H - About us
advertisement

1
Chemistry of C-C π-bonds
Lectures 5-8: Aromatic Chemistry
“I was sitting writing on my textbook, but the work did not progress; my thoughts were
elsewhere. I turned my chair to the fire and dozed. Again the atoms were gamboling
before my eyes. This time the smaller groups kept modestly in the background. My
mental eye, rendered more acute by the repeated visions of the kind, fitted together all
twining and twisting in snake-like motion. But look! What was that? One of the snakes
had seized hold of its own tail, and the form whirled mockingly before my eyes. As if by
a flash of lightning I awoke; and this time also I spent the rest of the night in working out
the consequences of the hypothesis.
Let us learn to dream, gentlemen, then perhaps we shall find the truth... But let us
beware of publishing our dreams till they have been tested by waking understanding”
Handouts will be available at:
http://msmith.chem.ox.ac.uk/teaching.html
Dr Martin Smith
Office: CRL 1st floor 30.087
Telephone: (2) 85103
Email: martin.smith@chem.ox.ac.uk
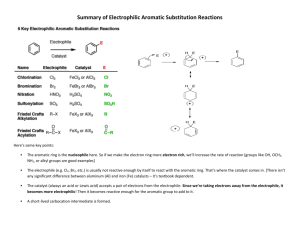
! Aromatic Chemistry.
2
Handout 2
! Benzene and general reactivity profile
! What is aromaticity?
! Resonance’ and molecular orbital explanation
! Typical reactivity – electrophilic aromatic substitution
! Mechanisms of electrophilic substitution – bromination as a worked example
! Nitration, Sulfonation
! Friedel Crafts Alkylation and Acylation
! Monosubstituted Benzenes
! Phenol – acidity
! Benzoic acid – preparation and acidity
! Aniline – preparation and basicity
! Reactions of Monosubstituted Benzenes
! Electrophilic Aromatic substution: ortho-, meta- and para! Substituent effects:
(i) ortho- and para- directing and ACTIVATING
(ii) ortho- and para- directing and DEACTIVATING
(iii) meta- directing and DEACTIVATING
! Reactions of Monosubstituted and Polysubstituted Arenes
Books:
! Substituents affect both rate and orientation
1. Organic Chemistry
! Designing synthetic routes
Clayden, Greeves, Wothers and Warren
! Multiple substitutions: effects of orientation (which group dominates?)
OUP, 2000/2012
! Transforming functional groups in aromatic chemistry
2. Aromatic Chemistry
! Generation and Stability
Malcolm Sainsbury,
! The SN1 reaction for aromatic compounds
Oxford Chemistry Primers,
! The Sandmeyer reaction (introduction of CN, Cl and Br)
OUP, 1992.
! Replacement with H (not as pointless as it appears!)
! Nucleophilic Aromatic Substitution
! An addition-elimination process (compare with conjugate addition-substitution)
! Substituent effects (which groups work and which ones don’t?).
! Real Examples:
Synthesis of Fluoxetine (Prozac)
Synthesis of Vancomycin
3
! Aromatic Chemistry
! Benzene
Flat
Benzene
All C-C bonds
the same length
isolated from
Coal Tar
! Typical Reaction
H
E
+
E+
+
H+
4
! Aromatic Chemistry
! Example: Halogenation
H
+
Br
Br
FeBr3
Br
+
HBr
The FeBr3 is required to increase the reactivity of the electrophile
! Compare with an isolated alkene in a bromination reaction: no catalyst is required
bromonium
cation
LUMO
σ* Br-Br
Br
HOMO
π on alkene
Br
Br
Br
SN 2
inversion
Br
Br
Conclusion: benzene is less reactive than an isolated (cyclic) alkene (why?)
5
! Aromatic Chemistry
! Benzene contains [4n+2] p electrons and is aromatic
also
drawn
as:
Benzene is an aromatic, conjugated system
Drawing the arrows above does not indicate the
actual movement of bonds
a continuous system
through overlap of
6 p-orbitals
The formation of a continuous π system through the overlap of six p-orbitals
is a stabilizing interaction
6
! Aromatic Chemistry
! How much is this ‘aromatic stability’ worth?
Examine hydrogenation – an
exothermic reaction (as the
products are thermodynamically
more stable than the starting
materials)
Predicted
'cyclic
hexatriene'
150 kJ mol-1
more stable
210
kJ mol-1
120
kJ mol-1
360
kJ mol-1
Conclusion: the cyclic
conjugated structure is
more stable
by about 150 kJmol-1
7
! Aromatic Chemistry
! Bromination gives a substitution rather than an addition product; mechanism?
