ARTICLES
GM1 structure determines SV40-induced membrane
invagination and infection
Helge Ewers1,12,14, Winfried Römer2,3,14, Alicia E. Smith1, Kirsten Bacia4,13, Serge Dmitrieff5, Wengang Chai6,
Roberta Mancini1, Jürgen Kartenbeck1,7, Valérie Chambon2,3, Ludwig Berland8,9, Ariella Oppenheim10,
Günter Schwarzmann11, Ten Feizi6, Petra Schwille4, Pierre Sens5, Ari Helenius1,15,16 and Ludger Johannes2,3,15
Incoming simian virus 40 (SV40) particles enter tight-fitting plasma membrane invaginations after binding to the carbohydrate
moiety of GM1 gangliosides in the host cell plasma membrane through pentameric VP1 capsid proteins. This is followed by
activation of cellular signalling pathways, endocytic internalization and transport of the virus via the endoplasmic reticulum to the
nucleus. Here we show that the association of SV40 (as well as isolated pentameric VP1) with GM1 is itself sufficient to induce
dramatic membrane curvature that leads to the formation of deep invaginations and tubules not only in the plasma membrane of
cells, but also in giant unilamellar vesicles (GUVs). Unlike native GM1 molecules with long acyl chains, GM1 molecular species
with short hydrocarbon chains failed to support such invagination, and endocytosis and infection did not occur. To conceptualize
the experimental data, a physical model was derived based on energetic considerations. Taken together, our analysis indicates
that SV40, other polyoma viruses and some bacterial toxins (Shiga and cholera) use glycosphingolipids and a common
pentameric protein scaffold to induce plasma membrane curvature, thus directly promoting their endocytic uptake into cells.
SV40 is a non-enveloped DNA virus of the polyoma family. The capsid is
45 nm in diameter, and composed of 72 icosahedrally organized VP1 pentamers1 that each bear five binding sites highly specific for GM1 (refs 2, 3),
its glycolipid receptor for infection4. Incoming SV40 virions attach to several GM1 molecules5,6 in the exoplasmic leaflet of the plasma membrane
and quickly become immobilized by the cortical actin cytoskeleton7,8.
Cholesterol-dependent entry7 occurs after kinase signalling7,9 via small,
tight-fitting indentations10, most of which are devoid of caveolin-1 (Cav-1;
ref. 11). Internalized vesicles are transported via microtubules to the smooth
endoplasmic reticulum12 where the protein folding and retrotranslocation
machineries are involved in SV40 export into the cytosol13 for infection.
How the binding of a virion to glycolipids in the exoplasmic leaflet
leads to cell entry and infection is not clear. Several other multivalent
glycolipid ligands are also internalized by clathrin-independent endocytosis14–16, suggesting that the reorganization of specific lipids into
membrane domains17,18 is important for the uptake process19–21. Indeed,
binding of the pentavalent cholera toxin to GM1 induces the formation
of membrane domains in vitro22, and multivalent binding is required for
efficient endocytosis23. By binding to up to 15 Gb3 glycolipid molecules,
Shiga toxin drives curvature changes of cell and model membranes24.
Whether multivalent binding and glycolipid structure mediate the process of cell infection by colloidal viral particles is not known.
Here, we investigate the role of the hydrocarbon chain structure of
the GM1 receptor molecule in SV40 endocytosis and infection. Based
on experimental work with cells and liposomal membranes and on
theoretical considerations, a physical model for the formation of SV40induced membrane invaginations is derived. Our results indicate that the
tight organization of GM1 molecules with specific hydrocarbon chain
structures is required for membrane mechanical processes leading to
endocytosis and infection by SV40.
RESULTS
Dependence of SV40 infection on GM1 hydrocarbon chain
structure
To test how critical the structure of the GM1 hydrocarbon chain is for cellular
uptake and infection, we took advantage of a mutant mouse melanoma cell
ETH Zurich, Institute of Biochemistry, HPM E, Schafmattstrasse 18, 8093 Zurich, Switzerland. 2Institut Curie, Centre de Recherche, Laboratoire Trafic, Signalisation
et Ciblage Intracellulaires, 75248 Paris Cedex 05, France. 3CNRS UMR144, France. 4Institute for Biophysics, TU Dresden, BIOTEC, Tatzberg 47‑51, D‑01307
Dresden, Germany. 5UMR Gulliver CNRS-ESPCI 7083, 10 rue Vauquelin, 75231 Paris Cedex 05, France. 6Glycosciences Laboratory, Imperial College London, Harrow,
Middlesex, HA1 3UJ, UK. 7M010, German Cancer Research Center (DKFZ), D‑69120 Heidelberg, Germany. 8Institut Curie, Centre de Recherche, Laboratoire PhysicoChimie, 75248 Paris Cedex 05, France. 9Université P. et M. Curie/CNRS UMR168, France. 10Department of Hematology, The Hebrew University-Hadassah Medical
School, Ein Kerem, Jerusalem 91120, Israel. 11LIMES, Membrane Biology and Lipid Biochemistry Unit c/o Kekulé-Institut für Organische Chemie und Biochemie
der Universitaet Bonn, 53123 Bonn, Germany. 12Current address: ETH Zurich, Laboratory for Physical Chemistry, HCI F, Wolfgang-Pauli Strasse 10, 8093 Zurich,
Switzerland. 13Current address: University of California at Berkeley, Department of Molecular and Cell Biology, Berkeley, CA 94720, USA.
14
These authors contributed equally to this work.
15
These authors contributed equally to this work.
16
Correspondence should be addressed to A.H. (e-mail: ari.helenius@bc.biol.ethz.ch).
1
Received 16 September 2009; accepted 24 November 2009; published online 20 December 2009; DOI: 10.1038/ncb1999
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
11
None
2 × C18:0
e
nt-GM1
80
60
40
20
Nocodazole
Latrunculin
Jasplakinolide
mβCD
Genistein
f
200
150
100
50
0
nt-
M1
C8-GM1
DO
-G
M1
-G
DP
M1
-G
DL
-G
C8
GM
nt-
M1
0
nt-GM1
1
Total
100
–
GM
120
1
Cells expressing T-ag
(percentage of control)
d
0
M1
None
2 × C18:1
20
-G
None
2 × C16:0
40
C8
None
2 × C12:0
60
Intracellular SV40
fluorescence
(percentage of control)
Sphingosine d18:1/d20:1 d20:1
Fatty acid
C18:0
C8:0
DP
DO
DS
Phosphatidylethanolamine
80
No GM1
O
O
GM95 nt-GM1
DL
100
CV-1
nt
C8
Ceramide
Internalized
Isoform
Lipid moiety
120
No SV40
Cells expressing T-ag
(percentage of control)
O
O
O
GM95
NHR
O
O
O
O
O
O
O
O
O
c
b
O P
O
O-
NHR
O
O P
O
O-
O
O P
O
O-
O
O
O
O
CH20
NH
O
OH
CH20
NH
O
OH
O P
O
O-
R
R
NHR
a
NHR
A RT I C L E S
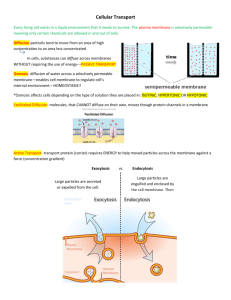
Figure 1 SV40 infection and endocytosis depend on GM1 hydrocarbon
chain structure. (a) Structures of nt-GM1 and the chemically synthesized
GM1 species used in this study. The native (nt-GM1) species is shown
on the left next to C8-GM1, which has an 8‑carbon short-chain fatty
acid. For other species, the GM1 pentasaccharide was attached to the
amino groups of phosphatidylethanolamine (PE) glycerophospholipid
species bearing different fatty acid chains: di-lauroyl-PE (DL-GM1),
di-palmitoyl-PE (DP-GM1), di-oleoyl-PE (DO-GM1) and di-stearoyl-PE
(DS-GM1). (b) Fluorescence microscopy images of Cy5-labelled SV40
(SV40–Cy5) incubated with GM1-deficient GM95 cells, GM95 cells that
were supplemented with nt-GM1, or CV-1 cells naturally expressing GM1.
(c) SV40 infection in GM95 cells that were supplemented or not with ntGM1, as indicated. nt-GM1-supplemented cells were mock treated (–) or
pre-incubated for 1 h with methyl‑β-cyclodextrin (mβCD, 5 mM), genistein
(0.1 mM), nocodazole (1 μM), latrunculin (0.1 μM) or jasplakinolide
(0.1 μM). Inhibitors were maintained during the experiment. Infection was
scored by immunofluorescence detection of nuclear SV40 T‑antigen (T-ag)
expression after Hoechst staining and data were normalized to expression
in nt-GM1-supplemented GM95 cells. Data are the mean ± s.d. of at least
three independent experiments, P < 0.01 for all inhibitors compared with
nt-GM1-supplemented cells (Student’s t-test). (d) SV40 infection in GM95
cells supplemented with the indicated GM1 species. Infection was scored
and plotted as in c. Data are the mean ± s.d., P < 0.01 for C8-GM1 and DLGM1 compared with nt-GM1 cells (Student’s t-test). (e) Confocal images of
atto‑488-biotin dual-labelled SV40-VLPs after 2 h incubation at 37ºC with
GM95 cells supplemented with nt-GM1 (left) or C8-GM1 (right). Total atto488 fluorescence of SV40-VLPs (top) and specific detection of intracellular
SV40-VLPs through the biotin label by indirect immunofluorescence
(bottom). (f) Quantification of intracellular SV40-biotin-VLPs after
immunofluorescence detection of internalized particles as in e. Fluorescence
intensity was normalized to that of cells supplemented with nt-GM1. Data
are the mean ± s.d., P < 0.001 (Student’s t-test). Scale bars, 10 μm.
line, GM95, that lacks all glucose-based glycolipids25. We incorporated GM1
species with differing lipid chain compositions26,27 into these cells. GM1 with
C18:1/C20:1 sphingosine molecules and C18:0 fatty acids (native, nt-GM1,
Fig. 1a) was used as a control. After GM95 cell reconstitution with nt-GM1,
the lipid was evenly distributed in the plasma membrane, as judged by the
homogeneous appearance of fluorescently labelled cholera toxin B‑subunit
(CTxB–FITC; Supplementary Information, Fig. S1a), another natural GM1
ligand28. This situation was similar to the one found on cells that naturally
express GM1 (data not shown). Fluorescence recovery after photobleaching
(FRAP) demonstrated that the GM1-bound CTxB–FITC was mobile in the
plane of the membrane (Supplementary Information, Fig. S1a). Moreover,
confocal microscopy showed that CTxB was efficiently internalized by endocytosis (data not shown). We concluded that nt-GM1 was integrated as a
functional component of the plasma membrane, as demonstrated previously
by electron spin resonance27.
