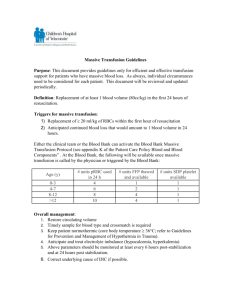
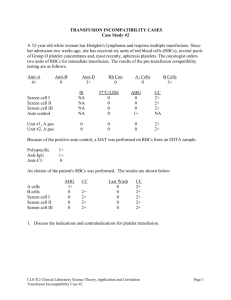
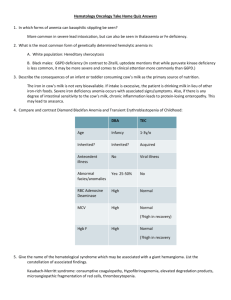
Assessment and Management of Patients With Hematologic Disorders
advertisement

Chapter 33 ● Assessment and Management of Patients With Hematologic Disorders LEARNING OBJECTIVES ● On completion of this chapter, the learner will be able to: 1. Describe the process of hematopoiesis. 2. Describe the processes involved in maintaining hemostasis. 3. Differentiate between the hypoproliferative and the hemolytic 4. 5. 6. 7. 8. 9. anemias and compare and contrast the physiologic mechanisms, clinical manifestations, medical management, and nursing interventions for each. Use the nursing process as a framework for care of patients with anemia. Compare the leukemias, their incidence, physiologic alterations, clinical manifestations, management, and prognosis. Use the nursing process as a framework for care of patients with acute leukemia. Use the nursing process as a framework for care of patients with lymphoma or multiple myeloma. Use the nursing process as a framework for care of patients with bleeding disorders. Identify therapies for blood disorders, including the nursing implications for the administration of blood and blood components. 867 868 U Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION nlike many other body systems, the hematologic system truly encompasses the entire human body. Patients with hematologic disorders can be quite challenging to nurses because they often have significant abnormalities in blood tests but few or no symptoms. It is therefore imperative that nurses have a good understanding of the pathophysiology of the patient’s condition and can make a thorough assessment that relies heavily on the interpretation of laboratory tests. It is equally important for the nurse to anticipate potential patient needs and to target nursing interventions accordingly. Because it is so important to the under- standing of most hematologic diseases, a basic appreciation of blood cells and bone marrow function is necessary. Anatomic and Physiologic Overview The hematologic system consists of the blood and the sites where blood is produced, including the bone marrow and the reticuloendothelial system (RES). Blood is a specialized organ that differs from other organs in that it exists in a fluid state. Blood is Glossary absolute neutrophil count (ANC): a mathematical calculation of the actual number of neutrophils in the circulation, derived from the total WBCs and the percentage of neutrophils counted in a microscope’s visual field; provides a rough indication of infection risk anemia: decreased RBC count anergy: diminished reactivity to antigens (transient or complete) angiogenesis: formation of new blood vessels, such as in a healing wound or in a malignant tumor angular cheilosis: cracking sore at corner of mouth aplasia: lack of cellular development (eg, of cells within the bone marrow) apoptosis: complex process of programmed cell death band cell: slightly immature neutrophil blast cell: primitive WBC cytokines: hormones produced by leukocytes that are vital to regulation of hematopoiesis, apoptosis, and immune responses D-dimer: test that measures fibrin breakdown; considered to be more specific than fibrin degradation products in the diagnosis of disseminated intravascular coagulation (DIC) differentiation: development of functions and characteristics that are different from those of the parent stem cell dysplasia: abnormal development (eg, of blood cells); size, shape and appearance of cells are altered ecchymosis: bruise erythrocyte: see RBC erythrocyte sedimentation rate (ESR): laboratory test that measures the rate of settling of RBCs; elevation is indicative of inflammation; also called the “sed rate” erythroid cells: broad term used in reference to any cell that is or will become a mature RBC erythropoiesis: process of formation of RBCs erythropoietin: hormone produced primarily by the kidney; necessary for erythropoiesis fibrin: filamentous protein; basis of thrombus and blood clot fibrinogen: protein converted into fibrin to form thrombus and clot granulocyte: granulated WBC (neutrophil, eosinophil, basophil); sometimes used synonymously with neutrophil granulocytopenia: fewer than normal granulocytes hematocrit: percentage of total blood volume consisting of RBCs hematopoiesis: complex process of the formation and maturation of blood cells hemoglobin: iron-containing protein of RBCs; delivers oxygen to tissues hemolysis: destruction of RBCs; can occur within or outside of the vasculature hemosiderin: iron-containing pigment derived from breakdown of hemoglobin hemostasis: intricate balance between clot formation and clot dissolution histiocytes: cells present in all loose connective tissue, capable of phagocytosis; part of the RES hyperplasia: abnormally increased proliferation of normal cells hypochromia: pallor within the RBC caused by decreased hemoglobin content left shift, or shift to the left: increased release of immature forms of WBCs from the bone marrow in response to need leukocyte: see WBC leukemia: uncontrolled proliferation of WBCs, often immature leukopenia: less than normal amount of WBCs in circulation lymphoid: pertaining to lymphocytes lymphocyte: form of WBC involved in immune functions lysis: destruction of cells macrocytosis: larger than normal RBCs macrophage: cells of the RES that are capable of phagocytosis mast cell: cells found in connective tissue involved in defense of the body and coagulation microcytosis: smaller than normal RBCs monocyte: large WBC that becomes a macrophage when it leaves the circulation and moves into body tissues myeloid: pertaining to nonlymphoid blood cells that differentiate into RBCs, platelets, monocytes and macrophages, neutrophils, eosinophils, basophils, and mast cells myelopoiesis: formation and maturation of cells derived from myeloid stem cell neutropenia: lower than normal number of neutrophils neutrophil: fully mature WBC capable of phagocytosis; primary defense against bacterial infection normochromic: normal RBC color, indicating normal amount of hemoglobin normocytic: normal size of RBC nucleated RBCs: immature form of RBC; portion of nucleus remains within the red cell; not normally seen in circulating blood oxyhemoglobin: combined form of oxygen and hemoglobin; found in arterial blood pancytopenia: abnormal decrease in WBCs, RBCs, and platelets petechiae: tiny capillary hemorrhages phagocytosis: process of ingestion and digestion of bacteria plasma: liquid portion of blood plasminogen: protein that is converted to plasmin to dissolve thrombi and clots platelet: thrombocyte; a cellular component of blood involved in blood coagulation poikilocytosis: variation in shape of RBCs polycythemia: excess RBCs RBC: red blood cell, erythrocyte; a cellular component of blood involved in the transport of oxygen and carbon dioxide red blood cell: see RBC reticulocytes: slightly immature RBCs, usually only 1% of total circulating RBCs reticuloendothelial system (RES): complex system of cells throughout body capable of phagocytosis serum: portion of blood remaining after coagulation occurs stem cell: primitive cell, capable of selfreplication and differentiation into myeloid or lymphoid stem cell thrombin: enzyme necessary to convert fibrinogen into fibrin clot thrombocyte: see platelet thrombocytopenia: lower than normal platelet count thrombocytosis: higher than normal platelet count WBC: white blood cells, leukocytes; cellular components of blood involved in defense of the body; subtypes include neutrophils, eosinophils, basophils, monocytes, and lymphocytes white blood cell: see WBC Chapter 33 Assessment and Management of Patients With Hematologic Disorders composed of plasma and various types of cells. Plasma is the fluid portion of blood; it contains various proteins, such as albumin, globulin, fibrinogen, and other factors necessary for clotting, as well as electrolytes, waste products, and nutrients. About 55% of blood volume is plasma. BLOOD The cellular component of blood consists of three primary cell types (Table 33-1): RBCs (red blood cells or erythrocytes), WBCs (white blood cells or leukocytes), and platelets (thrombocytes). These cellular components of blood normally make up 40% to 45% of the blood volume. Because most blood cells have a short life span, the need for the body to replenish its supply of cells is continuous; this process is termed hematopoiesis. The primary site for hematopoiesis is the bone marrow. During embryonic development and in other conditions, the liver and spleen may also be involved. Under normal conditions, the adult bone marrow produces about 175 billion RBCs, 70 billion neutrophils (mature form of a WBC), and 175 billion platelets each day. When the body needs more blood cells, as in infection (when WBCs are needed to fight the invading pathogen) or in bleeding (when more RBCs are required), the marrow increases its production of the cells required. Thus, under normal conditions, the marrow responds to increased demand and releases adequate numbers of cells into the circulation. The volume of blood in humans is approximately 7% to 10% of the normal body weight and amounts to 5 to 6 L. Circulating through the vascular system and serving as a link between body organs, the blood carries oxygen absorbed from the lungs and nutri- Table 33-1 • Blood Cells CELL TYPE MAJOR FUNCTION WBC (Leukocyte) Neutrophil Fights infection Essential in preventing or limiting bacterial infection via phagocytosis; average life span is 2 to 4 h Enters tissue as macrophage; highly phagocytic, especially against fungus; immune surveillance Involved in allergic reactions (neutralizes histamine); digests foreign proteins Contains histamine; integral part of hypersensitivity reactions Integral component of immune system Responsible for cell-mediated immunity; recognizes material as “foreign” (surveillance system) Responsible for humoral immunity; many mature into plasma cells to form antibodies Secretes immunoglobulin (Ig, antibody); most mature form of B lymphocyte Carries hemoglobin to provide oxygen to tissues; average life span is 120 days Fragment of megakaryocyte, not really a cell; provides basis for coagulation to occur; maintains hemostasis; average life span is 10 days Monocyte Eosinophil Basophil Lymphocyte T lymphocyte B lymphocyte Plasma cell RBC (Erythrocyte) Platelet (Thrombocyte) 869 ents absorbed from the gastrointestinal tract to the body cells for cellular metabolism. Blood also carries waste products produced by cellular metabolism to the lungs, skin, liver, and kidneys, where they are transformed and eliminated from the body. Blood also carries hormones, antibodies, and other substances to their sites of action or use. To function, blood must remain in its normally fluid state. Because blood is fluid, the danger always exists that trauma can lead to loss of blood from the vascular system. To prevent this, an intricate clotting mechanism is activated when necessary to seal any leak in the blood vessels. Excessive clotting is equally dangerous, because it can obstruct blood flow to vital tissues. To prevent this, the body has a fibrinolytic mechanism that eventually dissolves clots (thrombi) formed within blood vessels. The balance between these two systems, clot (thrombus) formation and clot (thrombus) dissolution or fibrinolysis, is called hemostasis. BONE MARROW The bone marrow is the site of hematopoiesis, or blood cell formation (Fig. 33-1). In a child all skeletal bones are involved, but as the child ages marrow activity decreases. By adulthood, marrow activity is usually limited to the pelvis, ribs, vertebrae, and sternum. Marrow is one of the largest organs of the body, making up 4% to 5% of total body weight. It consists of islands of cellular components (red marrow) separated by fat (yellow marrow). As the adult ages, the proportion of active marrow is gradually replaced by fat; however, in the healthy person, the fat can again be replaced by active marrow when more blood cell production is required. In adults with disease that causes marrow destruction, fibrosis, or scarring, the liver and spleen can also resume production of blood cells by a process known as extramedullary hematopoiesis. The marrow is highly vascular. Within it are primitive cells called stem cells. The stem cells have the ability to self-replicate, thereby ensuring a continuous supply of stem cells throughout the life cycle. When stimulated to do so, stem cells can begin a process of differentiation into either myeloid or lymphoid stem cells. These stem cells are committed to produce specific types of blood cells. Lymphoid stem cells produce either T or B lymphocytes. Myeloid stem cells differentiate into three broad cell types: RBCs, WBCs, and platelets. Thus, with the exception of lymphocytes, all blood cells are derived from the myeloid stem cell. A defect in the myeloid stem cell can cause problems not only with WBC production but also with RBC and platelet production. The entire process of hematopoiesis is highly complex. Research has identified many of the complex mechanisms involved, often at the molecular level. A thorough description of these processes is beyond the scope of this textbook; however, some mechanisms against which a specific treatment is targeted are briefly described in the relevant disease-specific sections of this chapter. BLOOD CELLS Red Blood Cells (RBCs) The normal RBC is a biconcave disk that resembles a soft ball compressed between two fingers (Fig. 33-2). It has a diameter of about 8 µm and is so flexible that it can pass easily through capillaries that may be as small as 2.8 µm in diameter. The RBC membrane is so thin that gases, such as oxygen and carbon dioxide, can easily diffuse across it; the disk shape provides a large surface area that facilitates the absorption and release of oxygen molecules. 870 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Physiology/Pathophysiology FIGURE 33-1 Hematopoiesis. Uncommitted (pluripotent) stem cells can differentiate into myeloid or lymphoid stem cells. These stem cells then undergo a complex process of differentiation and maturation into normal cells that are released into the circulation. The myeloid stem cell is responsible not only for all nonlymphoid white blood cells (WBCs) but also for the production of red blood cells (RBCs) and platelets. Each step of the differentiation process depends in part on the presence of specific growth factors for each cell type. When the stem cells are dysfunctional, they may respond inadequately to the need for more cells, or they may respond excessively, sometimes uncontrollably, as in leukemia. Adapted from Amgen, Inc., 1995, Thousand Oaks, CA. Chapter 33 Assessment and Management of Patients With Hematologic Disorders 871 RBCs Basophil (WBC) Platelet (thrombocyte) Neutrophil (WBC) B-Lymphocyte (WBC) Monocyte (WBC) FIGURE 33-2 Normal types of blood cells. Mature RBCs consist primarily of hemoglobin, which contains iron and makes up 95% of the cell mass. RBCs have no nuclei, and they have many fewer metabolic enzymes than do most other cells. The presence of a large amount of hemoglobin enables the RBC to perform its principal function, the transport of oxygen between the lungs and tissues. Occasionally the marrow releases slightly immature forms of RBCs, called reticulocytes, into the circulation. This occurs as a normal response to an increased demand for RBCs (as in bleeding) or in some disease states. The oxygen-carrying hemoglobin molecule is made up of four subunits, each containing a heme portion attached to a globin chain. Iron is present in the heme component of the molecule. An important property of heme is its ability to bind to oxygen loosely and reversibly. Oxygen readily binds to hemoglobin in the lungs and is carried as oxyhemoglobin in arterial blood. Oxyhemoglobin is a brighter red than hemoglobin that does not contain oxygen (reduced hemoglobin), which is why arterial blood is a brighter red than venous blood. The oxygen readily dissociates (detaches) from hemoglobin in the tissues, where the oxygen is needed for cellular metabolism. In venous blood, hemoglobin combines with hydrogen ions produced by cellular metabolism and thus buffers excessive acid. Whole blood normally contains about 15 g of hemoglobin per 100 mL of blood. ERYTHROPOIESIS Erythroblasts arise from the primitive myeloid stem cells in bone marrow. The erythroblast is a nucleated cell that, in the process of maturing within the bone marrow, accumulates hemoglobin and Eosinophil (WBC) gradually loses its nucleus. At this stage, the cell is known as a reticulocyte. Further maturation into an RBC entails the loss of the dark-staining material and slight shrinkage. The mature RBC is then released into the circulation. Under conditions of rapid erythropoiesis (RBC production), reticulocytes and other immature cells (eg, nucleated RBCs) may be released prematurely into the circulation. Differentiation of the primitive myeloid stem cell of the marrow into an erythroblast is stimulated by erythropoietin, a hormone produced primarily by the kidney. If the kidney detects low levels of oxygen (as would occur in anemia, in which fewer RBCs are available to bind oxygen, or in people living at high altitudes), the release of erythropoietin is increased. The increased erythropoietin then stimulates the marrow to increase production of RBCs. The entire process typically takes 5 days. For normal RBC production, the bone marrow also requires iron, vitamin B12, folic acid, pyridoxine (vitamin B6), protein, and other factors. A deficiency of these factors during erythropoiesis can result in decreased RBC production and anemia. Iron Stores and Metabolism. The average daily diet in the United States contains 10 to 15 mg of elemental iron; normally 0.5 to 1 mg of ingested iron is absorbed from the small intestine. The rate of iron absorption is regulated by the amount of iron already stored in the body and by the rate of RBC production. Additional amounts of iron, up to 2 mg daily, must be absorbed by women to replace blood lost during menstruation. Total body iron content in the average adult is approximately 3 g, most of which is present in 872 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION hemoglobin or in one of its breakdown products. Iron is stored in the small intestine as ferritin and in reticuloendothelial cells. When required, the iron is released into the plasma, binds to transferrin, and is transported into the membranes of the normoblasts (RBC precursor cells) within the marrow, where it is incorporated into hemoglobin. Iron is lost in the feces, either in bile, blood, or mucosal cells from the intestine. The concentration of iron in blood is normally about 75 to 175 µg/dL (13 to 31 µmol/L) for men and 65 to 165 µg/dL (11 to 29 µmol/L) for women. With iron deficiency, bone marrow iron stores are rapidly depleted; hemoglobin synthesis is depressed, and the RBCs produced by the marrow are small and low in hemoglobin. Iron deficiency in the adult generally indicates that blood has been lost from the body (eg, from bleeding in the gastrointestinal tract or heavy menstrual flow). In the adult, lack of dietary iron is rarely the sole cause of iron deficiency anemia. The source of iron deficiency should be investigated promptly, because iron deficiency in an adult may be a sign of bleeding in the gastrointestinal tract or colon cancer. Vitamin B12 and Folic Acid Metabolism. Vitamin B12 and folic acid are required for the synthesis of DNA in many tissues, but deficiencies of either of these vitamins have the greatest effect on erythropoiesis. Both vitamin B12 and folic acid are derived from the diet. Folic acid is absorbed in the proximal small intestine, but only small amounts are stored within the body. If the diet is deficient in folic acid, stores within the body quickly become depleted. Because vitamin B12 is found only in foods of animal origin, strict vegetarians may ingest little B12. Vitamin B12 combines with intrinsic factor produced in the stomach. The vitamin B–intrinsic factor complex is absorbed in the distal ileum. People who have had a partial or total gastrectomy may have limited amounts of intrinsic factor, and therefore the absorption of B12 may be diminished. The effects of either decreased absorption or decreased intake of B12 are not apparent for 2 to 4 years. Vitamin B12 and folic acid deficiencies are characterized by the production of abnormally large RBCs called megaloblasts. Because these cells are abnormal, many are sequestered (trapped) while still in the bone marrow, and their rate of release is decreased. Some of these cells actually die in the marrow before they can be released into the circulation. This results in megaloblastic anemia. RED BLOOD CELL DESTRUCTION The average life span of a normal circulating RBC is 120 days. Aged RBCs lose their elasticity and become trapped in small blood vessels, particularly in the spleen. They are removed from the blood by the reticuloendothelial cells, particularly in the liver and the spleen. As the RBCs are destroyed, their hemoglobin is largely recycled. Some hemoglobin also breaks down to form bilirubin and is secreted in the bile. Most of the iron is recycled to form new hemoglobin molecules within the bone marrow; small amounts are lost daily in the feces and urine and monthly in menstrual flow. White Blood Cells (WBCs) Leukocytes are divided into two general categories: granulocytes and lymphocytes. In normal blood, the total leukocyte count is 5000 to 10,000 cells per cubic millimeter. Of these, approximately 60% to 70% are granulocytes and 30% to 40% are lymphocytes. Primarily, WBCs protect the body against infection and tissue injury. GRANULOCYTES Granulocytes are defined by the presence of granules in the cytoplasm of the cell. Granulocytes are divided into three main subgroups, which are characterized by the staining properties of these granules (see Fig. 33-2). Eosinophils have bright-red granules in their cytoplasm, whereas the granules in basophils stain deep blue. The third and by far the most numerous cell in this class is the neutrophil, with granules that stain a pink to violet hue. Neutrophils are also called polymorphonuclear neutrophils (PMNs, or polys) or segmented neutrophils (segs). The nucleus of the mature neutrophil has multiple lobes (usually two to five) that are connected by thin filaments of nuclear material, a “segmented” nucleus; it is usually twice the size of an RBC. The somewhat less mature granulocyte has a single-lobed, elongated nucleus and is called a band cell. Ordinarily, band cells account for only a small percentage of circulating granulocytes, although their percentage can increase greatly under conditions in which neutrophil production increases, such as infection. An increased number of band cells is sometimes called a “left shift” or “shift to the left.” (Traditionally, the diagram of neutrophil maturation shows the stem cell on the left with progressive maturation stages toward the right, ending with a fully mature neutrophil on the right side. A shift to the left indicates that more immature cells are present in the blood than normally occurs.) Granulocyte production from the myeloid stem cell pool results in the gradual differentiation of these cells from a myeloid blast cell into a fully mature neutrophil. The process, called myelopoiesis, is highly complex and depends on many factors. These factors, including specific cytokines such as growth factors, are normally present within the marrow itself. As the blast cell matures, the cytoplasm of the cell changes in color (from blue to violet) and granules begin to form with the cytoplasm. The shape of the nucleus also changes. The entire process of maturation and differentiation takes about 10 days (see Fig. 33-1). Once the neutrophil is released into the circulation from the marrow, it stays there for only about 6 hours before it migrates into the body tissues to perform its function of phagocytosis (ingestion and digestion of bacteria and particles) (Fig. 33-3). Here, neutrophils last no more than 1 to 2 days before they die. The number of circulating granulocytes found in the healthy person is relatively constant, but in infection large numbers of these cells are rapidly released into the circulation. MONONUCLEAR WHITE BLOOD CELLS (AGRANULOCYTES) Monocytes. Monocytes (also called mononuclear leukocytes) are WBCs with a single-lobed nucleus and a granule-free cytoplasm— hence the term agranulocyte. In normal adult blood, monocytes account for approximately 5% of the total WBCs. Monocytes are the largest of the WBCs. Produced by the bone marrow, they remain in the circulation for a short time before entering the tissues and transforming into macrophages. Macrophages are particularly active in the spleen, liver, peritoneum, and the alveoli of the lungs. Lymphocytes. Mature lymphocytes are small cells with scanty cytoplasm. Immature lymphocytes are produced in the marrow from the lymphoid stem cells. A second major source of production is the cortex of the thymus. Cells derived from the thymus are known as T lymphocytes (or T cells); those derived from the marrow can also be T cells but are more commonly B lymphocytes (or B cells). Lymphocytes complete their differentiation and maturation primarily in the lymph nodes and in the lymphoid tissue of the intestine and spleen after exposure to a specific antigen. Mature lymphocytes are antigen-specific cells. Chapter 33 Assessment and Management of Patients With Hematologic Disorders 873 Physiology/Pathophysiology Neutrophil (WBC) Capillary Bacteria Endothelial cell FIGURE 33-3 Phagocytosis. When foreign matter (such as bacteria or dead tissue) comes in contact with the cell membrane of the neutrophil, the membrane surrounds and pinches off the area, leaving the membrane intact. Thus, the engulfed material is left in a vacuole within the neutrophil, where enzymes within the cell destroy the foreign material. FUNCTION OF WHITE BLOOD CELLS WBCs protect the body from invasion by bacteria and other foreign entities. The major function of neutrophils is phagocytosis (see Fig. 33-3). Neutrophils arrive at the site within 1 hour after the onset of an inflammatory reaction and initiate phagocytosis, but they are short-lived. An influx of monocytes follows; these cells continue their phagocytic activities for long periods as macrophages. This process constitutes a second line of defense for the body against inflammation and infection. Although neutrophils can often work adequately against bacteria without the need for excessive involvement with macrophages, macrophages are particularly effective against fungi and viruses. Macrophages also digest senescent (aging or aged) blood cells, such as RBCs, primarily within the spleen. The primary function of lymphocytes is to produce substances that aid in attacking foreign material. One group of lymphocytes (T lymphocytes) kills foreign cells directly or releases a variety of lymphokines, substances that enhance the activity of phagocytic cells. T lymphocytes are responsible for delayed allergic reactions, rejection of foreign tissue (eg, transplanted organs), and destruction of tumor cells. This process is known as cellular immunity. The other group of lymphocytes (B lymphocytes) is capable of differentiating into plasma cells. Plasma cells, in turn, produce immunoglobulin (Ig), or antibodies, which are protein molecules that destroy foreign material by several mechanisms. This process is known as humoral immunity. Eosinophils and basophils function in hypersensitivity reactions. Eosinophils are important in the phagocytosis of parasites. The increase in eosinophil levels in allergic states indicates that these cells are involved in the hypersensitivity reaction; their function there is to neutralize histamine. Basophils produce and store histamine as well as other substances involved in hypersensitivity reactions. The release of these substances provokes allergic reactions. Platelets (Thrombocytes) Platelets, or thrombocytes, are not actually cells. Rather, they are granular fragments of giant cells in the bone marrow called megakaryocytes. Platelet production in the marrow is regulated in part by the hormone thrombopoietin, which stimulates the production and differentiation of megakaryocytes from the myeloid stem cell. Platelets play an essential role in the control of bleeding. They circulate freely in the blood in an inactive state, where they nurture the endothelium of the blood vessels, maintaining the integrity of the vessel. When vascular injury does occur, platelets collect at the site and are activated. They adhere to the site of injury and to each other, forming a platelet plug that temporarily stops bleeding. Substances released from platelet granules activate coagulation factors in the blood plasma and initiate the formation of a stable clot composed of fibrin, a filamentous protein. Platelets have a normal life span of 7 to 10 days. PLASMA AND PLASMA PROTEINS After cellular elements are removed from blood, the remaining liquid portion is called plasma. More than 90% of plasma is water. The remainder consists primarily of plasma proteins, clotting factors (particularly fibrinogen), and small amounts of other substances such as nutrients, enzymes, waste products, and gases. If plasma is allowed to clot, the remaining fluid is called serum. Serum has essentially the same composition as plasma, except that fibrinogen and several clotting factors have been removed in the clotting process. Plasma proteins consist primarily of albumin and globulins. The globulins can be separated into three main fractions—alpha, beta, and gamma—each of which consists of distinct proteins 874 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION that have different functions. Important proteins in the alpha and beta fractions are the transport globulins and the clotting factors that are made in the liver. The transport globulins carry various substances in bound form around the circulation. For example, thyroid-binding globulin carries thyroxin, and transferrin carries iron. The clotting factors, including fibrinogen, remain in an inactive form in the blood plasma until activated by the clotting cascade. The gamma globulin fraction refers to the immunoglobulins, or antibodies. These proteins are produced by the well-differentiated lymphocytes and plasma cells. The actual fractionation of the globulins can be seen on a specific laboratory test (serum protein electrophoresis). Albumin is particularly important for the maintenance of fluid balance within the vascular system. Capillary walls are impermeable to albumin, so its presence in the plasma creates an osmotic force that keeps fluid within the vascular space. Albumin, which is produced by the liver, has the capacity to bind to several substances that are transported in plasma (eg, certain medications, bilirubin, some hormones). People with poor hepatic function may have low concentrations of albumin, with a resultant decrease in osmotic pressure and the development of edema. RETICULOENDOTHELIAL SYSTEM (RES) The RES is composed of special tissue macrophages, which are derived from monocytes. When released from the marrow, monocytes spend a short time in the circulation (about 24 hours) and then enter the body tissues. Within the tissues, the monocytes continue to differentiate into cells called macrophages, which can survive for months. Macrophages have a variety of important functions. They defend the body against foreign invaders (ie, bacteria and other pathogens) via phagocytosis. They remove old or damaged cells from the circulation. They stimulate the inflammatory process and present antigen to the immune system (see Chapter 50). Macrophages give rise to tissue histiocytes, including Kupffer cells of the liver, peritoneal macrophages, alveolar macrophages, and other components of the RES. Thus, the RES is a component of many other organs within the body, particularly the spleen, lymph nodes, lung, and liver. The spleen is the site of activity for most macrophages. Most of the spleen (75%) is made of red pulp; here the blood enters the venous sinuses through capillaries that are surrounded by macrophages. Within the red pulp are tiny aggregates of white pulp, consisting of B and T lymphocytes. The spleen sequesters newly released reticulocytes from the marrow, removing nuclear fragments and other materials (eg, denatured hemoglobin, iron) before the now fully mature RBC returns to the circulation. Although a minority of RBCs (less than 5%) is pooled in the spleen, a significant proportion of platelets (20%–40%) is pooled here. If the spleen is enlarged, a greater proportion of RBCs and platelets can be sequestered. The spleen is a major source of hematopoiesis in fetal life. It can resume hematopoiesis later in adulthood if necessary (eg, in bone marrow fibrosis). The spleen has important immunologic functions as well. It forms a substance that promotes the phagocytosis of neutrophils; it also forms the antibody IgM after exposure to antigen. HEMOSTASIS Hemostasis is the process of preventing blood loss from intact vessels and of stopping bleeding from a severed vessel. The prevention of blood loss from intact vessels requires adequate numbers of functional platelets. Platelets nurture the endothelium and thereby maintain the structural integrity of the vessel wall. Two processes are involved in arresting bleeding: primary and secondary hemostasis. In primary hemostasis, the severed blood vessel constricts. Circulating platelets aggregate at the site and adhere to the vessel and to one another. An unstable hemostatic plug is formed. For the coagulation process to be correctly activated, circulating inactive coagulation factors must be converted to active forms. This process occurs on the surface of the aggregated platelets at the site of vessel injury. The end result is the formation of fibrin, which reinforces the platelet plug and anchors it to the injury site. This process is termed secondary hemostasis (Fig. 33-4). The process of blood coagulation is highly complex. It can be activated by the intrinsic or the extrinsic pathway. Both pathways are needed for maintenance of normal hemostasis. Many factors are involved in the reaction cascade that forms fibrin. When tissue is injured, the extrinsic pathway is activated by the release from the tissue of a substance called thromboplastin. As the result of a series of reactions, prothrombin is converted to thrombin, which in turn catalyzes the conversion of fibrinogen to fibrin. Clotting by the intrinsic pathway is activated when the collagen that lines blood vessels is exposed. Clotting factors are activated sequentially until, as with the extrinsic pathway, fibrin is ultimately formed. Although the intrinsic pathway is slower, this sequence is probably most often responsible for clotting in vivo. Physiology/Pathophysiology Endothelial membrane injury Tissue injury Intrinsic pathway (slow) Extrinsic pathway (fast) Activates Factor X Prothrombin Thrombin Fibrinogen Fibrin (clot) anchors platelet plug to site of injury FIGURE 33-4 Secondary hemostasis. Based on the type of stimulus (injury to the endothelial membrane of a blood vessel or a tissue), one of two clotting pathways is initiated. The end result from either pathway is the conversion of prothrombin to thrombin. Thrombin is necessary for fibrinogen to be converted into fibrin, the stabilizing protein that anchors the fragile platelet plug to the site of injury to prevent further bleeding and permit the injured vessel or site to heal. Chapter 33 Assessment and Management of Patients With Hematologic Disorders As the injured vessel is repaired and again covered with endothelial cells, the fibrin clot is no longer needed. The fibrin is digested via two systems: the plasma fibrinolytic system and the cellular fibrinolytic system. The substance plasminogen is required to lyse (break down) the fibrin. Plasminogen, which is present in all body fluids, circulates with fibrinogen and is therefore incorporated into the fibrin clot as it forms. When the clot is no longer needed (eg, after an injured blood vessel has healed), the plasminogen is activated to form plasmin. Plasmin actually digests the fibrinogen, and the breakdown particles of the clot (fibrin degradation products) are released into the circulation. Through this system, clots are dissolved as tissue is repaired, and the vascular system returns to its normal baseline state. PATHOPHYSIOLOGY OF THE HEMATOLOGIC SYSTEM Most hematologic diseases reflect a defect in the hematopoietic, hemostatic, or RES systems. The defect can be quantitative (eg, increased or decreased production of cells), qualitative (eg, the cells that are produced are defective in their normal functional capacity), or both. Gerontologic Considerations In elderly patients, a common problem is decreased ability of the bone marrow to respond to the body’s need for blood cells (RBCs, WBCs, and platelets). This inability is a result of many factors, including diminished production of the growth factors necessary for hematopoiesis by stromal cells within the marrow or a diminished response to the growth factors (in the case of erythropoietin). When an elderly person needs more blood cells (eg, WBCs in infection, RBCs in anemia), the bone marrow may not be able to increase production of these cells adequately. Leukopenia (a decreased number of circulating WBCs) or anemia can result. In the elderly, the bone marrow may be more susceptible to the myelosuppressive effects of medications. Anemia is the most common hematologic condition affecting elderly patients; with each successive decade of life, the incidence of anemia increases. Anemia frequently results from iron deficiency (in the case of blood loss) or from a nutritional deficiency, particularly folate or B12 deficiency or protein-calorie malnutrition; it may also result from inflammation or chronic disease. Management of the disorder varies depending on the etiology. Therefore, it is important to identify the cause of the anemia rather than to consider it an inevitable consequence of aging. Elderly people with concurrent cardiac or pulmonary problems may not tolerate anemia very well, and a prompt, thorough evaluation is warranted. Assessment and Diagnostic Findings Many hematologic conditions cause few symptoms. Therefore, the use of extensive laboratory tests is often required to diagnose a hematologic disorder. For most hematologic conditions, continued monitoring via specific blood tests is required because it is very important to assess for changes in test results over time. HEMATOLOGIC STUDIES The most common tests used are the complete blood count (CBC) and the peripheral blood smear (Table 33-2). The CBC identifies the total number of blood cells (WBCs, RBCs, and platelets) as well as the hemoglobin, hematocrit (percentage of blood consisting of RBCs), and RBC indices. Because cellular 875 morphology (shape and appearance of the cells) is particularly important in most hematologic disorders, the physician needs to examine the blood cells involved. This process is referred to as the manual examination of the peripheral smear, which may be part of the CBC. In this test, a drop of blood is spread on a glass slide, stained, and examined under a microscope. The shape and size of the RBCs and platelets as well as the actual appearance of the WBCs provides useful information in identifying hematologic conditions. Blood for the CBC is typically obtained by venipuncture. BONE MARROW ASPIRATION AND BIOPSY The bone marrow aspiration and biopsy are crucial when additional information is needed to assess how an individual’s blood cells are being formed and to assess the quantity and quality of each type of cell produced within the marrow. These tests are also used to document infection or tumor within the marrow. Normal bone marrow is in a semifluid state and can be aspirated through a special large needle. In adults, bone marrow is usually aspirated from the iliac crest and occasionally from the sternum. The aspirate provides only a sample of cells. Aspirate alone may be adequate for evaluating certain conditions, such as anemia. However, when more information is required, a biopsy is also performed. Biopsy samples are taken from the posterior iliac crest; occasionally, an anterior approach is required. A marrow biopsy shows the architecture of the bone marrow as well as its degree of cellularity. Most patients need no more preparation than a careful explanation of the procedure, but for some very anxious patients, an antianxiety agent may be useful. It is always important for the physician or nurse to describe and explain to the patient the procedure and the sensations that will be experienced. The risks, benefits, and alternatives are also discussed. A signed informed consent is needed before the procedure is performed. Before aspiration, the skin is cleansed as for any minor surgery, using aseptic technique. Then a small area is anesthetized with a local anesthetic through the skin and subcutaneous tissue to the periosteum of the bone. It is not possible to anesthetize the bone itself. The bone marrow needle is introduced with a stylet in place. When the needle is felt to go through the outer cortex of bone and enter the marrow cavity, the stylet is removed, a syringe is attached, and a small volume (0.5 mL) of blood and marrow is aspirated. Patients typically feel a pressure sensation as the needle is advanced into position. The actual aspiration always causes sharp but brief pain, resulting from the suction exerted as the marrow is aspirated into the syringe; the patient should be forewarned about this. Taking deep breaths or using relaxation techniques often helps ease the discomfort. If a bone marrow biopsy is necessary, it is best performed after the aspiration and in a slightly different location, because the marrow structure may be altered after aspiration. A special biopsy needle is used. Because these needles are large, the skin is punctured first with a surgical blade to make a 3- or 4-mm incision. The biopsy needle is advanced well into the marrow cavity. When the needle is properly positioned, a portion of marrow is cored out, using a twisting or gentle rocking motion to free the sample and permit its removal within the biopsy needle. Patients feel a pressure sensation but should not feel actual pain. The nurse should instruct the patient to inform the physician if pain occurs so that additional anesthetic can be administered. The major hazard of either bone marrow aspiration or biopsy is a slight risk of bleeding and infection. The bleeding risk is somewhat increased if the patient’s platelet count is low or if the patient has been taking a medication (eg, aspirin) that alters platelet func- Unit 6 876 Table 33-2 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION • Frequently Used Laboratory Tests in Hematology TEST NORMAL RANGE Complete blood count (CBC) Red blood cells (RBCs) Hemoglobin (Hgb) M: 4.7–6.1 × 106 F: 4.2–5.4 × 106 M: 13.5–17.5 g/dL F: 11.5–15.5 g/dL DESCRIPTION INDICATIONS/COMMENTS General survey of bone marrow function; evaluates all three cell lines (WBCs, RBCs, platelets) Important to note changes over time; many hematologic conditions show changes in CBC long before patient becomes symptomatic Carries hemoglobin; survival time, 120 days Delivers O2 through circulation to body tissues and returns CO2 from tissues to lungs Indicates relative proportions of plasma and RBCs (volume of RBCs/L whole blood) Indicates size of RBCs; very useful in differentiating types of anemia Average concentration of Hgb in RBCs; independent of cell size Hematocrit (Hct) M: 40–52% F: 36–48% Mean corpuscular volume (MCV) Mean corpuscular hemoglobin concentration (MCHC) Red cell distribution width (RDW) Reticulocyte count 81–96 µm3 Platelets 150,000–400,000/mm3 White blood cells (WBCs) Differential 4,500–11,000/mm3 Percentages of various types of WBCs Total WBC count % of cell type × total WBC = absolute number of that cell type Prothrombin time (PT) Varies (compare with control), 11–12.5 sec Measure time elapsed until clot forms; measures extrinsic and common pathways 33–36 g/dL 11–14.5% 0.5–1.5% International normalized ratio (INR) 1.0 Standard warfarin (Coumadin) treatment, 2.0–3.0 INR; high-dose warfarin (Coumadin) treatment, 3.0–4.5 INR Partial thromboplastin time Varies (compare with control): 25–35 sec (PTT) Thrombin time (TT) Fibrinogen Varies (compare with control), 8–11 sec 170–340 mg/100 mL D-dimer 0–0.5µg/mL Measures degree of variation in size of RBCs Measure of marrow production of erythrocytes; 1% of RBC mass is produced daily (to replace the 1% of old cells that die) Total number of platelets in circulation; average life span, 7–10 days A standard method of measuring PT independent of the thromboplastin reagent used in the test; calculated by dividing the PT result by the mean normal PT Surface active agent added to plasma; measures time elapsed until clot forms; measures intrinsic and common pathways Tests conversion of fibrinogen to fibrin Measurement of fibrinogen concentration within plasma available for conversion to fibrin clot Measures the amount of fragments of fibrin when it is lysed (broken down); useful for distinguishing fibrinolysis from fibrinogenolysis Decreased in anemia; increased in polycythemia Usually three times the Hgb If < 80, cells are microcytic; if > 100, cells are macrocytic Indicates marrow’s response to anemia (when anemia is present, reticulocyte level should rise) Thrombocytopenia: < 20,000/mm3, serious; < 10,000/mm3, potentially life-threatening Left shift: bone marrow ↑ production of WBCs; more immature forms released into the bloodstream Increased in liver disease, disseminated intravascular coagulation (DIC), obstructive biliary disease, clotting factor depletion, warfarin (Coumadin) use Increased with anticoagulant excess and conditions that cause increased PT; decreased with insufficient anticoagulant and conditions that cause decreased PT Increased in clotting factor depletion, DIC, liver disease, biliary obstruction, circulating anticoagulants (heparin) Time to clot is inversely proportional to fibrinogen level Decreased in bleeding disorders, pregnancy, malignancy, inflammatory disease Increased with fibrinolytic activity, rheumatoid arthritis, ovarian cancer (with increased CA 125) (continued ) Chapter 33 Table 33-2 TEST Assessment and Management of Patients With Hematologic Disorders 877 • Frequently Used Laboratory Tests in Hematology (Continued) NORMAL RANGE DESCRIPTION INDICATIONS/COMMENTS Byproduct of fibrinolysis >40 µg/mL indicates DIC Essential in preventing/limiting bacterial infection; average life span: 2–4 hr If >8,000: infection, some inflammatory states, stress, steroids, other drugs, myeloproliferative disease Absolute neutrophil count (ANC) <500: increased risk for infection; ANC <100: infection certain (if neutropenia persists) <1,500: lymphopenia; >4,000: lymphocytosis; increased in convalescent phase after bacterial or viral infection, lymphoproliferative disease Increased in acute and chronic infection, inflammation, some myeloproliferative disorders, chronic myelomonocytic leukemia (CMML) Increased in allergic states, medications, parasites, chronic myeloid leukemia (CML), metastatic/ necrotic tumors Increase is very rare (CML) Fibrin degradation products <10 µg/mL (FDP) Neutrophils 40–75% (2,500–7,500/mm3) Lymphocytes 20–50% (1,500–5,500/mm3) Integral component of immune system Monocytes 1–10% (100–800/mm3) Enter tissue as macrophages; phagocytosis Eosinophils 0–6% (0–440/mm3) Involved in allergic reactions (neutralizes histamine); digest foreign proteins Basophils 0–2% (0–200/mm3) Contain histamine; integral part of hypersensitivity reactions tion. After the marrow sample is obtained, pressure is applied to the site for several minutes. The site is then covered with a sterile dressing. Most patients have no discomfort after a bone marrow aspiration, but the site of a biopsy may ache for 1 or 2 days. Warm tub baths and use of a mild analgesic (eg, acetaminophen) may be useful. Aspirin-containing analgesics should be avoided because they can aggravate or potentiate any bleeding that may occur. • Loss of RBCs—occurs with bleeding, potentially from any • Management of Hematologic Disorders Commonly encountered blood disorders are anemia, polycythemia, leukopenia and neutropenia, leukocytosis, lymphoma, myeloma, leukemia, and various bleeding and coagulation disorders. Nursing management of patients with these disorders requires skillful assessment and monitoring as well as meticulous care and teaching to prevent deterioration and complications. ANEMIA Anemia, per se, is not a specific disease state but a sign of an underlying disorder. It is by far the most common hematologic condition. Anemia, a condition in which the hemoglobin concentration is lower than normal, reflects the presence of fewer than normal RBCs within the circulation. As a result, the amount of oxygen delivered to body tissues is also diminished. There are many different kinds of anemia (Table 33-3), but all can be classified into three broad etiologic categories: • major source, such as the gastrointestinal tract, the uterus, the nose, or a wound Decreased production of RBCs—can be caused by a deficiency in cofactors (including folic acid, vitamin B12, and iron) required for erythropoiesis; RBC production may also be reduced if the bone marrow is suppressed (eg, by tumor, medications, toxins) or is inadequately stimulated because of a lack of erythropoietin (as occurs in chronic renal disease). Increased destruction of RBCs—may occur because of an overactive RES (including hypersplenism) or because the bone marrow produces abnormal RBCs that are then destroyed by the RES (eg, sickle cell anemia). A conclusion as to whether the anemia is caused by destruction or by inadequate production of RBCs usually can be reached on the basis of the following factors: • The marrow’s ability to respond to the decreased RBCs (as • • evidenced by an increased reticulocyte count in the circulating blood) The degree to which young RBCs proliferate in the bone marrow and the manner in which they mature (as observed on bone marrow biopsy) The presence or absence of end products of RBC destruction within the circulation (eg, increased bilirubin level, decreased haptoglobin level) Unit 6 878 Table 33-3 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION • Classification of Anemias TYPE OF ANEMIA LABORATORY FINDINGS Hypoproliferative (Resulting From Defective RBC Production) Iron deficiency Decreased reticulocytes, iron, ferritin, iron saturation, MCV; increased TIBC Vitamin B12 deficiency Decreased vitamin B12 level; (megaloblastic) increased MCV Folate deficiency Decreased folate level; increased MCV Decreased erythropoietin Decreased erythropoietin level; production (eg, from renal normal MCV and MCH; dysfunction) increased creatinine level Cancer /inflammation Normal MCV, MCH, normal or decreased erythropoietin level; increased % of iron saturation, ferritin level; decreased iron; TIBC Bleeding (Resulting From RBC Loss) Increased reticulocyte level; normal Bleeding from gastroHgb and Hct if measured soon intestinal tract, menorrhagia after bleeding starts, but levels (excessive menstrual flow), decrease thereafter; normal MCV epistaxis (nosebleed), initially but later decreases; detrauma creased ferritin and iron levels (later) Hemolytic (Resulting From RBC Destruction) Decreased MCV; fragmented Altered erythropoiesis (sickle RBCs; increased reticulocyte level cell anemia, thalassemia, other hemoglobinopathies) Increased MCV Hypersplenism (hemolysis) Increased spherocyte level Drug-induced anemia Increased spherocyte level Autoimmune anemia Fragmented red cells Mechanical heart valve– related anemia Hct, hematocrit; Hgb, hemoglobin concentration; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; RBCs, red blood cells; TIBC, total iron binding capacity. Classification of Anemias Anemia may be classified in several ways. The physiologic approach is to determine whether the deficiency in RBCs is caused by a defect in their production (hypoproliferative anemia), by their destruction (hemolytic anemia), or by their loss (bleeding). In the hypoproliferative anemias, RBCs usually survive normally, but the marrow cannot produce adequate numbers of these cells. The decreased production is reflected in a low reticulocyte count. Inadequate production of RBCs may result from marrow damage due to medications or chemicals (eg, chloramphenicol, benzene) or from a lack of factors necessary for RBC formation (eg, iron, vitamin B12, folic acid, erythropoietin). Hemolytic anemias stem from premature destruction of RBCs, which results in a liberation of hemoglobin from the RBC into the plasma. The increased RBC destruction results in tissue hypoxia, which in turn stimulates erythropoietin production. This increased production is reflected in an increased reticulocyte count, as the bone marrow responds to the loss of RBCs. The released hemoglobin is converted in large part to bilirubin; therefore, the bilirubin concentration rises. Hemolysis can result from an abnormality within the RBC itself (eg, sickle cell anemia, glucose-6-phosphate dehydrogenase [G-6-PD] deficiency) or within the plasma (eg, immune hemolytic anemias), or from direct injury to the RBC within the circulation (eg, hemolysis caused by mechanical heart valve). Chart 33-1 identifies the causes of hemolytic anemia. Clinical Manifestations Aside from the severity of the anemia itself, several factors influence the development of anemia-associated symptoms: • • • • • The speed with which the anemia has developed The duration of the anemia (ie, its chronicity) The metabolic requirements of the individual Other concurrent disorders or disabilities (eg, cardiopulmonary disease) Special complications or concomitant features of the condition that produced the anemia In general, the more rapidly an anemia develops, the more severe its symptoms. An otherwise healthy person can often tolerate as much as a 50% gradual reduction in hemoglobin without pronounced symptoms or significant incapacity, whereas the rapid loss of as little as 30% may precipitate profound vascular collapse in the same individual. A person who has been anemic for a very long time, with hemoglobin levels between 9 and 11 g/dL, usually has few or no symptoms other than slight tachycardia on exertion and fatigue. Chart 33-1 Causes of Hemolytic Anemias Inherited Hemolytic Anemia Abnormal hemoglobin Sickle cell anemia* Thalassemia* Red blood cell membrane abnormality Hereditary spherocytosis* Hereditary elliptocytosis Acathanthocytosis Stomatocytosis Enzyme deficiencies Glucose-6-phosphate dehydrogenase (G-6-PD) deficiency* Acquired Hemolytic Anemia Antibody-related Iso-antibody/transfusion reaction* Autoimmune hemolytic anemia (AIHA)* Cold agglutinin disease* Not antibody-related Red blood cell membrane defects Paroxysymal nocturnal hemoglobinuria (PNH) Liver disease Uremia Trauma Mechanical heart valve Microangiopathic hemolytic anemia Infection Bacterial Parasitic Disseminated intravascular coagulation (DIC)* Toxins Hypersplenism* *Discussed in text. Chapter 33 Assessment and Management of Patients With Hematologic Disorders Patients who customarily are very active or who have significant demands on their lives (eg, a single, working mother of small children) are more likely to have symptoms, and those symptoms are more likely to be pronounced than in a more sedentary person. A patient with hypothyroidism with decreased oxygen needs may be completely asymptomatic, without tachycardia or increased cardiac output, at a hemoglobin level of 10 g/dL. Similarly, patients with coexistent cardiac, vascular, or pulmonary disease may develop more pronounced symptoms of anemia (eg, dyspnea, chest pain, muscle pain or cramping) at a higher hemoglobin level than those without these concurrent health problems. Finally, some anemic disorders are complicated by various other abnormalities that do not result from the anemia but are inherently associated with these particular diseases. These abnormalities may give rise to symptoms that completely overshadow those of the anemia, as in the painful crises of sickle cell anemia. Assessment and Diagnostic Findings A variety of hematologic studies are performed to determine the type and cause of the anemia. In an initial evaluation, the hemoglobin, hematocrit, reticulocyte count, and RBC indices, particularly the mean corpuscular volume (MCV), are particularly useful. Iron studies (serum iron level, total iron-binding capacity [TIBC], percent saturation, and ferritin), as well as serum vitamin B12 and folate levels, are also frequently obtained. Other tests include haptoglobin and erythropoietin levels. The remaining CBC values are useful in determining whether the anemia is an isolated problem or part of another hematologic condition, such as leukemia or myelodysplastic syndrome (MDS). Bone marrow aspiration may be performed. In addition, other diagnostic studies may be performed to determine the presence of underlying chronic illness, such as malignancy, and the source of any blood loss, such as polyps or ulcers within the gastrointestinal tract. Complications General complications of severe anemia include heart failure, paresthesias, and confusion. At any given level of anemia, patients with underlying heart disease are far more likely to have angina or symptoms of heart failure than those without heart disease. Complications associated with specific types of anemia are included in the description of each type. Medical Management Management of anemia is directed toward correcting or controlling the cause of the anemia; if the anemia is severe, the RBCs that are lost or destroyed may be replaced with a transfusion of packed RBCs (PRBCs). The management of the various types of anemia is covered in the discussions that follow. NURSING PROCESS: THE PATIENT WITH ANEMIA Assessment The health history and physical examination provide important data about the type of anemia involved, the extent and type of symptoms it produces, and the impact of those symptoms on the patient’s life. Weakness, fatigue, and general malaise are common, as are pallor of the skin and mucous membranes (sclera, oral mucosa). 