BrH
Br
SLOW
Br
Br
FeBr3
breaks
aromaticity
Br
FAST
regains
aromaticity
! Stabilization of the cationic intermediate by delocalization (π-conjugation)
Br
Br
Br
Remember: These arrows do not imply actual movement of the bonds or electrons
or oscillation between these structures but rather that the charge is delocalized over
the whole system
8
! Aromatic Chemistry
! Evidence for the cationic intermediate [for reference, δC (benzene) = 128.5]
H
Less electron density (cation-like)
179
13C
NMR: o, p- carbons
very deshielded
52
136
Similar to benzene
187
Less electron density (cation-like)
Consistent with charge being distributed around the conjugated system
9
! Aromatic Chemistry
! Reaction Energy Profile
H
Br
TS 1
H
Br
TS 2
Br
H
Br
Activation
Energy
E
H
Intermediate
Starting
materials
Br
Products
reaction progress
10
! Aromatic Chemistry
! Transition states:
H
TS 1
Br
Br
A transition state (or transition structure, TS) is a nonisolable, non observable hypothetical state in between bond
forming and bond breaking
An intermediate can be observed (and often isolated!)
! Transition states and the Hammond postulate
We cannot directly look at the TS, so we make assumptions about what the TS looks like
based on the Hammond Postulate:
“If two states, as for example a transition state and an unstable intermediate, occur
consecutively during a reaction process and have nearly the same energy content,
their interconversion will only involve a small reorganisation of molecular structure.”
This is an elegant way of saying:
“the transition state (probably) looks like an intermediate close to it in energy”
11
! Electrophilic substitution reactions: Nitration (E = NO2)
! Electrophile is NO2+ [generated by the protonation of nitric acid]
O
O
N
O
H
OH
O
O
O
S
OH
O
N
O
N
OH2
H2O is a good
leaving group
O
Electrophile
O
N
O
H
H
SLOW
O
N
breaks
aromaticity
Aromatic
O
O
N
FAST
O
regains
aromaticity
Non-Aromatic
Aromatic
The nitro group is electron withdrawing so the product of the reaction
is less reactive than the starting materials
! Electrophilic substitution reactions: sulfonation (E = SO3H)
! Electrophile is SO3H+
O
HO
O
S
O
H
OH
O
O
O
S
S
HO
OH
H
O
-H2O
OH2
S
O
S
O
O
Electrophile
H
O
O
O
H
SLOW
breaks
aromaticity
O
H
O
S
OH
O
FAST
O
S
OH
regains
aromaticity
The sulfonyl group is electron withdrawing so the product of the reaction
is less reactive than the starting materials
12
13
! Electrophilic substitution reactions
! At high temperatures sulfonation is reversible
OH
Br
OH
SO3H
H2SO4
Br
This means
that we can use
the SO3H
group to direct
other groups
H
200˚C
SO3H
H
! Friedel Crafts Alkylation (R = alkyl)
R
Cl
AlCl3
R
H
Cl AlCl3
R
SLOW
breaks
aromaticity
Product MORE reactive than the starting material (SM): polyalkylation
The intermediate cation rearranges to the more stable cation
The reaction is catalytic in Lewis Acid (AlCl3 in this case)
R
14
! Friedel Crafts alkyation
! Rearrangement and polyalkylation
H
Cl
AlCl3
catalytic
Cl
1,2 hydride
shift
H
AlCl3
2˚ cation
more stable
effectively a
primary cation
SLOW
breaks
aromaticity
H
FAST
regains
aromaticity
plus other products of
polysubstitution and
rearrangement
major product
more reactive
than benzene
15
! Cation stabilization recap:
! Alkyl groups are electron-donating through hyperconjugation
(so the starting materials are more reactive than the products)
C-H σ bond
H
H
H
π-system
p orbitals
'hyperconjugation' or σ-conjugation one of the C-H bonds interacts with the π system
[C-H must be perpendicular to the plane of the ring for the C-H σ−orbital to overlap with the π system]
this means that alkyl groups are electron donating
and means that alkyl substituted benzenes are MORE reactive than unsubstituted benzenes
This is important in Friedel-Crafts alkylation
16
! Cation stabilization recap:
! 1. Hyperconugation
Cation Stability: 3˚ > 2˚ > 1˚
CH3
Planar Structure
minimize electron
repulsion
empty p orbital
H
H
H
H
H
H
H
H
2 x C-H σ bond
coplanar with
vacant p-orbital
! 2. p-conjugation –
H
3 x C-H σ bond
coplanar with
vacant p-orbital
[‘resonance’ is the shorthand description of delocalization]
also
drawn
as:
remember: the bonds
are not 'moving'
the cation is delocalized
over these three atoms
Essentially: filled (bonding) orbitals interacting with empty (non-bonding) ones
* This has a stabilizing effect *
17
! Friedel Crafts Acylation
! Mechanism:
Acylium
Cation
Cl
Al
O
Cl
O
O
Cl
Cl
R
Cl
R
AlCl3
R
O
R
O
O
R
This is equally as
effective as the
acid chloride
SLOW
anhydride
Product LESS reactive than the SM
O
R
O
No rearrangement - cation is stabilized
Clean monosubstitution
Requires superstoichoimetric AlCl 3
(complexation to oxygen in the product)
-HCl
FAST
H
R
18
! Friedel Crafts Acylation
! How to introduce alkyl groups on an aromatic ring (if FC alkylation does not work):
! Use Friedel-Crafts acylation and reduce the ketone functional group
Target:
Problem with FC alkylation:
AlCl3
Cl
plus other products
of rearrangement
and polyalkylation
O
O
AlCl3
NH2NH2
Cl
KOH, heat
clean
monoacylation
Wolff-Kishner
reaction
19
! Gatterman-Koch reaction
! Gatterman-Koch formylation (a special Friedel-Crafts type reaction):
Electrophile
AlCl3
O
C O
H
CuCl
HCl
O
H
-HCl
FAST
H C O
O
H
H
H
C
SLOW
O
Cl
This material is
unstable and so
cannot be used in a
F-C acylation
reaction to introduce
an aldehyde
! Monosubstituted benzenes so far:
Y
Y = Halogen, NO2, SO3H, Alkyl, acyl (aldehyde, ketone)
20
! Substituted benzenes
! Phenol (Y = OH)
Acidity: compare with non aromatic alcohol:
OH
OH
Phenol
an extremely
stable enol
OH
pKa = 10
pKa = 16
! Reminder (and brief aside): pKa is a measure of the position of the equilibrium between
an acid and its conjugate base
Most important factor in acid strength is the stability of the conjugate base A
So for a strong acid, the conjugate base A is stable,
the equilibrium lies over to the RHS and the pKa is low.
