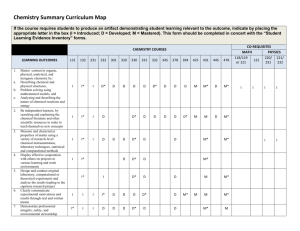
NPTEL-Module-1: Introduction to Bioorganic Chemistry Dr. S. S. Bag
advertisement

NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics NPTEL Phase – II (Syllabus Template) Course Title: Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Module 3: Designed Enediyne Model Systems: Introduction to Structural Features of Enedynes; Factors Affecting the Reactivity of Enediynes; Molecular Design of Enediyne Models; Various Synthetic Approaches to Acyclic/Cyclic Enediynes; Various Synthetic Aprroaches to Cyclic Enediynes; Synthesis of Dienediyne Core of NCS chromophore; β-Lactam as a Molecular Lock of Enediyne: Synthesis of β-Lactam Fused Enediynes; Enediynes with pH-Based Triggering Devices; Photoswitchable Enediynes; Biological Actions of Some Synthetic Models; Enediyne as a Scaffold for Peptidomimetics; Enediyne as Peptide Cleaving Agent. 3.1. Introduction to Structural Features of Enedynes The common structural motif among enediyne antibiotics is an enediyne moiety (Z-hexa-1,5diyn-3-ene), the conjugated system, found embedded within a 9- or 10-membered cyclic framework. The enediyne antibiotics have been divided into two subfamilies, including 9membered cyclic enediynes such as NCS, kedarcidin, LDM, maduropeptin and N1999A2 and 10-membered cyclic enediynes such as CAL, ESP, DYN, and shishijimicins A-C (Figure 1). 9-membered cyclic enediynes contain the chromophore containing the enediyne core and an apoprotein unit with noncovalent binding. The enediyne core of the chromophore is located in the center of the pocket and other substituents are arranged around the core. The enediyne core of the chromophore is the anticancer part, but the free chromophore is labile. The apoprotein is inactive in cleavage of DNA; however it plays an important role in drug action by stabilizing the labile chromophore. The apoprotein is believed to be resistant to proteases, protect the chromophore from deactivation and to deliver the enediyne to intracellular target DNA. Only N1999A2 is a non-protein 9-membered cyclic enediyne antibiotic and is stable in nature. The structures of 10-membered cyclic enediynes do not contain an apoprotein and are more stable than those of 9-membered cyclic enediynes. Figure 1. Presentation of three types of enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 1 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics In calicheamicins and dynemicins, a 3-ene-1, 5-diyne system is embedded in a 10-membered ring. These compounds belong to Type I enediynes. In Type II enediynes, the 3-ene-1, 5-diyne system is included in a 9-membered ring as in kedarcidin. There is another class of enediyne (Type III), in which a 9-membered cyclic dienediyne is present as in neocarzinostatin. The enediynes represent an ingenuity of nature’s work. It has been compared to a smart bomb equipped with: a) a delivery system which is responsible for a strong and specific complexation with DNA. This system is represented by the oligosaccharide unit as in calicheamicin and esperamicin; b) a warhead (the enediyne moiety) that is able to attack simultaneously the two complementary DNA strands, causing the lethal double strand cut; c) a safety catch or a locking device that prevents the enediyne from undergoing the diradical formation, by imposing a structural restraint to its reaction. In this way the warhead does not explode until a particular chemical event takes place (Table 1). In calichamicin this is represented by the enamine double bond; d) finally, a chemical trigger that mediates the removal of the safety catch and therefore unleashes the high reactivity of the enediyne. In calicheamicins the trigger is the trisulfide group (Figure 2). Table 1. General structural features of enediynes Structural Units Warhead: Locking Device Triggering Device Binding Device Structural Features/Functions Pictorial presentation The Enediyne. Stabilizes the enediyne from undergoing rearrangement It offers a mechanism by which locking is removed and enediynes become reactive Gives Specificity Joint initiative of IITs and IISc – Funded by MHRD Page 2 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Figure 2. Structural features of Calichiamycin. 3.2. Factors Affecting the Reactivity of Enediynes The process of generating benzenoid diradical in enedynes via the well known Bergman cyclization (BC), and/ or Myers−Saito cyclization (MSC) reaction is primarily responsible for the antitumor activity of naturally occurring enediynes, such as calicheamicins. Since the discovery, these two reactions are believed to be at the heart of antitumor activity naturally occurring enediynes. Therefore the factors that affect the kinetics of these reactions are mostly responsible for biological activity of enediynes. It is the inherent property of 9- or 10-membered monocyclic natural enediynyl systems to cyclize spontaneously at physiological temperature, while the acyclic enediynes undergo thermal cycloaromatization at elevated temperatures only (≥200 °C). To ensure the safe delivery of the enediyne molecule to the target before the enediyne functionality is activated toward diradical generation DNA cleavage by abstraction of H-atom from the sugar−phosphate backbone of DNA via a triggering mechanism Nature has incorporated locking devices into these systems. The triggering or activation process for the natural enediynes primarily involves a change of hybridization at a carbon center encompassing the enediyne moiety or an opening of the epoxide ring fused onto the enediyne in bi- or tricyclic fashion. This structural change is believed to lower the activation barrier for the cycloaromatization process either by bringing the terminal acetylenic carbon atoms (the c and d distance) closer or by minimizing the overall conformational restrictions. Both the factors (distance and strain) often act synergistically to drive the process of diradical generation (BC) at a physiologically relevant temperature. Joint initiative of IITs and IISc – Funded by MHRD Page 3 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Mainly the following factors play the important role in affecting/predicting the kinetics of BC. 1. 2. 3. 4. 5. Ring Size or c, d Distance (Distance theory) State of Hybridization (Strain Theory) Incorporation of Hetero Atom or Strained Ring System Metal ion Complexation Weak non-covalent Interactions The details of all the factors are described in module 1. Here the summary of the most important two factors are given below in Table 2. Table 2. Factors affecting kinetics of BC. Distance theory 1. Rate is proportional to the c,d-distance 3.20 Å -3.31 Å = Critical Distance. 2. It mainly relies on the ground-state configuration of the enediynes. 3. For Acyclic or Simple monocyclic systems, distance theory may be applied. 4. For bicyclic or strained systems it needs caution to apply the theory. Strain Theory 1. Rate is proportional to the strain energy difference between the TS and GS. 2. Strain theory is more general but difficult to estimate. 3. Other steric and stereoelectronic and factors also affects the rate. 4. The strain theory thus supports the consideration of the stereo-electronic factor for the design, synthesis, and study of the chemical and biological reactivity of synthetic mimics. Joint initiative of IITs and IISc – Funded by MHRD Page 4 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics DFT-based calculations on monocyclic enediynes showed that there is no predictive relationship between the alkyne carbon distance and the cyclization activation enthalpy. However, for similar monocyclic systems, a crude relationship does exist between the c and d distance and the kinetics of BC (Nicolaou's empirical critical distance of 3.31−3.20 Å for spontaneous cyclization at room temperature was extended to 3.40−2.90 Å). These calculations were in agreement with the argument that ring-strain effects may become more important in strained systems. It may be mentioned that BC leading to the formation of a diradical is an endothermic process. Dimethyl substitution at both of the alkyne termini increases the endothermicity of the reaction by 12 kcal mol-1. The greater the endothermicity, the slower is the kinetics of BC. In BC the orthogonal relationship is obeyed between the diradical and the aromatic π system. This ruled out the influence of a mesomeric effect upon the stability of the radical and hence the cyclization rate. Ortho effect on BC: Substituents in ortho position in benzannulatedenediynes exert a significant influence on BC kinetics (ortho effect) (Scheme 1). This interaction can be either destabilizing through steric or stabilizing through hydrogen-bonding/hyperconjugation/electron transfer. As for example acceleration of the BC rate is observed for X = OMe, NH 3+, NO2, and CF3. DFT-based calculations have shown that the ortho effect operates through the σ framework of the resulting neutral diradical intermediate. Scheme 1. Stabilizing/destabilizing effects of ortho-substituents on the kinetics of BC. Joint initiative of IITs and IISc – Funded by MHRD Page 5 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3. Molecular Design of Enediyne Models The cycloaromatization reaction of enediynes has created the door of never-ending opportunities for synthetic organic chemists for the development of strategies to synthesize model enediyne architectures. That is why field of enediyne chemistry is rapidly growing toward the design of novel anticancer antibiotic model systems with suitable triggering devices. Research in this field is mainly aim to enhanc the reactivity of designed enediyne toward BC as well as to lower the toxicity under suitable triggering conditions. As a result of tremendous research efforts, during the past 20 years, various enediyne models have been designed and reported that showed potent DNA cleaving activity. The basis of all of these designs can be classified into the following: 1. Consideration of Two Theory: Both the “distance theory” as well as “molecular strain theory” must be considered to design new enediynes as possible therapeutic agents. 2. Stability of Designed Enediyne: The designed enediyne should be sufficiently stable at biological temperature at which the enediyne must possess sufficient half-life (“decent half-life”) that enables full characterization of the molecule. The intrinsically reactive enediynes synthesized must have half-lives ranging from 10 to 36 h at the biological temperature of 37 °C. 3. Designing Acyclic Enediyne: Generally the acyclic enediynes are stable and do not undergo facile BC at biological temperature required to become an efficiently bioactive enediynes. Therefore the designed acyclic enediynes of sufficient stability should be converted to a cyclic framework which would undergo cycloaromatization (BC) at a lower temperature as compared to their progenitor. There are various strategies available exploiting which the stable acyclic enediynes can be converted to a comparatively more reactive cyclic enediynes. These are as follows: (a) Formation of a cyclic framework. (b) Conversion into a metal coordinated cyclic network (metallocycle) (c) Generation of an H-bonded cyclic architecture. (d) Creation of a pseudocyclic network via (i) electrostatic interaction, (ii) charge transfer interaction, (iii) - stacked cycle. (e) Attaching electron withdrawing groups might also lead to enhance the rate of BC. (f) Alternatively, isomerization of acyclic enediyne to a more reactive eneyne−allene system might allow the comparatively unreactive acyclic enediyne to undergo spontaneous MSC. Joint initiative of IITs and IISc – Funded by MHRD Page 6 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 4. Designing Cyclic Enediyne: In the case of design of cyclic enediyne the attractive strategy that is generally adopted is the generation of reactive cyclic enediynes from precursors, which is made ambiently stable by incorporation of small, strained ring systems. The activation/triggering process then operates for the removal of strain via the opening of small rings under various conditions leading to the generation of reactive cyclic enediyne that undergo facile cycloaromatisation to show potent DNA cleaving activity. There are several activation strategies which includes: (a) Oxidative activation of stable cyclic enediynes. (b) Organometal-mediated activation. (c) pH based triggering. (d) Changing the state of hybridization as is shown in Calicheamicin. (e) Photo chemical activation 5. Designing Macrocyclic Enediyne: For design of macrocyclicenediynes one should have option to convert macrocycle into systems of appropriate smaller size (e.g., 10membered ring via transanular reaction) that could react under ambient conditions. 6. DNA Binding Appendage: To design any enediyne capable of showing DNA cleavage activity, the enediyne must possess DNA binding appendage. All of these approaches are summarized in Scheme 2 and 5. Joint initiative of IITs and IISc – Funded by MHRD Page 7 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.1. Designing Strategy for Acyclic Enediynes Scheme 3.2: Design strategy of acyclic enediynes. Scheme 2. Design strategy for acyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 8 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.1.1. Structural Representation of Various Strategically Designed Acyclic Enediynes 3.3.1.1. 1. (a) Organometal/(b) Coenzyme/(c) Base mediated rearrangement Scheme 3. Structural representations of (a) Organometal/(b) Coenzyme/(c) Base mediated rearrangement in acyclic enediynes. 3.3.1.1. 2. (d) Acid mediated rearrangement/(e) photochemical/(f) metal ligation Scheme 4. Structural representations of various designs (d-acid mediated rearrangement; e- photochemical reaction; f-bidentate ligation to metal ion) of acyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 9 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.1.1. 3. (g, h) photochemical activation Scheme 5. Structural representations of various designs (g, h-photochemical activation) of acyclic enediynes. 3.3.1.2. Representative Examples of Various Strategically Designed Acyclic Enediynes 3.3.1.2.1. Examples of Metal-Ion-Induced Bergman Cyclization (Pathway “a”) Scheme 6. Metal-ion-induced BC of biscrown ether. Joint initiative of IITs and IISc – Funded by MHRD Page 10 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 7. Metal-ion-induced BC of bisphosphinoenediyne. Scheme 8. Hg-ion induced of BC bipyridylenediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 11 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 9. Metal-ion-induced BC of bis-sulfonamido and bis-aminoenediynes. Scheme 10. Metal-ion-induced BC of tetraamino enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 12 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 11. Metal-ion-induced BC of bissalicylaldimino enediynes. Scheme 12. Pd-mediated BC of Tetrahedral Pd-phosphino-enediyne complex. Joint initiative of IITs and IISc – Funded by MHRD Page 13 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 13. BC of Cu(I) and Cu(II) complexes of enediynes. 3.3.1.2.2. Examples of Pathway “b” Coenzyme mediated BC Scheme 14. Pyridoxal co-enzyme-mediated BC of acyclic enediyne. 3.3.1.2.3. Examples of Pathway “c”-Base mediated BC Scheme 15. Representative examples of various designs (base mediated BC). Joint initiative of IITs and IISc – Funded by MHRD Page 14 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.1.2.4. Examples of Pathway “d”-Acid Mediated BC Scheme 16. Representative examples of various designs (acid mediated BC). 3.3.1.2.5. Examples of Pathway “e”-Photochemical Mediated BC Scheme 17. Representative examples of various designs (Photochemical BC). Joint initiative of IITs and IISc – Funded by MHRD Page 15 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.1.2.6. Examples of Pathway “f”-Enediyne Cyclization Mediated by Organometallic Reagents Scheme 18. MSC mediated by formation of organometallic complex. Scheme 19. BC of acyclic aliphatic enediynes mediated by Rh-metal complexation and the mechanism. Joint initiative of IITs and IISc – Funded by MHRD Page 16 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 20. Organometallic reagent mediated BC. 3.3.1.2.7. Examples of Pathway “g”-Phochemical Activation Mediated BC Scheme 21. Phochemical activation mediated BC. 3.3.1.2.8. Examples of Pathway “h”-Photochemical mediated BC Scheme 22. Phochemical activation mediated BC. Joint initiative of IITs and IISc – Funded by MHRD Page 17 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.2. Designing Strategy for Cyclic Enediynes Scheme 23. Design strategy of cyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 18 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.2.1. Structural Representation of Various Strategically Designed Cyclic Enediynes: 3.3.2.1. 