Whereas Cy5-labelled SV40 failed to associate with untreated GM95
cells (Fig. 1b), it bound to nt-GM1-supplemented cells to a similar extent
as to wild-type murine cells (Fig. 1b). In contrast to untreated cells that
were totally resistant to infection, up to 76% of the cells supplemented with
nt-GM1 were infected, as indicated by T‑antigen expression. The level of
infection depended on the amounts of GM1 and SV40 used (Supplementary
Information, Fig. S1b, c). As in normal host cells, infection was inhibited by
extraction of cellular cholesterol (mβCD), inhibition of tyrosine kinase activity (genistein), disruption of microtubulules (nocodazole) and interference
with the dynamics of the actin cytoskeleton (jasplakinolide and latrunculin
A; Fig. 1c) 7,29. By supplementing cells with GM1, we could thus reconstitute
the normal, productive entry pathway of SV40 in GM95 cells.
Next, GM95 cells were supplemented with GM1 species with differing
tail structures. One was a GM1 molecule with short fatty acid chains
(C8), and the others (DL, DP and DO) were glycerophospholipids with
differing saturation levels and lengths of lipid chains to which the GM1
pentasaccharide was conjugated through the amino group of the phosphatidylethanolamine (Fig. 1a; Methods)27,30. By measuring CTxB–Cy5
binding to GM95 cells, we first determined the amount of each lipid
12
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
A RT I C L E S
a
e
SV40
FM dye
Merge
f
2
b
1
g
c
VLP-atto565
d
2
VLP-atto565
1
f1
CV-1
f2
g1
g2
Cav-1–/–
Figure 2 SV40 binding induces caveolin-independent membrane
invagination in cells. (a) Electron micrographs of CV‑1 cells that were
incubated for 7 min with SV40. Note the tight-fitting membrane under
SV40 particles. (b) Electron micrograph of polyomavirus VLPs after 30 min
incubation with cells. The VLPs line the lumen of tubular membrane
invaginations like beads on a string (arrowheads). Scale bars, 200 nm.
(c–d) Confocal images of fluorescence-labelled SV40-VLPs after 30 min
incubation with energy-depleted CV‑1 cells (c) or Cav-1–/– cells (d). Scale
bars, 5 μm. (e) Confocal images of fluorescence labelled SV40-VLPs (red)
and the membrane dye FM 43 FX (green) after 30 min incubation with
energy-depleted Cav-1–/– cells (top panels). Scale bar, 10 μm. A magnified
region of the cell is shown in the bottom panels. (f, g) TIRF microscopy
images of fluorescence-labelled SV40-VLPs a few minutes after binding
to the bottom surface of untreated (f) or energy-depleted (g) Cav‑1–GFPexpressing cells. Magnified regions of the cells are shown in the bottom
panel. Scale bars, 10 μm.
species needed to result in a level of binding comparable to that found on
cells supplemented with nt-GM1 (Supplementary Information, Fig. S1d).
To make sure that the different lipid species were properly inserted into
the plasma membrane, lateral CTxB–FITC mobility was confirmed
under all conditions by FRAP analysis (data not shown).
We then analysed the binding of SV40 to these reconstituted cells using
virus-like particles (VLPs), which are recombinant capsids composed
exclusively of VP1 proteins3,31,32. VLPs behave as intact virus with respect
to receptor binding and endocytosis, but are devoid of viral DNA as well
as internal capsid proteins. SV40-VLP binding was similar between cells
reconstituted with the different GM1 species (Supplementary Information,
Fig. S1e, f). However, only GM1 species with long acyl chains, DP‑ and
DO‑GM1, supported SV40 infection (Fig. 1d). The levels of infection were
88.9 ± 15.1% and 26.8 ± 7.4%, respectively, compared with nt-GM1supplemented cells. In contrast, infection in cells that were supplemented with
short chain species was close to background levels (Fig. 1d). We concluded
that a ceramide base structure was not an absolute requirement for glycolipid receptor function, but acyl chain length was critical.
To determine which step in the infectious entry programme required
a receptor lipid with long acyl chains, GM95 cells were supplemented
with either nt-GM1 or C8-GM1. The latter was chosen to represent a
non-permissive receptor species. Confocal microscopy after indirect
immunofluorescence labelling of internalized particles showed that only
nt-GM1 supported efficient endocytosis of the virus (Fig. 1e). SV40-VLP
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
13
A RT I C L E S
VLP
DOPC
Merge
a
nt-GM1
b
C8-GM1
c
DL-GM1
d
DP-GM1
e
DO-GM1
f
DS-GM1
Figure 3 SV40-induced membrane invagination on model membranes is
dependent on GM1 hydrocarbon chain structure. (a–f) Confocal sections
in the equatorial plane of GUVs made from a mixture of 68 mol% DOPC,
30 mol% cholesterol, 1 mol% BodipyFl‑C5-HPC (green) and 1 mol% of ntGM1 (a), C8-GM1 (b), DL‑GM1 (c), DP‑GM1 (d), DO‑GM1 (e) or DS‑GM1 (f).
Fluorescence-labelled SV40-VLPs are shown in red. Scale bars, 5 μm.
internalization in C8-GM1-supplemented cells was reduced to 34.4 ± 1.5%
when compared with nt-GM1 supplemented cells (Fig. 1f), probably representing particles taken up by non-infectious pathways.
Caveolin-independent membrane invagination in cells
A few minutes after binding to CV‑1 cells, transmission electron
microscopy showed SV40 particles in small invaginations that sometimes extended to enclose few virions (Fig. 2a) and in tight-fitting early
14
endocytic vesicles lacking sub-membrane, electron-dense material10,12.
The close contact between virus and the plasma membrane and the
formation of tight-fitting indentations differentiates SV40 and other
polyomavirus family members from most other viruses. When high
VLP doses of mouse polyomavirus, a closely related virus with a similar
structure33, ganglioside receptor 4,26 and endocytic pathway 8,34,35, were
used, even longer tubular structures were observed that extended from
the plasma membrane and contained a continuous row of particles
(Fig. 2b, arrowheads). These thin (50–60-nm diameter) tubules could
thus penetrate the cortical actin meshwork, which has a mesh size of
200–300 nm (ref. 36).
Electron microscopy images indicated an inward-directed budding
process, as if the virus particles directly participated in the induction of
membrane curvature by binding to gangliosides. To test this possibility, we
blocked active cellular processes, such as coat dynamics and membrane
traffic, in CV‑1 and HeLa cells by using metabolic inhibitors to deplete
energy. We hypothesized that this treatment would abolish abscission
of VLP-containing invaginations from the plasma membrane. Starting
20–30 min after addition to such cells, fluorescently labelled SV40-VLPs
were found to induce numerous tubular, virus-containing structures of
variable length that in some cases reached several microns into the cytoplasm and contained the majority of SV40 particles (Fig. 2c; Supplementary
Information, Fig. S2a). These tubules did not form in the absence of virus
(data not shown), and using the membrane dye FM‑1‑43 FX, it was found
that they were connected to the plasma membrane (Fig. 2e). Incubation of
SV40-VLPs with Cav-1-negative mouse embryonic fibroblasts led to the
formation of identical tubular structures (Fig. 2d). Furthermore, tubules in
wild-type cells did not colocalize with endogenous Cav-1 immunostaining
(Supplementary Information, Fig. S2c) and, even before tubule formation, SV40-VLPs rarely colocalized with Cav-1–GFP in normal (Fig. 2f)
or energy-depleted cells (Fig. 2g), as observed by total internal reflection
fluorescence (TIRF) microscopy. These findings are consistent with previously published immuno-electron microscopy experiments in which only
13–20% of viruses on the cell surface were associated with anti-Cav-1
gold37, and confocal microscopy experiments on Cav-1-expressing cells
in which only about 1 out of ten viruses associated with Cav-1, and few
entered together with Cav-1 (ref. 11). We concluded that SV40 induces the
formation of membrane invaginations without the help of active cellular
machinery or caveolar coats.
Interference with the actin cytoskeleton, depletion of cholesterol or
inhibition of tyrosine kinases in non energy-depleted cells also led to
SV40-VLP-induced tubule formation (Supplementary Information,
Fig. S2b), suggesting that these factors have a role in the scission process.
On the other hand, treatment of cells with the dynamin-inhibitor dynasore did not result in tubule formation (Supplementary Information,
Fig. S2b), indicating that the budding of SV40 binding-induced membrane invaginations is dynamin-independent, which is consistent with
previous reports11.
SV40 binding-induced membrane invaginations — properties
of GM1
To test whether SV40 binding to GM1 was sufficient to induce membrane invagination, we incubated fluorescently labelled VLPs with GUVs
made of a mixture of 1,2-dioleoylphosphatidylcholine (DOPC; 68 mol%;
spiked with 1 mol% BodipyFl‑C5-HPC), cholesterol (30 mol%) and
nt-GM1 (1 mol%). The VLPs bound to the GUVs, and within a few
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
A RT I C L E S
seconds membrane invaginations were formed that contained the VLPs
(Fig. 3a; Supplementary Information, Movie 1). These invaginations had
a similar morphology to those observed in cells and their formation did
not require the presence of cholesterol in the lipid mixture (data not
shown). Mouse polyomavirus-VLPs also induced tubular invaginations
on GUVs containing the ganglioside GD1a (Supplementary Information,
Fig. S3a), the polyomavirus cellular receptor 4,26,38.