The stronger the acid, the lower the pKa
O
F3C
pKa
OH
O
OH
OH
1
NO2
O2 N
5
O
7
O
EtO
OH
10
10
O
CH3OH
H2O
15.3
15.74
H
OEt
12
20
25
-
21
! Substituted benzenes
! Phenol (Y = OH)
OH
O
OH
O
-H+
-H+
+H+
+H+
1. Delocalization of the
charge via the aromatic ring
2. An inductive electron
withdrawing effect
this anion is localized on the
electronegative oxygen
! 1. ‘Delocalization’
O
O
The lone pair in a p-orbital on oxgen, which is
perpendicular to the plane of the ring, can interact
with the π−system.
22
! Substituted benzenes
! 1. ‘Delocalization’ (continued)
We can draw the interaction of the electrons on the oxygen
with the aromatic ring in a shorthand way as:
O
O
O
O
[Remember – the charge is not actually moving around the ring]
This gives us an indication that the charge is delocalized around the ring
! 2. Inductive
The aromatic substituent is sp2 hybridized (vs sp3 hybridized in cyclohexanol) and hence has
more ‘s’ character. The higher proportion of ‘s’ character means that the electrons see more
effective nuclear charge [cf radial probability functions].
Hence the aromatic sp2 carbon (in phenol) is more electronegative
than the sp3 carbon (in cyclohexanol) and therefore more electron-withdrawing.
23
! Substituted benzenes: Y = CO2H (benzoic acids)
! Preparation: (1) Oxidation of toluene
CO2H
KMnO4
CO2H
! Preparation: (2) Grignard reaction with CO2
A Grignard reagent
Br
Mg
dry Et 2O
Et2O is an
aprotic solvent
MgBr
O
C
O
then H +
CO 2H
! Substituted benzenes: Y = CO2H (benzoic acids), Y = NH2 (anilines)
! Benzoic acid pKa = 4.2 (compare with acetic acid CH3CO2H, pKa 4.8)
O
CO 2H
O
OH
CO 2
O
CH 3 is inductively electron
donating (by hyperconjugation)
The aryl ring is electron
withdrawing (sp2 carbon vs sp3 )
! Y = NH2 (anilines); Prepared by reduction of nitro compounds
NH2
NO2
Sn/HCl
or
Pd/H2 (g)
NH2
24
25
! Substituted benzenes: Y = NH2 (anilines)
! Basicity: Aniline is less basic than cyclohexylamine
NH 2
NH 3
-H+
NH 2
NH 3
-H+
+H+
+H+
pKaH = 10.7
pKaH = 4.6
! Two effects:
Delocalization
Inductive effect
NH2
N
NH2
NH2
H
H
The N lone pair is not perfectly
perpendicular to the aromatic ring
sp3
sp2
sp2 carbon is more electronegative
26
! How do substituents affect reactivity?
! More than one ‘position’ is available:
Y
Y
Y
Y
E
E+
E
"ortho"
"meta"
E
"para"
The nature of Y affects both orientation (o- vs m- vs p-) and rate of reaction
! 1. Ortho- and para- directing, and ACTIVATING groups
An ACTIVATING group means that the reaction goes faster than benzene
Typically: Y = alkyl, NH2, NR2 (R = alkyl), NHCOR, OH, OR, OCOR)
OMe
OMe
Br
OMe
OMe
The OMe group is
ACTIVATING (the reaction
goes 109 times faster than
it does with benzene) –
why?
Br
Br
Br
ortho
(minor)
meta
(not observed)
Br
para
(major)
27
! How do substituents affect reactivity?