1. (i-n) Small ring fused cyclic/stable cyclic enediyne/cyclic-1,4-diyne Scheme 24. Structural representations of various designs (i-photochermical, j-base mediated, k-hydride mediated) of cyclic enediynes. Scheme 25. Structural representations of various designs (l- Small ring cleavage by nucleophile, m- Oxidative activation, n-Reductive formation of enediyne) of cyclic enediynes. 3.3.2.1. 2. (o-q) Hybridisation/allylic rearrangement/organometallic activation Joint initiative of IITs and IISc – Funded by MHRD Page 19 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 26. Structural representations of various designs (o-Change of hybridization, p- Allylic rearrangement, q-Organometallic reagent induced activation) of cyclic enediynes. 3.3.2.1. 2. (r) Enediyne Macrocyle to reactive cyclic enediyne via trans annular reaction Scheme 27. Design strategy of macrocyclic enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 20 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.2.3.3. Representative Examples of Various Strategically Designed Cyclic Enediynes: 3.3.2.3.3.1. Examples of Pathway “i” Scheme 28. Representative examples of various designs (i-Photochemical removal of protecting group) of cyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 21 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.2.3.3.2. Examples of Pathway “j” and k Scheme 29. Representative examples of various designs (j-Base induced removal of protecting group, k -Hydride mediated removal of protecting group) of cyclic enediynes. 3.3.2.3.3.3. Examples of Pathway “l” Scheme 30. Representative examples of various designs (l-Small ring cleavage by nucleophile,) of cyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 22 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.2.3.3.3. Examples of Pathway “m” Scheme 31. Representative examples of various designs (m-Oxidative activation) of cyclic enediynes. 3.3.2.3.3.3. Examples of Pathway “n” Scheme 32. Representative examples of various designs (n-Reductive formation of enediyne,) of cyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 23 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.2.3.3.3. Examples of Pathway “o” Scheme 33. Representative examples of various designs (o-Change of hybridization) of cyclic enediynes. 3.3.2.3.3.3. Examples of Pathway “p” Scheme 34. Representative examples of various designs (p-Allylic rearrangement) of cyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 24 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.3.2.3.3.3. Examples of Pathway “q” Scheme 35. BC Mediated by Pentamethyl cyclopentadienyl ruthenium cation (A). 3.3.2.3.3.3. Examples of Pathway “r” Scheme 36. Representative example of (r)-transannular reaction of cyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 25 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.4. Various Synthetic Approaches to Acyclic/Cyclic Enediynes 3.4.1. Basic Strategies for Enediyne Synthesis Basic Strategies for Enediyne Synthesis Z X1 X2 + R X3 R1 (Z) R2 RLarge Y1 Z OR Y2 Scheme 37. Basic retrosynthetic strategies for enediyne synthesis. 3.4.2. Methodologies Involved for the Synthesis of Enediyne Various Methodologies for the Synthesis of Enediynes: Pd-Mediated Coupling Reactions R1 X + R2 R H X R R1 Pd-Cat, Cu-Cat Base R2 R 1. I2, CH2Cl2, 0 oC o 1 ZnCl2, Et2O, -45 C o , -50 C X 2. R1 Bu3Sn Cu(CN)Li 45-75% R1 = (CH3)2CO2Me X= Br, I 2. R2 X Pd(PPh3)4, CuI, Bu3Sn R1 BuNH2, Benzene, r.t OTBS R2 = R1 R2 Me SPh OH 80% Stracker, E. C.; Zweifel, G. Tetrahedron Lett. 1991, 32, 3329. 3 Steps R1 R2 R1 R3 I2, THF Bu3Sn 0 oC- r.t R1 R3 I R2 Magriotis, P. A.; Scott, M. E.; Kim, K. D. Tetrahedron Lett. 1991, 32, 6085. R4 R1 ZnCl R3 Pd(PPh3)4, THF R2 R2 R1 R1, R3 = H, R2 = Sliyl, R4 = H, Alkyl, Aryl, Silyl Scheme 38. Various methodologies for the synthesis of enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 26 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Various Methodologies for the Synthesis of Enediynes Hypervalent iodine R Z Z R H n-BuLi CuCN,DCM, -78 ~25 oC PPh3 I+Ph 2TfO- Z= CH2, O; R=TMS, t-Bu, Bu, Ph 40-69% R Stang, P. J.; Blume, T.; Zhdankin, V. V. Synthesis 1993, 35. Olefination: TBS TMS C H B CHO HO 1. R H 2. HO(CH2)2NH2 TBS H KH,Et2O, r.t 94~81% R R TMS TMS R= n-Bu, Ph Wang, K. K.; Wang, Z.; Gu, Y. G. Tetrahedron Lett. 1993, 34, 8391 Elimination Reaction ODMST R ODMST R OPMB o MsCl, Et3N, DCM, 0 C R R OPMB HO cis : trans > 99 :1 to 96 : 4 R=H, Me, Ph Shibuya, M.; Sakai, Y.; Naoe, Y. Tetrahedron Lett. 1995, 36, 897 Scheme 39. Various methodologies for the synthesis of enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 27 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Various Methodologies for the Synthesis of Enediynes Diels Alder reaction: R R O Cl NiCl2(dppp), THF, Malaic Anhydride, Cl -78 to 20 oC Toluene, 36% R O O R R = nPr Hopf, H.; Theurig, M. Angew. Chem., Int. Ed. Engl. 1994, 33, 1099. Retro-Diels Alder reaction: O KH, THF, 25 oC OH + 90% Bunnage, M. E.; Nicolaou, K. C. Angew. Chem., Int. Ed. Engl. 1996, 35, 4986. Norrish type II fragmentation: h, Benzene, Pyrex O O O O Ph 28 O 98% O O 29 Nuss, J. M.; Murphy, M. M. Tetrahedron Lett. 1994, 35, 37. Scheme 40. Various methodologies for the synthesis of enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 28 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Various Methodologies for the Synthesis of Enediynes Ramberg Backlund reaction: Ph2t-Bu- SiO Br 1)Na2S.Al2O3, DCM, EtOH 2) mCPBA, Et2O, -30°C Br 3) SO2Cl2, pyr, DCM, -78°C 4) mCPBA, Et2O, 25°C% Cl Ph2t-Bu- SiO OH 1) MeLi, THF, -78 °C SO2 Ph2t-Bu- SiO 2) TBAF, THF, 0 oC OH Ph2t-Bu- SiO Nicolaou, K. C.; Zuccarello, G.; Riemer, C.; Estevez, V. A.; Dai, W. M. J. Am. Chem. Soc. 1992, 114, 7360. Carbenoid coupling elimination: Co2(CO)8 LiHMDS, Co2(CO)8, HMPA/THF,- 450C 92% Co2(CO)8 a) Jones, G. B.; Huber, R. S.; Mathews, J. E. J. Chem. Soc., Chem. Commun. 1995, 1791. b) Huber, R. S.; Jones, G. B. Tetrahedron Lett. 1994, 35, 2655. Me N P R N Me Corey-Winter reaction: OH HO OH HO O 7 steps S O OH OH n n = 2, 3 n R =Me (-20 to -5 oC) R = Ph (20 to 25 oC) n = 2 (8%) n = 3 (48%) Semmelhack, M. F.; Gallagher, J. Tetrahedron Lett. 1993, 34, 4121 Oxidation reaction: Y Y TBSO TBSO Ar DDQ, Benzene X Ar = p-MeOC6H4 Ar X Maier, M. E.; Greiner, B. Liebigs Ann. Chem. 1992, 855. Scheme 41. Various methodologies for the synthesis of enediyne Joint initiative of IITs and IISc – Funded by MHRD Page 29 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.5. Various Synthetic Aprroaches to Cyclic Enediynes 3.5.1. Synthesis of 9-Membered Enediynes For the synthesis of the enediyne core analogue of 9-membered enediynes, various carbon carbon bond forming reactions are needed to be carried out. Because of the instinct instability of 9-membered ring enediyne, it is always necessary to prepare them in a suitably protected form like (a) the incorporation of epoxide as in found in kedarcidin chromophore or (b) the protection of one of the triple bond of the enediyne via complexation with metal or (c) incorporation of a strained bicyclic core. The general synthetic approaches to the bicyclic system represented by the following figure can be summarized as follows: General structure of 9-membered enediynes with a bicyclic core a b c e d i) Sonogashira coupling to form the bonds a, b and d. ii) Bond “e” can be formed by intramolecular aldol type condensation followed by elimination. iii) For the generation of NCS core, the double bond “c” is usually converted to epoxide before carrying out the intramolecular aldol reaction for ring closing. iv) The ring closing is preceded by complexation with cobalt. Joint initiative of IITs and IISc – Funded by MHRD Page 30 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.5.2. Magnus’ Approach Magnus' Approach to the Synthesis of 9-Membered Ring Enediyne O THF -78 oC to rt O H3O+ Co2(CO)8 Pd(PPh3)4, O CuI, n-BuNH2 O OR OR A Li HO TBSO OR H O TBSO OH O O O EtO O OEt Co(CO)3 Co EtO OEt (CO)3 Co(CO)3 Co (CO)3 H O O R=H R = TBS OR Bu2BOTf Et3N OR OBBu2 OBBu2 OR . NMMO O H Co(CO)3 OH Co (CO)3 1,4-CHD O O H OH H OR OBBu2 O . H OH OH H OBBu2 H Synthesis of A EtO2C EtO CHO EtO I PPh3 EtO CO2Et EtO I 1.2.14 i) DIBAL EtO ii) TBSOTf, Et3N EtO A I OTBS Scheme 42. Magnus’ approach to the synthesis of 9-membered ring enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 31 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.5.3. Grierson’s Approach Grierson's Approach to the Synthesis of 9-Membered Ring Enediyne O Co(CO)3 (OC)3Co H OTBDPS + CO2Me a Co(CO)3 (OC)3Co OTBDPS b OTBDPS CO2Me H (OC)3Co Co(CO)3 e OTBDPS CO2Me H OH OH H OH OH c OTBDPS Br d CO2Me Br H H OTHP Br OTHP Br Br H OH f g H OTHP S H H H OH OTHP OTHP Reagents and conditions: a) (Meo)MeAlCl, CH2Cl2, 40 oC, (91%); b) CAN, MeOH, 0 oC, (84%); c) DHP, (TMSO)2SO2, CH2Cl2, 0 oC, (87%); d) i. DIBAL-H/THF, 0 oC; ii. TBAF/THF, 0 oC; iii. CBr4, PPh3, 2,6-lutidine, MeCN, 0 oC, (3 steps, 83%); e) DIBAL-H/THF, 0 oC, (81%); f) Na2S / Al2O3, EtOH-CH2Cl2 (2:5), 0 oC, (90%); g) LIHMDS, HMPA, THF, -40 oC, (10%). Scheme 43. Grierson’s approach to the synthesis of 9-membered ring enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 32 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.5.4. Caddik’s Approach Michael addition of enediyne acetylide cyclopentenone .cobalt complexation acetal hydrolysis and aldol condensation the 9-membered framework (Scheme 44). Caddik's Approach to the Synthesis of 9-Membered Ring Enediyne PO H H TBSO OH TBSO OTBDPS a c O MeO MeO b P=H O O d P = COPh OTBDPS PhCO2H H TBSO H e O H + H OH H O H Co2(CO)6 R3 R3 = CH(OMe)2 R3 = CHO OTBDPS OH H H TBSO Co2(CO)6 OTBDPS PhCO2H H TBSO Co2(CO)6 H CO2Ph 1.2.32 OTBDPS H OTBDPS Br HO EtO OMe A OMe O Br TMS Br Br H Reagents and conditions: a) i. A, n- BuLi, THF, 0 oC, - 78 oC, 30 min., ii. Et2AlCl, rt, 1 h, iii. 1.2.27, (51%); b) PhCOCL, Pyr, DMAP, CH2Cl2, (89%); c) Co2(CO)8, CH2Cl2, 0 oC, (84%); d) TFA, CH2Cl2, (96%); e) n-Bu2OTf, Et3N, CH2Cl2, (68-75%). Scheme 44. Caddik’s approach to the synthesis of 9-membered ring enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 33 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.5.5. Hirama’s Approach Hirama 's Approach to the Synthesis of 9-Membered Ring Enediyne: Synthesis of seco analogues of 9-membered systems present in kedarcidin MOMO a, b OMOM c MOMO OMOM MOMO MOMO d MOMO MOMO O I HO I I HO HO 1.2.41 NHBoc NHBoc TBSO TBSO O e-g I O I l, m O h-k O O Cl N TBSO O NHBoc Cl O O TBSO r-t O MeO O TES OMOM OTES O TES NH OMe Cl N OMOM O 4/ O 4 1 OTES 2 8 NHBoc A O OAllyl O TBSO O HO Cl N O O I O O HO O TBSO O OMOM OTES O OMe i-PrO N q n-p O I O O Cl N OH TES OMOM OH B i-PrO MeO O TES OAllyl O OMe OH C Reagents and conditions: a) TPAP/NMO; b) I2/Py. (89%, in 2 steps); c) i-PrMgCl/CH2Cl2, 78 to 0 oC, THF, 6 h (69-71%); d) HClO4, THF(aq) (61%); e) (Cl3C)2C=O/Py; f) PPTS/2Butanone; g) TBSCl, imidazole. (77% in 3 steps); h) DIBAL-H, -80 oC; i) (MeO)MeCH=CH2; j) TBAF, 0 oC; k) DIAD/PPh3 (72%, in 4 steps); l)A, CsF, DMF, 60 oC; m) TBSCl. (90%, in 2 steps); n) 0.3 M KOH (aq); o) B, EDC.HCl, DMAP; p) TESCl (89%, in 3 steps); q) CuI, Pd2(dba)3.CHCl3, i-Pr2NEt, DMF, rt, 1 h (88 - 90%); r) TBSOTf; s) SiO2; t) C, HOAT, EDC.HCl (87%, in 3 steps). Scheme 45. Hirama’s approach to the synthesis of 9-membered ring enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 34 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.5.6. Basak’s Approach , Basak 's Approach to the Synthesis of 9-Membered Ring Enediyne Ts Ts N H (89%) OH Co(CO)3 H N (OC)3Co N H a b (50%) OMs OMs Co(CO)3 (OC)3Co e A (63%) c, d (OC)3Co Co(CO)3 N H Ts + (OC)3Co OMs c (OC)3Co (40%) Ts N H + Ts N H A OMs Co(CO)3 Ts CH3 (55%) c, d Co(CO)3 . d N Ts N Ts N Ts . H H N Ts + H (8%) I N Ts + I (64%) N Ts I (13%) Reagents and conditions: a) MsCl, NEt3, DCM, 0 oC; b) Co2(CO)8 (2.2 eq), DCM, 0 oC, 30 min; c) K2CO3, DMF, rt, 40 min; d) I2, THF, 0 oC, 1 h; e) Co2(CO)8 (1.1 eq), DCM, 0 oC, 5 min Scheme 46. Basak’s approach to the synthesis of 9-membered ring enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 35 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.6. Synthesis of Dienediyne Core of NCS chromophore The synthesis of parent dienediyne unit of NCS chromophore has been approached from three different angles looking at the simplicicity and feasibility of the reactions. The synthesis of actual NCS chromophore core unit has been reported by Myers and subsequently by Kobayashi. The targets are divided into the following categories: a) Category A: In this category the bicyclic dienediyne lacked the epoxy unit that is replaced by two saturated carbons. b) Category B: In category B one of the epoxy carbons in NCS chromophore has a hydroxy functionality which can be converted into an epoxy unit. c) Category C: In this category C, the dienediyne unit has the epoxy unit like the natural product. Below are the presentations of various synthetic approaches to these molecules. 3.6.1. Synthesis of Category A molecules: Wender’s Approach The strategy relied on the following steps a) addition of an acetylide to a halo cyclopentenone, b) a Sonogashira coupling to generate the diyne functionality and c) ring formation to generate the bicyclic system. d) The final cyclic network formation was carried out by a photochemical extrusion of SO 2 from a cyclic sulphone. e) After then β-elimination generated the target framework. Joint initiative of IITs and IISc – Funded by MHRD Page 36 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics The sequence of reactions is represented in Scheme 47. Wender's Retrosynthetic Approach to Dienediyne core (Category A) Y 1. Sonogashira coupling Elimination 2. Homologation of terminal alkyne Y OH Target A X Y Organometallic X addition to a carbonyl O + OH R + M Wender's Synthetic Approach to Dienediyne core (Category A) OH OH O a, b HO c, d Br HO Br OH e-g HO O S HO h i O Reagents and conditions: a) HCCCH2MgBr, Et2O, rt; b) EtMgBr, HMPA, Et2O, 50 oC; CH2O, Et2O, rt; c) PdCl2(PPh3)2, CuI, (i-Pr)2NH, HCCCH2OTBDPS, THF, rt; d) n-Bu4NF, THF, -50 oC to rt; e) MsCl, Et3N, CH2Cl2, -20 oC; f) Na2S, aq. EtOH, rt; g) m-CPBA, NaHCO3, CH2Cl2, 0 oC to rt; h) PhCOPh, MeCN/PhH (1:1), hn, rt; i) MsCl, DMAP, CH2Cl2. Scheme 47. Wender’s approach to the synthesis of dienediyne core (category A). Joint initiative of IITs and IISc – Funded by MHRD Page 37 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.6.2. Synthetic Approaches to category B molecules 3.6.2.1. Wender’s Approach In Wender’s appraoch to category A molecules the saturated carbons may not serve any useful purpose for the enediyne to show MSC and hence the biological activity. Thus this was modified wherein the earlier saturated carbon bears hydroxy group that might allow potential creation of the epoxide as in NCS. The molecules belonging to this category are designated by structure target A in the following scheme. Wender's Retrosynthetic Approach to Dienediyne core (Category B) OH HO OH HO OH Target A M X O HO OH I + + OH Wender's Synthetic Approach to Dienediyne core (Category B) OTBS OTBS HO a HO OH HO Br HO CHO OH d b, c Br Reagents and conditions: a) Et3N, MsCl, -78 oC; LiBr, (CH3)2CO, rt; b) 3:l:l HOAc/THF/H2O, rt; c) MnO2, rt; d) CrCl2, THF. Scheme 48. Wender’s approach to the synthesis of dienediyne core (category B). This approach follows the similar steps to the ones described for the synthesis of category A molecules. The final ring closure was carried out using a Nozaki-Hayashi reaction mediated by CrCl2 which worked nicely to afford the target molecule in high yield (Scheme 48). Joint initiative of IITs and IISc – Funded by MHRD Page 38 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.6.2.2. Myers’ Approach Myers adopted a different strategy for the synthesis of the bis-oxygenated dienediyne system (Scheme 49). The key step was the transannular reductive cyclization of the potassium salt of the tetrayne alcohol with Red-Al that afforded the bicyclic product in good yield (50-54%). It is important to mention that the proximal transannular cyclization was favoured over the distal mode. Myers' Synthetic Approach to Dienediyne core (Category B) HO OSPIT TIPSO OSPIT TIPSO 1. Cu(OAC)2, DBU, O2 TIPSO 2. Et3N.HF (Distal) OTIPS HO Red-Al (Proximal) HO OTIPS OTIPS TIPSO (Not obtained) TIPSO OTIPS Target Scheme 49. Myers’ approach to the synthesis of dienediyne core (category B). Joint initiative of IITs and IISc – Funded by MHRD Page 39 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.6.2.3. Magnus’ Approach In their synthesis of NCS chromophore analogue, Magnus et al. introduced an epoxide moiety on the ene part of acyclic enediyne. This was followed by cobalt complexation and final intramolecular aldol condensation (Scheme 50). Magnus' Synthetic Approach to Dienediyne core (Category B) OH OCOBut O TBSO TBSO a O OCOBut O TBSO b O OEtO OEt OEtO OEt EtO OEt O c t OCOBu O t OCOBu O e TBSO TBSO O Co OH CO3 + d Co(CO)3 Co(CO)3 O H TBSO OCOBut O O H Co (CO)3 TBSO f O H Co(CO)3 Co OH (CO)3 OCOBut O TBSO Co(CO)3 O EtO OEt Co (CO)3 OCOBut OCOBut TBSO O O + O H OH O H OH Reagents and conditions: a) i. (-)DET/3Å MS/Ti(OPri)4/t-BuOOH/CH2Cl2; ii. tBuCOCl/Et3N/DMAP/CH2Cl2 (67% overall, 1:1 diastereoisomers); b) CF3CO2H/THF/H2O, 5 oC (99%); c) Co2CO8/n-heptane, 25 oC (80%); d) CF3CO2H/CHCl3 (56%); e) n-Bu2BOTf/Et3N/CH2Cl2, -78 o to 0 oC (57%, ca. 1:1 diastereomars); f) I2/PhH, 25 oC (69% and 75%). Scheme 50. Magnus’ approach to the synthesis of dienediyne core (category B). Joint initiative of IITs and IISc – Funded by MHRD Page 40 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.6.3. Synthetic Approaches to category C molecules The synthesis of the actual core of NCS chromophore containing the epoxy functionality was done following two different strategies which are given below. 