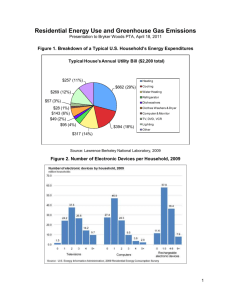
QbD implementation in Generic Industry: Overview and Case
advertisement

QbD implementation in Generic Industry: Overview and Case-Study Inna Ben-Anat, Ben Anat QbD Strategy Leader, Leader Teva Pharmaceuticals IFPAC JAN 2013 R&D Three Core Components of QbD and Generic Industry: How Do They Overlap Quality by Design Clearly Cl l d defining fi i the h iintended d d purpose off the future developed product and design this product to fit its purpose 2. Understanding what attributes of this product are critical so it (product) will keep serving g its intended p purpose p 1 1. The connectio T on is clear 1 1. Generic Industry 2. 3. Enhanced understanding ‘what’ impacting the critical quality attributes and ‘how’ (materials, process, packaging etc) ; define control strategies so that the intended 3. purpose of the product will reproducibly maintain i t i itits iintegrity t it Reproducibly R d ibl M Making ki “A d drug product d t that is comparable to brand/reference listed drug product in dosage form, strength route of administration strength, administration, quality and performance characteristics, and intended use" Providing uninterrupted supply of high quality and affordable medication to our patients Efficiency and Speed QbD for Generics: Finding the right balance between Speed, Efficiency and Excellence Overview of QbD (GPhA, May 2012) QbD Guide for Generics: Step 1-Product Design RLD Characterization Quality Target Product Profile Critical Quality Attributes GPhA/FDA CMC Workshop, May 2012 QbD Guide for Generics: Step 2 - What are the potential Risks Risk Assessment Defines the Development Strategy What are the Risks?... API Excipients Formulation and Process Equipment i Testing Packaging … How do we stay efficient Effective Prior Knowledge utilization and management Generic Industry has a lot of information and in-house knowledge available Data bases of pre pre-created created Ishikawa diagrams in order to harmonize and streamline the Risk Assessment process p Historical data-mining Historical Data Mining: Drug Layering of Pellets Example Example: Previously developed product, multiply batches are available for Data Mining: In-Process Pellets Assay vs. Fines Correlation Based on the found relationship relationship, Assay decreases ~0.6% with each % fines How do we control low % fines by process parameters (Drug Layering)… Layering) ‘All examples are for illustration purposes only’ Historical Data Mining: Drug Layering of Pellets Example Actual Processing Parameters from all available historical lots were collected and ‘data‘data-mined’ Partition per most critical factor affecting % Fines All Rows Count 31 LogWorth Difference Mean 3.666129 3 666129 1.6232558 1 6232558 1 98596 1.98596 Std Dev 2.4278793 Slit Temp Actual (°C) -max<74.3 Count 19 LogWorth Difference Mean 2.8973684 0.6352248 1.21364 Std Dev 2.1367027 Exhaust Temp-AVG<44.4 Count 11 Mean 2.3863636 Std Dev 2.1165362 Slit Temp Actual (°C) -max>=74.3 Count 12 Mean 4.8833333 Std Dev 2.4430061 1. Most Significant parameters affecting %Fines are Slit Temp and Exhaust Temp 2. Lower Slit Temperature (<74˚C)and lower Exhaust Temperatures (<44 (<44˚C) C) will generate less % Fines Exhaust Temp-AVG>=44.4 Count 8 Mean 3.6 Std Dev 2.0894223 Potential DOE Factors for future similar products/processes or for further process fine-tuning ‘All examples are for illustration purposes only’ QbD Guide for Generics: Step 3 - Plan the right/relevant Experiments Efficient and Informative DOE: CQAs= f (CPPs, CMAs) How do we stay efficient o Effective Prior Knowledge Utilization What do we vary and what do we fix? What target and range do we evaluate and why? What statistical model do we use and why? (Can we assess what interactions are most likely to occur? Can we assess what factors would have non linear relationship with the response?) o Modern DOE techniques for efficient yet powerful designs (DOptimum, I-Optimum) o Monte Carlo Simulations to assess the process robustness using historical data to assess expected variabilityy Let’s take a typical manufacturing process for tablets as an example to start with… Wet Granulation Fluid Bed Drying Milling Blending Compression How many potentially Critical Process Parameters do we need to assess? 5? 10? 