! For non-reversible reactions (those under kinetic control), the rate of reaction is dictated by
the activation energy
TS 1
TS 2
To predict reactivity we need
to look at the nature of the TS We can do this using the
Hammond Postulate:
E
Activation
energy
Intermediate
Y
E
Y
Starting
materials
Y
Products
E
o, m, p
reaction progress
The transition state looks like an intermediate close to it in energy
[so anything that stabilizes the intermediate also stabilizes the transition state]
28
! How do substituents affect reactivity?
! Therefore consider intermediates in this reaction:
! for the ortho- case
OMe
Br
Br
OMe
OMe
Br
Ortho: the intermediate carbocation
is stabilized by the OMe group
This leads to a LOWER energy TS
Therefore: rate of reaction in this
position is HIGHER
OMe
Br
-H+
OMe
Br
Br
29
! How do substituents affect reactivity?
! Therefore consider intermediates in this reaction:
! for the meta-case
HOMO
π C=C OMe
OMe
Br
OMe
OMe
Br
LUMO
Br-Br
Br
meta: the intermediate carbocation is
NOT stabilized by the OMe group
The TS is relatively higher in energy
Therefore: rate of reaction in this
position is LOWER
Br
-H+
OMe
Br
Br
30
! How do substituents affect reactivity?
! Therefore consider intermediates in this reaction:
! for the para- case
OMe
Br
OMe
OMe
OMe
Br
Br
Br
Br
para: the intermediate carbocation is
stabilized by delocalization involving
the OMe group
This leads to a LOWER energy TS
-H+
OMe
Therefore: rate of reaction in this
position is HIGHER
There is less steric hindrance in the
para position than in the ortho position
Br
31
! How do substituents affect reactivity?
! If we take all this and relate it to TS energy:
benzene
higher in
energy
ΔEa for
Benzene
TS 1
mhigher in
energy
TS 2
Therefore: more stable
intermediate formed faster, and
ortho- and para- products
predominate
E
ΔEa for
p-OMe
benzene
Intermediate
o-, psimilar in
energy
Starting
materials
Products
reaction progress
32
! How do substituents affect reactivity?
! 2. Ortho- and para- directing, and DEACTIVATING groups
A DEACTIVATING group means these reactions go slower than benzene
Typically: Y = F, Cl, Br, I (these groups ‘withdraw’ and ‘donate’ electrons)
Cl
Cl
Br
Cl
Cl
Br
Br
FeBr3
Br
ortho
(minor)
meta
(not observed)
Br
para
(major)
Halogens withdraw electrons via an inductive effect (this affects the rate)
and donate through the unsaturated system (this affects orientation
and is sometimes called a ‘mesomeric’ effect).
33
! How do substituents affect reactivity?
! Consider ortho-
FeBr 3
Cl
Br
Br
Cl
Cl
Cl
Br
Br
Br
Br 2
FeBr 3
A catalyst is needed (chlorobenzene
is less reactive than benzene due to
inductive effect of the Cl)
The Cl lone pair can conjugate with
the ring in the ortho and para
cases, but not in the meta case
Therefore: rate of reaction in o, p
positions is HIGHER than in m
-H+
Cl
Br
ortho and para
bromination
observed
34
! How do substituents affect reactivity?
! Reaction coordinate:
TS 1
mhigher in
energy
TS 2
o-, psimilar in
energy
E
benzene
lower in energy
Intermediate
Starting
materials
Products
reaction progress
Conclusions: Benzene is more reactive than chlorobenzene (Cl is electronegative)
Orientation a consequence of delocalization of lone pairs though π-system
35
! How do substituents affect reactivity?
! 2. Meta- directing, and DEACTIVATING groups
A DEACTIVATING group means these reactions go slower than benzene
Typically: NO2, SO3H, almost all carbonyl compounds (CO2H, CO2R, CHO, COR)
CO2Me
CO2Me
CO2Me
CO2Me
NO 2
NO 2
H 2SO4
HNO 3
NO 2
NO 2
MAJOR
! Consider ortho- and metaCO2Me
CO2Me
NO 2
NO 2
both very minor
CO2Me
CO2Me
NO 2
NO 2
Cation destabilized as next to the
electron withdrawing group
The cation is never next to the
electron withdrawing group
36
! How do substituents affect reactivity?
! 2. Meta- directing, and DEACTIVATING groups
o-, phigher in
energy
TS 1
TS 2
mlower in
energy
E
benzene
lower in
energy
Intermediate
Starting
materials
Products
reaction progress
Conclusions: Benzene reacts faster than these substrates as it is more electron-rich
o, p intermediate destabilized by EWG – meta favoured
37
! Real world examples:
! Designing a synthetic route: substituent effects are important for selectivity and efficiency
CO2H
NO2
CO2H
NO2
or
or
NO2
TARGET MATERIAL
All cheap and readily available
Which is the best starting material?