When the different GM1 species were incorporated into GUVs,
addition of SV40-VLPs led to efficient binding in all cases. However,
VLPs failed to induce invaginations in GUVs that contained the two
short chain species C8 and DL (Fig. 3b, c). In contrast, lipid species with
longer acyl chains (DP, DO and DS — di-stearoyl-GM1, which could
not be incorporated into cells at non-toxic concentrations) supported
the formation of invaginations, independently of their saturation status
(Fig. 3d–f). These observations demonstrated that the structure of receptor lipid tails was essential for the virus-induced formation of invaginations on GUVs, as observed for endocytosis and infectivity in cells.
SV40 binding-induced membrane invaginations — properties of
SV40
In virus particles and VLPs, the VP1 molecules provide a surface with
360 regularly spaced binding sites for the GM1 glycan moiety. To test
whether the number and colloidal topology of binding sites was critical for tubule formation, we expressed and purified recombinant VP1
pentamers that are incapable of assembly into VLP capsids39. When fluorescence-labelled and incubated with energy-depleted CV‑1 cells, such
VP1 proteins induced long invaginations (Fig. 4a). CTxB, whose five
GM1-binding sites are arranged in an identical geometry to that found
on the SV40 VP1 pentamer 2, also induced invaginations (Fig. 4b). In
contrast, an antibody against cell-bound GM1 failed to induce invaginations, even when crosslinked by a secondary antibody (Fig. 4c).
These results indicated that the spatial organization of the GM1binding sites was critical for tubule formation. Crosslinking of GM1
via antibodies did not allow tubule formation, which required the pentavalent organization of binding sites, as found in VP1 and CTxB molecules. This was confirmed on GUVs containing nt-GM1. VP1 and CTxB
induced invaginations (Fig. 4d, e), whereas the crosslinked anti-GM1
antibody did not (Fig. 4f). Interestingly, VP1-induced tubule formation
was also sensitive to lipid structure in that tubules failed to form on
GUVs that were made with C8-GM1 (Fig. 4g). This result is similar to
that observed for infection (Fig. 1d), endocytosis (Fig. 1e, f) and VLPinduced tubule formation on GUVs (Fig. 3a, b).
Some differences could be observed between isolated VP1 pentamers or CTxB proteins and intact, colloidal viral capsids. The time lag
between ligand addition to GUVs and the formation of invaginations
was much longer for isolated pentamers (minutes) than for SV40-VLPs
(seconds). When we repeated these experiments in nt-GM1 containing
GUVs that were made from a lipid mixture that generates a more rigid,
liquid-ordered (lo) phase (40.7 mol% brain SM, 13.6 mol% cholesterol,
40.7 mol% DOPC and 5 mol% GM1), SV40-VLPs were still able to induce
membrane invaginations (Fig. 5a). However, CTxB failed to form tubules
under these conditions. Furthermore, SV40-VLPs induced tubules on
GUVs with a high membrane tension (data not shown), in contrast to
CTxB (data not shown) and Shiga toxin B‑subunit24. We concluded that
the pre-curved colloidal organization of GM1-binding sites on the virion
was not necessary for membrane deformation and tubulation, but it made
a
CTxB
b
SV40-pentamers
c
Anti-GM1
d
CTxB
DHPC
Merge
e
SV40-pentamers
f
Anti-GM1
g
SV40-pentamers
C8-GM1
Figure 4 Induction of membrane invaginations by GM1-binding pentamer units.
(a–c) Confocal microscopy images of energy-depleted CV‑1 cells incubated for
30 min with fluorescence-labelled SV40 VP1 pentamers (a), CTxB (b) or an
anti-GM1 antibody crosslinked with a fluorescence-labelled secondary antibody
(c). (d–f), Confocal sections of equatorial planes of GUVs made from a mixture
of 68 mol% DOPC, 30 mol% cholesterol, 1 mol% BodipyFl‑C5-HPC (green)
and 1 mol% of nt-GM1. GUVs were incubated with fluorescence-labelled (red)
CTxB (d), SV40 VP1 pentamers (e) or an anti-GM1 antibody crosslinked with
a fluorescent secondary antibody (f). (g) Confocal section of a GUV made from
a mixture of 64 mol% DOPC, 30 mol% cholesterol, 1 mol% BodipyFl‑C5-HPC
(green) and 5 mol% C8-GM1. GUVs were incubated with fluorescence labelled
SV40 pentamers (red). Scale bars, 5 μm.
the process more efficient, enabling it to overcome high membrane tension and rigidity.
In a previous study, we found that the pentameric Gb3 glycolipid-binding Shiga toxin B‑subunit (STxB) can induce tubule formation in cells and
GUVs with kinetics similar to the kinetics of tubule induction by SV40
pentamer and CTxB24. Like SV40 capsids, STxB required glycosphingolipid receptor species with long acyl-chains for tubule formation. However,
for STxB-induced membrane tubulation, the Gb3 acyl-chains needed to
be unsaturated, suggesting a difference in the underlying physical mechanism between the spherical capsids and the pentameric proteins.
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
15
A RT I C L E S
a
SV40
DHPC
Merge
SV40-VLP
DiI
Merge
nt-GM1
f
d
VLP
C8-GM1
C8-GM1
Fluorescence
intensity
Radial distance
j
VP1
C8-GM1
Fluorescence
intensity
Radial distance
e
DO-GM1
i
DP-GM1
VP1
nt-GM1
Fluorescence
intensity
c
h
DL-GM1
Radial distance
k
Radial distance
DS-GM1
VLP
nt-GM1
Fluorescence
intensity
g
b
Figure 5 Clustering and domain formation analysis. (a) Confocal sections
in the equatorial plane of GUVs made from a mixture of 40.7 mol%
brain-sphingomyelin, 13.6 mol% cholesterol, 40.7 mol% DOPC, 1 mol%
BodipyFl‑C5-HPC (green) and 5 mol% of nt-GM1. Under these conditions, GUV
membranes are in a homogeneous liquid-ordered state. Fluorescence-labelled
SV40-VLPs are shown in red. Scale bar, 10 μm. (b–e) Confocal sections in the
equatorial plane of GUVs made from a mixture of 68 mol% DOPC, 30 mol%
cholesterol, 1 mol% BodipyFl‑C5-HPC (green) and 1 mol% of either nt-GM1
(b, c) or C8-GM1 (d, e). Fluorescence intensity profiles of the GUV membranes
are plotted on the right. Peaks indicate areas of relative enrichment of VLP or
VP1 fluorescence. (f–k) 3D projections of GUVs formed from a lipid mixture of
33 mol% stearoyl-sphingomyelin, 33 mol% DOPC and 33 mol% cholesterol
that undergoes phase separation. The lipid analogue DiI (pseudocoloured
green, middle columns) was incorporated into the lipid mixture at 0.1 mol%
to specifically label the liquid-disordered (ld) phase and the nt-GM1 or
variant-GM1 species were incorporated at 0.1 mol% to investigate the phase
preference of bound fluorescence-labelled SV40-VLPs (pseudocoloured red,
left columns). While in GUVs containing nt-GM1 (f), DP‑GM1 (j) and DS‑GM1
(k), bound SV40-VLPs localized to the unlabelled liquid-ordered (lo) phase, in
GUVs containing C8-GM1 (g), DL‑GM1 (h) or DO‑GM1 (i) bound SV40-VLPs
localized to the DiI-labelled ld phase as evident from the merged images (right
columns). Scale bars, 10 μm.
A physical model for SV40 binding-induced membrane
deformation
The formation of membrane tubules by the aggregation of membranebound monomers (which may either be small proteins such as toxins or
VP1 pentamers, or large spherical capsids, Fig. 6a) can be understood
from a generic physical model that couples the thermodynamics of phase
separation to the mechanics of membrane deformation (Supplementary
Information,Theoretical model). Tubule nucleation and growth is controlled by the free energy difference, ∆FN = Eneck – N(∆e + kTlog ϕ1),
between N species being either aggregated in a tubule or isolated on the
membrane. Here, Eneck is the energy of the toroidal neck connecting the
tubule to the membrane (Fig. 6b), and ∆e and kTlog ϕ1 are the energy
gain and entropy loss, respectively, when a species joins a growing tubule
(ϕ1 is the surface fraction, concentration × species area, of the isolated
species on the membrane and kT ~ 2.5 kJ mol–1 is the thermal energy).
Tubules can form if the energetic gain overcomes the entropic loss, which
requires a sufficiently high species density on the membrane: ϕ1 > e–∆e/kT.
Tubules nucleate in a typical time controlled by a free energy barrier ∆Fc
(Fig. 6b), which is strongly influenced by the cost of membrane deformation in the tubule neck (Supplementary Information, Theoretical
model). The insensitivity of spherical VLP-induced tubule formation
to membrane tension and other factors such as the lipid tail saturation, as
opposed to what is observed for individual proteins, stems directly from
the properties of the energy difference ∆e, as discussed below.
Because of their intrinsic curvature, large spherical VLPs imprint
a close to tubular shape onto the membrane to which they adhere.
Membrane tension can influence the virus-induced membrane deformation and tubulation only to the extent that it prevents the membrane
16
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
A RT I C L E S
wrapping around the capsids, which requires tensions in the order of the
high adhesion energy; about 10kT per GM1 molecule40, or 10–3 J m–2.