25? High Shear Wet Granulation: > 40 potential CPPs… High Shear Wet Granulation Fish-Bone Diagram CQAs >40… 40 Fluid Bed Drying: > 30 potential CPPs… Fluid Bed Drying Fish-Bone Diagram CQAs A Typical Manufacturing Process for Tablets… HS Wet Granulation Fluid Bed Drying Milling Blending Compression For a process involving the above unit operations we may end up with over 100 potential CPPs. g it? How do we manage Effective Knowledge Management ! Prior Knowledge Utilization Blending Unit Operation CQAs 4 critical variables are left for assessment, the rest are kept at constant and monitored Design Variable Prior Experience/Fixed Justify!! Effective Knowledge Management ! With efficient Prior Knowledge utilization, we can end up with 8-16 trials for Experimental Design- feasible! JMP® Statistical Software from SAS Main effects Interactions Prior Knowledge Efficient and Informative Design of Experiments • Brainstorming sessions will identify the design factors and their ranges, while previous knowledge should be effectively utilized to identify those and limit them to the most critical ones • While conducting DoE, all parameters that are not studied should be kept constant at their optimum fixed level (justify!) in order to eliminate the noise and additional variation and increase the effectiveness of the study • Prior to DoE execution, measurement’s system integrity and sensitivity must be verified • There is a lot to learn from every DoE: if a factor was found to have no effect, it can be used to minimize cost or increase robustness by having it set on convenient level DOE and Modeling: Process Robustness and Monte Carlo Simulation Monte Carlo Simulation: Predicted OOS Rate: ~0.02% Distribution of the predicted output Predicted OOS rate Estimated Process Variability Estimated Analytical Variability ‘All examples are for illustration purposes only’ QbD Guide for Generics: Step 4 - Define Control Strategies Questions to ask ourselves: 1. Did we evaluate the impact of CMAs and CPPs on CQAs? Did we find any interactions? What do they mean for us? 2. Do we have a robust and reproducible process? Do we know the impact of raw materials variability? Did we identify potential sources of variation? 3. Did we establish meaningful In Process and Release specifications? 4. Did we address scale-up p challenges? g 5. ………………………………………….. Case-Study IR Tablet Tablet, Dry Granulation Process Product Development Outline Analysis of the reference listed drug (RLD) Defining f Quality Q Target Product Profile f (QTPP) (Q ) Identification of Critical Quality Attributes (CQAs) potential risks related to Drug g Identification and evaluation of p Product Components (DS and Excipients – stability and compatibility), Formulation and Manufacturing Process, etc. Screening and optimization of formulation Development of a robust process (DOE for high risk parameters) Manufacture of the exhibit batch Establishment of control strategies QTPP Component Target Justification Dosage Form Tablet Administration Route Oral Dosage Design Immediate release tablet Strength X and Y mgs Bioequivalence AUC and Cmax match RLD under food Bioequivalent to RLD Appearance Both: Brown to orange elegant film coated tablet. Dimensions similar to RLD RLD. X mg: round; Y mg: oval Marketing g requirement; q Needed for patient acceptability Identity Positive for API Needed for labeled claim & therapeutic efficacy Assay 100% of label claim Needed for therapeutic efficacy Impurities Specified and unspecified impurities meet ICH Q3B. Needed to ensure safety Disintegration Comparable disintegration time as RLD in appropriate media at room temperature Pharmaceutical equivalence to RLD (possible route of administration as suspension) Content Uniformity AV <15.0 (tested by weight variation) Targeted for consistent clinical effectiveness Residual solvents Complies with USP <467> Regulatory requirement. Needed to ensure safety Dissolution USP Apparatus pp II, 50 rpm, p 1000 mL 0.1M HCl, 370C. NLT 85Q is dissolved in 45min Regulatory g y requirement q Stability NLT 24 month shelf life Needed for commercialization Container closure system HDPE bottles with Child Resistant (CR) Caps and appropriate desiccants , if required Needed for safety and commercial requirements Pharmaceutical equivalence to RLD CQAs CQA Justification Potentially affected by Assay Needed for therapeutic efficacy Process Impurity Needed to ensure safety Formulation & Process Content Uniformity Needed for therapeutic efficacy of each unit Formulation & Process Dissolution Presumptive qualification for in vivo release and therapeutic efficacy Formulation & Process Disintegration Needed to