879 Jaundice may be present in patients with megaloblastic anemia or hemolytic anemia. The tongue may be smooth and red (in iron deficiency anemia) or beefy red and sore (in megaloblastic anemia); the corners of the mouth may be ulcerated (angular cheilosis) in both types of anemia. Individuals with iron deficiency anemia may crave ice, starch, or dirt (known as pica); their nails may be brittle, ridged, and concave. The health history should include a medication history, because some medications can depress bone marrow activity or interfere with folate metabolism. An accurate history of alcohol intake, including the amount and duration, should be obtained. Family history is important, because certain anemias are inherited. Athletic endeavors should be assessed, because extreme exercise can decrease erythropoiesis and RBC survival in some athletes. A nutritional assessment is important, because it may indicate deficiencies in essential nutrients such as iron, vitamin B12, and folic acid. Children of indigent families may be at higher risk for anemia because of nutritional deficiencies. Strict vegetarians are also at risk for megaloblastic types of anemia if they do not supplement their diet with vitamin B12. Cardiac status should be carefully assessed. When the hemoglobin level is low, the heart attempts to compensate by pumping faster and harder in an effort to deliver more blood to hypoxic tissue. This increased cardiac workload can result in such symptoms as tachycardia, palpitations, dyspnea, dizziness, orthopnea, and exertional dyspnea. Heart failure may eventually develop, as evidenced by an enlarged heart (cardiomegaly) and liver (hepatomegaly) and by peripheral edema. Assessment of the gastrointestinal system may disclose complaints of nausea, vomiting (with specific questions as to the appearance of any emesis [eg, looks like “coffee grounds”]), melena or dark stools, diarrhea, anorexia, and glossitis (inflammation of the tongue). Stools should be tested for occult blood. Women should be questioned about their menstrual periods (eg, excessive menstrual flow, other vaginal bleeding) and the use of iron supplements during pregnancy. The neurologic examination is also important because of the effect of pernicious anemia on the central and peripheral nervous systems. Assessment should include the presence and extent of peripheral numbness and paresthesias, ataxia, poor coordination, and confusion. Finally, it is important to monitor relevant laboratory test results and to note any changes over time. Diagnosis NURSING DIAGNOSES Based on the assessment data, major nursing diagnoses for the anemic patient may include: • Activity intolerance related to weakness, fatigue, and general malaise • Imbalanced nutrition, less than body requirements, related to inadequate intake of essential nutrients • Ineffective tissue perfusion related to inadequate blood volume or hematocrit • Noncompliance with prescribed therapy COLLABORATIVE PROBLEMS/ POTENTIAL COMPLICATIONS Based on the assessment data, potential complications that may develop include: • Heart failure • Paresthesias • Confusion 880 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Planning and Goals The major goals for the patient may include increased tolerance of normal activity, attainment or maintenance of adequate nutrition, maintenance of adequate tissue perfusion, compliance with prescribed therapy, and absence of complications. Nursing Interventions MANAGING FATIGUE The most frequent symptom and complication of anemia is fatigue. This distressing symptom is too often minimized by health care providers. Fatigue is often the symptom that has the greater negative impact on the individual’s level of functioning and consequent quality of life. Patients describe the fatigue from anemia as oppressive. Fatigue can be significant, yet the anemia may not be severe enough to warrant transfusion. Fatigue can interfere with an individual’s ability to work, both inside and outside the home. It can harm relationships with family and friends. Patients often lose interest in hobbies and activities, including sexual activity. The distress from fatigue is often related to an individual’s responsibilities and life demands as well as the amount of assistance and support received from others. Nursing interventions can focus on assisting the patient to prioritize activities and to establish a balance between activity and rest that is realistic and feasible from the patient’s perspective. Patients with chronic anemia need to maintain some physical activity and exercise to prevent the deconditioning that results from inactivity. MAINTAINING ADEQUATE NUTRITION Inadequate intake of essential nutrients, such as iron, vitamin B12, folic acid, and protein can cause some anemias. The symptoms associated with anemia (eg, fatigue, anorexia) can in turn interfere with maintaining adequate nutrition. A healthy diet should be encouraged. Because alcohol interferes with the utilization of essential nutrients, the nurse should advise the patient to avoid alcoholic beverages or to limit their intake and should provide the rationale for this recommendation. Dietary teaching sessions should be individualized, including cultural aspects related to food preferences and food preparation. The involvement of family members enhances compliance with dietary recommendations. Dietary supplements (eg, vitamins, iron, folate, protein) may be prescribed as well. Equally important, the patient and family must understand the role of nutritional supplements in the proper context, because many forms of anemia are not the result of a nutritional deficiency. In such cases, excessive intake of nutritional supplements will not improve the anemia. A potential problem in individuals with chronic transfusion requirements occurs with the indiscriminate use of iron. Unless an aggressive program of chelation therapy is implemented, these individuals are at risk for iron overload from their transfusions alone. The addition of an iron supplement only exacerbates the situation. MAINTAINING ADEQUATE PERFUSION Patients with acute blood loss or severe hemolysis may have decreased tissue perfusion from decreased blood volume or reduced circulating RBCs (decreased hematocrit). Lost volume is replaced with transfusions or intravenous fluids, based on the symptoms and the laboratory findings. Supplemental oxygen may be necessary, but it is rarely needed on a long-term basis unless there is underlying severe cardiac or pulmonary disease as well. The nurse monitors vital signs closely; other medica- tions, such as antihypertensive agents, may need to be adjusted or withheld. PROMOTING COMPLIANCE WITH PRESCRIBED THERAPY For patients with anemia, medications or nutritional supplements are often prescribed to alleviate or correct the condition. These patients need to understand the purpose of the medication, how to take the medication and over what time period, and how to manage any side effects of therapy. To enhance compliance, the nurse can assist patients in developing ways to incorporate the therapeutic plan into their lives, rather than merely giving the patient a list of instructions. For example, many patients have difficulty taking iron supplements because of related gastrointestinal effects. Rather than seeking assistance from a health care provider in managing the problem, some of these patients simply stop taking the iron. Abruptly stopping some medications can have serious consequences, as in the case of high-dose corticosteroids to manage hemolytic anemias. Some medications, such as growth factors, are extremely expensive. Patients receiving these medications may need assistance with obtaining needed insurance coverage or with exploring alternatives for obtaining these medications. MONITORING AND MANAGING POTENTIAL COMPLICATIONS A significant complication of anemia is heart failure from chronic diminished blood volume and the heart’s compensatory effort to increase cardiac output. Patients with anemia should be assessed for signs and symptoms of heart failure. A serial record of body weights can be more useful than a record of dietary intake and output, because the intake and output measurements may not be accurate. In the case of fluid retention resulting from congestive heart failure, diuretics may be required. In megaloblastic forms of anemia, the significant potential complications are neurologic. A neurologic assessment should be performed for patients with known or suspected megaloblastic anemia. Patients may initially complain of paresthesias in their lower extremities. These paresthesias are usually manifested as numbness and tingling on the bottom of the foot, and they gradually progress. As the anemia progresses and damage to the spinal cord occurs, other signs become apparent. Position and vibration sense may be diminished; difficulty maintaining balance is not uncommon, and some patients have gait disturbances as well. Initially mild but gradually progressive confusion may develop. Evaluation EXPECTED PATIENT OUTCOMES Expected patient outcomes may include: 1. Tolerates activity at a safe and acceptable level a. Follows a progressive plan of rest, activity, and exercise b. Prioritizes activities c. Paces activities according to energy level 2. Attains and maintains adequate nutrition a. Eats a healthy diet b. Develops meal plan that promotes optimal nutrition c. Maintains adequate amounts of iron, vitamins, and protein from diet or supplements d. Adheres to nutritional supplement therapy when prescribed e. Verbalizes understanding of rationale for using recommended nutritional supplements f. Verbalizes understanding of rationale for avoiding nonrecommended nutritional supplements Chapter 33 Assessment and Management of Patients With Hematologic Disorders 3. Maintains adequate perfusion a. Has vital signs within baseline for patient b. Has pulse oximetry (arterial oxygenation) value within normal limits 4. Absence of complications a. Avoids or limits activities that cause dyspnea, palpitations, dizziness, or tachycardia b. Uses rest and comfort measures to alleviate dyspnea c. Has vital signs within baseline for patient d. Has no signs of increasing fluid retention (eg, peripheral edema, decreased urine output, neck vein distention) e. Remains oriented to time, place, and situation f. Ambulates safely, using assistive devices as necessary g. Remains free of injury h. Verbalizes understanding of importance of serial CBC measurements i. Maintains safe home environment; obtains assistance as necessary. Hypoproliferative Anemias 881 ever, few patients with suspected iron deficiency anemia undergo bone marrow aspiration. In many patients, the diagnosis can be established with other tests, particularly in patients with a history of conditions that predispose them to this type of anemia. There is a strong correlation between laboratory values measuring iron stores and levels of hemoglobin. After the iron stores are depleted (as reflected by low serum ferritin levels), the hemoglobin level falls. The diminished iron stores cause small RBCs. Therefore, as the anemia progresses, the MCV, which measures the size of the RBC, also decreases. Hematocrit and RBC levels are also low in relation to the hemoglobin level. Other laboratory tests that measure iron stores are useful but are not as consistent indicators as a low ferritin level, which reflects low iron stores. Typically, patients with iron deficiency anemia have a low serum iron level and an elevated TIBC, which measures the transport protein supplying the marrow with iron as needed (also referred to as transferrin). However, other disease states, such as infection and inflammatory conditions, can also cause a low serum iron level and TIBC with an elevated ferritin level. Therefore, the most reliable laboratory findings in evaluating iron deficiency anemia are the ferritin and hemoglobin values. IRON DEFICIENCY ANEMIA Iron deficiency anemia typically results when the intake of dietary iron is inadequate for hemoglobin synthesis. The body can store about one fourth to one third of its iron, and it is not until those stores are depleted that iron deficiency anemia actually begins to develop. Iron deficiency anemia is the most common type of anemia in all age groups, and it is the most common anemia in the world. More than 500 million people are affected, more commonly in underdeveloped countries, where inadequate iron stores can result from inadequate intake of iron (seen with vegetarian diets) or from blood loss (eg, from intestinal hookworm). Iron deficiency is also common in the United States. In children, adolescents, and pregnant women, the cause is typically inadequate iron in the diet to keep up with increased growth. However, for most adults with iron deficiency anemia, the cause is blood loss. In fact, in adults, the cause of iron deficiency anemia should be considered to be bleeding until proven otherwise. The most common cause of iron deficiency in men and postmenopausal women is bleeding (from ulcers, gastritis, inflammatory bowel disease, or gastrointestinal tumors). The most common cause of iron deficiency anemia in premenopausal women is menorrhagia (excessive menstrual bleeding) and pregnancy with inadequate iron supplementation. Patients with chronic alcoholism often have chronic blood loss from the gastrointestinal tract, which causes iron loss and eventual anemia. Other causes include iron malabsorption, as is seen after gastrectomy or with celiac disease. Clinical Manifestations Patients with iron deficiency primarily have the symptoms of anemia. If the deficiency is severe or prolonged, they may also have a smooth, sore tongue, brittle and ridged nails, and angular cheilosis (an ulceration of the corner of the mouth). These signs subside after iron-replacement therapy. The health history may be significant for multiple pregnancies, gastrointestinal bleeding, and pica (a craving for unusual substances, such as ice, clay, or laundry starch). Assessment and Diagnostic Findings The most definitive method of establishing the diagnosis of iron deficiency anemia is bone marrow aspiration. The aspirate is stained to detect iron, which is at a low level or even absent. How- Medical Management Except in the case of pregnancy, the cause of iron deficiency should be investigated. Anemia may be a sign of a curable gastrointestinal cancer or of uterine fibroid tumors. Stool specimens should be tested for occult blood. People 50 years of age or older should have a colonoscopy, endoscopy, or other examination of the gastrointestinal tract to detect ulcerations, gastritis, polyps, or cancer. Several oral iron preparations—ferrous sulfate, ferrous gluconate, and ferrous fumarate—are available for treating iron deficiency anemia. In some cases, oral iron is poorly absorbed or poorly tolerated, or iron supplementation is needed in large amounts. In these situations, intravenous or intramuscular administration of iron dextran may be needed. Before parenteral administration of a full dose, a small test dose should be administered to avoid the risk of anaphylaxis with either intravenous or intramuscular injections. Emergency medications (eg, epinephrine) should be close at hand.If no signs of allergic reaction have occured after 30 minutes, the remaining dose of iron may be administered. Several doses are required to replenish the patient’s iron stores. Nursing Management Preventive education is important, because iron deficiency anemia is common in menstruating and pregnant women. Food sources high in iron include organ meats (beef or calf’s liver, chicken liver), other meats, beans (black, pinto, and garbanzo), leafy green vegetables, raisins, and molasses. Taking iron-rich foods with a source of vitamin C enhances the absorption of iron. The nurse helps the patient select a healthy diet. Nutritional counseling can be provided for those whose usual diet is inadequate. Patients with a history of eating fad diets or strict vegetarian diets are counseled that such diets often contain inadequate amounts of absorbable iron. The nurse encourages patients to continue iron therapy as long as it is prescribed, although they may no longer feel fatigued. Because iron is best absorbed on an empty stomach, patients should be advised to take the supplement an hour before meals. Most patients can use the less expensive, more standard forms of ferrous sulfate. Tablets with enteric coating may be poorly 882 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Chart 33-2 • PATIENT EDUCATION Taking Iron Supplements Iron supplementation is usually given in oral form, typically as ferrous sulfate, or FeSO4. Many patients have difficulty tolerating iron supplements, primarily due to gastrointestinal toxicities (eg, nausea, abdominal discomfort, constipation). Here are some helpful guidelines for taking iron supplements: • Take iron on an empty stomach (1 hour before or 2 hours after a meal). Iron absorption is reduced with food, especially dairy products. • To prevent gastrointestinal distress, the following schedule may work better if more than one tablet a day is prescribed: Start with only one tablet per day for a few days, then increase absorbed and should be avoided. Other patients have difficulty taking iron supplements because of gastrointestinal side effects (primarily constipation, but also cramping, nausea, and vomiting). Some iron formulations are designed to limit gastrointestinal side effects by the addition of a stool softener or use of sustainedrelease formulations to limit nausea or gastritis. Specific patient teaching aids, such as the accompanying patient education guide (Chart 33-2), can assist patients with the use of iron supplements. If taking iron on an empty stomach causes gastric distress, the patient may need to take the iron supplement with meals. However, doing so diminishes iron absorption by as much as 50%, thus prolonging the time required to replenish iron stores. Antacids or dairy products should not be taken with iron, because they greatly diminish the absorption of iron. Polysaccharide iron complex forms that have less gastrointestinal toxicity are also available, but they are more expensive. Liquid forms of iron that cause less gastrointestinal distress are available. However, they can stain the teeth; patients should be instructed to take this medication through a straw, to rinse the mouth with water, and to practice good oral hygiene after taking this medication. Finally, patients should be informed that iron salts may color the stool dark green or black. However, iron replacement therapy does not cause a false-positive result on stool analyses for occult blood. Intramuscular supplementation is used infrequently. The volume of iron required may be excessive. The intramuscular injection causes some local pain and can stain the skin. These side effects are minimized by using the Z-track technique for administering iron dextran deep into the gluteus maximus muscle (buttock). Avoid vigorously rubbing the injection site after the injection. Because of the problems with intramuscular administration, the intravenous route is preferred for administration of iron dextran. ANEMIAS IN RENAL DISEASE The degree of anemia in patients with end-stage renal disease varies greatly, but in general patients do not become significantly anemic until the serum creatinine level exceeds 3 mg/100 mL. The symptoms of anemia are often the most disturbing of the patient’s symptoms. The hematocrit usually falls to between 20% and 30%, although in rare cases it may fall to less than 15%. The RBCs appear normal on the peripheral smear. This anemia is caused by both a mild shortening of RBC life span and a deficiency of erythropoietin (necessary for erythro- to two tablets per day, then three tablets per day. This method permits the body to adjust gradually to the iron. • Increase the intake of vitamin C (citrus fruits and juices, strawberries, tomatoes, broccoli), to enhance iron absorption. • Eat foods high in fiber to minimize problems with constipation. • Remember that stools will become dark in color. Polysaccharide iron complex forms are better tolerated but are more expensive. Liquid forms of iron supplementation may be better tolerated than solid forms, although they are more expensive. The liquid forms can discolor teeth. Use a straw or place the spoon at the back of the mouth to take the supplement; rinse mouth thoroughly afterward. poiesis). As renal function decreases, erythropoietin, which is produced by the kidney, also decreases. Because erythropoietin is also produced outside the kidney, some erythropoiesis does continue, even in patients whose kidneys have been removed. However, the amount is small and the degree of erythropoiesis is inadequate. Patients undergoing long-term hemodialysis lose blood into the dialyzer and therefore may become iron deficient. Folic acid deficiency develops because this vitamin passes into the dialysate. Therefore, patients who receive hemodialysis and who are anemic should be evaluated for iron and folate deficiency and treated appropriately. The availability of recombinant erythropoietin (epoetin alfa [Epogen, Procrit]) has dramatically altered the management of anemia in end-stage renal disease by decreasing the need for RBC transfusion, with its associated risks. Erythropoietin, in combination with oral iron supplements, can raise and maintain hematocrit levels to between 33% and 38%. This treatment has been successful with dialysis patients. Many patients report decreased fatigue, increased energy, increased feelings of well-being, improved exercise tolerance, better tolerance of dialysis treatments, and improved quality of life. Hypertension is the most serious side effect in this patient population when the hematocrit rapidly increases to a high level. Therefore, the hematocrit should be checked frequently when a patient with renal disease begins erythropoietin therapy. The dose of erythropoietin (epoetin alfa) should be titrated to the hematocrit. In some patients, the elevated hematocrit and associated hypertension may necessitate antihypertensive therapy. ANEMIA OF CHRONIC DISEASE The term “anemia of chronic disease” is a misnomer in that only the chronic diseases of inflammation, infection, and malignancy cause this type of anemia. Many chronic inflammatory diseases are associated with a normochromic, normocytic anemia (ie, the RBCs are normal in color and size). These disorders include rheumatoid arthritis; severe, chronic infections; and many cancers. It is therefore imperative that the “chronic disease” be diagnosed when this form of anemia is identified so that it can be appropriately managed. The anemia is usually mild to moderate and nonprogressive. It develops gradually over 6 to 8 weeks and then stabilizes at a hematocrit seldom less than 25%. The hemoglobin level rarely falls below 9 g/dL, and the bone marrow has normal cellularity Chapter 33 Assessment and Management of Patients With Hematologic Disorders with increased stores of iron as the iron is diverted from the serum (and thus is unavailable as a growth factor for invading pathogens). Erythropoietin levels are low, perhaps because of decreased production, and iron use is blocked by erythroid cells (cells that are or will become mature RBCs). A moderate shortening of RBC survival also occurs. Most of these patients have few symptoms and do not require treatment for the anemia. With successful treatment of the underlying disorder, the bone marrow iron is used to make RBCs and the hemoglobin level rises. APLASTIC ANEMIA Aplastic anemia is a rather rare disease caused by a decrease in or damage to marrow stem cells, damage to the microenvironment within the marrow, and replacement of the marrow with fat. It results in bone marrow aplasia (markedly reduced hematopoiesis). Therefore, in addition to severe anemia, significant neutropenia and thrombocytopenia (a deficiency of platelets) are also seen. 883 the diagnosis is established. Typical complications are infection and symptoms of anemia (eg, fatigue, pallor, dyspnea). Purpura (bruising) may develop later and should trigger a CBC and hematologic evaluation if these were not performed initially. If the patient has had repeated throat infections, cervical lymphadenopathy may be seen. Other lymphadenopathies and splenomegaly sometimes occur. Retinal hemorrhages are common. Assessment and Diagnostic Findings In many situations, aplastic anemia occurs when a medication or chemical is ingested in toxic amounts. However, in a few people, it develops after a medication has been taken at the recommended dosage. This may be considered an idiosyncratic reaction in those who are highly susceptible, possibly caused by a genetic defect in the medication biotransformation or elimination process. A bone marrow aspirate shows an extremely hypoplastic or even aplastic (very few to no cells) marrow replaced with fat. Pathophysiology Medical Management Aplastic anemia can be congenital or acquired, but most cases are idiopathic (ie, without apparent cause). Infections and pregnancy can trigger it, or it may be caused by certain medications, chemicals, or radiation damage (Chart 33-3). Agents that regularly produce marrow aplasia include benzene and benzene derivatives (eg, airplane glue). Certain toxic materials, such as inorganic arsenic and several pesticides (including DDT, which is no longer used or available in the United States), have also been implicated as potential causes. Various medications have been associated with aplastic anemia. It is presumed that the lymphocytes of patients with aplastic anemia destroy the stem cells and consequently impair the production of RBCs, WBCs, and platelets. Despite its severity, aplastic anemia can be successfully treated in most people. Potentially, those who are younger than 60 years of age, who are otherwise healthy, and who have a compatible donor can be cured of the disease by a bone marrow transplantaton (BMT) or peripheral stem cell transplantation (BSCT). In others, the disease can be managed with immunosuppressive therapy. A combination of antithymocyte globulin and cyclosporine is used most commonly. Immunosuppressants prevent the patient’s lymphocytes from destroying the stem cells. If relapse occurs (ie, the patient becomes pancytopenic again), reinstitution of the same immunologic agents may induce another remission. Corticosteroids are not very useful as an immunosuppressive agent, because patients with aplastic anemia appear particularly susceptible to the development of bone complications from corticosteroids (ie, aseptic necrosis of the head of the femur). Supportive therapy plays a major role in the management of aplastic anemia. Any offending agent is discontinued. The patient is supported with transfusions of RBCs and platelets as necessary. Death usually is caused by hemorrhage or infection. Clinical Manifestations The manifestations of aplastic anemia are often insidious. Complications resulting from bone marrow failure may occur before Chart 33-3 Substances Associated With Aplastic Anemia Analgesics Antiseizure agents (mephenytoin, triethadione*) Antihistamines Antimicrobials* Antineoplastic agents (alkylating agents, antitumor antibiotics, antimetabolites) Antithyroid medications Benzene* Chloramphenicol* Gold compounds* Heavy metals Hypoglycemic agents Insecticides Organic arsenicals* Phenylbutazone* Phenothiazines Sulfonamides* Sedatives *Most common. Nursing Management Patients with aplastic anemia are vulnerable to problems related to RBC, WBC, and platelet deficiencies. They should be assessed carefully for signs of infection and bleeding. Specific interventions are delineated in the sections on neutropenia and thrombocytopenia. MEGALOBLASTIC ANEMIAS In the anemias caused by deficiencies of vitamin B12 or folic acid, identical bone marrow and peripheral blood changes occur, because both vitamins are essential for normal DNA synthesis. In either anemia, the RBCs that are produced are abnormally large and are called megaloblastic RBCs. Other cells derived from the myeloid stem cell (nonlymphoid WBCs, platelets) are also abnor- 884 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION mal. A bone marrow analysis reveals hyperplasia (abnormal increase in the number of cells), and the precursor erythroid and myeloid cells are large and bizarre in appearance. Many of these abnormal RBCs and myeloid cells are destroyed within the marrow, however, so the mature cells that do leave the marrow are actually fewer in number. Thus, pancytopenia (a decrease in all myeloid-derived cells) can develop. In an advanced situation, the hemoglobin value may be as low as 4 to 5 g/dL, the WBC count 2,000 to 3,000/mm3, and the platelet count less than 50,000/mm3. Those cells that are released into the circulation are often abnormally shaped. The neutrophils are hypersegmented. The platelets may be abnormally large. The RBCs are abnormally shaped, and the shapes may vary widely (poikilocytosis). Because the RBCs are very large, the MCV is very high, usually exceeding 110 µm3. Pathophysiology FOLIC ACID DEFICIENCY Folic acid, a vitamin that is necessary for normal RBC production, is stored as compounds referred to as folates. The folate stores in the body are much smaller than those of vitamin B12, and they are quickly depleted when the dietary intake of folate is deficient (within 4 months). Folate is found in green vegetables and liver. Folate deficiency occurs in people who rarely eat uncooked vegetables. Alcohol increases folic acid requirements, and, at the same time, patients with alcoholism usually have a diet that is deficient in the vitamin. Folic acid requirements are also increased in patients with chronic hemolytic anemias and in women who are pregnant, because the need for RBC production is increased in these conditions. Some patients with malabsorptive diseases of the small bowel, such as sprue, may not absorb folic acid normally. VITAMIN B12 DEFICIENCY A deficiency of vitamin B12 can occur in several ways. Inadequate dietary intake is rare but can develop in strict vegetarians who consume no meat or dairy products. Faulty absorption from the gastrointestinal tract is more common. This occurs in conditions such as Crohn’s disease, or after ileal resection or gastrectomy. Another cause is the absence of intrinsic factor, as in pernicious anemia. Intrinsic factor is normally secreted by cells within the gastric mucosa; normally it binds with the dietary vitamin B12 and travels with it to the ileum, where the vitamin is absorbed. Without intrinsic factor, orally consumed vitamin B12 cannot be absorbed, and RBC production is eventually diminished. Even if adequate vitamin B12 and intrinsic factor are present, a deficiency may occur if disease involving the ileum or pancreas impairs absorption. Pernicious anemia, which tends to run in families, is primarily a disorder of adults, particularly the elderly. The abnormality is in the gastric mucosa: the stomach wall atrophies and fails to secrete intrinsic factor. Therefore, the absorption of vitamin B12 is significantly impaired. The body normally has large stores of vitamin B12, so years may pass before the deficiency results in anemia. Because the body compensates so well, the anemia can be severe before the patient becomes symptomatic. For unknown reasons, patients with pernicious anemia have a higher incidence of gastric cancer than the general population; these patients should have endoscopies at regular intervals (every 1 to 2 years) to screen for early gastric cancer. Clinical Manifestations Symptoms of folic acid and vitamin B12 deficiencies are similar, and the two anemias may coexist. However, the neurologic manifestations of vitamin B12 deficiency do not occur with folic acid deficiency, and they persist if B12 is not replaced. Therefore, careful distinction between the two anemias must be made. Serum levels of both vitamins can be measured. In the case of folic acid deficiency, even small amounts of folate will increase the serum folate level, sometimes to normal. Measuring the amount of folate within the RBC itself (red cell folate) is therefore a more sensitive test in determining true folate deficiency. After the body stores of vitamin B12 are depleted, patients may begin to show signs of the anemia. However, because the onset and progression of the anemia are so gradual, the body can compensate very well until the anemia is severe, so that the typical manifestations of anemia (weakness, listlessness, fatigue) may not be apparent initially. The hematologic effects of deficiency are accompanied by effects on other organ systems, particularly the gastrointestinal tract and nervous system. Patients with pernicious anemia develop a smooth, sore, red tongue and mild diarrhea. They are extremely pale, particularly in the mucous membranes. They may become confused; more often they have paresthesias in the extremities (particularly numbness and tingling in the feet and lower legs). They may have difficulty maintaining their balance because of damage to the spinal cord, and they also lose position sense (proprioception). These symptoms are progressive, although the course of illness may be marked by spontaneous partial remissions and exacerbations. Without treatment, patients can die after several years, usually from heart failure secondary to anemia. Assessment and Diagnostic Findings The classic method of determining the cause of vitamin B12 deficiency is the Schilling test, in which the patient receives a small oral dose of radioactive vitamin B12, followed in a few hours by a large, nonradioactive parenteral dose of vitamin B12 (this aids in renal excretion of the radioactive dose). If the oral vitamin is absorbed, more than 8% will be excreted in the urine within 24 hours; therefore, if no radioactivity is present in the urine (ie, the radioactive vitamin B12 stays within the gastrointestinal tract), the cause is gastrointestinal malabsorption of the vitamin B12. Conversely, if the urine is radioactive, the cause of the deficiency is not ileal disease or pernicious anemia. Later, the same procedure is repeated, but this time intrinsic factor is added to the oral radioactive vitamin B12. If radioactivity is now detected in the urine (ie, the B12 was absorbed from the gastrointestinal tract in the presence of intrinsic factor), the diagnosis of pernicious anemia can be made. The Schilling test is useful only if the urine collections are complete; therefore, the nurse must promote the patient’s understanding and ability to comply with this collection. Another useful, easier test is the intrinsic factor antibody test. A positive test indicates the presence of antibodies that bind the vitamin B12–intrinsic factor complex and prevent it from binding to receptors in the ileum, thus preventing its absorption. Unfortunately, this test is not specific for pernicious anemia alone, but it can aid in the diagnosis. Medical Management Folate deficiency is treated by increasing the amount of folic acid in the diet and administering 1 mg of folic acid daily. Folic acid is administered intramuscularly only for people with malab- Chapter 33 Assessment and Management of Patients With Hematologic Disorders sorption problems. With the exception of the vitamins administered during pregnancy, most proprietary vitamin preparations do not contain folic acid, so it must be administered as a separate tablet. After the hemoglobin level returns to normal, the folic acid replacement can be stopped. However, patients with alcoholism should continue receiving folic acid as long as they continue alcohol consumption. Vitamin B12 deficiency is treated by vitamin B12 replacement. Vegetarians can prevent or treat deficiency with oral supplements through vitamins or fortified soy milk. When, as is more common, the deficiency is due to defective absorption or absence of intrinsic factor, replacement is by monthly intramuscular injections of vitamin B12, usually at a dose of 1000 µg. The reticulocyte count rises within 1 week, and in several weeks the blood counts are all normal. The tongue improves in several days. However, the neurologic manifestations require more time for recovery; if there is severe neuropathy, the patient may never recover fully. To prevent recurrence of pernicious anemia, vitamin B12 therapy must be continued for life. ! NURSING ALERT Even when the anemia is severe, RBC transfusions should not be used because the patient’s body has compensated over time by expanding the total blood volume. Administration of blood transfusions to such patients, particularly those who are elderly or who have cardiac dysfunction, can precipitate pulmonary edema. If transfusions are required, the RBCs should be transfused slowly, with careful attention to signs and symptoms of fluid overload. Nursing Management Assessment of patients who have or are at risk for megaloblastic anemia includes inspection of the skin and mucous membranes. Mild jaundice may be apparent and is best seen in the sclera without using fluorescent lights. Vitiligo (patchy loss of skin pigmentation) and premature graying of the hair are often seen in patients with pernicious anemia. The tongue is smooth, red, and sore. Because of the neurologic complications associated with these anemias, a careful neurologic assessment is important, including tests of position and vibration sense. PROMOTING HOME AND COMMUNITY-BASED CARE The nurse needs to pay particular attention to ambulation and should assess the patient’s gait and stability as well as the need for assistive devices (eg, canes, walkers) and for assistance in managing daily activities. Of particular concern is ensuring safety when position sense, coordination, and gait are affected. Physical and occupational therapy referrals may be needed. If sensation is altered, patients need to be instructed to avoid excessive heat and cold. Because mouth and tongue soreness may restrict nutritional intake, the nurse can advise patients and families to prepare bland, soft foods and to eat small amounts frequently. The nurse also may explain that other nutritional deficiencies, such as alcohol-induced anemia, can induce neurologic problems. Patients must also be taught about the chronicity of their disorder and the necessity for monthly vitamin B12 injections even in the absence of symptoms. Many patients can be instructed to self-administer their injections. The gastric atrophy associated with pernicious anemia increases the risk of gastric carcinoma, so these patients need to understand that ongoing medical followup and screening are important. 