3.6.3.1. Myers’ Approach Myers et al. reported the synthesis of the target aglycon. The target was achieved from an epoxy alcohol (Scheme 51). Myers' Synthetic Approach to Dienediyne core (Category C) O H R1 R1 O H TMSO H CHO O TBSO R1 O H Cl R2 O R1 O O OH R2 O R2 j OR O R=H R = TES O R1 = l OH HO O O O OR H i HO m k H H H R = TES OH R1 O O HO h HO O O R2 O TBSO H g HO R1 TMSO H CHO HO R1 O HO H d TMSO H OH TBSO TBSO e, f R1 O O H b, c a TMSO R1 R2 O MeO R2 = O OH Me OH R=H Reagents and conditions: a) DIBAL, CH2Cl2, -78 oC (94%); b) (+) DET, Ti(Oi-Pr)4, TBHP, 3A MS, CH2Cl2, -30 oC, (98%); c) Dess-Martin, pyr, CH2Cl2, (97%); d) LiN(SiPhMe2)3, LiCl, THF, -78 oC, (85%); e) (ClCH2CO)2O, pyr, CH2Cl2, 0o C, (89%); f) Et3N.3HF, THF, 23 o C, (100%); g) 1.3.48, DCC, THF & n-PrNH2, -10 oC, (80%, two steps) h) p-TsOH, MeOH, 23 oC, (81%); i) CDI, THF, 0 to 23 oC; then (+)-CSA, CH3CN/H2O, (89%); j) TESOTf, 2,6lutidine, CH2Cl2, -78oC, (100%); k) Martine Sulfurane, CH2Cl2, 23 oC; l) Et3N.3 HF, THF, 0 oC, (78%), (two steps); m) PPh3, I2, imidazole, CH2Cl2, -10 oC, (71%). Scheme 51. Myers’ approach to the synthesis of dienediyne core (category C). Joint initiative of IITs and IISc – Funded by MHRD Page 41 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.6.3.2. Kobayashi’s Approach In Kobayashi’s approach the steps involved: (a) stereoselective intramolecular acetylide-aldehyde cyclization to form the C5-C6 bond. (b) installation of the epoxide. (c) Then the formation of naphthoate and carbonate. (d) ultimately the C8-C9 olefin was introduced by using the Martin sulfurane dehydration reaction to furnish the target aglycon. Kobayashi's Synthetic Approach to Dienediyne core (Category C) R1 h TESO f, g OTES OPiv MPMO TESO R1 R1 TESO OTES 5 6 O HO R1 OTES MPMO OH TESO 15% MPMO TESO O PivO MPMO TESO HO i, j OTES MPMO O OH TESO c-e a, b O + PivO O MPMO R1 TBDPSO I TBDPSO OH + OMs MPMO TESO R1 O 58% k OH OH O HO l, m OH n, o O MPMO OMe O O O O MPMO OMe OSET O OTES O p, q O OH OH O O O TESO OMe O O O O r, s O O O 9 8 OH O R1 = O HO OMe O O Reagents and conditions: a) (Ph3P)4Pd, CuI, iPr2NEt, DMF, (97%); b) TESOTf, 2,6lutidine, CH2Cl2, -70 oC, (100%); c) HCCCH2MgBr, toluene, -78 oC; d) TBAF, THF, (74%); e) TESOTf, 2,6-lutidine, CH2Cl2, -50 oC, (90%); f) DIBAL, Et2O, -78 oC, (89%); g) Dess-Martin periodinane, pyridine, CH2Cl2, (90%); h) LiN(TMS)2, CeCl3, THF, 35 to 0 oC, (73%); i) MsCl, Et3N, CH2Cl2, -20 oC; j) TBAF, THF, -35 oC; k) K2CO3, EtOH, (52%); l) 1.3.48, EDC.HCl, DMAP, CH2Cl2, 0 oC, (64%); m) 0.5% HF in CH3CN, 0 oC; n) carbonyldiimidazole, THF, (78%); o) TBAF, THF, 0 oC, (84%); p) DDQ, CH2Cl2H2O (20:1), (81%); q) TESOTf, 2,6-lutidine, CH2Cl2, -90 oC, (71%); r) Martin sulfurane dehydrating agent, CH2Cl2; s) TFA-THF-H2O (1:10:5), (61%). Scheme 52. Kobayashi’s approach to the synthesis of dienediyne core (category C). Joint initiative of IITs and IISc – Funded by MHRD Page 42 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.7. β-Lactam as a Molecular Lock of Enediyne: Synthesis of β-Lactam fused Enediynes 3.7.1. β-Lactam as Molecular Lock The first use of -lactam as a molecular lock was reported by Basak et al. and is demonstrated in -lactam fused bispropargyl sulphones. The bis-propargyl sulphones are found to be good DNA-cleaving agents. Bis-propargyl sulphones on isomerization afforded allenic sulphone which is a good Michael acceptor. Thus, DNA base attack it in a conjugate fashion. The positive charge developed on the DNA base ultimately resulting in a Maxam-Gilbert type cleavage (Scheme 53). An alternative mechanism, involving formation of diradicals via GarattBraverman rearrangement might also operate to show DNA cleavage potential. Similar catechol or alizarin based bis-propargyl sulphones readily isomerized to the allenic sulphones under the same conditions (Scheme 53). Mechanism of DNA cleavage by bis-propargyl sulphones O R S O R pH>7 O O S Nu-DNA Maxam-Gilbert R mechanism R R O S R O DNA cleavage 1.H2O 2. pH>7 O R O O S R O DNA cleavage Garratt-Braverman mechanism O S O Nu-DNA R R O R S R Scheme 53. Bis-propargyl sulphones’s isomerization to allenic sulphone and the DNA cleavage activity. Joint initiative of IITs and IISc – Funded by MHRD Page 43 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Basak et al. was successful in arresting the spontaneous isomerization of bis-propargyl sulphones to afforded allenic sulphone by fusing a -lactam ring onto its’ skeleton. Thus, the lactam ring acts as a molecular lock in stabilizing the bispropargyl sulphone even under basic conditions (Scheme 54). Arrested isomerization in -lactam fused bis-propargyl sulphones O O S H O O H O 1. mCPBA 2. NaHCO3 CH2Cl2 N O S H O O + N O S O N (Not Obtained) Spontaneous isomerization in -lactam fused bis-propargyl sulphones O S O 1. mCPBA 2. NaHCO3 CH2Cl2 O O S O O O O O S O O 1. mCPBA O O S O O O O + 2. NaHCO3 CH2Cl2 O O O O O O + S O S O O Scheme 54. Areested isomerization of bis-propargyl sulphones to allenic sulphone by fusing a -lactam ring. Banfi and Guanti have demonstrated that the -lactam ring can act as a locking device to stabilize the otherwise unstable 10-membered enediyne system. Opening of the -lactam ring unlock the system which enabled it to undergo facile BC. They have further developed a lactam fused system with a nucleophilic handle that under acidic conditions opens up the lactam ring to produce bicyclic reactive enediyne which ultimately undergoes facile BC. These examples demonstrats the ability of the azetidinone ring to act as a molecular lock (Scheme 55). -lactam as a molecular lock of reactive enediyne OR OH NHBoc OH N N OMeO OMeO H+ OH OH N NH2 NH O OMe OMe O R = OTBS R = NHBoc Scheme 55. -lactam as a locking device to stabilize unstable 10-membered enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 44 of 115 BC NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.7.2. Strategies for the Synthesis of β-Lactam Fused Enediynes There are three possible strategies for the synthesis of -lactam fused enediynes which are presented in Scheme 56. (i). (ii). (iii). Pathway a and b: In these two pathways the formation of enediyne takes place on to a -lactam ring. Pathway c: It involves the formation of -lactam on to an enediyne. The third alternative pathway involves (path d) involves the concerted formation of both the enediyne and -lactam rings. Strategy for the Synthesis of -Lactam Fused Enediynes R R a R c N N N O O O 1.4.24 O O d O b N N N R R R Scheme 56. Strategy for the design and synthesis of β-lactam fused enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 45 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.7.2.1. Guanti’s Approach for β-Lactam Fused Enediyne Guanti's Approach for -Lactam Fused Enediyne TMS OTBS OH a N O OMe O OMe OTBS O OH I e OH pH > 7 H OMe O OMe pH > 7 O OH . OH H OH H MeO O DNA . . OMe NH2 O OTBS OTBS N NH N OMeO OH OH OTBS O N O OMe d, e b-d OTBS N a-c N NHBoc OAc .. OH N . MeO OTBS OH NH NH NH OOO . OMe O OMe Reagents and conditios: a) Pd(PhCN)2Cl2, Reagents CuI, piperidine/THF; b) AgNO3, KCN, pyridine; b) HF, CH3CN/H2O, (96%); c) i. (83%); c) I2-morpholine, benzne, rt (95%); MsCl, Et3N, CH2Cl2, -30 oC; ii. NaN3, DMF, d) (COCl)2, DMSO, Et3N, CH2Cl2, (96%); e) 60oC, (87%); iii. Ph3P, THF/H2O, 8 h, rt; iv) CrCl2, NiCl2 cat, THF, rt (64%). Boc2O, Et3N, (85%); d) MeONa, MeOH, 0 oC; NH MeO . . . O and DNA Cleaved conditions: a) Ac2O, e) CF3CO2H, CH2Cl2. Scheme 57. Guanti’s approach for β-lactam fused enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 46 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.7.2.2. Basak’s Approach for β-Lactam Fused Enediyne Synthetic strategy involves: (a) an intramolecular N-alkylation (Scheme 58) (b) an intramolecular carbene insertion (Scheme 59). These two methods started with either the sensitive -lactam or the sensitive cyclic enediyne. (c) To bypass the problem associated with the instability of the starting core an intramolecular Kinugasa reaction was adopted by the same group in which process the two rings, -lactam and the cyclic enediyne are formed in a single step. (Scheme 60). Intramolecular N-alkylation Route to -Lactam Fused Enediyne NH O a + O NH b O NH c N O OH OH Cl Cl Reagents and conditions: a) Pd(PPh3)4, CuI, n-BuNH2, C6H6 (61%); b) TsCl, DMAP, CH2Cl2 (60%); c) K2CO3, KI, DMF, rt (40%). Scheme 58. Intramolecular N-alkylation route to β-Lactam fused enediyne. Intramolecular Carbene Insertion Route to -Lactam Fused Enediyne COOEt Et3N, CH2Cl2 NH PTSN3, K2CO3 N COOEt O COCl N2 COOEt Rh2(OAC)4/CH2Cl2, rt. N N O O COOEt Scheme 59. Intramolecular carbine insertion route to β-Lactam fused enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 47 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Intramolecular Kinugasa Reaction Route to -Lactam Fused Enediyne H n O Cu(I) O N Ph n NEt3 Ph N n=1 N n=1 n=1 Cu(I) H O NEt3 H O Ph N + H OH Scheme 60. Intramolecular Kinugasa reaction route to β-Lactam fused enediyne. Joint initiative of IITs and IISc – Funded by MHRD Ph H O N O N n=0 OH n + n=0 Ph H O O H H n=0 O Page 48 of 115 Ph NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.8. Enediynes with pH-Based Triggering Devises 3.8. 1. Introduction Tumor cells have acidic pH. The pH of these cells again becomes low when they are administered hyperglycaemic agents while the normal cells remain unaffected. Thus, greater selectivity against tumor cells is expected with enediynes triggered by acidic pH. This is the most striking phenomenon that led the scientists to design and synthesize tumor specific enediyne warheads. Although there is no apparent advantage if the triggering happens physiological pH (higher that the pH of tumor cell), several designed enediyneprodrugs were activated under mild basic condition. Some of these prodrugs showed excellent IC50 values against a number of cancer cell lines. Many of these showed selectivity against cancer cells. 3.8.2. Approaches for Enediynes with pH-Based Triggering Various strategies are adapted for pH-based triggering of enediynes which can be classified into the following five categories (Scheme 61). Examples of each category are subsequently described. Scheme 61. Strategies for designing enediynes with pH-based triggering devices. Joint initiative of IITs and IISc – Funded by MHRD Page 49 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.8.2.1. Category 1: Activation through Enediyne to Eneyne−Cumulene Conversion in Altered pH This strategy is one of the widely exploited strategies in pH-based triggering of enediynes. In their quest to develop new DNA-cleaving agents related to the neocarzinostatinchromophore, Toshima and co-workers have synthesized the cyclic sulfide A which upon oxidation with mCPBA produced the allenicsulfone B. Compound B when treated with a base, such as 1,8diazabicyclo[5.4.0]undec-7-ene (DBU), isomerized to the eneynecumulene C. The cumulene, being extremely reactive, underwent spontaneous MSC under ambient conditions to the diradical that was able to cause damage to double-stranded (ds) DNA (Scheme 62). Scheme 62. Base catalysed activation of cyclic sulfones via eneyne−cumulene conversion. They have also reported that the thia, oxa, or azaenediyne undergo MSC when subjected to weakly acidic or basic conditions. Under such pH values, the compound first isomerizes to the eneyne−allene and subsequently undergoes MSC to generate the toluene diradicals which have been shown to cleave ds DNA. Joint initiative of IITs and IISc – Funded by MHRD Page 50 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Shibuya et al. exploited the approach depicted under this category. They synthesized enediyne model compounds represented by A. Compound A produced the eneyne−allene B and ultimately generated toluene diradicals C via a reaction cascade triggered by hydrolysis of the malonyl ester group under basic conditions (Scheme 63). Scheme 63. Activation through eneyne−allene via decarboxylation. Enediyne models having electron-withdrawing groups were also designed and subsequently synthesized. These molecules, represented by F, upon treatment with TFA in the presence of 1,4cyclohexadiene (1,4-CHD) in benzene or MeOH at 37 °C afforded the phenol J as the only isolable product, thus indicating that cycloaromatization proceeded via a diradical pathway as shown in Scheme 64. Scheme 64. Activation through acid catalysed eneyne−allene conversion by lactonization. Joint initiative of IITs and IISc – Funded by MHRD Page 51 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Kerwin's group has prepared 4-aza-3-ene-1,6-diyne systems represented by structure A and demonstrated that these compounds possess powerful pH-dependent DNA-cleavage activity with some degree of cytosine specificity. The probable mechanism involves isomerization to the azaeneyne−allene system, which undergoes aza MSC to generate methyl pyridiniumdiradicals C (Scheme 65). The latter then cleaves the ds DNA, producing mainly single-strand cuts at a concentration of 100 μM. Another possible mechanism of DNA cleavage involves the alkylation pathway through the intermediacy of D or through the formation of carbene intermediate F via the ylide E (Scheme 65). Scheme 65. pH-dependent activation and DNA cleavage of Azaenediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 52 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.8.2.2. Category 2: Activation through Acid- or Base-Catalyzed Ring Opening In this strategy, the designed molecules have one special structural feature in common. The common feature is the cyclic unstable enediyne which has been made stable by the fusion of a locking device. The locking device is usually a small ring, such as an epoxide, a β-lactam, or even an isooxazoline ring. All of these molecules are equipped with pH-based triggering devices, which unlock them by opening the small ring. With the removal of strain, the molecules become active under ambient conditions. Inspired by the chemistry of the natural dynemicins, Nicolaouet et al. first reported the synthesis of a series of analogues in which the flow of electrons to open the epoxide ring was blocked by the incorporation of protecting groups in potential donors. In one such design, the pivaloyl group was used to protect the free phenolic -OH group to generate the