The idea: prepare target material (TM) in a clean, selective and efficient fashion
! Consider monosubstituted starting materials:
CO2H
CO2H group deactivating
m - directing
NO2
Me group activating
o, p - directing
NO2 group deactivating
m - directing
38
! Real world examples:
! Choice of starting material:
mono-nitration
(product is less electron-rich than
SM)
HNO 3
H 2SO4
para-position minor product though
statistically more dominant (steric
reasons)
NO 2
[note: there are
effectively twice as many
ortho- positions – a
statistical effect]
37% para59% ortho-
! The order of reactions in a synthetic sequence can be important
CO2H
CH3 group
o- & p- directing
ROUTE 1
OXIDIZE
NITRATE
NO2
NO2
NO2 group
m-directing
ROUTE 2
NO2
NO2
Which route
is best?
NO2
NITRATE
CO2H
OXIDIZE
NO2
39
! Real world examples:
! ROUTE 1 : Oxidation then nitration
CO2H group
m-directing
CO2H
KMnO4
NO2
HNO3
H2SO4
NO2
CO2H
CO2H
NO2
NO2
NO2
NO2 group
m-directing
UNDESIRED
Conclusion: This produces
a mixture of the materials
we want (and something
else that we don’t want)
NO2
TARGET
MATERIAL
! ROUTE 2 : Nitration then oxidation
CH3 group
o, p- directing
CO2H
NO2
HNO3
NO2
KMnO4
H2SO4
NO2
NO2 group
m- directing
NO2
NO2
the only product
we observe
Conclusion: this is the
optimum route – the
ORDER of steps is
important
40
! What about arenes with two or more groups?
! Which effects dominate?
Examine the effects of individual substituents: electronically first, then consider steric effects
Me
Me
Me
CO 2H
Br
Me
Cl
NO 2
CH3 groups
o-, p- directing
CH3: o-, pCl: o-, p-
NO 2: meta
CH3: ortho
CO 2H: meta
Br: ortho, para
41
! What about arenes with two or more groups?
! (i) substituents direct to conflicting positions
Broadly categorize substituents into 3 classes of decreasing effect
(1) STRONGLY activating and ortho- & para- directing (OH, OR, NH2 and NR2 groups)
(2) Alkyl groups and halogens
(3) All other meta- directors
If substituents are in ‘different’ classes, then the ‘higher numbered class’ dominates.
O
OMe
HN
O
NMe 2
H
F
F 3C
Me
OMe: o, p
F: o, p
OMe dominates F
para
NHAc: o, p
Me: o, p
NHAc dominates Me
ortho
MeO
OMe
NMe 2: o, p
CF3: m
NMe 2 dominates CF3
para
OMe: o, p
CHO: m
OMe dominates
para
All ortho- & para- directors generally dominate over meta-
42
! What about arenes with two or more groups?
! (i) If substituents are in the same class then it is to be expected that mixtures will be
produced (and hence that this is maybe not a good route to the proposed compound!)
CO 2Me
Cl
Me: o, p
Cl: o, p
MIXTURE
CO 2Me
CO 2Me: meta
MIXTURE
Important to remember that we can extend and modify these effects through
functional group interconversion reactions:
NO 2
NH 2
H 2, Pd
(or Sn/HCl)
Deactivating
meta-
NaNO 2
N
HCl (aq.), 0˚C
V. activated
o-, p-
SN1-like
(radicals)
N
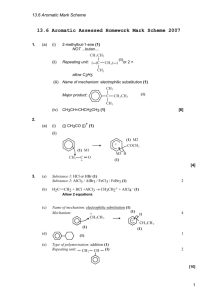
! Exam question (2007 long vacation)
43
44
! Exam question (2007 long vacation)
Part I
TS 1
TS 2
Activation
energy
Intermediate
E
“The transition state looks like
an intermediate close to it in
energy”
Y
Starting
materials
Y
Products
E
o, m, p
reaction progress
OH
1.Very activated
by conjugation
2. direct o,p
(conjugation)
Cl
1. deactivated
(inductive)
2. direct o,p
(conjugation)
CH3
1. activating
(inductive)
2. direct o, p
(hyperconjugation)
NO2
1. deactivated
(conjugation)
2. m- directing
(conjugation)
CF3
1. deactivated
(inductive)
2. m- directing
(inductive)
! Exam question (2007 long vacation)
45
46
! Exam question (2007 long vacation)
Part II (a)
H+
H
FAST
SLOW
F-
secondary
most stable
(hyperconjugation)
concentration of benzene must be
much higher than concentration of
alkylbenzene to outcompete donating
effect of alkyl group
(product is more reactive than the SM)
FeBr 3
Br
Br
H
FeBr 3
1,2 hydride shift
generate more stable
2˚ cation
! Exam question (2007 long vacation)
47
48
! Exam question (2007 long vacation)
part II (b)
NMe 2
NMe 2 a powerful donor - more
reactive than protonated form.
Therefore reacts faster even
though a minor component of
equilibrium
H
HNO 3
H 2SO4
Anilines are
weakly basic
OMe
NMe 2
More abundant but less
reactive anilinium.
Now not a good
donor/activator
Mostly inductively
withdrawing (deactivated)
meta- predominates
protonated in
strong acid
OMe
OMe group not basic
Not protonated in strong acid
Therefore: still activating
ortho- & para directing through conjugation
of the OMe group.