Individual pentameric proteins on the other hand are fully bound to the
membrane, whether in tubules or not. Membrane tubulation is in this
case not driven by protein–lipid adhesion, but is the result of protein
clustering due to the existence of a line tension µ ~ 0.5 pN between lipiddomains of different composition and ordering 41. Individual pentamers
or toxins should not be able to form tubules if the membrane tension
is larger than the ratio of the line tension to the particle size (γ ~ µ/a,
Supplementary Information, Theoretical model), which we estimate to
be in the order of 10–4 J m–2. Furthermore, the kinetics of tubule nucleation is largely controlled by the tension-dependent energy of the tubule
neck, and we expect tubule nucleation to be slow when the line energy
associated with membrane deformation is larger than the line tension
of the clustered domain (√ κγ > μ, with membrane bending rigidity in
the order of κ ~ 20kT, Supplementary Information, Theoretical model),
which we estimate to be about 10–5 J m–2 (or smaller when the membrane
is in the more rigid lo-state and Shiga toxin fails to induce membrane
invaginations)42.
These considerations explain a number of experimental findings. SV40VLPs can induce tubules even on GUVs with a membrane tension in the
order of 10–3 J m–2, close to the lysis tension of a bilayer. In contrast, individual VP1 pentamers and toxins only form tubules in relatively ‘floppy’
vesicles with tensions in the order of 10–5 J m–2. Furthermore, these observations clarify why several minutes are required for tubule nucleation by
pentamers and CTxB, which is similar to what is observed with the Shiga
toxin system24, in comparison to seconds with SV40 capsids.
The structure of the receptor tail can influence two important membrane properties: the degree of lipid ordering, related to the line tension
of glycolipid receptor-enriched membrane domains, and mechanical
properties, expressed by the bending rigidity and spontaneous curvature.
Whereas the former property is crucial to the aggregation process of both
individual proteins (pentamers or toxins) and spherical VLP capsids, the
latter should only influence the tubulation induced by individual proteins, as adhesion of VLP s onto membranes is by itself sufficient to create
strong curvature. Our findings show that long acyl chains are required
for the induction of tubules by both capsids and individual pentamers,
strongly suggesting that the effect is mediated by line tension. Indeed,
both SV40-VLPs and VP1 pentamers form lateral aggregates shortly
after binding to nt-GM1-containing GUVs (Fig. 5b, c), whereas such
aggregates were not detectable in C8-GM1-containing GUVs (Fig. 5d, e).
Receptor tail length should also have a strong influence on the nucleation
of membrane phase separation. In agreement with this prediction, SV40
was found in lo phase membranes when associated with saturated long
chain GM1 molecules, and in the ld phase when GUVs were made with
short chain GM1 species (Fig. 5f). Saturated receptor chains were found
to prevent membrane invagination by shiga toxin24, but did not modify
the ability of the capsid to induce tubules. A likely explanation is that tail
saturation strongly couples to spontaneous negative curvature, possibly
by amplifying a head group compaction and chain splaying effect. This
property is crucial for membrane tubulation induced by small proteins24,
but dispensable in the case of intrinsically curved spherical capsids.
The picture emerging from the arguments summarized above and
detailed in the Supplementary Information is that although pentamers
and capsids share a common aspect related to lipid organization and
line tension for their clustering, the mechanisms that drive membrane
a
Line energy
Neck energy
b
No tubule
∆FN
∆Fc
Neck energy
Nucleation
Nc
Tubule size N
Tubule
Figure 6 Physical parameters controlling membrane invagination by SV40.
(a) Sketch of membrane invaginations and tubules induced by large
spherical capsids (right) and by the aggregation of small proteins (left)
after multivalent binding to glycolipid receptors. The lipid receptors are
specifically enriched in the contact area and thereby create an interface
between the receptor-enriched membrane (lilac) and the adjacent
cellular membrane. In the case of the viral particle, it is the shape of the
membrane-bound surface that drives the formation of an indentation and
the minimization of neck energy promotes tubule formation. (b) Free energy
difference between a tubule containing N particles and N independent
particles. Tubules grow if this energy decreases with N. The energy of
forming a tubular membrane neck around the tubule provides a barrier (∆Fc)
that slows tubule nucleation. Tubules form if the driving force overcomes the
loss of entropy and the forces opposing membrane deformation.
deformation are different. Capsids imprint membrane curvature through
adhesion, whereas small proteins must promote the emergence of a spontaneously curved membrane organisation.
DISCUSSION
From our study it seems that, unlike other viruses that rely on celldriven processes, SV40 initiates its internalization process by inducing
membrane curvature itself from the extracellular side of the membrane
through multivalent binding of its VP1 pentamers to cell surface GM1.
The association with caveolae, which is occasionally observed for SV40
but is not essential for entry or infection11,37,43,44, probably reflects a
preferred localization of the virus to membrane environments with
appropriate curvature radii, and does not seem to have a role in the
invagination process described here. Indeed, while Cav-1 expression
does not influence the infection efficiency of SV40, the structure of the
hydrocarbon chain of GM1 does so significantly (Fig. 1). Hence, what we
describe here is the infectious pathway of SV40. In cellular and artificial
membranes, the binding of SV40 to GM1 alone suffices to induce the
formation of invaginations. The subsequent membrane scission reaction to form an endocytic vesicle depends on signal transduction and
active cellular fusion factors7,9,29. If this scission reaction does not occur,
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
17
A RT I C L E S
membrane invagination can proceed to the formation of tubules. The
capacity of the virus to induce membrane invaginations is shared with
other polyomaviruses and with some bacterial toxins (Shiga and cholera
toxins). As they all bear a remarkably similar pentameric organization
for association with glycan moieties of glycosphingolipids, we suggest
that they exploit a common mechanism that integrates precise requirements in the hydrocarbon chains of the glycolipid receptors and a compact organization of receptor binding sites within the pentameric protein
scaffolds. These pentamers serve as nanoscale lipid-clustering devices
for membrane mechanical processes leading to the coat-independent
formation of endocytic membrane invaginations. METHODS
Methods and any associated references are available in the online version
of the paper at http://www.nature.com/naturecellbiology/.
Note: Supplementary Information is available on the Nature Cell Biology website.
Acknowledgements
This work was funded by the Swiss SystemsX.ch initiative, grant LipidX‑2008/011
(to A.H.), the Human Frontier Science Program (to A.E.S., L.J. and P.S.), a FEBS
fellowship (to H.E.), a CNRS fellowship (to W.R.), the Israel Science Foundation
(Grant # 604/07 to A.O.), the Delegation Generale pour l’Armement (to L.B.), the
Wellcome Trust (to W.C. and T.F.), Deutsche Forschungsgemeinschaft (to G.S.) and
the European Regional Development Fund grant # 4212/04‑01 (to K.B. and P.S.).
H.E. thanks D. Choquet for his patience and support. The authors thank M. AbdEl-Latif for the preparation of VLPs and pentamers.
Author contributions
H.E. and A.E.S. designed and performed the experiments involving infectious SV40;
W.R. and V.C. designed and performed tubulation experiments in cells; A.E.S., J.K.
and R.M. performed electron microscopy experiments; W.R., L.B., K.B. and Pe.S.
designed and performed GUV experiments; W.C., G.S. and T.F. provided GM1
species; A.O. provided SV40 VLPs; S.D. and P.S. performed theoretical analysis;
H.E., P.S., L.J. and A.H. wrote the manuscript; A.H. and L.J. supervised the work.
Competing financial interests
The authors declare no competing financial interests.
Published online at http://www.nature.com/naturecellbiology/
Reprints and permissions information is available online at http://npg.nature.com/
reprintsandpermissions/
1. Liddington, R. C. et al. Structure of simian virus 40 at 3.8-A resolution. Nature 354,
278–284 (1991).
2. Neu, U., Woellner, K., Gauglitz, G. & Stehle, T. Structural basis of GM1 ganglioside
recognition by simian virus 40. Proc. Natl Acad. Sci. USA 105, 5219–5224 (2008).
3. Campanero-Rhodes, M. A. et al. N-glycolyl GM1 ganglioside as a receptor for simian
virus 40. J. Virol. 81, 12846–12858 (2007).
4. Tsai, B. et al. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J.
22, 4346–4355 (2003).
5. Kukura, P., Ewers, H., Mller, C., Renn, A., Helenius, A. & Sandoghdar, V. High-speed
nanoscopic tracking of the position and orientation of a single virus. Nature Methods
6, 923–927 (2009).
6. Ewers, H. et al. Label-free optical detection and tracking of single virions bound to their
receptors in supported membrane bilayers. Nano Lett. 7, 2263–2266 (2007).
7. Pelkmans, L., Puntener, D. & Helenius, A. Local actin polymerization and dynamin
recruitment in SV40-induced internalization of caveolae. Science 296, 535–539
(2002).
8. Ewers, H. et al. Single-particle tracking of murine polyoma virus-like particles on
live cells and artificial membranes. Proc. Natl Acad. Sci. USA 102, 15110–15115
(2005).
9. Richards, A. A., Stang, E., Pepperkok, R. & Parton, R. G. Inhibitors of COP-mediated
transport and cholera toxin action inhibit simian virus 40 infection. Mol. Biol. Cell 13,
1750–1764 (2002).
10.Hummeler, K., Tomassini, N. & Sokol, F. Morphological aspects of the uptake of simian
virus 40 by permissive cells. J. Virol. 6, 87–93 (1970).
11.Damm, E. M. et al. Clathrin- and caveolin‑1‑independent endocytosis: entry of simian
virus 40 into cells devoid of caveolae. J. Cell Biol. 168, 477–488 (2005).
18
12.Kartenbeck, J., Stukenbrok, H. & Helenius, A. Endocytosis of simian virus 40 into the
endoplasmic reticulum. J. Cell Biol. 109, 2721–2729 (1989).
13.Schelhaas, M. et al. Simian virus 40 depends on ER protein folding and quality control
factors for entry into host cells. Cell 131, 516–529 (2007).