885 MYELODYSPLASTIC SYNDROMES (MDS) The MDSs are a group of disorders of the myeloid stem cell that cause dysplasia (abnormal development) in one or more types of cell lines. The most common feature of MDS—dysplasia of the RBCs—is manifested as a macrocytic anemia; however, the WBCs (myeloid cells, particularly neutrophils) and platelets can also be affected. Although the bone marrow is actually hypercellular, many of the cells within it die before being released into the circulation. Therefore, the number of affected cells in the circulation is typically lower than normal. In addition to the quantitative defect (ie, fewer cells than normal), there is also a qualitative defect: the cells are not as functional as normal. The neutrophils have diminished ability to destroy bacteria by phagocytosis; platelets are less able to aggregate and are less adhesive than usual. The result of these qualitative defects is an increased risk for infection and bleeding, even when the actual number of circulating cells may not be excessively low. A significant proportion of MDS cases evolve into acute myeloid leukemia (AML); this type of leukemia tends to be nonresponsive to standard therapy. Primary MDS tends to be a disease of the elderly; more than 80% of patients with MDS are older than 60 years of age. Secondary MDS may occur at any age and results from prior toxic exposure to chemicals, including chemotherapeutic medications (particularly alkylating agents). Secondary MDS tends to have a poorer prognosis than does primary MDS. Clinical Manifestations The manifestations of MDS can vary widely. Many patients are asymptomatic, with the illness being discovered incidentally when a CBC is performed for other purposes. Other patients have profound symptoms and complications from the illness. Fatigue is often present, at varying levels. Neutrophil dysfunction renders the person at risk for infection; recurrent pneumonias are not uncommon. Because platelet function can also be altered, bleeding can occur. These problems may persist in a fairly steady state for months, even years. They may also progress over time; as the dysplasia evolves into a leukemic state, the complications increase in severity. Assessment and Diagnostic Findings The CBC typically reveals a macrocytic anemia; WBC and platelet counts may be diminished as well. Serum erythropoietin levels may be inappropriately low, as is the reticulocyte count. As the disease evolves into AML, more immature blast cells are noted on the CBC. Medical Management With the exception of allogeneic bone marrow transplantation (BMT), there is no known cure for MDS. Chemotherapy has been used, particularly in patients with more aggressive forms of the illness, typically with disappointing results (Deeg & Applebaum, 2000; Beran, 2000). However, patients with mild cytopenias (low blood counts) actually require no therapy. For most patients with MDS, transfusions of RBCs are required to control the anemia and its symptoms. These patients can develop significant problems with iron overload from the repeated transfusions; this problem can be diminished with prompt initiation of chelation therapy to remove the excess iron (see Nursing Management). In some patients, the use of erythropoietin can be successful in 886 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION reducing the need for transfusions and their attendant complications. Some patients may also require ongoing platelet transfusions to prevent significant bleeding. Infections need to be managed aggressively and promptly. Administration of growth factors, particularly granulocyte colony-stimulating factor (G-CSF), erythropoietin, or both, has been successful in increasing neutrophils and diminishing anemia in certain patients; however, these agents are expensive and the effect is lost if the medications are stopped. Because MDS tend to occur in elderly people, other chronic conditions may limit treatment options. Secondary MDS and MDS that evolves into AML tend to be much more refractory to conventional therapy for leukemia. Nursing Management Caring for patients with MDS can be challenging because the illness is unpredictable. As with other hematologic conditions, some patients (especially those with no symptoms) have difficulty perceiving that they have a serious illness that can place them at risk for life-threatening complications. At the other extreme, many patients have tremendous difficulty coping with the uncertain trajectory of the illness and fear that the illness will evolve into AML at a time when they are feeling very well physically. Patients with MDS need extensive instruction about infection risk, measures to avoid it, signs and symptoms of developing infection, and appropriate actions to initiate should such symptoms occur. Instruction should also be given regarding the risk for bleeding. Patients with MDS who are hospitalized may require neutropenic precautions. Laboratory values need to be monitored closely to anticipate the need for transfusion and to determine response to treatment with growth factors. Patients with chronic transfusion requirements usually benefit from a vascular access device for this purpose. Patients receiving growth factors or chelation therapy must be educated about these medications, their side effects, and administration techniques. Chelation therapy is a process that is used to remove excess iron acquired from chronic transfusions. Iron is bound to a substance, the chelating agent, and then excreted in the urine. Oral forms of chelating agents have not been successful (due to either diminished efficacy or excessive toxicity). Chelation therapy is most effective as a subcutaneous infusion administered over 8 to 12 hours; most patients prefer to do this at night. Because chelation therapy removes only a small amount of iron with each treatment, patients with chronic transfusion requirements (and iron overload) need to continue chelation therapy as long as the iron overload exists, potentially for the rest of their lives. Patients who are embarking on chelation therapy must be highly motivated and need instruction in the subcutaneous infusion technique, infusion pump maintenance, and side effect management. Local erythema at the injection site is the most common reaction and typically requires no intervention. Patients should have baseline and annual auditory and eye examinations, because hearing loss and visual changes can occur with treatment. producing new RBCs and releasing some of them into the circulation somewhat prematurely as reticulocytes. If the RBC destruction persists, the hemoglobin is broken down excessively; about 80% of the heme is converted to bilirubin, conjugated in the liver, and excreted in the bile. The mechanism of RBC destruction varies, but all types of hemolytic anemia share certain laboratory features: the reticulocyte count is elevated, the fraction of indirect (unconjugated) bilirubin is increased, and the supply of haptoglobin (a binding protein for free hemoglobin) is depleted as more hemoglobin is released. As a result, the plasma haptoglobin level is low. If the marrow cannot compensate to replace the RBCs (indicated by a decreased reticulocyte count), the anemia will progress. Hemolytic anemia has various forms. Among the inherited forms are sickle cell anemia, thalassemia and thalassemia major, G-6-PD deficiency, and hereditary spherocytosis. Acquired forms include autoimmune hemolytic anemia, nonimmune-mediated paroxysmal nocturnal hemoglobinuria, microangiopathic hemolytic anemia, and heart valve hemolysis, as well as anemias associated with hypersplenism. SICKLE CELL ANEMIA Sickle cell anemia is a severe hemolytic anemia that results from inheritance of the sickle hemoglobin gene. This gene causes the hemoglobin molecule to be defective. The sickle hemoglobin (HbS) acquires a crystal-like formation when exposed to low oxygen tension. The oxygen level in venous blood can be low enough to cause this change; consequently, the RBC containing (HbS) loses its round, very pliable, biconcave disk shape and becomes deformed, rigid, and sickle-shaped (Fig. 33-5). These long, rigid RBCs can adhere to the endothelium of small vessels; when they pile up against each other, blood flow to a region or an organ may be reduced (Hoffman, et al., 2000). If ischemia or infarction results, the patient may have pain, swelling, and fever. The sickling process takes time; if the RBC is again exposed to adequate amounts of oxygen (eg, when it travels through the pulmonary circulation) before the membrane becomes too rigid, it can revert to a normal shape. For this reason, the “sickling crises” are intermittent. Cold can aggravate Hemolytic Anemias In hemolytic anemias, the RBCs have a shortened life span; thus, the number of RBCs in circulation is reduced. Fewer RBCs result in decreased in available oxygen causes hypoxia, which in turn stimulates an increase in erythropoietin release from the kidney. The erythropoietin stimulates the bone marrow to compensate by FIGURE 33-5 blood cell. A normal red blood cell (upper left ) and a sickled red Chapter 33 Assessment and Management of Patients With Hematologic Disorders the sickling process, because vasoconstriction slows the blood flow. Oxygen delivery can also be impaired by an increased blood viscosity, with or without occlusion due to adhesion of sickled cells; in this situation, the effects are seen in larger vessels, such as arterioles. The HbS gene is inherited in people of African descent and to a lesser extent in people from the Middle East, the Mediterranean area, and aboriginal tribes in India. Sickle cell anemia is the most severe form of sickle cell disease. Less severe forms include sickle cell hemoglobin C (SC) disease, sickle cell hemoglobin D (SD) disease, and sickle cell beta-thalassemia. The clinical manifestations and management are the same as for sickle cell anemia. The term sickle cell trait refers to the carrier state for SC diseases; it is the most benign type of SC disease, in that less than 50% of the hemoglobin within an RBC is HbS. However, in terms of genetic counseling, it is still an important condition. If two people with sickle cell trait have children, the children may inherit two abnormal genes. These children will produce only HbS and therefore will have sickle cell anemia. Virtually any organ may be affected by thrombosis, but the primary sites involve those areas with slowed circulation, such as the spleen, lungs, and central nervous system. All the tissues and organs are constantly vulnerable to microcirculatory interruptions by the sickling process and therefore are susceptible to hypoxic damage or true ischemic necrosis. Patients with sickle cell anemia are unusually susceptible to infection, particularly pneumonia and osteomyelitis. Complications of sickle cell anemia include infection, stroke, renal failure, impotence, heart failure, and pulmonary hypertension. Table 33-4 summarizes the complications resulting from sickle cell anemia. SICKLE CELL CRISIS There are three types of sickle cell crisis in the adult population. The most common is the very painful sickle crisis, which results from tissue hypoxia and necrosis due to inadequate blood flow to a specific region of tissue or organ. Aplastic crisis results from infection with the human parvovirus. The hemoglobin level falls rapidly and the marrow cannot compensate, as evidenced by an absence of reticulocytes. Sequestration crisis results when other organs pool the sickled cells. Although the spleen is the most common organ responsible for sequestration in children, by 10 years of age most children with sickle cell anemia have had a splenic infarction and the spleen is then no longer functional (autosplenectomy). In adults, the common organs involved in sequestration are the liver and, more seriously, the lungs. Clinical Manifestations Symptoms of sickle cell anemia vary and are only somewhat based on the amount of HbS. Symptoms and complications result from chronic hemolysis or thrombosis. The sickled RBCs have a shortened life span. Patients are always anemic, usually with hemoglobin values of 7 to 10 g/dL. Jaundice is characteristic and is usually obvious in the sclerae. The bone marrow expands in childhood in a compensatory effort to offset the anemia, sometimes leading to enlargement of the bones of the face and skull. The chronic anemia is associated with tachycardia, cardiac murmurs, and often an enlarged heart (cardiomegaly). Dysrhythmias and heart failure may occur in adults. Table 33-4 887 ACUTE CHEST SYNDROME Acute chest syndrome is manifested by a rapidly falling hemoglobin level, tachycardia, fever, and bilateral infiltrates seen on the chest x-ray. These signs often mimic infection; in fact, recent studies have identified infection as a major cause of acute chest syndrome (Vichinsky, et al., 2000). Another common cause is pulmonary fat embolism. Increased secretory phospholipase A2 • Summary of Complications in Sickle Cell Anemia* ORGAN INVOLVED MECHANISMS* ASSESSMENT FINDINGS SYMPTOM Spleen Primary site of sickling → infarctions → ↓ phagocytic function of macrophages Infection Infarction → ↑ pulmonary pressure → pulmonary hypertension Infarction Autosplenectomy; ↑ infection (esp. pneumonia, osteomyelitis) Pulmonary infiltrate ↑ sPLA2† Abdominal pain; fever, signs of infection Chest pain; dyspnea CVA (cerebral vascular accident, brain attack) Hematuria; inability to concentrate urine; renal failure Tachycardia; cardiomegaly → heart failure Widening of medullary spaces and cortical thinning Osteosclerosis → avascular necrosis Jaundice and gallstone formation; hepatomegaly Skin ulcers; ↓ wound healing Weakness (if severe); learning difficulties (if mild) Dehydration Scarring, hemorrhage, retinal detachment Priapism → impotence ↓ Vision; blindness Lungs Central Nervous System Kidney Sickling → damage to renal medulla Heart Anemia Bone ↑ Erythroid production Liver Infarction of bone Hemolysis Skin and peripheral vasculature Eye ↑ Viscosity /stasis → infarction → skin ulcers Infarction Penis Sickling * Problems encountered in sickle cell anemia vary and are the result of a variety of mechanisms, as depicted in this table. Common physical findings and symptoms are also variable. † sPLA : Secretory phospholipase A , a laboratory test that can predict impending acute chest syndrome (see text). 2 2 Weakness, fatigue, dyspnea Ache Bone pain, especially hips Abdominal pain Pain Pain, impotence 888 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION concentration has been identified as a predictor of impending acute chest syndrome; the increased amounts of free fatty acids can cause increased permeability of the pulmonary endothelium and leakage of the pulmonary capillaries. Although this syndrome is potentially lethal, prompt intervention can result in a favorable outcome. Assessment and Diagnostic Findings The patient with sickle cell trait usually has a normal hemoglobin level, a normal hematocrit, and a normal blood smear. In contrast, the patient with sickle cell anemia has a low hematocrit and sickled cells on the smear. The diagnosis is confirmed by hemoglobin electrophoresis. Prognosis Patients with sickle cell anemia are usually diagnosed in childhood, because they become anemic in infancy and begin to have sickle cell crises at 1 or 2 years of age. Some children die in the first years of life, typically from infection, but the use of antibiotics and parent teaching have greatly improved the outcomes for these children. However, with current management strategies, the average life expectancy is still suboptimal, at 42 years. Young adults are often forced to live with multiple, often severe, complications from their disease. In some patients, the symptoms and complications diminish by 30 years of age; these patients live into the sixth decade or longer. At this time, there is no way to predict which patients will fall into this subgroup. Medical Management Treatment for sickle cell anemia is the focus of continued research (Steinberg, 1999). Many trials of medications that have antisickling properties are being conducted, as is research using antiadhesion treatment for vasoocclusive crises. However, aside from the equally important aggressive management of symptoms and complications, currently there are only three primary treatment modalities for sickle cell diseases: BMT, hydroxyurea, and longterm RBC transfusion. BMT offers the potential for cure for this disease. However, this treatment modality is available to only a small subset of the patient population, because of either the lack of a compatible donor or the severe organ (eg, renal, liver, lung) damage already present in the patient. PHARMACOLOGIC THERAPY Hydroxyurea (Hydrea), a chemotherapy agent, has been shown to be effective in increasing hemoglobin F levels in patients with sickle cell anemia, thereby decreasing the permanent formation of sickled cells. Patients who receive hydroxyurea appear to have fewer painful episodes of sickle cell crisis, a lower incidence of acute chest syndrome, and less need for transfusions (Ferster et al., 2001). However, whether hydroxyurea can prevent or reverse actual organ damage remains unknown. Side effects of hydroxyurea include chronic suppression of WBC formation, teratogenesis, and potential for later development of a malignancy. Patient response to the medication varies significantly. The incidence and severity of side effects are also highly variable within a dose range. Some patients have toxicity when receiving a very small dose (5 mg/kg per day), whereas others have little toxicity with a much higher dose (35 mg/kg per day). More research is needed to identify specific patient subgroups that are more likely to respond to this medication. TRANSFUSION THERAPY Chronic transfusions with RBCs have been shown to be highly effective in several situations: in an acute exacerbation of anemia (eg, aplastic crisis), in the prevention of severe complications from anesthesia and surgery, and in improving the response to infection (when it results in exacerbated anemia) (Ohene-Frempong, 2001). Chronic transfusions have also been shown to be effective in diminishing episodes of sickle cell crisis in pregnant women; however, these transfusions have not been shown to improve fetal survival. Transfusion therapy may be effective in preventing complications from sickle cell disease. Although controversial, some data support the use of chronic transfusions in patients with cerebral ischemic injury (as seen on magnetic resonance imaging [MRI] or Doppler studies) to prevent more severe injury (eg, CVA). More than 50% of asymptomatic patients have some cerebral ischemia documented by MRI. In a recent study (Adams, 2000), chronic transfusion with RBCs resulted in a 90% reduction of stroke in children at risk for this complication, as demonstrated by elevated blood viscosity on transcranial Doppler ultrasonography. Transfusions may also be useful in the management of severe cases of acute chest syndrome. The risk of complications from transfusion is important to consider. These risks include iron overload, which necessitates chronic chelation therapy (see MDS Nursing Management); poor venous access, which necessitates a vascular access device (and its attendant risk for infection or thrombosis); infections (hepatitis, human immunodeficiency virus [HIV]); and alloimmunization from repeated transfusions. Another complication from transfusion is the increased viscosity of blood before the concentration of hemoglobin S is reduced. Exchange transfusion (in which the patient’s own blood is removed and replaced via transfusion) may be performed to diminish the risk of increasing the viscosity excessively; the objective is to reduce the hematocrit to less than 30%, with transfusions supplying more than 80% of the patient’s blood volume. Finally, it is important to consider the significant financial cost of an aggressive transfusion and chelation program. Patients with sickle cell anemia require daily folic acid replacements to maintain the supply required for increased erythropoiesis from hemolysis. Infections must be treated promptly with appropriate antibiotics; infection remains a major cause of death in these patients. Acute chest syndrome is managed by prompt initiation of antibiotic therapy. Incentive spirometry has been shown to decrease the incidence of pulmonary complications significantly. In severe cases, bronchoscopy may be required to identify the source of pulmonary disease. Fluid restriction may be more beneficial than aggressive hydration. Corticosteroids may also be useful. Transfusions reverse the hypoxia and decrease the level of secretory phospholipase A2. Pulmonary function should be monitored regularly to detect pulmonary hypertension early, when therapy (hydroxyurea, transfusions, or transplantation) may have a positive impact. Because repeated blood transfusions are necessary, patients may develop multiple autoantibodies, making cross-matching difficult. In this patient population, a hemolytic transfusion reaction (see later discussion) may mimic the signs and symptoms of a sickle cell crisis. The classic distinguishing factor is that, with a hemolytic transfusion reaction, the patient becomes more anemic after being transfused. These patients need very close observation. Further transfusion is avoided if possible until the hemolytic process abates. If possible, the patient is supported with corticosteroids (Prednisone), intravenous immunoglobulin (IVIG; Gammagard, Sandoglobulin, Venoglobulin), and erythropoietin (Epogen, Procrit). Chapter 33 Assessment and Management of Patients With Hematologic Disorders 889 SUPPORTIVE THERAPY Supportive care is equally important. A significant issue is pain management. The incidence of painful sickle cell crises is highly variable; many patients have pain on a daily basis. The severity of the pain may not be enough to cause the patient to seek assistance from health care providers but severe enough to interfere with the ability to work and function within the family. Acute pain episodes tend to be self-limited, lasting hours to days. If the patient cannot manage the pain at home, intervention is frequently sought in the acute care setting, usually at an urgent care facility or emergency department. Adequate hydration is important during a painful sickling episode. Oral hydration is acceptable if the patient can maintain adequate amounts of fluids; intravenous hydration with dextrose 5% in water (D5W) or dextrose 5% in 0.25 normal saline solution (3 L/m2/24 hours) is usually required for sickle crisis. Supplemental oxygen may also be needed. The use of medication to relieve pain is important (see Chap. 13 for a discussion of pain management). Aspirin is very useful in diminishing mild to moderate pain; it also diminishes inflammation and potential thrombosis (due to its ability to diminish platelet adhesion). Nonsteroidal anti-inflammatory drugs (NSAIDs) are useful for moderate pain or in combination with opioid analgesics. Although no tolerance develops with NSAIDs, a “ceiling effect” does develop whereby an increase in dosage does not increase analgesia. NSAID use must be carefully monitored, because these medications can precipitate renal dysfunction. When opioid analgesics are used, morphine is the medication of choice for acute pain. Patient-controlled analgesia is frequently used. Chronic pain increases in incidence as the patient ages. Here, the pain is caused by complications from the sickling, such as avascular necrosis of the hip. With chronic pain management, the principal goal is to maximize functioning; pain may not be completely eliminated without sacrificing function. This concept may be difficult for patients to accept; they may need repeated explanations and support from nonjudgmental health care providers. Nonpharmacologic approaches to pain management are crucial in this setting. Examples include physical and occupational therapy, physiotherapy (including the use of heat, massage, and exercise), cognitive and behavioral intervention (including distraction, relaxation, and motivational therapy), and support groups. Working with patients who have multiple episodes of severe pain can be challenging. It is important for health care providers to realize that patients with sickle cell disease must face a lifelong experience with severe and unpredictable pain. Such pain is disruptive to the person’s level of functioning, including social functioning, and may result in a feeling of helplessness. Patients with inadequate social support systems may have more difficulty coping with chronic pain. are included in this assessment. If a sickle cell crisis is suspected, the nurse needs to determine whether the pain currently experienced is the same as or different than the pain typically encountered in crisis. Because the sickling process can interrupt circulation in any tissue or organ, with resultant hypoxia and ischemia, a careful assessment of all body systems is necessary. Particular emphasis is placed on assessing for pain, swelling, and fever. All joint areas are carefully examined for pain and swelling. The abdomen is assessed for pain and tenderness because of the possibility of splenic infarction. The respiratory system must be assessed carefully, including auscultation of breath sounds, measurement of oxygen saturation levels, and signs of cardiac failure, such as the presence and extent of dependent edema, an increased point of maximal impulse, and cardiomegaly (as seen on chest x-ray). The patient should be assessed for signs of dehydration by a history of fluid intake and careful examination of mucous membranes, skin turgor, urine output, and serum creatinine and blood urea nitrogen values. A careful neurologic examination is important to elicit symptoms of cerebral hypoxia. However, ischemic findings on MRI or Doppler studies may significantly precede the findings on the physical examination. MRI and Doppler studies are used for early diagnosis and may be more beneficial to improve patient outcome, because therapy can be initiated more promptly. Because patients with sickle cell anemia are so susceptible to infections, they are assessed for the presence of any infectious process. Particular attention is given to examination of the chest, long bones, and femoral head, because pneumonia and osteomyelitis are especially common. Leg ulcers, which may be infected and are slow to heal, are common. The extent of anemia (as measured by the hemoglobin level and the hematocrit) and the ability of the marrow to replenish RBCs (as measured by the reticulocyte count) should be monitored and compared with the patient’s baseline values. The patient’s current and past history of medical management should also be assessed, particularly chronic transfusion therapy, hydroxyurea use, and prior treatment for infection. NURSING PROCESS: THE PATIENT WITH SICKLE CELL CRISIS COLLABORATIVE PROBLEMS/ POTENTIAL COMPLICATIONS Based on the assessment data, potential complications may include: Patients in sickle cell crisis should be assessed for factors that could have precipitated the crisis, such as symptoms of infection or dehydration, or situations that promote fatigue or emotional stress. Assessment Patients are asked to recall factors that seemed to precipitate previous crises and measures they use to prevent and manage crises. Pain levels should always be monitored; a pain-rating scale, such as a 0-to-10 scale, best accomplishes this. The quality of the pain (eg, sharp, dull, burning), the frequency of the pain (constant versus intermittent), and factors that aggravate or alleviate the pain Diagnosis NURSING DIAGNOSES Based on the assessment data, major nursing diagnoses for the patient with sickle cell crisis may include: • Acute pain related to tissue hypoxia due to agglutination of sickled cells within blood vessels • Risk for infection • Risk for powerlessness related to illness-induced helplessness • Deficient knowledge regarding sickle crisis prevention • Hypoxia, ischemia, infection, and poor wound healing lead• • • • • • • • ing to skin breakdown and ulcers Dehydration Cerebrovascular accident (CVA, brain attack, stroke) Anemia Renal dysfunction Heart failure, pulmonary hypertension, and acute chest syndrome Impotence Poor compliance Substance abuse related to poorly managed chronic pain 890 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Planning and Goals The major goals for the patient are relief of pain, decreased incidence of crisis, enhanced sense of self-esteem and power, and absence of complications. Nursing Interventions MANAGING PAIN Acute pain during a sickle cell crisis can be severe and unpredictable. The patient’s subjective description and rating of pain on a pain scale must guide the use of analgesics, which are valuable in controlling the acute pain of a sickle crisis. Any joint that is acutely swollen should be supported and elevated until the swelling diminishes. Relaxation techniques, breathing exercises, and distraction are helpful for some patients. After the acute painful episode has diminished, aggressive measures should be implemented to preserve function. Physical therapy, whirlpool baths, and transcutaneous nerve stimulation are examples of such modalities. PREVENTING AND MANAGING INFECTION Nursing care focuses on monitoring the patient for signs and symptoms of infection. Prescribed antibiotics should be initiated promptly, and the patient should be assessed for signs of dehydration. If the patient is to take prescribed oral antibiotics at home, he or she must understand the need to complete the entire course of antibiotic therapy and must be able to identify a feasible administration schedule. PROMOTING COPING SKILLS This illness, because of its acute exacerbations that often result in chronic health problems, frequently leaves the patient feeling powerless and with decreased self-esteem. These feelings can be exacerbated by inadequate pain management. The patient’s ability to use normal coping resources of physical strength, psychological stamina, and positive self-esteem is dramatically diminished. Enhancing pain management can be extremely useful in establishing a therapeutic relationship based on mutual trust. Nursing care that focuses on the patient’s strengths rather than deficits can enhance effective coping skills. Providing the patient with opportunities to make decisions about daily care may increase the patient’s feelings of control. MINIMIZING DEFICIENT KNOWLEDGE Patients with sickle cell anemia benefit from understanding what situations can precipitate a sickle cell crisis and the steps they can take to prevent or diminish such crises. Keeping warm and maintaining adequate hydration can be very effective in diminishing the occurrence and severity of attacks. Avoiding stressful situations is more challenging. Group education may be more effective if it is carried out by members of the community who are from the same ethnic group as those with the disease. MONITORING AND MANAGING POTENTIAL COMPLICATIONS Management measures for many of the potential complications were delineated in previous sections. Other measures follow. Leg Ulcers Leg ulcers require careful management and protection from trauma and contamination. Referral to a wound care specialist may facilitate healing and assist with prevention. If leg ulcers fail to heal, skin grafting may be necessary. Scrupulous aseptic technique is warranted to prevent nosocomial infections. Priapism Leading to Impotence Male patients may develop sudden, painful episodes of priapism (persistent penile erection). The patient is taught to empty his bladder at the onset of the attack, exercise, and take a warm bath. If an episode persists longer than 3 hours, medical attention is recommended. Repeated episodes may lead to extensive vascular thrombosis, resulting in impotence. Chronic Pain and Substance Abuse Many patients have considerable difficulty coping with chronic pain and repeated episodes of sickle crisis. Those who feel they have little control over their health and the physical complications that result from this illness may find it difficult to understand the importance of complying with a prescribed treatment plan. Being nonjudgmental and actively seeking involvement from the patient in establishing a treatment plan are useful strategies. Some patients with sickle cell anemia develop problems with substance abuse. For many, this abuse results from inadequate management of acute pain during episodes of crisis. Some clinicians suggest that abuse may result from prescribing inadequate amounts of opioid analgesics for an inadequate time. The patient’s pain may never be adequately relieved, promoting mistrust of the health care system and (from the patient’s perspective) the need to seek care from a variety of sources when the pain is not severe. This cycle is best managed by prevention. Receiving care from a single provider over time is much more beneficial than receiving care from rotating physicians and staff in an emergency department. When crises do arise, the staff in the emergency department should be in contact with the patient’s primary health care provider so that optimal management can be achieved. Once the pattern of substance abuse is established, it is very difficult to manage, but continuity of care and establishing written contracts with the patient can be useful management strategies. PROMOTING HOME AND COMMUNITY-BASED CARE Teaching Patients Self-Care Because patients with sickle cell anemia are typically diagnosed as children, parents participate in the initial education. Based on the parents’ education, literacy, socioeconomic level, and interest, teaching focuses on the disease process (including some pathophysiology), treatment, and the assessment and monitoring skills for potential complications (see previous discussion). As the child ages, educational interventions with the child prepare the child to assume more responsibility for self-care. Vascular access device management and chelation therapy can be taught to most families. Follow-up and care for patients with vascular access devices may also need to be provided by nurses in an outpatient facility or by a home care agency. Continuing Care The illness trajectory of sickle cell anemia is highly varied, with unpredictable episodes of complications and crises. Care is often provided on an emergency basis, especially for some patients with pain management problems (see previous section). Nurses in all settings used by this patient population need to communicate regularly with each other. Patients need to learn which parameters are important for them to monitor and how Chapter 33 Assessment and Management of Patients With Hematologic Disorders to monitor them. Parameters should also be given as to when to seek urgent care. Evaluation EXPECTED PATIENT OUTCOMES Expected patient outcomes may include: 1. Control of pain a. Acute pain is controlled with analgesics b. Uses relaxation techniques, breathing exercises, distraction to help relieve pain 2. Is free of infection a. Has normal temperature b. Shows WBC count within normal range (5000 to 10,000/mm3) c. Identifies importance of continuing antibiotics at home (if applicable) 3. Expresses improved sense of control a. Participates in goal setting and in planning and implementing daily activities b. Participates in decisions about care 4. Increases knowledge about disease process a. Identifies situations and factors that can precipitate sickle cell crisis b. Describes lifestyle changes needed to prevent crisis c. Describes the importance of warmth, adequate hydration, and prevention of infection in preventing crisis 5. Absence of complications THALASSEMIA The thalassemias are a group of hereditary disorders associated with defective hemoglobin-chain synthesis. These anemias occur worldwide, but the highest prevalence is found in people of Mediterranean, African, and Southeast Asian ancestry (Hoffman et al., 2000). Thalassemias are characterized by hypochromia (an abnormal decrease in the hemoglobin content of RBCs), extreme microcytosis (smaller-than-normal RBCs), destruction of blood elements (hemolysis), and variable degrees of anemia. In thalassemia, the production of one or more globulin chains within the hemoglobin molecule is reduced. When this occurs, the imbalance in the configuration of the hemoglobin causes it to precipitate in the erythroid precursors or the RBCs themselves. This increases the rigidity of the RBCs and thus the premature destruction of these cells. The thalassemias are classified into two major groups according to the globin chain diminished: alpha and beta. The alphathalassemias occur mainly in people from Asia and the Middle East; the beta-thalassemias are most prevalent in Mediterranean populations but also occur in people from the Middle East or Asia. The alpha-thalassemias are milder than the beta forms and often occur without symptoms. The RBCs are extremely microcytic, but the anemia, if present, is mild. The severity of beta-thalassemia varies depending on the extent to which the hemoglobin chains are affected. Patients with mild forms have a microcytosis and mild anemia. If left untreated, severe beta-thalassemia (thalassemia major, or Cooley’s anemia) can be fatal within the first few years of life. If it is treated with regular transfusion of RBCs, patients may survive into their 20s and 30s. Patient teaching during the reproductive years should include pre-conception counseling about the risk of congenital thalassemia major. 891 Thalassemia Major Thalassemia major (Cooley’s anemia) is characterized by severe anemia, marked hemolysis, and ineffective erythropoiesis (production of RBCs). With early regular transfusion therapy, growth and development through childhood are facilitated. Organ dysfunction due to iron overload results from the excessive amounts of iron obtained through the RBC transfusions. Regular chelation therapy (eg, via subcutaneous deferoxamine) has reduced the complications of iron overload and prolonged the life of these patients. This disease is potentially curable by BMT if the procedure can be performed before damage to the liver occurs (ie, during childhood). GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY The abnormality in this disorder is in the G-6-PD gene; this gene produces an enzyme within the RBC that is essential for membrane stability. A few patients have inherited an enzyme so defective that they have a chronic hemolytic anemia; however, the most common type of defect results in hemolysis only when the RBCs are stressed by certain situations, such as fever or the use of certain medications. The disorder came to the attention of researchers during World War II, when some soldiers developed hemolysis while taking primaquine, an antimalarial agent. African Americans and people of Greek or Italian origin are those primarily affected by this disorder. The type of deficiency found in the Mediterranean population is more severe than that in the African Caribbean population, resulting in greater hemolysis and sometimes in life-threatening anemia. All types of G-6-PD deficiency are inherited as X-linked defects; therefore, many more men are at risk than women. In the United States, about 12% of African American males are affected. The deficiency is also common in those of Asian ancestry and in certain Jewish populations. Medications that have hemolytic effects for people with G-6-PD deficiency are oxidant drugs. These medications include antimalarial agents (eg, chloroquine [Aralen]), sulfonamides (eg, trimethoprim and sulfamethoxazole [Septra]), nitrofurantoin (eg, Macrodantin), common coal tar analgesics (including aspirin in high doses), thiazide diuretics (eg, hydrochlorothiazide [HydroDIURIL], chlorothiazide [Diuril]), oral hypoglycemic agents (eg, glyburide [Micronase], metformin [Glucophage]), chloramphenicol (Chloromycetin), and vitamin K (phytonadione [AquaMephyton]). In affected people, a severe hemolytic episode can result from ingestion of fava beans. Clinical Manifestations Patients are asymptomatic and have normal hemoglobin levels and reticulocyte counts most of the time. However, several days after exposure to an offending medication, they may develop pallor, jaundice, and hemoglobinuria (hemoglobin in the urine). The reticulocyte count rises, and symptoms of hemolysis develop. Special stains of the peripheral blood may then disclose Heinz bodies (degraded hemoglobin) within the RBCs. Hemolysis is often mild and self-limited. However, in the more severe Mediterranean type of G-6-PD deficiency, spontaneous recovery may not occur and transfusions may be necessary. Assessment and Diagnostic Findings The diagnosis is made by a screening test or by a quantitative assay of G-6-PD. 892 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Medical Management The treatment is to stop the offending medication. Transfusion is necessary only in the severe hemolytic state, which is more commonly seen in the Mediterranean variety of G-6-PD deficiency. Nursing Management The patient should be educated about the disease and given a list of medications to avoid. If hemolysis does develop, nursing interventions are the same as for hemolysis from other causes. HEREDITARY SPHEROCYTOSIS Hereditary spherocytosis is a relatively common (1 in 5,000 people) hemolytic anemia characterized by an abnormal permeability of the RBC membrane; this permits the cells to change into a spherical shape. These RBCs are destroyed prematurely in the spleen. The severity of this hemolytic anemia varies; jaundice can be intermittent, and splenomegaly (enlarged spleen) also can occur. Surgical removal of the spleen is the principal treatment for this disorder. IMMUNE HEMOLYTIC ANEMIA Hemolytic anemias can result from exposure of the RBC to antibodies. Alloantibodies (ie, antibodies against the host, or “self”) result from the immunization of an individual with foreign antigens (eg, the immunization of an Rh-negative person with Rh-positive blood). Alloantibodies tend to be large (IgM type) and cause immediate destruction of the sensitized RBCs, either within the blood vessel (intravascular hemolysis) or within the liver. The most common type of alloimmune hemolytic anemia in adults results from a hemolytic transfusion reaction. Autoantibodies are developed by an individual for varying reasons. In many instances, the person’s immune system is dysfunctional, so that it falsely recognizes its own RBCs as foreign and produces antibodies against them. This mechanism is seen in people with chronic lymphocytic leukemia (CLL). Another mechanism is a deficiency in suppressor lymphocytes, which normally prevent antibody formation against a person’s own antigens. Autoantibodies tend to be of the IgG type. The RBCs are sequestered in the spleen and destroyed by the macrophages outside the blood vessel (extravascular hemolysis). Autoimmune hemolytic anemias can be classified based on the body temperature involved when the antibodies react with the RBC antigen. Warm-body antibodies bind to RBCs most actively in warm conditions (37°C); cold-body antibodies react in cold (0°C). Most autoimmune hemolytic anemias are the warm-body type. Autoimmune hemolytic anemia is associated with other disorders in most cases (eg, medication exposure, lymphoma, CLL, other malignancy, collagen vascular disease, autoimmune disease, infection). In idiopathic autoimmune hemolytic states, the reason why the immune system produces the antibodies is not known. All ages and genders are equally vulnerable to this form, whereas the incidence of secondary forms is greater in people older than 45 years of age and in females. Clinical Manifestations Clinical manifestations can vary, and they usually reflect the degree of anemia. The hemolysis may be very mild, so that the patient’s marrow compensates adequately and the patient is asymptomatic. At the other extreme, the hemolysis can be so severe that the resul- tant anemia is life-threatening. Most patients complain of fatigue and dizziness. Splenomegaly is the most common physical finding, occurring in more than 80% of patients; hepatomegaly, lymphadenopathy, and jaundice are also common. Assessment and Diagnostic Findings The laboratory tests show a low hemoglobin level and hematocrit, most often with an accompanying increase in the reticulocyte count. RBCs appear abnormal; spherocytes are common. The serum bilirubin level is elevated, and if the hemolysis is severe, the haptoglobin level is low or absent. The Coombs test (also referred to as the direct antiglobulin test [DAT]), which detects antibodies on the surface of RBCs, shows a positive result. Medical Management Any possibly offending medication should be immediately discontinued. The treatment consists of high doses of corticosteroids (1 mg/kg per day) until hemolysis decreases. Corticosteroids decrease the macrophage’s ability to clear the antibody-coated RBCs. If the hemoglobin level returns toward normal, usually after several weeks, the corticosteroid dose can be lowered or, in some cases, tapered and discontinued. However, corticosteroids rarely produce a lasting remission. In severe cases, blood transfusions may be required. Because the antibody may react with all possible donor cells, careful blood typing is necessary, and the transfusion should be administered slowly and cautiously. Splenectomy (removal of the spleen) removes the major site of RBC destruction; therefore, splenectomy may be performed if corticosteroids do not produce a remission. If neither corticosteroid therapy nor splenectomy is successful, immunosuppressive agents may be administered. The two immunosuppressive agents most frequently used are cyclophosphamide (eg, Cytoxan), which has a more rapid effect but more toxicity, or azathioprine (Imuran), which has a less rapid effect but less toxicity. The synthetic androgen danazol (Cyclomen, Danocrine) can be useful in some patients, particularly in combination with corticosteroids. The mechanism for this success is unclear. If corticosteroids or immunosuppressive agents are used, the taper must be very gradual to prevent a rebound “hyperimmune” response and exacerbation of the hemolysis. Immunoglobulin administration is effective in about one third of patients, but the effect is transient and the medication is expensive. Transfusions may be necessary if the anemia is severe; it may be extremely difficult to cross-match samples of available units of RBCs with that of the patient. For patients with cold-antibody hemolytic anemia, treatment may not be required, other than to advise the patient to keep warm; relocation to a warm climate may be necessary. Nursing Management Patients may have great difficulty understanding the pathologic mechanisms underlying the disease and need repeated explanations in terms they can understand. Patients who have had a splenectomy should be vaccinated against pneumococcal infections (Pneumovax) and informed that they are permanently at greater risk for infection. Patients receiving long-term corticosteroid therapy, particularly those with concurrent diabetes or hypertension, need careful monitoring. They must understand the need for this medication and the importance of never abruptly discontinuing it. A written explanation and a tapering schedule should be provided, and adjustments based on hemoglobin levels Chapter 33 Assessment and Management of Patients With Hematologic Disorders should be emphasized. Similar teaching should be provided when immunosuppressive agents are used. Corticosteroid therapy is not without significant risk, and patients need to be monitored closely for complications. The short- and long-term complications of corticosteroid therapy are presented in Chart 33-4 and in Chap. 42. ! NURSING ALERT It can be difficult to cross-match blood when antibodies are present. If imperfectly cross-matched RBCs must be transfused, the nurse begins the infusion very slowly (10 to 15 mL over 20 to 30 minutes) and monitors the patient very closely for signs and symptoms of a hemolytic transfusion reaction. HEREDITARY HEMOCHROMATOSIS Hemochromatosis is a genetic condition in which iron is abnormally (excessively) absorbed from the gastrointestinal tract. The excessive iron is deposited in various organs, particularly the liver, Chart 33-4 893 myocardium, testes, thyroid, and pancreas. Eventually, the affected organs become dysfunctional. The actual incidence of hemochromatosis is unknown; however, hereditary hemochromatosis is diagnosed in 0.5% of the population in the United States (ie, 1 million people). Recent data suggest that this defect may be a common cause of diabetes (Schechter, et al., 2000). Because of to their natural loss of iron through menses, women are less affected than men. Because the accumulation of iron in body organs occurs gradually, there often is no evidence of tissue injury until middle age. Symptoms of weakness, lethargy, arthralgia, weight loss, and loss of libido are common. The skin may be hyperpigmented with melanin deposits (occasionally hemosiderin, an iron-containing pigment) and appears bronze in color. Cardiac dysrhythmias and cardiomyopathy can occur, with resulting dyspnea and edema. Endocrine dysfunction is manifested as hypothyroidism, diabetes mellitus, and hypogonadism (testicular atrophy, diminished libido, and impotence). A significant effect of hemochromatosis is the • PHARMACOLOGY Complications Associated with Corticosteroid Therapy Whenever a patient begins a course of corticosteroid therapy, the potential for complications is great. Dosing regimens vary widely, depending on the underlying hematologic condition and the patient’s response to the medication. For example, several chemotherapy protocols include high doses of corticosteroids for a period of several days. After that time, the medication is stopped abruptly without tapering the dosage. In other conditions, such as idiopathic thrombocytopenic purpura or hemolytic anemias, the corticosteroids are very carefully tapered to prevent a flare up of the underlying disease. With the exception of patients with preexisting conditions such as diabetes, hypertension, and osteoporosis, it is difficult to predict which complications will occur in a given patient. Patients who receive high doses of corticosteroids for longer than a few weeks should be screened for symptoms related to the potential complications listed here. If at all possible, patients who require long-term corticosteroid use should be switched to an alternate-day dosing schedule; this method may diminish the severity of complications that arise. Short-Term Complications Fluid and Electrolyte Complications Fluid retention Sodium retention Potassium loss Hypokalemic alkalosis Hypertension Endocrine Complications Decreased carbohydrate tolerance Diabetes mellitus Uncontrolled glucose levels in diabetes mellitus Neurologic Complications Headache Musculoskeletal Complications Muscle weakness Psychologic Complications Depression Euphoria Mood swings Insomnia Psychosis Long-Term Complications Endocrine Complications Decreased adrenocortical activity Decreased ability to respond to stress Decreased carbohydrate tolerance Decreased growth rate (children) Cushingoid state Menstrual irregularities Increased sweating Metabolic Complications Protein catabolism causing negative nitrogen balance Gastrointestinal Complications Gastritis Ulcerative esophagitis Peptic ulcer Pancreatitis Musculoskeletal Complications Decreased muscle mass Osteoporosis Vertebral compression fracture Pathologic fracture Aseptic necrosis of femoral and humeral heads Neurologic Complications Vertigo Increased intracranial pressure Seizures Ophthalmic Complications Cataract formation Glaucoma Exophthalmos Dermatologic Complications Impaired wound healing Ecchymoses Increased skin fragility Decreased skin thickness Petechiae Immunologic Complications Decreased response to infection Masked signs of (early stages of infection) Suppressed reaction to skin tests Increased risk for opportunistic infection (eg, Pneumocystis, herpes zoster) 894 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION development of hepatocellular carcinoma in one third of those affected. CBC values are typically normal. The most useful laboratory findings are an elevated serum iron level and high transferrin saturation (more than 60% in men, more than 50% in women). The definitive diagnostic test is a liver biopsy. Recently, a mutation in the HFE gene has been shown to occur in most patients with hereditary hemochromatosis (Gochee & Powell, 2001). Patients who are homozygous for the gene are at high risk for development of the disorder. Medical Management Therapy involves the removal of excess iron via therapeutic phlebotomy (removal of whole blood from a vein). Each unit of blood removed results in a decrease of 200–250 mg of iron. The objective typically is to reduce the serum ferritin to less than 50 µg/L and the transferrin saturation to 35% or less. To achieve this, a frequent phlebotomy schedule is required (1 to 2 units weekly), with a gradual reduction in frequency of phlebotomies over a 1- to 3-year period. After 1 to 3 years, the frequency of phlebotomy can be reduced to 1 unit of blood every several months to prevent reaccumulation of iron deposits. Removal of excess iron appears to diminish the severity of diabetes and skin hyperpigmentation; cardiac function also tends to improve. Nursing Management Patients with hemochromatosis often believe that it is important to limit their dietary intake of iron, although this management method has been shown to be very ineffective and need not be encouraged. However, it is important for these patients to avoid any additional insults to the liver, such as alcohol abuse. Serial screening tests for hepatoma are important; alpha-fetoprotein is used for this purpose. Other body systems should be monitored for signs of organ dysfunction, particularly the endocrine and cardiac systems. These systems should also be screened routinely for dysfunction so that appropriate management can be implemented quickly. Because patients with hemochromatosis require frequent phlebotomies, problems with venous access are common. Patients who are heterozygous for the HFE do not develop the disease but need to be counseled that they can transmit the gene to their children. The Polycythemias Polycythemia refers to an increased volume of RBCs. It is a term used when the hematocrit is elevated (to more than 55% in males, more than 50% in females). Dehydration (decreased volume of plasma) can cause an elevated hematocrit, but not typically to the level to be considered polycythemia. Polycythemia is classified as either primary or secondary. POLYCYTHEMIA VERA Polycythemia vera, or primary polycythemia, is a proliferative disorder in which the myeloid stem cells seem to have escaped normal control mechanisms. The bone marrow is hypercellular, and the RBC, WBC, and platelet counts in the peripheral blood are elevated. However, the RBC elevation is predominant; the hematocrit can exceed 60%. This phase can last for an extended period (10 years or longer). The spleen resumes its embryonic function of hematopoiesis and enlarges. Over time, the bone marrow may become fibrotic, with a resultant inability to produce as many cells (“burnt out” or spent phase). The disease evolves into myeloid metaplasia with myelofibrosis or AML in a significant proportion of patients; this form of AML is usually refractory to standard treatments (Hoffman, et al., 2000). The median survival time exceeds 15 years (Gruppo Italiano Studio Policitemia, 1995). Clinical Manifestations Patients typically have a ruddy complexion and splenomegaly (enlarged spleen). The symptoms result from the increased blood volume (headache, dizziness, tinnitus, fatigue, paresthesias, and blurred vision) or from increased blood viscosity (angina, claudication, dyspnea, and thrombophlebitis), particularly if the patient has atherosclerotic blood vessels. Another common and bothersome problem is generalized pruritus, which may be caused by histamine release due to the increased number of basophils. Erythromelalgia, a burning sensation in the fingers and toes, may be reported and is only partially relieved by cooling. Assessment and Diagnostic Findings Diagnosis is made by finding an elevated RBC mass (a nuclear medicine procedure), a normal oxygen saturation level, and an enlarged spleen. Other factors useful in establishing the diagnosis include elevated WBC and platelet counts. The erythropoietin level is not as low as would be expected with an elevated hematocrit; it is normal or only slightly low. Causes of secondary erythrocytosis should not be present (see later discussion). Complications Patients with polycythemia vera are at increased risk for thromboses resulting in a CVA (brain attack, stroke) or heart attack (MI); thrombotic complications are the most frequent cause of death. Bleeding is also a complication, possibly due to the fact that the platelets (often very large) are somewhat dysfunctional. The bleeding can be significant and can occur in the form of nosebleeds, ulcers, and frank gastrointestinal bleeding. Medical Management The objective of management is to reduce the high blood cell mass. Phlebotomy is an important part of therapy and can be performed repeatedly to keep the hematocrit within normal range. This is achieved by removing enough blood (initially 500 mL once or twice weekly) to deplete the patient’s iron stores, thereby rendering the patient iron deficient and consequently unable to continue to manufacture RBCs excessively. Patients need to be instructed to avoid iron supplements, including those within multivitamin supplements. If the patient has an elevated uric acid concentration, allopurinol (Zyloprim) is used to prevent gouty attacks. Antihistamines are not particularly effective in controlling itching. If the patient develops ischemic symptoms, dipyridamole (eg, Persantine) is sometimes used. Radioactive phosphorus (32P) or chemotherapeutic agents (eg, hydroxyurea [Hydrea]) can be used to suppress marrow function, but they may increase the risk for leukemia. Patients receiving hydroxyurea appear to have a lower incidence of thrombotic complications; this may result from a more controlled platelet count. The use of aspirin to prevent thrombotic complications is controversial. Lowdose aspirin is frequently used in patients with cardiovascular disease, but even this dose is often avoided in patients with prior bleeding, especially bleeding from the gastrointestinal tract. Aspirin Chapter 33 Assessment and Management of Patients With Hematologic Disorders is also useful in diminishing pain associated with erythromelalgia. Anagrelide (Agrylin) inhibits platelet aggregation and can also be useful in controlling the thrombocytosis associated with polycythemia vera. Interferon alfa-2b (Intron-A) has also been studied, but it may be difficult for patients to tolerate due to the frequent side effects experienced (Tefferi et al., 2000; Lengfelder, Berger, & Hehlmann, 2000). Nursing Management The nurse’s role is primarily that of educator. Risk factors for thrombotic complications should be assessed, and patients should be instructed regarding the signs and symptoms of thrombosis. Patients with a history of bleeding are usually advised to avoid aspirin and aspirin-containing medications, because these medications alter platelet function. Minimizing alcohol intake should also be emphasized to further diminish any risk for bleeding. For pruritus, the nurse may recommend bathing in tepid or cool water, along with applications of cocoa butter–based lotions and bath products. Chart 33-5 895 Causes of Neutropenia Decreased Production of Neutrophils • Aplastic anemia, due to medications or toxins • Metastatic cancer, lymphoma, leukemia • Myelodysplastic syndromes • Chemotherapy • Radiation therapy Ineffective Granulocytopoiesis • Megaloblastic anemia Increased Destruction of Neutrophils • Hypersplenism • Medication-induced* • Immunologic disease (eg, systemic lupus erythematosus [SLE]) • Viral disease (eg, infectious hepatitis, mononucleosis) • Bacterial infections *Formation of antibody to medication, leading to a rapid decrease in neutrophils. SECONDARY POLYCYTHEMIA Secondary polycythemia is caused by excessive production of erythropoietin. This may occur in response to a reduced amount of oxygen, which acts as a hypoxic stimulus, as in cigarette smoking, chronic obstructive pulmonary disease, or cyanotic heart disease, or in nonpathologic conditions such as high altitude. It can also result from certain hemoglobinopathies in which the hemoglobin has an abnormally high affinity for oxygen (eg, hemoglobin Chesapeake). Secondary polycythemia can also occur from neoplasms (eg, renal cell carcinoma) that stimulate erythropoietin production. Medical Management Management of secondary polycythemia may not be necessary; when it is, it involves treating the primary problem. If the cause cannot be corrected (eg, by treating the renal cell carcinoma or improving pulmonary function), therapeutic phlebotomy may be necessary in symptomatic patients to reduce blood viscosity and volume. Leukopenia and Neutropenia Leukopenia, a condition in which there are fewer WBCs than normal, results from neutropenia (diminished neutrophils) or lymphopenia (diminished lymphocytes). Even if other types of WBCs (eg, monocytes, basophils) are diminished, their numbers are too few to reduce the total WBC count significantly. Lymphopenia (lymphocytes less than 1500/mm3) can result from ionizing radiation, long-term use of corticosteroids, uremia, some neoplasms (eg, breast and lung cancers, advanced Hodgkin’s disease), and some protein-losing enteropathies (in which the lymphocytes within the intestines are lost). intestinal tract and skin are common endogenous sources). The risk for infection is based not only on the severity of the neutropenia (low neutrophil count), but also on the duration of the neutropenia. The actual number of neutrophils, known as the absolute neutrophil count (ANC), is determined by a simple mathematical calculation using data obtained from the CBC and differential test (Chart 33-6). The risk of infection increases proportionately with the decrease in neutrophil count. The risk is significant when the ANC is less than 1000, is high when it is less than 500, and is almost certain when it is less than 100. The risk Chart 33-6 Calculating the Absolute Neutrophil Count (ANC) ANC = Total WBC count × (% neutrophils + % bands) 100 Normally, the neutrophil count is greater than 2000/mm3. The actual (or absolute) neutrophil count (ANC) is calculated using the above formula. For example, if the total white blood cell (WBC) count is 3000/mm3 with 72% neutrophils and 3% bands, the ANC would be calculated as follows: ANC = This result is not indicative of neutropenia, because the ANC is greater than 2000 despite the low total WBC count (3000/mm3). Conversely, in the following example, neutropenia is evident despite a normal WBC count (5500/mm3) with 8% neutrophils and 0% bands: ANC = NEUTROPENIA Neutropenia (neutrophils less than 2000/mm3) results from decreased production of neutrophils or increased destruction of these cells (Chart 33-5). Neutrophils are essential in preventing and limiting bacterial infection. A patient with neutropenia is at increased risk for infection, both exogenous and endogenous (the gastro- 3000 (72 + 3) = 2250 100 5500 (8 + 0) = 440 100 Here, the ANC is severely low (440) despite the normal total WBC count (5500/mm3). When evaluating neutropenia, it is important to calculate the ANC and not to rely solely on the total WBCs and percentage of neutrophils alone. 