HNO 3
H 2SO4
direct o,p
! Exam question (2007 long vacation)
49
50
! Exam question (2007 long vacation)
Part III (a)
N
NH 2
NH 2
D + (DCl, D 2O)
D
N
D
D
D
0-5˚C, H 2O
heat
NH 2 activating
o-, p- directing
NaNO 2, H 2SO4
D
D
(what about protonation on N?)
the N-acetyl derivative is a less reactive
and non-basic alternative
CuBr
heat
O
O
H
D
D
D
H
MgBr
N
D
Br
D
D
Mg, Et 2O
D
D
D
51
! Exam question (2007 long vacation)
Part III (b)
Sn/HCl
HNO3
H2SO4
NH2
NO2
O
Cl
LiAlH4
HN
HN
O
52
! Diazonium salts
! Diazonium salt generation:
NaNO2 + HCl
HNO2 + NaCl
N
O + H2O
H
H
NH 2
HCl
N
N
O
N
N
OH
H
N
O
If HCl is used, we often get some chlorobenzene formed
So the other option is to use H 2SO4 (less nucleophillic counterion)
Diazo salts are stable in solution below about 0˚C
N
N
Cl
53
! Diazonium salts
! Effectively the SN1 reaction for aromatic compounds (cation NOT stable!)
N
OH 2
N
warm
OH
H + /H2O
N 2: World's best
leaving group
sp2 cation not stabilized
by delocalization
loss of N 2
entropically
favoured
Compare with SNAr reaction in the next lecture.
! We can use this principle to introduce other functional groups (such as iodine)
N
N
KI
H2O
I
54
! Diazonium salts
! Iodide introduction probably a radical mechanism
Remember: single
headed ('fish-hook')
arrows indicate the
movement of a
single electron
N
N
N
N
I
I
I
I
I
I
I
I
I
I2
one electron
two electrons
N
N
N
N
I
I
chain process
continues
55
! Diazonium salts
! Fluorine introduction (the Balz-Schiemann reaction)
N
NH2
N
BF4-
F
Mechanism:
probably via
fluoride trapping
onto an aryl cation
-N2
NaNO2
HF, BF3
[it is generally demanding to introduce fluorine onto an aromatic ring]
! (iii) The Sandmeyer reaction (to introduce Cl, Br CN)
N
NH2
N
NaNO2
Cl-
X
Cu
X
aq. HCl
X = Cl, Br, CN
56
! Diazonium salts
! Sandmeyer reaction mechanism
N
N
X
N
Cu
X
N
+ N 2 (g)
Copper is
oxidized
X
Cu(I)
Cu
X
Cu(II)
Copper is
reduced
X
Cu
Recycle - catalytic in Cu
X
Cu(I)
57
! Diazonium salts
! Replacement by ‘H’
not a good way to make benzene, but useful for directing other groups, though an
outdated way to achieve this – better methods available
N
NH 2
Br
N
Br
NaNO 2
aq. HCl
HO 2C
Br
NH 2 used to direct
orientation of
bromination
Br
O
Cl
H
Br
HO 2C
-N2 (g)
Br
Br
HO 2C
Br
O
P
H
H
H
Br
Br
HO 2C
Br
Br
58
! Nucleophilic Aromatic Substitution
! SNAr (substitution nucleophilic aromatic)
O
O
N
OH –
N
O
O
HO
F
Overall: substitution on an aromatic ring – what is the mechanism?
! Mechanistic considerations: I. Cannot be SN2
C-F σ*
F
NO2
F
NO2
SN2 requires access to s* orbital of C-F bond (which is buried inside the aromatic ring)
Therefore nucleophile (HO-) cannot get ‘anti’ to the requisite C-F bond
59
! Nucleophilic Aromatic Substitution
! Mechanistic considerations: II. Unlikely to be SN1 (compare with diazo compounds!)
O
N
O
SN1
O
N
X
O
F
Carbocation would be in an sp2 orbital (and would not be stabilized by the aromatic ring)
Compare with other cations we have seen:
H
H
H
Cation not stabilized
by delocalization
tertiary carbocation
stabilized by
hyperconjugation
Allylic cation
delocalized through
pi-conjugation
60
! Nucleophilic Aromatic Substitution
! Mechanism: an addition-elimination reaction
O
N
O
HO-
N
O
O
HO
F
O
N
HO
O
HO
F
HO
N
O
O
N
O
O
HO
F
F
These arrows indicate that the intermediate anion is delocalized
Reminder: there is no actual movement of electrons
or oscillation between these structures
Remember: SN2 reactions at sp2 centres (including aromatic rings) are very rare
For nucleophilic aromatic substitution an Electron-Withdrawing Group (EWG) is
required ortho or para to the leaving group on the aromatic ring.
61
Nucleophilic Aromatic Substitution
Our shorthand structures indicate that the charge is delocalized around the ring but
is centred on the ‘ortho’ and ‘para’ positions – is there evidence for this?