enediyne A. Base-promoted hydrolysis produced the free phenolic form B, which is capable of promoting the epoxide ring opening via the flow of electrons followed by BC to give diradical D which can cleave DNA (Scheme 66). Scheme 66. Base-mediated deprotection and activation of dynemicin model enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 53 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Similar to dynemicin, electrons can also flow from the ring nitrogen if its lone pair is free. On that basis, the enediyne F was synthesized by Nicolaou and co-workers. The interesting feature of this molecule is that the nucleophilic N atom is made non-nucleophilic by protection with a 2-(phenylsulphonyl) ethoxy carbonyl group. The group falls off upon treatment with mild bases, thus leading to the generation of free amine G the lone pair of which is being free flows toward the direction of epoxide, which then opens up. With the release of strain, the resulting compound I shows BC under ambient conditions (Scheme 67). The produced diradical J have DNA-cleaving activity at micromolar concentrations, resulting in the formation of both relaxed and linear forms of DNA. Scheme 67. Base-catalyzed β-elimination and activation of Dynemicin model enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 54 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics The sensitivity of epoxide rings under acidic conditions was exploited by Unno and coworkers in their designed dynemicin analogues. Thus, several novel analogues designated by structure K were made, and their DNA-cleaving potential under acidic conditions was evaluated. It was demonstrated that the size and electronic character of the substituents (R1 and R2) at the C9 position critically influenced the DNA-cleaving ability of the synthesized enediynes. The compounds represented by K were shown to undergo BC under acidic conditions (Scheme 68). A similar cascade of reactions involving ring opening and BC may operate in cancer cells because they are sufficiently acidic with pH less than 7. Scheme 68. Acid-catalyzed ring opening of epoxide and activation of enediyne. From previous discussions, it is clear that, among the various small ring systems, Nature has picked up the epoxide ring to lock the unstable enediynes, with the reason being the easy unlocking of these systems by opening of the strained epoxide ring by an acid-catalyzed process or opening because of an inward flow of electrons. Joint initiative of IITs and IISc – Funded by MHRD Page 55 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.8.2.3. Category 3: Activation through Salt Formation The rationale behind the activation upon protonation (salt formation) of an enediyne with a basic functionality near the enediyne moiety lies in the fact that the electron-withdrawing effect (−I or electron transfer) of the protonated species lowers the repulsion between the in-plane alkyne π orbitals (Koga−Morokuma hypothesis). By electron withdrawal the protonated enediyne lowers the singlet−triplet gap and thus favors the triplet state. The triplet state is proven to be a better hydrogen abstractor than a singlet. Thus, the triplet state produces the cleavage of DNA. Computational analysis supports this which revealed that a singlet diradical abstract hydrogen much in a slower rate compared to the triplet diradical. The prediction has been confirmed for the singlet didehydroanthracenediradical, for which the hydrogen-abstraction rate from 2-propanol was found to be reduced by 2−3 orders of magnitude relative to phenyl or 9-anthryl radicals. For a singlet ground-state biradical to show radical-like chemistry, e.g., hydrogen abstraction, it must add extra energy to scale up the singlet−triplet gap. The singlet lies below the triplet because it is stabilized and, accordingly, one has to pay back the stabilization energy to reach a TS where the two electrons in the nonbonded molecular orbitals (NBMOs) are uncoupled to reach the triplet state. It can also be summarized that an increase in the electron density in the intervening σ bonds can increase the through-bond coupling and hence increase the singlet−triplet splitting. Conversely, a decrease in electron density will decrease the coupling and hence decrease the singlet−triplet gap. Thus, the in-plane lone pair of the nitrogen atom in the 2,5-didehydropyridine diradical lies antiperiplanar to the σ bonds coupling the NBMOs and therefore could donate electron density. However, when the nitrogen is protonated, the effect is reversed. This has been confirmed by ab initio computed singlet−triplet gaps, where the didehydropyridine with its lone pair shows a much larger singlet−triplet gap in comparison to the protonated form. The protonated azaenediyne liesg mostly in the triplet state and hence should be a better hydrogenatom abstractor. The first strategy is reflected in the works from the research group of Alabugin et al., as well as from Basak’s group. Alabugin et al. has reported that the rate of BC in benzannulatedenediynes can be tuned by varying the electronic nature of the ortho substituents. The most striking of them is the acceleratory effect on BC kinetics when an amino group at the ortho position is protonated B (Scheme 69) (The ortho effects on BC has been described in Module 1). Scheme 69. Ortho effect on protonation on the rate of BC of benzannulated enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 56 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Such acceleration of the BC rate upon protonation has also been reported by Basak et al. in the case of 2,6-diamino pyridine-based enediynes E. The latter was prepared by double Nalkylation of the bis-sulfonamide B. Deprotection using PhSH under basic conditions (K2CO3) gave the free amine E. The reactivity of the enediyne E and its salts F(X), F(Y), F(Z) with acids of various pKa values (X-Z) was studied by DSC, which indicated the lowering of the onset temperature for BC upon salt formation. Interestingly, the extent of lowering was shown to depend upon the degree of salt formation, which was monitored by 1H nuclear magnetic resonance (NMR) studies. The greater the degree of salt formation, higher the lowering of the onset temperature for BC (Scheme 70). Scheme 70. Reactivity of pyridine diamine based enediyne upon salt formation. Another set of example of salt formation and enhancement of rate of BC is shown by the 10membered azaenediyne shown in Scheme 71. Reactivity of Free Amine Salt from 10-Membered Azaenediyne O H N S O NO2 O H N S O MsCl, Et3N, 0 °C OH O N S O NO2 K2CO3, DMF, r.t OMs PhSH, K2CO3, NO2 pH = 8 NH DMF N H H r.t. BC Scheme 71. Synthesis and reactivity of free amine from 10-membered azaenediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 57 of 115 H N H NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics The cyclization chemistry of C, N-dialkynyl imine azaenediyne A in which one of the ene carbons is replaced by nitrogen was first reported by Kerwin's group. They have reported that the ultimate product of BC, (G), could only be isolated if there was a small amount of picric acid. Computational studies have shown that the singlet diradical (F), which is stabilized by the pyridine N lone pair, is a poor hydrogen-atom abstractor as compared to the triplet diradical (C), which is mainly the species generated from the protonated form of azaenediyne E (Scheme 72). Scheme 72. Reactivity of azaenediyne under acidic pH. Joint initiative of IITs and IISc – Funded by MHRD Page 58 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.8.2.4. Category 4: In Situ Generation of Enediynes through Allylic Rearrangement/β-Elimination The drive for this type of design was derived from the chemistry of an artifact of the natural enediyne maduropeptin chromophore. The compound A was obtained during isolation of the parent enediyne using methanol. An intramolecular nucleophilic attack in an SN2' fashion generates the reactive enediyne functionality B from compound A. Compound B then undergoes BC and shows DNA-damaging property (Scheme 73). This overall cascade of reactions represents an emerging strategy for the design of the enediyne prodrug. Scheme 73. Triggering of Maduropeptin artifact under acidic pH. An intermolecular allylic rearrangement strategy for enediyne generation from the prodrug was adopted by Dai et al. They designed and synthesized the alcohol A, which, upon treatment with nucleophiles, such as ethanol or water, in acidic medium, underwent a S N2' reaction to generate the reactive enediyne system B. Compound B then underwent BC under ambient conditions, and the resulting diradical C was found to cleave ds DNA (Scheme 74) at millimolar concentrations, producing relaxed DNA. In Situ Generation of Enediyne by Allylic Rearrangement Under Acidic pH OR OR CSA EtOH HO OR H2O A ROH DNA H OR BC EtO Ph OR Ph EtO EtO Ph B Ph C D Cleaved DNA Ph Dai, W. M.; Fong, K. C.; Lau, C. W.; Zhau, L.; Hamaguchi, W.; Nishimoto, S-i. J. Org. Chem. 1999, 64, 682. Scheme 74. In situ generation of enediyne by allylic rearrangement under acidic pH. Joint initiative of IITs and IISc – Funded by MHRD Page 59 of 115 H E NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics The synthesis of a number of enediyne prodrugs possessing free hydroxymethyl groups on the exocyclic double bond has been reported by Dai et al. These compounds as represented by A cause single-strand cleavage in circular supercoiled DNA at a pH of 8.5. A mechanism based on allylic rearrangement to form a putative epoxyenediyne C has been proposed. Cleavage of DNA may have taken place via both BC pathways, involving hydrogen abstraction by the diradical and alkylation of the DNA base followed by a Maxam−Gilbert-type reaction (Scheme 75). In Situ Generation of Enediyne via Allyllic Rearrangement and DNA Cleavage under Basic pH OR OR X OR -X Ph Ph Allyl Cation Formation Ph O Si O Si O pH = 8.5 BC pH = 8.5 pH = 8.5, DNA pH = 8.5 Maxam-Gilbert-type OR OR OR X Ph Ph O Ph Ph HO HO DNA DNA HO DNA X = OH, OAc, OCH2OMe Cleaved DNA Cleaved DNA Cleaved DNA Scheme 75. In situ generation of enediyne by allylic rearrangement under acidic pH. Below is an example of base mediated MSC (Scheme 76). Activation of Enediyne under Basic pH OMOM Ar OMOM Myers-Saito Cyclizaiton Ar KOH Ar OMOM OMOM Ar EtOH CO2Et CO2Et CO2Et OMe C OMe OMe CO2Et CO2Et OEt OMe OTDS Ar= Shibuya, M.; Naoe, Y.; Bando, M.; Nemoto, H. Tetrahedron Lett. 1998, 39, 2361. Scheme 76. Activation of enediyne toward MSC under basic pH. Joint initiative of IITs and IISc – Funded by MHRD Page 60 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Takahashi et al. prepared the cyclic 1,5-diyne derivative A equipped with a good leaving group in one of the two propargylic carbons separating the alkynes. Treatment with a base-like DBU converted B via β-elimination to the 10-membered enediyne C, which then smoothly underwent BC under ambient conditions (Scheme 77). Scheme 77. In situ generation of enediyne via β-elimination under basic pH. 3.8.2.5. Category 5: Activation through Acid-Catalyzed Enol to Keto Tautomerism On the basis of the way of activation of calicheamicins and esperamicins via conjugate addition that converts bridgehead sp3 carbon into a sp2 carbon, Semmelhack and co-workers104 have designed an enediyne system (A-B, F) in which a double bond is present at the bridgehead in the form of an enol. Conversion of the enol into the keto form (C, G) under acidic conditions removes the bridgehead double bond and triggers the molecule toward undergoing BC. It is interesting to note that similar acid treatment of the enolic enediyne system (J-K) generated the ketone L, which is, however, stable under ambient conditions (Scheme 78). The reason for this anomalous behavior (less reactivity) of the substituted ketone is likely to be steric effect exerted by the two –R groups. Scheme 78. Triggering enedyne via acid-catalyzed enol−keto tautomerization. Joint initiative of IITs and IISc – Funded by MHRD Page 61 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9. Photoswitchable Enediynes 3.9.1. Introduction Recently photodynamic therapy (PDT) has become attracted clinical attention for selective targeting and damaging of tumor cells that are quite localized and yet metastasized. The photodynamic therapy (PDT) combines a drug and a light of particular wavelength. The light used for PDT can come from a laser or other source. In this process the light is directed through an optical fiber to deliver light at the appropriate localized area. One important issue in PDT is the penetration ability of the light through the tissue. That is why PDT is less effective toward large tumors. The use of light of a longer wavelength increases the penetration ability. Photo triggering method is not only interesting but important because this would allow one to develop target-specific chemotherapeutic agents. There is no precedence of this type of triggering of enediynes in Nature. Therefore researchers of the field of enediyne have taken the advantage of light triggering concept to design and synthesized photo-triggerable enediynes which can be activated by light of a higher wavelength. Few such examples are given in the following sections to address the efforts made to generate light-mediated triggering devices in enediyne chemistry. Joint initiative of IITs and IISc – Funded by MHRD Page 62 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9.2. Strategies for Designing Enediynes with Photo-Triggering Devices Literature reports revealed a wide variety of available strategies to trigger enediynes by photochemical means. Some of these strategies involve Bergman Cyclization induced by irradiation. Other strategies use photoirradiation wherein a structural change occurs in the prodrug. Upon change in structure the enediyne is activated toward thermal BC. All of these strategies can be classified according to the flow chart shown below in Scheme 79. Scheme 79. Strategies for photo-activation of enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 63 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9.2.1. Category 1: Use of Enediynes Activated toward Photo-Bergman Cyclization Turro, Evanzahav, and Nicolaou were the first to show that enediynes also undergo cycloaromatization upon photo-irradiation and they were able to produce products similar to that obtained in a thermal BC. The process is now popularly known as Photo-Bergman cyclization (photo-BC). They have reported the formation of naphthalene derivatives B when an isopropanol solution of n-propyl- or n-phenyl-substituted enediyne A was irradiated. However, in addition, products resulting from photoreduction of one of the alkynes were also formed (compounds C and D) (Scheme 80). Scheme 80. Photo-induced BC. Unlike the thermal counterpart, photo-BC is not as versatile and the quantum yield as well as the actual isolated yield of the cyclized product are usually low. However, certain enediynes can show facile photo-BC, depending upon the nature of substituents at the alkyne termini or the electronic nature of the ring fused onto the enediyne. The use of different modes of energy transfer is another approach to improve the efficiency of photo-BC. As a result of tremendous research efforts, several novel enediynes were synthesized that could be activated toward BC upon irradiation. The photo-BC is an efficient and symmetry-allowed process when the photochemical excitation involves the in-plane p-orbitals (Figure 3). This is likely to be the case, because such excitation promotes an electron from the molecular orbital (MO) that is C 1−C6-antibonding to a MO, which is C1−C6-bonding. Thus, the excitation should increase the extent of C 1−C6 bonding Joint initiative of IITs and IISc – Funded by MHRD Page 64 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics at a relatively early reaction stage. However, the in-plane excitation requires much more energy than the excitation of the out-of-plane orbitals and is thus difficult to access experimentally. But, the efficiency of the photochemical BC can be increased by decreasing the energy gap between the in-plane frontier orbitals. This can be achieved by putting the enediyne framework in a cyclic cage. Decreasing the C1−C6 distance destabilizes the occupied MO in