14.Torgersen, M. L., Skretting, G., van Deurs, B. & Sandvig, K. Internalization of cholera
toxin by different endocytic mechanisms. J. Cell Sci. 114, 3737–3747 (2001).
15.Sandvig, K. et al. Pathways followed by ricin and Shiga toxin into cells. Histochem.
Cell Biol. 117, 131–141 (2002).
16.Gilbert, J. et al. Ganglioside GD1a restores infectibility to mouse cells lacking functional
receptors for polyomavirus. J. Virol. 79, 615–618 (2005).
17.Jacobson, K., Mouritsen, O. G. & Anderson, R. G. Lipid rafts: at a crossroad between
cell biology and physics. Nature Cell Biol. 9, 7–14 (2007).
18.Hancock, J. F. Lipid rafts: contentious only from simplistic standpoints. Nature Rev.
Mol. Cell Biol. 7, 456–462 (2006).
19.Mayor, S. & Pagano, R. E. Pathways of clathrin-independent endocytosis. Nature Rev.
Mol. Cell Biol. 8, 603–612 (2007).
20.Lencer, W. I. & Saslowsky, D. Raft trafficking of AB5 subunit bacterial toxins. Biochim.
Biophys. Acta 1746, 314–321 (2005).
21.Parton, R. G. & Richards, A. A. Lipid rafts and caveolae as portals for endocytosis: new
insights and common mechanisms. Traffic 4, 724–738 (2003).
22.Hammond, A. T. et al. Crosslinking a lipid raft component triggers liquid ordered-liquid
disordered phase separation in model plasma membranes. Proc. Natl Acad. Sci. USA
102, 6320–6325 (2005).
23.Wolf, A. A. et al. Attenuated endocytosis and toxicity of a mutant cholera toxin with
decreased ability to cluster ganglioside GM1 molecules. Infect. Immun. 76, 1476–
1484 (2008).
24.Romer, W. et al. Shiga toxin induces tubular membrane invaginations for its uptake
into cells. Nature 450, 670–675 (2007).
25.Ichikawa, S., Nakajo, N., Sakiyama, H. & Hirabayashi, Y. A mouse B16 melanoma
mutant deficient in glycolipids. Proc. Natl Acad. Sci. USA 91, 2703–2707 (1994).
26.Smith, A. E., Lilie, H. & Helenius, A. Ganglioside-dependent cell attachment and endo‑
cytosis of murine polyomavirus-like particles. FEBS Lett. 555, 199–203 (2003).
27.Schwarzmann, G., Hoffmann-Bleihauer, P., Schubert, J., Sandhoff, K. & Marsh, D.
Incorporation of ganglioside analogues into fibroblast cell membranes. A spin-label
study. Biochemistry 22, 5041–5048 (1983).
28.Merritt, E. A. et al. Crystal structure of cholera toxin B-pentamer bound to receptor
GM1 pentasaccharide. Protein Sci. 3, 166–175 (1994).
29.Dangoria, N. S., Breau, W. C., Anderson, H. A., Cishek, D. M. & Norkin, L. C.
Extracellular simian virus 40 induces an ERK/MAP kinase-independent signalling
pathway that activates primary response genes and promotes virus entry. J. Gen. Virol.
77 ( Pt 9), 2173–2182 (1996).
30.Chai, W., Stoll, M. S., Galustian, C., Lawson, A. M. & Feizi, T. Neoglycolipid technology:
deciphering information content of glycome. Methods Enzymol. 362, 160–195 (2003).
31.Sandalon, Z. & Oppenheim, A. Self-assembly and protein-protein interactions between
the SV40 capsid proteins produced in insect cells. Virology 237, 414–421 (1997).
32.Kosukegawa, A. et al. Purification and characterization of virus-like particles and pen‑
tamers produced by the expression of SV40 capsid proteins in insect cells. Biochim.
Biophys. Acta 1290, 37–45 (1996).
33.Yan, Y., Stehle, T., Liddington, R. C., Zhao, H. & Harrison, S. C. Structure determination
of simian virus 40 and murine polyomavirus by a combination of 30-fold and 5-fold
electron-density averaging. Structure 4, 157–164 (1996).
34.Richterova, Z. et al. Caveolae are involved in the trafficking of mouse polyomavirus virions
and artificial VP1 pseudocapsids toward cell nuclei. J. Virol. 75, 10880–10891 (2001).
35.Gilbert, J. M. & Benjamin, T. L. Early steps of polyomavirus entry into cells. J. Virol.
74, 8582–8588 (2000).
36.Kusumi, A. et al. Paradigm shift of the plasma membrane concept from the two-dimen‑
sional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of
membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34, 351–378 (2005).
37.Stang, E., Kartenbeck, J. & Parton, R. G. Major histocompatibility complex class I mol‑
ecules mediate association of SV40 with caveolae. Mol. Biol. Cell 8, 47–57 (1997).
38.Stehle, T., Yan, Y., Benjamin, T. L. & Harrison, S. C. Structure of murine polyoma‑
virus complexed with an oligosaccharide receptor fragment. Nature 369, 160–163
(1994).
39.Roitman-Shemer, V., Stokrova, J., Forstova, J. & Oppenheim, A. Assemblages of simian
virus 40 capsid proteins and viral DNA visualized by electron microscopy. Biochem.
Biophys. Res. Commun. 353, 424–430 (2007).
40.Turnbull, W. B., Precious, B. L. & Homans, S. W. Dissecting the cholera toxin-gan‑
glioside GM1 interaction by isothermal titration calorimetry. J. Am. Chem. Soc. 126,
1047–1054 (2004).
41.Baumgart, T., Hess, S. T. & Webb, W. W. Imaging coexisting fluid domains in biomem‑
brane models coupling curvature and line tension. Nature 425, 821–824 (2003).
42.Windschiegl, B. et al. Lipid reorganization induced by Shiga toxin clustering on planar
membranes. PLoS ONE 4, e6238 (2009).
43.Pelkmans, L., Kartenbeck, J. & Helenius, A. Caveolar endocytosis of simian virus 40
reveals a new two-step vesicular-transport pathway to the ER. Nature Cell Biol. 3,
473–483 (2001).
44.Anderson, H. A., Chen, Y. & Norkin, L. C. Bound simian virus 40 translocates to cave‑
olin-enriched membrane domains, and its entry is inhibited by drugs that selectively
disrupt caveolae. Mol. Biol. Cell 7, 1825–1834 (1996).
nature cell biology VOLUME 12 | NUMBER 1 | JANUARY 2010
© 2010 Macmillan Publishers Limited. All rights reserved.
METHODS
DOI: 10.1038/ncb1999
METHODS
Cell culture and Reagents. GM95 are a ceramide-glucosyl-transferase-deficient
mouse melanoma cell line, and therefore do not express glucose-based gangliosides25. Cav‑1–/– cells are embryonic fibroblasts derived from a Cav‑1-knockout
mouse45. CV‑1 and murine 3T6 Swiss albino fibroblasts were obtained from the
American Type Culture Collection. All cells were kept in DMEM supplemented
with 10% FCS and 4 mM Glutamax (culture medium) and maintained at 37 ºC in
5% CO2. Di-oleoyl-phosphatidylcholine, cholesterol, sphingomyelin and nt-GM1
were from Avantilipids. Fluorescence-labelled CTxB and transferrin, DiIC18,
DiOC18, Bodipy-FL‑C5-DHPC and latrunculin A were from Invitrogen. The
anti-Cav‑1 antibody was rabbit N20 anti-Cav‑1 from Santa Cruz.
GM1 glycolipids. The native (nt) GM1 (Avanti Polar Lipids) had either an 18 or
20 carbon-atom sphingosine base that was monounsaturated near the headgroup
(C18:1, C20:1), and a fully saturated 18 carbon-atom fatty acid chain (C18:0) as
specified by the supplier and verified by electrospray mass spectrometry. For
greater flexibility in design, the GM1 pentasaccharide was coupled to phosphatidylethanolamine (PE). Di-oleoyl-PE, which has two monounsaturated C18 chains
within the plane of the bilayer (C18:1), di-lauroyl-PE which has two short saturated acyl chains (C12:0), di-palmitoyl-PE (C16:0) and di-stearoyl-PE (C18:0)
were used to yield DO‑GM1, DL‑GM1, DP‑GM1 and DS‑GM1, respectively.
Preparation of neoglycolipids. Neoglycolipids of the GM1 pentasaccharides
(Accurate Chemicals) were prepared with four PEs, 1,2-dilauroyl-sn-glycero‑3phosphoethanol amine (DLPE, Sigma), 1,2-dipalmitoyl-sn-glycero‑3-phosphoethanolamine (DPPE, Sigma), 1,2-dioleoyl-sn-glycero‑3-phosphoethanolamine
(DOPE, Fluka) and 1,2-distearoyl-sn-glycero‑3-phosphoethanolamine (DSPE,
Sigma) essentially as described previously 30. Briefly, water (5 μl), phospholipid
(100 μl, 5 μg μl–1 in CHCl3/MeOH 1:1, by vol) and freshly prepared tetrabutylammonium cyanoborohydride solution (20 μl, 20 μg μl–1 in MeOH) were added to
100 nmol of lyophilized pentasaccharide. The reaction mixture was incubated at
60 °C for 60 h. The neoglycolipid products were analysed by high-performance
thin-layer chromatography (TLC), purified using silica cartridges (Waters SepPak, 500 mg silica) and quantified based on orcinol-hexose response30 using the
neoglycolipid of lactoneotetraose as a standard.
Mass spectrometric analyses of glycolipids and neoglycolipids. Molecular
masses of the neoglycolipids of GM1 pentasaccharide were determined by
matrix-assisted desorption/ionization mass spectrometry on a TOF SPEC 2E
instrument (Micromass) using 2,4,6-trihydroxyacetophenone as the matrix.