For Anion (often called a ‘Meisenheimer’ complex)
H
Consistent with
an anion at
these centres
NH 2
78
13C
132
NMR: o, p- carbons
very shielded
76
3
In both cases the ionic charge is localized almost exclusively to the ortho and para positions
Implication: groups to stabilize the anionic intermediates in SNAr reactions
MUST be on these carbons
62
! Addition-Elimination reactions
! Compare with other addition-eliminations: (i) conjugate substitution of an amine
O
[written as ArNH2]
O
O
Cl
EtO
OEt
heat
+
EtO
OEt
NH 2
EtO
O
NH
Amine nucleophiles
prefer 1,4 addition
Cl
ArNH2
Overall additionelimination
mechanism
O
EtO
EtO
-OEt
O
O
Ac
H
N
O
EtO
EtO
H
Cl
OEt
N
H
Cl
ArNH2
63
! Addition-Elimination reactions
! (ii) conjugate substitution of an alcohol
O
MeOH
Cl
H
O
MeOH
Ph
MeO
Ph
Overall additionelimination
mechanism
-HCl
Me
Cl
O
H
proton transfer
O
Ph
(this could be interor intramolecular)
Me
Cl
O
H
O
Ph
64
! Addition-Elimination reactions
! Example of SNAr
RSH
SR
Cl
Cl
N
RSH
O
SR
-Cl
Cl
Base
Cl
N
O
O
Cl
N
O
Only the ortho
chlorine is lost –
the meta one is
retained
O
O
! Further confirmation: isolation of an intermediate (!)
O
MeN
NO 2
O
N
O
H
O
MeN
MeN
O
NO 2
HO -
O
N
O
NO 2
O
N
O
A stable molecule
(structure confirmed by
X-ray crystallography)
(1) Nucleophile is
intramolecular, and so
cannot ‘escape’
(2) Alkoxides are not
great leaving groups
65
! Addition-Elimination reactions
! Which (EWG) groups can accelerate nucleophilic aromatic substitution?
So far we have seen the NO2 group – but other groups can also function in this regard.
O
FAST
SLOW
F
F
O
O
Nu
Nu
Nu
So any group that can stabilize the negative charge in the intermediate can
facilitate the reaction – so carbonyl groups are effective too
66
! Addition-Elimination reactions
! Nucleophilic aromatic substitution is generally fastest when the leaving group is fluoride.
O
SLOW
RDS
F
[breaks
aromaticity]
F
O
O
FAST
[restores
aromaticity]
Nu
Nu
Nu
Rate F > Cl > Br > I [compare with SN2: Rate I > Br > Cl > F]
The rate-determining step is attack of the nucleophile on the aromatic ring
as this breaks the aromaticity.
The second step, involving loss of the leaving group and restoration of aromaticity, is fast.
Implication: we need an electronegative leaving group
Electronegative F polarizes s-bond and inductively withdraws electron density from the
high energy anionic intermediate
Note that the reaction is bimolecular in the RDS and therefore: Rate = k [substrate] [Nu-]
67
! Application in the real world
! Synthesis of Fluoxetine ‘Prozac’ - serotonin uptake inhibitor for treatment of depression
F
NHMe
HO
+
Ph
F3C
Me2NAc
NHMe
O
NaH
Ph
F3C
NaH
FAST
F
F
F3C
NHMe
O
Ph
CF3 is a powerful electron
withdrawing group
SLOW
NHMe
O
F3C
Ph
Anion stabilized by EWG
68
! Application in the real world
! Example of SNAr in the synthesis of a complex molecule: Synthesis of Vancomycin
O
sugar
O
HO
H
N
O
O
O
Cl
O
H
N
H H
O
NH
Cl
N
H
O
HO
OH
OH
H
N
O
NH2
HO2C
OH
O
N
H
NHMe
69
! Vancomycin and antibiotic resistance
O
sugar
Cl
O
HO
O
H
N
O
H
O
Cl
O
H
N
H
O
NH
N
H
O
OH
H
N
O
Glycopeptide antibiotic
O
Isolated from
Nocarida orientalis in soil
samples in mid-50s
NHMe
N
H
NH2
HO2C
OH
OH
HO
! Timeline
Sulfonamides
Penicillin
discovered
Erythromycin
(macrolides)
β-lactams
(penicillin,
cephalosporins)
1928
Vancomycin
1952
1932
1940's
Quinolones
Linezolid
(a new class!)
1962
1956
2000
A penicillin derivative – methicillin – was used in preference to Vancomycin (toxicity), but now
Vancomycin is ‘last line of defence’ to treat methicillin-resistant staphlococcus aureus [MRSA].
70
! Application in the real world
! Example of SNAr in the synthesis of a complex molecule: Synthesis of Vancomycin
Fluorine orthoto nitro group
Bond made
by SN Ar
Phenol
(pKa 10)
Cl
NO 2
OR
F
HO
OR
OH
O 2N
H 2NOC
O
H
N
H
O
NH
HO
Na 2CO 3
H
N
NHR
H O
OH
MeO
Na 2CO3
NH
OMe
OMe
F
SLOW
N
O
NHR
H O
OH
O N O
F
O
OR
Cl
O
OR
FAST
OR
O
Cl
O
H
N
H
N
H
O
H 2NOC
OMe
OMe
MeO
OR
O
Cl
OR
OR
71
! Recap of lecture 4
! 1. Nucleophilic additions to alkenes can be under Kinetic or Thermodynamic control
KCN (cat.)