which the interaction between the end orbitals is antibonding. At the same time decrease in the C1−C6 distance the empty MO has got stabilization which has favorable interactions between the end orbitals. With a decrease in the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), photo-BC is favored. Figure 3. (a) Frontier molecular orbital diagrams involved in photo-BC. (b) Comparison between the antiaromatic π−π* interaction pattern and the antiaromatic TS for the [2s + 2s] cycloaddition involved in photo-BC. (c) Effect of locking the enediyne moiety in a cycle on the energy gap between the frontier in-plane MOs. Joint initiative of IITs and IISc – Funded by MHRD Page 65 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Funk and Williams have designed the pyrene-based enediyne which was tethered to an aminoalkyl side chain to provide affinity for DNA (Scheme 43). This, upon irradiation in the presence of pBR 322 DNA, caused cleavage of the DNA. In this process both forms II and III of DNA at micromolar concentrations were observed. This is the first example of an enediynebased DNA photocleaver. Scheme 81. Photo-activated pyrenyl enediyne. Hirama et al. have reported the photo-BC of several non-benzenoid enediynes. They have reported the photo-BC of 1,2-diakynyl cyclopentene. Usually, the yield of cyclized product was low ( 3%), except for the dipropynyl derivative in which case the cyclized product was isolated in yields up to 71%. No photoreduction products were observed unlike in the case of the benzenoid counterpart (Scheme 82). The 10-membered ring enediyne upon similar irradiation with a low-pressure Hg-lamp produced the cycloaromatized product along with the acyclic enediyne. The latter was generated via retro-BC (Scheme 82). It may be noted that such a retroBC product was not isolated under the thermal conditions. Scheme 82. Photo-BC of non-benzenoid cyclic enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 66 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Russell et al. have reported facile photo-BC of pyrimidine-based enediynes A in isopropanol solution to quinazoline (X = H or OH) (Scheme 83). It is interesting to note that the corresponding ketone C failed to undergo photo-BC. However, compound C give cyclization products only under thermal conditions. This clearly demonstrated different activation parameters controlling the kinetics of BC under thermal and photochemical conditions. Scheme 83. Pho-induced reactivity of pyrimidine-based enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 67 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Apart from their DNA-cleaving property, the enediynes have been shown to possess proteincleavage ability. This is one of the mechanism via which microorganisms producing enediynes protect themselves through the sacrifice of a protein that is secreted by the organism itself. Jones et al. have showed the likely mechanism of protein cleavage and synthesized photo-activated enediynes to study their protein-cleaving ability. The mechanism of protein cleavage involved the formation of radical B at the captodatively stabilized α carbon. This radical then reacts with molecular oxygen to form the peroxo radical E. This can undergo strand scission or cross-linking to provide G and H, the cleaved protein (Scheme 84). Scheme 84. Protein degradation pathways enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 68 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Support for the proposed mechanism depicted in Scheme 84 came from the cycloaromatization reaction of an enediyne in the presence of labeled amino acids, such as dideuteriated glycine (A) (Scheme 85). When the diradical is formed upon BC, the deuterium got abstracted by the diradical to form compound D. The isolation of the dimerization product F and the amide H together with the deuteriated aldehyde I could be explained on the basis of the formation of glycyl radical E. Scheme 85. Fate of the diradical produced after BC in the presence of glycine-d2. Conjugates of photo-activated enediynes and amino acids were also synthesized by Jones's group. These conjugated assemblies readily underwent photo-BC upon irradiation, thus making them potential agents for photodynamic therapy. The photo-activated enediyne bearing amino acid upon irradiation was found to cause degradation of bovine serum albumin, histone, and an estrogen receptor (Scheme 86). These observations on protein degradation by enediynes open up a new application of enediynes as chemical proteases. Scheme 86. Photo-activation of enediyne−amino acid conjugates. Joint initiative of IITs and IISc – Funded by MHRD Page 69 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Peterson et al. has reported the synthesis and reactivity of imidazole-fused cyclic enediynes toward photo-induced BC. The more conformationally rigid analogues gave higher yields of cycloaromatized products upon irradiation at ambient temperature. The bicyclic analogue shown in Scheme 87 was shown to undergo photo-BC to produce the cycloaromatized product and consequently to induce single-strand breaks in supercoiled DNA at micromolar concentrations. Scheme 87. Photo-activation of imidazole-fused enediynes. Photo-irradiation of bis(phenylethynyl) sulfide in hexane in the presence of 1,4-CHD produced 3,4-diphenyl-thiophene through the presumed intermediacy of the 2,5didehydrothiophene diradical (Scheme 88). This constitutes the first example of a 5-membered ring cycloaromatization exploiting the aromaticity of heterocyclic rings, such as thiophene. Scheme 88. Photo-BC of diethynyl sulfide. Joint initiative of IITs and IISc – Funded by MHRD Page 70 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9.2.2. Category 2: Activation of Enediynes via Photo-induced Electron Transfer Alabugin et al. have shown that bistetrafluoropyridinyl ethynyl benzenes I A-D, a class of photo-activated benzenes, smoothly undergo a novel C1−C5 photochemical cyclization to provide isomeric indenes II A-D and III A-D (Scheme 89). In this process the cycloaromatization follows a different mechanism than that operating in normal BC. The key step in this is the photo-induced electron transfer from 1,4-CHD to the singlet excited state of the enediyne. The presence of strongly electron-withdrawing tetrafluoropyridinyl (TFP) substituents renders the photo-induced electron transfer from 1,4-CHD to the singlet state of enediyne highly exothermic. Unlike the cyclization of neutral enediynes, the C 1−C5 cyclization of enediyne radical anions H leads to an intermediate L stabilized by resonance involving cyclopentadienyl anions, which makes the cyclization mode possible (Scheme 89). This is inferred after calculating the energies of the starting enediyne, the TS, and the radical product formed by photochemical cyclization. It is also to be noted that the enediyne−lysine conjugate E has been shown to possess some degree of sequence-selective binding and cleavage of DNA. Scheme 89. Photo-activation of fluoropyridinyl enediyne and the mechanism for the formation of indenes. Joint initiative of IITs and IISc – Funded by MHRD Page 71 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9.2.3. Category 3: Photo-excitation of Enediyne Metal Complexes via Ligandto-Metal or Metal-to-Ligand Charge Transfer Photodynamic therapy relies on the use of longer excitation wavelengths for the drugs to be used. This will ensure enhanced tissue penetration by near-infrared (IR) photons. Therefore, two possible approaches can be adopted to shift the excitation wavelength to a longer region (beyond λ > 600 nm) required for photo-BC. The first choice of design involves the synthesis of enediynes with extended π conjugation. However this is synthetically challenging and may suffer from solubility problems. The alternative strategy is the use of long-wavelength electronic transition with considerable absorptivity that can be achieved via metal-to-ligand charge transfer (MLCT) process within compounds where both the metal oxidation state and donor/acceptor redox potentials have been properly chosen. This idea was utilized demonstrated with design of a novel vanadium (V) metalloenediyne compound A of a 4,5-bis(phenylethynyl) benzene 1,2-diol ligand by Zaleski et al. The metalloenediyne exhibited strong ligand-to-metal charge transfer (LMCT) transitions in the nearIR region because of low redox potentials of the high valent vanadium center and the easily oxidizable metal-binding motif. Differential Scanning Calorimetry (DSC) and resonance Raman spectroscopy showed that these LMCTs can be successfully used to photothermally activate the metalloenediyne toward BC. Thus, upon laser excitation at 785 or 1064 nm compound A in the solid state become photothermally activated toward BC to produce the insoluble polymeric material C. The compound was inert to BC upon electronic excitation in the UV spectral region (Scheme 90) suggesting the necessity of excitation at very long wave length. Scheme 90. Photo-excitation of enediyne via ligand-to-metal charge transfer (LMCT). Joint initiative of IITs and IISc – Funded by MHRD Page 72 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Photo-BC can also be prompted by MLCT. Zaleski et al. have reported that photolysis of copper complexes of Cu(bpod)2PF6 and/or Cu(bpod)2(NO3)2 (A) (bpod = cis-1,8-bis(pyridine-3oxy)oct-4-ene-2,6-diyne) yielded BC of bound ligands. In contrast, the uncomplexed ligand and Zn(bpod)2(CH3COO)2 compound (E) were photochemically inert under the same conditions (Scheme 91). The observed BC of the compounds A has been ascribed because of MLCT. Moreover, the intermediates produced upon photolysis are capable of degrading both pUC19 plasmid DNA as well as a 25 base pair double-stranded oligonucleotide via C-4' hydrogen-atom abstraction in micromolar concentrations. Photo-excitation of Enediyne via Metal-to-Ligand Charge Transfer O O N Cu N N O O > 395 nm N N O B N (a) > 395 nm, 1,4-CHD or 2-propanol, CH3CN N (b) O DMF·H2O, CH2Cl2. N O A EDTA, Cu N 1,4-CHD or 2-propanol, CH3CN O O N O S O Cu N EDTA, DMF·H2O, CH2Cl2. C O N N N O S = Solvent O D S OCOMe O O N N Zn N N O O E OCOMe Scheme 91. Photo-excitation of enediyne via metal-to-ligand charge transfer (MLCT). Joint initiative of IITs and IISc – Funded by MHRD Page 73 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics The above example depicted nicely how a metal ion can facilitate photo-BC (Scheme 91). An interesting variation whereby the photo-BC is completely shut down upon metal complexation is provided in the next example (Scheme 92). Thus, the parent benzene-fused enediyne A undergoes photo-BC upon irradiation to produce the cycloaromatized product C via the diradical B. However, the ruthenium complex of A (D) did not produce any cyclized product upon similar photo-irradiation. This fact thus, highlights the importance of the electronic effect in controlling the BC kinetics (Scheme 92). The reluctance of compound D to undergo cycloaromatization (relative to A) is most likely due to decreased aromaticity in the incipient 1,4-diradical, which would be generated from D. Scheme 92. Shut down of photo-BC of benzene-fused enediyne upon complexation with Ru- metal. Joint initiative of IITs and IISc – Funded by MHRD Page 74 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9.2.4. Category 4: Photo-activation of Locked or Acyclic Enediyne The basis of this design strategy relies on the use of photo-cleavable protecting groups. This group masks the nucleophilic character of an amine or a phenolic hydroxyl group. The protecting group falls off upon photolysis and then liberates the free amine or phenol. Then, the free amine or the phenol allows flow of electrons which results in the opening of a strained ring-like epoxide. Thus, the strain is released. As a result, the enediynes get activated toward BC under ambient conditions. This idea was best demonstrated in the design of a model compound reported by Nicolaou et al. (Scheme 93). Thus, the compound A under photolytic conditions got converted into the diol D via the intermediacy of the quinone−methide C formed by epoxide ring opening. With the release of strain, the compound D underwent BC. The addition of an external nucleophile, such as EtOH or EtSH, led to the isolation of the cycloaromatized product. Scheme 93. Photodeprotection and activation of the dynemicin model. Joint initiative of IITs and IISc – Funded by MHRD Page 75 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Another design based on similar idea was devised by Wender et al. who synthesized a 5nitroveratryloxy (N-Voc)-protected dynemicin analogue E (Scheme 94). Compound E is photochemically activable dynemicin analogue that underwent cycloaromatization upon irradiation with wavelengths greater than 300 nm. The ability of this compound to DNA cleavage was also demonstrated. It was also observed that the enediyne E is also activated toward thermal BC by treatment with acid, which opens up the epoxide. Thus, the molecule is equipped with a dual triggering mechanism, pH as well as light. Activation of the Dynemicin Model by Photodeprotection MeO NO2 MeO O N O O H H H N O hn (365 nm) N HO THF-MeOH HO H E MeO HO H H MeOH HO H F G AcCl, THF, MeOH NO2 MeO O O N HO H DNA H MeO Cl HO H K H H N HO H H N HO H H N HO MeO MeO HO H J H Cleaved DNA HO H HO H I H Scheme 94. Photodeprotection and activation of the dynemicin model. The design of molecules with an acyclic framework to be activated in a similar way is based on the fact that cyclic 10-membered azaenediyne spontaneously cyclizes under ambient conditions with a decent half-life (t1/2 = 36 h at 30 °C). Taking the idea from this fact, a new design strategy has been adopted in which an acyclic enediyne having an amino group in one arm of the enediyne is protected in the form of N-Voc or β-lactam. Cleavage of N-Voc or opening of the β-lactam ring releases the nucleophilic nitrogen free which then undergoes intramolecular attack to close the cycle. The latter with appropriate size then undergoes BC. The resulting diradical in this process shows DNA cleavage or antibacterial property. Thus, Basak et al. designed an acyclic enediyne molecule with a photo-cleavable amine protecting group that is stable at biological temperatures but is convertible to a cyclic 10membered enediyne after a triggering reaction. This in situ generated cyclic enediyne could then be capable of showing DNA cleavage activity upon BC (Scheme 95). Thus, they have synthesized the enediyne A, where the photocleavable protecting group satisfies two criteria: (a) it suppresses the nucleophilicity of the nitrogen, and (b) it is removed by photolysis. The compound A upon irradiation at 365 nm was able to induce single-strand cleavage of plasmid DNA. It is also to be mentioned that the ketone A also caused partial DNA damage under nonirradiating conditions. However, the efficiency of cleavage was 2.5 times less compared to that observed for the in situ generated cyclic enediyne C which could be produced after deprotection Joint initiative of IITs and IISc – Funded by MHRD Page 76 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics of the protecting group of A and an intramolecular ring closure by nucleophilic attack by free – NH2 to the ketone. This ruled out the cleavage via the Maxam−Gilbert mechanism as the major pathway. Scheme 95. Photodeprotection and the mechanism of DNA cleavage of enediyne containing heteroatom “N”. Lane 1: DNA; lane 2: DNA + 5.082 (10 μM) (24 h) + hν (365 nm); lane 3: DNA + 5.082 (10 μM) (24 h). Joint initiative of IITs and IISc – Funded by MHRD Page 77 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9.2.5. Category 5: Activation Cyclopropenone/Diazoketone of the Prodrug via Photolysis of In this design strategy, a reactive enediyne system is protected with a group that arrests the spontaneous BC to occur at ambient conditions of a monocyclic enediyne. Although cobalt carbonyl complexation of the acetylenic moiety is a commonly used protecting group, the difficulty in the removal of such a group under photochemical conditions prohibited its use. Popik et al. have used cyclopropenone as a photocleavable protecting group to lock a benzenefused enediyne system A. Irradiation led to cheletropic removal of carbon monoxide