ensure patient compliance (suspension) Formulation & Process Formulation: Initial Risk Assessment and studies conducted Filler DP CQA Assay type & amount Low Formulation Attribute Disintegrant Lubricant Glid t Glidant type type amount & amount & amount Low Low Low Coating C ti formulation Low Impurities Low Low Low Low Low Content Uniformity y Low Medium Low Low Low Dissolution Medium Low High Medium Low Disintegration Medium Low High Low Low Vary type & amount (control strategy: optimized and fixed)) Fix on high level based on prior knowledge Vary type & amount (control t t optimized ti i d and d fixed) fi d) strategy: Process Scheme Pharmacy Compression II (cores) Mixing I Mixing IV & V C Cosmetic ti C Coating ti Milling I (De-lumping) Milling II Mixing II+III Compression I (slugs) Initial Risk Assessment: Process Unit Operations DP CQA Mixing I Milling I Mixing II+III Medium (De-lumping) Medium Compression I (Slugs) Low Low Impurities Low Medium Low Medium Content Uniformity Low Medium Medium Medium Dissolution L Low M di Medium L Low Hi h High Disintegration Low Low Low High y Assay Unit Operations-cont'd Operations cont'd DP CQA Mixing IV+V Compression II Coating Low Low (Tablets) Low Low Impurities Medium Low Low Medium Content Uniformity Low Low Low Low Dissolution High Low High Medium Disintegration High Low High Medium y Assay Milling II Process Optimization DOE Based on prior knowledge, previous experience and initial feasibility studies, the most potentially critical process parameters were chosen for further evaluation in DOE study. Additional parameters were set at their h i optimum i fifixed d constant llevell iin order d to reduce d uncontrolled ll d noise i and variability (13 runs including 2 centers, D-Optimum Design using JMP software from SAS) Unit Operation Compression I (Slugs) Milling II Compression II (Tablets) DOE Factors Levels Used -1 0 +1 Compression force Low Medium High Compression speed Low Medium High Mill type Quadro Quad o NA Frewitt e tt Mill screen 0.6 NA 0.8 Compression force Low Medium High Compression speed Low Medium High Responses 1. 2. Slug weight /RSD Slug hardness 1. 2. 3. PSD S Bulk & tap density Hausner ratio/Flow 1. 2. 3. 4. 5. Assay & impurities Dissolution Content Uniformity Disintegration time Tablet Hardness Prediction Profilers: Factors/Responses relationship-% on PAN (Fines) Interaction: Mill screen impact is low for Frewitt Type Mill Prediction Profilers: Factors/Responses relationship (Dissolution) DOE Model Prediction vs. actual Exhibit Batch data Hausner Ratio 1.31 Model Predicted Value 1.28 % Fines 19% 17% Dissolution-T1 Dissolution T1 AVG (N=6) Dissolution-T1 RSD (N=6) Dissolution-T3 AVG (N=6) UoC RSD (N=10) 36% 36% 10.1% 8.8 % 69% 68 % 1.69 % 1.45 % Selected Response Exhibit Batch Value Good Correlation between Values p predicted by y DOE Model & Actual Responses Process-Risk Mitigation, 1/2 Unit Operations DP CQA Mixing I Assay Controlled by mixing time/speed Milling I Controlled by S Screen size Low Low (Was found not critical) Low Controlled by Screen size Dissolution Low Low (Was found not critical) Disintegration Low Impurities p CU Low Mixing II+III Low Compression I (Slugs) Low Low Low (Was found not critical) Controlled by mixing time/speed Low (Was found not critical) Low Low Controlled by slug hardness Process-Risk Mitigation, 2/2 Unit Operations-cont'd DP CQA y Assay Milling II Mixing IV+V Compression II (Tablets) Coating Low Low Low Low Low (Was found not critical) Impurities Low (Was found not critical) Low Low Content Uniformity Low Low Low Low Controlled by mill type/ mill screen Low Controlled by core hardness and compression speed Controlled by fixed coating level Dissolution Disintegration g Low Summary Despite all of the challenges, the Generics Industry acknowledges p gQ QbD is the way y forward,, g gaining g that implementing o Enhanced product and process understanding- robust products and processes o Id Identification tifi ti and d control t l off sources off variationi ti f t and faster d efficient tech transfers, greater process capability Efficient utilization of prior knowledge is a key to successful QbD implementation in generics Real change will come if and when o The risk/cost benefits are realized o Playing field is leveled o FDA review of the applications shows the benefits of QbD 32 -“What “Wh should h ld I do d next?” ?” -“Create Create an action plan, Adopt the Big Q Concept Concept” Juran on Quality by Design