandes PM (2010) High hydrostatic pressure and the cell membrane: stress response of Saccharomyces cerevisiae. Ann NY Acad. Sci. 1189: 127132. Breitkreutz, A., Choi, H., Sharom, J.R., Boucher, L., Neduva, V., Larsen, B., Lin, Z.Y., Breitkreutz, B.J., Stark, C., Liu, G., et al. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043-1046. Breslow, D.K. (2013) Sphingolipid homeostasis in the endoplasmic reticulum and beyond. Cold Spring Harb Perspect Biol 5, a013326.013321-013316. Breslow, D.K., Collins, S.R., Bodenmiller, B., Aebersold, R., Simons, K., Shevchenko, A., Ejsing, C.S., and Weissman, J.S. (2010) Orm family proteins mediate sphingolipid homeostasis. Nature 463, 1048-1053. Breslow, D.K., and Weissman, J.S. (2010) Membranes in balance: mechanisms of sphingolipid homeostasis. Mol Cell 40, 267-279. Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC (1993) An osmosensing signal transduction pathway in yeast. Science 259: 1760-1763. Brewster JL, Gustin MC (2014) Hog1: 20 years of discovery and impact. Sci. Signal. 7: re7.1re.7.10. 1 Brown, D.A., and Rose, J.K. (1992). Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533-544. Brügger, B. (2014). Lipidomics: analysis of the lipid composition of cells and subcellular organelles by electrospray ionization mass spectrometry. Annu Rev Biochem 83, 79-98. Buhaescu, I., and Izzedine, H. (2007). Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 40, 575-584. Bullard, T.A., Hastings, J.L., Davis, J.M., Borg, T.K., and Price, R.L. (2007). Altered PKC expression and phosphorylation in response to the nature, direction, and magnitude of mechanical stretch. Can J Physiol Pharmacol 85, 243-250. Bultynck, G., Heath, V.L., Majeed, A.P., Galan, J.M., Haguenauer-Tsapis, R., and Cyert, M.S. (2006). Slm1 and slm2 are novel substrates of the calcineurin phosphatase required for heat stress-induced endocytosis of the yeast uracil permease. Mol Cell Biol 26, 4729-4745. Burkard, M.E., Randall, C.L., Larochelle, S., Zhang, C., K.M., S., Fisher, R.P., and Jallepalli, P.V. (2007). Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Nat Acad Sci USA 104, 438398 4388. Burke, D., Amberg, D., and Strathern, J. (2005). Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual (Cold Spring Harbor Laboratory Press). Byrne, K.P., and Wolfe, K.H. (2005). The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res 15, 14561461. Casamayor, A., Torrance, P.D., Kobayashi, T., Thorner, J., and Alessi, D.R. (1999). Functional counterparts of mammalian protein kinases PDK1 and SGK in budding yeast. Curr Biol 9, 186197. Casanovas, A., Sprenger, R.R., Tarasov, K., Ruckerbauer, D.E., Hannibal-Bach, H.K., Zanghellini, J., Jensen, O.N., and Ejsing, C.S. (2015). Quantitative analysis of proteome and lipidome dynamics reveals functional regulation of global lipid metabolism. Chem Biol 22, 412425. Chantranupong L, Wolfson RL, Sabatini DM (2015) Nutrient-sensing mechanisms across evolution. Cell 161: 67-83. Chen, P., Lee, K.S., and Levin, D.E. (1993). A pair of putative protein kinase genes (YPK1 and YPK2) is required for cell growth in Saccharomyces cerevisiae. Mol Gen Genet 236, 443-447. Chen, P.W., Fonseca, L.L., Hannun, Y.A., and Voit, E.O. (2013). Coordination of rapid sphingolipid responses to heat stress in yeast. PLoS Comput Biol 9, e1003078. Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF (2007) Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. USA 104: 2193-2198. Chiarini F, Evangelisti C, McCubrey JA, Martelli AM (2015) Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol. Sci. 36: 124-135. Chichili, G.R., and Rodgers, W. (2009). Cytoskeleton-membrane interactions in membrane raft structure. Cell Mol Life Sci 66, 2319-2328. Clark, M.R. (2011). Flippin' lipids. Nat Immunol 12, 373-375. Clay, L., Caudron, F., Denoth-Lippuner, A., Boettcher, B., Buvelot Frei, S., Snapp, E.L., and Barral, Y. (2014). A sphingolipid-dependent diffusion barrier confines ER stress to the yeast mother cell. Elife 3, e01883. Cohen P, Knebel A (2006) KESTREL: a powerful method for identifying the physiological substrates of protein kinases. Biochem J. 393: 1-6. 99 Colabardini, A.C., Brown, N.A., Savoldi, M., Goldman, M.H., and Goldman, G.H. (2013). Functional characterization of Aspergillus nidulans ypkA, a homologue of the mammalian kinase SGK. PLoS One 8, e57630. Cooper, G.M., and Hausman, R.E. (2013). The cell : a molecular approach, 6th edn (Sunderland, Mass.: Sinauer Associates). Corthals GL1, Aebersold R, Goodlett DR (2005) Identification of phosphorylation sites using microimmobilized metal affinity chromatography. Methods Enzymol. 405: 66-81. Cowart, L.A., Okamoto, Y., Pinto, F.R., Gandy, J.L., Almeida, J.S., and Hannun, Y.A. (2003). Roles for sphingolipid biosynthesis in mediation of specific programs of the heat stress response determined through gene expression profiling. J Biol Chem 278, 30328-30338. Cuadrado A, Nebreda AR (2010) Mechanisms and functions of p38 MAPK signalling. Biochem J. 429: 403-417. Cuenda A, Rousseau S (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 1773: 1358-1375. Cullen PJ (2007) Signaling mucins: the new kids on the MAPK block. Crit. Rev. Eukaryot. Gene Expr. 17: 241-257. Cybulski, N., and Hall, M.N. (2009). TOR complex 2: a signaling pathway of its own. Trends Biochem Sci 34, 620-627. da Silveira Dos Santos, A.X., Riezman, I., Aguilera-Romero, M.A., David, F., Piccolis, M., Loewith, R., Schaad, O., and Riezman, H. (2014). Systematic lipidomic analysis of yeast protein kinase and phosphatase mutants reveals novel insights into regulation of lipid homeostasis. Mol Biol Cell 25, 3234-3246. Daquinag, A., Fadri, M., Jung, S.Y., Qin, J., and Kunz, J. (2007). The yeast PH domain proteins Slm1 and Slm2 are targets of sphingolipid signaling during the response to heat stress. Mol Cell Biol 27, 633-650. Das, A., Slaughter, B.D., Unruh, J.R., Bradford, W.D., Alexander, R., Rubinstein, B., and Li, R. (2012). Flippase-mediated phospholipid asymmetry promotes fast Cdc42 recycling in dynamic maintenance of cell polarity. Nat Cell Biol 14, 304-310. de Nadal, E., Ammerer, G., and Posas, F. (2011). Controlling gene expression in response to stress. Nat Rev Genet 12, 833-845. de Nadal, E., Casadomé, L., and Posas, F. (2003). Targeting the MEF2-like transcription factor Smp1 by the stress-activated Hog1 mitogen-activated protein kinase. Mol Cell Biol 23, 229-237. 100 de Nobel H, van Den Ende H, Klis FM (2000) Cell wall maintenance in fungi. Trends Microbiol. 8: 344-345. deHart, A.K., Schnell, J.D., Allen, D.A., and Hicke, L. (2002). The conserved Pkh-Ypk kinase cascade is required for endocytosis in yeast. J Cell Biol 156, 241-248. Denis, V., and Cyert, M.S. (2002). Internal Ca2+ release in yeast is triggered by hypertonic shock and mediated by a TRP channel homologue. J Cell Biol 156, 29-34. Dephoure N, Gould KL, Gygi SP, Kellogg DR (2013) Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol. Biol. Cell 24: 535-542. Devaux, P.F. (1991). Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30, 1163-1173. Devaux, P.F., Herrmann, A., Ohlwein, N., and Kozlov, M.M. (2008). How lipid flippases can modulate membrane structure. Biochim Biophys Acta 1778, 1591-1600. Dickson, R.C. (2008). Thematic review series: sphingolipids. New insights into sphingolipid metabolism and function in budding yeast. J Lipid Res 49, 909-921. Ding J, Huang X, Zhang L, Zhao N, Yang D, Zhang K (2009) Tolerance and stress response to ethanol in the yeast Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 85: 253-263. Dodou E, Treisman R (1997) The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol. Cell. Biol. 17: 1848-1859. Douglass, A.D., and Vale, R.D. (2005). Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell 121, 937-950. Doyon Y, Côté J (2004) The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 14: 147-154. Duskova M, Borovikova D, Herynkova P, Rapoport A, Sychrova H (2015) The role of glycerol transporters in yeast cells in various physiological and stress conditions. FEMS Microbiol. Lett. 362: 1-8. Dustin, M.L., and Groves, J.T. (2012). Receptor signaling clusters in the immune synapse. Annu Rev Biophys 41, 543-556. Efeyan A, Comb WC, Sabatini DM (2015) Nutrient-sensing mechanisms and pathways. Nature 517: 302-310. Engelberg D, Perlman R, Levitzki A (2014) Transmembrane signaling in Saccharomyces 101 cerevisiae as a model for signaling in metazoans: state of the art after 25 years. Cell. Signal. 26: 2865-2878. Engelman DM (2005) Membranes are more mosaic than fluid. Nature 438: 578-580. Eggeling, C., Ringemann, C., Medda, R., Schwarzmann, G., Sandhoff, K., Polyakova, S., Belov, V.N., Hein, B., von Middendorff, C., Schönle, A., et al. (2009). Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 457, 1159-1162. Ejsing, C.S., Sampaio, J.L., Surendranath, V., Duchoslav, E., Ekroos, K., Klemm, R.W., Simons, K., and Shevchenko, A. (2009). Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci U S A 106, 2136-2141. Fagerberg, L., Jonasson, K., von Heijne, G., Uhlén, M., and Berglund, L. (2010). Prediction of the human membrane proteome. Proteomics 10, 1141-1149. Fahy, E., Sud, M., Cotter, D., and Subramaniam, S. (2007). LIPID MAPS online tools for lipid research. Nucleic Acids Res 35, W606-612. Fairn, G.D., Schieber, N.L., Ariotti, N., Murphy, S., Kuerschner, L., Webb, R.I., Grinstein, S., and Parton, R.G. (2011). High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J Cell Biol 194, 257-275. Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20: 301-305. Foster, L.J., De Hoog, C.L., and Mann, M. (2003). Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc Natl Acad Sci U S A 100, 5813-5818. Futerman, A.H., and Hannun, Y.A. (2004). The complex life of simple sphingolipids. EMBO Rep 5, 777-782. Fraenkel DG (2011) Yeast Intermediary Metabolism. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 434pp. Free SJ (2013) Fungal cell wall organization and biosynthesis. Adv. Genet. 81: 33-82. Friant S, Lombardi R, Schmelzle T, Hall MN, Riezman H (2001) Sphingoid base signaling via Pkh kinases is required for endocytosis in yeast. EMBO J. 20: 6783-6792. Gandhavadi, M., Allende, D., Vidal, A., Simon, S.A., and McIntosh, T.J. (2002). Structure, composition, and peptide binding properties of detergent soluble bilayers and detergent resistant rafts. Biophys J 82, 1469-1482. Gibson, D.G., Young, L., Chuang, R.Y., Venter, J.C., Hutchison, C.A., and Smith, H.O. (2009). 102 Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343345. Gleason, C.E., Frindt, G., Cheng, C.J., Ng, M., Kidwai, A., Rashmi, P., Lang, F., Baum, M., Palmer, L.G., and Pearce, D. (2015). mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J. Clin. Invest. 125, 117-128. Gomar-Alba M, Alepuz P, del Olmo M (2013) Dissection of the elements of osmotic stress response transcription factor Hot1 involved in the interaction with MAPK Hog1 and in the activation of transcription. Biochim. Biophys. Acta 1829: 1111-1125. Graves PR, Haystead TA (2003) A functional proteomics approach to signal transduction. Recent Prog. Horm. Res. 58: 1-24. . Green, M.R., and Sambrook, J. (2012). Molecular cloning : a laboratory manual, 4th edn (Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press). Guan, X.L., and Wenk, M.R. (2006). Mass spectrometry-based profiling of phospholipids and sphingolipids in extracts from Saccharomyces cerevisiae. Yeast 23, 465-477. Guimaraes RS, Delorme-Axford E, Klionsky DJ, Reggiori F (2015) Assays for the biochemical and ultrastructural measurement of selective and nonselective types of autophagy in the yeast Saccharomyces cerevisiae. Methods 75: 141-150. Guiney, E.L., Goldman, A.R., Elias, J.E., and Cyert, M.S. (2015). Calcineurin regulates the yeast synaptojanin Inp53/Sjl3 during membrane stress. Mol Biol Cell 26, 769-785. Gwinn, D.M., Shackelford, D.B., Egan, D.F., Mihaylova, M.M., Mery, A., Vasquez, D.S., Turk, B.E., and Shaw, R.J. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30, 214-226. Ha TS, Xia R, Zhang H, Jin X, Smith DP (2014) Lipid flippase modulates olfactory receptor expression and odorant sensitivity in Drosophila. Proc. Natl. Acad. Sci. USA 111: 7831-7836. Han J, Lee JD, Bibbs L, Ulevitch RJ (1994) A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265: 808-811. Hanson, B.A., and Lester, R.L. (1980). The extraction of inositol-containing phospholipids and phosphatidylcholine from Saccharomyces cerevisiae and Neurospora crassa. J Lipid Res 21, 309-315. Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD (2013) A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 41: 906-912. Hedfalk, K., Bill, R.M., Mullins, J.G., Karlgren, S., Filipsson, C., Bergstrom, J., Tamás, M.J., Rydström, J., and Hohmann, S. (2004). A regulatory domain in the C-terminal extension of the 103 yeast glycerol channel Fps1p. J Biol Chem 279, 14954-14960. Heidler, S.A., and Radding, J.A. (1995). The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob Agents Chemother 39, 2765-2769. Heitman J, Movva NR, Hall MN (1991) Targets for cell cycle arrest by the immuno-suppressant rapamycin in yeast. Science 253: 905-909. Henry SA, Kohlwein SD, Carman GM (2012) Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190: 317-349. Hermoso, M., Olivero, P., Torres, R., Riveros, A., Quest, A.F., and Stutzin, A. (2004). Cell volume regulation in response to hypotonicity is impaired in HeLa cells expressing a protein kinase Calpha mutant lacking kinase activity. J Biol Chem 279, 17681-17689. Hersen, P., McClean, M.N., Mahadevan, L., and Ramanathan, S. (2008). Signal processing by the HOG MAP kinase pathway. Proc Natl Acad Sci U S A 105, 7165-7170. Hillenmeyer, M.E., Fung, E., Wildenhain, J., Pierce, S.E., Hoon, S., Lee, W., Proctor, M., St Onge, R.P., Tyers, M., Koller, D., et al. (2008). The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362-365. Hohmann, S. (2002). Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev 66, 300-372. Hohmann S (2009) Control of high osmolarity signalling in the yeast Saccharomyces cerevisiae. FEBS Lett. 583: 4025-4029. Hohmann, S. (2015). An integrated view on a eukaryotic osmoregulation system. Curr Genet. (E-pub ahead of print, 8 Febr. 2015] Holt, L.J., Hutti, J.E., Cantley, L.C., and Morgan, D.O. (2007). Evolution of Ime2 phosphorylation sites on Cdk1 substrates provides a mechanism to limit the effects of the phosphatase Cdc14 in meiosis. Mol Cell 25, 689-702. Hua, Z., Fatheddin, P., and Graham, T.R. (2002). An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol Biol Cell 13, 3162-3177. Huang J, Manning BD (2009) A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 37: 217-222. Huang K-J, Lan C-Y, Igo MM (1997) Phosphorylation stimulates the cooperative DNA-binding properties of the transcription factor OmpR. Proc. Natl. Acad. Sci. USA 94: 2828–2832. 104 Hutti, J.E., Shen, R.R., Abbott, D.W., Zhou, A.Y., Sprott, K.M., Asara, J.M., Hahn, W.C., and Cantley, L.C. (2009). Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell 34, 461-472. Iida, K., Teng, J., Tada, T., Saka, A., Tamai, M., Izumi-Nakaseko, H., Adachi-Akahane, S., and Iida, H. (2007). Essential, completely conserved glycine residue in the domain III S2-S3 linker of voltage-gated calcium channel alpha1 subunits in yeast and mammals. J Biol Chem 282, 2565925667. Ikeda, M., Kihara, A., and Igarashi, Y. (2006). Lipid asymmetry of the eukaryotic plasma membrane: functions and related enzymes. Biol Pharm Bull 29, 1542-1546. Jendretzki A, Wittland J, Wilk S, Straede A, Heinisch JJ (2011) How do I begin? Sensing extracellular stress to maintain yeast cell wall integrity. Eur. J. Cell Biol. 90: 740-744. Jenkins, G.M., Richards, A., Wahl, T., Mao, C., Obeid, L., and Hannun, Y. (1997). Involvement of yeast sphingolipids in the heat stress response of Saccharomyces cerevisiae. J Biol Chem 272, 32566-32572. Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL (2015) Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347: 194-198. Jiang, J.C., Kirchman, P.A., Zagulski, M., Hunt, J., and Jazwinski, S.M. (1998). Homologs of the yeast longevity gene LAG1 in Caenorhabditis elegans and human. Genome Res 8, 1259-1272. Jünger MA, Aebersold R (2014) Mass spectrometry-driven phosphoproteomics: patterning the systems biology mosaic. Wiley Interdiscip. Rev. Dev. Biol. 3: 83-112. Kageyama-Yahara, N., and Riezman, H. (2006). Transmembrane topology of ceramide synthase in yeast. Biochem J 398, 585-593. Kamada, Y., Fujioka, Y., Suzuki, N.N., Inagaki, F., Wullschleger, S., Loewith, R., Hall, M.N., and Ohsumi, Y. (2005). Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol Cell Biol 25, 7239-7248. Karlgren S, Filipsson C, Mullins JG, Bill RM, Tamás MJ, Hohmann S (2004) Identification of residues controlling transport through the yeast aquaglyceroporin Fps1 using a genetic screen. Eur. J. Biochem. 271: 771-779. Kihara, A., Mitsutake, S., Mizutani, Y., and Igarashi, Y. (2007). Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1phosphate. Prog Lipid Res 46, 126-144. Kim, S., Fyrst, H., and Saba, J. (2000). Accumulation of phosphorylated sphingoid long chain bases results in cell growth inhibition in Saccharomyces cerevisiae. Genetics 156, 1519-1529. 105 Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163-275. Kinoshita, E., Kinoshita-Kikuta, E., and Koike, T. (2009). Separation and detection of large phospho-proteins using Phos-tag SDS-PAGE. Nat. Protoc. 4, 1513–1521. Kippenberger, S., Loitsch, S., Guschel, M., Müller, J., Knies, Y., Kaufmann, R., and Bernd, A. (2005). Mechanical stretch stimulates protein kinase B/Akt phosphorylation in epidermal cells via angiotensin II type 1 receptor and epidermal growth factor receptor. J Biol Chem 280, 30603067. Kliegman, J.I., Fiedler, D., Ryan, C.J., Xu, Y.F., Su, X.Y., Thomas, D., Caccese, M.C., Cheng, A., Shales, M., Rabinowitz, J.D., et al. (2013). Chemical genetics of rapamycin-insensitive TORC2 in S. cerevisiae. Cell Rep 5, 1725-1736. Kobayashi, S.D., and Nagiec, M.M. (2003). Ceramide/long-chain base phosphate rheostat in Saccharomyces cerevisiae: regulation of ceramide synthesis by Elo3p and Cka2p. Eukaryot Cell 2, 284-294. Koch, A., and Hauf, S. (2010). Strategies for the identification of kinase substrates using analogsensitive kinases. Eur. J. Cell Biol. 89, 184-193. Kock C, Dufrêne YF, Heinisch JJ (2015) Up against the wall: is yeast cell wall integrity ensured by mechanosensing in plasma membrane microdomains? Appl. Environ. Microbiol. 81: 806-811 Kono, K., Saeki, Y., Yoshida, S., Tanaka, K., and Pellman, D. (2012). Proteasomal degradation resolves competition between cell polarization and cellular wound healing. Cell 150, 151-164. Kornberg RD (2005) Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 30: 235-239. Kornberg, R.D., and McConnell, H.M. (1971) Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry 10, 1111-1120. Krick R, Henke S, Tolstrup J, Thumm M (2008) Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy 4: 896-910. Lam, H.Y., Kim, P.M., Mok, J., Tonikian, R., Sidhu, S.S., Turk, B.E., Snyder, M., and Gerstein, M.B. (2010). MOTIPS: automated motif analysis for predicting targets of modular protein domains. BMC Bioinformatics 11, 243. Lam, M.H., Snider, J., Rehal, M., Wong, V., Aboualizadeh, F., Drecun, L., Wong, O., Jubran, B., Li, M., Ali, M., et al. (2015). A comprehensive membrane interactome mapping of Sho1p reveals Fps1p as a novel key player in the regulation of the HOG pathway in S. cerevisiae. J. 106 Mol. Biol. 427: 2088-2103. Lamming DW, Sabatini DM (2013) A central role for mTOR in lipid homeostasis. Cell Metab. 18: 465-469. Laplante, M., and Sabatini, D.M. (2012). mTOR signaling in growth control and disease. Cell 149, 274-293. Laviad, E.L., Kelly, S., Merrill, A.H., and Futerman, A.H. (2012). Modulation of ceramide synthase activity via dimerization. J Biol Chem 287, 21025-21033. Lee, J., Reiter, W., Dohnal, I., Gregori, C., Beese-Sims, S., Kuchler, K., Ammerer, G., and Levin, D.E. (2013). MAPK Hog1 closes the S. cerevisiae glycerol channel Fps1 by phosphorylating and displacing its positive regulators. Genes Dev 27, 2590-2601. Lee, Y.J., Huang, X., Kropat, J., Henras, A., Merchant, S.S., Dickson, R.C., and Chanfreau, G.F. (2012a). Sphingolipid signaling mediates iron toxicity. Cell Metab 16, 90-96. Lee, Y.J., Jeschke, G.R., Roelants, F.M., Thorner, J., and Turk, B.E. (2012b) Reciprocal phosphorylation of yeast glycerol-3-phosphate dehydrogenases in adaptation to distinct types of stress. Mol Cell Biol 32, 4705-4717. Levin, D.E. (2011). Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics 189, 1145-1175. Li, Z., Vizeacoumar, F.J., Bahr, S., Li, J., Warringer, J., Vizeacoumar, F.S., Min, R., Vandersluis, B., Bellay, J., Devit, M., et al. (2011). Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol 29, 361-367. Liao, H.C., and Chen, M.Y. (2012). Target of rapamycin complex 2 signals to downstream effector yeast protein kinase 2 (Ypk2) through adheres-voraciously-to-target-of-rapamycin-2 protein 1 (Avo1) in Saccharomyces cerevisiae. J Biol Chem 287, 6089-6099. Lin CH, MacGurn, JA, Chu T, Stefan CJ, Emr SD (2008) Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 135, 714-725. Lin, D., Yin, X., Wang, X., Zhou, P., and Guo, F.B. (2013). Re-annotation of protein-coding genes in the genome of Saccharomyces cerevisiae based on support vector machines. PLoS One 8, e64477.64471-64476. Linding, R., Jensen, L.J., Ostheimer, G.J., van Vugt, M.A., Jørgensen, C., Miron, I.M., Diella, F., Colwill, K., Taylor, L., Elder, K., et al. (2007). Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415-1426. Liu, K., Zhang, X., Lester, R.L., and Dickson, R.C. (2005). The sphingoid long chain base phytosphingosine activates AGC-type protein kinases in Saccharomyces cerevisiae including 107 Ypk1, Ypk2, and Sch9. J Biol Chem 280, 22679-22687. Liu, M., Huang, C., Polu, S.R., Schneiter, R., and Chang, A. (2012). Regulation of sphingolipid synthesis through Orm1 and Orm2 in yeast. J Cell Sci 125, 2428-2435. Liu, X., Zhang, M.I., Peterson, L.B., and O'Neil, R.G. (2003). Osmomechanical stress selectively regulates translocation of protein kinase C isoforms. FEBS Lett 538, 101-106. Loewith, R., and Hall, M.N. (2011). Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189, 1177-1201. Loewith, R., Jacinto, E., Wullschleger, S., Lorberg, A., Crespo, J.L., Bonenfant, D., Oppliger, W., Jenoe, P., and Hall, M.N. (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10, 457-468. Luyten, K., Albertyn, J., Skibbe, W.F., Prior, B.A., Ramos, J., Thevelein, J.M., and Hohmann, S. (1995). Fps1, a yeast member of the MIP family of channel proteins, is a facilitator for glycerol uptake and efflux and is inactive under osmotic stress. EMBO J 14, 1360-1371. Lynch-Day MA, Klionsky DJ (2010) The Cvt pathway as a model for selective autophagy. FEBS Lett. 584: 1359-1366. Macek B, Mann M, Olsen JV (2009) Global and site-specific quantitative phosphoproteomics: principles and applications. Annu. Rev. Pharmacol. Toxicol. 49: 199-221. MacGurn, J.A., Hsu, P.C., Smolka, M.B., and Emr, S.D. (2011). TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147, 1104-1117. Mah, A.S., Elia, A.E., Devgan, G., Ptacek, J., Schutkowski, M., Snyder, M., Yaffe, M.B., and Deshaies, R.J. (2005). Substrate specificity analysis of protein kinase complex Dbf2-Mob1 by peptide library and proteome array screening. BMC Biochem 6, 22. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, García-Echeverría C (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7: 1851-1863. Malinsky J, Opekarová M, Tanner W (2010) The lateral compartmentation of the yeast plasma membrane. Yeast 27: 473-478. Malinsky J, Opekarová M, Grossmann G, Tanner W (2013) Membrane microdomains, rafts, and detergent-resistant membranes in plants and fungi. Annu. Rev. Plant Biol. 64: 501-529. Mandala, S.M., Thornton, R., Tu, Z., Kurtz, M.B., Nickels, J., Broach, J., Menzeleev, R., and Spiegel, S. (1998). Sphingoid base 1-phosphate phosphatase: a key regulator of sphingolipid 108 metabolism and stress response. Proc Natl Acad Sci, USA 95, 150-155. Manning, B.D., Tee, A.R., Logsdon, M.N., Blenis, J., and Cantley, L.C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10, 151-162. Manno, S., Takakuwa, Y., and Mohandas, N. (2002). Identification of a functional role for lipid asymmetry in biological membranes: Phosphatidylserine-skeletal protein interactions modulate membrane stability. Proc Natl Acad Sci U S A 99, 1943-1948. Masui K, Cavenee WK, Mischel PS (2014) mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol. Metab 25: 364-373. McClean, M.N., Hersen, P., and Ramanathan, S. (2009). In vivo measurement of signaling cascade dynamics. Cell Cycle 8, 373-376. McLachlin DT, Chait BT (2001) Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr. Opin. Chem. Biol. 5: 591-602. Mok, J., Kim, P.M., Lam, H.Y., Piccirillo, S., Zhou, X., Jeschke, G.R., Sheridan, D.L., Parker, S.A., Desai, V., Jwa, M., et al. (2010). Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal 3, ra12. Mollapour, M., and Piper, P.W. (2007). Hog1 mitogen-activated protein kinase phosphorylation targets the yeast Fps1 aquaglyceroporin for endocytosis, thereby rendering cells resistant to acetic acid. Mol Cell Biol 27, 6446-6456. Momoi, M., Tanoue, D., Sun, Y., Takematsu, H., Suzuki, Y., Suzuki, M., Suzuki, A., Fujita, T., and Kozutsumi, Y. (2004). SLI1 (YGR212W) is a major gene conferring resistance to the sphingolipid biosynthesis inhibitor ISP-1, and encodes an ISP-1 N-acetyltransferase in yeast. Biochem J 381, 321-328. Monier, S., Dietzen, D.J., Hastings, W.R., Lublin, D.M., and Kurzchalia, T.V. (1996). Oligomerization of VIP21-caveolin in vitro is stabilized by long chain fatty acylation or cholesterol. FEBS Lett 388, 143-149. Monier, S., Parton, R.G., Vogel, F., Behlke, J., Henske, A., and Kurzchalia, T.V. (1995). VIP21caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Mol Biol Cell 6, 911-927. Muir A, Ramachandran S, Roelants FM, Timmons G, Thorner J (2014) TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. eLIFE 3: e03779.1-e03779.34. Mukai H (2003) The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J. Biochem. 133: 17-27. 109 Mulet, J.M., Martin, D.E., Loewith, R., and Hall, M.N. (2006). Mutual antagonism of target of rapamycin and calcineurin signaling. J Biol Chem 281, 33000-33007. Muro, E., Atilla-Gokcumen, G.E., and Eggert, U.S. (2014). Lipids in cell biology: how can we understand them better? Mol Biol Cell 25, 1819-1823. Nagiec, M.M., Nagiec, E.E., Baltisberger, J.A., Wells, G., Lester, R.L., and Dickson, R.C. (1997). Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. J Biol Chem 9809-9817. Nagiec, M.M., Skrzypek, M., Nagiec, E.E., Lester, R.L., and Dickson, R.C. (1998). The LCB4 (YOR171c) and LCB5 (YLR260w) genes of Saccharomyces encode sphingoid long chain base kinases. J Biol Chem 273, 19437-19442. Nair U, Cao Y, Xie Z, Klionsky D (2010) Roles of the lipid-binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J. Biol. Chem. 285: 11476-11488. Niles, B.J., Joslin, A.C., Fresques, T., and Powers, T. (2014). TOR complex 2-Ypk1 signaling maintains sphingolipid homeostasis by sensing and regulating ROS accumulation. Cell Rep. 6: 541-552. Niles, B.J., Mogri, H., Hill, A., Vlahakis, A., and Powers, T. (2012). Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by target of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc Natl Acad Sci U S A 109, 1536-1541. Nita-Lazar A, Saito-Benz H, White FM (2008) Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics 8: 4433-4443. O'Donnell, A.F., Huang, L., Thorner, J., and Cyert, M.S. (2013). A calcineurin-dependent switch controls the trafficking function of alpha-arrestin Aly1/Art6. J Biol Chem 288, 24063–24080. Orlean P (2012) Architecture and biosynthesis of the Saccharomyces cerevisiae cell wall. Genetics 192: 775-818. Panchaud N, Péli-Gulli MP, De Virgilio C (2013) SEACing the GAP that nEGOCiates TORC1 activation: evolutionary conservation of Rag GTPase regulation. Cell Cycle 12: 2948-2952. Papinski, D., and Kraft, C. (2014). Atg1 kinase organizes autophagosome formation by phosphorylating Atg9. Autophagy 10: 1338-1340. Paulsel, A.L., Merz, A.J., and Nickerson, D.P. (2013). Vps9 family protein Muk1 is the second Rab5 guanosine nucleotide exchange factor in budding yeast. J Biol Chem 288, 18162-18171. Pearce, L.R., Komander, D., and Alessi, D.R. (2010). The nuts and bolts of AGC protein kinases. 110 Nat Rev Mol Cell Biol 11, 9-22. Piper PW, Truman AW, Millson SH, Nuttall J (2006) Hsp90 chaperone control over transcriptional regulation by the yeast Slt2(Mpk1)p and human ERK5 mitogen-activated protein kinases (MAPKs). Biochem. Soc. Trans. 34: 783-785. Potthoff MJ, Olson EN (2007) MEF2: a central regulator of diverse developmental programs. Development 134: 4131–4140. Reinke A, Anderson S, McCaffery JM, Yates J 3rd, Aronova S, Chu S, Fairclough S, Iverson C, Wedaman KP, Powers T (2004) TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol. Chem. 279: 14752-14762. Remize, F., Barnavon, L., and Dequin, S. (2001). Glycerol export and glycerol-3-phosphate dehydrogenase, but not glycerol phosphatase, are rate limiting for glycerol production in Saccharomyces cerevisiae. Metab Eng 3, 301-312. Rennefahrt, U.E., Deacon, S.W., Parker, S.A., Devarajan, K., Beeser, A., Chernoff, J., Knapp, S., Turk, B.E., and Peterson, J.R. (2007). Specificity profiling of Pak kinases allows identification of novel phosphorylation sites. J Biol Chem 282, 15667-15678. Rispal, D., Eltschinger, S., Stahl, M., Vaga, S., Bodenmiller, B., Abraham, Y., Filipuzzi, I., Movva, N.R., Aebersold, R., Helliwell, S.B., et al. (2015). Target of rapamycin complex 2 regulates actin polarization and endocytosis via multiple pathways. J Biol Chem. Roelants, F.M., Baltz, A.G., Trott, A.E., Fereres, S., and Thorner, J. (2010). A protein kinase network regulates the function of aminophospholipid flippases. Proc Natl Acad Sci U S A 107, 34-39. Roelants, F.M., Breslow, D.K., Muir, A., Weissman, J.S., and Thorner, J. (2011). Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 108, 19222-19227. Roelants, F.M., Su, B.M., von Wulffen, J., Ramachandran, S., Sartorel, E., Trott, A.E., and Thorner, J. (2015). Protein kinase Gin4 negatively regulates flippase function and controls plasma membrane asymmetry. J Cell Biol 208, 299-311. Roelants, F.M., Torrance, P.D., Bezman, N., and Thorner, J. (2002). Pkh1 and Pkh2 differentially phosphorylate and activate Ypk1 and Ykr2 and define protein kinase modules required for maintenance of cell wall integrity. Mol Biol Cell 13, 3005-3028. Roelants FM, Torrance PD, Thorner J (2004) Differential roles of PDK1- and PDK2phosphorylation sites in the yeast AGC kinases Ypk1, Pkc1 and Sch9. Microbiology 150: 32893304. 111 Roy, J., and Cyert, M.S. (2009). Cracking the phosphatase code: docking interactions determine substrate specificity. Sci Signal 2, re9.1-7. Roy, J., Li, H., Hogan, P.G., and Cyert, M.S. (2007). A conserved docking site modulates substrate affinity for calcineurin, signaling output, and in vivo function. Mol Cell 25, 889-901. Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH (1994) RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78: 35-43. Sachs, J.N., and Engelman, D.M. (2006). Introduction to the membrane protein reviews: the interplay of structure, dynamics, and environment in membrane protein function. Annu Rev Biochem 75, 707-712. Sadowski, I., Breitkreutz, B.J., Stark, C., Su, T.C., Dahabieh, M., Raithatha, S., Bernhard, W., Oughtred, R., Dolinski, K., Barreto, K., et al. (2013). The PhosphoGRID Saccharomyces cerevisiae protein phosphorylation site database: version 2.0 update. Database (Oxford) 2013, bat026. Saito, H., and Posas, F. (2012). Response to hyperosmotic stress. Genetics 192, 289-318. Saito, K., Fujimura-Kamada, K., Hanamatsu, H., Kato, U., Umeda, M., Kozminski, K.G., and Tanaka, K. (2007). Transbilayer phospholipid flipping regulates Cdc42p signaling during polarized cell growth via Rga GTPase-activating proteins. Dev Cell 13, 743-751. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14: 1296-1302. Sartorel, E., Barrey, E., Lau, R.K., and Thorner, J. (2015). Plasma membrane aminoglycerolipid flippase function is required for signaling competence in the yeast mating pheromone response pathway. Mol Biol Cell 26, 134-150. Schmelzle, T., Helliwell, S.B., and Hall, M.N. (2002). Yeast protein kinases and the RHO1 exchange factor TUS1 are novel components of the cell integrity pathway in yeast. Mol Cell Biol 22, 1329-1339. Schmick M, Kraemer A, Bastiaens PI (2015) Ras moves to stay in place. Trends Cell Biol. 25: 190-197. Schorling, S., Vallée, B., Barz, W.P., Riezman, H., and Oesterhelt, D. (2001). Lag1p and Lac1p are essential for the acyl-CoA-dependent ceramide synthase reaction in Saccharomyces cerevisiae. Mol Biol Cell 12, 3417-3427. Sengupta, S., Lorente-Rodríguez, A., Earnest, S., Stippec, S., Guo, X., Trudgian, D.C., Mirzaei, H., and Cobb, M.H. (2013). Regulation of OSR1 and the sodium, potassium, two chloride 112 cotransporter by convergent signals. Proc Natl Acad Sci U S A 110, 18826-18831. Sharifpoor, S., van Dyk, D., Costanzo, M., Baryshnikova, A., Friesen, H., Douglas, A.C., Youn, J.Y., VanderSluis, B., Myers, C.L., Papp, B., et al. (2012). Functional wiring of the yeast kinome revealed by global analysis of genetic network motifs. Genome Res 22, 791-801. Sharom, F.J. (2011). Flipping and flopping--lipids on the move. IUBMB Life 63, 736-746. Shevchenko, A., and Simons, K. (2010). Lipidomics: coming to grips with lipid diversity. Nat Rev Mol Cell Biol 11, 593-598. Shima J, Takagi H (2009) Stress-tolerance of baker's-yeast (Saccharomyces cerevisiae) cells: stress-protective molecules and genes involved in stress tolerance. Biotechnol. Appl. Biochem. 53: 155-164. Shimada, K., Filipuzzi, I., Stahl, M., Helliwell, S.B., Studer, C., Hoepfner, D., Seeber, A., Loewith, R., Movva, N.R., and Gasser, S.M. (2013). TORC2 signaling pathway guarantees genome stability in the face of DNA strand breaks. Mol Cell 51, 829-839. Shimobayashi M, Hall MN (2014) Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 15(: 155-162. Shimobayashi, M., Oppliger, W., Moes, S., Jenö, P., and Hall, M.N. (2013). TORC1-regulated protein kinase Npr1 phosphorylates Orm to stimulate complex sphingolipid synthesis. Mol Biol Cell 24, 870-881. Shore P, Sharrocks AD (1995) The MADS-box family of transcription factors. Eur. J. Biochem. 229: 1-13. Sikorski, R.S., and Hieter, P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19-27. Simons, K., and Sampaio, J.L. (2011). Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol 3, a004697. Simons, K., and van Meer, G. (1988). Lipid sorting in epithelial cells. Biochemistry 27, 61976202. Singer, S.J., and Nicolson, G.L. (1972). The fluid mosaic model of the structure of cell membranes. Science 175, 720-731. Sokolov, S., Knorre, D., Smirnova, E., Markova, O., Pozniakovsky, A., Skulachev, V., and Severin, F. (2006). Ysp2 mediates death of yeast induced by amiodarone or intracellular acidification. Biochim Biophys Acta 1757, 1366-1370. Soltwisch, J., Kettling, H., Vens-Cappell, S., Wiegelmann, M., Müthing, J., and Dreisewerd, K. 113 (2015). Mass spectrometry imaging with laser-induced post-ionization. Science 348, 211-215. Sopko, R., Huang, D., Preston, N., Chua, G., Papp, B., Kafadar, K., Snyder, M., Oliver, S.G., Cyert, M., Hughes, T.R., et al. (2006). Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 21, 319-330. Spector, A.A., and Yorek, M.A. (1985). Membrane lipid composition and cellular function. J Lipid Res 26, 1015-1035. Spiegel, S., and Milstien, S. (2003). Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4, 397-407. Spira, F., Mueller, N.S., Beck, G., von Olshausen, P., Beig, J., and Wedlich-Söldner, R. (2012). Patchwork organization of the yeast plasma membrane into numerous coexisting domains. Nat Cell Biol 14, 640-648. Stathopoulos-Gerontides, A., Guo, J.J., and Cyert, M.S. (1999). Yeast calcineurin regulates nuclear localization of the Crz1p transcription factor through dephosphorylation. Genes Dev 13, 798-803. Sun, Y., Miao, Y., Yamane, Y., Zhang, C., Shokat, K.M., Takematsu, H., Kozutsumi, Y., and Drubin, D.G. (2012). Orm protein phosphoregulation mediates transient sphingolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55-PP2A pathways. Mol Biol Cell 23, 2388-2398. Sun, Y., Taniguchi, R., Tanoue, D., Yamaji, T., Takematsu, H., Mori, K., Fujita, T., Kawasaki, T., and Kozutsumi, Y. (2000). Sli2 (Ypk1), a homologue of mammalian protein kinase SGK, is a downstream kinase in the sphingolipid-mediated signaling pathway of yeast. Mol Cell Biol 20, 4411-4419. Swinnen, E., Wilms, T., Idkowiak-Baldys, J., Smets, B., De Snijder, P., Accardo, S., Ghillebert, R., Thevissen, K., Cammue, B., De Vos, D., et al. (2014). The protein kinase Sch9 is a key regulator of sphingolipid metabolism in Saccharomyces cerevisiae. Mol Biol Cell 25, 196-211. Tabuchi M, Audhya A, Parsons AB, Boone C, Emr SD (2006) The phosphatidylinositol 4,5biphosphate and TORC2 binding proteins Slm1 and Slm2 function in sphingolipid regulation. Mol. Cell. Biol. 26, 5861-5875. Tamás, M.J., Karlgren, S., Bill, R.M., Hedfalk, K., Allegri, L., Ferreira, M., Thevelein, J.M., Rydström, J., Mullins, J.G., and Hohmann, S. (2003). A short regulatory domain restricts glycerol transport through yeast Fps1p. J Biol Chem 278, 6337-6345. Tamás, M.J., Luyten, K., Sutherland, F.C., Hernandez, A., Albertyn, J., Valadi, H., Li, H., Prior, B.A., Kilian, S.G., Ramos, J., et al. (1999). Fps1p controls the accumulation and release of the compatible solute glycerol in yeast osmoregulation. Mol Microbiol 31, 1087-1104. 114 Tanaka K, Tatebayashi K, Nishimura A, Yamamoto K, Yang HY, Saito H (2014) Yeast osmosensors Hkr1 and Msb2 activate the Hog1 MAPK cascade by different mechanisms. Sci. Signal. 7: ra21.1Tatebe, H., Morigasaki, S., Murayama, S., Zeng, C.T., and Shiozaki, K. (2010). Rab-family GTPase regulates TOR complex 2 signaling in fission yeast. Curr Biol 20, 1975-1982. Tatebayashi K, Yamamoto K, Nagoya M, Takayama T, Nishimura A, Sakurai M, Momma T, Saito H (2015) Osmosensing and scaffolding functions of the oligomeric four-transmembrane domain osmosensor Sho1. Nat. Commun. 6: 6975Thorsen, M., Di, Y., Tängemo, C., Morillas, M., Ahmadpour, D., Van der Does, C., Wagner, A., Johansson, E., Boman, J., Posas, F., et al. (2006). The MAPK Hog1p modulates Fps1pdependent arsenite uptake and tolerance in yeast. Mol Biol Cell 17, 4400-4410. Thumkeo D, Watanabe S, Narumiya S (2013) Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 92: 303-315. Togo, T., Krasieva, T.B., and Steinhardt, R.A. (2000). A decrease in membrane tension precedes successful cell-membrane repair. Mol Biol Cell 11, 4339-4346. Truman AW, Millson SH, Nuttall JM, King V, Mollapour M, Prodromou C, Pearl LH, Piper PW (2006) Expressed in the yeast Saccharomyces cerevisiae, human ERK5 is a client of the Hsp90 chaperone that complements loss of the Slt2p (Mpk1p) cell integrity stress-activated protein kinase. Eukaryot. Cell 5: 1914-1924. Umekawa, M., and Klionsky, D.J. (2012). The cytoplasm-to-vacuole targeting pathway: a historical perspective. Int J Cell Biol 2012, 142634. Vallée, B., and Riezman, H. (2005). Lip1p: a novel subunit of acyl-CoA ceramide synthase. EMBO J 24, 730-741. van der Rest ME, Kamminga AH, Nakano A, Anraku Y, Poolman B, Konings WN (1995) The plasma membrane of Saccharomyces cerevisiae: structure, function, and biogenesis. Microbiol Rev. 59: 304-322. van Meer, G., Voelker, D.R., and Feigenson, G.W. (2008). Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112-124. Vincent, F., Duquesnes, N., Christov, C., Damy, T., Samuel, J.L., and Crozatier, B. (2006). Dual level of interactions between calcineurin and PKC-epsilon in cardiomyocyte stretch. Cardiovasc Res 71, 97-107. Vlahakis A, Graef M, Nunnari J, Powers T (2014) TOR complex 2-Ypk1 signaling is an essential positive regulator of the general amino acid control response and autophagy. Proc. Natl. Acad. Sci. USA 111: 10586-10591. 115 Walther, T.C., Aguilar, P.S., Fröhlich, F., Chu, F., Moreira, K., Burlingame, A.L., and Walter, P. (2007). Pkh-kinases control eisosome assembly and organization. EMBO J 26, 4946-4955. Walther TC, Brickner JH, Aguilar PS, Bernales S, Pantoja C, Walter P (2006) Eisosomes mark static sites of endocytosis. Nature 439: 998-1003. Wedaman KP, Reinke A, Anderson S, Yates J 3rd, McCaffery JM, Powers T (2003) Tor kinases are in distinct membrane-associated protein complexes in Saccharomyces cerevisiae. Mol. Biol. Cell 14: 1204-1220. Westfall, P.J., Patterson, J.C., Chen, R.E., and Thorner, J. (2008). Stress resistance and signal fidelity independent of nuclear MAPK function. Proc Natl Acad Sci U S A 105, 12212-12217. Williamson, P., and Schlegel, R.A. (1994). Back and forth: the regulation and function of transbilayer phospholipid movement in eukaryotic cells. Mol Membr Biol 11, 199-216. Wu, C., Yosef, N., Thalhamer, T., Zhu, C., Xiao, S., Kishi, Y., Regev, A., and Kuchroo, V.K. (2013). Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513-517. Wullschleger, S., Loewith, R., Oppliger, W., and Hall, M.N. (2005). Molecular organization of target of rapamycin complex 2. J Biol Chem 280, 30697-30704. Yaffe, M.B., Leparc, G.G., Lai, J., Obata, T., Volinia, S., and Cantley, L.C. (2001). A motifbased profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol 19, 348-353. Yates JR, Ruse CI, Nakorchevsky A (2009) Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 11: 49-79. Zacharias, D.A., Violin, J.D., Newton, A.C., and Tsien, R.Y. (2002). Partitioning of lipidmodified monomeric GFPs into membrane microdomains of live cells. Science 296, 913-916. Zaman, S., Lippman, S.I., Schneper, L., Slonim, N., and Broach, J.R. (2009). Glucose regulates transcription in yeast through a network of signaling pathways. Mol Syst Biol 5, 245. Zanolari, B., Friant, S., Funato, K., Sütterlin, C., Stevenson, B.J., and Riezman, H. (2000). Sphingoid base synthesis requirement for endocytosis in Saccharomyces cerevisiae. EMBO J 19, 2824-2833. Zaremberg, V., and McMaster, C.R. (2002). Differential partitioning of lipids metabolized by separate yeast glycerol-3-phosphate acyltransferases reveals that phospholipase D generation of phosphatidic acid mediates sensitivity to choline-containing lysolipids and drugs. J Biol Chem 277, 39035-39044. 116 Zarubin, T., and Han, J. (2005). Activation and signaling of the p38 MAP kinase pathway. Cell Res 15, 11-18. Zhao, H., Michelot, A., Koskela, E.V., Tkach, V., Stamou, D., Drubin, D.G., and Lappalainen, P. (2013). Membrane-sculpting BAR domains generate stable lipid microdomains. Cell Rep 4, 1213-1223. Zheng, Z., and Zou, J. (2001). The initial step of the glycerolipid pathway: identification of glycerol 3-phosphate/dihydroxyacetone phosphate dual substrate acyltransferases in Saccharomyces cerevisiae. J Biol Chem 276, 41710-41716. Zhou Y, Hancock JF (2015) Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta 1853: 841-849. Zimmermann, C., Santos, A., Gable, K., Epstein, S., Gururaj, C., Chymkowitch, P., Pultz, D., Rødkær, S.V., Clay, L., Bjørås, M., et al. (2013). TORC1 inhibits GSK3-mediated Elo2 phosphorylation to regulate very long chain fatty acid synthesis and autophagy. Cell Rep 5, 1036-1046. Ziółkowska, N.E., Karotki, L., Rehman, M., Huiskonen, J.T., and Walther, T.C. (2011). Eisosome-driven plasma membrane organization is mediated by BAR domains. Nat Struct Mol Biol 18, 854-856. 117 Appendix: A study of places to walk around Berkeley and the Bay Area Introduction As I was deciding where to go for graduate school, I was extremely fortunate to come across “Berkeley’s Longest Paths or, why I took so long to graduate”, an appendix from Alexei Efros’ thesis (Efros, 2003). At the time I was reading (and quite enamored with) a whole bunch of stuff on pedestrianism and its effect on thinking and mental processes. In particular, I was interested in the links between walking and various philosophical schools from Aristotle to Rousseau, especially as artfully and exuberantly detailed in Rebecca Solnit’s book Wanderlust. Thus, Dr. Efros’ appendix and the promise of living in a place with some space to walk about and think (and yeah, also, the excellent scientists) ultimately sent me to Berkeley. Dr. Efros prescribed many interesting walks and I followed him around to many beautiful places in town and in the near East Bay. However, my favorite recommendation of his stemmed from the obvious observation that his list was by no means exhaustive. Thus, he recommended (in an excellent pun on math textbook speak) that it would be “[…] an enjoyable exercise to the reader to discover others”. So, I spent quite a bit of time (ssshhh, don’t tell my advisor!) finding some other beautiful places, and decided to write my favorites down. I admit I have been somewhat reticent to write this appendix - does Berkeley really need another thesis appendix describing walks? Well, if Hollywood can make as many sequels as they want, I think I can make one small one as well. I’ll focus on the two things I have the most experience with and can offer the most insight into: (1) walking around Berkeley at night and (2) day hikes accessible by public transportation. As with Dr. Efros, my experience is quite limited and this guide is not complete in any sense. I hope it spurs you the reader onto your own investigations. Lastly, I’d like to note that these recommendations will be loose descriptions, not detailed instructions or guides. I highly recommend having a good map of Berkeley, such as the Berkeley Path Wanderers Association map (http://berkeleypaths.org/maps/) or access to Google Maps to sort out the details. Night walks “[…] fretted vault of heaven by night gives you the foil of the infinite, making your petty exploit a brave adventure” Stephen Graham, The Gentle Art of Tramping “Out at night, you understand that wildness is not only a permanent property of land – it is also a quality which can settle on a place with a snowfall, or with the close of day” Robert Macfarlane, The Wild Places Berkeley has many excellent places to walk at night. These make for great after work walks. When I’ve felt stuck on projects and alienated with the normal routine, the darkness of these night walks always transported me a thousand miles away from ordinary lab life with its cares and worries. I’ve always felt like I was coming back into life after a much need long break or 118 adventure, even though these walks are only an hour long give or take. I hope you the reader find them similarly useful. Panoramic Way tree tunnel Panoramic Way is a beautiful road that runs up the ridge between Strawberry and Claremont Canyons. Claremont Canyon drops off dramatically on the south side of the road, giving amazing views of Berkeley blending into Oakland. As such, parking along the lower stretches of this road and making out is a favorite past time of the younger set of Berkeley residents. However, an even more amazing experience is to be had further up the road and away from the legions of lovebirds. Walk up to Memorial Stadium, and keep walking south along Stadium Rim Way until you get to Orchard Lane. Take these steps up to Arden Road and hang a left until you get to Arden Path. Follow this path up to Panoramic Way. Walk along this road until you get to the Upper Fire Trail. Take the very steep fire trail (commonly known as “the Connector”) up until it rejoins Panoramic Way. Exit onto this road, and walk the road a bit until it forks. Take the lower road on the right. As you walk along this narrow and potholed road, you should find yourself in a tree tunnel. If you pick a moonless night (which I highly recommend), you’ll have the impression of being in a perfectly black tunnel miraculously suspended above the city. It is quite disorienting. The light at the end of the tunnel will be downtown Oakland blazing away. Traffic along CA-24 moves like a snake in the foreground. As you continue, it feels quite a bit like you’re walking out of the tunnel into the air right above town. Your own personal turn as Harrison Ford in The Fugitive. Hopefully, Tommy Lee Jones isn’t chasing you with a gun though. As you exit the tree tunnel, make sure to take a look down the slope to your right. Someone has made two giant banana slug statues. You’ll see them down there, perpetually sliming their way up. Just take Panoramic Way and Dwight Way past the lovers in their cars and back into town. Welcome back to normal life. Creepy redwood forest Sometimes the best way to get out of a funk is to scare the hell out of yourself (or at least creep yourself out a little bit). The Berkeley fire trails in Strawberry Canyon are an amazing place to walk at night. During the day, they are packed with people and it is a fun park to go people watching. During the night, the trails become deserted and walking here then takes you far away from the normal routine and places you on adventure in a wild forest. It’s a great place and time to see animals from deer to the occasional tree climbing fox! It’s also just spooky enough to pull you out of everyday life. There is plenty to explore out there, but here is one route I recommend. Walk up to the stadium, and take Centennial Drive up past Strawberry Canyon Pool. The fire trail heads up from a small parking lot on the right. Take the trail up and keep your eyes open. After ~10 minutes of walking up the trail, the trail bends and on the left there is a small plaque marking the entrance to the redwood grove. Take one of the many trails heading up into the grove on the left. The redwood grove is beautiful and uncrowded at night, except for the occasional fellow walker or illegal Side-O mountain biker. The stars and moon disappear under the canopy of the trees as you head further in, giving the grove a dark and spooky feel. I highly recommend bringing a flashlight if you get easily spooked. The upper exit of the grove puts you back at the fire trail near the top of the Connector. You can take the Connector down and head back into town using the Orchard or Arden steps. Or you can head from here to the Panoramic Way tree tunnel described above. 119 Atlas and Scott Newhall Paths The view from Atlas and Scott Newhall Paths cannot be beat! To get there, take Cedar Street to La Loma Avenue and then head north on La Loma. Take a left onto Glendale Avenue and then follow the Glendale Paths east up the hill. After the Glendale Paths end, take Arcade Avenue. Then cross Grizzly Peak Boulevard. Atlas Path heads east from the left hand side of Grizzly Peak. After about half an hour of non-stop uphill walking, there is a bench on Atlas Path that provides a much needed rest, along with panoramic views from Richmond to the Golden Gate to San Francisco. Keep going up Atlas Path and make a left hand turn on Hill Road. Follow this road about 2 minutes until it dead ends. Scott Newhall Path (named after the famed San Francisco Chronicle newspaperman) starts from the right hand side of the road and provides again beautiful views as you walk along. From here take Hill Road down and then many routes are available back to campus. Northside Berkeley paths loop This walk loops together several of my favorite north Berkeley paths. Take Spruce Street up north. Take a right on Easter Path shortly before Marin Avenue and follow this path up. It ends in Cragmont Park, which has good climbing (rock and tree) and views onto the Bay (and none of the crowds of Indian Rock). From here, follow Regal Road east and take a left onto Pinnacle Path. This path has a very interesting ceramic mosaic wall. It is especially beautiful at night, where the tiles reflect and twinkle the light of your flashlight. From here, take a right onto Keeler Avenue. This ends in Keeler Path, which winds through Remillard Park and leaves you feeling on a trail in the forest far from town, if only for a second. When this path ends, take an immediate right onto Sterling Path, which has dramatic views down into Berkeley and the Bay. From here, Bret Hart Road and Bret Hart Way lead to Euclid Avenue and back to campus. Garber Park This walk leads through Garber Park and the local south Berkeley environs. It feels super secret and leads through a lovely little park filled with many ferns. To get there, walk east on Claremont Avenue past the Claremont Hotel. It’s the giant ostentatious white building, you can’t miss it. It’ll feel like you’re walking past something out of The Great Gatsby. Keep on going east, and a park will rise steeply on the right hand side of the road. There is a trail that heads up the hillside and a little ~1 mile loop trail that goes through the park. Take the loop around and it ends at Evergreen Lane. Take Evergreen Lane west where it ends in Evergreen Path. This path goes to Alvarado Place which leads to the Shortcut Path. Shortcut Path leads back to Tunnel Road, which you can then follow west back towards campus. Public transit assisted hikes “The end of automania would save open spaces, encourage wiser land use, and contribute greatly to ending suburban sprawl. It would lead to the building of more compact, sensitively planned communities in the future—and it would prompt many cities to build quick, quiet, and convenient modes of transportation ranging from bicycle paths to mass transit systems” Stewart Udall, “The Last Traffic Jam”, The Atlantic 120 One of the great things about the Bay Area is that it is easily possible to get to beautiful parks far out of town by public transit alone. Many cities have great public transit systems, except when you want to leave town to vacation. Then it becomes necessary to have a car. Berkeley is quite the exception and it is possible to get far out of town, and go camping even, all without a car. This unique aspect of public transit is an exceptionally good community resource, and I have been glad to take advantage of it. I hope for its continued existence. Here are some of my favorite day trips and the currently available transit options to get there. Redwood Regional Park to Berkeley A very nice ~6-7 hour trip is to follow the hills from Oakland’s Redwood Regional Park back to Strawberry Canyon and Berkeley. To get to Redwood Park, take BART to Fruitvale Station. A nice pre-walk treat can be found at Fruitvale. Powderface at Fruitvale BART serves excellent coffee and beignets. From Fruitvale BART, ride AC Transit bus #39 east into the hills. Take the stop at Skyline Boulevard and Crestmont Drive. Cross the street and you are in Redwood Regional Park. There are many trail options now to head back north towards Berkeley. The most direct is to take Dunn Trail to the Skyline National Recreational Trail and follow this north to Berkeley. The Skyline Trail is a beautiful trail that follows the spine of the Oakland/Berkeley hills. As you head north from Redwoods Park, you’ll cross through much varied scenery. From the damp and forested Huckleberry Botanical Preserve, to the dry and windswept grasses as you cross over the CA-24 Caldecott Tunnel into Tilden Park. The trail is well marked and easy to follow. Many options for exploration and variation on the standard trail exist. An excellent detailed description of the Skyline Trail and links to various resources can be found here: http://gurmeet.net/hiking/hikes/East_Bay_Skyline_National_Recreation_Trail.html. Once you have followed the Skyline Trail back to Tilden you might think you’re going crazy as you hear the faint sounds of a stream train whistle. You have not. The park operates a small model train near Vollmer Peak. It is a nice break to stop and ride the train. From here, you can take any of the Tilden Park paths back towards Berkeley. A more direct route is to cross Grizzly Peak Boulevard and where trailheads exist for the fire trails leading back into either Claremont or Strawberry Canyons. All in all, it is quite the adventure, and a very satisfying feeling to bring yourself home under your own power. Another small note. AC Transit #39 runs right alongside Joaquin Miller Park before arriving at Redwoods Park. This is another nice park to explore. It also hosts a permanent orienteering course. The maps and directions can be downloaded from here: http://baoc.org/wiki/Maps/Permanent_Courses. This makes for another nice afternoon walk and adds some adventure while sharpening up your map and compass skills. Lake Chabot to Redwood Park It is possible to go even further afield in the Oakland hills using public transit. Rather than starting in Redwood Park, it is possible to take public transit to Lake Chabot Regional Park. To get to Lake Chabot, take BART to the Coliseum stop. Exit here and take AC Transit #46L to the stop at Dunkirk Avenue and Golf Links Road. A short walk along Golf Links Road will take you to a staging area with access to the many trails of Lake Chabot. A very enjoyable day long loop is to take the Goldenrod Trail down to the end of Lake Chabot and the start of Grass Valley Creek. If you are into fishing, and have the requisite permits, this trail takes you right into Bass 121 Cove at Lake Chabot. I’ve never caught a bass here, but I can say for sure there are some catfish there. After visiting Lake Chabot, cross over the creek and take the Columbine Trail back. This trail follows the creek and eventually links with the Grass Valley Trail. This trail is quite beautiful and follows a long grassy valley, as the name would imply. It is particularly beautiful in the summer, where as far as you can see there is only yellow grass in the foreground, green hillsides on the left and blue sky everywhere else. The entire world in three colors. This trail connects to the MacDonald Trail further north, which can then be followed to the Golden Spike Trail in Redwood Park. This trail can then be followed to an AC Transit #39 bus stop, which takes you back to BART and Berkeley. Briones and Layfayette Ridge A favorite trip of mine is out to Briones Regional Park. This is a bucolic park with rolling green hills, and it is a particularly nice place for full moon hikes (and dark and clear enough to see meteor showers). The park also hosts another of the Bay Area Orienteering permanent courses as well. One of the nicest things about Briones, is that BART can get you there in about an hour. Take BART out to the Lafayette stop. Then walk east on Deer Hill Road before making a left hand turn on Elizabeth Street. Follow this to the end, and you will end up at the park on the Lafayette Ridge Trail. If you Google image search this trail, you’ll see a trail rolling endlessly along a ridge of green grassy hills. Mount Diablo stands off in the distance. Being there is every bit as awesome as it sounds. The whole ridge trail is quite long and makes an excellent morning to afternoon trip. Unfortunately, as far as I’ve ever looked, there are no other transit options out there beyond the BART station (I know, life is so hard sometimes), so this trip is an out and back. Head back down to the Lafayette BART station when finished exploring. Mount Tamalpais to the Marin Headlands I found out about the Marin Transit coaches when I was a third year student, and I felt like I was let in on a great secret. These buses will take you all along the west edge of Marin from Stinson Beach to Point Reyes, all for only $2. This makes it possible to explore the huge park system and beautiful beaches stretching from the Golden Gate all the way to Tomales Bay. Marin Transit #61 takes you through Mount Tamalpais, to Muir Woods along the way, and down to Stinson Beach. This bus makes it possible to do many trips in the Mount Tam area. Some examples. Take the bus to Stinson beach to spend the morning relaxing, before walking up Mount Tam in the afternoon and catching the bus home at Muir Woods. Circumambulate Mount Tam with Gary Snyder’s poem in hands. The possibilities are many. I’ll describe a slightly longer tour (~12 miles) that has become my favorite. Take BART to San Francisco at catch Golden Gate Transit #70 bus to the Donahue and Terners stop. From here take Marin Transit #61 to Pantoll Station. From Pantoll, walk south on the Coast View Trail. At times I’ve started from Pantoll above the fog line, and steadily descended below it. Quite an experience. Take the Heather Cutoff and Redwood Creek Trail into Muir Beach. If you’re feeling snackish, the Pelican Inn offers nice desserts and a bar. Great for a break. From Muir Beach, follow the Coast Trail into the Marin Headlands. This trail hugs the coast with its dramatic cliffs falling into the Pacific. Quite fetching. Keep following the Coast Trail (or one of many variations that are possible) into the Rodeo Beach/Fort Cronkhite area. From here, take the MUNI #76X bus back into San Francisco. Please note, as of now, the MUNI #76X only runs weekends. 122 Grand adventure from Point Reyes to Mount Tamalpais Lastly, Marin Transit #68 will take you to the visitor center at Point Reyes National Seashore. I’ve still never gotten over how great that is. Point Reyes is one of the most beautiful places in California. Using #68 bus, it is possible to do many day trips in this area. Point Reyes also has many excellent camping sites and one excellent hostel, makeing overnight trips and further exploration quite easy. I leave all of this for you to explore. Instead, I will describe one of my favorite trips. This is a very long (~20 mile) trek from Point Reyes to Pantoll Station at Mount Tam. It requires a bit of commitment and perhaps an overnight stay at Pantoll Station if you miss the last bus home. For the challenge, you’ll be rewarded with a trip across much varied and striking scenery. From the Point Reyes Visitor center, walk south along the Rift Zone and Olema Valley Trails. You’ll pass many miles and sag ponds in the Olema Trough, where two tectonic plates abut each other. Take a left and cross Highway 1 and go up to the Bolinas Ridge Trail on either the Randall or McCurdle Trails. From here, continue south until you reach the FairfaxBolinas Road. Cross this road and head south along the Coastal Trail. This trail traverses steep grassy hills that fall hundreds of feet to Bolinas below, a thin brown stripe edged into the huge endless hills. Follow this trail to Pantoll. Along the way, you’ll pass a knocked over tree that strikingly has sent all of its available branches skyward. If you ever need inspiration to keep going when stuff isn’t going your way, there it is (I know this is gross anthropomorphization but I don’t care). If you’re lucky enough, you might see some hang gliders launching from the hills above you. If you’re really lucky, maybe even a fox. You’ll be sad when it’s over and you’re at Pantoll. From here, you can catch the #61 coach back to town. Or if you’re lucky and you missed the last bus, you’ll have to camp at Pantoll and wait for the next one in the morning. They have a no-reservations, never filled camp site specifically for hikers. So, you needn’t worry about having a place to stay at Pantoll. As of now, it is only $5 to stay in the hiker site. References Efros, A.A. (2003). Data-driven approaches for texture and motion (University of California, Berkeley), pp. viii, 83 leaves. 123