with consequent generation of enediyne B, which then underwent BC. It may be noted that the acetylene-protected enediyne A is thermally stable, showing no sign of decomposition even after heating at 84 °C for 7 days. They have also demonstrated that the p-quinonoid cyclopropenonecontaining enediyne prodrug A can be activated by photolysis via a single- or two-photon transfer to the enediyne. The latter undergoes BC at 40 °C with a half-life of 88 h (Scheme 96). Scheme 96. Photochemical in situ generation of enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 78 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Similar to the MSC, eneyne−ketones can also undergo similar cyclization chemistry to form a phenoxy diradical. The latter reaction also takes place under ambient conditions similar to the MSC. On the basis of this Saito et al. synthesized the diazoketone B via Sonogashira coupling followed by the reaction with diazomethane or methyldiazomethane with the acid chloride A. The resulting diazoketone, upon photo-irradiation (high-pressure Hg-lamp), rearranges into the ketene D. The ketene is conjugated to the eneyne system. Therefore it undergoes cycloaromatization to produce the phenoxy diradical E which was shown to induce cleavage of ds plasmid DNA (pBR322) (Scheme 97). Scheme 97. Photo-activation through conversion of eneyne−ketene. Joint initiative of IITs and IISc – Funded by MHRD Page 79 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.9.2.6. Category 6: Activation via Photo-isomerization of Azobenzene-Based Enediynes and Sulfones Conformational changes can bring about significant perturbation in the kinetics of BC. Previously, König et al. have shown that (for a bipyridyl enediyne) a decrease in the distance between the two acetylenic arms undergoing covalent connection (c and d distance) upon complexation to mercury(II) brings about a remarkable increase in the reactivity toward BC. It is thus reasonable to think that a similar conformational change might be achieved if a group capable of switching between E and Z configurations is incorporated in an enediyne moiety (Scheme 98). Thus, Basak et al. designed azo-based enediyne systems represented by the general structure A. These molecules should exist in the thermally stable E isomer. Photoisomerization to the Z isomer B is expected to bring down the c and d distance (Scheme 98), which should lead to an increase in reactivity. Scheme 98. Rationale behind triggering through E−Z isomerization and phototriggering of azo enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 80 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 99. Synthesis of nonaromatic azo enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 81 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics With this idea, the cyclic enediyne A, containing a stable E-azo moiety (azoenediynes) was synthesized and reported by Basak et al. The key step is the double N-alkylation to form the cyclic network (Scheme 99). The stable E-azoenediynes A and G upon irradiation with longwavelength UV isomerize to the Z compounds F and H respectively, which can be thermally reisomerized to the corresponding E compounds (Scheme 100). Reactivity studies toward BC using DSC predictably indicated higher reactivity for the Z isomers. These studies provide a novel way to modulate the reactivity of enediynes under thermal or photochemical conditions. With an appropriately sized enediyne, there could be a possibility of inducing BC upon irradiation under ambient conditions. Scheme 100. Photochemical trans-cis isomerization of azo enediynes and thermal reactivity thereof. Joint initiative of IITs and IISc – Funded by MHRD Page 82 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Encouraged by the above trans-cis isomerization of azo enediynes in effecting a change in BC kinetics, a novel cyclic bispropargyl sulfone A containing stable E-azo moiety has also been synthesized by Basak et al. (Scheme 101). Scheme 101. Synthesis of azo bispropargyl sulfones. Joint initiative of IITs and IISc – Funded by MHRD Page 83 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics The compound A upon irradiation with long UV (350 nm) isomerized to the Z compounds B, which can be thermally re-isomerized to the E compounds. The E isomer A upon treatment with Et3N equilibrates to the mono-allenic sulfone C. The formation of bisallene E was not seen during the base treatment. On the other hand, a similar treatment of the corresponding Z isomer forms the monoallene D first, which subsequently further isomerized to the unstable bisallene F which finally underwent decomposition presumably via the Garratt−Braverman pathway (Scheme 102). Incubation with plasmid DNA also indicated higher DNA-cleavage efficiency ( 2.5 times) for the Z isomer. Scheme 102. Photochemical trans-cis isomerization of azo bis-propargyl sulphones their reactivity thereof. Joint initiative of IITs and IISc – Funded by MHRD Page 84 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics A similar increase in DNA-cleavage activity of a related sulfone with an extra conjugation was also observed for the Z isomer Q as compared to the E analogue P (Schemes 103). Scheme 103. Synthesis of unsaturated bispropargyl sulfones. Joint initiative of IITs and IISc – Funded by MHRD Page 85 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Dai et al. have earlier reported efficient photo-inducible DNA-cleaving ability of propargylic sulfone conjugated to the anthraquinone moiety (Scheme 104). From DNA-cleavage studies using different anthraquinone-based sulfones, it was concluded that appropriate spatial arrangement between the activated allenic sulfone and nucleobase, which is derived from an efficient intercalation, leads to a substantial amount of DNA cleavage via alkylation of the nucleobase and photo-induced one-electron oxidation of guanine bases. Scheme 104. Anthraquinone-based propargyl sulfone. Joint initiative of IITs and IISc – Funded by MHRD Page 86 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.10. Biological Actions of Some Synthetic Models In general two types of DNA cleavages occur- (a) the single- and (b) double-strand cuts (Figure 4a). In a typical experiment to check the DNA-cleaving activity a supercoiled plasmid DNA is used. Single-strand cleavage means the breakage or nicking of only one of the two strands, which enables the DNA to relax. On the other hand, a linear form will result if cuts are produced on complimentary sites (or close to complimentary sites) of both of the strands. The mobilities of all three forms in a gel under electric field are different and hence can be easily identified via gel electrophoresis technique (Figure 4d). Calicheamicin show its potential DNA cleaving activity at nanomolar concentrations (1 pg μL -1). However, all the reported synthetic enediynes show lower DNA cleavage activity compared to natural calichiamicin. These synthetic enediynes show their potency at micromolar concentrations. Thus, in comparison to the natural enediynes, the cleavage efficiency of designed enediynes is a moderate. Figure 4. (a) Single- and double-strand DNA cuts. (b) Supercoiled plasmid DNA. (c) Relaxed plasmid DNA. (d) Gel electrophoresis pattern of various forms of plasmid DNA [form I: supercoiled; form II: nicked, single-stranded (ss) cleavage; form III: linear, doublestranded (ds) cleavage]. Joint initiative of IITs and IISc – Funded by MHRD Page 87 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.10. 1. Biological Activity of Basak’s β-Lactam Fused Enediyne Under basic conditions (pH = 8), the -lactam fused enediyne as is shown in Figure 5 showed cleavage of supercoiled plasmid DNA (pBR 322) at concentrations of 50 mol. No such cleavage was observed under neutral conditions. In alkaline condition, the -lactam hydrolyses to produce the amine, which is predominantly present in the protonated form and is the actual DNA cleaving agent. The result clearly showed the potential of the -lactam fused enediyne as a cytotoxic agent. Interaction of Supercoiled DNA and -Lactam Fused Enediyne N BC and DNA Cleavage NH2 O H H EtOOC COOEt 1 CO2 2 3 Form I Form II Figure 5. Interaction of supercoiled DNA (in Tris-acetate buffer, pH 8.0) and enediyne in acetonitrile. Lanes 1: DNA; Lanes 2: DNA + enediyne 1.4.46 (50 mmol); Lanes 3: DNA. Joint initiative of IITs and IISc – Funded by MHRD Page 88 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.10. 2. Biological Activity of Basak’s Azo Bispropargyl Sulfones The amino enediyne shown in the figure below which is an intermediate for the synthesis of Basak’s β-lactam-fused enediyne by the carbene insertion route, is able to cleave double strand DNA at a pH of 8.0. Both the double-strand cut (linear form) as well as the nicked form was seen using pBR 322 plasmid in micromolar concentrations range (Figure 6A). Only the nicked form of cleaved DNA was seen at similar concentrations while pBlueScript SK+ plasmid DNA was used (Figure 6B). The protonated form was proposed as the DNA-damaging agent. The positively charged nitrogen is able to withdraw electrons from the enediyne π- framework leading to lowering the activation barrier for the generation of benzene diradical via BC. It is also important to note that the perturbation of the singlet−triplet barrier in the diradical via through-bond or through-space could also be responsible for such DNA cleavage activity. It was also demonstrated that the protonated amine in the form of tosylate salt also undergoes BC at 30 °C with a half-life of 30 days. Interaction of Supercoiled DNA and the Precursor Amino Enediyne of -Lactam Fused Enediyne pH = 7.8 NH (A) 1 2 Form II Form III NH2 BC and DNA Cleavage (B) 1 2 Form II Form I Form I With Supercoiled Plasmid DNA (pBR 322) With Supercoiled Plasmid DNA pBlueScript SK+ Figure 6. Interaction of supercoiled plasmid DNA (A) pBR 322 and (B) pBlueScript SK+ in Tris-acetate buffer at pH 8.0 and enediyne shown in acetonitrile. Agarose (0.7%) gel electrophoresis using ethidium bromide stain-Lane 1: DNA; Lane 2: DNA + enediyne (50 μmol). Joint initiative of IITs and IISc – Funded by MHRD Page 89 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.10.2. Biological Activity of Basak’s Azo Bispropargyl Sulfones The cyclic Z-azo-bispropargyl sulfone showed higher DNA-cleavage efficiency ( 2.5 times) than the corresponding E isomer presumably via the Garratt−Braverman pathway (Figure 7). Interaction of Supercoiled DNA with Azo Bis-Propargyl Sulphones N N N O N O O h 350 nm O Kcis N N O Ktrans S O O Et3N A B O S N O Et3N N O O O C S O O D DNA Cleavage X C 1 N N C DNA Cleavage via Garratt-Braverman 2 O 3 N S O N Form II O O O O C C E Form I C C S O O F O S O With Supercoiled Plasmid DNA pBlueScript SK+ Figure 7. DNA-cleavage experiment of compounds A and B in TAE buffer (pH 8.5) with 7 μL DNA of 0.4 μm/bp concentration. Lane 1: control DNA + CH3CN (10 μL); Lane 2: DNA + Z-sulfone B (0.02 mM) in 5 μL CH3CN; Lane 3: DNA + E-sulfone A (0.02 mM) in 5 μL CH3CN. Joint initiative of IITs and IISc – Funded by MHRD Page 90 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.10.3. Biological Activity of Basak’s Unsaturated Azo Bispropargyl Sulfones A similar increase in DNA-cleavage activity of a related sulfone with an extra conjugation was also observed for the Z isomer as compared to the E analogue (Figure 8). Interaction of Supercoiled DNA with Unsaturated Azo Bis-Propargyl Sulphones N N N N O O h 350 nm O O DNA Cleavage O S O Trans azo- Cis azo1 2 S O O 3 Form II Form I With Supercoiled Plasmid DNA Figure 8. DNA-cleavage experiment of trans- and cis- unsaturated azo-bis-propargyl sulphone in TAE buffer (pH 8.5), and 7 μL of plasmid DNA (0.4 μm/bp). Lane 1: control DNA + CH3CN (10 μL); Lane 2: DNA + E-sulfone (0.02 mM) in 5 μL CH 3CN; Lane 3: DNA + Z-sulfone (0.02 mM) in 5 μL CH 3CN. Joint initiative of IITs and IISc – Funded by MHRD Page 91 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.10.4. Biological Activity of Popik’s In-situ Generated Enediyne An evaluation of the photogenerated nine-membered ring enediyne B, towards nuclease activity was carried out using supercoiled plasmid DNA cleavage assays. Three forms of this DNA: native (RF I), circular relaxed (RF II, produced by single-strand cleavage), and linear (RF III, formed by scission of both strand in close proximity) are readily separated by the agarose gel electrophoresis. Aqueous solution (5 mM) of cyclopropenone A was irradiated with low-pressure mercury lamp to produce reactive enediyne B. A solution of φX174 supercoiled circular DNA (10 ng/μL) in TE buffer was added to photolysate and incubated for 16 h at 25 °C. Photochemical in Situ Generation of Enediyne and the DNA cleavage O A h (300 nm) Photo Bergman -CO Cyclization OH B OH C OH DNA DNA Cleavage 1 2 3 4 5 Form II Form III Form I Figure 9. Cleavage of φX174 plasmid DNA by the photogenerated enediyne B. Lane 1: DNA alone; Lane 2 and 4: DNA + cyclopropenone precursors A in Dark; Lane 3 and 5: DNA + irradiated solution of A. (Pandithavidana, D. R.; Poloukhtine, A.; Popik, V. V. J. Am. Chem. Soc. 2009, 131, 351.). Incubation of the DNA with cyclopropenone precursor A (lanes 2 and 4, Figure 9) does not induce any detectable DNA cleavage. The photogenerated enediyne B, on the other hand, induces substantial single strand cleavage of φX174 DNA (RF II), while linearized form (RF III) becomes prominent only at higher (5 mM) concentration of the cleaving agent (lanes 3 and 5, Figure 9). Integration of fluorescence of bands on the gel shown in Figure 9, allowed the authors to evaluate the relative abundance of the native, circular, and linearized forms of φX174 DNA. Thus, incubation of the DNA with the 1 mM of enediyne B produces 45% of single strand cleavage (RF II) and less than 5% of the double strand cleavage (RF III, lane 3, Figure 9). At 5 mM concentration of B, DNA-cleavage efficiency increases to 67% and 10%, respectively (lane 5, Figure 9). Joint initiative of IITs and IISc – Funded by MHRD Page 92 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.10.5. Biological Activity of Rashel’s Pyrimidine Based Enediyne Compounds A and B cleave double-stranded DNA which is shown in Figure 10 Both of them showed this property when incubating with ΦX174 dsDNA for 70 h at 40 °C. The ketone B showed significant DNA nicking (Form II) at concentrations as low as 40 μM and nearly complete nicking at 4 mM. The alcohol A showed almost no reactivity at 40 μM but was able to nick DNA at 4 mM. While no double-strand (ds) cleavage (Form III) was observed at the lower concentrations, both A and B show slight ds cleavage at 4 mM. DNA Cleavage Activity of Pyrimidine Based Enediyne OCH3 OH N H3CO N 1 3 4 5 6 7 8 Form II O O Thermal BC N N 2 DNA A OCH3 H3CO OH Photo BC Form III Form I DNA Cleavage B Figure 10. Supercoiled DNA interaction ΦX174. DNA was incubated for 70 h at 40 °C with compounds A and B in buffer (TE, pH 7.6) and analyzed by electrophoresis (1% agarose gel, ethinium bromide stain). Lanes 1−3: A (4000, 400, and 40 μM); Lane 4: DNA control; Lane 5: DNA control + restriction enzyme, Dra I; Lanes 6−8: B (4000, 400, and 40 μM). Photochemical DNA cleavage ability was also demonstrated for A and B (hν, 40 °C, 3 h). In this case, compound A showed superior DNA cleavage ability. At 40 μM, compound B showed significant DNA single-strand cleavage. But, compound B showed no discernible activity. At higher concentrations (4000 μM), both compounds showed signs of double-strand scission, again with A giving the more complete reaction. Compounds A and B both showed anticancer activity against human leukemic lymphoblasts of the CCRF-CEM cell line (log-phase cultures) (IC50 values of ≈1.25 μM). Joint initiative of IITs and IISc – Funded by MHRD Page 93 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11. Enediyne as a Scaffold for Peptidomimetics 3.11.1. Introduction: Z-Enediyne as Scaffold for Peptidomimetics Design During the past decade, the construction and investigation of expanded acetylenic chromophores has become a central area of chemical research. It has been fueled by the availability of new synthetic methods, in particular Pd(0)-catalyzed cross-coupling reactions, the discovery of the antitumor activity of a series of natural compounds possessing