896 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION of developing infection increases with the length of time during which neutropenia persists, even if it is fairly mild. Conversely, even a severe neutropenia may not result in infection if the duration of the neutropenia is brief, as is often seen after chemotherapy (Chart 33-7). against the invading infectious organisms, broad-spectrum antibiotics are initiated as soon as the samples for culture are obtained, although the medications may be changed after culture and sensitivity results become available. Nursing Management Clinical Manifestations There are no definite symptoms of neutropenia until the patient becomes infected. Routine CBC with differential tests, such as those obtained after chemotherapy treatment, can reveal neutropenia before the onset of infection. Medical Management Treatment of the neutropenia varies depending on its cause. If the neutropenia is medication induced, the offending agent needs to be stopped, if possible. Treatment of an underlying neoplasm can temporarily make the neutropenia worse, but with bone marrow recovery treatment may improve it. Corticosteroids may be used if the cause is an immunologic disorder. The use of growth factors such as G-CSF or granulocyte/macrophage colony-stimulating factor (GM-CSF) can be effective in increasing neutrophil production when the cause of the neutropenia is decreased production. Withholding or reducing the dose of chemotherapy or radiation therapy may be required when the neutropenia is caused by these treatments; however, in the case of potentially curative therapy, administration of growth factor is considered to be preferable, so that the maximum antitumor effect can be achieved. Should the neutropenia be accompanied by fever, the patient is automatically considered to be infected and usually is admitted to the hospital. Cultures of blood, urine, and sputum should be obtained, as well as a chest radiograph. To ensure adequate therapy Nurses in all settings have a crucial role in assessing the severity of neutropenia and in preventing and managing infectious complications. Patient teaching is equally important, particularly in the outpatient setting, so that the patient can implement appropriate self-care measures and know when and how to seek medical care (Chart 33-8). Patients at risk for neutropenia should have blood drawn for CBCs; the frequency is based on the suspected severity and duration of the neutropenia. Nurses need to be able to calculate the ANC (see Chart 33-6) and to assess the severity of neutropenia and the risk for infection. Chart 33-9 identifies nursing interventions related to neutropenia. Leukocytosis and the Leukemias The term leukocytosis refers to an increased level of WBCs in the circulation. Typically, only one specific cell type is increased. Usually, because the proportions of several types of WBCs are small (eg, eosinophils, basophils, monocytes), only an increase in neutrophils or lymphocytes can be great enough to elevate the total WBC count. Although leukocytosis can be a normal response to increased need (eg, in acute infection), the elevation in WBCs should decrease as the need decreases. A prolonged or progressively increasing elevation in WBCs is abnormal and should be evaluated. A significant cause for persistent leukocytosis is malignancy. Hematopoiesis is characterized by a rapid, continuous turnover of cells. Normally, production of specific blood cells Chart 33-7 Risk Factors for Development of Infection and Bleeding in Patients with Hematologic Disorders Risk for Infection Severity of neutropenia: Risk of infection is proportional to duration and severity of neutropenia Duration of neutropenia: Increased duration leads to increased risk of infection Nutritional status: Decreased protein stores lead to decreased immune response and anergy Deconditioning: Decreased mobility leads to decreased respiratory effort, leading to increased pooling of secretions Lymphocytopenia; disorders of lymphoid system (chronic lymphocytic leukemia [CLL], lymphoma, myeloma): Decreased cell-mediated and humoral immunity Invasive procedures: Break in skin integrity leads to increased opportunity for organisms to enter blood system Hypogammaglobulinemia: Decreased antibody formation Poor hygiene: Increased organisms on skin, mucous membranes Poor dentition; mucositis: Decreased endothelial integrity leads to increased opportunity for organisms to enter blood system Antibiotic therapy: Increased risk for superinfection, often fungal Certain medications: See text Risk for Bleeding Severity of thrombocytopenia: Risk increases when platelet count decreases; usually not a significant risk until platelet count is lower than 20,000/mm3; lower than 50,000/mm3 when invasive procedure performed Duration of thrombocytopenia: Risk increases when duration increases (eg, risk is less when duration is transient after chemotherapy than when duration is permanent with poor marrow production) Sepsis: Mechanism unknown; appears to cause increased platelet consumption Increased intracranial pressure (eg, vomiting/coughing): Increased blood pressure leads to rupture of blood vessels Liver dysfunction: Decreased synthesis of clotting factors Renal dysfunction: Decreased platelet function Dysproteinemia: Protein coats surface of platelet, leading to decreased platelet function; protein causes increased viscosity, which leads to increased stretching of capillaries and thus increased bleeding Alcohol abuse: Suppressive effect on marrow leads to decreased platelet production, decreased platelet function; decreased liver function, resulting in decreased production of clotting factors Splenomegaly: Increased platelet destruction; spleen traps circulating platelets Concurrent medications: See text Chapter 33 Assessment and Management of Patients With Hematologic Disorders 897 Chart 33-8 Home Care Checklist • The Patient at Risk for Infection At the completion of the home care instruction, the patient or caregiver will be able to: • Describe consequences of alterations in neutrophils, lymphocytes, immunoglobulins, or their sources. • Verbalize the rationale for being at risk for infection. • Identify signs and symptoms of infection. • Demonstrate how to monitor for signs of infection. • Describe to whom, how, and when to report signs of infection. • Identify appropriate behaviors to take to prevent infection: – Maintain good hand hygeine technique, total body hygiene, and skin integrity. – Avoid fresh flowers, plants, garden work (soil), bird cages, and litter boxes. – Avoid fresh salads and unpeeled fruits or vegetables. – Maintain a high-calorie, high-protein diet, with fluid intake of 3000 mL (unless fluids are restricted). – Avoid people with infections, crowds. – Perform deep breathing, use incentive spirometer every 4 hr while awake. – Provide adequate lubrication with gentle vaginal manipulation during sexual intercourse; avoid anal intercourse. • Describe appropriate actions to take should infection occur. from their stem cell precursors is carefully regulated according to the body’s needs. If the mechanisms that control the production of these cells are disrupted, the cells can proliferate to an excessive, potentially dangerous degree. Hematopoietic malignancies are often classified according to the cells involved. Leukemia, literally “white blood,” is a neoplastic proliferation of one particular cell type (granulocytes, monocytes, lymphocytes, or megakaryocytes). The defect originates in the hematopoietic stem cell, the myeloid, or the lymphoid stem cell. The lymphomas are neoplasms of lymphoid tissue, usually derived from B lymphocytes. Multiple myeloma is a malignancy of the most mature form of B lymphocyte, the plasma cell. The common feature of the leukemias is an unregulated proliferation of WBCs in the bone marrow. In acute forms (or late stages of chronic forms), the proliferation of leukemic cells leaves little room for normal cell production. There can also be a proliferation of cells in the liver and spleen (extramedullary hematopoiesis). With acute forms, there can be infiltration of other organs, such as the meninges, lymph nodes, gums, and skin. The cause of leukemia is not fully known, but there is some evidence that genetic influence and viral pathogenesis may be involved. Bone marrow damage from radiation exposure or from chemicals such as benzene and alkylating agents (eg, melphalan [Alkeran]) can cause leukemia. The leukemias are commonly classified according to the stem cell line involved, either lymphoid or myeloid. They are also classified as either acute or chronic, based on the time it takes for symptoms to evolve and the phase of cell development that is halted (ie, with few WBCs differentiating beyond that phase). In acute leukemia, the onset of symptoms is abrupt, often occurring within a few weeks. WBC development is halted at the blast phase, so that most WBCs are undifferentiated or are blasts. Acute leukemia progresses very rapidly; death occurs within weeks to months without aggressive treatment. In chronic leukemia, symptoms evolve over a period of months to years, and the majority of WBCs produced are mature. Chronic leukemia progresses more slowly; the disease trajectory can extend for years. Patient Caregiver ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ACUTE MYELOID LEUKEMIA (AML) AML results from a defect in the hematopoietic stem cell that differentiates into all myeloid cells: monocytes, granulocytes (neutrophils, basophils, eosinophils), erythrocytes, and platelets. All age groups are affected; the incidence rises with age, with a peak incidence at age 60 years. AML is the most common nonlymphocytic leukemia. The prognosis is highly variable and is not consistently based on patient or disease variables. Patients with AML have a potentially curable disease. However, patients who are older or have a more undifferentiated form of AML tend to have a worse prognosis. Those who have preexisting MDS or who had previously received alkylating agents for cancer (secondary AML) have a much worse prognosis; the leukemia tends to be more resistant to treatment, resulting in a much shorter duration of remission. With treatment, these patients survive an average of less than 1 year, with death usually a result of infection or hemorrhage. Patients receiving supportive care also usually survive less than 1 year, again dying from infection or bleeding. Clinical Manifestations Most of the signs and symptoms evolve from insufficient production of normal blood cells. Fever and infection result from neutropenia, weakness and fatigue from anemia, and bleeding tendencies from thrombocytopenia. The proliferation of leukemic cells within organs leads to a variety of additional symptoms: pain from an enlarged liver or spleen, hyperplasia of the gums, and bone pain from expansion of marrow. Assessment and Diagnostic Findings The disorder develops without warning, with symptoms occurring over a period of weeks to months. CBC results show a decrease in both erythrocytes and platelets. Although the total leukocyte count can be low, normal, or high, the percentage of normal cells is usually vastly decreased. A bone marrow analysis shows an ex- 898 Chart 33-9 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Neutropenia Precautions Nursing Diagnosis Risk for infection secondary to impaired immunoincompetence due to: • Diminished neutrophil count (see below) secondary to bone marrow invasion or hypocellularity secondary to medications • Dysfunctional neutrophils (eg, secondary to myelodysplastic syndrome [MDS]) • Dysfunctional or diminished lymphocytes • Hypogammaglobulinemia • Diminished immune response or anergy • Malnutrition • Surgery or invasive procedures • Antibiotic therapy (increased risk for superimposed infection) – Neutropenic, infected patients often do not exhibit the classic signs of inflammation/infection (ie, redness, cloudiness of any drainage); the only initial sign may be fever (and it often occurs later in the infectious process with neutropenia). – Skin and mucous membranes are the body’s first line of defense against infection; loss of endothelial cell integrity allows organisms to enter the blood and lymph system. Assessment Patient Assess the following areas thoroughly every shift or visit (with spot checks throughout shift if hospitalized) and notify physician of any signs of infection or worsening of status: • Skin: Check for tenderness, edema, breaks in skin integrity, moisture, drainage, lesions (especially under breasts, axillae, groin, skin folds, bony prominences, perineum); check all puncture sites (eg, intravenous sites) for signs and symptoms of inflammation/infection. • Oral mucosa: Check for moisture, lesions, color (check palate, tongue, buccal mucosa, gums, lips, oropharynx). • Respiratory: Check for presence of cough, sore throat; auscultate breath sounds. • Gastrointestinal: Check for abdominal discomfort/distention, nausea, change in bowel pattern; auscultate bowel sounds. • Genitourinary: Check for dysuria, urgency, frequency; check urine for color, clarity, odor. • Neurologic: Check for complaints of headache, neck stiffness, visual disturbances; assess level of consciousness, orientation, behavior. • Temperature: Check every 4 hr or every visit; call primary health care provider if temperature is >38°C (>101°F), fever is unresponsive to acetaminophen, or patient shows a decline in hemodynamic status. Diagnostic Studies • Monitor complete blood count (CBC) and differential daily (especially absolute neutrophil count [ANC], lymphocyte count). • Call physician if ANC is <1000, significantly different from previous count, or whenever patient becomes symptomatic (eg, febrile). • Monitor globulin, albumin, total protein levels. • Monitor all culture and sensitivity reports. • Monitor radiology reports. Nursing Interventions Environment and Staff • Thorough hand hygiene must be done by everyone before entering patient’s room each and every time. cess of immature blast cells (more than 30%). AML can be further classified into seven different subgroups, based on cytogenetics, histology, and morphology (appearance) of the blasts. The actual prognosis varies somewhat between subgroups, but the clinical course and treatment differ substantially with only one subtype, acute promyelocytic leukemia (APL, or AML-M3). Patients with this leukemia often have significantly more problems with bleed- • Allow no one with a cold or sore throat to care for the patient or to enter room, or come in contact with patient at home. • Care for neutropenic patients before caring for other patients (as much as possible). • Use private room for patient if ANC is <1000. • Allow no fresh flowers (stagnant water). • Change water in containers every shift (include O2 humidification systems every 24 hr). • Ensure room is cleaned daily. Dietary • Provide low microbial diet. • Eliminate fresh salads and unpeeled fresh fruits or vegetables. Patient • Avoid suppositories, enemas, rectal temperatures. • Practice deep breathing (with incentive spirometer) every 4 hr while awake. • Ambulate; wear high-efficiency particulate air (HEPA) filter mask if neutropenia is severe. • Prevent skin dryness with water-soluble lubricants, especially in high-risk areas (eg, lips, corners of mouth, elbows, feet, bony prominences). Hygiene • Provide meticulous total body hygiene daily (preferably with antimicrobial solution), including perineal care after every bowel movement. • Provide thorough oral hygiene after meals and every 4 hr while awake; warm saline, or salt and soda solution, is effective; avoid use of lemon-glycerine swabs, commercial mouthwashes, and hydrogen peroxide. Intravenous (IV) Therapy • Do not use plastic cannulas for peripheral IVs when ANC is <500 (if possible per agency); a central vascular access device is preferred for long-term or intensive IV therapy. • Inspect IV sites every shift; monitor closely for any discomfort; erythema may not be present. • Maintain meticulous IV site care. • Cleanse skin with antimicrobial solution before venipuncture (unless patient is allergic). • Moisture-vapor–permeable dressings are permissible with strict adherence to institutional protocol. • Change IV tubing per institution policy, using aseptic technique. • Administer antimicrobial agents on time. Expected Patient Outcomes • Patient demonstrates an absence of infection as evidenced by an absence of fever, chills, inflammation, drainage, cough, dyspnea, sore throat, dysuria, or urinary frequency. • Patient demonstrates an absence of infection as evidenced by the presence of vital signs within normal limits, including intact neurologic status and intact skin. Duration of Evaluation Until patient is no longer neutropenic and any infection is resolved. ing, in that they have underlying coagulopathy and a higher incidence of disseminated intravascular coagulation (DIC). Complications Complications of AML include bleeding and infection, the major causes of death. The risk of bleeding correlates with the level of Chapter 33 Assessment and Management of Patients With Hematologic Disorders platelet deficiency (thrombocytopenia). The low platelet count can result in ecchymoses (bruises) and petechiae (pinpoint red or purple hemorrhagic spots on the skin). Major hemorrhages also may develop when the platelet count drops to less than 10,000/mm3. The most common sites of bleeding are gastrointestinal, pulmonary, and intracranial. For undetermined reasons, fever and infection also increase the likelihood of bleeding. Because of the lack of mature and normal granulocytes, patients with leukemia are always threatened by infection. The likelihood of infection increases with the degree and duration of neutropenia; neutrophil counts that persist at less than 100/mm3 make the chances of systemic infection extremely high. As the duration of severe neutropenia increases, the patient’s risk for developing fungal infection also increases. Medical Management The overall objective of treatment is to achieve complete remission, in which there is no detectable evidence of residual leukemia remaining in the bone marrow. Attempts are made to achieve remission by the aggressive administration of chemotherapy, called induction therapy, which usually requires hospitalization for several weeks. Induction therapy typically involves high doses of cytarabine (Cytosar, Ara-C) and daunorubicin (DaunoXome) or mitoxantrone (Novantrone) or idarubicin (Idamycin); sometimes etoposide (VP-16, VePesid) is added to the regimen. The choice of agents is based on the patient’s physical status and history of prior antineoplastic treatment. The aim of induction therapy is to eradicate the leukemic cells, but this is often accompanied by the eradication of normal types of myeloid cells. Thus, the patient becomes severely neutropenic (an ANC of 0 is not uncommon), anemic, and thrombocytopenic (a platelet count of less than 10,000/mm3 is common). During this time, the patient is typically very ill, with bacterial, fungal, and occasionally viral infections, bleeding, and severe mucositis, which causes diarrhea and a marked decline in the ability to maintain adequate nutrition. Supportive care consists of administering blood products (RBCs and platelets) and promptly treating infections. The use of granulocytic growth factors, either G-CSF (filgrastim [Neupogen]) or GM-CSF (sargramostim [Leukine]), can shorten the period of significant neutropenia by stimulating the bone marrow to produce leukocytes more quickly; these agents do not appear to increase the risk of producing more leukemic cells. When the patient has recovered from the induction therapy (ie, the WBC and platelet counts have returned to normal and any infection has resolved), the patient typically receives consolidation therapy (postremission therapy). The goal of consolidation therapy is to eliminate any residual leukemia cells that are not clinically detectable, thereby diminishing the chance for recurrence. Multiple treatment cycles of various agents are used, usually containing some form of cytarabine (eg, Cytosar, Ara-C). Frequently, the patient receives one cycle of treatment that is almost the same, if not identical, to the induction treatment but uses lower dosages (therefore resulting in less toxicity). Despite the aggressive use of chemotherapy, the likelihood of remaining in remission for a prolonged period is not great. About 70% of patients with AML experience a relapse (Hiddemann & Buchner, 2001). A recent study of long-term survival of patients with AML found that only 11% survived 10 years or longer (Micallef et al., 2001). Another aggressive treatment option is bone marrow transplantation (BMT) or peripheral blood stem cell transplantation 899 (PBSCT). When a suitable tissue match can be obtained, the patient embarks on an even more aggressive regimen of chemotherapy (sometimes in combination with radiation therapy), with the treatment goal of destroying the hematopoietic function of the patient’s bone marrow. The patient is then “rescued” with the infusion of the donor stem cells to reinitiate blood cell production. Patients who undergo PBSCT transplantation have a significant risk for problems with infection, potential graft-versus-host disease (in which the donor’s lymphocytes [graft] recognize the patient’s body as “foreign” and set up reactions to attack the “foreign” host), and other complications. PBSCT has been shown to cure AML in 25% to 50% of patients who are at high risk for relapse or who have relapsed (Radich & Sievers, 2000). Recent advances in understanding of the molecular biology of myeloid blast cells have resulted in a new therapeutic option. After the uncommitted stem cell differentiates into a myeloid stem cell, it expresses a specific antigen on the cell surface, called CD33. It appears that 90% of blast cells found in AML express CD33; normal hematopoietic stem cells do not express this antigen (Radich & Sievers, 2000). Armed with that discovery, researchers developed a monoclonal antibody to target cells with the CD33 antigen. The anti-CD33 antibody is linked to a potent antitumor antibiotic, calicheamicin; this medication is called gemtuzumab ozogamicin (Mylotarg). When administered, the anti-CD33 antibody binds to cells with CD33 antigens, and the calicheamicin causes cell death. Normal myeloid and megakaryocyte precursors have the CD33 antigen, so the Mylotarg destroys them. Patients develop severe neutropenia and thrombocytopenia after receiving this medication. Nonetheless, Mylotarg shows promise as an effective agent against AML. In elderly patients, it appears to be somewhat less toxic than conventional induction therapy regimens. Another important option for the patient to consider is supportive care alone. In fact, supportive care may be the only option if the patient has significant comorbidity, such as extremely poor cardiac, pulmonary, renal, or hepatic function. In such cases, aggressive antileukemia therapy is not used; occasionally, hydroxyurea (eg, Hydrea) may be used briefly to control the increase of blast cells. Patients are more commonly supported with antimicrobial therapy and transfusions as needed. This treatment approach provides the patient with some additional time at home; however, death frequently occurs within months, typically from infection or bleeding. COMPLICATIONS OF TREATMENT The massive leukemic cell destruction from chemotherapy results in release of electrolytes and fluids within the cell into the systemic circulation. Increases in uric acid levels, potassium, and phosphate are seen; this process is referred to as tumor lysis syndrome (see Chap. 16). The increased uric acid and phosphorus levels make patients vulnerable to renal stone formation and renal colic, which can progress to acute renal failure. Hyperkalemia and hypocalcemia can lead to cardiac dysrhythmias, hypotension, neuromuscular effects such as muscle cramps, weakness, spasm/tetany, confusion, and seizure. Patients require a high fluid intake, alkalization of the urine, and prophylaxis with allopurinol to prevent crystallization of uric acid and subsequent stone formation. Gastrointestinal problems may result from the infiltration of abnormal leukocytes into the abdominal organs and from the toxicity of the chemotherapeutic agents. Anorexia, nausea, vomiting, diarrhea, and severe mucositis are common. Because of the profound myelosuppressive effects of chemotherapy, significant neutropenia and thrombocytopenia typically result in serious infection and increased risk for bleeding. 900 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Nursing Management Nursing management of the patient with acute leukemia is discussed at the end of the leukemia section in this chapter. CHRONIC MYELOID LEUKEMIA Chronic myeloid leukemia (CML) arises from a mutation in the myeloid stem cell. Normal myeloid cells continue to be produced, but there is a preference for immature (blast) forms. Therefore, a wide spectrum of cell types exists within the blood, from blast forms through mature neutrophils. Because there is an uncontrolled proliferation of cells, the marrow expands into the cavities of long bones (eg, the femur), and cells are also formed in the liver and spleen (extramedullary hematopoiesis), resulting in enlargement of these organs that is sometimes painful. In 90% to 95% of patients with CML, a section of DNA is found to be missing from chromosome 22 (the Philadelphia chromosome [Ph1]); it is, in fact, translocated onto chromosome 9. The specific location of these changes is on the BCR gene of chromosome 22 and the ABL gene of chromosome 9. When these two genes fuse (BCR-ABL gene), they produce an abnormal protein (a tyrosine kinase protein) that causes WBCs to divide rapidly. This BCR-ABL gene is present in virtually all patients with this disease. CML is uncommon in people younger than 20 years of age, but the incidence increases with age (median age, 40 to 50 years). Patients diagnosed with CML in the chronic phase have an overall median life expectancy of 3 to 5 years. During that time, they have very few symptoms and complications from the disease itself. Problems with infections and bleeding are rare. However, once the disease transforms to the acute phase (blast crisis), the overall survival time rarely exceeds several months. Clinical Manifestations The clinical picture of CML varies. Many patients are asymptomatic, and leukocytosis is detected by a CBC performed for some other reason. The WBC count commonly exceeds 100,000/mm3. Patients with extremely high WBC counts may be somewhat short of breath or slightly confused due to decreased capillary perfusion to the lungs and brain from leukostasis (the excessive amount of WBCs inhibits blood flow through the capillaries). Patients may complain of an enlarged, tender spleen. The liver may also be enlarged. Some patients have somewhat insidious symptoms, such as malaise, anorexia, and weight loss. Lymphadenopathy is rare. There are three stages in CML: chronic, transformation, and accelerated or blast crisis. Patients have more symptoms and complications as the disease progresses. Medical Management Advances in understanding of the pathology of CML at a molecular level have led to dramatic changes in its medical management. An oral formulation of a tyrosine kinase inhibitor, imatinib mesylate (Gleevec) works by blocking signals within the leukemia cells that express the BCR-ABL protein, thus preventing a series of chemical reactions that cause the cell to grow and divide (Tennant, 2001; Goldman & Melo, 2001). Gleevec appears to be more useful in the chronic phase of the illness. In clinical trials, it has been generally well tolerated. Antacids and grapefruit juice may limit drug absorption, and large doses of acetaminophen can cause hep- atotoxicity. The long-term effects of Gleevec, its impact on survival, and the optimal length of treatment are being determined. Conventional therapy depends on the stage of disease. In the chronic phase, the expected outcome is correction of the chromosomal abnormality (ie, conversion of the malignant stem cell population back to normal). Agents that have been used successfully for this purpose are interferon-alfa (Roferon-A) and cytosine, often in combination. These agents are administered daily as subcutaneous injections. This therapy is not benign; many patients cannot tolerate the profound fatigue, depression, anorexia, mucositis, and inability to concentrate. A less aggressive therapeutic approach focuses on reducing the WBC count to a more normal level, but does not alter cytogenetic changes. This goal can be achieved by using oral chemotherapeutic agents, typically hydroxyurea (eg, Hydrea) or busulfan (eg, Myleran). In the case of an extreme leukocytosis at diagnosis (eg, WBC count higher than 300,000/mm3), a more emergent treatment may be required. In this instance, leukopheresis (in which the patient’s blood is removed and separated, with the leukocytes withdrawn, and the remaining blood returned to the patient) can temporarily reduce the number of WBCs. An anthracycline chemotherapeutic agent (eg, daunomycin) may also be used to bring the WBC count down quickly to a safer level, where more conservative therapy can be instituted. The transformation phase can be insidious, but it marks the process of evolution (or transformation) to the acute form of leukemia (blast crisis). In the transformation phase, the patient may complain of bone pain and may report fevers (without any obvious sign of infection) and weight loss. Even with chemotherapy, the spleen may continue to enlarge. The patient may become more anemic and thrombocytopenic; an increased basophil level is detected by the CBC. Despite its being a myeloid stem cell disease, CML will transform in up to 25% of patients to resemble not AML, but acute lymphoid leukemia (ALL), with lymphoidappearing blasts (Derderian et al., 1993). Transformation into the acute phase can be gradual or rapid. In the more acute form of leukemia (blast crisis), treatment may resemble induction therapy for acute leukemia, using the same medications as for AML or ALL. Patients whose disease evolves into a “lymphoid” blast crisis are more likely to be able to reenter a chronic phase after induction therapy. For those whose disease evolves into AML, therapy is largely ineffective in achieving a second chronic phase. Life-threatening infections and bleeding occur frequently in this phase. CML is a disease that can potentially be cured with BMT or PBSCT. Patients who receive such transplants while still in the chronic phase of the illness tend to have a greater chance for cure than those who receive them in the acute phase. The transplantation procedure may now be considered for otherwise healthy patients who are younger than 70 years of age. ACUTE LYMPHOCYTIC LEUKEMIA ALL results from an uncontrolled proliferation of immature cells (lymphoblasts) derived from the lymphoid stem cell. The cell of origin is the precursor to the B lymphocyte in approximately 75% of ALL cases; T-lymphocyte ALL occurs in approximately 25% of ALL cases. The BCR-ABL translocation (see earlier discussion) is found in 20% of ALL blast cells. ALL is most common in young children, with boys affected more often than girls; the peak incidence is 4 years of age. After age 15 years, ALL is relatively uncommon. Increasing age appears to be associated with diminished survival (Nachman, 1999). Because of improvements in therapy for ALL, more than 80% of children survive at least Chapter 33 Assessment and Management of Patients With Hematologic Disorders 5 years. Even if relapse occurs, resumption of induction therapy can often achieve a second complete remission. Moreover, BMT may be successful even after a second relapse. Clinical Manifestations Immature lymphocytes proliferate in the marrow and crowd the development of normal myeloid cells. As a result, normal hematopoiesis is inhibited, resulting in reduced numbers of leukocytes, erythrocytes, and platelets. Leukocyte counts may be either low or high, but there is always a high proportion of immature cells. Manifestations of leukemic cell infiltration into other organs are more common with ALL than with other forms of leukemia and include pain from an enlarged liver or spleen, bone pain, and headache and vomiting (because of meningeal involvement). 901 in use). In the early stage, an elevated lymphocyte count is seen and can exceed 100,000/mm3. Because the lymphocytes are small, they can easily travel through the small capillaries within the circulation, and the pulmonary and cerebral complications of leukocytosis (as seen with myeloid leukemias) typically are not found in CLL. Lymphadenopathy occurs as the lymphocytes are trapped within the lymph nodes. The nodes can become very large and are sometimes painful. Hepatomegaly and splenomegaly then develop. In later stages, anemia and thrombocytopenia may develop. Treatment is typically initiated in the later stages; earlier treatment does not appear to increase survival. Autoimmune complications can also occur at any stage, as either autoimmune hemolytic anemia or idiopathic thrombocytopenic purpura (ITP). In the autoimmune process, the RES destroys the body’s own RBCs or platelets. Medical Management The expected outcome of treatment is complete remission. Lymphoid blast cells are typically very sensitive to corticosteroids and to vinca alkaloids; therefore, these medications are an integral part of the initial induction therapy. Because ALL frequently invades the central nervous system, prophylaxis with cranial irradiation or intrathecal chemotherapy (eg, methotrexate [Folex]) or both is an integral part of the treatment plan. Treatment protocols for ALL tend to be complex, using a wide variety of chemotherapeutic agents. They often include a maintenance phase, when lower doses of medications are given for up to 3 years. Despite the complexity, treatment can be provided in the outpatient setting in some circumstances until severe complications develop. Infections are common, especially viral infections. The use of corticosteroids to treat ALL increases the patient’s susceptibility to infection. Patients with ALL tend to have a better response to treatment than patients with AML do. BMT or PBSCT offers a chance for prolonged remission or even cure if the illness recurs after therapy. Clinical Manifestations Many patients are asymptomatic and are diagnosed incidentally during routine physical examinations or during the course of treatment for another disease. An increased lymphocyte count (lymphocytosis) is always present. The RBC and platelet counts may be normal or, in later stages of the illness, decreased. Enlargement of lymph nodes (lymphadenopathy) is common; it can be severe and sometimes painful. The spleen can also be enlarged (splenomegaly). Patients with CLL can develop “B symptoms,” a constellation of symptoms including fevers, drenching sweating (especially at night), and unintentional weight loss. These patients have defects in their humoral and cell-mediated immune systems; therefore, infections are common. The defect in cellular immunity is evidenced by an absent or decreased reaction to skin sensitivity tests (eg, Candida, mumps), which is known as anergy. Problems with life-threatening infections are common. Viral infections, such as herpes zoster, can become widely disseminated. Medical Management Nursing Management Nursing management of the patient with acute leukemia is discussed at the end of the leukemia section in this chapter. CHRONIC LYMPHOCYTIC LEUKEMIA (CLL) CLL is a common malignancy of older adults; two thirds of all patients are older than 60 years of age at diagnosis. It is the most common form of leukemia in the United States and Europe, affecting more than 120,000 people, but is rarely seen in Asia. The average survival time for patients with CLL ranges from 14 years (early stage) to 2.5 years (late stage). Pathophysiology CLL typically derives from a malignant clone of B lymphocytes (T-lymphocyte CLL is rare). In contrast to the acute forms of leukemia, most of the leukemia cells in CLL are fully mature. It appears that these cells can escape apoptosis (programmed cell death), with the result being an excessive accumulation of the cells in the marrow and circulation. The antigen CD52 is prevalent on the surface of many of these leukemic B cells. The disease is classified into three or four stages (two classification systems are In early stages, CLL may require no treatment. When symptoms are severe (drenching night sweats, painful lymphadenopathy), or when the disease progresses to later stages (with resultant anemia and thrombocytopenia), chemotherapy with corticosteroids and chlorambucil (Leukeran) is often used. Other useful agents include cyclophosphamide (eg, Cytoxan), vincristine (eg, Oncovin), and doxorubicin (eg, Adriamycin). A significant number of patients who do not respond to these medications have achieved remission with fludarabine (Fludara), and this medication is increasingly being used as front-line therapy. The major side effect of fludarabine is prolonged bone marrow suppression, manifested by prolonged periods of neutropenia, lymphopenia, and thrombocytopenia. Patients are then at risk for such infections as Pneumocystis carinii, Listeria, mycobacteria, herpes viruses, and cytomegalovirus (CMV). The monoclonal antibody rituximab (Rituxan) also has efficacy in CLL therapy. It is often used in combination with other chemotherapeutic medications. Research has shown that the monoclonal antibody alemtuzumab (Campath) targets the CD52 antigen commonly found on CLL cells and that it is effective in clearing the marrow and circulation of these cells without affecting the stem cells. Because CD52 is present on both B and T lymphocytes, patients receiving alemtuzumab are at significant risk for infection; prophylactic use of antiviral agents and Unit 6 902 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION antibiotics (eg, trimethoprim and sulfamethoxazole [Septra]) is important and needs to continue for a minimum of 2 months after the patient stops treatment. Because bacterial infections are common in patients with CLL, intravenous treatment with immunoglobulin may be given to selected patients. NURSING PROCESS: THE PATIENT WITH ACUTE LEUKEMIA Assessment Although the clinical picture varies with the type of leukemia involved as well as the treatment implemented, the health history may reveal a range of subtle symptoms reported by the patient before the problem is manifested by findings on physical examination. Weakness and fatigue are common manifestations, not only of the leukemia but also of the resulting complications of anemia Chart 33-10 and infection. If the patient is hospitalized, the assessments should be performed daily, or more frequently as warranted. Because the physical findings may be subtle initially, a thorough, systematic assessment incorporating all body systems is essential. For example, a dry cough, mild dyspnea, and diminished breath sounds may indicate a pulmonary infection. However, the infection may not be seen initially on the chest x-ray. The lack of neutrophils delays the inflammatory response against the pulmonary infection, and it is the inflammatory response that causes the x-ray changes. The platelet count can become dangerously low, leaving the patient at risk for significant bleeding. The specific body system assessments are delineated in the neutropenic precautions and bleeding precautions, found in Charts 33-9 and 33-10, respectively. When serial assessments are performed, current findings are compared with previous findings to evaluate improvement or worsening. The nurse also must closely monitor the results of laboratory studies. Flow sheets and spreadsheets are particularly useful in Bleeding Precautions Nursing Diagnosis Potential bleeding* and injury secondary to thrombocytopenia/altered coagulation due to: • Malignant invasion in bone marrow • Bone marrow suppression resulting from chemotherapy (particularly alkylators, antitumor antibiotics, antimetabolites) and radiation therapy • Hypersplenism • Disseminated intravascular coagulation (DIC) • Altered coagulation Assessment Patient Assess the following areas thoroughly every shift or visit (with spot checks throughout the shift if patient is hospitalized), and notify physician if there is new onset of the following and/or worsening of status: • Integument: Petechiae (usually located on trunk, thighs), ecchymoses or hematomas, conjunctival hemorrhages, bleeding gums, bleeding at puncture sites (venipuncture, lumbar puncture, bone marrow) • Cardiovascular: Hypotension, tachycardia, complaints of dizziness, epistaxis • Pulmonary: Respiratory distress, tachypnea • Gastrointestinal: Hemoptysis, abdominal distention, rectal bleeding • Genitourinary: Vaginal or urethral bleeding • Neurologic: Headache, blurred vision, mental status changes Laboratory Tests • Monitor complete blood count (CBC), platelets daily (at least); coagulation panel. • Notify physician if platelet count is <10,000/mm3 or if count has changed significantly from previous count (including coagulation), or whenever patient becomes symptomatic. • Ensure patient’s blood was human leukocyte antigen (HLA) typed before transfusions or chemotherapy begins if admitted for induction therapy (eg, for acute leukemia). • Obtain 1-hour posttransfusion platelet count if warranted. • Test all urine, emesis, stools for occult blood. *Serious hemorrhage is unusual in mildly thrombocytopenic patients in absence of local lesions (peptic ulcer, bleeding from hemorrhoids, cystitis). Nursing Interventions Prevent Complications • Avoid aspirin and aspirin-containing medications or other medications known to inhibit platelet function, if possible. • Do not give intramuscular injections. • Do not insert indwelling catheters. • Take no rectal temperatures; do not give suppositories, enemas. • Use stool softeners, oral laxatives to prevent constipation. • Use smallest possible needles when performing venipuncture. • Apply pressure to venipuncture sites for 5 min or until bleeding has stopped. • Permit no flossing of teeth and no commercial mouthwashes. • Use only soft-bristled toothbrush for mouth care. • Use only toothettes for mouth care if platelet count is <10,000/mm3, or if gums bleed. • Lubricate lips with water-soluble lubricant every 2 hr while awake. • Avoid suctioning if at all possible; if unavoidable, use only gentle suctioning. • Discourage vigorous coughing or blowing of the nose. • Use only electric razor for shaving. • Pad side rails as needed. • Prevent falls by ambulating with patient as necessary. Control Bleeding • Apply direct pressure. • For epistaxis, position patient in high Fowler’s position; apply ice pack to back of neck and direct pressure to nose. • Notify physician for prolonged bleeding (eg, unable to stop within 10 min). • Administer platelets, fresh frozen plasma, packed red blood cells, as prescribed. Evaluation and Expected Patient Outcomes • Patient demonstrates an absence of bleeding as evidenced by absence of spontaneous petechiae, ecchymoses, epistaxis, hemoptysis, bleeding gums, conjunctival hemorrhage, vaginal bleeding, hematuria, guaiac positive stool, blurred vision, orthostatic hypotension, and prolonged bleeding from puncture sites. • Patient demonstrates an absence of bleeding as evidenced by the presence of vital signs within normal limits and intact neurologic status. Chapter 33 Assessment and Management of Patients With Hematologic Disorders 903 tracking the WBC count, ANC, hematocrit, platelet, and