HCN,80˚C
O
KCN (cat.)
HCN, 0˚C
O
HO CN
NC
Thermodynamic
Kinetic
If the reaction is reversible,
the conjugate addition
product predominates
(strong C=O bond is
retained)
! 2. Reactions can be dominated by charge or by orbital control
HOMO
Nu:
Nu:
O
O
LUMO
High energy HOMO on Nu:
close in energy to LUMO
on C=O
72
! Recap of lecture 4
! 3. Hard and Soft Nucleophiles and Electrophiles
Kinetic and
Thermodynamic
O
Bu
Kinetic
BuMgBr
O
BuMgBr
Bu
OH
1% CuCl
Soft Nucleophile
Orbital controlled
Hard Nucleophile
Charge controlled
Hard nucleophiles are
generally small and
charged [ie; HO-, Cl-, H-]
Soft nucleophiles are
generally uncharged, less
electrophilic [ie: I-, RS-,
RSH, RPH]
! 2. Reactions can be dominated by charge or by orbital control
O
O
O
Cl
All 1,2 (direct)
addition
R
NO2
NR2
All 1,4 (conjugate)
addition
More reactive nucleophiles
and electrophiles prefer
direct addition.
Less reactive nucleophiles
and electrophiles prefer
conjugate addition.
73
! Recap of lecture 5
! 1. Benzene possesses aromatic stability
also
drawn
as:
Benzene is an aromatic, conjugated system
The 'aromatic stability' is worth about 150kJ mol-1
a continuous system
through overlap of
6 p-orbitals
! 2. Benzene reacts with reactive electrophiles to give substitution products
O
N
O
H
O
N
O
O
N
O
Proceeds via cationic intermediate
{as seen in Friedel Crafts, halogenation, nitration, sulfonation}
74
! Recap of lecture 5
! 3. Friedel-Crafts alkylation is not usually a useful reaction (rearrangement and
polyalkylation result)…
AlCl3
Cl
minor
major
MORE
reactive
than SM
plus other
further
substituted
products
! 4…….so we use Friedel-Crafts acylation instead
O
AlCl3
O
Cl
NH2NH2
KOH, heat
less reactive
than SM
WolffKishner
reaction
75
! Recap of lecture 6
! 1. The Hammond Postulate helps us make assumptions about what the transition state looks like
TS 1
TS 2
The intermediate is closer in energy to
the TS than to the SM and therefore
more resembles the TS than the SM
H
Br
E
Therefore:
anything that stabilizes the intermediate
will stabilize the TS
H
Intermediate
Br
Starting
materials
Products
Reaction progress
! 2. Substituents on a ring affect the rate of reaction with electrophiles….
O
N
O
very deactivated
by conjugation
[CN, CO2R, SO3R]
Cl
deactivated
inductive withdrawal
[Halogens]
CH3
activating
inductive effect
[alkyl groups]
OR
Very activated
by conjugation
[O and N]
76
! Recap of lecture 6
! 3. ....and also affect orientation
O
N
O
Cl
meta only
[CN, CO2R, SO3R]
o, p direction
donation by conjugation
[Halogens]
CH3
OR
some o, p direction
[alkyl groups]
o, p directing
by conjugation
[O and N]
! 4. We can rationalize this through considering delocalization in the intermediate(s)
Disfavoured - Minor product
CO2Me
CO2Me
NO 2
NO 2
Favoured - Major product
CO2Me
CO2Me
NO 2
NO 2
cation next to EWG - destabilized
cation is never next to EWG -
77
! Recap of lecture 7
! 1. When designing a synthetic route we need to consider the order of steps
CO2H
CO2H
CO2H
NO2
NO2
NO2
1. Oxidize
1. Nitrate
2. Nitrate
2. Oxidize
NO2
NO2
NO2
NO2
Mixture of products
Single Product
! 2. When considering different or conflicting groups (orientation and activation), we can
generally say that ortho- and para- directors win over meta and also that OR and NR2 groups
are generally dominant over everything else.
OMe activates and
directs o- & pF deactivates and
directs o- and p-
O
OMe
H
CHO deactivates
and directs m-
F
MeO
OMe
both OMe groups
activate and direct
o- and p-
78
! Recap of lecture 7
! 3. Diazo compounds are easily made and transformed – an aromatic SN1 reaction
NH2
NaNO2
N
N
OH
H2O, warm
HCl (aq.), 0˚C
sp2 cation not stabilized
by delocalization
Stable at 0-5˚C
! 4. We can also do radical reactions with diazonium salts (the Sandmeyer Reaction).
N
N Cl
NH2
NaNO2
X
Cu
X
aq. HCl
X = Cl, Br, CN