reactive Zenediyne -chromophores, and the need for new nanoscale molecules and polymeric materials that exhibit unusual electronic and optical functions and properties. The enediyne antitumor antibiotics are appreciated for their novel molecular architecture, their remarkable biological activity and their fascinating mode of action and many have spawned considerable interest as anticancer agents in the pharmaceutical industry. Of equal importance to these astonishing properties, the enediynes also offer a distinct opportunity to study the unparalleled biosyntheses of their unique molecular scaffolds and what promises to be unprecedented modes of selfresistance to highly reactive natural products. Elucidation of these aspects should unveil novel mechanistic enzymology, and may provide access to the rational biosynthetic modification of enediyne structure for new drug leads, the construction of enediyne overproducing strains and eventually lead to an enediyne combinatorial biosynthesis program. Joint initiative of IITs and IISc – Funded by MHRD Page 94 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.2. Enediynyl Amino Acid AS β-Sheet Nucleator As already mentioned, Enediynes have drawn unprecedented interest amongst the scientific community because of their cytotoxic activity and possible use as anticancer drug. All studies so far have been concentrated on their synthesis and evaluation of chemical as well as biological activity. The special structural feature of Z-enediynes is the type of reverse-turn associated with the two acetylenic arms. One can consider making enediynyl amino acid containing peptides, which may be forced to adopt typical conformational motifs. Figure 11. The solution conformation of the enediynyl peptides This structural motif, for the first time in enediyne chemistry, was used, in our laboratory, as a possible scafolld for peptide secondary structure mimetic. Specially, we were interested in designing and synthesis of the -sheet mimetic by incorporating the enediynyl amino acid into a peptide chain. We thought that this enediynyl motif can act as nucleator and thus may induce in adopting the sheet which constitutes a well-studied subset of the reverse turn and is a common feature in biologically active peptides and globular proteins. The sheet capping turns are widely believed to act as a molecular recognition site for many biological processes. Bag and Basak et al. have incorporated the enediynyl amino acid A into peptides B-D (Figure 11) and then found out the conformational preferences by NMR and CD-measurements. Circular Dichroism (CD) spectra of the fully protected peptides and the generally higher (T) values for the chemical shifts of and N-H’s reveales that the peptides adapt a significant proportion of -sheet like conformation. However, the results also indicated the presence of other conformations as well, specially the -NH as being intramolecularly Hbonded. The variable temperature NMR experiments indicate that the conformation resembling -sheet capping type motif is more predominant (Figure 11). Joint initiative of IITs and IISc – Funded by MHRD Page 95 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.3. Enediynyl Pentapeptide AS β-Turn Peptidomimetics 3.11.3.1. Introduction An enediynyl pentapeptide A (Figure 12) in which a novel enediynyl -amino acid acts as fluorophoric reporter was designed and synthesized by Basak et al. The reason behind the design was two fold: a) to use the Z-enediyne moiety as a nucleator for turn peptidomimetic and b) to exploit and elaborate further, the intrinsic florophoric properties of this moiety simultaneaously. This was the first report of application of the intrinsic fluorophoric property of Z-enediyne in monitoring binding process of metal ions and colloidal gold nano-particles. Figure 12. The enediynyl pentapeptide. Joint initiative of IITs and IISc – Funded by MHRD Page 96 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.3.2. Synthesis of the Target Enediynyl Pentapeptide A proline residue was chosen because prolines and its derivatives are extremely important in synthetic as well as biological chemistry and its known ability to induce turns. The chiral auxiliary oxazaborolidine and the many of the protease inhibitors required for management of AIDS are modeled after proline. The various structural motifs like -turn or -bend are often found where a polypeptide chain often abruptly reverses its direction as were revealed from the discussions above. Proline residues are frequently found in -turns because the peptide bonds involving the imino nitrogen of proline readily assume the cis-configuration, a form that is particularly amenable to a tight turn. One extremely important enzyme that cleaves the peptide bond involving proline [actually Tyr (Phe)-Proline] is HIV-protease, which cleaves the long polypeptide chain into the functional oligomers. Various proline containing peptidomimetics have been made that are promising candidates as anti AIDS-drugs. Another class of drugs where proline plays an important role is in the case of management of hypertension (ACE inhibitors). Proline or modified Proline containing natural products with interesting biological activities are also well known. These include Kainic acid and (-) Detoxinine. The synthesis of the enediynyl pentapeptide in the protected form is given below (Scheme 105). Scheme 105. Synthesis of Novel enediynyl pentapeptide A. Joint initiative of IITs and IISc – Funded by MHRD Page 97 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.3.3. Evidence of β-Turn Structural Motif The pentapetide A exists predominantly in -turn structural motif as revealed by variable temperature. NMR and CD spectroscopy. In the presence of transition metal ions and gold nanoparticles, the fluorescence intensity of the peptide got enhanced with remarkable quantum yield with the Z-enediynyl -amino acid acting as a fluorophoric reporter. The interesting photophysical behaviors with alkali and alkaline earth metal ions are also reported. Figure 13. The enediynyl pentapeptide and its CD spectra. The secondary structure of peptide A was estimated by recording its CD spectrum in methanol, which showed a strong maximum at ∼198 nm followed by several broad minima at ∼205, 212, and 222 nm, indicating that the peptide predominantly adopts a β-turn like structure (Figure 13) at least in the solvent used for the study. The peptide secondary structure estimation using CD estima program shows a 60% turn like structure, the existence of which implies the possible presence of intramolecular H-bond between the peptide strands on the two arms of the enediyne framework. This could be assessed by determining the variation of chemical shifts of the various NHs with temperature in DMSO-d6 in which all the four NHs exhibited different chemical shifts. Interestingly, the turn like structure is more or less maintained in the presence of Ca2+ ions. Of the four amide NH’s, one alanine NH and the NH belonging to the enediynyl amino acid exhibited Δδ/ΔT values that are within the Kessler limit of −3 ppb/K, indicating strong intramolecular H-bonding and supported the predominant turn like structure of the peptide. The appearance of a crosspeak for the hydrogens attached to C-2 and C-11 in NOESY spectrum also provided further evidence for the turn like conformation of the two peptide arms of the enediyne backbone. The H-bonded conformation was also supported by the semi-empirical AM1 geometry optimization. Joint initiative of IITs and IISc – Funded by MHRD Page 98 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.3.4. β-Turn Mimitic Enediyne as A Fluorescence Chemosensor Ever since the discovery of naturally occurring enediynes, all efforts have been drawn towards the finding analogous rationally designed molecules having excellent antitumor activities with less toxicity as well as evaluation of the parameters controlling the Bergman cyclization (BC). The photophysical properties of this class of molecules have so far been restricted to the study of BC under photoirradiation. Because of the presence of special structural features, in which a double bond is flanked between two acetylenes in a cis fashion, it occurred to us that, the enediynes might serve as a probe for fluorescent spectroscopy, for example to study their binding with DNA or sensing of metal ions. As a matter of fact, the photo physical properties of this class of molecules have not been explored to a large extent except for the photo-Bergman Cyclization. Enediynes with metal coordinated architectures might show interesting photophysical properties, esspecially upon complexation a change in the absorption / fluorescence behaviour of the enediynes might occur. This change might allow one to use the enediyne scaffold in fluorescence based sensing of ions that is of great interest as sensors in biomedical research and in molecular information processing. 3.11.3.5. Fluorescence Photophysical Behavior in Bare and Complexed form of the Enediynyl Pentapeptide The peptide itself in TFE showed emission at max 380 nm when excited at 320 nm with a large Stoke shift (60 nm), a characteristic emission of enediyne moiety. A significant changes in fluorescence intensity was observed upon addition of metal ions. Transition metal ions all caused enhancement of fluorescence intensity with the extent of enhancement depending upon the nature of the metal ions added. Although the precise nature of interactions between the peptide and the metal ions are not known, it is possible that the intrinsic turn like structure allows the accommodation of the metal ion for binding. Generally, transition metal ions are strong quenchers of fluorescence. However, in this case the enhancement of fluorescence demonstrated the ability of a enediynyl peptide to show fluorescence enhancement upon complexation to various transition metal ions similar to cryptand-based fluorophores. One possible explanation behind the enhancement of fluorescence may be that the distance and orientation of the metal ion entering the cavity, in relation to the -system is such that the spin-orbit coupling which facilitates the S-T intersystem crossing is minimum. In other words the metal ion-fluorophore communication is much less than the metal-receptor interaction. Joint initiative of IITs and IISc – Funded by MHRD Page 99 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics A good correlation between fluorescence quantum yield and their complex formation constants was observed (Table 3). The order of K and is Co(II) Zn(II) > Cu(II)> Ni(II). Fluorescence titration experiment with Cu2+ ion showed that the peptide binds more than one Cu2+ ion. For the other metal ions, a linear plot revealed 1:1 complex formation. Table 3. Summary of complex formation constants and fluorescence Quantum Yields in TFE at 298K The pentapeptide also showed an enhancement of fluorescence intensity upon complexation with Au(0)-nano particle in dry THF solvent. nano particle was increased. The gold nanoparticles are nonfluorescent and enediyne in THF is moderately fluorescent with an emission maximum at 372 nm. The peptide bound to gold nanoparticles exhibit strong emission bands at 375 nm and the intensity increases as the concentration of nano particles increases. The red-shift in the emission peaks parallels the shift in absorption bands. These new electronic transitions of the enediyne chromophore become allowed as the N-of amide functional groups binds strongly to the gold particle. No such spectral shifts or enhanced emission could be seen when we added a THF solution of enediynyl peptide containing tetraoctyl ammonium bromide and treated with NaBH4. The fluorescence quantum yield of surface-bound peptide is as high as 0.32 at a gold concentration of 30 x 10 -5 mM. On the basis of the absorption, steady-state emission, it is concluded that surface binding has a significant effect on the fluorescence yield, but it has no observable effect on the intersystem crossing efficiency and thus the enhancement of fluorescence was observed (Figure 14). Figure 14. Probable mode of fluorescence enhancement by gold nano particle Joint initiative of IITs and IISc – Funded by MHRD Page 100 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics The observed increase in the fluorescence yield reflects the suppression of the nonradiative decay processes upon adsorption of peptide on to the surface of gold nanoparticles. The photoinduced electron transfer between the lone pair of N-of amide functional groups and the enediyne moiety competes with the radiative and nonradiative decay of the singlet excited state. Upon binding of the N-lone pairs to the gold surface, the electron-donating ability of the N is decreased and this in turn suppresses the electron transfer from its lone pair to the enediyne moiety (Figure 14). A similar chelation-enhanced fluorescence has been reported earlier by Czarnick and co-workers and de Silva and co-workers. By binding metal cations to amine functional groups of probes (e.g., anthracene), they were able to demonstrate the suppression of intramolecular quenching and thereby enhancement of fluorescence was observed. 3.11.4. Cyclic Enediyne Amino Acids as β-Turn Peptidomimetic 3.11.4.1. Introduction Looking at constrained peptidomimetics and their structure-function relationships, it is reasonable to think that incorporation of an enediyne framework into a cyclic peptide scaffold might offer useful structures with defined and characteristic secondary structures such as the βturn mimetics. Moreover, cyclic enediynyl amino acids may have potential affinity for biological targets such as receptors or transcriptional regulators thereby promoting nicking and degradation. Such interactions could also provide information as to the location of ligand binding by inspection of where the target is cleaved. With this idea in mind, Allen B. Reitz et al. have investigated the incorporation of benzofused enediynes into 10- and 12-membered cyclic αamino acids 1 and dipeptide 2, respectively, and examined their reactivity and conformational preferences (Figure 15). Figure 15. Structures of cyclic enediynyl amino acids. Joint initiative of IITs and IISc – Funded by MHRD Page 101 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.4.2. Synthesis of 10-Membered Cyclic Enediynyl Amino Acids The synthesis of 10-membered cyclic enediynyl amino acid 1 was started with Sonogashira coupling of propargyl glycine (2) to 1,2-diiodobenzene (3) in DME solvent using ammonia as base. The produced alkyne 4 was coupled in a second Sonogashira reaction with propargyl alcohol to form 1,2-diynylbenzene 6 in 94% yield (Scheme 106). Intramolecular Mitsunobu reaction of 6 at 0 °C afforded the final enediyne 1 in 90% yield. Compound 4 and 1,4cyclohexadiene (100 mol equiv) were subjected to microwave irradiation in DMF when the cyclized product 7 was produced in 84% yield after 10 min at 120 °C (Scheme 106 ). Scheme 106. Synthesis of 10-membered cyclic enediyne amino acid 1. The cyclic 10-membered ring enediynyl amino acid 1 is stable for several months at room temperature in the solid state. The single-crystal X-ray structure analysis revealed that the tosyl group adopts an endo-orientation placing it above the enediyne core which was also observed in solution as was revealed from a NOESY NMR experiment in CDCl3. The amino acid 1 was also found to cleave double-stranded DNA when incubated with supercoiled DNA at 37 °C for a period of 24 h at concentrations 10 μM and higher, with many fragments being observed at the highest concentrations. Joint initiative of IITs and IISc – Funded by MHRD Page 102 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.4.3. Synthesis of 12-Membered Cyclic Enediynyl Amino Acids/Peptide The synthesis of dipeptide 2 is depicted below in Scheme 107. Scheme 107. Synthesis of 12-membered ring enediynyl peptide 2. The cyclic enediynyl dipeptide 2 did not undergo the Bergman cyclization possibly because the terminal alkynes are too far apart (3.86 Å contrary to the distance required for spontaneous Bergman cyclization of 2.9–3.3 Å.) (Scheme 107). Upon irradiation with light, regioselective reduction of peptide 2 provided Z-olefin 13 instead of expected product 12. In the photoreduction process