creatinine levels, hepatic function tests, and electrolyte levels. Culture results need to be reported immediately so that appropriate antimicrobial therapy can begin or be modified. with the diagnosis and prognosis, positive body image, and an understanding of the disease process and its treatment. Diagnosis PREVENTING OR MANAGING INFECTION AND BLEEDING The nursing interventions related to diminishing the risk for infection and for bleeding are delineated in Charts 33-9 and 33-10. NURSING DIAGNOSES Based on the assessment data, major nursing diagnoses for the patient with acute leukemic may include: • Risk for infection and bleeding • Risk for impaired skin integrity related to toxic effects • • • • • • • • • • • • • • • • of chemotherapy, alteration in nutrition, and impaired mobility Impaired gas exchange Impaired mucous membranes due to changes in epithelial lining of the gastrointestinal tract from chemotherapy or prolonged use of antimicrobial medications Imbalanced nutrition, less than body requirements, related to hypermetabolic state, anorexia, mucositis, pain, and nausea Acute pain and discomfort related to mucositis, WBC infiltration of systemic tissues, fever, and infection Hyperthermia related to tumor lysis and infection Fatigue and activity intolerance related to anemia and infection Impaired physical mobility due to anemia and protective isolation Risk for excess fluid volume related to renal dysfunction, hypoproteinemia, need for multiple intravenous medications and blood products Diarrhea due to altered gastrointestinal flora, mucosal denudation Risk for deficient fluid volume related to potential for diarrhea, bleeding, infection, and increased metabolic rate Self-care deficit due to fatigue, malaise, and protective isolation Anxiety due to knowledge deficit and uncertain future Disturbed body image related to change in appearance, function, and roles Grieving related to anticipatory loss and altered role functioning Potential for spiritual distress Deficient knowledge about disease process, treatment, complication management, and self-care measures COLLABORATIVE PROBLEMS/ POTENTIAL COMPLICATIONS Based on the assessment data, potential complications that may develop include: • • • • • • Infection Bleeding Renal dysfunction Tumor lysis syndrome Nutritional depletion Mucositis Planning and Goals The major goals for the patient may include absence of complications and pain, attainment and maintenance of adequate nutrition, activity tolerance, ability for self-care and to cope Nursing Interventions MANAGING MUCOSITIS Although emphasis is placed on the oral mucosa, it is important to realize that the entire gastrointestinal mucosa can be altered, not only by the effects of chemotherapy but also from prolonged administration of antibiotics. Assessment of the oral mucosa must be thorough; therefore, dentures must be removed. Areas to assess include the palate, buccal mucosa, tongue, gums, lips, oropharynx, and the area under the tongue. In addition to identifying and describing lesions, the color and moisture of the mucosa should be noted. Oral hygiene is very important to diminish the bacteria within the mouth, maintain moisture, and provide comfort. Soft-bristled toothbrushes should be used until the neutrophil and platelet counts become very low; at that time, sponge-tipped applicators should be substituted. Lemon-glycerin swabs and commercial mouthwashes should never be used because the glycerin and alcohol within them are extremely drying to the tissues. Simple rinses with saline (or saline and baking soda) solutions are inexpensive but effective in cleaning and moistening the oral mucosa. Because the risk of yeast or fungal infection in the mouth is great, other medications are often prescribed, such as chlorhexidine rinses (eg, Peridex) or clotrimazole troches (eg, Mycelex). The nurse reminds the patient about the importance of these medications to enhance adherence to the therapeutic regimen. Chlorhexidine rinses may discolor the teeth. To diminish perineal–rectal complications, it is important to cleanse the perineal–rectal area thoroughly after each bowel movement. Women are instructed to cleanse the perineum from front to back. Sitz baths are a comfortable method of cleansing; the perineal–anal region and buttocks must be carefully dried afterward to minimize the chance of excoriation. Stool softeners should be used to increase the moisture of bowel movements; however, the stool texture must be monitored so that the softeners can be decreased or stopped if the stool becomes too loose. IMPROVING NUTRITIONAL INTAKE The disease process can increase, and sepsis further increases, the patient’s metabolic rate and nutritional requirements. Nutritional intake is often reduced because of pain and discomfort associated with stomatitis. Mouth care before and after meals and administration of analgesics before eating can help increase intake. If oral anesthetics are used, the patient must be warned to chew with extreme care to avoid inadvertently biting the tongue or buccal mucosa. Nausea should not be a major contributing factor, because recent advances in antiemetic therapy are highly effective. However, nausea can result from antimicrobial therapy, so some antiemetic therapy may still be required after the chemotherapy has been completed. Small, frequent feedings of foods that are soft in texture and moderate in temperature may be better tolerated. Low-microbial diets are typically prescribed (avoiding uncooked fruits or vegetables and those without a peelable skin). Nutritional supplements are frequently used. Daily body weights (as well as in- 904 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION take and output measurements) are useful in monitoring fluid status. Calorie counts are useful, as are more formal nutritional assessments. Parenteral nutrition is often required to maintain adequate nutrition. EASING PAIN AND DISCOMFORT Recurrent fevers are common in acute leukemia; at times, they are accompanied by shaking chills, which can be severe (rigors). Myalgias and arthralgias can result. Acetaminophen is typically given to decrease fever, but it does so by increasing diaphoresis. Sponging with cool water may be useful, but cold water or ice packs should be avoided because the heat cannot dissipate from constricted blood vessels. Bedclothes need frequent changing as well. Gentle back and shoulder massage may provide comfort. Stomatitis can also cause significant discomfort. In addition to oral hygiene practices, patient-controlled analgesia can be effective in controlling the pain (see Chap. 13). Because patients with acute leukemia require hospitalization for extensive nursing care (either during induction or consolidation therapy or during resultant complications), sleep deprivation frequently results. Nurses need to implement creative strategies that permit uninterrupted sleep for at least a few hours while still administering necessary medications on time. With the exception of severe mucositis, less pain is associated with acute leukemia than with many other forms of cancer. However, the amount of psychologic suffering that the patient must endure can be immense. Patients greatly benefit from active listening. DECREASING FATIGUE AND DECONDITIONING Fatigue is a common and oppressive problem. Nursing interventions should focus on assisting the patient to establish a balance between activity and rest. Patients with acute leukemia need to maintain some physical activity and exercise to prevent the deconditioning that results from inactivity. Use of a high-efficiency particulate air (HEPA) filter mask can permit the patient to ambulate outside the room despite severe neutropenia. Although many patients lack the motivation to use them, stationary bicycles within the room can also be used. At a minimum, patients should be encouraged to sit up in a chair while awake rather than staying in bed; even this simple activity can improve the patient’s tidal volume and enhance circulation. Physical therapy can also be beneficial. MAINTAINING FLUID AND ELECTROLYTE BALANCE Febrile episodes, bleeding, and inadequate or overly aggressive fluid replacement can alter the patient’s fluid status. Similarly, persistent diarrhea, vomiting, and long-term use of certain antimicrobial agents can cause significant deficits in electrolytes. Intake and output need to be measured accurately, and daily weights should also be monitored. The patient should be assessed for signs of dehydration as well as fluid overload, with particular attention to pulmonary status and the development of dependent edema. Laboratory test results, particularly electrolytes, blood urea nitrogen, creatinine, and hematocrit, should be monitored and compared with previous results. Replacement of electrolytes, particularly potassium and magnesium, is commonly required. Patients receiving amphotericin or certain antibiotics are at increased risk for electrolyte depletion. IMPROVING SELF-CARE Because hygiene measures are so important in this patient population, they must be performed by the nurse when the patient cannot do so. However, the patient should be encouraged to do as much as possible, to preserve mobility and function as well as selfesteem. Patients may have negative feelings, even disgust that they can no longer care for themselves. Empathetic listening is helpful, as is realistic reassurance that these deficits are temporary. As the patient recovers, it is important to assist him or her to resume more self-care. Patients are usually discharged from the hospital with a central vascular access device (eg, Hickman catheter, PICC), and most patients can care for the catheter with adequate instruction and practice under observation. MANAGING ANXIETY AND GRIEF Being diagnosed with acute leukemia can be extremely frightening. In many instances, the need to begin treatment is emergent, and patients have little time to process the fact that they have the illness before making decisions about therapy. Providing emotional support and discussing the uncertain future are crucial. The nurse also needs to assess how much information patients want to have regarding the illness, its treatment, and potential complications. This desire should be reassessed at intervals, because needs and interest in information change throughout the course of the disease and treatment. Priorities must be identified so that procedures, assessments, and self-care expectations are adequately explained even to those who do not wish extensive information. Many patients become depressed and begin to grieve for the losses they feel, such as normal family functioning, professional roles and responsibilities, and social roles, as well as physical functioning. Nurses can assist patients to identify the source of the grief and encourage them to allow time to adjust to the major life changes produced by the illness. Role restructuring, in both family and professional life, may be required. Again, when possible, permitting patients to identify options and to take time making significant decisions regarding such restructuring is helpful. Discharge from the hospital can also provoke anxiety. Although most patients are extremely eager to go home, they may lack confidence in their ability to manage potential complications and to resume their normal activity. Close communication between nurses across care settings can reassure patients that they will not be abandoned. ENCOURAGING SPIRITUAL WELL-BEING Because acute leukemia is a serious, potentially life-threatening illness, the nurse may offer support to enhance the patient’s spiritual well-being. The patient’s spiritual and religious practices should be assessed and pastoral services offered. Throughout the patient’s illness, it is important that the nurse assist the patient to maintain hope. However, that hope should be realistic and will certainly change over the course of the illness. For example, the patient may initially hope to be cured, but with repeated relapses and a change to terminal care the same patient may hope for a quiet, dignified death. MONITORING AND MANAGING POTENTIAL COMPLICATIONS Nursing interventions for potential complications were described previously. PROMOTING HOME AND COMMUNITY-BASED CARE Teaching Patients Self-Care Most patients cope better when they have an understanding of what is happening to them. Based on their education, literacy level, and interest, teaching of patient and family should focus on Chapter 33 Assessment and Management of Patients With Hematologic Disorders 905 the disease (including some pathophysiology), its treatment, and certainly the significant risk for infection and bleeding (Charts 33-8 and 33-11) that results. Management of a vascular access device can be taught to most patients or family members. Follow-up and care for the devices may also need to be provided by nurses in an outpatient facility or by a home care agency or a health care providerv. another option be sought, family members who may feel guilty that they could not keep the patient at home will require support from the nurse. Continuing Care. Shortened hospital stays and outpatient care have significantly altered care for patients with acute leukemia. In many instances, when the patient is clinically stable but still requires parenteral antibiotics or blood products, these procedures can be performed in an outpatient setting. Nurses in these various settings must communicate regularly. Patients need to learn which parameters are important for them to monitor, and how to monitor them. Specific instructions need to be given as to when the patient should seek care from the physician or a health care provider. Patients and their families need to have a clear understanding of the disease and the prognosis. The nurse acts as an advocate to ensure that this information is provided. When patients no longer respond to therapy, it is important to respect their choices about treatment, including measures to prolong life and other end-of-life measures. Advance directives and living wills provide patients with some measure of control during terminal illness. Many patients in this stage still choose to be cared for at home, and families often need support when considering this option. Coordination of home care services and instruction can help to alleviate anxiety about managing the patient’s care in the home. As the patient becomes weaker, the caregivers must assume more of the patient’s care. In addition, caregivers often need to be encouraged to take care of themselves, allowing time for rest and accepting emotional support. Hospice staff can assist in providing respite for family members as well as care for the patient. Patients and families also need assistance to cope with changes in their roles and responsibilities. Anticipatory grieving is an essential task during this time (see Chap. 17). In patients with acute leukemia, death typically occurs from infection or bleeding. Family members need to have information about these complications and the measures to take should either occur. Many family members cannot cope with the care required when a patient begins to bleed actively. It is important to delineate alternatives to keeping the patient at home. Should 1. Shows no evidence of infection 2. Experiences no bleeding 3. Has intact oral mucous membranes a. Participates in oral hygiene regimen b. Reports no discomfort in mouth 4. Attains optimal level of nutrition a. Maintains weight with increased food and fluid intake b. Maintains adequate protein stores (albumin) 5. Reports satisfaction with pain and discomfort levels 6. Has less fatigue and increased activity 7. Maintains fluid and electrolyte balance 8. Participates in self-care 9. Copes with anxiety and grief a. Discusses concerns and fears b. Uses stress management strategies appropriately c. Participates in decisions regarding end-of-life care 10. Absence of complications Evaluation EXPECTED PATIENT OUTCOMES Expected patient outcomes may include: AGNOGENIC MYELOID METAPLASIA (AMM) Agnogenic myeloid metaplasia (AMM), also known as myelofibrosis, is a chronic myeloproliferative disorder that arises from neoplastic transformation of an early hematopoietic stem cell. The disease is characterized by marrow fibrosis or scarring, splenomegaly, extramedullary hematopoiesis (typically spleen, liver, or both), leukocytosis and thrombocytosis, and anemia. Some patients have suppressed WBC and platelet counts as well as anemia (pancytopenia). Patients with AMM have increased angiogenesis (formation of new blood vessels) within the marrow. Early forms of blood cells (including nucleated RBCs and megakaryocyte fragments) are frequently found in the circulation. AMM is a disease of the elderly, with a median age at diagnosis of 60 to 65 years. Survival time varies from as little as 1 year to more than 30 years; the average is 4 to 5 years (Anderson, Hamblin, & Traynor, 1999). Heart failure, complications of marrow failure, and transformation to AML are the common causes of death. Chart 33-11 Home Care Checklist • The Patient at Risk for Bleeding At the completion of the home care instruction, the patient or caregiver will be able to: • Describe the source and function of platelets and clotting factors. • Verbalize the rationale for being at risk for bleeding. • Identify medications and other substances to avoid (eg, aspirin-containing medications, alcohol). • Demonstrate how to monitor for signs of bleeding. • Describe to whom, how, and when to report signs of bleeding. • Notify health care professional before having dental work. • Describe appropriate ways to prevent bleeding (avoid use of suppositories, enemas, tampons; avoid constipation, vigorous sexual intercourse, anal sex; use only electric razor for shaving and a soft-bristled toothbrush for teeth). • Demonstrate appropriate actions to take should bleeding occur. Patient Caregiver ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ 906 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Medical Management Medical management is directed toward palliation, reducing symptoms related to cytopenias, splenomegaly, and hypermetabolic state. Although one third of anemic patients respond to the combination of an androgen plus a corticosteroid, the primary treatment remains RBC transfusion. Because of the prolonged requirement for RBC transfusion, iron overload is a common problem. Iron chelation therapy should be initiated for those individuals in whom survival is expected to exceed a few years (Anderson Hamblin, Traynor, 1999). Hydroxyurea is often used to control high WBC and platelet counts and to reduce the size of the spleen. Splenic irradiation or splenectomy may also be used to control the massive splenomegaly that can develop. However, both modalities render the patient at significant risk for development of infection. BMT may be a useful treatment modality in younger, otherwise healthy individuals. thought to be of immature lymphoid origin. It is the pathologic hallmark and essential diagnostic criterion for Hodgkin’s disease. However, the tumor is very heterogeneous and may actually contain few Reed-Sternberg cells. Repeated biopsies may be required to establish the diagnosis. Hodgkin’s disease is customarily classified into five subgroups based on pathologic analyses that reflect the natural history of the malignancy and suggest the prognosis. For example, when lymphocytes predominate, with few Reed-Sternberg cells and minimal involvement of the lymph nodes, the prognosis is much more favorable than when the lymphocyte count is low and the lymph nodes are virtually replaced by tumor cells of the most primitive type. The majority of patients with Hodgkin’s disease have the types currently designated “nodular sclerosis” or “mixed cellularity.” The nodular sclerosis type tends to occur more often in young women, at an earlier stage but with a worse prognosis than the mixed cellularity subgroup, which occurs more commonly in men and causes more constitutional symptoms but has a better prognosis. Nursing Management The extent of splenomegaly can be profound in patients with AMM, with enlargement of the spleen that extends to the pelvic rim. This condition is extremely uncomfortable to the patient and can severely limit nutritional intake. Analgesics are often ineffective. Methods to reduce the spleen’s size are usually more effective in controlling pain. Splenomegaly, coupled with a hypermetabolic state, results in weight loss (often severe) and muscle wasting. Patients benefit from very small, frequent meals of foods that are high in calories and protein. Weakness, fatigue, and altered body image are other significant problems. Energy conservation methods and active listening are important nursing interventions. Patients need to be educated about signs and symptoms of infection as well as appropriate interventions when an infection is suspected. The Lymphomas The lymphomas are neoplasms of cells of lymphoid origin. These tumors usually start in lymph nodes but can involve lymphoid tissue in the spleen, the gastrointestinal tract (eg, the wall of the stomach), the liver, or the bone marrow. They are often classified according to the degree of cell differentiation and the origin of the predominant malignant cell. Lymphomas can be broadly classified into two categories: Hodgkin’s disease and non-Hodgkin’s lymphoma (NHL). HODGKIN’S DISEASE Hodgkin’s disease is a relatively rare malignancy that has an impressive cure rate. It is somewhat more common in men than women and has two peaks of incidence: one in the early 20s and the other after 50 years of age. Unlike other lymphomas, Hodgkin’s disease is unicentric in origin in that it initiates in a single node. The disease spreads by contiguous extension along the lymphatic system. The cause of Hodgkin’s disease is unknown, but a viral etiology is suspected. In fact, fragments of the EpsteinBarr virus have been found in 40% to 50% of patients; this occurs more commonly in the younger patient population (Weiss, 2000). There is a familial pattern associated with Hodgkin’s disease: firstdegree relatives have a higher-than-normal frequency of the disease. There is no increased incidence documented for non-blood relatives (eg, spouses). The malignant cell of Hodgkin’s disease is the Reed-Sternberg cell, a gigantic tumor cell that is morphologically unique and is Clinical Manifestations Hodgkin’s disease usually begins as a painless enlargement of one or more lymph nodes on one side of the neck. The individual nodes are painless and firm but not hard. The most common sites for lymphadenopathy are the cervical, supraclavicular, and mediastinal nodes; involvement of the iliac or inguinal nodes or spleen is much less common. A mediastinal mass may be seen on chest x-ray; occasionally, the mass is large enough to compress the trachea and cause dyspnea. Pruritus is common; it can be extremely distressing, and the cause is unknown. Approximately 20% of patients experience brief but severe pain after drinking alcohol (Cavalli, 1998). The pain is usually at the site of the Hodgkin’s disease; again, the cause is unknown. All organs are vulnerable to invasion by Hodgkin’s disease. The symptoms result from compression of organs by the tumor, such as cough and pulmonary effusion (from pulmonary infiltrates), jaundice (from hepatic involvement or bile duct obstruction), abdominal pain (from splenomegaly or retroperitoneal adenopathy), or bone pain (from skeletal involvement). Herpes zoster infections are common. A cluster of constitutional symptoms has important prognostic implications. Referred to as “B symptoms,” they include fever (without chills), drenching sweats (particularly at night), and unintentional weight loss of more than 10%. “B symptoms” are found in 40% of patients and are more common in advanced disease. A mild anemia is the most common hematologic finding. The WBC count may be elevated or decreased. The platelet count is typically normal, unless the tumor has invaded the bone marrow, suppressing hematopoiesis. The erythrocyte sedimentation rate (ESR) and the serum copper level are used by some clinicians to assess disease activity. Patients with Hodgkin’s disease have impaired cellular immunity, as evidenced by an absent or decreased reaction to skin sensitivity tests (eg, Candida, mumps). Assessment and Diagnostic Findings Because many manifestations are similar to those occurring with infection, diagnostic studies are performed to rule out an infectious origin for the disease. The diagnosis is made by means of an excisional lymph node biopsy and the finding of the Reed-Sternberg cell. Once the diagnosis is confirmed and the histologic type is established, it is necessary to assess the extent of the disease, a process referred to as staging. Chapter 33 Assessment and Management of Patients With Hematologic Disorders During the health history, the nurse should assess for any “B symptoms.” Physical examination requires a careful, systematic evaluation of the lymph node chains, as well as the size of the spleen and liver. A chest x-ray and a CT scan of the chest, abdomen, and pelvis are crucial to identify the extent of lymphadenopathy within these regions. Laboratory tests include CBC, platelet count, ESR, and liver and renal function studies. A bone marrow biopsy is performed if there are signs of marrow involvement, and some physicians routinely perform bilateral biopsies. Bone scans may be performed to identify any involvement in these areas. A staging laparotomy and lymphangiography are no longer considered mandatory, primarily because of the accuracy of CT. Medical Management The general intent in treating Hodgkin’s disease, regardless of stage, is cure. Treatment is determined primarily by the stage of the disease, not the histologic type; however, extensive research is ongoing to target treatment regimens to histologic subtypes or prognostic features. Traditionally, early Hodgkin’s disease was treated by a staging laparotomy followed by radiation therapy. Recent data show improved results and decreased complications with a short course (2 to 4 months) of chemotherapy followed by radiation therapy in certain subsets of early-stage disease (IA and IIA); patients with early-stage disease and good prognostic features may receive radiation therapy alone (Hoppe et al., 2000). Combination chemotherapy, for example with doxorubicin (Adriamycin), bleomycin (Blenoxane), vinblastine (Velban), and dacarbazine (DTIC), referred to as ABVD, is now the standard treatment for more advanced disease (stages III and IV and all B stages). Radiation therapy is still very useful for patients with extensive adenopathy (often termed bulky disease). In this group, residual disease often persists after the chemotherapy treatment is finished; radiation therapy to the areas of remaining adenopathy has been shown to improve survival. Even when Hodgkin’s disease does recur, the use of high doses of chemotherapeutic agents, followed by autologous BMT or stem cell transplantation (PBSCT), can be very effective in controlling the disease and extending survival time. Long-Term Complications of Therapy Much is now known about the long-term effects of chemotherapy and radiation therapy, primarily from the large numbers of people who were cured of Hodgkin’s disease by these treatments. The various complications of treatment are listed in Chart 33-12. Risk factors for other cancers should be assessed, and long-term surveillance is crucial. The potential development of a second malignancy is obviously of concern to patients, and this potential should be addressed with the patient when treatment decisions are made. However, it is important to consider that Hodgkin’s disease is curable. Revised treatment approaches are aimed at diminishing the risk for complications without sacrificing the potential for cure. Chart 33-12 907 Potential Long-Term Complications of Therapy for Hodgkin’s Disease Immune dysfunction Herpes infections (zoster and varicella) Pneumococcal sepsis Acute myeloid leukemia (AML) Myelodysplastic syndromes (MDS) Non-Hodgkin’s lymphoma Solid tumors Thyroid cancer Thymic hyperplasia Hypothyroidism Pericarditis (acute or chronic) Cardiomyopathy Pneumonitis (acute or chronic) Avascular necrosis Growth retardation Infertility Impotence Dental caries lignant lymphoid cells occurs unpredictably, and true localized disease is uncommon. Lymph nodes from multiple sites may be infiltrated, as may sites outside the lymphoid system (extranodal tissue). The incidence of NHL has increased dramatically over the past decade; it is now the fourth most common type of cancer diagnosed in the United States and the fifth most common cause of cancer death (Greenlee, Hill-Horton, Murray, & Thun, 2001; Zelenetz et al., 2000). The incidence increases with each decade of life; the average age at diagnosis is 50 to 60 years. Although no common etiologic factor has been identified, there is an increased incidence of NHL in people with immunodeficiencies or autoimmune disorders, viral infections (including Epstein-Barr virus and HIV), or exposure to pesticides, solvents, or dyes. Prognosis varies greatly among the various types of NHL. Long-term survival (more than 10 years) is commonly achieved in low-grade, localized lymphomas. Even with aggressive disease forms, cure is possible in at least one third of patients who receive aggressive treatment. Clinical Manifestations Symptoms are highly variable, reflecting the diverse nature of these diseases. With early-stage disease, or with the types that are considered more indolent, symptoms may be virtually absent or very minor, and the illness typically is not diagnosed until it progresses to a later stage, when the patient is more symptomatic. At these stages (III or IV), lymphadenopathy is noticeable. One third of patients have “B symptoms” (recurrent fever, drenching night sweats, and unintentional weight loss of 10% or more). NON-HODGKIN’S LYMPHOMAS (NHLs) Assessment and Diagnostic Findings The NHLs are a heterogeneous group of cancers that originate from the neoplastic growth of lymphoid tissue. As in CLL, the neoplastic cells are thought to arise from a single clone of lymphocytes; however, in NHL, the cells may vary morphologically. Most NHLs involve malignant B lymphocytes; only 5% involve T lymphocytes. In contrast to Hodgkin’s disease, the lymphoid tissues involved are largely infiltrated with malignant cells. The spread of these ma- The actual diagnosis of NHL is categorized into a highly complex classification system based on histopathology, immunophenotyping, and cytogenetic analyses of the malignant cells. The specific histopathologic type of the disease has important prognostic implications. Treatment also varies and is based on these features. Indolent (less aggressive) types tend to have small cells and are distributed in a follicular pattern. Aggressive types tend to have 908 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION large or immature cells distributed through the nodes in a diffuse pattern. Staging, also an important factor, is typically based on data obtained from CT scans, bone marrow biopsies, and occasionally cerebrospinal fluid analysis. The stage is based on the site of disease and its spread to other sites. For example, in stage I disease, only one area of involvement is detected; thus, stage I disease is highly localized and may respond well to localized therapy (eg, radiation therapy). In contrast, stage IV disease is detected in at least one extranodal site. Although low-grade lymphomas may not require treatment until the disease progresses to a later stage, historically they have also been relatively unresponsive to treatment in that most therapeutic modalities did not improve overall survival. More aggressive types of NHL (eg, lymphoblastic lymphoma, Burkitt’s lymphoma) require prompt initiation of chemotherapy; however, these types tend to be more responsive to treatment. Medical Management Treatment is based on the actual classification of disease, the stage of disease, prior treatment (if any), and the patient’s ability to tolerate therapy. If the disease is not an aggressive form and is truly localized, radiation alone may be the treatment of choice. With aggressive types of NHL, aggressive combinations of chemotherapeutic agents are given even in early stages. More intermediate forms are commonly treated with combination chemotherapy and radiation therapy for stage I and II disease. The biologic agent interferon has been approved for the treatment of follicular lowgrade lymphomas, and an antibody to CD20, rituximab (Rituxan), has been effective in achieving partial responses in patients with recurrent low-grade lymphoma. Studies of this agent in combination with conventional chemotherapy have demonstrated an improvement in survival as well (Coiffier, 2002; Emmanouilides et al., 2000; Petryk & Grossbard, 2000). Central nervous system involvement is also common with some aggressive forms of NHL; in this situation, cranial radiation or intrathecal chemotherapy is used in addition to systemic chemotherapy. Treatment after relapse is controversial. BMT or PBSCT may be considered for patients younger than 60 years of age (See Chap. 16). Nursing Management Most of the care for patients with Hodgkin’s disease or NHL is performed in the outpatient setting, unless complications occur (eg, infection, respiratory compromise due to mediastinal mass). For patients who require treatment, chemotherapy and radiation therapy are most commonly used. Chemotherapy causes systemic side effects (eg, myelosuppression, nausea, hair loss, risk for infection), whereas the side effects from radiation therapy are specific to the area being irradiated. For example, patients receiving abdominal radiation therapy may experience nausea and diarrhea but not hair loss. Regardless of the type of treatment, all patients may experience fatigue. The risk of infection is significant for these patients, not only from treatment-related myelosuppression but also from the defective immune response that results from the disease itself. Patients need to be taught to minimize the risks for infection, to recognize signs of possible infection, and to contact the health care professional should such signs develop (see Chart 33-8). Many lymphomas can be cured with current treatments. However, as survival rates increase, the incidence of second malignancies, particularly AML or MDS, also increases. Therefore, survivors should be screened regularly for the development of second malignancies. Lymphoma is a highly complex constellation of diseases. When caring for the patient with lymphoma, it is extremely important to know the specific disease type, stage of disease, treatment history, and current treatment plan. MULTIPLE MYELOMA Multiple myeloma is a malignant disease of the most mature form of B lymphocyte, the plasma cell. It is not classified as a lymphoma. Plasma cells secrete immunoglobulins, proteins necessary for antibody production to fight infection. Pathophysiology In myeloma, the malignant plasma cells produce an increased amount of a specific immunoglobulin that is nonfunctional. Functional types of immunoglobulin are still produced by nonmalignant plasma cells, but in lower-than-normal quantity. The specific immunoglobulin secreted by the myeloma cells is detectable in the blood or urine and is referred to as the monoclonal protein, or M protein. This protein serves as a useful marker to monitor the extent of disease and the patient’s response to therapy. It is measured by serum or urine protein electrophoresis. Moreover, the patient’s total protein level is typically elevated, again due to the production of M protein. Malignant plasma cells also secrete certain substances to stimulate the creation of new blood vessels to enhance the growth of these clusters of plasma cells; this process is referred to as angiogenesis. Occasionally the plasma cells infiltrate other tissue, in which case they are referred to as plasmacytomas. Plasmacytomas can occur in the sinuses, spinal cord, and soft tissues. Median survival time is 3 to 5 years. Death usually results from infection. Clinical Manifestations The classic presenting symptom of multiple myeloma is bone pain, usually in the back or ribs. Bone pain is reported by two thirds of all patients at diagnosis. Unlike arthritic pain, the bone pain associated with myeloma increases with movement and decreases with rest; patients may report that they have less pain on awakening but the pain intensity increases during the day. In myeloma, a substance secreted by the plasma cells, osteoclast activating factor, as well as other substances (eg, interleukin-6 [IL-6]) are involved in stimulating osteoclasts. Both mechanisms appear to be involved in the process of bone breakdown. Thus, lytic lesions as well as osteoporosis may be seen on bone x-rays. (They are not well visualized on bone scans.) The bone destruction can be severe enough to cause fractures, including spinal fractures, which can impinge on the spinal cord and result in spinal cord compression. It is this bone destruction that causes significant pain. ! NURSING ALERT Any elderly patient whose chief complaint is back pain, and who has an elevated total protein level, should be evaluated for possible myeloma. If the bone destruction is fairly extensive, excessive ionized calcium is lost from the bone and enters the serum; patients may therefore become hypercalcemic (frequently manifested by excessive thirst, dehydration, constipation, altered mental status, confusion, and perhaps coma). Renal failure may also be seen; the configuration of the circulating immunoglobulin molecule (particularly the shape of lambda light chains) can damage the renal tubules. Chapter 33 Assessment and Management of Patients With Hematologic Disorders As more and more malignant plasma cells are produced, the marrow has less space for RBC production, and the patient can become anemic. This anemia is also caused to a great extent by a diminished production of erythropoietin (a glycoprotein necessary for RBC production) by the kidney. Patients may complain of fatigue and weakness due to the anemia. In the late stage of the disease, a reduced number of WBCs and platelets may also be seen because the bone marrow is infiltrated by malignant plasma cells. When plasma cells secrete excessive amounts of immunoglobulin, particularly IgA, the serum viscosity can be elevated. Hyperviscosity may be manifested by bleeding from the nose or mouth, headache, blurred vision, paresthesias, or heart failure. Assessment and Diagnostic Findings Finding an elevated monoclonal protein spike in the serum (via serum protein electrophoresis) or urine (via urine protein electrophoresis) or light chain in the urine (sometimes referred to as Bence Jones protein) is considered to be a major criterion in the diagnosis of multiple myeloma. The presence of lytic bone lesions on x-ray aids in the diagnosis, as does the presence of anemia or hypercalcemia. The diagnosis of myeloma can be confirmed by bone marrow biopsy; the presence of sheets of plasma cells is the hallmark diagnostic criterion. Because the infiltration of the marrow by these malignant plasma cells is not uniform, the extent of plasma cells may not be increased in a given sample (a false-negative result). Gerontologic Considerations The incidence of multiple myeloma increases with age; the disease rarely occurs in patients younger than 40 years of age. Because of the increasing older population, more patients are seeking treatment for this disease. BMT or PBSCT is an option that can prolong remission and potentially cure some patients. However, it is unavailable to most because of age limitations. Back pain, which is often a presenting symptom in this disease, should be closely investigated in elderly patients. Medical Management There is no cure for multiple myeloma. Even BMT or PBSCT is considered by most authorities to extend remission rather than provide a cure. However, for many patients, it is possible to control the illness and maintain their level of functioning quite well for several years or longer. Chemotherapy is the primary treatment; corticosteroids, particularly dexamethasone (Decadron), are especially effective and are often combined with other agents (such as melphalan (Alkeran), cyclophosphamide (Cytoxan), doxorubicin (Adriamyein), vincristine (Oncouin), and BCNU (Carmustine). Radiation therapy is very useful in strengthening a specific bone lesion, particularly one at risk for bone fracture or spinal cord compression. It is also useful in relieving bone pain and reducing the size of plasma cell tumors that occur outside the skeletal system. However, because it is a nonsystemic form of treatment, it does not diminish the source of the bone problems (ie, the production of malignant plasma cells). Therefore, radiation therapy is typically used with systemic treatment such as chemotherapy. The biologic agent alpha-interferon has been used successfully to maintain remission in selected types of myeloma, particularly IgA type; however, its role in prolonging survival is controversial. Newer forms of bisphosphonates, such as pamidronate (Aredia) and zoledronic acid (Zometa), have been shown to strengthen bone 909 in this disease (by diminishing the secretion of osteoclast activating factor) (Terpos et al., 2000), controlling bone pain and potentially preventing bone fracture. They are also effective in managing and preventing hypercalcemia. Some evidence suggests that bisphosphonates may actually have activity against the myeloma cells themselves by inhibiting a growth factor necessary for myeloma cell survival (Berenson, 2001) (see later discussion). When patients manifest signs and symptoms of hyperviscosity, plasmapheresis may be used to lower the immunoglobulin level. Symptoms may be more useful than serum viscosity levels in determining the need for this intervention. Recent advances in the understanding of the process of angiogenesis have resulted in new therapeutic options. The sedative thalidomide (Thalomid), initially used as an antiemetic, has significant antimyeloma effects. It inhibits cytokines necessary for new vascular generation, such as, vascular endothelial growth factor (VEGF) and for myeloma cell growth and survival, such as IL-6 and tumor necrosis factor), by boosting the body’s immune response against the tumor and by creating favorable conditions for apoptosis of the myeloma cells. Thalidomide is effective in refractory myeloma and in “smoldering” disease states, and may prevent progression to a more active state. Thalidomide is not a typical chemotherapeutic agent and has a unique side effect profile. Fatigue, dizziness, constipation, rash, and peripheral neuropathy are commonly encountered; myelosuppression is not (Goldman, 2001). Thalidomide is contraindicated in pregnancy because of associated severe birth defects. Nursing Management Pain management is very important in this patient population. NSAIDs can be very useful for mild pain, or in combination with opioid analgesics. However, care needs to be taken, because NSAIDs can cause renal dysfunction. Patients need to be educated about activity restrictions (eg, lifting no more than 10 pounds, use of proper body mechanics). Braces are occasionally needed to provide support to the spinal column. Patients also need to be instructed about the signs and symptoms of hypercalcemia. Maintaining mobility and hydration is important to diminish exacerbations of this complication; however, the primary cause is the disease itself. Renal function should also be monitored closely. Renal failure can become severe, and dialysis may be needed. Maintaining high urine output (3 L/day) can be very useful in preventing this complication. Because antibody production is impaired, infections, particularly bacterial infections, are common and can be life-threatening. Patients need to be instructed in appropriate infection prevention measures (see Chart 33-8) and should be advised to contact their health care provider immediately if they have a fever or other signs and symptoms of infection. Patients should receive Pneumovax and flu vaccines. Prophylactic antibiotics are sometimes used. Intravenous gamma globulin (IVIG) can be useful for patients with recurrent infections. Bleeding Disorders Normal hemostatic mechanisms can control bleeding from vessels and prevent spontaneous bleeding. The bleeding vessel constricts and platelets aggregate at the site, forming an unstable hemostatic plug. Circulating coagulation factors are activated on the surface of these aggregated platelets, forming fibrin, which anchors the platelet plug to the site of injury. 