Their carbohydrate sequence and lipid identity were verified by electrospray
tandem mass spectrometry (ES-MS/MS) with collision-induced dissociation
(CID)46, carried out on a Q‑TOF instrument (Micromass). ES‑CID-MS/MS
nitrogen was used as desolvation and nebulizer gas at a flow rate of 250 l h–1
and 15 l h–1, respectively. Source temperature was 80 ºC and the desolvation
temperature 150 °C. The capillary voltage was maintained at 3 kV and cone
voltage at 50 V. Product-ion spectra were obtained from CID with argon as the
collision gas at a pressure of 1.7 bar. The collision energies for fragmentation
were 60–65 V. For the analysis, samples were dissolved in CHCl3/MeOH/H2O
25:25:8, at a concentration of 25 pmol μl–1, 5 μl of which were loop-injected. The
same solvent was used as mobile phase and delivered by a Harvard syringe pump
(Harvard Apparatus) at a flow rate of 5 μl min–1.
SV40 virus purification. The protocols for virus purification were based on
those described previously 47. Briefly, monkey kidney CV‑1 cells were cultured in
complete medium: Dulbecco’s Modified Eagle’s Medium (DMEM from Gibco)
supplemented with 10% fetal calf serum (FCS, LabForce), 4 mM GlutaMAX, and
50 mM HEPES (Gibco) at 37 °C in 5% CO2. Forty T175 flasks of sub-confluent
CV‑1 cells were infected with SV40 at a multiplicity of infection (MOI) of 0.01.
Cells were cultured in complete medium for 14 days. To collect virus, cells were
put through three freeze-thaw cycles, and then centrifuged at 10,000g for 10 min
at 4 °C. Virus-containing supernatant (30 ml) was loaded on a 10-ml cushion of
CsCl (1.4 g ml–1) in 10 mM HEPES at pH 7.4. Following centrifugation at 76,000g
for 3 h at 4 °C in a SW28 rotor (Beckman), the banded virus in the CsCl cushion
was collected. The density of the CsCl fraction containing SV40 was checked
and the fraction was adjusted to a CsCl density of 1.34 g ml–1 in 10 mM HEPES
at pH 7.4. Following equilibrium centrifugation at 100,000g for 16 h at 4 °C in a
70.1 Ti rotor (Beckman), the lower virus band was isolated, and dialysed against
50 mM HEPES at pH 8.0, 150 mM NaCl and 1 mM CaCl2 (virus buffer). The
purified infectious virus was stored in aliquots at –80°C.
Virus-like particle (VLP) Purification. SV40-VLPs were generated as described
previously 32 from lysates of SF9 cells that expressed SV40 VP1. Lysates contained
fully assembled SV40-VLPs as well as assembly intermediates and unassembled
VP1 protein. To purify the fully assembled VLPs, lysates were clarified by centrifugation for 30 min at 10,000g in an Eppendorf microfuge. The clarified supernatant (0.5 ml) was centrifuged at 4 °C for 2.5 h at 160,000g using a SW41Ti rotor
(Beckman) through a 5–20% (w/v) linear sucrose gradient with a 0.5 ml 60%
(w/w) sucrose cushion in 10 mM HEPES at pH 8.0, 200 mM NaCl and 1 mM
CaCl2. After fractionation, 0.5 ml fractions were analysed by transmission electron
microscopy following negative staining, and fractions with homogenous intact
particle populations were pooled. The purified particles were dialysed against
virus buffer and stored at –80°C. In later experiments, the VLPs were collected
from the medium of baculovirus-infected Sf9 following cell lysis, 5 days postinfection, as follows: intact cells and cell debris were removed by centrifugation
at 6,000g for 10 min. The supernatant was further clarified at 17,000g for 20 min.
VLPs were precipitated at 80,000g for 3 h. The VLP pellet was suspended in 0.5 M
NaCl, purified by ultrafiltration and treated as above.
Labelling of SV40-VLPs. Cy‑5 (Amersham Biosciences), atto‑565 (atto-tec),
FITC (Sigma) or biotin (Pierce) were covalently coupled to SV40-VLPs in 0.2 M
NaHCO3 at pH 8.2 using a 10-fold molar excess of the dye relative to VP1 protein,
and resulting in less than 200 fluorophore and/or biotin molecules per VLP as
determined according to the manufacturer’s instructions. Unbound dye was
removed by chromatography with a Nap‑5 column (Amersham Biosciences).
The modified SV40-VLPs were able to bind, enter and traffic within CV‑1 cells
to the same extent as non-modified infectious SV40 as described previously 6
(data not shown).
Supplementation of GM95 cells. Lipid supplementation was as described previously 48, adapted from a protocol49. GM95 cells were grown in Lab-tek 8‑well
coverglass culture chambers for three days. Cells were washed with serum-free
culture medium and incubated in 200 μl of serum-free culture medium in the
presence of nt-GM1 (19 μM), C8-GM1 (7 μM), DL-GM1 (1.25 μM), DO‑GM1
(1.25 μM) or DP‑GM1 (10 μM). Because lipid incorporation into cellular membranes is strongly influenced by its hydrocarbon chains, lipid-addition conditions were standardized to allow equivalent levels of plasma membrane GM1 as
detected by CTxB plasma membrane staining, as detailed below. Cells were then
cultured at 37 °C in 5% CO2 for 48 h to allow uptake and incorporation of the
glycolipids. To remove unincorporated glycolipids, the cells were then cooled to
4 ºC, and washed with DMEM supplemented with 25% FCS, 4 mM GlutaMAX
and 50 mM HEPES at pH 7.4.
Quantification of membrane-incorporated GM1. After lipid addition with each
GM1 species, live cells were incubated for 15 min with 1 μg ml–1 CTxB–Cy5
in phosphate buffered saline, washed twice with phosphate buffered saline and
imaged with a Zeiss LSM 510 confocal microscope. Settings were adjusted such
that the strongest staining observed throughout the experiment just saturated a
few pixels to ensure a high dynamic range. Confocal slices were taken 2 μm above
the substrate and the fluorescence in a line perpendicular to the cell membrane
was quantified. The highest intensity pixels in the peaks representing the cellular
plasma membrane on both ‘sides’ of the cell were compared, to yield unambiguous
quantitative binding data.
Analysis of SV40 infection. For infection assays, cells were incubated with infectious SV40 virus at an MOI of 200 (as judged by plaque assay in reference host
CV1 cells) in inoculation media or with inoculation medium only as a negative
control, in 200 μl for 2 h at 37 ºC. Cells were then washed with serum-containing
phenol-red free culture medium to remove unbound virus and were maintained
in phenol-red free culture medium at 37 ºC in 5% CO2 for an additional 18 h. Cells
were fixed with paraformaldehyde 4% (w/v) and viral T‑antigen expression was
immuno-detected by epi-fluorescence microscopy to identify SV40-infected cells.
Hoechst (Sigma) staining of the nuclei was used to determine the total number of
cells per field. With the virus load of MOI = 178, 72.1% of 3T6 cells were infected
nature cell biology
© 2010 Macmillan Publishers Limited. All rights reserved.
METHODS
DOI: 10.1038/ncb1999
(data not shown). The cell infection rate in the nt-GM1-supplemented GM95 cell
line was 76.6% for a MOI = 200. Infection was normalized to the rate in nt-GM1supplemented GM95 cells.
Analysis of SV40 infection in the presence of drugs. After lipid addback, cells
were incubated for 1 h with 0.1 mM genistein, 0.1 μM latrunculin A, 0.1 μM
jasplakinolide, 1 μM nocodazole, 5 mM methyl‑β-cyclodextran or DMSO in
inoculation medium. SV40 was added in inoculation medium supplemented with
the respective drugs at an MOI of 200. After 20 h of incubation at 37ºC, cells were
processed and quantified as indicated above.
Fluorescence recovery after photobleaching (FRAP). For FRAP, we bound
0.5 μg ml–1 of FITC–CTxB to supplemented cells for 15 min, washed with PBS
and photobleached a circular area of 2.7-μm in diameter by 10 iterations of
the 488 nm laser of a Zeiss LSM 510 confocal microscope set to 6 A at 100%
output. Fluorescence recovery was imaged at 0.1% laser output for 20 consecutive frames.
Energy depletion of cells and tubule formation. Cellular energy was depleted by
incubating HeLa cells in PBS++ supplemented with 10 mM 2‑deoxy‑D-glucose and
10 mM NaN3 for 30 min at 37 °C. Residual ATP levels were 2.1% under energyblock conditions, similar to a previous report50. HeLa, CV‑1 and Cav-1–/– cells
were depleted in cellular energy for 30 min at 37 °C and further incubated with
SV40-VLPs (25–250 μg ml–1) and SV40-Pentons (10 μg ml–1) for 1 h at 37 °C in
energy depletion medium enriched in BSA (0.1%). Cells were fixed in PFA 4% at
37 °C and imaged on a Leica SP2 confocal microscope.
Preparation of giant unilamellar vesicles (GUVs). GUVs were grown using the
electroformation technique as previously described24, 51. Lipid mixtures containing the different GM1 species were prepared in chloroform/methanol with the
appropriate lipid ratio. Vesicles were grown in a solution of sucrose. The size of
observed vesicles ranged between 5 and 50 μm. GUVs were transferred into a
chamber containing various concentrations of labelled SV40-VLPs, SV40 pentamers, Py-VLPs, CTxB or anti-GM1 antibody in PBS buffer for invagination
experiments. The difference in density between the outside (PBS) and the inside
of the vesicles (sucrose) led to their sedimentation. Phase-separated GUVs were
prepared in a flow chamber and labelled. SV40-VLPs were added to VLP buffer
(10 mM HEPES at pH 6.8, 133 mM NaCl and 2 mM CaCl2, osmolality adjusted to
330 mOsm kg–1, with sucrose). Live-cell imaging of cells or GUVs was performed
using a Zeiss LSM 510 confocal microscope. Observations were made at room
temperature and/or at 37 °C.