generation of radicals were proposed. Thus, it was observed that the enediynyl peptide 2 lead to non-specific protein degradation when incubated with bovine serum albumin (BSA) and irradiated the mixture at ambient temperature for a period of 1 h (at 1.0 μM). Joint initiative of IITs and IISc – Funded by MHRD Page 103 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.11.4.4. Peptide β-Turn Conformation Adopted by 12-Membered Cyclic Enediynyl Peptide 2 Single crystal X-ray structure analysis of cyclic enediynyl dipeptide 2 revealed a reversed turn structure with hydrogen bonding between the carbonyl on the benzyloxycarbonyl protecting (-Cbz) group and -NHs on both the N-methyl amide terminus and the bridging internal amide (Figure 16). A β-turn will fall into a particular defined class if three of the four backbone torsional angles do not deviate more than 30 °C and the other not more than 45 °C from ideal values. Evaluation of the torsional angles of 2 indicated that it adopts a Type II β-turn conformation in the solid state. The Type II β-turn comprises of about 13% of β-turns among structures in the Protein Data Bank. Dihedral Angles i + 1 i+1 i + 2 i+2 Ideal Type II b-Turn -60 o 120 o 80 o 0o Enediyne 2 -50.4o 132.6o 58o 29o Figure 16. Structural adoption (Similar to its X-ray structure of enediyne 2), Various torsional parameters revealed a Type II β-turn conformation in the solid state possessed by enediyne 2. Joint initiative of IITs and IISc – Funded by MHRD Page 104 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12. Enediyne as Peptide Cleaving Agent 3.12.1. Introduction: Self-Decomposition of C-1027 Chromophore Some naturally occurring enediynes were found to have proteolytic activity. As for example both the naturally occurring kedarcidin and the related enediyne neocarzinostatin (NCS) are chromoproteins and capable of causing damage to histones its enediyne warhead. Esperamicin also has proteolytic and antitumour activity via the involvement of its enediyne moiety. It is believed that the diradicals generated from the reactive enediyne core via Bergman cycloaromatization abstract hydrogen from proteins producing a C-centered radical intermediate. The generated C-centered radical intermediate, in aerobic condition, react with molecular oxygen. The resulting peroxy radical ultimately is responsible to cleave the peptide. Alternatively, the peptide radical may cross-link or form an adduct with another radical source. The apoprotein class of enediyne antibiotics, such as neocarzinostatin, kedarcidin, and maduropeptin, C-1027 are composed of a highly reactive enediyne chromophore in complex with an apoprotein. The apoprotein (10489 Da) is a single polypeptide chain of 110 amino acid residues cross-linked by two disulfide bonds. The enediyne chrophores are bound noncovalently in a cleft of the apoprotein via hydrophobic interaction force. Studies revealed that the apoprotein stabilizes the radical-generating enediyne chromophore in C-1027 by tight binding. Though there involved tight binding, yet the chromophore is released to react upon reaching its target DNA in the cell nucleus. However, the ways in which apoprotein modulates and deliver the highly reactive enediyne chromophore in biological target was unclear. Considering the fact that the C-1027 constantly generates the benzene diradical even when bound to the apoprotein, hydrogen abstraction from the apoprotein by the diradical then is expected to lead to its facile decomposition before reaching its biological target. However the studies showed that the hydrogen abstraction is geometry unfavorable. Therefore, the apoprotein kinetically stabilizes the enediyne moiety by positioning the diradical with low accessibility to hydrogen of apoprotein. Thus, self-decomposition of the chromophore−protein complex is halted. Thus, the apoprotein appears to function both as a stabilizer and as an effective carrier/ delivery system. Although, the apoprotein stabilizes the enediyne core from BC to occure, C1027 is known to undergo aging i.e. the apoprotein is unable to completely inhibit the radical to abstract proton from its backbone leading to decomposition of apoprotein. Several studies thus showed that suppressing the self-decomposition pathway provides a basis for enhanced properties for C-1027. The self-decomposition pathway of C-1027 was studied in detail. It is now well accepted that the p-benzyne B is in equilibrium with chromophore A and abstracts the hydrogen of Gly96 to generate the Gly radical C in the first step. Then, the Gly96 radical (C) produces the peroxy radical D upon reacting with oxygen. This is that peroxy radical which is responsible for peptide cleavage via dioxetane intermediate E. The whole process is shown below in Scheme 108. Joint initiative of IITs and IISc – Funded by MHRD Page 105 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 108. The mechanism of self-decomposition pathway of chromoprotein of C-1027. Mass spectroscopy data of the peptide fragments. Joint initiative of IITs and IISc – Funded by MHRD Page 106 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12.2. Mechanism of Peptide Cleavage by Enediyne Cleavageof Peptide by enediyne: Generation and Fate of C-Centered Peptide Radicals H N O Protein Cleavage by Hirama's Pathway O RO O 3 O2 Aerobic H N O R O Enediyne H H N H N O H N O RO O O R O + O O HO O R 3 H N O2 O Protein Cleavage by Jones Pathway O R O RO NH2 + O O Figure 17. Mechanism of peptide cleavage by enediyne and the generation and fate of C-centered peptide radicals. 3.12.3. Basak’s Design Strategy of Peptide Cleaving Enediyne Inspired by the self-decomposition pathway of C-1027 several research groups come forward for the synthesis of peptide/protein cleaving enediynes. Basak et al. synthesized a novel enediyne –peptide hybrid with peptide cleavage activity at tailor-made positions. In their design, they used a molecular scaffold where the basic template contains two long parallel chains in the cisoid form. One of these is a polypeptide chain while the other carries the enediyne frame connected to the template via a suitable linker (Figure 18). The cisoid conformation of these two chains is essential to bring the enediyne moiety closer to the peptide chain. In order to achieve such an arrangement, they selected linkers that can participate in hydrogen bonding with the peptide chain. The purpose is to cut the peptide chain selectively via the aid of the 1,4-diyl radicals. Figure 18. Design strategy of peptide cleaving enediyne. Joint initiative of IITs and IISc – Funded by MHRD Page 107 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12.3.1. Example of Basak’s Peptide Cleaving Enediynes Based upon consideration of H-bonded enediyne containing scaffolds Basak et al. have designed and synthesized a novel class of peptide–enediyne conjugate A and B (Figure 19) and demonstrated their selective cleavage ability of the peptide moiety maintaining selectivity in the degradation pathway by mass spectrometric studies. Enediyne–Peptide Hybrid with Peptide Cleavage Activity O O O N H O N N H O O H N O N H O O Ph H N O H N NHBoc N H O O O NHBoc N H O B O O O O O N N H O N H O N N H N O O O O O N N C O H N N H A N N O O N H H N O O O H N OCH2Ph Ph O O N H D H N O Figure 19. Examples of enediyne–peptide hybrids for peptide cleavage. Joint initiative of IITs and IISc – Funded by MHRD Page 108 of 115 O OCH2Ph Ph NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12.3.2. Synthesis of a Representative Peptide Cleaving Enediynes A and B NHBoc I NHBoc (b) (a) I 1 OH 2 Br O 6 O N O O O N H (g) N O 10 11 NHBoc NH2.TFA N H N O N H O O O O O O H N (i) O O (h) N H O A NHBoc N O Ph H N O H N N H O O 9 N O OH O O O N H (f) O OCHPh2 O O NH2 5 (e) O 8 N3 4 O N NHBoc (c) OMs 3 (d) N NHBoc O N H O O H N O B NHBoc N H O O O OH O HO 7 O N H Ph O H N O N H NHBoc 12 HO O O N H O H N O N H NHBoc 13 Reagents and conditions: (a) MsCl, Et3N, DCM, 0 oC, 15 min, 96%; (b) NaN3, DMF, rt. 7 h, 67%; (c) PPh3, THF, H2O, rt, 10 h, 70%; (d) 7, Cs2CO3, DMF, rt, 10 h, 59%; (e) TFA, anisole, DCM,0 oC to rt, 1 h; (f) 5, EDC.HCl, DMAP, DCM, rt, 10 h, 50%; (g) TFA, DCM, 0 oC to rt, 1.5 h; (h) 12, EDC.HCl, DMAP, DCM, 0 oC to rt, 10 h, 41%; (i) 13, EDC.HCl, DMAP, DCM, 0 oC to rt, 12 h, 39%. Scheme 109. Synthesis of a peptide cleaving enediynes A and B. Joint initiative of IITs and IISc – Funded by MHRD Page 109 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12.3.3. Study of Peptide Cleavage by Basak’s Designed Enediynes Scheme 110. Mass fragmentation pattern for α-H-abstraction at C-3 by enediyne-peptide hybrid A. Joint initiative of IITs and IISc – Funded by MHRD Page 110 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics Scheme 111. Possible fragmentation (that was not observed) for α-H-abstraction at C-2 by enediyne-peptide hybrid A. Scheme 112. Abstraction of α-H from third glycine unit of the pentapeptide chain of by enediyne-peptide hybrid B. Joint initiative of IITs and IISc – Funded by MHRD Page 111 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12.4. Protein Degradation by Jones’ Photoactivated Enediynes (Jones, G. B. et al. J. Org. Chem. 2005, 70, 9789.) 3.12.4.1. Design Strategy In their design Jones et al. considered the following criteria: (a) the atom-transfer chemistry of enediyne-derived diyls with peptide under both thermal and photochemical activation conditions (b) the impact that hydrophilic functionality on abstraction efficiency (c) considerable flexibility would need to be built into the system for application to protein targets. (d) Their design involves the derivatization of the alkyne termini of the enediyne with appropriate protein recognition functions (e) The derivatization also takes care the tuning of photoactivity of the Bergman cycloaromatization. (f) Most photoactivated enediynes studied have the vinyl moiety embedded in an arene. In their design they concentrated on the photochemical activation to as a function of ringstrain and electronic effects. (Figure 20). Figure 20. Structures of Jones’ photoactivable enediynes. Joint initiative of IITs and IISc – Funded by MHRD Page 112 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12.4.2. Synthesis of Jones’ photoactivable enediynes Ph O Ph TIPS Ph (a) (b) OH O O 1 H 2 4 Ph (c) Ph H N O (d) Ph H N COOH O 6 COOH P Ph OH I O 3 H N Ph ClH.H2N 5 COOMe O R COOH QI R = Ph, QII R = COOH QIII R = CH2COOH Reagents and Conditions: (a) (i) NaBH4, EtOH, (ii) PPh3, Br2, DCM, (iii) LiHMDS, HMPA, THF, (iv) TBAF, THF; (b) (i) 3, Pd(PPh3)4, CuI, Et3N, (ii) LiOH, THF, H2O; (c) (i) 5, EDCI, DMF, (ii) LiOH, THF, H2O; (d) h, 450W, 3h, i-PrOH Scheme 113. Synthesis and photo-Bergman cyclization of enediyne-peptide hybrids. Joint initiative of IITs and IISc – Funded by MHRD Page 113 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics 3.12.4.3. Applications (Protein Degradation) of Jones’ photoactivable enediynes A. Interaction with Bovine Serum Albumin (BSA) Photochemical activation (450 W, 3 h) of the enediyne-peptide hybrid P (Figure 20) gave arene product 6 in appreciable yield, together with unreacted starting material. They have also studied the photochemically induced modification of BSA and lysozyme by photolyzing compound P in aqueous buffer and using 6 and enediynes 4 as controls. In the case of 66 kD protein BSA, subsequent analysis (SDS−PAGE) showed some evidence of enediyne-induced degradation with one principal fragment ( 40 kDa) visible (<5% relative to parent) when a 5:1 ratio of enediyne P/BSA was employed. Encouraged by this initial finding, they have also synthesized enedynes QI-QIII and studied their protein degradation ability. Compound QII and QIII showed enhanced affinity for BSA when photolysis was conducted. Significant cleavage into two principal fragments (31 and 35 kDa) was observed. In the case of the 14 kDa protein lysozyme, dimerization to form a species with m/z 28.6 kDa was observed when a mixture of enediyne P and lysozyme were irradiated that suggests the possibility of intermolecular diyl-mediated cross-coupling. B. Interaction with Histone H1 Naturally occurring chromoproteins kedarcidin, C-1027, and neocarzinostatin, were able to show proteolytic activity. In the case of kedarcidin, studies suggested that the highly acidic apoprotein was capable of inducing selective degradation of histone H1. Following these findings, Jones et al. have prepared an acidic enediyne−peptide conjugate to determine if enediyne-mediated histone degradation could be achieved. To investigate this, enediyne containing the triaspartic acid derivative R was also synthesized (R, Figure 20) studied for their interaction with Histone H1. Binding of this agent to a variety of histones revealed that affinity, as expected, was greater for H1 versus others. Irradiation of triaspartate-enediyne hybrid in the presence of histone H1 (21.5 kDa) led to degradation of the protein into principal components in the 8−11 kDa range. The application of a designed photoactivated enediyne with near micromolar affinity for a biological target is highly interesting and suggests even greater affinity might be attainable by coupling the photowarhead to the ligand with a complimentary protein receptor. Joint initiative of IITs and IISc – Funded by MHRD Page 114 of 115 NPTEL – Chemistry – Bio-Organic Chemistry of Natural Enediyne Anticancer Antibiotics C. Interaction with Human Estrogen Receptor α Jones et al. have also synthesized enediyne-lipophilic steroidal ligands hybrid molecule (S, Figure 20) to study the capability of degrading the human estrogen receptor (hER). In their design, they have utilized estrone wherein the enediyne functionality was introduced at the 17α position of estrone. The hybrid underwent smooth photocycloaromatization to give complete conversion to cycloaromatized product within 12 h. Irradiation in the presence of recombinant hER (66 kDa) resulted in marked degradation to produce two discrete fragments within 12 h (31 and 35 kDa) . The ability to induce on demand degradation of a nuclear receptor using a lipophilic warhead is of significance and underscores the potential use of such entities as molecular reagents. 4.13. Selected References 1. Basak, A.; Mandal, S.; Bag, S. S. Chem. Rev. 2003, 10, 4077 and references therein. 2. Kar, M.; Basak, A. Chem. Rev. 2007, 107, 2861–2890 and references therein. 3. Basak, A.; Roy, S.; Roy, B.; Basak, A. Curr. Top. Med. Chem. 2008, 8, 487 and references therein. 4. (a)Basak, A.; Mitra, D.; Kar, M.; Biradha, K. Chem.Commun. 2008, 3067-9. (b) Mitra, D.; Banerjee, D. R.; Das, A. K.; Basak, A. Bioor. Med. Chem. Lett. 2010, 20, 6831. (c) Mitra, D.; Kar, M.; Pal, R.; Basak, A. Bioor. Med. Chem. Lett. 2007, 1007. (d) Basak, A.; Roy, S.; Das, S.; Hazra, A.; Ghosh, S.; Jha, S. Chem.Commun. 2007, 622. (e) Roy, S. K.; Basak, A. Chem. Commun. 2006, 1646. (f) Basak, A.; Mandal, S. Tetrahedron Lett. 2002, 43, 4241. (g) Basak, A.; Rudra, K.; Bag, S. S.; Basak, A. J. Chem. Soc. Perkin Trans. 1 2002,1805. (h) Basak, A.; Mandal, S.; Das, A. K.; Bertolosi, V. Med. Chem. Lett. 2002, 12, 873. (i) Basak, A.; Bag, S. S.; Rudra, K.; Burman, J.; Dutta, S. Chem. Lett. 2002, 710. (j) Basak, A.; Rudra, K.; Bag, S. S.; J. Chem. Soc. Perkin Trans 2002, 1, 1805. (k) Pal, R.; Basak, A. Chem. Commun.2006, 2992. 5. (a) Biggins, J. B.; Onwueme, K. C.; Thorson, J. S. Science 2003, 301, 1537. (b) Singh, S.; Hager, M. H.; Zhang, C.; Griffith, B. R.; Lee, M. S.; Hallenga, K.; Markley, J. L.; Thorson, J. S. Acs. Chem. Biol. 2006, 1, 451–460. 6. Hirama, M.; Akiyama, K.; Tanaka, T.; Noda, T.; Iida, K.-i.; Sato, I.; Hanaishi, R.; Fukuda-Ishisaka, S.; Ishiguro, M.; Otani, T.; Leet, J. E. J. Am. Chem. Soc. 2000, 122, 720-721. 7. Usuki, T.; Inoue, M.; Hirama, M.; Tanaka, T. J. Am. Chem. Soc. 2004, 126, 3022. 8. Roy, S.; Basak, A. Chem. Commun. 2010, 2283. 9. Basak, A.; Bag, S.S.; Bdour, H. M. M. Chem. Commun. 2003, 1, 2614–2615. 10. Fouad, F. S.; Wright, J. M.; Plourde II, G.; Purohit, A. D.; Wyatt, J. K.; El-Shafey, A.; Hynd, G.; Crasto, C. F. ; Lin, Y.; Jones, G. B. J. Org. Chem. 2005, 70, 97899797. 11. Du,Y.; Creighton, C. J.; Yan, Z.; Gauthier, D. A.; Dahl, J. P.; Zhao, B.; Belkowski, S. M.; Reitz, A. B. Bioorg. Med. Chem. 2005, 13, 5936–5948 Joint initiative of IITs and IISc – Funded by MHRD Page 115 of 115