910 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION The failure of normal hemostatic mechanisms can result in bleeding, which is severe at times. This bleeding is commonly provoked by trauma, but in certain circumstances it can occur spontaneously. When the source is platelet or coagulation factor abnormalities, the site of spontaneous bleeding can be anywhere in the body. When the defect is caused by vascular abnormalities, the site of bleeding may be more localized. Some patients have defects in more than one hemostatic mechanism simultaneously. In a variety of situations, the bone marrow may be stimulated to increase platelet production (thrombopoiesis). The increased production may be a reactive response, as in a compensatory response to significant bleeding, or a more general response to increase hematopoiesis, as in iron deficiency anemia. Sometimes, the increase in platelets does not result from increased production but from a loss in platelet pooling within the spleen. The spleen typically holds about one third of the circulating platelets at any time. If the spleen is lost (eg, splenectomy), the platelet reservoir is also lost, and an abnormally high amount of platelets enter the circulation. In time, the rate of thrombopoiesis slows to reestablish a more normal platelet level. contact sports. The skin is observed for petechiae and ecchymoses (bruises) and the nose and gums for bleeding. Hospitalized patients may be monitored for bleeding by testing all drainage and excreta (feces, urine, emesis, and gastric drainage) for occult as well as obvious blood. Outpatients are often given fecal occult blood screening cards to detect occult blood in stools. PRIMARY THROMBOCYTHEMIA Primary thrombocythemia (also called essential thrombocythemia) is a stem cell disorder within the bone marrow. A marked increase in platelet production occurs, with the platelet count consistently greater than 600,000/mm3. Platelet size may be abnormal, but platelet survival is typically normal. Occasionally, the platelet increase is accompanied by an increase in RBCs or WBCs or both; however, these cells are not increased to the extent that they are in polycythemia vera, CML, or myelofibrosis. Although the exact cause is unknown, primary thrombocythemia is similar to other myeloproliferative disorders, particularly polycythemia vera. Unlike the other myeloproliferative disorders, however, it rarely evolves into acute leukemia. Clinical Manifestations Signs and symptoms of bleeding disorders vary depending on the type of defect. A careful history and physical examination can be very useful in determining the source of the hemostatic defect. Abnormalities of the vascular system give rise to local bleeding, usually into the skin. Because platelets are primarily responsible for stopping bleeding from small vessels, patients with platelet defects develop petechiae, often in clusters; these are seen on the skin and mucous membranes but also occur throughout the body. Bleeding from platelet disorders can be severe. Unless the platelet disorder is severe, bleeding can often be stopped promptly when local pressure is applied; it does not typically recur when the pressure is released. In contrast, coagulation factor defects do not tend to cause superficial bleeding, because the primary hemostatic mechanisms are still intact. Instead, bleeding occurs deeper within the body (eg, subcutaneous or intramuscular hematomas, hemorrhage into joint spaces). External bleeding diminishes very slowly when local pressure is applied; it often recurs several hours after pressure is removed. For example, severe bleeding may start several hours after a tooth extraction. Risk factors for bleeding are provided in Chart 33-7. Medical Management Management varies based on the underlying cause of the bleeding disorder. If bleeding is significant, transfusions of blood products are indicated. The specific blood product used is determined by the underlying defect. In specific situations in which fibrinolysis is excessive, hemostatic agents such as aminocaproic acid (Amicar) can be used to inhibit this process. This agent must be used with caution, because excessive inhibition of fibrinolysis can result in thrombosis. Nursing Management Patients who have bleeding disorders or who have the potential for development of such disorders as a result of disease or therapeutic agents must be taught to observe themselves carefully and frequently for bleeding. They need to understand the importance of avoiding activities that increase the risk of bleeding, such as Clinical Manifestations Many patients with primary thrombocythemia are asymptomatic; the illness is diagnosed as the result of finding an elevated platelet count on a CBC. Symptoms, when they do occur, result primarily from hemorrhage or vasoocclusion in the microvasculature. Symptoms may occur more when the platelet count exceeds 1 million/ mm3. However, symptoms do not always correlate with the extent to which the platelet count is elevated. Thrombosis is common and can be either arterial or venous; major thromboses occur in 15% to 40% of these patients (Jantunen et al., 2001). Because these platelets can be dysfunctional, minor or major hemorrhage can also occur. Bleeding from the mucous membranes of the nose and mouth is common, and significant gastrointestinal bleeding is also possible. Bleeding typically does not occur until the platelet count exceeds 1 million/mm3. Vasoocclusive manifestations are most frequently seen in the form of erythromelalgia. The toxic effects of platelet substances include painful burning, warmth, and redness in a localized distal area of the extremities. Neurologic manifestations may also be seen, such as numbness, tingling, and visual disturbance; these occlusive manifestations can progress to stroke and seizure and, less commonly, to myocardial infarction. The spleen may be enlarged, but usually not to a significant extent. Assessment and Diagnostic Findings The diagnosis of primary thrombocythemia is made by ruling out other potential disorders. Iron deficiency should be excluded, because a reactive increase in the platelet count often accompanies this deficiency. The myeloproliferative disorders (CML, polycythemia vera) should also be excluded. Examination of the CBC shows markedly abnormal platelets. Analysis of the bone marrow (by aspiration and biopsy) shows a marked increase in megakaryocytes (platelet precursors) and is useful in excluding CML as a possible cause for the elevated platelet count. The disease, which affects men and women equally, tends to occur in late middle age. The median survival time exceeds 10 years. No data reliably predict the development of complications. Risk factors for the development of thrombotic complications are Chapter 33 Assessment and Management of Patients With Hematologic Disorders age greater than 65 years, prior thrombotic events, and long duration of thrombocytosis. Major bleeding tends to occur when the platelet count is very high. Medical Management The management of primary thrombocythemia is highly controversial. The risk of significant thrombotic or hemorrhagic complications may not be increased until the platelet count exceeds 1 million/mm3 (Briere & Guilmin, 2001). A careful assessment of other risk factors, such as history of peripheral vascular disease, history of tobacco use, atherosclerosis, and prior thrombotic events, should be used in making the decision as to when to initiate therapy. In younger patients with no risk factors, low-dose aspirin therapy may be sufficient to prevent thrombotic complications; however, the use of aspirin can increase the risk for hemorrhagic complications and may be considered a contraindication in patients with a history of gastrointestinal bleeding. The neurologic symptoms (eg, headache and erythromelalgia) and visual symptoms of primary thrombocytopenia can be relieved by low-dose aspirin. More aggressive measures may be required in older patients and in those with concurrent risk factors. Hydroxyurea (eg, Hydrea), a chemotherapeutic medication, is effective in lowering the platelet count. It is taken orally and causes minimal side effects other than dose-related leukopenia. However, its potential for leukogenesis is in question. The medication anagrelide (Agrylin) is more specific in lowering the platelet count than is hydroxyurea, but it has more side effects. Severe headaches cause many patients to stop taking the medication. Tachycardia and chest pain may also occur, and anagrelide is contraindicated in patients with concurrent cardiac problems. Interferon-alfa-2b (eg, Intron-A) has been shown to lower platelet counts by an unknown mechanism. The medication is administered subcutaneously at varying frequency, commonly three times per week. Significant side effects, such as fatigue, weakness, memory defects, dizziness, anemia, and liver dysfunction, limit its usefulness. Rarely, the occlusive symptoms are so great that the platelet count must be reduced immediately. Platelet pheresis (see later discussion) can reduce the amount of circulating platelets, but only transiently. The extent by which symptoms and complications (eg, thromboses) are reduced remains unclear. Nursing Management Patients with primary thrombocythemia need to be instructed about the accompanying risks of hemorrhage and thrombosis. Patients should be informed about signs and symptoms of thrombosis, particularly the neurologic manifestations, such as visual changes, numbness, tingling, and weakness. Risk factors for thrombosis should be assessed, and measures to diminish risk factors (particularly cessation of tobacco use) should be encouraged. Patients receiving aspirin therapy should be informed about the increased risk for bleeding. Patients who are at risk for bleeding should be instructed about medications that can alter platelet function, such as aspirin, NSAIDs, and alcohol. Patients receiving interferon therapy should be taught to self-administer the medication and manage side effects. SECONDARY THROMBOCYTOSIS Increased platelet production is the primary mechanism of secondary, or reactive, thrombocytosis. The platelet count is above 911 normal, but, in contrast to primary thrombocythemia, an increase above 1 million/mm3 is rare. Platelet function is normal; the platelet survival time is normal or decreased. Symptoms associated with hemorrhage or thrombosis are rare. Many disorders can cause a reactive increase in platelets, including chronic inflammatory disorders, iron deficiency, malignant disease, acute hemorrhage, and splenectomy (see previous discussion of primary thrombocythemia). Treatment is aimed at the underlying disorder. With successful management, the platelet count usually returns to normal. THROMBOCYTOPENIA Thrombocytopenia (low platelet level) can result from various factors: decreased production of platelets within the bone marrow, increased destruction of platelets, or increased consumption of platelets. Causes and treatments are summarized in Table 33-5. Clinical Manifestations Bleeding and petechiae usually do not occur with platelet counts greater than 50,000/mm3, although excessive bleeding can follow surgery or other trauma. When the platelet count drops below Table 33-5 • Causes and Management of Thrombocytopenia CAUSE Decreased Production Hematologic malignancy, especially acute leukemias Myelodysplastic syndromes (MDS): metastatic involvement of bone marrow from solid tumors Aplastic anemia Megaloblastic anemia Toxins Medications Infection (esp. septicemia, viral infection, tuberculosis) Alcohol Chemotherapy Increased Destruction Due to Antibodies Idiopathic thrombocytopenic purpura Lupus erythematosus Malignant lymphoma Chronic lymphocytic leukemia (CLL) Medications Due to Infection Bacteremia Postviral infection Sequestration of platelets in an enlarged spleen Increased Consumption Disseminated intravascular coagulation (DIC) MANAGEMENT Treat leukemia; platelet transfusion Treat MDS; platelet transfusion Treat solid tumor Treat underlying condition Treat underlying anemia Remove toxin Stop medication Treat infection Refrain from alcohol consumption Delay or decrease dose; growth factor; platelet transfusion Treat condition Treat CLL and/or treat as ITP Stop medication Treat infection If thrombocytopenia is severe, splenectomy may be needed Treat underlying condition triggering DIC; administer heparin, EACA, blood products 912 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION 20,000/mm3, petechiae can appear, along with nose and gingival bleeding, excessive menstrual bleeding, and excessive bleeding after surgery or dental extractions. When the platelet count is less than 5000/mm3, spontaneous, potentially fatal central nervous system or gastrointestinal hemorrhage can occur. If the platelets are dysfunctional due to disease (eg, MDS) or medications (eg, aspirin), the risk of bleeding may be much greater even when the actual platelet count is not significantly reduced. Assessment and Diagnostic Findings A platelet deficiency that results from decreased production (eg, leukemia, MDS) can usually be diagnosed by examining the bone marrow via aspiration and biopsy. When platelet destruction is the cause of thrombocytopenia, the marrow shows increased megakaryocytes (the cells from which the platelets originate) and normal or even increased platelet production as the body attempts to compensate for the decreased platelets in circulation. Another cause of thrombocytopenia is sequestration. Approximately one third of the circulating platelets are within the spleen, and a greatly enlarged spleen results in increased sequestration of platelets. ingests the platelets, destroying them. The body attempts to compensate for this destruction by increasing platelet production within the marrow. Clinical Manifestations Many patients have no symptoms, and the low platelet count (often less than 20,000/mm3, and less than 5000/mm3 is not uncommon) is an incidental finding. Common physical manifestations are easy bruising, heavy menses, and petechiae on the extremities or trunk. Patients with simple bruising or petechiae (“dry purpura”) tend to have fewer complications from bleeding than those with bleeding from mucosal surfaces, such as the gastrointestinal tract (including the mouth) and pulmonary system (eg, hemoptysis), which is termed “wet purpura.” Patients with wet purpura have a greater risk for intracranial bleeding than do those with dry purpura. Despite low platelet counts, the platelets are young and very functional. They adhere to endothelial surfaces and to one another, so spontaneous bleeding does not always occur. Assessment and Diagnostic Findings Medical Management The management for secondary thrombocytopenia is usually treatment of the underlying disease. If platelet production is impaired, platelet transfusions may raise the platelet count and stop bleeding or prevent spontaneous hemorrhage. If excessive platelet destruction occurs, transfused platelets will also be destroyed, and the platelet count will not rise. The most common cause of excessive platelet destruction is ITP (see the following discussion). In some instances splenectomy can be a useful therapeutic intervention, but often it is not a therapeutic option, for example in patients in whom the enlarged spleen is due to portal hypertension related to excessive alcohol consumption. Nursing Management The interventions for a patient with thrombocytopenia are delineated in Chart 33-10. IDIOPATHIC THROMBOCYTOPENIC PURPURA (ITP) ITP is a disease that affects people of all ages, but it is more common among children and young women. There are two forms of ITP: acute and chronic. The acute form, which occurs predominately in children, often appears 1 to 6 weeks after a viral illness. This form is self-limited; remission often occurs spontaneously within 6 months. Occasionally, corticosteroids are needed for a brief time. Chronic ITP is often diagnosed by exclusion of other causes of thrombocytopenia. Pathophysiology Although the precise cause remains unknown, viral infections sometimes precede ITP in children. Occasionally medications such as sulfa drugs can induce ITP. Other conditions, such as systemic lupus erythematosus (SLE) or pregnancy, can also induce ITP. Anti-platelet autoantibodies that bind to the patient’s platelets are found in the blood of patients with ITP. When the platelets are bound by the antibodies, the RES or tissue macrophage system Patients may have an isolated decrease in platelets (less than 20,000/mm3 is common), but they may also have an increase in megakaryocytes (platelet precursors) within the marrow, as detected on bone marrow aspirate. Medical Management The primary goal of treatment is a safe platelet count. Because the risk of bleeding typically does not increase until the platelet count is lower than 10,000/mm3, patients whose counts exceed 30,000 to 50,000/mm3 may be carefully observed without additional intervention. However, if the count is lower than 20,000/mm3, or if bleeding occurs, the goal is to improve the patient’s platelet count, rather than to cure the disease. Treatment for ITP usually requires several approaches. If the patient is taking a medication that is known to cause ITP (eg, quinine, sulfacontaining medications), that medication must be stopped immediately. The mainstay of short-term therapy is the use of immunosuppressive agents. The immunosuppressants block the binding receptors on macrophages so that the platelets are not destroyed. Prednisone is the agent typically used (at a dose of 1 mg/kg), and it is effective in about 75% of patients. Cyclophosphamide (eg, Cytoxan) and azathioprine (Imuran) can also be used, and dexamethasone (eg, Decadron) may be effective. Platelet counts rise within a few days after institution of corticosteroid therapy; this effect takes longer with azathioprine. Because of the associated side effects, patients cannot take high doses of corticosteroids indefinitely. It is not unusual for the platelet count to drop once the corticosteroid dose is tapered. Some patients can be successfully maintained on low doses of prednisone (eg, 2.5 to 10 mg every other day). Intravenous gamma globulin (IVIG) is also commonly used to treat ITP. It is effective in binding the receptors on the macrophages; however, high doses (1 g/kg for 2 days) are required, and the drug is very expensive. Splenectomy is an alternative treatment but results in a normal platelet count only 50% of the time; however, many patients can maintain a “safe” platelet count of more than 30,000/mm3 after removal of the spleen. Even those who do respond to splenectomy may have recurrences of severe thrombocytopenia months or years later. Patients who Chapter 33 Assessment and Management of Patients With Hematologic Disorders have splenectomy are permanently at risk for sepsis; these patients should receive Pneumovax, Haemophilus influenzae B, and meningococcal vaccines, preferably 2 to 3 weeks before the splenectomy is preferred. Pneumovax vaccine should be repeated at 5- to 10-year intervals. Other options for management include use of the chemotherapy agent vincristine (Oncovin). Vincristine appears to work by blocking the receptors on the macrophages and therefore inhibiting platelet destruction; it may also stimulate thrombopoiesis. Some data support the efficacy of certain monoclonal antibodies (eg, rituximab) in increasing platelet counts, but more research is needed (Stasi, Pagano, Stipa, & Amadori, 2001; Saleh et al., 2000). Another approach to the management of chronic ITP involves the use of anti-D (eg, WinRho) in patients who are Rh(D)-positive. The actual mechanism of action is unknown. One theory is that the anti-D binds to the patient’s RBCs, which are in turn destroyed by the body’s macrophages. While the macrophages destroy the anti-D/RBC complex, they are not able to destroy platelets. Anti-D produces a transient decreased hematocrit and increased platelet count in many, but not all, patients with ITP. Anti-D appears to be most effective in children with ITP and least effective in patients who have undergone splenectomy. Despite the extremely low platelet count, platelet transfusions are usually avoided. Transfusions tend to be ineffective because the patient’s anti-platelet antibodies bind with the transfused platelets, causing them to be destroyed. Platelet counts can actually drop after platelet transfusion. Occasionally, transfusion of platelets may protect against catastrophic bleeding in patients with severe wet purpura. Epsilon-aminocaproic acid (EACA; Amicar) may be useful for patients with significant mucosal bleeding refractory to other treatments. Nursing Management Nursing care for these patients should include an assessment of the patient’s life style to determine the risk of bleeding from activity. A careful medication history should also be obtained, including use of over-the-counter medications, herbs, and nutritional supplements. The nurse must be alert for sulfa-containing medications and medications that alter platelet function (eg, medications that contain aspirin or other NSAIDs). The nurse should assess for any history of recent viral illness and reports of headache or visual disturbances (which could be initial symptoms of intracranial bleeding). Patients who are admitted to the hospital with wet purpura and low platelet counts should have a neurologic assessment incorporated into their routine vital sign measurements. No intramuscular injections or rectal medications should be administered, and rectal temperature measurements should not be performed, because they can stimulate bleeding. Patient teaching should address signs of exacerbation of disease (petechiae, ecchymoses); how to contact appropriate health care personnel; the name and type of medication inducing ITP (if appropriate); current medical treatment (medications, tapering schedule if relevant, side effects); and the frequency of monitoring the platelet count. Patients should be instructed to avoid all agents that interfere with platelet function. The patient should avoid constipation, the Valsalva maneuver (eg, straining at stool), and flossing of the teeth. Electric razors should be used for shaving, and soft-bristled toothbrushes should replace stiff-bristled ones. Patients should also be counseled to refrain from vigorous sexual intercourse when the platelet count is less than 10,000/mm3. Patients who are receiving chronic corticosteroids are at risk for 913 complications including osteoporosis, proximal muscle wasting, cataract formation, and dental caries (see Chart 33-4). Bone mineral density should be monitored, and these patients may benefit from calcium and vitamin D supplementation and bisphosphonate therapy to prevent significant bone disease. PLATELET DEFECTS Quantitative platelet defects are relatively common (thrombocytopenia), but qualitative defects can also occur. With qualitative defects, the number of platelets may be normal, but the platelets do not function normally. Platelet function is most commonly evaluated by the bleeding time; however, this test is a crude measurement at best. An important functional platelet disorder is that induced by aspirin. Even small amounts of aspirin reduce normal platelet aggregation, and the prolonged bleeding time lasts for several days after aspirin ingestion. Although this does not cause bleeding in most people, patients with a coagulation disorder (eg, hemophilia) or thrombocytopenia can have significant bleeding after taking aspirin, particularly if invasive procedures or trauma has occurred. NSAIDs can also inhibit platelet function, but the effect is not as prolonged as with aspirin (about 5 days versus 7 to 10 days). Other causes of platelet dysfunction include end-stage renal disease, possibly from metabolic products affecting platelet function; MDS; multiple myeloma (due to abnormal protein interfering with platelet function); cardiopulmonary bypass; and other medications and substances (Chart 33-13). Clinical Manifestations Bleeding may be mild or severe. Its extent is not necessarily correlated with the platelet count or with tests that measure coagulation (prothrombin time [PT], partial thromboplastin time [PTT]). Ecchymoses are common, particularly on the extremities. Patients with platelet dysfunction may be at risk for significant bleeding after trauma or invasive procedures (eg, biopsy, dental extraction). Medical Management If the platelet dysfunction is caused by medication, use of the offending medication should be stopped, if possible, particularly when bleeding occurs. If platelet dysfunction is marked, bleeding can often be prevented by transfusion of normal platelets before invasive procedures. Amniocaproic acid (EACA; Amicar) may be required to prevent significant bleeding after such procedures. Nursing Management Patients with significant platelet dysfunction need to be instructed to avoid agents that can diminish platelet function, such as certain over-the-counter medications, herbs, nutritional supplements, and alcohol. They also need to be assisted to serve as their own advocates and to inform their health care providers (including dentists) of the underlying condition before any invasive procedure is performed, so that appropriate steps can be initiated to diminish the risk of bleeding. Bleeding precautions should be initiated as appropriate (see Chart 33-10). HEMOPHILIA Two inherited bleeding disorders—hemophilia A and hemophilia B—are clinically indistinguishable, although they can be distinguished by laboratory tests. Hemophilia A is caused by a 914 Unit 6 Chart 33-13 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION • PHARMACOLOGY Medications and Substances That Impair Platelet Function Anesthetic Agents Local anesthetics Halothane Antibiotics Beta-lactam antibiotics Penicillins Cephalosporins Nitrofurantoin Sulfonylureas Anticoagulation Agents Heparin Fibrinolytic agents Anti-inflammatory Agents (Nonsteroidal) Aspirin Ibuprofen Naproxen Antineoplastic Agents BCNU Daunorubicin Mithramycin Cardiovascular Drugs Beta-blockers Calcium channel blockers Isosorbide Nitroglycerine Nitroprusside Quinidine Medications That Increase Platelet CAMP Dipyridamole Prostacycline Theophylline Food and Food Additives Caffeine Chinese black tree fungus Clove Cumin Ethanol Fish oils Garlic Onion extract Turmeric Plasma Expanders Dextrans Hydroxyethyl starch Psychotropic Agents Tricyclic antidepressants Phenothiazines Miscellaneous Antihistamines Clofibrate Furosemide Heroin Contrast agents Ticlopidine Vitamin E Herbal Supplements Feverfew Ginger Ginko Ginseng Kava kava genetic defect that results in deficient or defective factor VIII; hemophilia B (also called Christmas disease) stems from a genetic defect that causes deficient or defective factor IX. Hemophilia is a relatively rare disease; hemophilia A, which occurs in 1 of every 10,000 births, is three times more common than hemophilia B. Both types of hemophilia are inherited as X-linked traits, so almost all affected people are males; females can be carriers but are almost always asymptomatic. The disease is recognized in early childhood, usually in the toddler age group. However, patients with mild hemophilia may not be diagnosed until they experience severe trauma (eg, a high-school football injury) or surgery. Hemophilia occurs in all ethnic groups. Clinical Manifestations The disease, which can be severe, is manifested by hemorrhages into various parts of the body. Hemorrhage can occur even after minimal trauma. The frequency and severity of the bleeding depend on the degree of factor deficiency as well as the intensity of the precipitating trauma. For example, patients who have a mild factor VIII deficiency (ie, 6% to 50% of normal levels) rarely develop hemorrhage spontaneously; hemorrhage tends to occur secondary to trauma. In contrast, spontaneous hemorrhages, particularly hemarthroses and hematomas, can frequently occur in patients with severe factor VIII deficiency (ie, less than 1% of normal levels). These patients require frequent factor replacement therapy. About 75% of all bleeding in patients with hemophilia occurs into joints. The most commonly affected joints are the knees, elbows, ankles, shoulders, wrists, and hips. Patients often note pain in a joint before they are aware of swelling and limitation of motion. Recurrent joint hemorrhages can result in damage so severe that chronic pain or ankylosis (fixation) of the joint occurs. Many patients with severe factor deficiency are crippled by the joint damage before they become adults. Hematomas can be superficial or deep hemorrhages into muscle or subcutaneous tissue. With severe factor deficiency, they can occur without known trauma and progressively extend in all directions. When the hematomas occur within muscle, particularly in the extremities, peripheral nerves can be compressed. Over time, this compression results in decreased sensation, weakness, and atrophy of the area involved. Spontaneous hematuria and gastrointestinal bleeding can occur. Bleeding is also common in other mucous membranes, such as the nasal passages. The most dangerous site of hemorrhage is in the head (intracranial or extracranial). Any head trauma requires prompt evaluation and treatment. Surgical procedures typically result in excessive bleeding at the surgical site. Because clot formation is poor, wound healing is also poor. Such bleeding is most commonly associated with dental extraction. Medical Management In the past, the only treatment for hemophilia was infusion of fresh frozen plasma, which had to be administered in such large quantities that patients experienced fluid volume overload. Now factor VIII and factor IX concentrates are available to all blood banks. Recombinant forms of these factors have been made available and may diminish the use of factor concentrates. Patients are given concentrates when they are actively bleeding or as a preventive measure before traumatic procedures (eg, lumbar puncture, dental extraction, surgery). The patient and family are taught how to administer the concentrate intravenously at home at the first sign of bleeding. It is crucial to initiate treatment as soon as possible so that bleeding complications can be avoided. A few patients eventually develop antibodies to the concentrates, so their factor levels cannot be increased. Treatment of this problem is extremely difficult and often unsuccessful. Aminocaproic acid (EACA; Amicar) is a fibrinolytic enzyme inhibitor that can slow the dissolution of blood clots that do form; it is very effective as an adjunctive measure after oral surgery. It is also useful in treating mucosal bleeding. Another agent, desmopressin (eg, DDAVP), induces a transient rise in factor VIII levels; the mechanism for this response is unknown. In patients with mild forms of hemophilia A, desmopressin is extremely useful, significantly reducing the amount of blood products required. However, desmopressin is not effective in patients with severe factor VIII deficiency. Nursing Management Most patients with hemophilia are diagnosed as children. They often require assistance in coping with the condition because it is chronic, places restrictions on their lives, and is an inherited disorder that can be passed to future generations. From childhood, patients are helped to accept themselves and the disease and to identify the positive aspects of their lives. They are encouraged to be Chapter 33 Assessment and Management of Patients With Hematologic Disorders 915 self-sufficient and to maintain independence by preventing unnecessary trauma that can cause acute bleeding episodes and temporarily interfere with normal activities. As they work through their feelings about the condition and progress to accepting it, they can assume more and more responsibility for maintaining optimal health. Patients with mild factor deficiency may not be diagnosed until adulthood if they do not experience significant trauma or surgery as children. These patients need extensive teaching about activity restrictions and self-care measures to diminish the chance of hemorrhage and complications of bleeding. The nurse should emphasize safety at home and in the workplace. Patients with hemophilia are instructed to avoid any agents that interfere with platelet aggregation, such as aspirin, NSAIDs, herbs, nutritional supplements, and alcohol. This restriction applies to over-the-counter medications such as cold remedies. Dental hygiene is very important as a preventive measure because dental extractions are so hazardous. Applying pressure may be sufficient to control bleeding resulting from minor trauma if the factor deficiency is not severe. Nasal packing should be avoided, because bleeding frequently resumes when the packing is removed. Splints and other orthopedic devices may be useful in patients with joint or muscle hemorrhages. All injections should be avoided; invasive procedures (eg, endoscopy, lumbar puncture) should be minimized or performed after administration of appropriate factor replacement. Patients with hemophilia should be encouraged to carry or wear medical identification. During hemorrhagic episodes, the extent of bleeding must be assessed carefully. Patients who are at risk for significant compromise (eg, bleeding into the respiratory tract or brain) warrant close observation and systematic assessment for emergent complications (eg, respiratory distress, altered level of consciousness). If the patient has had recent surgery, the nurse frequently and carefully assesses the surgical site for bleeding. Frequent vital sign monitoring is needed until the nurse is certain that there is no excessive postoperative bleeding. Analgesics are commonly required to alleviate the pain associated with hematomas and hemorrhage into joints. Many patients report that warm baths promote relaxation, improve mobility, and lessen pain. However, during bleeding episodes, heat, which can accentuate bleeding, is avoided; applications of cold are used instead. Although recent technology (ie, the formulation of heatsolvent or detergent-treated factor concentrates) has rendered factor VIII and IX preparations free from viruses such as HIV and hepatitis, many patients have already been exposed to these infections. These patients and their families may need assistance in coping with the diagnosis and the consequences of these infections. Between 15% and 50% of patients with hemophilia A and between 1% and 3% of patients with hemophilia B develop antibodies (inhibitors) to factor concentrates, complicating factor replacement management (Lusher, 2000; White, Greenwood, Escobar, & Frelinger, 2000). These patients may require plasmapheresis or concurrent immunosuppressive therapy, particularly in the setting of significant bleeding. Patients with severe factor deficiency should be screened for antibodies, particularly before major surgery. essary for platelet adhesion at the site of vascular injury. Although synthesis of factor VIII is normal, its half-life is shortened; therefore, factor VIII levels commonly are mildly low (15% to 50% of normal). VON WILLEBRAND’S DISEASE VITAMIN K DEFICIENCY Von Willebrand’s disease, a common bleeding disorder affecting males and females equally, is usually inherited as a dominant trait. The disease is caused by a deficiency of von Willebrand factor (vWF), which is necessary for factor VIII activity. vWF is also nec- The synthesis of many coagulation factors depends on vitamin K. Vitamin K deficiency is typical in malnourished patients, and some antibiotics decrease the intestinal flora that produce vitamin K, depleting vitamin K stores. Administration of vitamin K ( phyton- Clinical Manifestations Patients commonly have nosebleeds, excessively heavy menses, bleeding from cuts, and postoperative bleeding, although they do not suffer from massive soft tissue or joint hemorrhages. As the laboratory values fluctuate, so does the bleeding. For example, a careful history of prior bleeding may show little problem with postoperative bleeding on one occasion but significant bleeding from a dental extraction at another time. Assessment and Diagnostic Findings Laboratory test results show a normal platelet count but prolonged bleeding time and slightly prolonged PTT. These defects are not static, and laboratory test results can vary widely within the same patient over time. Medical Management Both the factor deficiency and the platelet impairment can be corrected by administration of cryoprecipitate, which contains factor VIII, fibrinogen, and factor XIII (or fresh frozen plasma, if cryoprecipitate is unavailable). Replacement continues for several days to ensure correction of the factor VIII deficiency; up to 7 to 10 days of treatment may be necessary after major surgery. Desmopressin (DDAVP), a synthetic vasopressin analog, can be used to prevent bleeding associated with dental or surgical procedures or to manage mild bleeding after surgery. Desmopressin provides a transient increase in factor VIII coagulant activity and may also correct the bleeding time. It can be administered as an intravenous infusion or intranasally. With major surgery or invasive procedures, both desmopressin and cryoprecipitate may be needed to prevent hemorrhage. Acquired Coagulation Disorders LIVER DISEASE With the exception of factor VIII, most blood coagulation factors are synthesized in the liver. Therefore, hepatic dysfunction (due to cirrhosis, tumor, or hepatitis; see Chap. 39) can result in diminished amounts of the factors needed to maintain coagulation and hemostasis. Prolongation of the PT, unless it is caused by vitamin K deficiency, may indicate severe hepatic dysfunction. Although minor bleeding is common (eg, ecchymoses), these patients are also at risk for significant bleeding, related especially to trauma or surgery. Transfusion of fresh frozen plasma may be required to replace clotting factors and to prevent or stop bleeding. Patients may also have life-threatening hemorrhage from peptic ulcers or esophageal varices. In these cases, replacement with fresh frozen plasma, PRBCs, and platelets is usually required. 916 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION azdione [eg, Mephyton], either orally or as a subcutaneous injection) can correct the deficiency quickly; adequate synthesis of coagulation factors is reflected by normalization of the PT. COMPLICATIONS OF ANTICOAGULANT THERAPY Anticoagulants are used in the treatment or prevention of thrombosis. These agents, particularly warfarin or heparin, can result in bleeding. If the PT or PTT is longer than desired and bleeding has not occurred, the medication can be stopped or the dose decreased. Vitamin K is administered for warfarin toxicity. Protamine sulfate is rarely needed for heparin toxicity, because the half-life of heparin is very short. With significant bleeding, fresh frozen plasma replaces the vitamin K–dependent coagulation factors. Other complications of anticoagulant therapy are discussed in Chapter 31. DISSEMINATED INTRAVASCULAR COAGULATION (DIC) DIC is not a disease but a sign of an underlying condition. DIC may be triggered by sepsis, trauma, cancer, shock, abruptio placentae, toxins, or allergic reactions (Chart 33-14). It is potentially life-threatening. Pathophysiology In DIC, the normal hemostatic mechanisms are altered so that a massive amount of tiny clots forms in the microcirculation. Initially, the coagulation time is normal. However, as the platelets and clotting factors are consumed to form the microthrombi, coagulation fails. Thus, the paradoxical result of excessive clotting is bleeding. The clinical manifestations of DIC are reflected in the organs, which are affected either by excessive clot formation (with resultant ischemia to all or part of the organ) or by bleeding. The bleeding is characterized by low platelet and fibrinogen levels; prolonged PT, PTT, and thrombin time; and elevated fibrin degradation products (D-dimers) (Table 33-6). The mortality rate can exceed 80% of patients who develop DIC. Identification of patients who are at risk for DIC and recognition of the early clinical manifestations of this syndrome can result in earlier medical intervention, which may improve the prognosis. However, the primary prognostic factor is the ability to treat the underlying condition that precipitated DIC. Clinical Manifestations Patients with DIC may bleed from mucous membranes, venipuncture sites, and the gastrointestinal and urinary tracts. The bleeding can range from minimal occult internal bleeding to profuse hem- Chart 33-14 Risk Factors for Disseminated Intravascular Coagulation (DIC) Sepsis Obstetric complications Acute hemolysis (eg, transfusion reaction) Trauma Shock Cancer (especially prostate cancer and acute promyelocytic leukemia) Allergic reactions orrhage from all orifices. Patients may also develop organ dysfunction, such as renal failure and pulmonary and multifocal central nervous system infarctions as a result of microthromboses, macrothromboses, or hemorrhages. During the initial process of DIC, the patient may have no new symptoms, the only manifestation being a progressive decrease in the platelet count. As the thrombosis becomes more extensive, the patient exhibits signs and symptoms of thrombosis in the organs involved. Then, as the clotting factors and platelets are consumed to form these thrombi, bleeding occurs. Initially the bleeding is subtle, but it can develop into frank hemorrhage. Signs and symptoms depend on the organs involved and are listed in Table 33-7. Medical Management The most important management issue is treating the underlying cause of the DIC. Until the cause is controlled, the mechanism for DIC will persist. A second goal is to correct the secondary effects of tissue ischemia by improving oxygenation, replacing fluids, correcting electrolyte imbalances, and administering vasopressor medications. If serious hemorrhage occurs, the depleted coagulation factors and platelets may be replaced to reestablish the potential for normal hemostasis and thereby diminish bleeding. Cryoprecipitate is given to replace fibrinogen and factors V and VII; fresh frozen plasma is administered to replace other coagulation factors. A controversial method to interrupt the thrombosis process is the use of heparin infusion. Heparin may inhibit the formation of microthrombi and thus permit perfusion of the organs (skin, kidneys, or brain) to resume. Heparin is typically reserved for the patient in whom thrombotic manifestations predominate or in whom extensive blood component replacement fails to halt the hemorrhage or increase fibrinogen and other clotting levels. When heparin is administered, bleeding may actually worsen initially until the thrombotic process is interrupted. Consumed platelets and clotting factors need to be replaced. The effectiveness of heparin can best be determined by observing for normalization of the plasma fibrinogen concentration and diminishing signs of bleeding. NURSING PROCESS: THE PATIENT WITH DISSEMINATED INTRAVASCULAR COAGULATION (DIC) Assessment Nurses need to be aware of patients who are at risk for DIC. Sepsis and acute promyelocytic leukemia are the most common causes of DIC. Patients need to be assessed thoroughly and frequently for signs and symptoms of thrombi and bleeding and monitored for any progression of these signs (see Table 33-7). Diagnosis NURSING DIAGNOSES Based on the assessment data, major nursing diagnoses for the patient with DIC may include the following: • Risk for deficient fluid volume related to bleeding • Risk for impaired skin integrity related to ischemia or bleeding • Potential for excess fluid volume related to excessive blood/ factor component replacement • Ineffective tissue perfusion related to microthrombi • Anxiety and fear of the unknown and possible death Chapter 33 Table 33-6 Assessment and Management of Patients With Hematologic Disorders 917 • Laboratory Values Commonly Found in Disseminated Intravascular Coagulation (DIC)* TEST FUNCTION EVALUATED NORMAL RANGE Platelet count Prothrombin time (PT) Partial thromboplastin time (PTT) Thrombin time (TT) Fibrinogen D-dimer Fibrin degradation products (FDPs) Euglobulin clot lysis Platelet number Extrinsic pathway Intrinsic pathway Clot formation Amount available for coagulation Local fibrinolysis Fibrinolysis Fibrinolytic activity CHANGES IN DIC 3 150,000–450,000/mm 11–12.5 sec 23–35 sec 8–11 sec 170–340 mg/dL 0–250 ng/mL 0–5 µg/mL ≥2 hours ↓ ↑ ↑ ↑ ↓ ↑ ↑ ≤1 hour *Because DIC is a dynamic condition, the laboratory values measured will change over time. Therefore, a progressive increase or decrease in a given laboratory value is likely to be more important than the actual value of a test at a single point in time. COLLABORATIVE PROBLEMS/ POTENTIAL COMPLICATIONS Collaborative problems include the clinical conditions that precipitated the DIC. Based on the assessment data, potential complications may include: • • • • • • Renal failure Gangrene Pulmonary embolism or hemorrhage Altered level of consciousness Acute respiratory distress syndrome Stroke Planning and Goals Major patient goals include maintenance of hemodynamic status, maintenance of intact skin and oral mucosa, mainte- Table 33-7 nance of fluid balance, maintenance of tissue perfusion, enhanced coping, and absence of complications (see Plan of Nursing Care). Nursing Interventions See Plan of Nursing Care: The Patient with Disseminated Intravascular Coagulation. MONITORING AND MANAGING POTENTIAL COMPLICATIONS Despite aggressive measures, the lack of renal perfusion may result in acute renal failure, sometimes necessitating dialysis. Placement of a large-bore dialysis catheter is extremely hazardous in this patient population and should be accompanied by adequate platelet and plasma transfusions. • Recognizing Thrombosis and Bleeding in Disseminated Intravascular Coagulation (DIC)* SIGNS AND SYMPTOMS OF MICROVASCULAR THROMBOSIS SIGNS AND SYMPTOMS OF MICROVASCULAR AND FRANK BLEEDING Integumentary system (skin) ↓ Temperature, sensation; ↑ pain; cyanosis in extremities, nose, earlobes; focal ischemia, superficial gangrene Circulatory system Respiratory system ↓ Pulses; capillary filling time > 3 sec Hypoxia (secondary to clot in lung); dyspnea; chest pain with deep inspiration; ↓ breath sounds over areas of large embolism Gastric pain; “heartburn” Petechiae, including periorbital and oral mucosa; bleeding: gums, oozing from wounds, previous injection sites, around catheters (IVs, tracheostomies); epistaxis; diffuse ecchymoses; subcutaneous hemorrhage; joint pain Tachycardia High-pitched bronchial breath sounds; tachypnea; ↑ consolidation; signs and symptoms of acute respiratory distress syndrome Hematomesis (heme⊕† NG output) melana (heme⊕ stools → tarry stools → bright-red blood from rectum) retroperitoneal bleeding (abdomen firm and tender to palpation; distended; ↑ abdominal girth) Hematuria SYSTEM Gastrointestinal system Renal system Neurologic system ↓ Urine output; ↑ creatinine, ↑ blood urea nitrogen ↓ Alertness and orientation; ↓ pupillary reaction; ↓ response to commands; ↓ strength and movement ability Anxiety; restlessness; ↓ mentation, altered level of consciousness; headache; visual disturbances; conjunctival hemorrhage *Note: Signs of microvascular thrombosis are the result of an inappropriate activation of the coagulation system, causing thrombotic occlusion of small vessels within all body organs. As the clotting factors and platelets are consumed, signs of microvascular bleeding appear. This bleeding can quickly extend into frank hemorrhage. Treatment must be aimed at the disorder underlying the DIC; otherwise, the stimulus for the syndrome will persist. †heme⊕, positive for hemoglobin Plan of Nursing Care The Patient With Disseminated Intravascular Coagulation (DIC) Nursing Interventions Rationale Expected Outcomes Nursing Diagnosis: Potential for fluid volume deficit related to bleeding Goals: Hemodynamic status maintained Urine output ≥30 mL/hr 1. Avoid procedures/activities that can increase intracranial pressure (eg, coughing, straining to have a bowel movement). 2. Monitor vital signs closely, including neurologic checks: a. Monitor hemodynamics b. Monitor abdominal girth c. Monitor urine output 3. Avoid medications that interfere with platelet function if possible (eg, ASA, NSAIDs, beta-lactam antibiotics). 4. Avoid rectal probes, rectal medications. 5. Avoid IM injections. 6. Monitor amount of external bleeding carefully a. Monitor number of dressings, % of dressing saturated; time to saturate a dressing is more objective than “dressing saturated a moderate amount.” b. Monitor suction output, all excreta c. Monitor pad counts in menstruating females. d. Females may receive progesterone to prevent menses. 7. Use low pressure with any suctioning needed. 8. Administer oral hygiene carefully. a. Avoid lemon-glycerine swabs, hydrogen peroxide, commercial mouthwashes. b. Use sponge-tipped swabs, salt/baking soda (bicarbonate of soda) mouth rinses. 9. Avoid dislodging any clots, including those around IV sites and injection sites. 1. Prevents intracranial bleeding. • Level of consciousness (LOC) stable • CVP 5–12 cm H2O, systolic BP ≥ 2. Identifies signs of hemorrhage/shock quickly. • • • • • • • • 3. Decreases problems with platelet aggregation and adhesion. 4. Decreases chance for rectal bleeding. 5. Decreases chance for intramuscular bleeding. 6. 70 mm Hg Urine output ≥ 30 mL/hour Decreased bleeding Decreased oozing Decreased ecchymoses Amenorrhea Absence of oral and bronchial bleeding Oral mucosa clean, moist, intact Absence of bleeding a. Provides accurate, objective assessment of extent of bleeding. b. Identifies presence of or quantifies extent of bleeding. c. Quantifies extent of bleeding d. Decreases chance for gynecologic source of hemorrhage. 7. Prevents excessive trauma that could cause bleeding. 8. Prevents excessive trauma that could cause bleeding. Glycerin and alcohol (in commercial mouthwashes) will dry mucosa, increasing risk for bleeding. 9. Prevents excessive bleeding at sites. Nursing Diagnosis: Potential for impaired skin integrity secondary to ischemia or bleeding Goals: Skin integrity remains intact; oral mucosa remains intact 1. Assess skin, with particular attention to bony prominences, skin folds. 2. Reposition carefully; use pressure-reducing mattress. 3. Perform careful skin care every 2 hr, emphasizing dependent areas, all bony prominences, perineum. 4. Use lamb’s wool between digits, around ears, as needed. 5. Use prolonged pressure after injection or procedure when such measures must be performed (at least 5 min) 6. Administer oral hygiene carefully (see above). 1. Prompt identification of any area at risk for skin breakdown or showing early signs of breakdown can facilitate prompt intervention and thus prevent complications. 2–4. Meticulous skin care and use of measures to prevent pressure on bony prominences decrease the risk of skin trauma. • Skin integrity remains intact; skin is warm, and of normal color • Oral mucosa is intact, pink, moist, without bleeding 5. Initial platelet plug is very unstable and easily dislodged, which can lead to increased bleeding. 6. Metriculous care to decreased trauma, bleeding, and risk of infection. (continued) 918 Chapter 33 Assessment and Management of Patients With Hematologic Disorders 919 Plan of Nursing Care The Patient With Disseminated Intravascular Coagulation (DIC) (Continued) Nursing Interventions Rationale Expected Outcomes Nursing Diagnosis: Potential for fluid volume excess Goals: Absence of edema; absence of rales; Intake not greater than output 1. Auscultate breath sounds every 2–4 hr. 2. Monitor extent of edema 3. Monitor volume of IVs, blood products; decrease volume of IV medications if possible. 4. Administer diuretics as prescribed 1. Crackles can develop quickly. 2. Fluid may extend beyond intravascular system. 3. Helps prevent fluid overload. • • • • Breath sounds clear Absence of edema Intake does not exceed output Weight stable 4. Decreases fluid volume. Nursing Diagnosis: Potential for diminished tissue perfusion secondary to microthrombi Goals: Neurologic status remains intact; absence of hypoxemia; peripheral pulses remain intact; skin integrity remains intact; urine output remains ≥30 mL/hr 1. Assess neurologic, pulmonary, integumentry systems. 2. Monitor response to heparin therapy. 3. Assess extent of bleeding. 4. Monitor fibrinogen levels. 5. Stop ⑀-aminocaproic acid (EACA) if symptoms of thrombosis occur (see Table 33-7). 1. Initial signs of thrombosis can be subtle. • Arterial blood gases, O2 saturation, pulse 2. Response to heparin is most accurately reflected in fibrinogen level. 3. Objective measurements of all sites of bleeding are crucial to accurately assess extent of blood loss. 4. Response to heparin is most accurately reflected in fibrinogen level. 5. EACA should be used only in setting of extensive hemorrhage not responding to replacement therapy. • • • • oximetry, LOC within normal limits. Breath sounds clear Absence of edema Intake does not exceed output Weight stable Nursing Diagnosis: Potential for fear of unknown and possible death Goal: Fears verbalized/identified; maintain realistic hope 1. Identify previous coping mechanisms, if possible: a. Encourage patient to use them as appropriate. 2. Explain all procedures and rationale for these in terms patient and family can understand. 3. Assist family in supporting patient. 4. Use services from behavioral medicine, chaplain as needed. 1. Identifying previous stressful situations can aid in recall of successful coping mechanisms. • Previously used coping strategies identified and tried, to extent patient is able to do so • Patient indicates understanding of procedures and situation as condition permits 2. Decreased knowledge and uncertainty can increase anxiety. 