Fluorescence analysis of clustering of VLP and VP1 on GUVs. Radial analysis
of fluorescence of VLP and VP1 at the equatorial plane of GUVs was performed
with the oval profile plot of software package ImageJ. After masking the fluorescent
signal only at the membrane, a circular region of interest was defined surrounding the vesicle. Starting from the centre of the circular region, we defined n = 360
equally spaced radial sections and added the pixel intensity values for the respective
conditions: SV40-VLPs and SV40 VP1 pentamers with C8-GM1 and nt-GM1.
The standard deviation is defined as
the nth point, and x the mean.
√Σ
(x – x)2
where x is the value of
(n – 1)
Preparation for electron microscopy. Cell were fixed in 2.5% glutaraldehyde
(0.05 M sodium cacodylate, pH 7.2, 50 mM KCl, 1.25 mM MgCl2 and 1.25 mM
CaCl2) for 20 min at room temperature followed by 1 h in 2% OsO4. After washing in distilled water cells were stained with 0.5% uranyl acetate for 18 h. Further
sample preparation was according to standard protocols.
TIRF microscopy of SV40 on HeLa Cav-1–GFP cells. HeLa cells stably expressing Cav-1–GFP were plated in 8-well coverglass (Nunc) and imaged in PBS, 0.2%
BSA. TIRF microscopy was performed in the light microscopy facility of the ETH
Zurich (LMC) on an inverted Leica DMI 6000B system with a 100× 1.47NA
objective. Atto‑565-labelled SV40-VLPs were added to the media and imaged
right after binding to cells in TIRF mode with 90-nm penetration depth of both
the 488 nm and the 561 nm laser lines. Time sequences were acquired to ensure
detection of colocalization.
45.Drab, M. et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293, 2449–2452 (2001).
46.Chai, W. et al. Analysis of chain and blood group type and branching pattern of sialylated
oligosaccharides by negative ion electrospray tandem mass spectrometry. Anal. Chem.
78, 1581–1592 (2006).
47.Cole, C. N. & Cozen, S. D. in Polyomaviridae: the viruses and their replication (ed.
Knipe, D. M. & Howley, P. M. ) 2141–2229 (Lippincott-Raven Publishers, 2001).
48.Feizi, T., Stoll, M. S., Yuen, C. T., Chai, W. & Lawson, A. M. Neoglycolipids: probes of
oligosaccharide structure, antigenicity, and function. Methods Enzymol. 230, 484–519
(1994).
49.Facci, L. et al. Promotion of neuritogenesis in mouse neuroblastoma cells by exogenous
gangliosides. Relationship between the effect and the cell association of ganglioside
GM1. J. Neurochem. 42, 299–305 (1984).
50.Zha, X. et al. Sphingomyelinase treatment induces ATP-independent endocytosis. J.
Cell Biol. 140, 39–47 (1998).
51.Mathivet, L., Cribier, S. & Devaux, P. F. Shape change and physical properties of giant
phospholipid vesicles prepared in the presence of an AC electric field. Biophys. J. 70,
1112–1121 (1996).
nature cell biology
© 2010 Macmillan Publishers Limited. All rights reserved.
s u p p l e m e n ta r y i n f o r m at i o n
Ewers et al., Supplementary Figure 1
a
b
0s
19.2 s
45.4 s
92.2 s
100
80
60
40
20
0
20
66
200
Multiplicity of Infection
d
100
60
40
20
0
0.1
1
10
GM1 input [µM]
nt-GM1
C8-GM1
GM95
GM1
no GM1
nt-GM1
C8-GM1
DP-GM1
DO-GM1
f
DL-GM1
e
CTxB binding
Fluorescence Intensity
(AU)
80
GM95
Figure S1 Reconstitution of SV40 infection in GM95 cells. (a) Confocal
time-series of intensity images of FITC-labeled CTxB bound to nt-GM1 in
supplemented GM95 cells. CTxB-FITC fluorescence is photobleached. The
fluorescence recovery within the photobleached area indicates exchange
with unbleached molecules by lateral diffusion in the plane of the plasma
membrane. Scale bar is 10 µm. (b) GM95 cells were supplemented with
nt-GM1 and infected with SV40 at varying multiplicities of infection (MOIs).
SV40 infection was scored by immunofluorescence detection of SV40
T-antigen expression in the nucleus. n = 3 (c) Different concentrations of
the nt-GM1 and C8-GM1 species were supplemented to GM95 cells and
cells were infected with SV40. Shown are the absolute GM1 concentrations
in µM used for supplementation. The standard concentrations used in this
C8
DLPE
DPPE DOPE
SV40 binding
Fluorescence Intensity
(AU)
c
Cells expressing T-ag (%)
31.8 s
25.5 s
Cells expressing T-ag (%)
DOI: 10.1038/ncb1999
GM1
C8
DLPE
DPPE DOPE
work that yield equivalent binding of CTxB are highlighted in red. (d-f)
Cell surface levels of GM1 in supplemented GM95 cells. GM95 cells were
supplemented with the various GM1 species as indicated and fluorescencelabeled CTxB (d) or SV40 VLPs (e, images, f, quantification) were bound.
Cells were imaged in a Zeiss LSM 510 confocal microscope. All conditions
were imaged under identical settings and the membrane-associated
fluorescence was quantified in arbitrary units (AU). Shown are averages of
experiments with 95% confidence interval and total average. No significant
difference in binding of CTxB or SV40 between any species and nt-GM1 was
found (t-test). GM95 cells supplemented with all GM1 species bound CTxB
and SV40 significantly better than unsupplemented cells (p < 0.01, t-test).
Scale bars are 5 µm.
www.nature.com/naturecellbiology
1
© 2010 Macmillan Publishers Limited. All rights reserved.
s u p p l e m e n ta r y i n f o r m at i o n
a
b
control
methyl-ß-cyclodextrin
genistein
nocodazole
latrunculin A
jasplakinolide
dynasore
c
1
1
2
2
Figure S2 Tubule formation in cells. a, Cav -/- MEF cells were depleted of
cellular energy and thereafter incubated with fluorescence-labelled SV40
VLPs for 30 min at 37ºC. Cells were then fixed and imaged on a Leica SP2
confocal microscope. Shown are 6 confocal slices from a single cell (right),
a maximal-intensity z-projection of all (37) slices (bottom left) and an x-z
rotation of the cell (top left). Scale bars are 10 µm. b, HeLa cells were treated
with inhibitors as indicated (methyl-ß-cyclodextrin, 5 mM, 1 h; genistein,
100 µM, 20 min; latrunculinA, 0.1 µM, 20 min; jasplakinolide, 0.1 µM,
2
1 h; nocodazole, 1 µM, 1 h; dynasore, 80 µm, 30 min) and subsequently
incubated with fluorescence-labeled SV40-VLPs for 30 min at 37ºC in
presence of inhibitors. Cells were then fixed and imaged on a Leica SP2
confocal microscope. Areas of interest are enlarged in the bottom left.
Scale bar is 10 µm. c, CV-1 cells were energy depleted and incubated with
fluorescence labeled SV40 VLPs (red) for 1h. Cells were then fixed and
immunostained for caveolin-1 (green) and imaged. Shown are a cell (left) and
two regions of interest with SV40 containing tubules. Scale bar is 10 µm.
www.nature.com/naturecellbiology
© 2010 Macmillan Publishers Limited. All rights reserved.
s u p p l e m e n ta r y i n f o r m at i o n
Ewers et al., Supplementary Figure 3
a
Py
DHPC
Overlay
SV40 VLP
DHPC
Overlay
b
Figure S3 Membrane tubulation by Py and membrane tubulation in low
membrane tension vesicles. a, Shown is an equatorial confocal slice of
a GUV made from a lipid mixture containing 68 mol% DOPC, 30 mol%
cholesterol, 1 mol% GD1a and 1 mol% Bodipy-FL-C5-HPC (pseudocolored
green). Fluorescence-labeled Py-VLPs (pseudocolored red) were added and
the GUVs were imaged on a Zeiss LSM 510 confocal microscope. Scale bar
is 10 µm. b, Shown is an equatorial slice of a GUV made from a lipid mixture
containing 64 mol% DOPC, 30 mol% cholesterol, 5 mol% GM1 and 1 mol%
Bodipy-FL-C5-HPC (pseudocolored green). Alpha-hemolysin (0.1 mg/mL)
was added to reduce membrane tension. Fluorescence-labelled SV40-VLPs
(pseudocolored red) were added and the GUVs were imaged on a Zeiss LSM
510 confocal microscope. Scale bar is 5 µm.
www.nature.com/naturecellbiology
3
© 2010 Macmillan Publishers Limited. All rights reserved.
s u p p l e m e n ta r y i n f o r m at i o n
Supplementary Movie Legends
Movie S1 Confocal time-series of fluorescence labeled SV40-VLPs (red) bound to a GUV made from a mixture of 1,2-dioleoylphosphatidylcholine (DOPC; 68
mol%; spiked with 1 mol% Bodipy-FL-C5-HPC), cholesterol (30 mol%) and 1 mol% nt-GM1. Shown is an equatorial section of a GUV in a time-series with a
frame-rate of ~ 2 Hz. Field of view is 45 µm by 45 µm.
4
www.nature.com/naturecellbiology
© 2010 Macmillan Publishers Limited. All rights reserved.
Supplementary Information
Theoretical model
We review some theoretical concepts that link aggregation of proteins or virus-like
particles (VLPs) to the formation of membrane tubules.
Tubule statistics.