3. Family can be useful in assisting patient to use coping strategies and to maintain hope. 4. Additional professional intervention may be necessary, particularly if previous coping mechanisms are maladaptive or ineffective. Spiritual dimension should be supported. Evaluation See the Plan of Nursing Care for evaluation and expected outcomes for the patient with DIC. THROMBOTIC DISORDERS As in many bleeding disorders, several conditions can alter the balance within the normal hemostasis process and cause exces- sive thrombosis. Abnormalities that predispose a person to thrombotic events include decreased clotting inhibitors within the circulation (which enhances coagulation), altered hepatic function (which may decrease production of clotting factors or clearance of activated coagulation factors), lack of fibrinolytic enzymes, and tortuous vessels (which promote platelet aggregation). Thrombosis can be caused by more than one predisposing factor. Several conditions can result from thrombosis, such as myocardial infarction (see Chap. 28), cerebral vascular accident 920 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION (CVA, brain attack, or stroke; see Chap. 62), and peripheral arterial occlusion (see Chap. 31). Several inherited or acquired deficiency conditions, including hyperhomocystinemia, antithrombin III (AT III) deficiency, Protein C deficiency, activated Protein C (APC) resistance, factor V Leiden, and Protein S deficiency can predispose a patient to repeated episodes of thrombosis; they are referred to as hypercoagulable states or thrombophilia. Table 33-8 delineates these disorders, their abnormal laboratory values, and the need for family testing. Thrombosis requires anticoagulation therapy. The duration of therapy varies with the location and extent of the thrombosis, precipitating events (eg, trauma, immobilization), and concurrent risk factors (eg, use of oral contraceptives, tortuous blood vessels, history of thrombotic events). HYPERHOMOCYSTINEMIA Increased plasma levels of homocystine are a significant risk factor not only for venous thrombosis (eg, deep venous thrombosis [DVT], pulmonary embolism) but also for arterial thrombosis (eg, stroke, myocardial infarction). This disorder can be heredi- Table 33-8 DISORDER • Hypercoaguable States ABNORMAL LABORATORY VALUE* Inherited Disorders (Family Testing Necessary) Hyperhomocysteinemia Homocystine ↑ after methionine load Antithrombin III (AT III) AT III ↓ deficiency Protein C deficiency Protein C activity ↓ (must be measured off warfarin [Coumadin]) Activated protein C (APC) Must be measured off anticoagulant; resistance <2× prolongation of PTT when APC added. Patients with APC resistance have a smaller increase in clotting time than normal (ie, the prolongation of clotting time is less than normal). Positive Factor V Leiden Protein S activity ↓; must be meaProtein S deficiency sured off warfarin (Coumadin) Dysfibrinogenemia ↑ thrombin time; ↑ reptilase time; ↓ functional fibrinogen; often requires special fibrinogen assays Acquired Disorders (Family Testing Unnecessary) Positive Anticardiolipin antibody Cancer Positive Lupus anticoagulant Homocystine ↑ after methionine Hyperhomocysteinemia load AT III ↓ AT III Deficiency + Hamm’s test; acid hemolysis Paroxysmal nocturnal hemoglobinuria Varied, depending on disorder Myeloproliferative disorders Varied, depending on disorder Nephrotic syndrome Varied, depending on disorder Cancer chemotherapy *Protein C and protein S are vitamin K–dependent proteins. Warfarin (Coumadin) interferes with the hepatic synthesis of vitamin K-dependent factors, which may decrease levels of protein C or protein S; therefore, protein C and protein S should be measured while the patient is off warfarin. tary, or it can result from a nutritional deficiency of folic acid and, to a lesser extent, of vitamin B12 and B6, because these vitamins are cofactors in homocystine metabolism. For unknown reasons, people who are elderly, have renal failure, or smoke tobacco may also have elevated levels of homocystine in the absence of nutritional deficiencies of these vitamins. Although a simple fasting measurement of plasma homocystine can serve as a useful screening test, people with heterozygous defects in this gene and those who are vitamin B6 deficient may have normal or minimally elevated levels. A much more sensitive method involves obtaining a second measurement 4 hours after the patient consumes methionine; the prevalence of hyperhomocystinemia is twice as great when this method is used. In hyperhomocystinemia, the endothelial lining of the vessel walls is denuded; this can precipitate unnecessary thrombus formation. Recent studies have determined that this disorder is much more common than previously thought. In a long-term epidemiologic study on nurses’ health (Rimm et al., 1998), women who used dietary supplements with folic acid and vitamin B6 were found to have a lower incidence of thrombotic conditions such as DVT. Patients who are found to have hyperhomocystinemia should receive folic acid, B6, and/or B12 supplements and should be instructed in the rationale for their use to enhance compliance. ANTITHROMBIN III DEFICIENCY Antithrombin is a protein that inhibits thrombin and certain coagulation factors. AT III deficiency is an extremely common hereditary condition that can cause venous thrombosis, particularly when the level is less than 60% of normal. Patients with AT III deficiency can develop venous thrombosis as young adults; by 50 years of age, two thirds of patients with AT III deficiency have developed a venous thrombosis. The most common sites for thrombosis are the deep veins of the leg and the mesentery. Recurrent thrombosis often occurs. There is an increased resistance to heparin anticoagulation, so these patients may require greater amounts of heparin to achieve adequate anticoagulation. Patients with ATIII deficiency should be encouraged to have their family members tested for the deficiency. PROTEIN C DEFICIENCY Protein C is an enzyme that, when activated, inhibits coagulation. When levels of Protein C are deficient, the risk of thrombosis increases, and thrombosis can often occur spontaneously. Protein C deficiency is at least as prevalent as AT III deficiency, and people who are Protein C–deficient can develop thrombosis early in life, as early as 15 years of age. Warfarin-induced skin necrosis is a rare but significant complication of anticoagulation management in patients with Protein C deficiency (Hoffman et al., 2000). This complication appears to result from progressive thrombosis in the capillaries within the skin; the extent of the necrosis can be extreme. ACTIVATED PROTEIN C RESISTANCE AND FACTOR V LEIDEN MUTATION Activated protein C (APC) resistance is a common condition that can occur with other hypercoagulable states. APC is an anticoagulant, and resistance to APC increases the risk for venous thrombosis. A molecular defect in the factor V gene has been identified in Chapter 33 Assessment and Management of Patients With Hematologic Disorders most (90%) of those with APC resistance; this defect is called factor V Leiden mutation. It has been identified as the most common cause of inherited hypercoagulability in Caucasians, but its incidence appears to be much lower in other ethnic groups. Factor V Leiden mutation synergistically increases the risk for thrombosis in patients with other risk factors (eg, use of oral contraceptives, hyperhomocystinemia, increased age). It does not appear that the use of postmenopausal hormone therapy in women increases the risk for thrombotic events as does the use of oral contraceptives; the dose of estrogen in the former situation is much lower than in the latter. People who are homozygous for the factor V Leiden mutation are at extremely high risk for thrombosis. PROTEIN S DEFICIENCY Protein S is another natural anticoagulant normally produced in the liver. APC requires Protein S to inactivate certain clotting factors. When the level of Protein S is deficient, this inactivation process is diminished, and the risk for thrombosis can be increased. Like patients with Protein C deficiency, those with Protein S deficiency have a greater risk for recurrent venous thrombosis at a young age, as young as 15 years. ACQUIRED THROMBOPHILIA Antibodies to phospholipids are common, acquired causes for thrombophilia (hypercoagulable states). The most common antibodies present against phospholipids are either lupus or anticardiolipin antibodies. Both of these antibodies can be transient, resulting from infection or certain medications. Most thrombotic events are venous, but arterial thrombosis can occur in up to one third of the cases. Patients who persistently test positive for either antibody and who have had a thrombotic event are at significant risk for recurrent thrombosis (greater than 50%). Recurrent thromboses tend to be of the same type—that is, venous thrombosis after an initial venous thrombosis, arterial thrombosis after an initial arterial thrombosis. Another common acquired cause for thrombophilia is cancer. Specific types of stomach, pancreatic, lung, and ovarian cancers are most commonly associated with thrombophilia. The type of thrombosis that results is unusual. Rather than deep vein thrombosis or pulmonary embolism, the thrombosis occurs in unusual sites, such as the portal, hepatic, or renal vein or the inferior vena cava. Migratory superficial thrombophlebitis or nonbacterial thrombotic endocarditis can also occur. In these patients, anticoagulation can be difficult to manage in that the thrombosis can progress despite standard amounts of anticoagulation. Medical Management The primary method of treating thrombotic disorders is anticoagulation. However, in thrombophilic conditions, when to treat (prophylaxis or not) and how long to treat (lifelong or not) can be controversial. Anticoagulation therapy is not without risks; the most significant risk is bleeding. Risks of anticoagulation therapy are identified in Chapter 31. The most common anticoagulant medications are identified in the following section. PHARMACOLOGIC THERAPY Along with administering anticoagulant therapy, concerns include minimizing any risk factors that predispose a patient to 921 thrombosis. When risk factors (eg, immobility after surgery, pregnancy) cannot be avoided, prophylactic anticoagulation may be necessary. Unfractionated Heparin Therapy. Heparin is a naturally occurring anticoagulant that enhances AT III and inhibits platelet function. To prevent thrombosis, heparin is typically given as a subcutaneous injection, two or three times daily. To treat thrombosis, heparin is usually administered intravenously. The therapeutic effect of heparin is monitored by serial measurements of the activated partial prothrombin time; the dose is adjusted to maintain the range at 1.5 to 2.5 times the laboratory control. Oral forms are being evaluated, but their absorption remains variable (Money & York, 2001). A significant potential complication of heparin-based therapy is heparin-induced thrombocytopenia (HIT). Antibodies are formed within the body against the heparin complex. The actual incidence of HIT is unknown, but it is thought to occur in as many as 5% patients receiving heparin (Kelton, 1999). Whereas most patients remain asymptomatic, a significant proportion of those individuals with serologic HIT develop actual thrombocytopenia. A decline in platelet count typically develops after 5 to 8 days of heparin therapy, and the platelets can drop significantly, although in most instances the level stays higher than 50,000/mm3. These patients are at increased risk for thrombosis, either venous or arterial, and the thrombosis can range from DVT to myocardial infarction, CVA (brain attack, stroke), and ischemic damage to an extremity necessitating amputation. The risk for development of HIT appears to be increased when heparin is used at higher concentrations (ie, therapeutic versus prophylactic dosage) and with preexisting comorbidity, such as underlying cardiac disease. Low-Molecular-Weight Heparin Therapy. Low molecular-weight heparin (LMWH; eg, Dalteparin, Enoxaparin) is a special form of heparin that has a more selective effect on coagulation. Based on its biochemical properties, LMWH has a longer half-life and a less variable anticoagulant response than does standard heparin. These differences permit LMWH to be safely administered only once or twice daily, without the need for laboratory monitoring for dose adjustments. The incidence of HIT is much lower when LMWH is used. In certain conditions, the use of LMWH has allowed anticoagulation therapy to be moved entirely to the outpatient setting. Many cases of uncomplicated DVT are being managed outside the hospital setting. LMWH is also being increasingly used as “bridge therapy” when patients receiving anticoagulation therapy (warfarin) require an invasive procedure (eg, biopsy, surgery). In this situation, warfarin is stopped and LMWH is used in its place until the procedure is completed. After the procedure, warfarin therapy is resumed. LMWH is discontinued after a therapeutic level of warfarin is achieved. Warfarin (Coumadin) Therapy. Coumarin anticoagulants (warfarin; eg, Coumadin) are antagonists of vitamin K and therefore interfere with the synthesis of vitamin K–dependent clotting factors. Coumarin anticoagulants bind to albumin, are metabolized in the liver, and have an extremely long half-life. Typically, a patient is initially treated with both heparin (either the unfractionated form or LMWH) and warfarin. When the international normalized ratio (INR) reaches the desired therapeutic range, the heparin is stopped. The dosage required to maintain the thera- 922 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION peutic range (typically using an INR of 2.0 to 3.0) varies widely among patients and even within the same patient. Frequent monitoring of the INR is extremely important so that the dosage of warfarin can be adjusted as needed. Warfarin is affected by many medications; consultation with a pharmacist is important to assess the extent to which concurrently administered medications, herbs, and nutritional supplements may interact with warfarin. It is also affected by many foods, so patients need dietary instruction and may benefit from consultation with a dietitian when receiving warfarin therapy. See Chart 33-15 for a listing of agents that interact with warfarin. Nursing Management Patients with thrombotic disorders should avoid activities that promote circulatory stasis (eg, immobility, crossing the legs). Exercise, especially ambulation, should be performed frequently throughout the day, particularly during long trips by car or plane. Chart 33-15 • PHARMACOLOGY Agents That Interact with Warfarin (Coumadin) Although warfarin (Coumadin), an anticoagulant medication, is commonly used to treat and prevent thrombosis, many drug–drug and drug–food interactions are associated with its use. A careful medication history (including over-the-counter medications, herbs, and other substances, such as vitamins and minerals) is important when oral anticoagulation therapy is prescribed. Consultation with a pharmacist is recommended to assess the extent to which concurrent medications may affect the anticoagulant and for appropriate dosage adjustments. The following list contains a few examples of agents that interact with warfarin. Agents That Inhibit Warfarin Function Glutethimide Barbiturates Griseofulvin Carbamazepine Haloperidol Cholestyramine Oral contraceptives Corticosteroids Phenytoin Digitalis Rifampin Estrogens Spironolactone Ethanol Agents That Potentiate Warfarin Function Gingko Acetaminophen Allopurinol Ginseng Amiodarone Vitamin C (in very large doses) Anabolic steroids Vitamin E (in very large doses) Anti-inflammatory agents Isoniazid Antimalarial agents Mefenamic acid Aspirin Methotrexate Broad-spectrum antibiotics Metronidazole Chloral hydrate Oral hypoglycemic agents Chloramphenicol Oxyphenbutazone Cimetidine Phenytoin Colchicine Probenecid Clofibrate Propylthiouracil Chlorpromazine Quinidine Danazol Quinine Disulfiram Salicylates Ethacrynic acid Sulfinpyrazone Feprazone Sulfonamides (long-acting) Herbal medicines Thyroxine Feverfew Triclofos Garlic Tricyclic antidepressants Medications that alter platelet aggregation, such as low-strength aspirin, may be prescribed. Some patients require life-long therapy with anticoagulants such as warfarin (eg, Coumadin). Patients with thrombotic disorders, particularly those with thrombophilia, should be assessed for concurrent risk factors for thrombosis and should avoid concomitant risk factors if possible. For example, use of tobacco and nicotine products exacerbates the problem and should be avoided. Just as for other conditions, patients with thrombotic disorders, particularly thrombophilia, should know the name of their specific condition and understand its significance. In many instances, younger patients with thrombophilia may not require prophylactic anticoagulation; however, with concomitant risk factors (eg, pregnancy), increasing age, or subsequent thrombotic events, prophylactic or lifelong anticoagulation therapy may be required. Being able to provide the health care provider with an accurate health history can be extremely useful and can help guide the selection of appropriate therapeutic interventions. Patients with hereditary disorders should be encouraged to have their siblings and children tested for the disorder. When patients with thrombotic disorders are hospitalized, frequent assessments should be performed for signs and symptoms of beginning thrombus formation, particularly in the legs (DVT) and lungs (pulmonary embolism). Ambulation or range-of-motion exercises as well as the use of elastic compression stockings should be initiated promptly to decrease stasis. Prophylactic anticoagulants are commonly prescribed. Therapies for Blood Disorders SPLENECTOMY The surgical removal of the spleen (splenectomy) is sometimes necessary after trauma to the abdomen. Because the spleen is very vascular, severe hemorrhage can result if the spleen is ruptured. Under such circumstances, splenectomy becomes an emergency procedure. Splenectomy is also a possible treatment for other hematologic disorders. For example, an enlarged spleen may be the site of excessive destruction of blood cells. If the destruction is lifethreatening, surgery may be lifesaving. This is the case in autoimmune hemolytic anemia or ITP when these disorders do not respond to more conservative measures, such as corticosteroid therapy. Some patients with severe anemia due to inherited RBC defects (eg, thalassemia) may also benefit from splenectomy. In general, the mortality rate after splenectomy is low. Laparoscopic splenectomy can be used in selected patients, with a resultant decrease in the postoperative morbidity rate. Complications that may result from surgery are atelectasis, pneumonia, abdominal distention, and abscess formation. Although young children are at the highest risk after splenectomy, all age groups are vulnerable to overwhelming lethal infections and should receive pneumovax before undergoing this surgical procedure if possible. Patients are instructed to seek prompt medical attention if even relatively minor symptoms of infection occur. Often, patients with high platelet counts have even higher counts after splenectomy— more than 1 million/mm3—which can predispose them to serious thrombotic or hemorrhagic problems. This increase is, however, transient. Chapter 33 Assessment and Management of Patients With Hematologic Disorders THERAPEUTIC APHERESIS Apheresis is a Greek word meaning separation. In therapeutic apheresis (or pheresis), blood is taken from the patient and passed through a centrifuge, where a specific component is separated from the blood and removed (Table 33-9). The remaining blood is then returned to the patient. The entire system is closed, so the risk of bacterial contamination is extremely low. When platelets or WBCs are removed, the decrease in these cells within the circulation is temporary. However, the temporary decrease provides a window of time until suppressive medications (eg, chemotherapy) can have therapeutic effects. Sometimes plasma is removed rather than blood cells—typically so that specific, abnormal proteins within the plasma will be transiently lowered until a long-term therapy can be initiated. Apheresis is also used to obtain larger amounts of platelets from a donor than can be provided from a single unit of whole blood. A unit of platelets obtained in this way is equivalent to six to eight units of platelets obtained from six to eight separate donors via standard blood donation methods. Platelet donors can have their platelets apheresed as often as every 14 days. WBCs can be obtained similarly, typically after the donor has received growth factors (G-CSF, GM-CSF) to stimulate the formation of additional WBCs and thereby increase the WBC count. The use of these growth factors also stimulates the release of stem cells within the circulation. Apheresis is used to harvest these stem cells (typically over a period of several days) for use in PBSCT (peripheral blood stem cell transplant; see Chap. 16). THERAPEUTIC PHLEBOTOMY Therapeutic phlebotomy is the removal of a certain amount of blood under controlled conditions. Patients with elevated hematocrits (eg, those with polycythemia vera) or excessive iron absorption (eg, hemochromatosis) can usually be managed by periodically removing 1 unit (about 500 mL) of whole blood. Eventually this process can produce iron deficiency, leaving the patient Table 33-9 unable to produce as many RBCs. The actual procedure for therapeutic phlebotomy is similar to that for blood donation (see later discussion). BLOOD AND BLOOD COMPONENT THERAPY A single unit of whole blood contains 450 mL of blood and 50 mL of an anticoagulant. A unit of whole blood can be processed and dispensed for administration. However, it is more appropriate, economical, and practical to separate that unit of whole blood into its primary components: RBCs, platelets, and plasma (WBCs are rarely used; see later discussion). Because the plasma is removed, a unit of RBCs (packed RBCs, PRBCs) is very concentrated (hematocrit, approximately 70%). Each component must be processed and stored differently to maximize the longevity of the viable cells and factors within it; each individual blood component has a different storage life. PRBCs are stored at 4°C. With special preservatives, they can be stored safely for up to 42 days before they must be discarded. In contrast, platelets must be stored at room temperature because they cannot withstand cold temperatures, and they last for only 5 days before they must be discarded. To prevent clumping, platelets are gently agitated while stored. Plasma is immediately frozen to maintain the activity of the clotting factors within; it lasts for 1 year if it remains frozen. Plasma can be further pooled and processed into blood derivatives, such as albumin, immune globulin, factor VIII, and factor IX. Table 33-10 describes each blood component and how it is commonly used. SPECIAL PREPARATIONS Factor VIII concentrate (antihemophilic factor) is a lyophilized, freeze-dried concentrate of pooled fractionated human plasma. It is used in treating hemophilia A. Factor IX concentrate (prothrombin complex) is similarly prepared and contains factors II, • Types of Apheresis* PROCEDURE PURPOSE EXAMPLES OF CLINICAL USE Platelet pheresis Remove platelets Leukapheresis Remove WBCs (can be specific to neutrophils or lymphocytes) Erythrocytapheresis (RBC exchange) Plasmapheresis (plasma exchange) Remove RBCs Stem cell harvest Remove circulating stem cells Extreme thrombocytosis, essential thrombocythemia (temporary measure); single-donor platelets transfusion Extreme leukocytosis (eg, AML, CML) (very temporary measure); harvest WBCs for transfusion RBC dyscrasias (eg, sickle cell disease); RBCs replaced via transfusion Hyperviscosity syndromes; treatment for some renal and neurologic diseases (eg, Goodpasture’s syndrome, Guillain-Barré) Transplantation (donor harvest or autologous) Remove plasma proteins 923 *Therapeutic apheresis can be used to treat a wide variety of conditions. When it is used to treat a disease that causes an increase in a specific cell type with a short life in circulation (ie, WBCs, platelets), the reduction in those cells is temporary. However, this temporary reduction permits a margin of safety while waiting for a longer-lasting treatment modality (eg, chemotherapy) to take effect. Apheresis can also be used to obtain stem cells for transplantation, either from a matched donor (allogenic) or from the patient (autologous). AML, acute myeloid leukemia; CML, chronic myeloid leukemia; RBC, red blood cell; WBC, white blood cell. 924 Table 33-10 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION • Blood and Blood Components Commonly Used in Transfusion Therapy* COMPOSITION INDICATIONS AND CONSIDERATIONS Whole blood Cells and plasma, hematocrit about 40% Packed red blood cells (PRBCs) RBCs with little plasma (hematocrit about 75%); some platelets and WBCs remain Platelets—random Platelets (5.5 × 1010 platelets/unit) Plasma; some RBCs, WBCs Platelets—single donor Platelets (3 × 1011 platelets /unit) 1 unit is equivalent to 6–8 units of random platelets Plasma; all coagulation factors Complement Neutrophils (>1 × 1010/unit); lymphocytes; some RBCs and platelets Lymphocytes (number varies) Volume replacement and oxygen-carrying capacity; usually used only in significant bleeding (> 25% blood volume lost) ↑ RBC mass Symptomatic anemia: platelets in the unit are not functional; WBCs the unit may cause reaction and are not functional Bleeding due to severe ↓ platelets Prevent bleeding when platelets <5,000–10,000/mm3 Survival ↓ in presence of fever, chills, infection Repeated treatment → ↓ survival due to alloimmunization Used for repeated treatment: ↓ alloimmunization risk by limiting exposure to multiple donors Plasma (FFP) Granulocytes (pheresed) Lymphocytes (WBCs) (apheresed) Cryoprecipitate Antihemophilic factor (AHF) Factor IX concentrate Factor IX complex Bleeding in patients with coagulation factor deficiencies; plasmapheresis Severe neutropenia in selected patients; controversial Stimulate graft-versus-disease effect Fibrinogen ≥ 150 mg/bag, AHF (VIII:C) 80–110 units /bag, von Willebrand factor; fibronectin Factor VIII von Willebrand’s disease Hypofibrinoginemia Hemophilia A Hemophilia A Factor IX Factor II, VII, IX, X Hemophilia B (Christmas disease) Hereditary factor VII, IX, X deficiency; Hemophilia A with factor VII inhibitors Hypoproteinemia; burns; volume expansion by 5% to ↑ blood volume; 25% → ↓ hematocrit Hypogammaglobulinemia (in CLL, recurrent infections); ITP; primary immunodeficiency states ATIII deficiency with or at risk for thrombosis Albumin Albumin 5%, 25% Intravenous gamma globulin Antithrombin III concentrate (AT III) IgG antibodies AT III (trace amounts of other plasma proteins) * The composition of each type of blood component is described as well as the most common indications for using a given blood component. RBCs, platelets, and fresh frozen plasma are the blood products most commonly used. When transfusing these blood products, it is important to realize that the individual product is always “contaminated” with very small amounts of other blood products (eg, WBCs mixed in a unit of platelets). This contamination can cause some difficulties, particularly isosensitization, in certain patients. AHF, antihemophilic factor; CLL, chronic lymphocytic leukemia; ITP, idiopathic thrombopenic purpura. VII, IX, and X. It is used primarily for treatment of factor IX deficiency (hemophilia B). Factor IX concentrate is also useful in treating congenital factor VII and factor X deficiencies. Plasma albumin is a large protein molecule that usually stays within vessels and is a major contributor to plasma oncotic pressure. This protein is used to expand the blood volume of patients in hypovolemic shock and, rarely, to increase the concentration of circulating albumin in patients with hypoalbuminemia. Immune globulin is a concentrated solution of the antibody IgG; it contains very little IgA or IgM. It is prepared from large pools of plasma. The intravenous form (IVIG) is used in various clinical situations to replace inadequate amounts of IgG in patients who are at risk for recurrent bacterial infection (eg, those with CLL, those receiving BMT or PBSCT). IVIG, in contrast to all other fractions of human blood, cells, or plasma, are able to survive being subjected to heating at 60°C (140°F) for 10 hours to free them of viral contaminants. Procuring Blood and Blood Products BLOOD DONATION To protect both the donor and the recipients, all prospective donors are examined and interviewed before they are allowed to donate their blood. The intent of the interview is to assess the general health status of the donor and to identify risk factors that might harm a recipient of the donor’s blood. Donors should be in good health and without any of the following: • A history of viral hepatitis at any time in the past, or a his• • tory of close contact with a hepatitis or dialysis patient within 6 months A history of receiving a blood transfusion or an infusion of any blood derivative (other than serum albumin) within 6 months A history of untreated syphilis or malaria, because these diseases can be transmitted by transfusion even years later. A Chapter 33 • • • • • • • • • • • Assessment and Management of Patients With Hematologic Disorders person who has been free of symptoms and off therapy for 3 years after malaria may be a donor. A history or evidence of drug abuse in which substances were self-injected, because many intravenous/injection drug users are hepatitis carriers and because the risk for human immunodeficiency virus (HIV) is high in this group A history of possible exposure to HIV; the population at risk includes people who engage in anal sex, people with multiple sexual partners, intravenous/injection drug users, sexual partners of people at risk for HIV, and people with hemophilia A skin infection, because of the possibility of contaminating the phlebotomy needle, and subsequently the blood itself A history of recent asthma, urticaria, or allergy to medications, because hypersensitivity can be transferred passively to the recipient Pregnancy within 6 months, because of the nutritional demands of pregnancy on the mother A history of tooth extraction or oral surgery within 72 hours, because such procedures are frequently associated with transient bacteremia A history of exposure to infectious disease within the past 3 weeks, because of the risk of transmission to the recipient Recent immunizations, because of the risk of transmitting live organisms (2-week waiting period for live, attenuated organisms; 1 month for rubella; 1 year for rabies) A history of recent tattoo, because of the risk of blood-borne infections (eg, hepatitis, HIV) Cancer, because of the uncertainty about transmission of the disease A history of whole blood donation within the past 56 days Potential donors should be asked whether they have consumed any aspirin or aspirin-containing medications within the past 3 days. Although aspirin use does not render the donor ineligible, the platelets obtained would be dysfunctional and therefore not useful. Aspirin does not affect the RBCs or plasma obtained from the donor. All donors are expected to meet the following minimal requirements: • Body weight should exceed 50 kg (110 pounds) for a stan- • • • • dard 450-mL donation. Donors weighing less than 50 kg donate proportionately less blood. People younger than 17 years of age are disqualified from donation. The oral temperature should not exceed 37.5°C (99.6°F). The pulse rate should be regular and between 50 and 100 beats per minute. The systolic arterial pressure should be 90 to 180 mm Hg, and the diastolic pressure should be 50 to 100 mm Hg. The hemoglobin level should be at least 12.5 g/dL for women and 13.5 g/dL for men. Directed Donation At times, friends and family of a patient wish to donate blood for that person. These blood donations are termed directed donations. These donations are not any safer than those provided by random donors, because directed donors may not be as willing to identify themselves as having a history of any of the risk factors that disqualify a person from donating blood. 925 Standard Donation Phlebotomy consists of venipuncture and blood withdrawal. Standard precautions are used. Donors are placed in a semirecumbent position. The skin over the antecubital fossa is carefully cleansed with an antiseptic preparation, a tourniquet is applied, and venipuncture is performed. Withdrawal of 450 mL of blood usually takes less than 15 minutes. After the needle is removed, donors are asked to hold the involved arm straight up, and firm pressure is applied with sterile gauze for 2 or 3 minutes or until bleeding stops. A firm bandage is then applied. Donors remain recumbent until they feel able to sit up, usually within a few minutes. Donors who experience weakness or faintness should rest for a longer period. Donors then receive food and fluids and are asked to remain another 15 minutes. Donors are instructed to leave the dressing on and to avoid heavy lifting for several hours, to avoid smoking for 1 hour, to avoid drinking alcoholic beverages for 3 hours, to increase fluid intake for 2 days, and to eat a healthy meals for 2 weeks. Specimens from this donated blood are tested to detect infections and to identify the specific blood type (see later discussion). Autologous Donation A patient’s own blood may be collected for future transfusion; this method is useful for many elective surgeries where the potential need for transfusion is high (eg, orthopedic surgery). Preoperative donations are ideally collected 4 to 6 weeks before surgery. Iron supplements are prescribed during this period to prevent depletion of iron stores. Occasionally, erythropoietin (epoetin-alfa [Epogen, Procrit]) is given to stimulate erythropoiesis to ensure that the donor’s hematocrit remains high enough to be eligible for donation. Typically, 1 unit of blood is drawn each week; the number of units obtained varies with the type of surgical procedure to be performed (ie, the amount of blood anticipated to be transfused). Phlebotomies are not performed within 72 hours of surgery. Individual blood components can also be collected. The primary advantage of autologous transfusions is the prevention of viral infections from another person’s blood. Other advantages include safe transfusion for patients with a history of transfusion reactions, prevention of alloimmunization, and avoidance of complications in patients with alloantibodies. The policy of the American Red Cross requires autologous blood to be transfused only to the donor. If the blood is not required, it can be frozen until the donor needs it in the future (for up to 10 years). The blood is never returned to the general donor supply of blood products to be used by someone else. The disadvantage of autologous donation is that it may be performed even when the likelihood that the anticipated procedure will necessitate a transfusion is small. Needless autologous donation is expensive, takes time, and uses resources inappropriately. Moreover, in an emergency situation, the autologous units available may be inadequate, and the patient may still require additional units from the general donor supply. Contraindications to donation of blood for autologous transfusion are acute infection, severely debilitating chronic disease, hemoglobin level less than 11 g/dL, hematocrit less than 33%, unstable angina, and acute cardiovascular or cerebrovascular disease. A history of poorly controlled epilepsy may be considered a contraindication in some centers. Patients with cancer may donate for themselves. 926 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Intraoperative Blood Salvage This transfusion method is initiated before or after induction of anesthesia. About 1 or 2 units of blood are removed from the patient through a venous or arterial line and simultaneously replaced with a colloid or crystalloid solution. The blood obtained is then reinfused after surgery (Kreimeier & Messmer, 2002). The advantage of this method is that the patient loses fewer RBCs during surgery, because the added intravenous solutions dilute the concentration of RBCs and lower the hematocrit. Patients who are at risk for myocardial injury, however, should not be further stressed by hemodilution. ies to the virus (Korman, Leparc & Benson, 2001). This testing significantly shortens the “window” of inability to detect HIV and HCV from a donated unit, further ensuring the safety of the blood. Blood is also screened for CMV; if it tests positive for CMV, it can still be used, except in recipients who are negative for CMV and who are immunocompromised (eg, BMT or PBSCTrecipients). Equally important to viral testing is accurate determination of the blood type. More than 200 antigens have been identified on the surface of RBC membranes. Of these, the most important for safe transfusion are the ABO and Rh systems. The ABO system identifies which sugars are present on the membrane of an individual’s RBCs: A, B, both A and B, or neither A nor B (type O). To prevent a significant reaction, the same type of RBCs should be transfused. Previously, it was thought that in an emergency situation in which the patient’s blood type was not known, type O blood could be safely transfused. This practice is no longer advised by the American Red Cross. The Rh antigen (also called D) is present on the surface of RBCs in 85% of the population (Rh-positive). Those who lack the D antigen are called Rh-negative. RBCs are routinely tested for the D antigen as well as ABO. Patients should receive PRBCs with a compatible Rh type. COMPLICATIONS OF BLOOD DONATION TRANSFUSION Excessive bleeding at the donor’s venipuncture site is sometimes caused by a bleeding disorder in the donor but more often results from a technique error: laceration of the vein, excessive tourniquet pressure, or failure to apply enough pressure after the needle is withdrawn. Fainting is common after blood donation and may be related to emotional factors, a vasovagal reaction, or prolonged fasting before donation. Because of the loss of blood volume, hypotension and syncope may occur when the donor assumes an erect position. A donor who appears pale or complains of faintness should immediately lie down or sit with head lowered below the knees; he or she should be observed for another 30 minutes. Anginal chest pain may be precipitated in patients with unsuspected coronary artery disease. Seizures can occur in donors with epilepsy, although the incidence is very low. Both angina and seizures require further medical evaluation. Many people have the misconception that donating blood can cause AIDS and other infections. Potential donors need to be educated that the equipment used in donation is sterile, a closed system, and not reusable; they are at no risk for acquiring such infections from donating blood. Administration of blood and blood components requires knowledge of correct administration techniques and possible complications. It is very important to be familiar with the agency’s policies and procedures for transfusion therapy. Methods for transfusing blood components are presented in Charts 33-16 and 33-17. Potential complications of transfusion follow. BLOOD PROCESSING Pretransfusion Assessment Samples of the unit of blood are always taken immediately after donation so that the blood can be typed and tested. Each donation is tested for antibodies to HIV 1 and 2, hepatitis B core antibody (anti-HBc), hepatitis C virus (HCV), and human T-cell lymphotropic virus, type I (anti-HTLV-I/II). The blood is also tested for hepatitis B surface antigen (HbsAG) and for syphilis. Negative reactions are required for the blood to be used, and each unit of blood is labeled to certify the results. A new testing method, using nucleic acid amplification testing (NAT), has increased the ability to detect the presence of HCV and HIV infection, because it directly tests for genomic nucleic acids of the virus itself, rather than for the presence of antibod- PATIENT HISTORY Patient history is an important component of the pretransfusion assessment to determine the history of previous transfusions as well as previous reactions to transfusion. The history should include the type of reaction, its manifestations, the interventions required, and whether any preventive interventions were used in subsequent transfusions. It is important to assess the number of pregnancies a woman has had, because an increased number can increase her risk for reaction due to antibodies developed from exposure to fetal circulation. Other concurrent health problems should also be noted, with careful attention to cardiac, pulmonary, and vascular disease. This transfusion method provides replacement for patients who are unable to donate before surgery and for those undergoing vascular, orthopedic, or thoracic surgery. During a surgical procedure, blood lost into a sterile cavity (eg, hip joint) is suctioned into a cell-saver machine. The RBCs are washed, often with saline solution, and then returned to the patient as an intravenous infusion. Salvaged blood cannot be stored, because bacteria cannot be completely removed from the blood. Hemodilution Setting Although most blood transfusions are performed in the acute care setting, patients with chronic transfusion requirements often can receive transfusions in other settings. Free-standing infusion centers, ambulatory care clinics, a physician’s office, and even the home may be appropriate settings for transfusion. Typically, patients who need chronic transfusions but are otherwise stable physically are appropriate candidates for outpatient therapy. Verification and administration of the blood product are performed much as in a hospital setting. Although most blood products can be transfused in the outpatient setting, the home is typically limited to transfusions of PRBCs and factor components (eg, factor VIII for patients with hemophilia). Chart 33-16 Transfusion of Packed Red Blood Cells (PRBCs) Preprocedure 1. Confirm that the transfusion has been prescribed. 2. Check that patient’s blood has been typed and cross-matched. 3. Verify that patient has signed a written consent form per institution policy. 4. Explain the procedure to the patient. Instruct patient in signs and symptoms of transfusion reaction (itching, hives, swelling, shortness of breath, fever, chills). 5. Take patient’s temperature, pulse, respiration, and blood pressure to establish a baseline for comparing vital signs during transfusion. 6. Use hand hysgeine and wear gloves in accordance with Standard Precautions. 7. Use a 20-gauge or larger needle for placement in a large vein. Use special tubing that contains a blood filter to screen out fibrin clots and other particulate matter. Do not vent the blood container. Procedure 1. Obtain the PRBCs from the blood bank after the intravenous line is started. (Institution policy may limit release to only 1 unit at a time.) 2. Double-check the labels with another nurse or physician to make sure that the ABO group and Rh type agree with the compatibility record. Check to see that the number and type on the donor blood label and on the patient’s chart are correct. Check the patient’s identification by asking the patient’s name and checking the identification wristband. 3. Check the blood for gas bubbles and any unusual color or cloudiness. (Gas bubbles may indicate bacterial growth. Abnormal color or cloudiness may be a sign of hemolysis.) 4. Make sure PRBC transfusion is initiated within 30 min after removal of the PRBCs from the blood bank refrigerator. 5. For first 15 minutes, run the transfusion slowly—no faster than 5 mL/min. Observe the patient carefully for adverse effects. If no adverse effects occur during the first 15 min, increase the flow rate unless the patient is at high risk for circulatory overload. 6. Monitor closely for 15–30 min to detect signs of reaction. Monitor vital signs at regular intervals per institution policy; compare results with baseline measurements. Increase frequency of measurements based on patient’s condition. Observe the patient frequently throughout the transfusion for any signs of adverse reaction, including restlessness, hives, nausea, vomiting, torso or back pain, shortness of breath, flushing, hematuria, fever, or chills. Should any adverse reaction occur, stop infusion immediately, notify physician, and follow the agency’s transfusion reaction standard. 7. Note that administration time does not exceed 4 hr because of the increased risk for bacterial proliferation. 8. Be alert for signs of adverse reactions: circulatory overload, sepsis, febrile reaction, allergic reaction, and acute hemolytic reaction. 9. Change blood tubing after every 2 units transfused, to decrease chance of bacterial contamination. Postprocedure 1. Obtain vital signs and compare with baseline measurements. 2. Dispose of used materials properly. 3. Document procedure in patient’s medical record, including patient assessment findings and tolerance to procedure. 4. Monitor patient for response to and effectiveness of the procedure. Note: Never add medications to blood or blood products; if blood is too thick to run freely, normal saline may be added to the unit. If blood must be warmed, use an in-line blood warmer with a monitoring system. Chart 33-17 Transfusion of Platelets or Fresh Frozen Plasma (FFP) Preprocedure 1. Confirm that the transfusion has been prescribed. 2. Verify that patient has signed a written consent form per institution policy. 3. Explain the procedure to the patient. Instruct patient in signs and symptoms of transfusion reaction (itching, hives, swelling, shortness of breath, fever, chills). 4. Take patient’s temperature, pulse, respiration, and blood pressure to establish a baseline for comparing vital signs during transfusion. 5. Wash hands and wear gloves in accordance with Standard Precautions. 6. Use a 22-gauge or larger needle for placement in a large vein, if possible. Use appropriate tubing per institution policy (platelets often require different tubing from that used for other blood products). Procedure 1. Obtain the platelets or FFP from the blood bank (only after the intravenous line is started.) 2. Double-check the labels with another nurse or physician to make sure that the ABO group matches the compatibility record (not usually necessary for platelets; here only if compatible platelets are ordered). Check to see that the number and type on the donor blood label and on the patient’s chart are correct. Check the patient’s identification by asking the patient’s name and checking the identification wristband. 3. Check the blood product for any unusual color or clumps (excessive redness indicates contamination with larger amounts of red blood cells). 4. Make sure platelets or FFP units are administered immediately after they are obtained. 5. Infuse each unit as fast as patient can tolerate to diminish platelet clumping during administration. Observe the patient carefully for adverse effects, including circulatory overload. Decrease rate of infusion if necessary. 6. Observe the patient closely throughout the transfusion for any signs of adverse reaction, including restlessness, hives, nausea, vomiting, torso or back pain, shortness of breath, flushing, hematuria, fever, or chills. Should any adverse reaction occur, stop infusion immediately, notify physician, and follow the agency’s transfusion reaction standard. 7. Monitor vital signs at end of transfusion per institution policy; compare results with baseline measurements. 8. Flush line with saline after transfusion to remove blood component from tubing. Postprocedure 1. Obtain vital signs and compare with baseline measurements. 2. Dispose of used materials properly. 3. Document procedure in patient’s medical record, including patient assessment findings and tolerance to procedure. 4. Monitor patient for response to and effectiveness of procedure. A platelet count may be ordered 1 hr after platelet transfusion to facilitate this evaluation. Note: FFP requires ABO but not Rh compatibility. Platelets are not typically cross-matched for ABO compatibility. Never add medications to blood or blood products. 927 928 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION PHYSICAL ASSESSMENT A systematic physical assessment and measurement of baseline vital signs are important before transfusing any blood product. The respiratory system should be assessed, including careful auscultation of the lungs and for use of accessory muscles. Cardiac system assessment should include careful inspection for any edema as well as other signs of cardiac failure (eg, jugular venous distention). The skin should be observed for rashes, petechiae, and ecchymoses. The sclera should be examined for icterus. In the event of a possible transfusion reaction, a comparison of findings can help differentiate between types of reactions. Patient Teaching Reviewing the signs and symptoms of a potential transfusion reaction is crucial for patients who have not received a transfusion before. Even for those patients who have received prior transfusions, a brief review of signs and symptoms of potential transfusion reactions is advised. Signs and symptoms of a possible reaction include fever, chills, respiratory distress, low back pain, nausea, pain at the intravenous site, or anything “unusual.” Although a thorough review is very important, it is also important to reassure the patient that the blood is carefully tested against the patient’s own blood (cross-matched) to diminish the likelihood of any untoward reaction. Such assurance can be extremely beneficial in allaying anxiety. Similarly, it can be useful to mention again the very low possibility of contracting HIV from the transfusion; this fear persists among many people. Transfusion Complications Any patient who receives a blood transfusion may develop complications from that transfusion. When explaining the reasons for the transfusion, it is important to include the risks and benefits and what to expect during and after the transfusion. Patients must be informed that the supply of blood is not completely risk-free although it has been tested carefully. Nursing management is directed toward preventing complications, promptly recognizing complications if they develop, and promptly initiating measures to control any complications that occur. The following sections describe the most common or potentially severe transfusion-related complications. FEBRILE, NONHEMOLYTIC REACTION The nonhemolytic reaction, caused by antibodies to donor WBCs that are still present in the unit of blood or blood component, is the most common type of transfusion reaction, accounting for more than 90% of reactions. It occurs more frequently in patients who have had previous transfusions (exposure to multiple antigens from previous blood products) and in Rh-negative women who have borne Rh-positive children (exposure to an Rhpositive fetus raises antibody levels in the mother). These reactions occur in 1% of PRBC transfusions and 20% of platelet transfusions. More than 10% of patients with a chronic transfusion requirement develop this type of reaction. NURSING RESEARCH PROFILE 33-1 Perspectives of Recipients of Blood Transfusions Fitzgerald, M., Hodgkinson, B., & Thorp, D. (1999). Blood transfusion from the recipient’s perspective. Journal of Clinical Nursing 8(5), 593–600. Purpose The process of informed consent requires that information be provided to the patient before consent and implies that a dialogue between the patient and physicians and nurses occurs during this process. This study stemmed from previous studies examining transfusion practices in a large teaching hospital in Australia; the studies revealed that little is known about how much patients understood about the transfusion process. The purpose of this study was to more closely examine the meaning of patients’ experiences with blood transfusions. Study Sample and Design The study design employed interpretive phenomenology, a qualitative analysis process that allowed researchers to identify meanings that may have been hidden in common actions. A convenience sample of 19 patients was interviewed; subjects were asked to discuss their experience of receiving a blood transfusion, beginning with the time they were told about it. Subjects received transfusions for a variety of clinical conditions, primarily surgery, cancer, and emergency situations. The interviews were tape recorded and transcribed verbatim. The data were analyzed and themes were identified. Findings Three broad themes were identified and more closely analyzed: information regarding the transfusion, reaction to receiving a transfusion, and care received during the transfusion. The focus of many of the interviews was on the information process. Physicians’ explanations regarding the purpose of the transfusion were typically brief and were focused on providing factual information, particularly before the patient’s signing of the consent for the procedure. Nurses’ explanations continued as the procedure progressed but tended to focus only on potential reactions. Neither physicians nor nurses tended to encourage the patient to express his or her concerns, nor sought information from the patient. Few patients could actually recall any of the factual information presented to them. Fear of infection, particularly human immunodeficiency virus (HIV) infection, was present but was not excessive. Subjects verbalized understanding that the risk of acquiring HIV from their transfusion was very remote. Most subjects were told that the transfusion would make them feel better; in reality (and likely due to the severity of their illness/injury) such was not the case. Beyond the initial procedure by the nurse of double-checking the blood product for transfusion against the patient’s identity and hanging the blood, patients did not notice any difference in the nursing care they received during the transfusion. Nursing Implications Findings from this study have important implications for nurses, particularly concerning the information process. Despite the focus of both physicians and nurses on providing information, patients demonstrated that they did not comprehend it well. Informational needs vary, as do learning styles. It is crucial that nurses assess their patients’ level of understanding about the entire transfusion process. Not having the opportunity to deliberate on the information provided and to express concern was a common theme from the study subjects. Nurses need to make the time for patients to express their concerns and verbalize their feelings. This need exists not only for those receiving their first transfusion but also for those who have long-term transfusion requirements. Chapter 33 Assessment and Management of Patients With Hematologic Disorders The diagnosis of a febrile, nonhemolytic reaction is made by excluding other potential causes, such as a hemolytic reaction or bacterial contamination of the blood product. The signs and symptoms of a febrile, nonhemolytic transfusion reaction are chills (absent to severe) followed by fever (more than 1°C elevation). The fever typically begins within 2 hours after the transfusion is begun. Although not life-threatening, the fever and particularly the chills and muscle stiffness can be frightening to the patient. These reactions can be diminished, even prevented, by further depleting the blood component of donor WBCs; this is accomplished by a leukocyte reduction filter. The blood product may be filtered during processing, which achieves better results but is more expensive, or during the actual transfusion by adding the filter to the blood administration tubing. Antipyretics can be given to prevent fever, but routine premedication is not advised because it can mask the beginning of a more serious transfusion reaction. ACUTE HEMOLYTIC REACTION The most dangerous, and potentially life threatening, type of transfusion reaction occurs when the donor blood is incompatible with that of the recipient. Antibodies already present in the recipient’s plasma rapidly combine with antigens on donor RBCs, and the RBCs are hemolyzed (destroyed) in the circulation (intravascular hemolysis). The most rapid hemolysis occurs in ABO incompatibility. This reaction can occur after transfusion of as little as 10 mL of RBCs. Rh incompatibility often causes a less severe reaction. The most common causes of acute hemolytic reaction are errors in blood component labeling and patient identification that result in the administration of an ABO-incompatible transfusion. Symptoms consist of fever, chills, low back pain, nausea, chest tightness, dyspnea, and anxiety. As the RBCs are destroyed, the hemoglobin is released from the cells and excreted by the kidneys; therefore, hemoglobin is present in the urine (hemoglobinuria). Hypotension, bronchospasm, and vascular collapse may result. Diminished renal perfusion results in acute renal failure, and DIC may also occur. The reaction must be recognized promptly and the transfusion discontinued immediately. Blood and urine specimens must be obtained and analyzed for evidence of hemolysis. Treatment goals include maintaining blood volume and renal perfusion and preventing and managing DIC. Acute hemolytic transfusion reactions are preventable. Meticulous attention to detail in labeling blood samples and blood components and identifying the recipient cannot be overemphasized. ALLERGIC REACTION Some patients may develop urticaria (hives) or generalized itching during a transfusion. The cause of these reactions is thought to be a sensitivity reaction to a plasma protein within the blood component being transfused. Symptoms of an allergic reaction are urticaria, itching, and flushing. The reactions are usually mild and respond to antihistamines. If the symptoms resolve after administration of an antihistamine (eg, diphenhydramine [eg, Benadryl]), the transfusion may be resumed. Rarely, the allergic reaction is severe, with bronchospasm, laryngeal edema, and shock. These reactions are managed with epinephrine, corticosteroids, and pressor support, if necessary. 929 Giving the patient antihistamines before the transfusion may prevent future reactions. For severe reactions, future blood components are washed to remove any remaining plasma proteins. Leukocyte filters are not useful, because the offending plasma proteins can pass through the filter. CIRCULATORY OVERLOAD If too much blood infuses too quickly, hypervolemia can occur. This condition can be aggravated in patients who already have increased circulatory volume (eg, those with heart failure). PRBCs are safer to use than whole blood. If the administration rate is sufficiently slow, circulatory overload may be prevented. For patients who are at risk for, or already in, circulatory overload, diuretics are administered after the transfusion or between units of PRBCs. Patients receiving fresh frozen plasma or even platelets may also develop circulatory overload. The infusion rate of these blood components must also be titrated to the patient’s tolerance. Signs of circulatory overload include dyspnea, orthopnea, tachycardia, and sudden anxiety. Neck vein distention, crackles at the base of the lungs, and a rise in blood pressure can also occur. If the transfusion is continued, pulmonary edema can develop, as manifested by severe dyspnea and coughing of pink, frothy sputum. If fluid overload is mild, the transfusion can often be continued after slowing the rate of infusion and administering diuretics. However, if the overload is severe, the patient is placed in an upright position with the feet in a dependent position, the transfusion is discontinued, and the physician is notified. The intravenous line is kept patent with a very slow infusion of normal saline solution or a saline or heparin lock device to maintain access to the vein in case intravenous medications are necessary. Oxygen and morphine may be needed for severe dyspnea. BACTERIAL CONTAMINATION The incidence of bacterial contamination of blood components is very low; however, administration of contaminated products puts the patient at great risk. Contamination can occur at any point during procurement or processing. Many bacteria cannot survive in the cold temperatures used to store PRBCs (platelets are at greater risk for contamination because they are stored at room temperature), but some organisms can survive cold temperatures. Preventive measures include meticulous care in the procurement and processing of blood components. When PRBCs or whole blood is transfused, it should be administered within a 4-hour period, because warm room temperatures promote bacterial growth. A contaminated unit of blood product may appear normal, or it may have an abnormal color. The signs of bacterial contamination are fever, chills, and hypotension. These signs may not occur until the transfusion is complete, occasionally not until several hours after the transfusion. If the condition is not treated immediately with fluids and broad-spectrum antibiotics, shock can occur. Even with aggressive management, including vasopressor support, the mortality rate is high. As soon as the reaction is recognized, any remaining transfusion is discontinued and the intravenous line is kept open with normal saline solution. The physician and the blood bank are notified, and the blood container is returned to the blood bank for testing and culture. Septicemia is treated with intravenous fluids and antibiotics; corticosteroids and vasopressors also may be necessary. 930 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION TRANSFUSION-RELATED ACUTE LUNG INJURY This is a potentially fatal, idiosyncratic reaction that occurs in fewer than 1 in 5000 transfusions. Plasma antibodies (usually in the donor’s plasma) that are present in the blood component stimulate the recipient’s WBCs; aggregates of these WBCs form and occlude the microvasculature within the lungs. This lung injury is manifested as pulmonary edema; it can occur within 4 hours after the transfusion. Signs and symptoms include fever, chills, acute respiratory distress (in the absence of other signs of left ventricular failure, such as elevated central venous pressure), and bilateral pulmonary infiltrates. Aggressive supportive therapy (oxygen, intubation, diuretics) may prevent death. DELAYED HEMOLYTIC REACTION Delayed hemolytic reactions usually occur within 14 days after transfusion, when the level of antibody has been increased to the extent that a reaction can occur. The hemolysis of the RBCs is extravascular, via the RES, and occurs gradually. Signs and symptoms of a delayed hemolytic reaction are fever, anemia, increased bilirubin level, decreased or absent haptoglobin, and possibly jaundice. Rarely is there hemoglobinuria. Generally, these reactions are not dangerous, but it is useful to recognize them, because subsequent transfusions with blood products containing these antibodies may cause a more severe hemolytic reaction. However, recognition is also difficult, because the patient may not be in a health care setting to be tested for this reaction, and even if the patient is hospitalized, the reaction may be too mild to be recognized clinically. Because the amount of antibody present can be too low to detect, it is difficult to prevent delayed hemolytic reactions. The reaction is usually mild and requires no intervention. DISEASES TRANSMITTED BY BLOOD TRANSFUSION Despite the advances in donor screening and blood testing, certain diseases can still be transmitted by transfusion of blood components. The diseases in Chart 33-18 are examples of this phenomenon. COMPLICATIONS OF LONG-TERM TRANSFUSION THERAPY The complications that have been described represent a real risk for any patient any time a unit of blood is administered. However, patients with long-term transfusion therapy (eg, those with MDS, thalassemia, sickle cell anemia) are at greater risk for infection transmission and for becoming more sensitized to donor antigens, simply because they are exposed to more units of blood and, consequently, more donors. Iron overload is a complication unique to those individuals with long-term PRBC transfusions. A summary of complications associated with long-term transfusion therapy is depicted in Table 33-11. Iron Overload. One unit of PRBCs contains 250 mg of iron. Patients with chronic transfusion requirements can quickly acquire more iron than they can use, leading to iron overload. Over time, the excess iron deposits in the tissues and can cause Chart 33-18 Diseases Transmitted by Blood Transfusion Hepatitis (Viral Hepatitis B, C) • Greater risk from pooled blood products and blood of paid donors than from volunteer donors • Screening test detects most hepatitis B and C • Transmittal risk estimated at 1:10,000 AIDS (HIV and HTLV) • Donated blood screened for antibodies to HIV • Transmittal risk estimated at 1:670,000 • People with high-risk behaviors (multiple sex partners, anal sex, intravenous/injection drug use) and people with signs and symptoms that suggest AIDS should not donate blood Cytomegalovirus (CMV) • Transmittal risk greater for premature newborns with CMV antibody-negative mothers and for immunocompromised recipients who are CMV-negative (eg, those with acute leukemia, organ or tissue transplant recipients). • Blood products rendered “leukocyte-reduced” help reduce transmission of virus. Graft-Versus-Host Disease (GVHD) • Occurs only in severely immunocompromised recipients (eg, Hodgkin’s disease, bone marrow transplantation). • Transfused lymphocytes engraft in recipient and attack host lymphocytes or body tissues; signs and symptoms are fever, diffuse reddened skin rash, nausea, vomiting, diarrhea. • Preventive measures include irradiating blood products to inactivate donor lymphocytes (no known radiation risks to transfusion recipient) and processing donor blood with leukocyte reduction filters. Creutzfeldt-Jakob Disease (CJD) • Rare, fatal disease causing irreversible brain damage • No evidence of transmittal by transfusion, but hemophiliacs and others are concerned that transmittal is possible • All blood donors must be screened for positive family history of CJD. • Potential donors who spent 6 months or more in the United Kingdom (or Europe) from 1980 to 1996 cannot donate blood; blood products from a donor who develops CJD are recalled. organ damage, particularly in the liver, heart, testes, and pancreas. Promptly initiating a program of iron chelation therapy (eg, with deferoxamine [Desferal]) can prevent end-organ damage from iron toxicity (Giardina & Grady, 1995). NURSING MANAGEMENT FOR TRANSFUSION REACTIONS If a transfusion reaction is suspected, the transfusion must be immediately stopped and the physician notified. A thorough patient assessment is crucial, because many complications have similar signs and symptoms. The following steps are taken to determine the type and severity of the reaction: • Stop the transfusion. Maintain the intravenous line with • normal saline solution through new intravenous tubing, administered at a slow rate. Assess the patient carefully. Compare the vital signs with those from the baseline assessment. Assess the patient’s respiratory status carefully. Note the presence of adventitious breath sounds, use of accessory muscles, extent of dyspnea Chapter 33 Table 33-11 Infection Iron overload Transfusion reaction Assessment and Management of Patients With Hematologic Disorders 931 • Common Complications Resulting from Long-Term PRBC Transfusion Therapy* MANIFESTATION MANAGEMENT Hepatitis (B,C) May immunize against hepatitis B; give alpha-interferon for hepatitis C; monitor hepatic function WBC filters to protect against CMV Prevent by chelation therapy Cytomegalovirus (CMV) Heart failure Endocrine failure (diabetes, hypothyroidism, hypoparathyroidism, hypogonadism) Sensitization Febrile reactions Diminish by RBC phenotyping, using WBC-filtered products Diminish by using WBC-filtered products * Patients with long-term transfusion therapy requirements are at risk not only for the transfusion reactions discussed in the text but also for the complications noted above. In many cases, the use of WBC-filtered (eg, leukocyte-poor) blood products is standard for patients who receive long-term PRBC transfusion therapy. An aggressive chelation program initiated early in the course of therapy can prevent problems with iron overload. PRBC, packed red blood cells; WBC, white blood cell; RBC, red blood cell. • • • (if any), and changes in mental status, including anxiety and confusion. Note any chills, diaphoresis, complaints of back pain, urticaria, and jugular vein distention. Notify the physician of the assessment findings, and implement any orders obtained. Continue to monitor the patient’s vital signs and respiratory, cardiovascular, and renal status. Notify the blood bank that a suspected transfusion reaction has occurred. Send the blood container and tubing to the blood bank for repeat typing and culture. The identifying tags and numbers are verified. If a hemolytic transfusion reaction or bacterial infection is suspected, the nurse should do the following: • Obtain appropriate blood specimens from the patient. • Collect a urine sample as soon as possible for a hemoglobin determination. • Document the reaction, according to the institution’s policy. PHARMACOLOGIC ALTERNATIVES TO BLOOD TRANSFUSIONS Pharmacologic agents to stimulate production of one or more types of blood cells by the marrow are commonly used. Chart 33-19 presents examples of such pharmacologic agents. Researchers continue to seek a blood substitute that is practical and safe. Blood substitutes previously tried have not been successful. However, newer blood substitutes focus solely on oxygen delivery, as an RBC substitute (Rabinovici, 2001). Current blood substitutes in clinical trials have distinct advantages and disadvantages compared with human RBCs. They are manufactured hemoglobin solutions that can be sterilized without destroying the blood substitute. They require no refrigeration and appear to have a long shelf-life (possibly 1 year, versus little more than 1 month for PRBCs). Perhaps more importantly, they require no cross-matching, because there is no RBC membrane to interact with antibodies in the recipient’s serum. The most significant disadvantage stems from the blood substitutes extremely short life within human circulation—approximately 1 day, instead of the 30-day life span of a conventionally trans- fused RBC. Therefore, the use of these products would likely be limited to situations in which the need is short-term (eg, surgery, trauma). Finally, the blood substitutes are likely to be extremely expensive. PERIPHERAL BLOOD STEM CELL TRANSPLANTATION (PBSCT) AND BONE MARROW TRANSPLANTATION (BMT) PBSCT and BMT are therapeutic modalities that offer the possibility of cure for some patients with hematologic disorders such as severe aplastic anemia, some forms of leukemia, and thalassemia. Because most hematologic disease states arise from some form of bone marrow dysfunction, an autologous transplantation (receiving one’s own stem cells) is not as common an option as is allogeneic transplantation. A patient receives intensive chemotherapy (sometimes with radiation therapy as well), with the goal being complete ablation of the patient’s bone marrow. Stem cells from the donor (ideally, from a matched sibling), or actual marrow from the donor, is then infused into the patient using a process similar to an RBC transfusion. The stem cells travel to the marrow and slowly begin the process of resuming hematopoiesis. The advantage of autotransplantation is the reduced likelihood of complications and mortality; however, the risk of relapse is also higher. A relatively new strategy is based on transplantation for adoptive cell therapy using certain immune mechanisms derived from the donor’s lymphocytes (Slavin et al., 2001; Margolis, Borrello, & Flinn, 2000). In nonmyeloablative stem cell or marrow transplantation, also referred to as a “minitransplant,” the conditioning regimen involves much less myelosuppression than in conventional regimens, rendering the patient immunosuppressed but for a shorter period of time. Consequently, the procedure is less toxic to the patient, and there is a significant decrease in morbidity. After the deconditioning regimen (ie, during the time the patient is immunosuppressed), the allotransplantation is performed, using either marrow or stem cells. The goal is for the donor’s lymphocytes to react against any residual malignant cells within the patient and destroy them. This process is typically augmented by 932 Chart 33-19 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Pharmacologic Alternatives to Blood Transfusions Growth Factors Recombinant technology has provided a means to produce hematopoietic growth factors necessary for the production of blood cells within the bone marrow. By increasing the body’s production of blood cells, transfusions and complications resulting from diminished blood cells (eg, infection from neutropenia or transfusions) may be avoided. However, the successful use of growth factors requires functional bone marrow. Erythropoietin Erythropoietin (epoetin alpha [eg, Epogen, Procrit]) is an effective alternative treatment for patients with chronic anemia secondary to diminished levels of erythropoietin, as in chronic renal disease. This medication stimulates erythropoiesis. It also has been used for patients who are anemic from chemotherapy or zidovudine (AZT) therapy and for those who have diseases involving bone marrow suppression, such as myelodysplastic syndrome (MDS). The use of erythropoietin can also enable a patient to donate several units of blood for future use (eg, preoperative autologous donation). The medication can be administered intravenously or subcutaneously, although plasma levels are better sustained with the subcutaneous route. Side effects are rare, but erythropoietin can cause or exacerbate hypertension. If the anemia is corrected too quickly or is overcorrected, the elevated hematocrit can cause headache and, potentially, seizures. These adverse effects are rare except for patients with renal failure. Serial complete blood counts (CBCs) should be performed to evaluate the response to the medication. The dose and frequency of administration are titrated to the hematocrit. Granulocyte-Colony Stimulating Factor (G-CSF) G-CSF (filgrastim [Neupogen]) is a cytokine that stimulates the proliferation and differentiation of myeloid stem cells; a rapid increase in neutrophils is seen within the circulation. G-CSF is effective in improving transient but severe neutropenia after chemotherapy or in some forms of MDS. It is particularly useful in preventing bacterial infections that would be likely to occur with neutropenia. G-CSF is administered subcutaneously on a daily basis. The primary side effect is bone pain; this probably reflects the increase in hematopoiesis within the marrow. Serial CBCs should be performed to evaluate the response to the medication and to ensure that the rise in white blood cells is not excessive. The effect of G-CSF on myelopoiesis is short; the neutrophil count drops once the medication is stopped. Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) GM-CSF (sargramostim [Leukine]) is a cytokine that is naturally produced by a variety of cells, including monocytes and endothelial cells. It works either directly or synergistically with other growth factors to stimulate myelopoiesis. GM-CSF is not as specific to neutrophils as is G-CSF; thus, an increase in erythroid (RBC) and megakaryocytic (platelet) production may also be seen. GM-CSF serves the same purpose as G-CSF. However, it may have a greater effect on macrophage function and therefore may be more useful against fungal infections, whereas G-CSF may be better used to fight bacterial infections. GM-CSF is also administered subcutaneously. Side effects include bone pain, fevers, and myalgias. Thrombopoietin Thrombopoietin (TPO) is a cytokine that is necessary for the proliferation of megakaryocytes and subsequent platelet formation. Clinical studies have demonstrated efficacy of TPO in the setting of chemotherapy-induced thrombocytopenia with few side effects (Vadhan-Raj, 2000). Further studies are ongoing to assess the efficacy of TPO in other, more chronic conditions associated with thrombocytopenia (Kuter, 2000). infusion of the donor’s lymphocytes as well (referred to as donor lymphocyte infusion, or DLI). If relapse occurs, repeated DLI has been effective in reestablishing remission in many patients. This approach has great promise, particularly in the setting of hematologic malignancy, and may provide a mechanism to increase the utility of transplantation for more patients than is possible with conventional methods. Success of transplantation depends on tissue compatibility and the patient’s tolerance of the immunosuppression that results from the ablative therapy. Patients require intensive nursing care that is directed toward preventing infection and assessing for early signs and symptoms of complications. One common complication involves the formation of lymphocytes that respond to their new host (ie, the patient) as foreign and mount a reaction against the body. This process, known as graftversus-host disease (GVHD), can involve the skin, gastrointestinal tract, and liver and can be life-threatening. In hematologic malignancies, some GVHD is actually desirable in that the donor lymphocytes can also mount a reaction against any lingering tumor cells; this process is referred to as graft-versusmalignancy. GVHD is a significant complication in nonmyeloablative transplantation therapy, as well as in conventional allotransplantation. Late complications (occurring more than 100 days after transplantation) are frequent; these patients, particularly those who receive an allogeneic transplant, require careful follow-up for years after transplantation. (See Chap. 16 for further information on GVHD) ? Critical Thinking Exercises 1. You are working in a hematology-oncology clinic. The laboratory reports a critical study result for one of your patients with CLL: the reticulocyte count is 25%. What other laboratory results would be important to review or consider? The patient is profoundly anemic; does this support your original thinking and problem solving? What medical treatment orders would you anticipate? What nursing interventions would be appropriate? 2. You are caring for a young adult patient who has had repeated hospitalizations for sickle cell crisis. What factors should be assessed to determine the patient’s education, coping, and pain management needs? What is important for the patient’s discharge plan? 3. You are caring for a patient diagnosed with leukemia. The family members are very concerned about the patient’s risk for infection at home. What assessments will assist you to determine the patient’s risk of developing an infection at home? What instructions should you give about decreasing the risks for infection? How would you alter your interventions if the family members are not fluent in English? 4. You are caring for a patient who is septic and is now receiving a transfusion of 2 units of PRBCs. The patient’s temperature spikes to 38.5°C after half of the second unit has been transfused. What are the possible causes of the fever? What are the appropriate nursing interventions? Chapter 33 Assessment and Management of Patients With Hematologic Disorders REFERENCES AND SELECTED READINGS Books Abrams, A. C. (2001). Clinical drug therapy: Rationales for nursing practice. Philadelphia: Lippincott Williams & Wilkins. American Association of Blood Banks. (2002). Blood transfusion therapy: A physician’s handbook (7th ed.). Bethesda, MD: Author. Anderson, K. C., & Ness, P. M. (Eds.). (2000). Scientific basis of transfusion medicine: Implications for clinical practice. Philadelphia: W. B. Saunders. Blumenthal, M., Goldberg, A., & Brinkman, J. (Eds.). (2000). Herbal medicine. Newton, MA: Integrative Medicine Communications. Hoffman, R., Benz, E. J., Shattil, S. J., Furie, B., Cohen, H. J., Silberstein, L. E., et al. (Eds.). (2000). Hematology: Basic principles and practice (3rd ed.). New York: Churchill Livingstone. Lee, G., Foerster, J., Lukens, J., Paraskevas, F., Rogers, G. (1999). Wintrobe’s clinical hematology (10th ed.). Baltimore: Williams & Wilkins. Mintz, P. D. (Ed.). (1999). Transfusion therapy: Clinical principles and practice. Bethesda, MD: American Association of Blood Banks Press. Rieger, P. T. (Ed.). (2001). Biotherapy: A comprehensive overview (2nd ed.). Boston: Jones and Bartlett. Schechter, G., Berliner, N., & Telen, M. J. (Eds.). (2000). Hematology 2000. [Monograph]. San Francisco: American Society of Hematology Educational Program Book. Schechter, G., Hoffman, R., & Schrier, S. (Eds.). (1999). Hematology. [Monograph]. New Orleans: American Society of Hematology Educational Program Book. Skeel, R. (Ed.). (1999). Handbook of cancer chemotherapy (5th ed.). Philadelphia: Lippincott Williams & Wilkins. Westphal, R. G. (1996). Handbook of transfusion medicine (3rd ed.). Washington, DC: American Red Cross Blood Services. Wilkes, G. M., Ingwersen, K., & Barton-Burke, M. (2000). 2000 Oncology nursing drug handbook. Boston: Jones and Bartlett. Winningham, M. L., & Barton-Burke, M. (Eds.). (2000). Fatigue in cancer: A multidimensional approach. Boston: Jones and Bartlett. Journals Asterisks indicate nursing research articles. Aaronson, L. S., Teel, C. S., Cassmeyer, V., Neuberger, G. B., Pallikkathayil, L., Pierce, J., Press, A. N., Williams, P. D., & Wingate, A. (1999). Defining and measuring fatigue. Image: The Journal of Nursing Scholarship, 31(1), 45–50. Adams, R. J. (2000). Lessons from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study. Journal of Child Neurology, 15(5), 344–349. Alcoser, P. W., & Burchetts, S. (1999). Bone marrow transplantation: Immune system suppression and reconstitution. American Journal of Nursing, 99(6), 26–30. Anderson, K., Hamblin, T. J., & Traynor, A. (1999). Management of multiple myeloma today. Seminars in Hematology, 36, (1 Suppl. 3), 3–8. Andrews, N. C. (1999). Disorders of iron metabolism. New England Journal of Medicine, 341(26), 1986–1995. Applebaum, F. R., Baer, M. R., Carabisi, M. H., Coutre, S. E., Erba, H. P., Estey, E., Glenn, M. J., Kraut, E. H., Maslak, P., Millenson, M., Miller, C. B., Saba, H. I., Stone, R., Tallman, M. S.; National Comprehensive Cancer Network. (2000). NCCN practice guidelines for acute myelogenous leukemia. Oncology, 14(11A), 53–61. *Baker, F., Zabora, J., Pollard, A., & Wingard, J. (1999). Reintegration after bone marrow transplantation. Cancer Practice, 7(4), 190–197. Barbui, T., & Finazzi, G. (1999). Clinical parameters for determining when and when not to treat essential thrombocytopenia. Seminars in Hematology, 36, (1 Suppl. 2), 14–18. Bensinger, W., Martin, P. J., Storer, B., Clift, R., Forman, S. J., Negrin, R., Kashyap, A., Flowers, M. E., Lilleby, K., Chauncey, T. R., Storb, R., & Appelbaum, F. R. (2001). Transplantation of bone marrow as compared with peripheral-blood cells from HLA-identical relatives in patients with hematologic cancers. New England Journal of Medicine, 344(3), 175–181. 933 Beran, M. (2000). Intensive chemotherapy for patients with high-risk myelodysplastic syndrome. International Journal of Hematology, 72(2), 139–150. Berman, E., Clift, R. A., Copelan, E. A., Emanuel, P. D., Erba, H. P., Glenn, M. J., Greenberg, P. L., Jones, R. J., O’Brien, S., Saba, H. I., Schilder, R., Snyder, D. S., Soiffer, R. J., Tallman, M. S., Wetzler, M., Ravansi-Kashani, F., Kantarjian, H., Talpaz, M. (2000). NCCN practice guidelines for chronic myelogenous leukemia. Oncology, 14(11A), 229–240. Berenson, J. (2001). New advances in the biology and treatment of myeloma bone disease. Seminars in Hematology, 38(2 Suppl. 3), 15–20. Bick, R. L. (1999). Low molecular weight heparins in the outpatient management of venous thromboembolism. Seminars in Thrombosis and Hemostasis, 25 (Suppl. 3), 97–99. Bradbury, M., & Cruickshank, J. P. (2000). Blood transfusion: Crucial steps in maintaining safe practice. British Journal of Nursing, 9(3), 134–138. Briere, J., & Guilmin, F. (2001). Management of patients with essential thrombocythemia: Current concepts and perspectives. Pathologiebiologie, 49(2), 178–183. Brown, P. (2001). Transfusion medicine and spongiform encephalopathy. Transfusion, 41(4), 433–436. Burney, K. (2000). Tips for timely management of febrile neutropenia. Oncology Nursing Forum, 27(4), 617–618. Carella, A. M., Cavaliere, M., Lerma, E., Ferrara, R., Tedeschi, L., Romanelli, A., Vinci, M., Pinotti, G., Lambelet, P., Loni, C., Verdiani, S., De Stefano, F., Valbonesi, M., & Corsetti, M. T. (2000). Autografting followed by nonmyeloablative immunosuppressive chemotherapy and allogeneic peripheral-blood hematopoietic stem-cell transplantation as treatment of resistant Hodgkin’s disease and non-Hodgkin’s lymphoma. Journal of Clinical Oncology, 18(23), 3918–3924. Carella, A. M., Giralt, S., & Slavin, S. (2000). Low intensity regimens with allogeneic hematopoietic stem cell transplantation as treatment of hematologic neoplasia. Haematologica, 85(3), 304–313. Cassileth, P. A., Harrington, D. P., Appelbaum, F., Lazarus, H. M., Rowe, J. M., Paietta, E., Willman, C., Hurd, D. D., Bennett, J. M., Blume, K. G., Head, D. R., & Wiernik, P. H. (1998). Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in first remission. New England Journal of Medicine, 339(23), 1649–1656. Cavalli, F. (1998). Rare syndromes in Hodgkin’s disease. Annals of Oncology, 9, (Suppl. 5), S109–S113. Cervantes, F. (2001). Prognostic factors and current practice in treatment of myelofibrosis with myeloid metaplasia: An update anno 2000. Pathologie-biologie, 49(2), 148–152. Champlin, R., Khouri, I., Komblau, S., Molidrem, J., & Giralt, S. (1999). Reinventing bone marrow transplantation: Nonmyeloablative preparative regimens and induction of graft vs malignancy effect. Oncology, 13(5), 621–628. Cheson, B., Horning, S., Coiffier, B., Shipp, M., Fisher, R., Conners, J., Lister, T., Vose, J., Grillo-Lopez, A., Hagenbeek, A., Cabanillas, F., Klippensten, D., Hiddemann, W., Castellino, R., Harris, N., Armitage, J., Carter, W., Hoppe, R., & Canellos, G. (1999). Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. Journal of Clinical Oncology, 17(4), 1244–1253. Coiffer, B. (2002). Rituximab in the treatment of diffuse large B-cell lymphomas. Seminars in Oncology, 29(1 Suppl. 2), 30–35. Corwin, E. J. (2000). Understanding cytokines. Part I: Physiology and mechanisms of action. Biologic Research for Nursing, 2(1), 30–40. Corwin, E. J. (2000). Understanding cytokines. Part II: Implications for nursing research and practice. Biologic Research for Nursing, 2(1), 41–48. Creteur, J., Sibbald, W., & Vincent, J. L. (2000). Hemoglobin solutions: Not just red blood cell substitutes. Critical Care Medicine, 28(8), 3025–3034. Derderian, P. M., Kantarjian, H. M., Talpaz, M., O’Brien, S., Cork, A., Estey, E., Pierce, S., & Keating, M. (1993). Chronic myelogenous 934 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION leukemia in the lymphoid blastic phase: Characteristics, treatment response, and prognosis. American Journal of Medicine, 94(1), 69–74. Deagle, J., & Kelaher, N. (1999). Deep-vein thrombosis: The shift to outpatient care. Nursing Times, 95(46), 48–49. *de Carvalho, E. C., Goncalves, P. G., Bontempo, A. M., & Sloer, V. (2000). Interpersonal needs expressed by patients during bone marrow transplantation. Cancer Nursing, 23(6), 462–467. Deeg, H. J. & Applebaum, F. R. (2000). Hematopoietic stem cell transplantation in patients with myelodysplastic syndrome. Leukemia Research, 24(8), 653–663. Delmore, B. A., Hansen, D., Mooney, K. A., Paplanus, L. M., & Sutton, P. R. (2000). An anticoagulation pathway for quality management. Applied Nursing Research, 13(2), 105–110. Druker, B., Talpaz, M., Resta, D., Peng, B., Buchdinger, E., Ford., J., Lydon, N., Kantargian, H., Capdeville, R., Ohno-Jones, S., & Sawyers, C. L. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. New England Journal of Medicine, 344(14), 1031–1037. *Duquette-Petersen, L., Francis, M., Dohnalek, L., Skinner, R., & Dudas, P. (1999). The role of protective clothing in infection prevention in patients undergoing autologous bone marrow transplantation. Oncology Nursing Forum, 26(8), 1319–1324. Elalamy, I., Lecrubier, C., Horellou, M. H., Conard, J., & Samama, M. M. (2000). Heparin-induced thrombocytopenia: Laboratory diagnosis and management. Annals of Medicine, 32, (Suppl. 1), 60–67. Ely, E. W., & Bernard, G. R. (1999). Transfusions in critically ill patients. New England Journal of Medicine, 340(6), 467–468. Emmanouilides, C., Rosen, P., Telatar, M., Malone, R., Bosserman, L., Menco, H., Patel, R., Barstis, J., & Grody, W. (2000). Excellent tolerance of rituximab when given after mitoxantrone/cyclophosphamide: An effective and safe combination for indolent nonHodgkin’s lymphoma. Clinical Lymphoma, 1(2), 146–151. Engstrom, C. A., & Sarkodee-Adoo, C. (1998). The molecular biology of lymphoma. Seminars in Oncology Nursing, 14(4), 256–261. Fareed, J., Hoppensteadt, D. A., & Bick, R. L. (2000). An update on heparins at the beginning of the new millennium. Seminars in Thrombosis and Hemostasis, 26, (Suppl. 1), 5–21. Ferster, A., Tahriri, P., Vermylen, C., Sturbois, G., Corazza, F., Fondu, P., Devalck, C., Dresse, M., Feremans, W., Hunninck, K., Toppet, M., Philippet, P., Van Geet, C., & Sariban, E. (2001). Five years of experience with hydroxyurea in children and young adults with sickle cell disease. Blood, 97(11), 3628–3632. *Fitzgerald, M., Hodgkinson, B., & Thorp, D. (1999). Blood transfusion from the recipient’s perspective. Journal of Clinical Nursing, 8(5), 593–600. George, J. N., Kojouri, K., Perdue, J. J., & Vesely, S. K. (2000). Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Seminars in Hematology, 37(3), 290–298. Giardina, P. J. & Grady, R. W. (1995). Chelation therapy in betathalassemia: The benefits and limitations of desferrioxamine. Seminars in Hematology, 32(4), 304–312. Gilbert, H. S. (1999). Historical perspective on the treatment of essential thrombocythemia and polycythemia vera. Seminars in Hematology, 36, (1 Suppl. 2), 19–22. Gobel, B. H. (1999). Disseminated intravascular coagulation. Seminars in Oncology Nursing, 15(3), 174–182. Gochee, P. A. & Powell, L. W. (2001). What’s new in hemochromatosis. Current Opinion in Hematology, 8(2), 98–104. Goldman, D. A. (2001). Thalidomide use: Past history and current implications for practice. Oncology Nursing Forum, 28(3), 471–477. Goldman, J., & Melo, J. (2001). Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. New England Journal of Medicine, 344(14), 1084–1086. Gorman, K. (1999). Sickle cell disease. American Journal of Nursing, 99(3), 38–43. Greenberg, P., Baer, M., Bennett, J., Bloomfield, C., Deeg, H., J., Erba, H. P., et al. (2000). NCCN practice guidelines for myelodysplastic syndromes, Version 2000. In The Complete Library of NCCN Guidelines [CD-ROM]. Rockledge, PA. National Comprehensive Cancer Network. Greenlee, R. T., Hill-Harmon, M., Murray, T., & Thun, M. (2001). Cancer statistics, 2001. CA: A Cancer Journal for Clinicians, 51(1), 15–36. Gruppo Italiano Studio Policitemia (GISP). (1995). Polycythemia vera: The natural history of 1213 patients followed over 20 years. Annals of Internal Medicine, 123(9), 656–664. Hainsworth, J., Burris, H., Morrissey, L., Litchy, S., Scullin, D., Bearden, J., Richards, P., & Greco, F. (2000). Rituximab monoclonal antibody as initial systemic therapy for patients with low-grade non-Hodgkin lymphoma. Blood, 95(10), 3052–3056. Hays, K., & McCartney, S. (1998). Nursing care of the patient with chronic lymphocytic leukemia. Seminars in Oncology, 25(1), 75–79. Heaney, M. L., & Golde, D. W. (1999). Myelodysplasia. New England Journal of Medicine, 340(21), 1649–1660. Hehlmann, R. (2000). Interferon alpha in the treatment of polycythemia vera. Annals of Hematology, 79(3), 103–109. Hiddemann, W., & Buchner, T. (2001). Current status and perspectives of therapy for acute myeloid leukemia. Seminars in Hematology, 38, (3 Suppl. 6), 3–9. Hoelzer, D., & Gokbuget, N. (2000). New approaches to acute lymphoblastic leukemia in adults: Where do we go? Seminars in Oncology, 27(5), 540–559. Hu, K., & Yahalom, J. (2000). Radiotherapy in the management of plasma cell tumors. Oncology, 14(1), 101–108. Huffstutler, S. Y. (2000). Adult anemia. Advance for Nurse Practitioners, 8(3), 89–91. Jantunen, R., Juvonen, E., Ikkala, E., Okansen, K., Antilla, P., & Ruutu, T. (2001). The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Annals of Hematology, 80(2), 74–78. Josting, A., & Diehl, V. (2001). Early-stage Hodgkin’s disease. Current Oncology Reports, 3(3), 279–284. Kanis, J. A., & McCloskey, E. V. (2000). Bisphosphonates in multiple myeloma. Cancer, 88, (12 Suppl.), 3022–3032. Kelton, J. G. (1999). The clinical management of heparin-induced thrombocytopenia. Seminars in Hematology, 36, (1 Suppl. 1), 17–21. Kelton, J. G., & Bussel, J. B. (2000). Idiopathic immune thrombocytopenic purpura: An update. Seminars in Hematology, 37(3), 219–221. Korman, M. T., Leparc, G., & Benson, K. (2001). Nucleic acid amplification testing: The new infectious disease testing method for donor blood. Available at: http://www.moffitt.usf.edu/pubs/ccj/v6n5/ dept5.htm. Accessed June 28, 2001. Kornblith, A. B., Herndon, J. E. II, Silverman, L. R., Demakos, E. P., Holland, J. F., et al. (1998). The impact of 5-azacytidine on the quality of life of patients with myelodysplastic syndrome (MDS) treated in a randomized phase III trial of the cancer and leukemia group B (CALGB). Proceedings of the American Society of Clinical Oncology, 17, Abstract 189. Kosits, C., Callaghan M. (2000). Rituximab: A new monoclonal antibody therapy for non-Hodgkin’s lymphoma. Oncology Nursing Forum, 27(1), 51–59. Kreimeier, U., & Messmer, K. (2002). Perioperative hemodilution. Trannsfusions and Apheresis Science, 27(1), 59–72. Kuter, D. J. (2000) Future directions with platelet growth factors. Seminars in Hematology, 37, (2 Suppl. 4), 41–49. Lengfelder, E., Berger, U., & Hehlmann, R. (2002). Interferon alpha in the treatment of polycythemia vera. Annals of Hematology, 79(3): 103–109. Loney, M., & Chernecky, C. (2000). Anemia. Oncology Nursing Forum, 27(6), 951–962. Lusher, J. M. (2000). Inhibitor antibodies to factor VIII and factor IX: Management. Seminars in Thrombosis and Hemostasis, 26(2), 179–188. Major, P., Lortholary, A., Hon, J., Abdi, E., Mills, G., Menssen, H. D., Yunus, F., Bell, R., Body, J., Quebe-Fehling, E., & Seaman, J. (2001). Zolendronic acid is superior to pamidronate in the treatment of tumor-induced hypercalcemia: A pooled analysis of two randomized, controlled clinical trials. Journal of Clinical Oncology, 19(2), 558–567. Marcaccio, M. J. (2000). Laparoscopic splenectomy in chronic idiopathic thrombocytopenic purpura. Seminars in Hematology, 37(3), 267–274. Chapter 33 Assessment and Management of Patients With Hematologic Disorders Margolis, J., Borrello, I., & Flynn, I. W. (2000). New approaches to treating malignancies with stem cell transplantation. Seminars in Oncology, 27(5), 524–530. McCullough, J. (2000). Current issues with platelet transfusion in patients with cancer. Seminars in Hematology, 37, (2 Suppl. 4), 3–10. Micallef, I. N., Rohatiner, A. Z., Carter, M., Boyle, M., Slater, S., Amess, J. A., & Lister, T. A. (2001). Long-term outcome of patients surviving for more than ten years following treatment for acute leukemia. British Journal of Haematology, 113(2), 443–445. Michiels, J. J., Barbui, T., Finazzi, G., Fuchtman, S. M., Kutti, J., Rain, J. D., et al. (2000). Diagnosis and treatment of polycythemia vera and possible future study designs of the PVSG. Leukemia and Lymphoma, 36(3–4), 239–253. Mitka, M. (2001). FDA wants more restrictions on donated blood. Journal of the American Medical Association, 286(4), 408. Money, S. R., & York, J. W. (2001). Development of oral heparin therapy for prophylaxis and treatment of deep venous thrombosis. Cardiovascular Surgery, 9(3), 211–218. Murphy, S. (1999). Diagnostic criteria and prognosis in polycythemia vera and essential thrombocythemia. Seminars in Hematology, 36, (1 Suppl. 2), 9–13. NCCN practice guidelines for Hodgkin’s disease. National Comprehensive Cancer Network. Oncology, 13(5A), 78–110. Ohene-Frempong, K. (2001). Indications for red cell transfusion in sickle cell disease. Seminars in Hematology, 38, (1 Suppl. 1), 5–13. O’Rourke, M. E., & High, K. (2000). Diagnosing graft versus host disease. Clinical Journal of Oncology Nursing, 4(1), 47–48. Osterbor, A. (2000). The role of recombinant human erythropoietin in the management of anaemic cancer patients: Focus on haematological malignancies. Medical Oncology, 17, (Suppl 1), S17–S22. Patt, J., & Morrison, S. (2001). National Cancer Institute resources for patients and their caregivers. Cancer Practice, 9(5), 257–261. *Pederson, C., & Parren, L. (1999). Pain in adult recipients of blood or marrow transplant. Cancer Nursing, 22(6), 397–407. Petrykk, M., & Grossbard, M. L. (2000). Rituximab therapy of B-cell neoplasms. Clinical Lymphoma, 1(3), 186–194. Rabinovici, R. (2001). The status of hemoglobin-based red cell substitutes. Israel Medical Association Journal, 3(9), 691–697. Radich, J., & Sievers, E. (2000). New developments in the treatment of acute myeloid leukemia. Oncology, 14(11A), 125–131. Reed, W., Walker, P., Haddix, T., & Perkins, H. A. (2000). Acute anemic events in sickle cell disease. Transfusion, 40(3), 267–273. Rimm, E. B., Willett, W. C., Hu, F. B., Sampson, L., Colditz, G. A., Manson, J. E., Hennekens, C., & Stampfer, M. J. (1998). Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA, 279(5), 359–364. Rust, D., Simpson, J., & Lister, J. (2000). Nutritional issues in patients with severe neutropenia. Seminars in Oncology Nursing, 16(16), 152–162. Saleh, M. N., Gutheil, J., Moore, M., Bunch, P. W., Butler, J., Kunkel, L., Grillo-Lopez, A. J., & LoBuglio, A. F. (2000). A pilot study of the anti-CD20 monoclonal antibody rituximab in patients with refractory immune thrombocytopenia. Seminars in Oncology, 27, (6 Suppl. 12), 99–103. Scaradavou, A. (2000). Splenectomy-sparing, long-term maintenance with anti-D for chronic immune (idiopathic) thrombocytopenic purpura: The New York Hospital experience. Seminars in Hematology, 37, (1 Suppl. 1), 42–44. Seligsohn, U., & Lubetsky, A. (2001). Genetic susceptibility to venous thrombosis. New England Journal of Medicine, 344(16), 1222–1231. Shapiro, A. D. (2000). Platelet function disorders. Haemophilia, 6, (Suppl. 1), 120–127. Shelton, B. K. (1999). Sepsis. Seminars in Oncology Nursing, 15(3), 209–221. Sievers, E., Appelbaum, F., Speilberger, R. T., Forman, S., Flowers, D., Smith, F., Shannon-Dorcy, K., Berger, M. S., & Bernstein, I. D. (1999). Selective ablation of acute myeloid leukemia using antibodytargeted chemotherapy: A phase I study of anti-CD33 calicheamicin immunoconjugate. Blood, 93(11), 3678–3684. 935 Silverstein, M. R., & Tefferi, A. (1999). Treatment of essential thrombocythemia with anagrelide. Seminars in Hematology, 36, (1 Suppl. 2), 23–25. Singhal, S., Mehta, J., Desikan, R., Ayers, D., Roberson, P., Eddlemon, P., Munshi, N., Anaissie, E., Wilson, C., Dhodapkar, M., Zeddis, J., & Barlogie, B. (1999). Antitumor activity of thalidomide in refractory multiple myeloma. New England Journal of Medicine, 341(21), 1565–1571. Slavin, S., Or, R., Aker, M., Shapira, M. Y., Panigrahi, S., Symeonidis, A., Cividalli, G., & Nagler, A. (2001). Nonmyeloablative stem cell transplantation for the treatment of cancer and life-threatening nonmalignant disorders: Past accomplishments and future goals. Cancer Chemotherapy and Pharmacology, 48, (Suppl. 1), S79–S84. *Smith, L. H., & Besser, S. G. (2000). Dietary restrictions for patients with neutropenia: A survey of institutional practices. Oncology Nursing Forum, 27(3), 515–520. Stampfer, M. J., Hu, F. B., Manson, J. E., Rimm, E. B., & Willett, W. C. (2000). Primary prevention of coronary heart disease in women through diet and lifestyle. New England Journal of Medicine, 343(1), 16–22. Stasi, R., Pagano, A., Stipa, E., Amadori, S. (2001). Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood, 98(4), 952–957. Steinberg, M. H. (1999). Management of sickle cell disease. New England Journal of Medicine, 340(13), 1021–1030. Tefferi, A., Fonseca, R., Pereira, D. L., & Hoagland, H. C. (2001). A long-term retrospective study of young women with essential thrombocythemia. Mayo Clinic Proceedings, 76(1), 22–28. Tefferi, A., Solberg, L. A., & Silverstein, M. N. (2000). A clinical update in polycythemia vera and essential thrombocythemia. American Journal of Medicine, 109(2), 141–149. Tennant, L. (2001). Chronic myelogenous leukemia: An overview. Clinical Journal of Oncology Nursing, 5(5), 218–219. Terpos, E., Palermos, J., Tsinos, K., Anargyrou, K., Viniou, N., Papassavas, P., Meletis, J., & Yataganas, X. (2000). Effect of pamidronate administration on markers of bone turnover and disease activity in multiple myeloma. European Journal of Haematology, 65, 331–336. Tesch, H., Sieber, M., & Diehl, V. (2001). Treatment of advanced stage Hodgkin’s disease. Oncology, 60(2), 101–109. Thomas, M. L. (1998). Anemia and quality of life in cancer patients: Impact of transfusion and erythropoeitin. Medical Oncology, 15, (Suppl. 1), S13–S18. Thomas, M. L. (1998). Quality of life and psychsocial adjustment in patients with myelodysplastic syndromes. Leukemia Research, 22, (Suppl. 1), S41–S47. Traynor, A. E., & Noga, S. J. (2001). NCCN: multiple myeloma. Cancer Control, 8, (6 Suppl. 2), 78–87 Vadhan-Raj, S. (2000). Clinical experience with recombinant human thrombopoietin in chemotherapy-induced thrombocytopenia. Seminars in Hematology, 37, (2 Suppl. 4), 28–34. Viale, P. H. (1999). Management of thromboembolism in patients with cancer. Oncology Nursing Forum, 26(10), 1625–1632. Vichinsky, E. P., Neumayr, L. D., Earles, A. N., Williams, R., Lennett, E. T., Dean, D. D., Nickerson, B., Orringer, E., McKie, V., Bellevue, R., Daeschner, C., Manci, E. A. (2000). Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. New England Journal of Medicine, 342(25), 1855–1865. Wazny, L. D., & Ariano, R. E. (2000). Evaluation and management of drug-induced thrombocytopenia in the acutely ill patient. Pharmacotherapy, 20(3), 292–307. Weiss, L. M. (2000). Epstein-Barr virus and Hodgkin’s disease. Current Oncology Reports 2(2), 199–204. White, G. C., Greenwood, R., Escobar, M., & Frelinger, J. A. (2000). Hemophilia factor VIII therapy: Immunological tolerance. Haematologica, 85, (10 Suppl.), 113–116. Young, N. S. (2000). Hematopoietic cell destruction by immune mechanisms in acquired aplastic anemia. Seminars in Hematology, 37(1), 3–14. 936 Unit 6 CARDIOVASCULAR, CIRCULATORY, AND HEMATOLOGIC FUNCTION Young, N. S. (1999). Acquired aplastic anemia. JAMA, 282(3), 271–278. Zaidi, A. A., & Vesole, D. H. (2001). Multiple myeloma: An old disease with new hope for the future. CA: A Cancer Journal for Clinicians, 51(5), 273–285. Zelenetz, A., & Hoppe, R. T. (2001). NCCN: Non-Hodgkin’s lymphomas. Cancer Control, 8(6 Suppl. 2), 102–113. RESOURCES AND WEBSITES Alternative Medicine Foundation, Inc., 5411 W. Cedar Lane, Suite 205A, Bethesda, MD 20814; 301-340-1960; http://www.amfoundation.org. American Association of Blood Banks (AABB), 8101 Glenbrook Road, Bethesda, MD 20814-2749; 301-907-6977; http://www.aabb.org. American Cancer Society, 1599 Clifton Rd., N.E., Atlanta, GA 30329; 800-227-2345; http://www.cancer.org. American Hemochromatosis Society, 777 E. Atlantic Ave., PMB Z-363, Delray Beach, FL 33483-5352; 1-888-655-4766; http://www. americanhs.org. American Pain Society, 4700 W. Lake Ave., Glenview, IL 60025; 1-847375-4715; http://www.ampainsoc.org. American Red Cross, 1730 E Street NW, Washington, DC 20006; 1-202-639-3520; http://www.redcross.org. Aplastic Anemia and MDS International Foundation, PO Box 613, Annapolis, MD 21404; 1-800-747-2820; http://www.aplastic.org. Blood and Marrow Transplant Newsletter, 1985 Spruce Ave., Highland Park, IL 60036. International Myeloma Foundation, 12650 Riverside Drive, Suite 206, North Hollywood, CA 91607; 1-800-452-2873; http://www. myeloma.org. Leukemia and Lymphoma Society, 1311 Mamaroneck Ave. White Plains, NY 10605; 1-800-955-4572 http://www.leukemialymphoma.org. Myelodysplastic Syndromes Foundation, PO Box 477, 464 Main St., Crosswicks, NJ 08515; 1-800-637-0839; http://www.mdsfoundation.org. National Association of Vascular Access Networks, 11417 S. 700 East, Suite 205, Draper, UT 84020; 888-576-2826; http://www. navannet.org. National Cancer Institute Cancer Information Service, 31 Center Drive, MSC 2580, Building 31, Room 10A16, Bethesda, MD 20892-2580; 1-800-4-CANCER; http://www.nci.nih.gov. National Hemophilia Foundation, 116 W. 32nd St., 11th Floor, New York, NY 10001; 800-424-2634; http://www.hemophilia.org. National Marrow Donor Program, Suite 500, 3001 Broadway St. N.E., Minneapolis, MN 55413; 800-627-7692; http://www.marrow.org. Office of Dietary Supplements, National Institutes of Health, 6100 Executive Blvd., Rm 3B0l, MSC 7517, Bethesda, Maryland 208927517; 301-435-2920; http://ods.od.nih.gov. Oncology Nursing Society, 501 Holiday Dr., Pittsburgh, PA 152202749; http://www.ons.org. Sickle Cell Disease Association of America, Inc., 200 Corporate Pointe, Suite 495, Culver City, CA 90230-8727; http://www.sicklecell disease.org.