The thermally driven aggregation of membrane-bound monomers (proteins or capsids) is
controlled by the free energy difference ΔFN between an aggregate containing N
monomers and N isolated monomers freely diffusing on the membrane1:
" F N = Eneck # N(kT log $1 + "e) (1)
where Eneck is the aggregate's interfacial energy (e.g. the energy of the membrane neck
!
connecting a tubular aggregate to the main membrane). The free energy includes the loss
of monomer entropy upon aggregation that varies as the log of the monomer
concentration (φ1 is the surface fraction occupied by isolated monomers, and kT is the
thermal energy), and the energy gain (per monomer) Δe between an isolated monomer
and a monomer in an aggregate. Aggregation occurs for a critical density of monomers
φ*1 for which the free energy is a decreasing function of the aggregate size. The free
energy difference typically shows an energy-barrier ΔFc for short tubules of critical size
Nc. This energy barrier is due to the neck energy and is influenced both the line tension
between membrane domains of different composition and by the energy of deformation
of the membrane neck (see Figure 4). Aggregate nucleation occurs after an average
nucleation time τc that varies exponentially with the height of the barrier2
*
1
" ~e
!
#
$e
kT
; %c ~ e
$Fc
kT
(2)
1
© 2010 Macmillan Publishers Limited. All rights reserved.
Energetics.
The energy difference (per monomer) in and out of a tubule can be decomposed in three
parts: Δe = Δeadhes + Δecomp + Δedeform, including respectively the adhesion energy between
membrane and monomer, the energy from inhomogeneities of membrane composition
(e.g. receptor enrichment near the monomer), and the energy of membrane deformation.
Two size-scales are important in the problem: the size of the monomer a, and the radius
of the tubule R. For complete viral capsids a ~ R (see Figure 2b), while for individual
pentamers (or toxins), a << R (a ~R/5 for a virus of 50 nm diameter containing 72
pentamers). A third length-scale may play an important role. It is the inverse spontaneous
curvature C0 of the membrane adhered to the monomer, which is asymmetrically enriched
in receptors. The energy scales of the problem are given by the membrane-monomer
adhesion energy w (of order to 2 kT per GM1 receptor3), the membrane bending rigidity
κ, and the membrane tension γ. Since membrane binding occurs via specific receptors,
membrane composition-gradients near the sites adhered to monomers cause a line-energy
proportional to a line tension µ (the cost of creating an interface of unit length between
different membrane compositions) which increases with the degree of membrane order
near the monomer. Expected values for these parameters are given in Table 1:
energy scale for
physical parameter
-3
capsid
(radius a = 5 nm)
(radius R = 50 nm)
Adhesion energy
W = 10 J/m
wπa = 20 kT
w4πR2 = 2000 kT
Bending rigidity
κ = 20 kT
2πκ a2/R2 = 5 kT
8πκ = 500 kT
Line tension
µ = 0.4 pN = 0.1 kT/nm
µ2πa = 3 kT
µ2πR = 15 kT
Surface tension
γ = 10-6 – 10-3 J/m2
γπa2 = 0.02 – 20 kT
γ4πR2 = 2 – 2000 kT
"# = 0.1 – 2 pN
2πa "# = 2-11 kT
2πR "# = 70 – 350kT
Deformation line tension
Membrane decay length
!
λ=
2
monomer
2
" / # = 300 – 10 nm a/λ = 0.02 – 0.5
!
!
!
Table 1: Numerical values of the parameters
2
© 2010 Macmillan Publishers Limited. All rights reserved.
R/λ = 0.1 -3
Aggregation of small monomers.
Small monomers of size a are entirely covered by membrane, whether located to a tubule
or not, hence the adhesion energy does not play a role: Δeadhes = 0. If neither monomers
nor the bound membrane possess intrinsic curvature, monomers initially aggregate into
circular domains (as seen for Shiga toxin4). This aggregation process results only from
the balance between line energy and entropy, and is characterized by the free energy
difference (valid for N >> 1)
Flat domains: " F N = 2#µa N $ N(2#µa + kT log %1) (3)
Domains should steadily grow beyond a nucleation size Nc, which corresponds to the
!
maximum of this energy. The typical delay time to cross this energy barrier τc increases
exponentially with the energy barrier:
"c = "De
with
"D =
#aµ Nc
kT
%
(2
#µa
Nc = '
*
& 2#µa + kT log $1 ) (4)
2
#a!
10%4
~
D$ 1
$1 seconds as the typical time for a diffusion-limited encounter
between two proteins (for a diffusion coefficient D ~ 1 µm2/s). The nucleation size and
*
#2 $µa / kT
~ 4% , below which no
! the delay time diverge at the critical concentration "1 = e
aggregate can form at equilibrium. For larger concentrations, the delay time drops very
fast for increasing concentration, and is shorter than 1 sec for φ1 > 6%.
!
Line tension alone can force a membrane domain to curve with a radius of curvature of
order 4πκ/µ (~ 1 µm, see Table 1)5. The membrane tubules observed in this work and in4
have much higher curvature (R ~ 50 nm), and must rely on another mechanism. It is
likely to be spontaneous curvature, either of the monomer itself, or of the membrane
bound to it. The formation of a tubule is costly in terms of membrane tension, increasing
3
© 2010 Macmillan Publishers Limited. All rights reserved.
the energy by an amount γπa2 per monomer. The free energy Eq. (1) for tubules thus
reads:
2
Tubular domains: " F N = Eneck # N(2$µa # $a % + kT log &1) (5)
where the neck energy includes both compositional and deformation energy. In its
!
simplest form, valid for λ << R and N >> 1, it is Eneck " 2#a N (µ + $% /2) up to a hemispherical aggregate shape (N ~ (R/a)2) and Eneck ≅ constant for well-formed tubules. In
Eq.(5), we have neglected the energy of membrane deformation of individual proteins
!
gained when protein aggregates (Supplementary Figure 11), which is smaller than the
surface tension contribution by a factor at least of order (a/R)2 << 16.
The critical nucleation size and time are now given by:
"c = "De
#a( µ + $% / 2) Nc
kT
2
(
#a(µ + $% /2) +
Nc = *
2
) 2#µa + kT log &1 ' #a % , (6)
*
# ( $a
This defines
a critical monomer concentration for tubule formation "1 = e
!
-4
2
%2µa )/ kT
,
2
which cannot be reached for γ > 2µ/a ~ (~10 J/m ). At smaller membrane tensions,
tubules can form principally, but the nucleation time might be prohibitively long due to
!
the deformation energy of the neck, which starts playing an important role for γ > 4µ2/κ ~
10-5 J/m2.
Aggregation of Virus-Like-Particles.
The degree of membrane wrapping around a spherical particle (the ratio of the adhered
'2
#$2
w = w " & " C0 ) 7
( .
2%R
area over the colloid surface area) is of order w / " , with
Membrane deformation is driven by the adhesion energy, and is prevented under high
!
!
4
© 2010 Macmillan Publishers Limited. All rights reserved.
tension: γ > w . The existence of spontaneous membrane curvature can help the
deformation by increasing the effective adhesion energy w , but is not required. The
formation
of membrane tubules following colloid aggregation does not necessitate large
!
membrane deformation, since the membrane wrapped around a VLP is already curved.
!
The main limitation to tubule formation by VLPs should thus be that the membrane
actually wraps a sizable fraction of a VLP. Once this is achieved, colloid aggregation can
be driven by the membrane line tension, or by curvature-mediated attraction between
colloids5. This sets the maximal tension to be in the order of the binding energy γ < w
~10-3 J/m2. One can also expect that very low membrane tension would favor fullywrapped, isolated capsids rather than tubular aggregates. We have tested this prediction
by permeabilizing GUVs by addition of α-hemolysin resulting in extremely floppy
vesicles indicating strongly reduced membrane tension. However under these conditions
membrane tubulation could still be observed suggesting that residual membrane tension
was preventing full wrapping of SV40-VLPs (Supplementary Fig. 3b).
Theoretical predictions.
The discussion above shows that aggregation of small toxins and pentamers is promoted
by line tension, but tubulation requires spontaneous curvature and should be highly
sensitive to the level of membrane tension. Tubules cannot form in vesicles or cells with
γ > 2µ/a ~10-4 J/m2 and their nucleation should be slow for γ > 4µ2/κ ~10-5 J/m2.
Aggregation of capsids is also expected to be driven by line tension, but does not require
spontaneous curvature, and can proceed under conditions of much higher membrane
tension γ < w ~10-3 J/m2, because the adhesion energy provides the driving force for
membrane deformation. These predictions are a good qualitative agreement with our
observations. Indeed, the line tension is strongly dependent on differences of lipid order.
Long GM1 acyl chains are known to promote the liquid-ordered (lo) membrane phase,
and are required for the lateral aggregation of and subsequent membrane tubulation by
both capsids and toxins. Saturation of the lipid tails, on the other hand, is thought to
modify the spontaneous curvature of the membrane (i.e. via the “lipid compaction”
5
© 2010 Macmillan Publishers Limited. All rights reserved.
mechanism described in4), and is required for tubulation by toxins, but not by capsids.
Finally, high membrane tension can prevent toxin-induced tubulation, but does not seem
to affect capsid-induced tubulation.
Supplementary References
1.
2.
3.
4.
5.
6.
7.
Foret, L. & Sens, P. Kinetic regulation of coated vesicle secretion. Proc Natl Acad
Sci U S A 105, 14763-8 (2008).
Kramers, H. A. Brownian motion in a field of force and the diffusion model of
chemical reactions. Physica (Utrecht) 7, 284-304 (1940).
Turnbull, W. B., Precious, B. L. & Homans, S. W. Dissecting the Cholera ToxinGanglioside GM1 Interaction by Isothermal Titration Calorimetry. J Am Chem
Soc 126, 1047-54 (2004).
Romer, W. et al. Shiga toxin induces tubular membrane invaginations for its
uptake into cells. Nature 450, 670-5 (2007).
Lipowsky, R. Budding of membranes induced by intramembrane domains. J Phys
II 2 (1992).
Turner, M. S. & Sens, P. Gating-by-tilt of mechanically sensitive membrane
channels. Phys Rev Let 93, 118103 (2004).
Deserno, M. Elastic deformation of a fluid membrane upon colloid binding. Phys
Rev E 69, 031903 (2004).
6
© 2010 Macmillan Publishers Limited. All rights reserved.